

Abstract
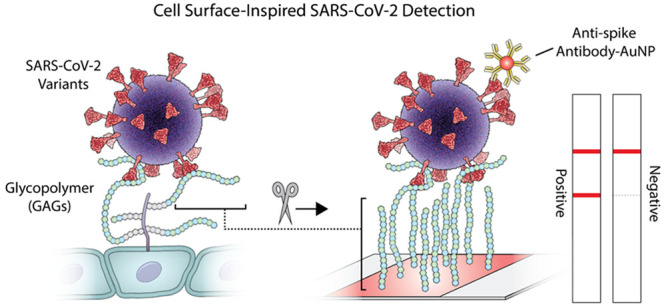
Inspired by the role of cell-surface glycoproteins as coreceptors for pathogens, we report the development of GlycoGrip: a glycopolymer-based lateral flow assay for detecting SARS-CoV-2 and its variants. GlycoGrip utilizes glycopolymers for primary capture and antispike antibodies labeled with gold nanoparticles for signal-generating detection. A lock-step integration between experiment and computation has enabled efficient optimization of GlycoGrip test strips which can selectively, sensitively, and rapidly detect SARS-CoV-2 and its variants in biofluids. Employing the power of the glycocalyx in a diagnostic assay has distinct advantages over conventional immunoassays as glycopolymers can bind to antigens in a multivalent capacity and are highly adaptable for mutated strains. As new variants of SARS-CoV-2 are identified, GlycoGrip will serve as a highly reconfigurable biosensor for their detection. Additionally, via extensive ensemble-based docking simulations which incorporate protein and glycan motion, we have elucidated important clues as to how heparan sulfate and other glycocalyx components may bind the spike glycoprotein during SARS-CoV-2 host-cell infection. GlycoGrip is a promising and generalizable alternative to costly, labor-intensive RT-PCR, and we envision it will be broadly useful, including for rural or low-income populations that are historically undertested and under-reported in infection statistics.
Short abstract
The cell surface is covered with a “sugary” glycocalyx matrix. We use this knowledge to develop a sensor that can detect SARS-CoV-2 and its rapidly emerging variants.
1. Introduction
The glycocalyx, a dense “sugary” matrix that coats epithelial tissue cells, is responsible for cell–cell adhesion, extracellular communication, growth factor monitoring, defense against exogenous pathogens, and much more.1,2 The major components of the glycocalyx include enzymatic glycoproteins, glycolipids, and proteoglycans. Proteoglycans are heavily glycosylated membrane proteins whose glycan components are mainly glycosaminoglycans (GAGs), or long, linear polysaccharides with repeating units of a uronic acid sugar and an amino sugar.3
Heparan sulfate (HS) proteoglycans are the most abundant component of the epithelial glycocalyx, making up 50–90% of the total sugar composition, followed by chondroitin sulfate (CS).4 HS is made up of repeating dimeric units of a uronic acid (either glucuronic or iduronic acid) and N-acetylglucosamine. CS consists of repeating dimeric units of glucuronic acid and N-acetylgalactosamine. Stereochemical composition (i.e., proportions of glucuronic acid versus iduronic acid) and sulfation rates in HS and CS vary greatly depending on tissue types, as well as other physiological conditions such as healthy or diseased tissue status.5−9 Viral pathogens often hijack glycocalyx receptor trafficking and signal transduction mechanisms to facilitate entry into host cells (Figure 1).10−12 An important example of this is SARS-CoV-2 (severe acute respiratory coronavirus 2) viral replication cycle and its resultant disease known as COVID-19 (coronavirus disease 2019).
Figure 1.

Graphical illustration of (A) virus interaction with GAGs on the cell surface and (B) on the GlycoGrip lateral flow (LF) biosensor for detecting SARS-CoV-2. The sample is deposited on the sample pad and migrates toward the conjugate. The conjugated antibodies bind the virus and migrate to the test line, where the bound target analyte is captured by the glycopolymers. Possible results and interpretation of the test are shown below.
SARS-CoV-2 is a member of the betacoronavirus genus within the Coronaviridae family of viruses; it is a lipid-enveloped, positive-sense, single-stranded RNA virus studded with approximately 30 structural S or “spike” glycoproteins (Figure 2). The spike is a homotrimeric protein composed of many interworking domains. Two of these domains are particularly important to this work: the receptor binding domain (RBD, residues 330–530) and the N-terminal domain (NTD, residues 13–296) (Figure 2A). Furthermore, the spike surface is heavily shielded with 66 N-linked glycans and a varying number of O-glycans.13,14 SARS-CoV-2 spike’s main function is to incite the membrane fusion process by binding to angiotensin converting enzyme 2 (ACE2), situated on the surface of ciliated lung epithelial cells.15−19 To bind ACE2, the spike must be in an “open” conformation, with at least one RBD in the “up” state (Figure 2A, Figure S1).20,21 In particular, the RBD moves into the “up” state to reveal the receptor binding motif (the RBM), the spike region that makes direct contact with ACE2. Recent works have described the role of the spike’s glycans in facilitating and stabilizing the conformational transition from down RBDs to up RBDs15,19,22 thereby facilitating host-cell invasion. Intriguingly, glycocalyx glycopolymers may also help facilitate SARS-CoV-2 invasion: Skidmore and co-workers identified a GAG binding site on the spike RBD,23 and Esko and co-workers have illustrated that spike binding to HS in the glycocalyx facilitates interaction with ACE2, and incubation with heparin (HEP) induces an increase in spike populations with up versus down RBDs.15,19,22,24−32 Furthermore, other works by Linhardt and co-workers,33 Fadda and co-workers,34 Wade and co-workers,35 and Gandhi and co-workers36 have posited HEP binding sites on the spike surface (Figure 2B).
Figure 2.

(A) Molecular representation of SARS-CoV-2 spike in the “1-up” conformational state. The spike protein is represented with red, salmon, and light pink surfaces. Spike glycan atoms are shown with light blue licorice representation. (top) From a top-down view the “closed” to “open”/“1-up” RBD conformational change required for host-cell invasion. The spike’s S1 domain is highlighted in red surface representation, while the spike’s S2 domain is in salmon surface representation. The spike’s RBD and NTD are outlined for reference. (B) Molecular representation of the SARS-CoV-2 spike in closed conformation depicting literature proposed HEP binding sites. Green surface: the “RBD patch”, a site proposed by Skidmore and co-workers23 and supported by Esko and co-workers to have high affinity for heparin.27 Pink surface: the “RBD cleft” a site proposed by Fadda and co-workers to have high affinity for polysaccharides.34 Red surface: the furin cleavage site.37−39 Light blue surface: the fusion peptide site proposed by Linhardt et al.33 Yellow surface: the connecting ridge proposed by Wade et al.35 Purple surface: the NTD site identified by Schuurs et al.36
GAGs overall have been underappreciated as potential bioreceptors in biosensors due to their complexity, heterogeneity in sulfation patterns, and variable binding specificity (i.e., one GAG can bind many analytes) compared to highly targeted antibodies.11,12,40,41 Biologically, HS and CS serve as cellular staging grounds: they bind many analytes while the targeted coreceptors find optimal orientation on the cell surface.42 We argue that GAG’s abilities to bind multiple analytes—inspired by glycobiology—can be leveraged to design highly generalizable sensors for the sensitive detection of viruses and viral antigens, and we applied this approach for sensing SARS-CoV-2 and its variants in a lateral flow strip-based assay (LFSA) (Figure 1B).
LFSA is an attractive platform for detecting viruses, especially in limited resource settings, due to its simplicity, low cost, and rapid signal generation. Typically, “sandwich-type” detection using lateral flow (LF) strips utilize two bioreceptors that bind to the target molecule simultaneously.43−46 One of the bioreceptors is usually immobilized on the nitrocellulose membrane surface to capture the target analyte, while the other bioreceptor is typically labeled with reporter molecules (e.g., gold nanoparticles, fluorescent dyes, enzymes, etc.) to signal the formation of a sandwich complex in the presence of the analyte. Using traditional bioreceptors, such as antibodies and aptamers as immobilizing agents in LFSA design, requires these receptors to be screened and optimized, as well as necessitating that LF strips be reconfigured, for every desired viral analyte and potential mutant strains. Furthermore, in the case of LFSA sandwich-type detection, pairs of antibodies or aptamers must be screened for both capturing and reporting, which delays sensor design and development.47,48 In contrast, GAGs could serve as universal capture agents for various viral analytes including mutant strains, resulting in an easily generalizable LF platform. This could greatly reduce the assay’s antibody screening and optimization time, which typically takes anywhere from 12 to 16 weeks.49,50 This could enable fast adaptation of GAG-based LF strips for current and emerging viruses and provide cheap and simple ways to administer viral antigen tests for critically undertested communities during times of global health crises. In the case of COVID-19, nucleic acid detection methods, such as RT-PCR, are the gold standard in viral testing. However, there is a significant time gap between testing and obtaining results due to the PCR testing capacity limitations. Additionally, areas without accessible RT-PCR testing capabilities are predominantly lower income and/or rural, making this a public health priority as well. Thus, self-administered antigen-based rapid testing would be able to fill the time and resource gaps of the nucleic acid-based testing method for monitoring and containment.51−53
Using GAGs as capture probes in LF biosensors introduces two major design challenges: (1) Due to their molecular flexibility, specific binding mechanisms between GAGs and the spike protein are largely unknown, and experimentally determined bound structures remain elusive and (2) due to their highly heterogeneous composition—i.e., varying stereochemical ratios, sulfation rates, and chain lengths—it is challenging to optimize sensitive and selective LF strips in a robust and reproducible fashion. To address these issues herein, we have integrated rigorous computational methods and extensive experimental system development to create GlycoGrip: a highly sensitive and selective LF strip biosensor for a rapid detection of SARS-CoV-2 spike protein (Figure 1B). Our GlycoGrip LF biosensor is inspired by eons old, biological signaling technology: the glycocalyx. It utilizes GAGs as primary bioreceptors anchored to a test trip to capture the spike protein, while antispike monoclonal antibodies labeled with gold nanoparticles (AuAb) are used as reporters. In the presence of the virus, GAGs and AuAb cobind to the target virus forming the “sandwich” ternary complex and generating color on both the test and control lines in under 30 min (Figure 1B, “positive”). In the absence of target virus, only color on the control line emerges as AuAb are captured by the control anti-IgG antibodies (Figure 1B, “negative”).
Through a recursive feedback loop of experiments and simulations, we have fine-tuned our GlycoGrip sensor and elucidated key mechanisms of how polysaccharides, specifically HS, bind spike during SARS-CoV-2 host-cell invasion. Our computational methods uniquely integrated fully flexible, ensemble-based docking procedures considering the entirety of the spike head, with a complete spike glycoprofile, and modeled several GAGs including HS, HEP, CS, and dextran sulfate (DEX, a synthetic HEP analog). Our GlycoGrip technology is uniquely tailored to capture and detect SARS-CoV-2 and its rapidly emerging variants and could be applied to detect other pathogenic proteins.
2. Results and Discussion
2.1. Establishing Glycocalyx Polymers as Capture Agents for Spike Binding
The concept of taking advantage of GAGs’ ability to bind spike in a multivalent manner and employ them as surface-anchored capture agents for detecting SARS-CoV-2 serves as the basis for our work. Skidmore and co-workers were the first to show, via circular dichroism, surface plasmon resonance, and molecular modeling, a clear GAG binding site on the spike RBD.23 Furthermore, they illustrate that SARS-CoV-2 cellular invasion could be inhibited by introduction of exogenous heparin. Esko and co-workers elaborated on this by showing that HS is a necessary coreceptor for SARS-CoV-2 viral infection, positing that HEP inhibition is caused by “distraction” from cellular HS.27 Esko and co-workers showed that binding of SARS-CoV-2 spike to ACE2 in mammalian cell lines is drastically reduced upon introduction of heparan lyase, and the presence of ACE2 alone on a mammalian cell surface is not sufficient for SARS-CoV-2 host-cell invasion, suggesting that HS is required.27,54 Interestingly, Fadda and co-workers,34 through investigation of evolutionary loss of a glycan at position N370, posit that a cleft at the tip of the RBD is uniquely suited for binding oligosaccharides. The computational modeling of Skidmore and Esko focused attention on interactions between HS/HEP and a patch along the spike’s RBD,23,27 whereas Wade and co-workers,35 Linhardt and co-workers,33 and Gandhi and co-workers36 extended their search ranges to include the entire spike head. Wade and co-workers identified a cleft connecting the RBD and furin cleavage site that can be stably occupied by long chain GAGs.35 Linhardt and co-workers identified several binding sites of interest including a site located near the fusion peptide.26 Gandhi and co-workers identified an otherwise unidentified site on the NTD.36 Additionally, Boons and co-workers29 and Desai and co-workers55 have conducted extensive mappings of HS stereochemistry and sulfation patterns to identify key “heparin-like” motifs optimal for targeting the SARS-CoV-2 spike glycoprotein. Taken together, these works indicate that the structure of SARS-CoV-2 spike may have evolved to be uniquely tailored for binding glycocalyx GAGs.
2.1.1. Modeling GAG Chemical Diversity
We first sought to identify spike-GAG binding sites to exploit for our sensor. We used an ensemble-based docking protocol that includes docking of multiple GAG identities into various well-sampled/varied protein and glycan conformations and unbiased searching of the entire spike head. In addition to the most abundant GAGs on the cell surface, HS and CS,4 low molecular weight HEP has been shown to bind effectively to the SARS-CoV-2 spike, as well as induce the RBD down (where the RBM is shielded, thus cannot bind ACE2) to RBD up (RBM exposed, thus ready to bind ACE2) conformational change.26,27,29,35 To sample sufficient GAG diversity, we modeled HEP, HS, CS, and DEX to identify which GAG would best capture the SARS-CoV-2 spike on an LF test strip (Figure 3A). Cellular HS is incredibly heterogeneous in both uronic acid identity and degree of sulfation.23,26,27,29,56−59 To better match HS considered in our LFSA design and testing (where HS was purchased from Sigma and reported to be only 5–7% sulfation by mass), we constructed 6-O-sulfated heparan sulfate, from here on referred to as our H6S model. Although this model does not capture the full heterogeneity of cellular HS, it does better reflect the low sulfation rate of HS considered experimentally in this work. As docking long polysaccharide chains is intractable—due to combinatorial enumeration of rotational degrees of freedom—we chose to model dimeric (n = 1) and tetrameric (n = 2) forms of each of our four candidates. Dimeric forms were included to capture highly localized interactions, while tetrameric forms were included to elucidate slightly longer-range effects, i.e., inaccessibility for longer polysaccharide chains. We used MatrixDB60−63 to build dimeric and tetrameric HEP and H6S and CHARMM-GUI64−67 to build CS and low sulfated (∼6% sulfated) DEX. (See Materials and Methods for a complete chemical description of each molecule and Figure 3A for ChemDraw images.)
Figure 3.

(A) Molecular representation of each GAG candidate considered in this work: HEP, H6S (heparan sulfate sulfated at the 6-O position), CS, and DEX. All four GAGs were modeled in both dimeric (n = 1) and tetrameric (n = 2) forms. (B) Molecular representations (side and top views) of spike head (gray ribbons) with spike glycans (light blue, licorice atoms). Colored spheres shown on the spike head illustrate the centers of mass of each GAG binding site as predicted by our ensemble-based docking studies. We predicted over 12 800 spike–GAG binding modes and clustered these binding modes into 17 distinct binding sites. These sites were each ranked by our binding site importance score. Spheres are colored to indicate the relative importance of each site according to our binding site importance score: red spheres indicating relatively important sites, and blue spheres indicating less important sites. (C) Molecular snapshots of binding site B, corresponding to a supersite formed between the RBD patch and the RBD cleft, for each protein/glycan conformation used in ensemble-based docking. Four spike (protein and glycan) conformations were used in docking studies to incorporate a degree of spike flexibility, and the degree of conformational diversity can be observed in these zoom-in images of site B. (D) Experimentally calculated binding affinities between spike and tested GAGs (steady-state analysis of BLI data to determine KD values). (E) Molecular representation of our constructed trivalent spike-hep40mer model.
2.1.2. Accounting for Spike Conformations and Surface Accessibility for Binding
To incorporate protein and glycan flexibility in our docking protocol, we docked all GAG models (eight total molecules) into four different spike conformations extracted from Casalino et al.’s trajectories (Figure 3B–C).15 We used accessibility of the furin-cleavage site as a metric to identify four conformationally unique frames to serve as receptor structures for docking. The polybasic furin-cleavage site (spike residues 674–685) is one of the most flexible regions of the spike protein (Figures S2–4) and postulated to bind a myriad of physiological cofactors.32,35,37 Therefore, we selected four spike conformations (conformations 1–4 in Figures 3C and S3–4) based on their degree of accessibility to the furin cleavage site (Table S1, see Supporting Information Methods for calculation details). Furthermore, given each of these GAGs is highly flexible, we thoroughly sampled polysaccharide rotational degrees of freedom by predicting 400 poses per ligand and protein conformation pair (see Supporting Information Methods for complete details). At 8 GAG models, four protein conformations, and 400 predicted poses per pair, this resulted in a total of 12 800 resultant binding modes.
2.1.3. Selecting Favorable Spike–Gag Binding Sites
To organize our 12 800 predicted binding poses into discernible binding “sites,” we clustered them by their centers of mass (Figure S5, Table S2, see SI Methods). From clustering, we determined 17 distinct spike–GAG binding sites (Figure 3B). For easy reference, we have indexed the sites by letter, A–Q: see Scheme S1, Figure S6, and SI Methods for complete details on our docking and analytical clustering methods; and for a complete listing of protein and glycan residues in each binding site, please see Table S2. We then derived a “binding site importance score” (eq 1) to describe the “importance” of each predicted binding site.
| 1 |
Out of the 17 unique binding sites, three sites (J, O, and Q) were omitted from further analysis as they are highly buried within the spike and would not be accessible to long-chain GAGs (Scheme S1, Figure S7). From the remaining 14 surface binding sites, we identified 6 novel binding sites (F, G, K, L, M, N) and validated 8 previously identified binding sites (A, B, C, D, E, H, I, P) (Figure S8). Sites C, B, and D correspond to a “supersite” formed between the RBD patch23,27 and RBD cleft34 sites (C, B, and D are analogous sites centered on one of each spike protomers), sites E and H correspond to the connecting ridge posited by Wade and co-workers35 which connects the RBD supersite down to the furin-cleavage site, site I is similar to the NTD site proposed by Gandhi and co-workers.36 Our results support the importance of these sites for GAG binding and reaffirm the need to focus on these regions when studying the role of HS in SARS-CoV-2 host-cell invasion mechanism. Novel sites F, L, and M have yet to be described in the literature, but they represent an alternative binding mode for long-chain GAGs on the spike surface. As shown in Figure 2B, the connecting ridge connects the RBD sites (green and pink) down to the furin cleavage site along a ridge formed between the left protomer’s RBD and the right protomer’s NTD. Sites M and L represent sites that could, instead, be used to connect the RBD supersite down to the furin cleavage site between a ridge formed by the right protomer’s RBD and the left protomer’s NTD (Figure S9), a path similar to that predicted by Schuurs et al.36 Thus, there are potentially two distinct surface paths along which a long GAG could bind to interact with both the RBD supersite and the furin cleavage site. This finding highlights the importance of GAGs’ multivalent binding mode in attaching to the spike surface. For all 14 surface binding sites, we estimated their relative “importance” through our binding site importance score (eq 1). Sites B, D, E, F, L, and M, all have relatively high importance with scores 0.9, 0.6, 0.6, 0.7, 0.8, and 0.9, respectively, (see Table S3–S7 for all docking results), indicating a high likelihood for GAG binding at these sites.
2.1.4. Experimental Characterization of Spike–GAG Binding Affinities by Biolayer Interferometry
Our ensemble-based docking indicated that all dimer and tetrameric GAGs bound with relatively similar predicted binding energies in each binding site (Tables S2–S6). To better discriminate their affinity to spike, we turned to biolayer interferometry. As the glycocalyx composition is heterogeneous with respect to GAG identity and GAG length, we were interested to evaluate the binding affinity of various GAGs at different chain lengths to the spike: 15 and 27 kDa HEP (HEP15, HEP27), 15 kDa HS (HS15), 25 kDa CS (CS25), and 5 and 50 kDa DEX (DEX5, DEX50). As shown in Figure 3D, all GAGs, at all chain lengths, bound to spike with relatively high affinity. HS15 and CS25 exhibited the highest binding affinities out of all GAGs tested, with KD values of 16.7 nM [95% CI; 8–29 nM] and 17.9 nM [95% CI; 5–43 nM], respectively. These are followed in order of KD by HEP27, DEX5, HEP15, and DEX50 (Figure S10, Table S8 for a summary of all BLI results).68−70 As HS15 shows the highest affinity for spike and considering the ubiquitous presence of HS in the glycocalyx, coexisting around ACE2, it is possible the SARS-CoV-2 spike sequence has undergone selective pressure with respect to binding ACE2 as well as HS. In the context of our sensor construction, although all BLI-tested GAGs showed high affinity for SARS-CoV-2 and could serve as capture agents, we chose to focus on HS and HEP in our current device.
2.1.5. Constructing a Long-Chain HEP–Spike Model
Although using short (e.g., dimeric and tetrameric) GAG models was necessary to conduct extensive ensemble-based docking protocols, these short GAGs do not fully capture the extent of steric hindrance and torsional constraints that would arise as SARS-CoV-2 spike approaches a long-chain GAG either in an LF test strip or during host cell invasion. Thus, we used our list of highly populated binding sites and literature sites to guide the construction of a more relevant computational model of spike-GAG binding (Figure 3E). We used octameric HEP units as a molecular “thread” to “sew” continuously through binding sites B, H, and down to the furin cleavage site, ultimately connecting all these sites with a 40-meric HEP (hep40mer) molecule (SI Methods). Given the symmetrical nature of spike, we repeated this process for all three spike protomers to illustrate the multivalent GAG-binding potential of spike under biological and in vitro assay conditions (Figure 3E). In this current work, our model serves to connect our docking results, at the atomic (molecular-) scale, to our experimental results and the biological context, at the macro-scale.
Taken together, our computational models and biolayer interferometry measurements reveal that glycocalyx-derived GAGs are a promising class of molecules to act as capture agents for SARS-CoV-2, as they have high affinity for its spike protein and can bind the spike in multiple binding sites, with various spike conformations, at high valency.
2.2. Mechanistic Insights: GAGs Adapt to Spike Conformation
Interestingly, the iterative process of computation and experiments for identifying spike-GAG binding has enabled us to unravel mechanistic insights into SARS-CoV-2 host-cell invasion. Accounting for protein and glycan flexibility via ensemble-based docking elucidated two key mechanistic hypotheses: (1) GAGs are likely to bind in multiple compensatory modes to accommodate changes in spike conformation and (2) the spike’s glycans compete with GAGs for certain binding sites on the spike surface yet stabilize other GAG–spike interactions.
2.2.1. GAGs Can Adapt Multiple Binding Modes, Adjusting to Spike Conformations
As mentioned, we predict site B to be a hotspot for GAG binding; according to our binding site importance score it is our no. 1 ranked site overall, and it is relatively accessible over the course of 1.8 μs (Figure S7). Furthermore, B sits at crucial spot for the spike: “behind” the RBM, and at the interface between the RBD and the neighboring protomer’s NTD (Figure S11). Esko posits interactions between low molecular weight HEP and this site could induce transition from a down to up RBD.27 Fadda elaborates this site has high affinity for neutral or negatively charged oligosaccharides.34 Additionally, Casalino et al.’s simulations illustrate that the RBM is one of the most flexible regions in the spike head, second only to the furin cleavage site (Figure S2).15 The RBM’s/site B’s conformational diversity is exemplified clearly in Figure 3C. Despite drastic differences in protein and glycan topography around the RBM, site B is highly populated in all four protein conformations for all GAG models (Table S3–7). Thus, we hypothesize that site B may accommodate multiple interconverting GAG binding modes. The differing binding modes at site B may reveal how HEP facilitates the RBD’s transition to the up state: by alleviating tight interfacial interactions between the RBD and NTD and by remaining bound despite drastic RBD conformational changes (Figure S11).
2.2.2. Spike Glycans Can Compete with GAGs or Stabilize GAGs at Surface Binding Sites
Some GAG–spike binding sites, most notably site E, are entirely unoccupied by GAGs in specific spike conformations but highly populated in others. Our analysis reveals that spike glycans may compete with, or stabilize GAG binding at the spike surface. In spike conformations 1 and 3, site E is highly populated for all GAGs. When considering data from only spike conformations 1 and 3, site E is ranked no. 1, above site B, according to our binding site importance score. However, in spike conformations 2 and 4, site E was shown to be completely inaccessible to GAG binding: 0 binding modes for all modeled molecules (Table S4, S6). Careful inspection of this site reveals that in spike conformations 2 and 4 the glycan at N331B directly occupies site E, whereas in conformations 1 and 3 the N331B glycan moves away from site E (Figure S12). Notably, while N331B moves away from site E in conformations 1 and 3, it is still neighboring site E and thus can provide stabilizing hydrogen bonding interactions to GAGs at this site (Figure S12E). Thus, glycan N331B serves to shield GAGs from binding at site E, but when site E becomes available, N331B, N122C, and N165C serve to stabilize GAGs bound at this site. This analysis suggests yet another role for glycans in the spike priming process. Casalino et al. have shown that glycans can shield key spike antigenic regions from recognition by potent antibodies, but glycans N165 and N234 can also prop up and stabilize the RBD in the up state.15 Furthermore, Sztain et al. have shown that N343 facilitates movement to the up state by “pushing” the RBD up through hydrophobic interactions with RBD residues.19 Here, we show that spike glycans serve a dual role: they can both shield the spike surface and stabilize GAGs that make it to the spike surface.
2.3. NTD is Ideal for Cobinding Spike Using GAG-Bound Test Strips
Next, to generate a robust sandwich-based lateral flow assay, we tested a few spike-binding proteins and antibodies for their ability to cobind spike with surface-bound GAGs. We experimentally screened the following candidates: (1) REGN10933 (RBD Ab), one-half of the powerful REGN-COV2 antibody cocktail which binds to a subregion of the spike RBM,71 (2) ACE2, the enzyme responsible for binding to spike RBM, which initiates host cell invasion,72,73 and (3) an NTD binding antibody (NTD Ab) (Figure 4A). As the NTD binding antibody we have used here does not have a specific known epitope, for our computational modeling we have chosen to use the 4A8 NTD binding antibody as a structural stand-in.74 Initial experimental screening revealed that all candidates form the sandwich-style complex with GAGs when exposed to spike protein for 30 min (Figure 4B). To investigate which candidate would generate a positive signal in a shorter detection time, we reduced the incubation time to 5 min. Interestingly, using NTD Ab resulted in, by far, the most intense LFSA signal compared to RBD Ab and ACE2 after 5 min, suggesting that the RBD site might be the most ideal for cobinding spike with GAGs (Figure 4B). To explore this result at the atomic scale, we constructed simple spatial models of these complexes: (1) the RBD antibody (REGN10933) bound to a “1-RBD-up” spike with three heparin-40mer molecules (3xhep40mer) also bound (Figure S13A),71 (2) ACE2 bound to a “1-RBD-up” spike with 3xhep40mer also bound (Figure S13B),72 and finally (3) three NTD antibodies (4A8) bound to an “all-RBDs-down” spike with 3xhep40mer also bound (Figure 4C).74 From these spatial models, even in the 3xhep40mer–spike complexes, binding of REGN10933, ACE2, or 4A8 could all be theoretically accommodated. Thus, static models alone could not explain why NTD antibodies would provide a much higher LFSA signal relative to RBD-binding biomolecules (REGN10933 and ACE2). None of the predicted binding sites, nor our long-range HEP model, nor the literature proposed sites, overlap with the REGN10933 epitope, the RBM/ACE2 binding domain, or the 4A8 epitope (as illustrated by Figures S13A,B and 2C, respectively). Therefore, we did not suspect that choice of antibody would have such dramatic impact on the intensity of LFSA signal.
Figure 4.

(A) Schematic illustration of our screening of various signaling probe candidates on HEP-based LF strip and (B) corresponding screening result with 30 and 5 min incubation. (C) Alignment of hep40mer and NTD Ab to the spike. (D) Computational model of NTD and RBD epitopes (4A8 and REGN10933, respectively) along with the ACE2 binding motif. (E) Accessible surface area calculated from the RBD epitope (REGN10933), ACE2 binding motif, and NTD epitope (4A8). Dark blue bars indicate the size of the interface area as seen in Cryo-EM structures for the RBD, ACE2, and NTD binding footprints (6XDG, 6M17, and 7C2L, respectively). p values < 0.05 (*), 0.01 (**), and 0.001 (***) determined using a two-way ANOVA with Tukey’s post hoc test.
To investigate why an NTD binding antibody might be more suitable than an RBD binding antibody for generating strong LFSA signals in our device, we utilized Casalino et al.’s simulations15 to interrogate the relative accessibilities of epitopes for REGN10933, ACE2, and 4A8 via accessible surface area (ASA) calculations (Figure 4D,E). We used the Shrake–Rupley algorithm75 to calculate ASA for each of these antibody’s epitopes (Figure 4E) in both the closed and open spike conformations. We also calculated the size of a “reference” interface from cryo-EM structures, to estimate the required exposed surface area for a successful binding event. To calculate the cryo-EM reference interface sizes, we used the Protein Data Bank structures for each antibody bound to the spike (PDB ID’s as follows: 6XDG for REGN10933, 6M17 for ACE2, and 7C2L for 4A8). We then removed the antibody binding partner and calculated surface area of the same interface with the Shrake–Rupley algorithm, as done for Casalino et al.’s simulations.
The resulting ASA plots reveal a high degree of protein self-shielding and glycan shielding exhibited by the two RBD based epitopes (REGN10933 and RBM/ACE2), whereas the NTD provides a consistently exposed epitope for antibody binding (Figure 4E). The reference values for REGN10933 and ACE2 epitopes are out of range of the simulation-visited surface areas for both closed and open spike conformations, with a more pronounced effect in the closed states. In contrast, the NTD epitope reference value is well within the range of simulation-visited surface areas for all probe radii, regardless of closed/open spike conformations. This analysis indicates that the NTD epitope is easily and consistently accessed by AuNP-NTD antibodies, making it a superior choice for LFSA, both for specificity and accessibility. In contrast, many antigenic regions of the RBD are sequestered within the spike trefoil and only become accessible after the down to up transition is triggered.76−78 This is likely due to selective pressure on the spike which has driven sequence and structural changes to hide the spike RBD antigenic regions to potentially limit exposure until close-range interactions can occur between the RBD and ACE2.27 Conversely, NTD structure and accessibility is not affected by spike conformation and thus there are always 3 NTD epitopes available. Even in the best case for an RBD binding antibody, where a spike has begun moving into an open conformation, there may be only 1 or 2 RBDs in the up conformation. Our analysis of Casalino et al.’s simulations provide a reasonable explanation for our observed LFSA results. Taken together, these results indicate that an NTD Ab, of the Abs tested, is an ideal partner for spike recognition with GlycoGrip test strips.
2.4. Sensor Optimization: Spike and GAG Binding Is Driven by Electrostatics and Tuned by Hydrogen Bonding
Recent work has posited that binding of HEP and other GAGs to the spike is driven by electrostatic interactions between negatively charged GAGs and positively charged patches on the spike surface.26,27,34,35 Further exploring the nature of spike–GAG binding will allow us to better optimize conditions for sensitive and specific sensing.
2.4.1. Spike Glycans Tune Its Surface Electrostatics, Shielding Electrostatically, and Sterically
Past works that have commented on the electrostatic potential of the spike surface have ignored electrostatic contributions of spike glycans.27,33 To better elucidate the spike’s surface electrostatic profile, we have calculated electrostatic potential (ESP) maps for wild type (WT) spike with the adaptive Poisson–Boltzmann solver (APBS) including and excluding glycan contributions. When considering closed spike surface alone (i.e., without including glycans, Figure S14A), we indeed see large positively charged regions. These regions tend to contour interfacial regions between protomers. For example, in the closed spike conformation (Figure S14A) there is a large positive region extending from the top of the spike head, between the RBD and neighboring protomer’s NTD, down along this interface between protomers, and then toward the furin cleavage site below the NTD. As expected, these positive regions are postulated by Skidmore, Esko, Linhardt, and Wade as primary sites for long-chain GAG interactions with spike, as shown in Figure 3B.23,26,27,35 Interestingly, when glycans are included in ESP maps (Figure S14B), the positive regions remain but they are not as pronounced. Glycans are decorated primarily with electron-rich hydroxyl groups and surround positively charged spike surface regions with significant electron density. Our electrostatic maps underscore the need to include glycans at every step of glycoprotein investigation, as glycans may shield a protein surface sterically or electrostatically. If GAG binding to the spike is dominated by electrostatic interactions, accounting for the electrostatic nature of glycans is important as electron dense glycans may compete with negatively charged GAGs. This pattern of an electron-poor protein surface crowded by electron-dense glycans is also observed in the open spike conformation (Figure 5A, S15). Without considering glycans, this putative long-range binding motif—starting at the RBD, running between the RBD and neighboring NTD, down along the protomer interface, and finally to the furin-cleavage site—is evident. In fact, with the exposure of the RBD in the open spike conformation, the positively charged surface on the RBD becomes more evident and extends to the top of the up RBD. But as in the closed structure, upon including glycans, one can see these positive protein surfaces are crowded by electron-rich glycans (Figure S15B).
Figure 5.

(A(i)) Electrostatic potential map (ESP) of 1-up spike without glycans included for simple illustration. ESP is illustrated on a range from −4 to +4 kT/e. The RBD supersite is highlighted with an orange dashed line. (A(ii)) Rotated viewpoint of electrostatic potential map of 1-up spike. Again, ESP is illustrated on a range from −4 to +4 kT/e and the RBD HS supersite is highlighted with an orange dashed line. (A(iii)) Close up view of spike RBD (surface representation) and bound HEP octamer (hep8mer, licorice representation). Both spike RBD and hep8mer are colored according to their corresponding electrostatic potentials (ranging from −4 to +4 kT/e). (A(iv)) Close-ups of key interactions between hep8mer (licorice representation, orange carbon atoms) and spike RBD (gray cartoon representation) facilitated by R346, N354, K356, and R357. (B) Computational calculation of binding energy of hep8mer to spike RBD over a range of implicit solvent ionic strengths. (C) BLI results of HEP to spike in three different concentrations of NaCl (10, 75, and 150 mM). (D) Response of the lateral flow test in different ionic concentrations (10, 75, and 150 mM). (E) Screening results of HS15, HEP15, HEP27, CS25, DEX5, and DEX50 using LFSA. p values <0.05 (*), 0.01 (**) and 0.001 (***) determined using a one-way ANOVA with Tukey’s post hoc test.
2.4.2. Solution Ionic Strength Modulates GAGs and Spike Binding
As encounter complex formation and electrostatically driven binding affinities can be modulated by solution ionic strength, we computationally predicted the binding affinity of HEP to spike RBD over a range of ionic concentrations using APBS (see SI Methods).79−82 These results show a clear trend: binding affinity between HEP and spike RBD decreases with increasing solution ionic strength (Figure 5B). BLI results confirm our computational predictions: HEP binding affinity to spike dramatically decreased so that KD values could not be determined under the concentration range of spike tested as ionic strength increased (Figure 5C, S16). We have also tested the effect of ionic strength on binding affinity between spike and the NTD antibody. The measured binding affinity between NTD Ab and spike slightly decreased (changed from 52 to 83 nM) as NaCl concentration decreased (Figure S17), suggesting that the Ab binding affinity is much less dominated by electrostatics. Next, we monitored the intensity of LFSA signals under three NaCl concentrations (10, 75, and 150 mM). As shown in Figure 5D, 10 mM NaCl solution conditions resulted in a 4.7 times more intense signal compared to 150 mM solution conditions (p < 0.05). Thus, lower ionic strength yields a higher signal intensity in our LFSA device.
2.4.3. Electrostatics is Not Everything: HS Binds with Higher Affinity to Spike than HEP
As mentioned, the prevailing hypothesis in HEP/HS interaction with SARS-CoV-2 spike is that binding is electrostatically driven.23,25−29,33,35,83 One intriguing result of ours complicates this, otherwise, straightforward spike–GAG electrostatic binding model. Our BLI measurements show that HS15 binds to spike with a greater affinity (16.7 nM KD) than the highly sulfated/highly charged HEP15 of the same molecular weight (215 nM KD). Due to the varied nature of the spike electrostatic surface—i.e., large, positively charged patches obscured by electron dense or electroneutral glycans—this likely indicates that, while electrostatics is a major initial driving force of spike–GAG binding, it is not the only driving force. In fact, at close range, hydrogen bonding interactions and appropriate moderation of electrostatics likely allows HS to bind with higher affinity to spike than highly charged HEP. Additionally, due to the highly charged nature of HEP, the spike–HEP interaction may incur a higher desolvation penalty than the less negatively charged HS.84−86
2.5. HS and HEP are Optimal Glycocalyx-Inspired LFSA Capture Agents
Applying our optimized conditions elucidated by computational and experimental investigations thus far—i.e., use of NTD conjugated AuNP and low ionic strength conditions—we compared test strips employing selected glycopolymers as capture probes (Figure 5E). HS15 showed the highest signal intensity, in agreement with our BLI results illustrating HS binding spike with the highest affinity (lowest KD). However, BLI affinity alone could not explain relative LFSA signal intensity trends seen for our tested GAGs, i.e., higher intensity shown for HS15 compared to HEP15 and HEP27, and low signal intensity from CS25, DEX50, and DEX5. Several factors can impact LFSA signal intensity such as individual GAG and antibody binding affinities, formation of sandwich-type complex between the target and both bioreceptors, and the adsorption efficiency of each GAG onto the nitrocellulose membrane. To assess the role of these factors, we have conducted ELISA assays using our GAGs as primary capture probes. HS15, HEP15, HEP27, and DEX5 showed stronger intensities than CS25 and DEX50 which suggest that CS25 and DEX50 may not pair well with the NTD Ab for sandwich-type binding (Figure S18). This might be due to their length or stereochemical differences such as sulfation pattern or branching of polysaccharides. Interestingly, short chain length DEX5 exhibited lower signal intensity on LFSA than could be expected from both BLI and ELISA results. We hypothesize that this may be due to the negatively charged nature of the nitrocellulose membrane repelling negatively charged GAGs during strip preparation.87 To check this hypothesis, we have immobilized biotinylated HS15, HEP15, and DEX5 onto the nitrocellulose membrane and flowed streptavidin-coated AuNP. HS15 exhibited a strong band on the nitrocellulose membrane, HEP15 showed a weak band, and for DEX5 no bands were observed (Figure S19). Considering that HS15 (approximately 0.8 sulfate per disaccharide) is less negatively charged than HEP15 (approximately 2.3 sulfate per disaccharide) and DEX5 (average 1.9 sulfate per glucosyl residue), this result indicates that adsorption of negatively charged GAGs on nitrocellulose membrane could be hindered due to repulsion.88 This also might explain why HS15 show higher intensity on LFSA than HEP15 (Figure 5E) as there may be a difference in the number of GAGs absorbed on the surface. Thus, we expect that further optimization of the adsorption conditions of GAGs onto the nitrocellulose membrane would improve the sensing performance.
Since both HS and HEP showed robust and rigorous LFSA bands, we tested the analytical performance of both. The presence of spike protein was detectable as low as 78 ng/reaction (3.13 μg/mL, 25 μL), and the detectable range was 78–1250 ng/reaction (3.13–50 μg/mL) with the naked eye for both HEP (Figure 6A) and HS (Figure S20) based LFSA. The limit of the detections (LODs) for HS and HEP were similar (Figure 6A, Figure S20). Despite our BLI indications that HS may be a better spike binder, SPR measurements indicate that binding affinity of HS and HEP are similar (Figure S21), suggesting that the LOD is most likely dictated by the NTD binding affinity, as confirmed by immunofluorescence (Figure S22, S23). Since HEP LFSA signals provide a larger range of observably intense signals to the naked eye, its binding affinity was comparable to HS (Figure S21) and it is more cost-effective than HS, which is certainly important when looking to mass produce testing kits for viral outbreaks, we continued to optimize our GlycoGrip biosensor with HEP as the surface-anchored GAG.89
Figure 6.

(A) Analytical performance of HEP15 based GlycoGrip LF biosensor in buffer conditions. (B) Selectivity of HEP15 based GlycoGrip LF biosensor was tested with different counter targets: SARS-CoV, MERS-CoV, ACE2, human serum albumin (HSA), and bovine serum albumin (BSA). (C) Schematic illustration of the signal enhancement using HRP and AEC. (D) Analytical performance of the signal enhanced GlycoGrip LF biosensor in human saliva conditions. p values < 0.05 (*), 0.01 (**) and 0.001 (***) determined using a one-way ANOVA with Tukey’s post hoc test.
2.6. GlycoGrip is a Rapid, Sensitive, Stable, and Selective Assay for the Detection of SARS-CoV-2
We tested the selectivity of our GlycoGrip biosensor against related betacoronavirus spike glycoproteins (SARS-CoV spike and MERS-CoV spike S1 domain), as well as biologically relevant proteins likely to be found in patient samples (ACE2, bovine serum albumin, BSA, and human serum albumin, HSA). As shown in Figure 6B, positive bands were observed only when GlycoGrip LF strips were treated with SARS-CoV-2 spike, whereas treatment with other betacoronavirus spike proteins and biologically relevant “distractors” did not indicate positive test results (Video S1). Moreover, when tested with a mixture of SARS-CoV-2, SARS-CoV, and MERS-CoV spike glycoproteins, band intensity was similar to the pure SARS-CoV-2 spike band. These results suggest that our GlycoGrip LF biosensor can selectively detect the SARS-CoV-2 in more complex solutions, potentially minimizing the possibility of undesirable false positive test results.
To further enhance the sensitivity of our GlycoGrip LF biosensor, we incorporated a reporter system (horseradish peroxidase (HRP) and 3-amino-9-ethylcarbazole (AEC)) (Figure 6C). NTD Ab tethered with multiple HRP enzymes were used as a signaling probe to enhance the sensitivity by catalytic reaction. Through the enzymatic reaction, water-insoluble red colored chromogenic products were released on the test line which enhances the signal intensity. The LOD with this enzymatic signal enhancement mechanism was estimated to be 19.5 ng/reaction (0.78 μg/mL, 25 μL), enhanced 4-fold compared with unamplified results (Figure S24).
2.7. GlycoGrip Detects Spike in Human Saliva Samples
GlycoGrip can serve as a rapid test whereby samples can be self-collected from one’s own saliva in a simple, noninvasive fashion without the need for specialized equipment or personnel. The benefits of this are 2-fold: (1) LFSA tests have the potential to reach a wider testing population and (2) removing the specialized personnel requirement reduces extra cost and eliminates direct contact between infected and noninfected persons. Moreover, collecting saliva samples as opposed to nasal samples has a higher likelihood of indicating positively for both symptomatic and asymptomatic SARS-CoV-2 carriers, as nasal collection has shown high variability in sample integrity due to sampling procedure differences on an individual basis.90,91 To test GlycoGrip performance in complex media, we introduced a range of spike concentrations into human saliva samples. Sensing proteins in complex fluid such as saliva can be challenging as it contains many other biomolecules that could limit or compete with spike-GAG binding interactions. In the context of sensing SARS-CoV-2, it has been recently reported that glycoproteins in saliva, such as mucin proteins (MUC7, MUC5B) and neutrophil defensins, may bind to and interact with the spike.92 Thus, these complex glycoproteins may then compete with HEP or Ab for binding sites on the spike surface.92 Remarkably, the LOD of our GlycoGrip in saliva samples was 78 ng/reaction (3.13 μg/mL, 25 μL) comparable to buffer conditions (LOD: 19.5 ng/reaction) (Figure 6D, see Tables S9 and S10 for a summary of GlycoGrip LF results and comparison with reported spike protein detection LF). These results indicate the strong feasibility of applying GlycoGrip technology to clinical samples.
In addition to sensitivity and selectivity, sensor stability is a vital factor for sensor distribution and storage. To test GlycoGrip’s stability, we stored same-day-fabricated GlycoGrip sensor strips in a plastic bag with desiccant at room temperature and tested signal intensity after varying lengths of storage (0, 11, and 47 days). We saw no significant decrease in the signal intensity over 47 days, which indicates that our GlycoGrip LF biosensor is stable for at least 47 days and most likely much longer (Figure S25).
2.8. GlycoGrip Detects the Presence of SARS-CoV-2 Spike Variants
New strains of SARS-CoV-2 began emerging as early as summer of 2020, and at the time of this publication, the World Health Organization has highlighted four variants of concern (Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1), Delta (B.1.617.2), and Omicron (B.1.1.529) and two variants of interest (Lambda (C.37) and Mu (B.1.621)).93,94 These variants exhibit key mutations in the spike protein (as well as mutations in other SARS-CoV-2 structural and nonstructural proteins) that are postulated to directly translate to increased infectivity and/or increased immune-system evasion ability. To ensure our GlycoGrip technology would signal positively for patients infected with these emerging SARS-CoV-2 strains, we have tested variant full spikes with our LFSA technology.
2.8.1. Characteristic Mutations of Alpha, Beta, and Delta Spike Variants Do Not Interfere with GAG Binding Sites
We hoped to assess at the molecular scale whether variations in spike sequence and structure, as seen in emerging SARS-CoV-2 variants, could impact binding to HEP on the LFSA test strip. While, at the time of this publication, there are several cryo-EM structures of Alpha, Beta, and Delta SARS-CoV-2 spikes in the Protein Data Bank,95,96 unfortunately, each of these structures contain unresolved regions corresponding to highly flexible loops. Thus, we constructed computational models of the Alpha, Beta, and Delta spike variants from our refined closed WT spike structure. We then aligned our trivalent spike-hep40mer complex to these variant structures and visually inspected for overlap between mutation points with proposed hep40mer binding regions (Figures 7A, S26). From these structures, we observed no overlap or clash between HEP and mutations characterizing Alpha and Beta spike variants. This suggests that GAGs will still likely capture Alpha and Beta spikes under LFSA conditions, as we have proposed in the WT case. In the case of the Delta spike, interestingly, we observed there are several mutations (L452R, T478K, and P681R) that increase the number of positively charged residues along the posited HEP binding cleft. Furthermore, WT spike protein and its glycans constitute an overall absolute charge of −32, while the overall charge of the Delta spike protein, with the same glycoprofile, is −17. Thus, the Delta spike exhibits a drastic increase in total charge as a result of changes in sequence.
Figure 7.

(A) Trivalent hep40mer aligned to the Delta spike variant. Light green van der Waals spheres represent single point mutation sites, black van der Waals spheres represent locations of NTD deletion sites, and blue van der Waals spheres represent mutations along the hep40mer binding site L452K, T478R, and P681R. (B) BLI results of the HEP to spike proteins (wild type, Alpha (B.1.1.7), Beta (B.1.351), and Delta (B.1.617.2)). (C) Response of the Alpha, Beta, and Delta variants in the GlycoGrip LF biosensor. Statistical analysis was performed using one-way ANOVA with Tukey’s post hoc test.
As discussed, past work has highlighted the importance of large positively charged regions on the spike surface.26,27,35 To ensure mutation points in spike variants do not disrupt these positive patches, we have also calculated electrostatic potential maps for Alpha, Beta, and Delta spikes. While there are some differences in surface electrostatic potential compared to WT (Figures S14, S15 versus Figures S27–S29), noticeably in the trefoil region between neighboring RBDs, our postulated hep40mer binding region remains largely positively charged at the protein surface for Alpha, Beta, and Delta spike variants. This again supports that binding of HEP to spike could be electrostatically driven, and these electrostatic interactions are not affected by the key mutations in new spike variants.97 We hypothesize that an evolutionary advantage exists to maintain the spike’s ability to bind glycocalyx polymers. This is supported by the fact that in Alpha and Beta spike variants there exist no single point mutations along the putative HEP binding cleft. Additionally, the Delta spike variant exhibits three mutations to positive residues along the putative HEP binding cleft (L452R, T478K, P681R) which likely potentiate the ability of spike to bind to negatively charged GAGs. Finally, although we have predicted a long-chain model for HEP binding on the spike surface based on docking studies with the WT, we note that our results indicate that there are at least 14 sites on the WT spike surface where GAGs can bind. Thus, if new spike variants emerge with mutations that interfere with our proposed long-chain HEP binding site, there is still potential for GAGs binding to spike via other long-chain modes according to our proposed multisite binding model.
2.8.2. Biolayer Interferometry Confirms Alpha, Beta, and Delta Spike Variants Bind to HEP
To confirm this in silico prediction, i.e., that GlycoGrip can bind and signal positively for emerging SARS-CoV-2 spike variants, we measured binding affinity of HEP15 to full-length trimeric Alpha, Beta, and Delta spikes with BLI (Figure 7B, S30). Alpha, Beta, and Delta spikes all bound to HEP15 with comparable binding affinity to WT spike, despite their characteristic point mutations and deletions. This result supports both our posited hep40mer binding mode and our electrostatic potential maps: HEP binding is not perturbed by mutations exhibited in the SARS-CoV-2 Alpha, Beta, and Delta strains. This result underscores the power of using GAGs as the capture probe for SARS-CoV-2 spike sensing. To complete the profile of our sandwich-style LFSA detector, we measured the binding affinity between NTD Ab and each spike variant. Binding affinities of variant spikes to the NTD Ab were decreased compared to the WT spike (Figure S31); however, this is to be expected as each variant exhibits characteristic mutations in the NTD. From all variants, the Alpha variant showed the lowest binding affinity, most likely due to two key deletions in the NTD which are characteristic of the Alpha variant, one of which is within the N2 loop and one within the N3 loop, with both loops being key for antibody recognition. However, these results reflect another key feature of GlycoGrip: modularization. Since the choice of capture probe and signaling antibody are decoupled in GlycoGrip’s design, selecting a new signaling antibody largely does not impact the performance of GAGs to capture analytes.
2.8.3. Alpha, Beta, and Delta Spike Variant Detection on GlycoGrip Strips
To confirm the sandwich-type binding of HEP15 and NTD Ab to variant strains of SARS-CoV-2 spike via an orthogonal methodology, we performed ELISA using full-length trimeric Alpha, Beta, and Delta spikes (Figure S32). Alpha, Beta, and Delta spikes all bound to HEP15 and NTD Abs in a sandwich-type complex with the highest signal intensity shown for the Delta variant spike. Encouraged by our BLI and ELISA results, we then tested Alpha, Beta, and Delta spikes with our GlycoGrip LF test strip. As shown in Figure 7C, all variants exhibited positive bands on the LF test line corresponding to the ELISA, and the currently circulating highly infectious Delta variant can be detected, demonstrating the universality of using glycopolymers as viral capture agents (Video S2). Thus, our GlycoGrip LF could be rapidly adaptable to newly emerging SARS-CoV-2 variants which is an important aspect of point-of-care sensing platforms for viral pathogen detection.
3. Conclusions
We have harnessed the power of the glycocalyx and its glycosaminoglycans to serve as capturing agents within an LFSA “sandwich” binding assay to develop our GlycoGrip sensor. Our rigorously applied lock-step integration of wet lab and computational experiments allowed us to optimize conditions for GlycoGrip and provide mechanistic insights into GAG and spike binding interactions. We have demonstrated the first use of GlycoGrip for detecting wild type SARS-CoV-2 spike as well as the newly emerging variants, Alpha, Beta, and Delta. We have shown that SARS-CoV-2 spikes can be detected on GlycoGrip LF strips, and due to specificity of the chosen NTD-based signaling antibody, we saw no cross-reactivity to SARS-CoV, MERS-CoV, HSA, or ACE2 in buffer or in human saliva. We have also seen that optimizing solution ionic strength and GAG length can enhance LFSA signals, along with traditional signal enhancement systems.
In addition to sensor design, we used our extensive ensemble-based docking results to provide biologically relevant mechanistic insights into SARS-CoV-2 host cell invasion mediated by spike-GAG binding. We have confirmed literature proposed sites as well as identified six novel GAG binding sites on the spike surface. Collectively, a clear picture emerges: GAGs in the glycocalyx bind tightly to spike, at multiple sites, and with potential for multivalent long-chain GAG binding. Our work also highlights the advantages of modeling glycans when studying spike dynamics and interactions. We predict spike glycans may play a role in shielding the spike surface from incoming GAGs, but once GAGs reach the surface, glycans are likely to support GAGs via hydrogen bonding and van der Waals interactions.
The power of GlycoGrip lies in its modularity and generalizability. Many pathogens—including viruses, bacteria, and parasites—exploit the glycocalyx for cell adhesion. Thus, these pathogens, and/or their characteristic antigens, have the potential to be captured by GAGs on a GlycoGrip strip. To achieve a selective detection, one simply needs to optimize an appropriate signaling antibody that will pair with GAGs. The need for one antibody instead of two (as required for constructing traditional sandwich-type LFSA sensors) will significantly shorten the screening time47,48 when applied toward a new pathogen or variant, as well as drastically lowering the cost, potentially 10-fold, as compared to current LFSA technologies.98,99
GlycoGrip has remarkable promise as a widespread tool for capturing and detecting current and emerging viruses. While SARS-CoV-2 was of preeminent concern at the time of writing this manuscript, GlycoGrip can be easily extended for rapid screening and detection of future pathogenic infections. Loss of life prevention in public health crises requires quick detection and disease containment. We specifically note that traditionally medically underserved, and therefore undertested, populations have the hardest time identifying communal outbreaks because they lack access to RT-PCR, a technique that requires highly skilled laboratory staff and expensive instrumentation. In the case of the SARS-CoV-2 spike, we have shown that GlycoGrip can detect rapidly emerging variants. This not only speaks to the generalizability of GlycoGrip but also to its robust longevity over the course of a real-time sustained global health crisis. One could envision GlycoGrip as a synthetic glycocalyx able to trap pathogenic antigens and, coupled with antibodies, used to test patients within minutes.
In summary, we have retooled the glycocalyx, an essential component of the host cell surface, into a rapid and sensitive biosensor for viral antigens. GlycoGrip is a novel, biologically inspired, generalizable assay that has the potential to be inexpensive to manufacture, easy to distribute, simple to operate, and effective.
4. Materials and Methods
4.1. Computational Methods
4.1.1. SARS-CoV-2 Spike and Sulfated Polysaccharide Docking
In this work we modeled heparin (HEP, specifically dimeric LIdoA2S-α(1–4)-DGlcNS6S-αOH and tetrameric LIdoA2S-α(1–4)-DGlcNS6S-α(1–4)-LIdoA2S-α(1–4)-DGlcNS6S-αOH), 6-O sulfated heparan sulfate (H6S, specifically dimeric LIdoA-α(1–4)-DGlcNAc6S-αOH and tetrameric LIdoA-α(1–4)-DGlcNAc6S-α(1–4)-LIdoA-α(1–4)-DGlcNAc6S-αOH), chondroitin sulfate (CS, specifically dimeric DGlcA2S-β(1–3)-DGalNAc4S6S-βOH and tetrameric DGlcA2S-β(1–3)-DGalNAc4S6S-β(1–3)-DGlcA2S-β(1–3)-DGalNAc4S6S-βOH), dextran sulfate, (DEX, specifically dimeric DGlc-α(1–6)-DGlc2S4S-αOH and tetrameric DGlc-α(1–6)-DGlc2S4S-α(1–6)-DGlc-α(1–6)-DGlc2S4S-αOH) as potential binding partners for the SARS-CoV-2 spike. H6S was chosen as a defined sequence for modeling and docking as it captures the 5–7% sulfation rate reported by Sigma-Aldrich while also capturing potentially important interactions facilitated by the 6-O sulfation position reported by several groups.23,26,27,29,56,59 As docking of long chain polysaccharides to large protein structures is combinatorially intractable, we modeled small dimer and tetramer structures of each sulfated polysaccharide. Dimeric sulfated polysaccharides were modeled with the intention of capturing highly localized interactions, while tetrameric sulfated polysaccharides were modeled with the intention of capturing steric hindrance effects encountered with larger substrates. HEP and H6S dimeric and tetrameric structures were constructed with MatrixDB,60−63 CS, and DEX dimeric and tetrameric structures were built with CHARMM-GUI Glycan Builder.64−67
To predict potential locations of sulfated polysaccharide binding to spike, we conducted extensive unbiased docking with AutoDock Vina.100,101 Using exposure of the furin cleavage site as a metric to detect conformational changes, we selected four spike coordinates from Casalino et al. closed spike trajectories (https://amarolab.ucsd.edu/covid19.php). These spike structures were prepared as described by Casalino et al.15
Two structures must be well-defined in any docking protocol: the receptor/macromolecule to be docked into and the ligands/small molecule to be docked. To avoid biasing docking results to only certain regions of the spike, for each of the four spike structures, we defined the “receptor” to be any location on/within the trimeric spike head (residues 13 to 1140 of chains A, B, and C). To define these receptors in AutoDock Vina, we generated grids centered on the trimeric spike head, with large enough dimensions to encompass the entire head, and with default grid spacings. To characterize the structural diversity of each of these protein structures we have calculated the root-mean-square deviation (RMSD) between chains for each spike subdomain of interest (Supporting Information Results, Table S1). We prepared all molecules (dimers and tetramers of HEP, H6S, CS, and DEX) with AutoDockTools and all polysaccharide torsions were treated flexibly with AutoDock Vina conformational sampling and scoring function. For complete details of receptor grid coordinates, grid values, and all other docking settings, see input scripts included with shared docking output files in the Supporting Information.
To identify as many binding sites as possible, we conducted 20 replicates of each docking procedure and requested AutoDock return 20 predicted binding modes per docking replicate—where one docking procedure would entail docking one dimer or tetramer sulfated polysaccharide to each spike conformation. As a result, we predicted 400 binding modes for each dimeric or tetrameric sulfated polysaccharide in each spike conformation, resulting in 12 800 predicted binding modes total: 400 binding modes per molecule, by 8 molecules (dimeric and tetrameric versions of each sulfated polysaccharide), by 4 protein conformations, is 12 800 total. To parse all 12 800 predicted binding modes into discernible binding “sites”, we clustered all these resultant poses by their centers of mass using k-means clustering through python’s scikitlearn. A knee/elbow locator algorithm was used to identify the optimal number of clusters.102 We then derived a binding site “importance” metric to rank binding sites according to average binding score and relative population in that site. The top “important” binding sites were then inspected by eye through Visual Molecular Dynamics (VMD)103 to determine important binding factors governing each of these sites.
4.1.2. SARS-CoV-2 Spike and Hep40mer Model System
The fully glycosylated SARS-CoV-2 spike model used in our hep40mer modeling is based on an experimental cryo-EM structure of the spike in the closed state where all RBDs are in the down conformation (PDB: 6VXX).104 To improve the accuracy of our model, fully resolved RBD and NTD loops were incorporated from another closed spike structure (PDB: 7JJI).105 We note that utilization of the 7JJI structure in its entirety was not ideal as this structure is known to be more compact than 6VXX due to the presence of a fatty acid ligand resolved in the RBD.105 The complete glycoprofile was replicated from Casalino et al.15 Protonation state assignment was performed for spike glycoprotein with complete glycans modeled with stand-alone PropKa (so that glycan atoms could be considered during calculation),106,107 histidine protonation states were assigned via PropKa through Schrödinger’s Protein Preparation Wizard.106−108 Protonation states of all titratable residues were then compared to those assigned in Casalino et al.15 for consistency. To propose a long-range, HEP binding site along the spike surface, we considered both literature-proposed binding sites, as well as proposed binding sites from our own docking simulations. We considered only surface sites, and the most highly ranked sites were prioritized for inclusion in long-range binding mode construction. To generate relaxed HEP conformations for building hep40mer, we conducted 6 replicates of 50 ns of NVT equilibrium molecular dynamics simulations of hep8mer in a water box with NAMD.109,110 From the resultant 300 ns of HEP simulation, we clustered those frames according to conformation. 95% of all hep8mer coordinates from these simulations could be clustered according to six conformational clusters. Hep8mer coordinates representing the frame closest to each cluster center were used as hep8mer units to fill necessary coordinates between docked poses predicted from AutoDock Vina. Molefacture, a VMD based modeling tool, was used to ensure there were no clashes between protein atoms and ligand atoms.
4.1.3. Accessible Surface Area (ASA) Analysis
ASA analysis was done using the measure sasa command built-in to VMD103 along with extra protocol established by Casalino et al.15 The ASA analyses were performed by considering the antigenic regions in the NTD (residues 143–153 and 245–259) and the RBD (residues 403–406, 416–422, 453–456, 473–478, and 484–498). Additionally, ASA analysis was performed on the canonical RBM/ACE2 binding site (residues 437–508). Calculated ASAs are shown for two probe radii: 7.2 and 18.6 Å.111 The reference interface areas were calculated from cryo-EM structures as follows: REGN10933 antibody bound to spike-RBD (PDB: 6XDG(71)), 4A8 NTD antibody bound to spike-RBD (PDB: 7C2L(74)), ACE2 bound to spike-RBD (PDB: 6M17(72)).
4.1.4. System Construction for Ionic Concentration Effect Monitoring
To investigate the effect of ionic concentration on HEP binding affinity, a HEP octamer (hep8mer) was docked to the RBD of an RBD/ACE2 complex (PDB: 6M17) using Schrödinger’s Induced Fit Docking protocol.112−114 This cryo-EM structure was prepared by removing the B0AT1 dimer chaperone coordinates manually with VMD,103 and N-glycans were added on the ACE2 and RBD as done in the work of Barros et al.20 The ACE2/RBD/hep8mer construct was inserted into a lipid bilayer patch of 225 Å × 225 Å with a composition similar to that of mammalian cell membranes (56% POPC, 20% CHL, 11% POPI, 9% POPE, and 4% PSM).115 The resulting system was then embedded into an orthorhombic box of explicit TIP3P waters.116 The system was ionized with Na+/Cl– ions at 150 mM for all simulations, unless otherwise specified. All-atom MD simulations were performed on the Frontera supercomputer at the Texas Advanced Supercomputing Center (TACC) using NAMD 2.14109,110 and CHARMM36m all-atom additive force fields.66,117,118 Minimization and equilibration were performed in four steps. In the first step, while keeping all the coordinates fixed but the lipid tails, the system was subjected to an initial minimization of 10 000 steps using the conjugate gradient energy approach, followed by an NVT equilibration of 0.5 ns at 1 fs/step, where the temperature was gradually increased from 10 to 310 K. In the second step, positional constraints on lipids head, water and ions were lifted, and the system was NPT-equilibrated for 0.5 ns at 1.01325 bar and 310 K with the protein, glycans, and hep8mer harmonically restrained using a spring constant of 1 kcal/mol/Å2. Then, the restraints on protein and glycan atoms were removed and the equilibration was extended by 10 ns. Next, restraints on hep8mer atoms were removed to allow the entire system to equilibrate for an additional 10 ns. Finally, MD simulation production runs were performed and 3 replicas of ∼500 ns each were collected (Figure S33).
APBS was used to estimate binding affinity between hep8mer and spike RBD at varying ionic concentrations. Binding affinity was calculated according to an appropriate thermodynamic cycle by calculating binding energy in a homogeneous reference medium (dielectric constant = 4) and then by calculating the solvation free energy difference between the homogeneous reference state and nonhomogenous target state (dielectric constant = 78). (see https://apbs.readthedocs.io/en/latest/using/examples/binding-energies.html and the Supporting Information for a complete listing of all APBS options used in this work). Binding energies were calculated for RBD-hep8mer complexes in the following NaCl concentrations: 0.0, 0.01, 0.025, 0.05, 0.075, 0.10, 0.125, 0.150, 0.175, 0.200 M.
4.2. Experimental Methods
4.2.1. Materials
The same source for each of the following GAGs is used throughout our set of experiments including BLI, ELISA, and LF assays. HEP15 (B9806, porcine mucosa) and HS15 (H7640, bovine kidney) were purchased from Sigma-Aldrich. CS25 (CS-Biotin-25k, porcine cartilage), DEX5 (DES-Biotin-5k), and DEX50 (DES-Biotin-50k) were purchased from HAWORKS. HEP27 (HP-207, porcine mucosa) was purchased from Creative PEGWorks. Human serum albumin (A3782), sucrose (S0389), AEC staining kit (AEC101), and streptavidin coated gold nanoparticles (53134) were purchased from Sigma-Aldrich. Biotin-PEG3-amine (BG-17) was purchased from G-Biosciences. Tween 20 (J20605-AP) was purchased from Thermo Fisher Scientific. Sodium chloride (BDH9286) was purchased from VWR. Bovine serum albumin (105033) was purchased from MP biomedicals. Gold nanoparticles (15703-20) were purchased from Ted Pella Inc. N-Terminal domain binding antibody (LT-2000) and HRP modified N-terminal domain binding antibody (LT2010) were purchased from Leinco Technologies. Receptor domain binding (RBD) antibody (Clone REGN10933; CPC511B) was purchased from Cell Sciences. Rabbit Anti-Human IgG (ab6715) and Goat Anti-Mouse IgG (ab6708) were purchased from Abcam. Mouse Anti6x-His Tag Monoclonal Antibody Alexa Fluor 488 (MA1–21315A488) and Goat Anti-Human Alexa 594 Antibody (A-11014) were purchased from Thermo Fisher Scientific. Human saliva pooled from human donors (991-05-P) was purchased from LEE Biosolutions. Nitrocellulose membrane (FF120HP), sample pad (Whatman CF4 dipstick pad), and absorbent pad (Whatman standard 17) were purchased from Cytiva. SARS-CoV-2 spike protein (40589-V08H4), SARS-CoV S1 (40150-V05H1), MERS-CoV S1 (40069-V08H), HRP modified antihuman antibody (10702-T16-H) were purchased from Sino Biological. SARS-CoV-2 Alpha (B.1.1.7) spike (10796-CV-100), SARS-CoV-2 Beta (B.1.351) spike (10786-CV-100), and SARS-CoV-2 Delta (B.1.617.2) spike (10878-CV-100) were purchased from R&D systems. Fc tagged human ACE2 (AC2-H5257) were purchased from Acro Biosystems. Streptavidin modified BLI biosensor tips (18-5019) and antihuman IgG Fc Capture (AHC) BLI biosensor tips (18-5060) were purchased from Sartorious. Immu-Mount (9990402) was purchased through Fisher Scientific.
4.2.2. Biolayer Interferometry
To measure the binding affinities of polysaccharides, biolayer interferometry (BLI) was used. Polysaccharide modified tips were prepared by the streptavidin–biotin methods. Streptavidin tips were functionalized with 1 mg/mL of biotin-polysaccharides (40 μL) in a kinetic buffer (10 mM HEPES, 10 mM NaCl, 0.05% Tween 20, pH 7.4) for 180 s. Polysaccharides modified tips were incubated with various concentrations of spike proteins from 0 to 500 nM in a kinetic buffer for 400 s. Then, dissociation was measured for 500 s. Dissociation constants (KD) were analyzed with steady-state analysis using the HT 11.1 software provided with instruments. In case of NTD antibody (NTD Ab), antihuman IgG Fc capture (AHC) tips were functionalized with 5 μg/mL of NTD Ab in a kinetic buffer, and the same measurement procedure was applied. For comparison study of salt effects, kinetic buffers containing different NaCl concentrations (75 mM, 150 mM) were used.
4.2.3. Preparation of Streptavidin Modified Polysaccharides
To immobilize the polysaccharides into nitrocellulose membranes, polysaccharides were conjugated to streptavidin by biotin–streptavidin interaction. Biotin modified polysaccharides were conjugated to streptavidin (1 mg/mL) with molar ratio of 4:1 (polysaccharides: streptavidin). After incubation for 1 h at room temperature, the mixture solutions were purified to remove excess polysaccharides by using the amicon filter (30, 50, and 100k) depending on the size of the polysaccharides.
4.2.4. Preparation of Antibody Modified Gold Nanoparticles
For naked-eye detection, antibodies were conjugated to gold nanoparticles (AuNP) as a signaling probe. To prepare antibody-AuNP conjugates, NTD antibody (5 μL of 1 mg/mL), RBD antibody (5 μL of 1 mg/mL), ACE2 (10 μL of 0.63 mg/mL) were each added to 1 mL of AuNP (10 nm) with 0.1 mL of borate buffer (0.1 M, pH 8.5). After 1 h incubation at room temperature, BSA (100 μL of 10 mg/mL) was introduced and incubated for 30 min to reduce the nonspecific adsorption by blocking the surface of the gold nanoparticles. Then, the mixture solution was centrifuged at 22 000g and 4 °C for 45 min. Supernatant was removed and AuNP solution was resuspended in 1 mL of BSA (1 mg/mL). Centrifugation and suspension process was repeated twice. Finally, antibody-AuNP conjugate was stored in the storage buffer (10 mM HEPES, 10 mM NaCl, 1 mg/mL BSA, pH 7.4) at 4 °C.
For signal enhancement testing, gold nanoparticles were modified with horseradish peroxidase (HRP) conjugated NTD Ab (NTD Ab-HRP). To prepare the gold nanoparticle modified with NTD-HRP (NTD-HRP-AuNP), 10 μL of NTD Ab-HRP (0.5 mg/mL) was added to the 1 mL of AuNP (10 nm) with 0.1 mL of borate buffer (0.1 M, pH 8.5). Then, the same procedure was utilized to prepare the NTD-HRP-AuNP.
4.2.5. Preparation of Polysaccharide Based Lateral Flow Strip Biosensor
Figure 1 shows the general design of the polysaccharides based lateral flow strip biosensor. Polysaccharides conjugated with streptavidin (1 mg/mL) and rabbit antihuman IgG (1 mg/mL) were dispensed on the nitrocellulose membrane (FF120HP.). Dispensed nitrocellulose membrane was dried at 65 °C for 3 min. After drying, nitrocellulose membrane was blocked with a blocking buffer (1% BSA, 0.05% Tween 20 in 10 mM HEPES, 10 mM NaCl, pH 7.4). Finally, the sample pad (Whatman CF4 dipstick pad) and the absorbent pad (Whatman standard 17) were assembled onto the nitrocellulose membrane. Assembled strips were stored at room temperature with desiccant before use.
4.2.6. Screening Optimal Epitope and Buffer Using Lateral Flow Assay
To screen the optimal antibody for the sandwich-type detection of spike protein that will work along with polysaccharides, two antibodies which bind to different epitopes of spike protein (i.e., N-terminal domain binding antibody (NTD Ab) and receptor binding domain binding antibody (RBD Ab)) and ACE2 receptor were tested. The lateral flow strip for screening optimal epitope was prepared as described in the previous section. The dipstick method was used for testing all the lateral flow strips using 96 well plates. For the comparison study, 625 ng of SARS-CoV-2 spike was incubated with each signaling probe (20 nM) in the kinetic buffer (10 mM HEPES, 10 mM NaCl, 0.05% Tween 20, pH 7.4) for 5 or 30 min at room temperature. Mixture solutions were loaded to the 96 well plate and prepared lateral strips were dipped for 20 min. After 20 min, red signals were observed by the naked eye and smartphone camera. Signals were quantitatively analyzed by ImageJ software.
To test the effect of the NaCl on lateral flow assay, lateral flow strips, signaling probes, and SARS-CoV-2 spike were prepared by using HEPES buffers containing different concentrations of NaCl (10, 75, and 150 mM). A 25 μL portion of SARS-CoV-2 spike (25 μg/mL) and 25 μL of signaling probes (20 nM) prepared in HEPES buffers containing different concentrations of NaCl (10, 75, and 150 mM) were incubated for 5 min. Then, the previously described dipstick method was used for the lateral flow assay.
4.2.7. Selectivity and Sensitivity Analysis
For the selectivity test, 25 μL of each of the proteins SARS-CoV spike (50 μg/mL), MERS-CoV spike (50 μg/mL), ACE2 (50 μg/mL), human serum albumin (50 mg/mL), bovine serum albumin (50 mg/mL), and the mixture of SARS-CoV spike (25 μg/mL), MERS-CoV spike (25 μg/mL), and SARS-CoV-2 spike (25 μg/mL) were incubated with 25 μL of signaling probe (NTD Ab-AuNP; 20 nM) for 5 min. Then, resulting solutions were loaded to the 96 well plate and lateral flow strip were dipped for 20 min.
For sensitivity test, 25 μL of various concentrations of spike (0, 0.39, 0.78, 1.56, 3.13, 6.25, 12.5. 25, 50 μg/mL) were incubated with 25 μL of signaling probe (NTD Ab-AuNP; 20 nM) for 5 min. Then, the same procedure of dipstick method was used for the lateral flow assay. The test line signals were quantitatively analyzed by ImageJ software. The limit of the detection (LOD) was calculated by using blank +3 standard deviations.
4.2.8. Signal Enhancement Analysis
For signal enhancement tests, a mixture of NTD Ab-AuNP and NTD Ab-HRP-AuNP were used as a signaling probe. The molar ratio of the mixture and reaction time was optimized (Figure S34), and a 1:1 molar ratio of NTD Ab-AuNP and NTD Ab-HRP-AuNP with 15 min reaction times were chosen for the signal enhancement testing. The assay was conducted in the buffer and spiked-in-human saliva condition. In the case of the buffer, 25 μL of various concentrations of spike (0, 0.05, 0.10, 0.20, 0.39, 0.78, 1.56, 3.13, 6.25, 12.5. 25, 50 μg/mL) were incubated with 25 μL of signaling probe mixture (20 nM of NTD Ab-AuNP and NTD Ab-HRP-AuNP) for 5 min. Resulting solutions were loaded to the 96 well plate and prepared lateral strips were dipped for 20 min. Subsequently, 100 μL of AEC solution was introduced to enhance the signal for 15 min. For human saliva conditions, various concentrations of SARS-CoV-2 spike spiked in 1/50 diluted human saliva were used as a testing sample following the same test procedure, which was used in buffer conditions. The test line signals were quantitatively analyzed by ImageJ software. The limit of the detection (LOD) was calculated by using blank +3 standard deviations.
4.2.9. Detection of Mutant Strain (SARS-CoV-2 Alpha, Beta, and Delta)
For the mutant strain testing, 25 μL of each proteins SARS-CoV-2 spike (50 μg/mL), Alpha strain spike (50 μg/mL), Beta strain spike (50 μg/mL), and Delta strain spike (50 μg/mL) were incubated with 25 μL of signaling probe (NTD Ab-AuNP; 20 nM) for 5 min. Then, resulting solutions were loaded to the 96 well plate and lateral flow strip were dipped for 20 min. The test line signals were quantitatively analyzed by ImageJ software.
4.2.10. Immunofluorescence Staining of Heparin and Heparin-Sulfate Surfaces
For immunofluorescence, APTES coated coverslips were incubated with streptavidin (200 nM) for 10 min prior to a fixation step (2.5% glutaraldehyde, 2.0% paraformaldehyde in PBS) for another 15 min. Samples were washed three times for 2 min with PBS (3 × 2:00). Surfaces were then incubated with biotin–heparin and biotin–heparan sulfate (800 nM) in PBS for 30 min. Samples were washed again with PBS (3 × 2:00). Samples were then blocked with bovine serum albumin (4% in PBS) for another 30 min. After blocking, samples were washed three times using the kinetic buffer (10 mM HEPES, 10 mM NaCl, 0.05% Tween 20, pH 7.4). Spike protein (50 nM) was incubated with Mouse Anti 6x-His Tag Antibody conjugated with Alexa Fluor 488 (1 μg/mL) for 1 h. Surfaces were washed with kinetic buffer (3 × 2:00) and were then incubated with mixture of spike-Ab for 1 h. Samples were then incubated with NTD Ab (1 μg/mL) in lateral flow assay buffer for 1 h. Finally, surfaces were incubated with Antihuman Alexa 594 in PBS for 1 h. Samples were washed with PBS and mounted onto glass slides using Immu-Mount. Samples were then imaged on a Ziess 710 confocal microscope.
Data analysis was done using FIJI. Prior to processing, immunofluorescence images were first blurred using a Gaussian threshold (diameter: 2 pixels) and a rolling pin filter for background subtraction (50 pixels). Protein locations were then identified through an automatic threshold using either a max entropy or triangle algorithm. Single randomly bright pixels were then removed using the “analyze particles” function to remove particles smaller than 0.5 μm2. Proteins with both NTD Ab and His-Tag Ab binding were then found using the “AND” function in the “image calculator” function of FIJI. Particles were then analyzed on all three channels (NTD, His-Tag, and Combined) to determine the percentage of particles displaying both NTD and His-Tag signals.
4.2.11. Immobilization and Binding of GAGs to Spike Proteins
Nunc maxisorp flat bottom 96 well plates were coated with streptavidin (200 nM; 50 μL) overnight at 4 °C. The plates were blocked with 2% BSA for 1 h and biotinylated GAGs (800 nM; 50 μL) were added to the plates for 1 h. Unbound GAGs were thoroughly washed with 200 μL of 1xPBST (0.05% Tween 20) for three times. Spike proteins (100 nM; 50 μL) diluted in the kinetic buffer (10 mM HEPES, 10 mM NaCl, 0.05% Tween 20, pH 7.4) were added to the plate and incubated for 1 h. Unbound spike protein were washed three times with 1xPBST and NTD Abs (2 μg/mL; 50 μL) diluted in the kinetic buffer were added for sandwich-type binding. Unbound NTD Abs were washed three times with 1×PBST and incubated with 50 μL each of 0.1 μg/mL antihuman-HRP (Sino Biological, 10702-T16-H) for 30 min at room temperature. The wells were washed thoroughly 5 times with 200 μL of 1×PBST. Finally, 100 μL of TMB substrate (Thermo Fisher Scientific, 34028) was added to each well to develop color. The reaction was stopped by adding 50 μL of stop solution (Thermo Fisher Scientific, N600) and absorbance was measured at 450 nm.
4.2.12. Immobilization and Binding of Heparin to Variant Spike Proteins
Nunc maxisorp flat bottom 96 well plates were coated with streptavidin (200 nM; 50 μL) overnight at 4 °C. The plates were blocked with 2% BSA for 1 h and biotinylated heparin (800 nM; 50 μL) was added to the plates for 1 h. Unbound GAGs were thoroughly washed three times with 200 μL of 1×PBST (0.05% Tween 20). Variant spike proteins along with wild-type spike protein (100 nM; 50 μL) diluted in the kinetic buffer (10 mM HEPES, 10 mM NaCl, 0.05% Tween 20, pH 7.4) were added to the plate and incubated for 1 h. Unbound spike protein was washed three times with 1×PBST and NTD Abs (2 μg/mL; 50 μL) diluted in the kinetic buffer were added for sandwich-type binding. Unbound NTD Abs were washed three times with 1×PBST and incubated with 50 μL each of 0.1 μg/mL antihuman-HRP (Sino Biological, 10702-T16-H) for 30 min at room temperature. The wells were washed thoroughly 5 times with 200 μL of 1×PBST. Finally, 100 μL of TMB substrate (Thermo Fisher Scientific, 34028 Thermo Fisher Scientific, 34028) was added to each well to develop color. The reaction was stopped by adding 50 μL of stop solution (Thermo Fisher Scientific, N600), and absorbance was measured at 450 nm.
4.2.13. Surface Plasmon Resonance (SPR)
To measure the binding affinity of HEP15 and HS15, a single cycle kinetic analysis method was used. Briefly, biotinylated HEP15 and HS15 were immobilized to a streptavidin (SA) chip by flowing 0.2 mg/mL of HEP15 and HS15 in kinetic buffer (10 mM HEPES, 10 mM NaCl, 0.05% Tween 20, pH 7.4) to the flow cell of the SA chip at a flow rate of 10 μL/min for 60 s. Various concentrations (10, 25, 50, 100, 200, 500 nM) of wild type spike protein samples were prepared by serial dilution in kinetic buffer. Spike protein samples were sequentially injected at a flow rate of 30 μL/min for 120 s without regeneration and dissociation was measured at the end using the same buffer for 6 min. Binding affinity was calculated using the steady-state analysis method.
4.2.14. Comparing Adsorption of GAGs onto the Nitrocellulose Membrane
To test the adsorption of HS15, HEP15, and DEX5 onto the nitrocellulose membrane, biotinylated HS15, HEP15, and DEX5 (140 μM; 1 μL) in lateral flow assay buffer (10 mM HEPES, 10 mM NaCl, pH 7.4) was dispensed to the nitrocellulose membrane (FF120HP). Dispensed nitrocellulose membrane was dried at 65 °C for 3 min. After drying, sample pad (Whatman CF4 dipstick pad) and the absorbent pad (Whatman standard 17) were assembled onto the nitrocellulose membrane. HS15, HEP15, and DEX5 adsorbed strip was dipped into the streptavidin coated AuNP (1 nM; 50 μL) for 5 min. Resulting image was analyzed with ImageJ.
Safety Statement
There are no unexpected, new, and/or significant hazards or risks associated with the reported work.
Acknowledgments
F.L.K. and M.A.R., would like to thank Robert Konecny and J. Andrew McCammon for insightful discussions about the use of APBS. F.L.K., M.A.R., and R.E.A. would like to thank Elisa Fadda, Sebastien Vidal, and Jeff Esko for their vital insights in glycobiology. F.L.K., M.A.R., L.C., and R.E.A. would like to thank TACC Frontera for their unwavering support of the Amaro Lab’s SARS-CoV-2 related efforts and their dedication to answering our many and varied emails. BLI measurements taken at the UNC Macromolecular Interactions Facility are supported in part by P30 CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center funded by the National Cancer Institute. Confocal was done at Microscopy Services Laboratory, Department of Pathology and Laboratory Medicine, which is supported in part by P30 CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We would like to thank Peter Harris for the graphical art incorporated into this manuscript.
Glossary
Abbreviations
- RBD
receptor binding domain
- RBM
receptor binding motif
- NTD
N-terminal domain
- ACE2
angiotensin converting enzyme 2
- HEP
heparin, (modeled in this work as dimeric LIdoA2S-α(1–4)-DGlcNS6S-αOH and tetrameric LIdoA2S-α(1–4)-DGlcNS6S-α(1–4)-LIdoA2S-α(1–4)-DGlcNS6S-αOH)
- H6S
6-O sulfated heparan sulfate (modeled in this work as dimeric LIdoA-α(1–4)-DGlcNAc6S-αOH and tetrameric LIdoA-α(1–4)-DGlcNAc6S-α(1–4)-LIdoA-α(1–4)-DGlcNAc6S-αOH)
- HS
used to refer to cellular heparan sulfate or heparan sulfate purchased from Sigma-Aldrich with no defined sequence
- DEX
dextran sulfate (modeled in this work as dimeric DGlc-α(1–6)-DGlc2S4S-αOH and tetrameric DGlc-α(1–6)-DGlc2S4S-α(1–6)-DGlc-α(1–6)-DGlc2S4S-αOH)
- CS
chondroitin sulfate (modeled in in this work as dimeric DGlcA2S-β(1–3)-DGalNAc4S6S-βOH and tetrameric DGlcA2S-β(1–3)-DGalNAc4S6S-β(1–3)-DGlcA2S-β(1–3)-DGalNAc4S6S-βOH)
- HEP15
fifteen kDa heparin
- HEP27
twenty-seven kDa heparin
- HS15
fifteen kDa heparan sulfate
- CS27
twenty-seven kDa chondroitin sulfate
- DEX5
five kDa dextran sulfate
- DEX50
fifty kDa dextran sulfate
- APBS
adaptive Poisson–Boltzmann solver
- LFSA
lateral-flow strip assay
- MD
molecular dynamics simulations
- ESP
electrostatic potential
- LOD
limit of detection
- BSA
bovine serum albumin
- HSA
human serum albumin
- HRP
horseradish peroxidase
- AEC
3-amino-9-ethylcarbazole
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.1c01080.
Author Contributions
⊥ S.H.K., F.L.K., and M.A.R. contributed equally. S.H.K. and R.F. designed all experiments and conducted all assays. F.L.K., M.A.R., L.C., and R.E.A. designed all computational simulations and analyses. F.L.K. and M.A.R. conducted all computational simulations and analyses. F.L.K., M.A.R., and L.C. constructed and designed all computational model images. F.L.K. produced computational plots and tables. F.L.K. and L.C. constructed ACE2, spike RBD, and hep8mer simulations used in APBS calculations. S.H.K., F.L.K., and M.A.R. wrote the manuscript with input from all authors. M.J.P. conducted confocal studies and provided lung biology insights and edited the paper. C.S. provided inspiration for utilizing multiple protein structures as receptor structures during docking simulations. R.E.A. and R.F. provided foundational guidance on experimental design and analyses.
The authors acknowledge the Texas Advanced Computing Center (TACC) at The University of Texas at Austin for providing HPC resources that have contributed to the research results reported within this paper (http://www.tacc.utexas.edu). C.S., R.E.A., and R.F. acknowledge a Research Corporation for Science Advancement (COVID Initiative grant no. 27350) award. R.F. also acknowledges support from the North Carolina Policy Collaboratory at the University of North Carolina at Chapel Hill with funding from the North Carolina Coronavirus Relief Fund established and appropriated by the North Carolina General Assembly, NSF RAPID (DMS-2028758) and the UNC Institute for Convergent Science Director’s Machete Award. R.E.A. acknowledges support from NIH GM132826, NSF RAPID MCB-2032054, and a UC San Diego Moores Cancer Center 2020 SARS-COV-2 seed grant. M.A.R. is supported by NIH T32 EB009380.
The authors declare the following competing financial interest(s): A patent was submitted on this work.
Notes
In conjunction with the data sharing philosophy initiated by Amaro and Mulholland et al.,119,120 all structures relating to simulations described herein will be made freely available to download on the AmaroLab Web site and through the COVID-19 Molecular Structure and Therapeutics Hub (https://covid.molssi.org). Additionally, in this current work we have utilized trajectories published by Casalino et al.15 for ensemble-based docking and accessible surface area analysis. The following files are available at https://amarolab.ucsd.edu/covid19.php: structures (psf/pdb formats) of all docking results described herein (8 GB, tar.gz) Spike_gag_docking_results_amarolab.tar.gz; APBS input files for all computational calculations done herein including docking and electrostatic potential and binding energy calculations (3 GB, tar.gz) Spike_apbs_files_amarolab.tar.gz.
Supplementary Material
References
- Reitsma S.; Slaaf D. W.; Vink H.; van Zandvoort M. A. M. J.; oude Egbrink M. G. A. The Endothelial Glycocalyx: Composition, Functions, and Visualization. Pfluegers Arch. 2007, 454 (3), 345–359. 10.1007/s00424-007-0212-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möckl L. The Emerging Role of the Mammalian Glycocalyx in Functional Membrane Organization and Immune System Regulation. Frontiers in Cell and Developmental Biology 2020, 8, 253. 10.3389/fcell.2020.00253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinbaum S.; Tarbell J. M.; Damiano E. R. The Structure and Function of the Endothelial Glycocalyx Layer. Annu. Rev. Biomed. Eng. 2007, 9 (1), 121–167. 10.1146/annurev.bioeng.9.060906.151959. [DOI] [PubMed] [Google Scholar]
- Essentials of Glycobiology, 3rd ed.;Varki A., Cummings R. D., Esko J. D., Stanley P., Hart G. W., Aebi M., Darvill A. G., Kinoshita T., Packer N. H., Prestegard J. H., et al. ; Cold Spring Harbor Laboratory Press: Cold Spring Harbor (NY), 2017. [PubMed] [Google Scholar]
- Feyzi E.; Saldeen T.; Larsson E.; Lindahl U.; Salmivirta M. Age-Dependent Modulation of Heparan Sulfate Structure and Function. J. Biol. Chem. 1998, 273 (22), 13395–13398. 10.1074/jbc.273.22.13395. [DOI] [PubMed] [Google Scholar]
- Han X.; Sanderson P.; Nesheiwat S.; Lin L.; Yu Y.; Zhang F.; Amster I. J.; Linhardt R. J. Structural Analysis of Urinary Glycosaminoglycans from Healthy Human Subjects. Glycobiology 2020, 30 (3), 143–151. 10.1093/glycob/cwz088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vongchan P.; Warda M.; Toyoda H.; Toida T.; Marks R. M.; Linhardt R. J. Structural Characterization of Human Liver Heparan Sulfate. Biochim. Biophys. Acta, Gen. Subj. 2005, 1721 (1), 1–8. 10.1016/j.bbagen.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Warda M.; Toida T.; Zhang F.; Sun P.; Munoz E.; Xie J.; Linhardt R. J. Isolation and Characterization of Heparan Sulfate from Various Murine Tissues. Glycoconjugate J. 2006, 23 (7), 555–563. 10.1007/s10719-006-7668-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casu B.; Garg H. G.; Linhardt R. J.; Hales C. A. Structure and Active Domains of Heparin. Chem. Biol. Heparin Heparan Sulf. 2005, 1–28. 10.1016/B978-008044859-6/50002-2. [DOI] [Google Scholar]
- Koehler M.; Delguste M.; Sieben C.; Gillet L.; Alsteens D. Initial Step of Virus Entry: Virion Binding to Cell-Surface Glycans. Annu. Rev. Virol. 2020, 7 (1), 143–165. 10.1146/annurev-virology-122019-070025. [DOI] [PubMed] [Google Scholar]
- Ströh L. J.; Stehle T. Glycan Engagement by Viruses: Receptor Switches and Specificity. Annu. Rev. Virol. 2014, 1 (1), 285–306. 10.1146/annurev-virology-031413-085417. [DOI] [PubMed] [Google Scholar]
- Thompson A. J.; de Vries R. P.; Paulson J. C. Virus Recognition of Glycan Receptors. Curr. Opin. Virol. 2019, 34, 117–129. 10.1016/j.coviro.2019.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y.; Allen J. D.; Wrapp D.; McLellan J. S.; Crispin M. Site-Specific Glycan Analysis of the SARS-CoV-2 Spike. Science (80-.). 2020, 369 (6501), 330 LP–333. 10.1126/science.abb9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shajahan A.; Supekar N. T.; Gleinich A. S.; Azadi P. Deducing the N- and O-Glycosylation Profile of the Spike Protein of Novel Coronavirus SARS-CoV-2. Glycobiology 2020, 30 (12), 981–988. 10.1093/glycob/cwaa042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casalino L.; Gaieb Z.; Goldsmith J. A.; Hjorth C. K.; Dommer A. C.; Harbison A. M.; Fogarty C. A.; Barros E. P.; Taylor B. C.; McLellan J. S.; et al. Beyond Shielding: The Roles of Glycans in the SARS-CoV-2 Spike Protein. ACS Cent. Sci. 2020, 6 (10), 1722–1734. 10.1021/acscentsci.0c01056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmer D.; Gilbert M.; Borman R.; Clark K. L. Quantitative MRNA Expression Profiling of ACE 2, a Novel Homologue of Angiotensin Converting Enzyme. FEBS Lett. 2002, 532 (1–2), 107–110. 10.1016/S0014-5793(02)03640-2. [DOI] [PubMed] [Google Scholar]
- Tipnis S. R.; Hooper N. M.; Hyde R.; Karran E.; Christie G.; Turner A. J. A Human Homolog of Angiotensin-Converting Enzyme. J. Biol. Chem. 2000, 275 (43), 33238–33243. 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- Hamming I.; Timens W.; Bulthuis M.; Lely A.; Navis G.; van Goor H. Tissue Distribution of ACE2 Protein, the Functional Receptor for SARS Coronavirus. A First Step in Understanding SARS Pathogenesis. J. Pathol. 2004, 203 (2), 631–637. 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztain T.; Ahn S.-H.; Bogetti A. T.; Casalino L.; Goldsmith J. A.; Seitz E.; McCool R. S.; Kearns F. L.; Acosta-Reyes F.; Maji S.; et al. A Glycan Gate Controls Opening of the SARS-CoV-2 Spike Protein. Nat. Chem. 2021, 13, 963. 10.1038/s41557-021-00758-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros E. P.; Casalino L.; Gaieb Z.; Dommer A. C.; Wang Y.; Fallon L.; Raguette L.; Belfon K.; Simmerling C.; Amaro R. E. The Flexibility of ACE2 in the Context of SARS-CoV-2 Infection. Biophys. J. 2021, 120 (6), 1072–1084. 10.1016/j.bpj.2020.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdecchia P.; Cavallini C.; Spanevello A.; Angeli F. The Pivotal Link between ACE2 Deficiency and SARS-CoV-2 Infection. Eur. J. Intern. Med. 2020, 76, 14–20. 10.1016/j.ejim.2020.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman M. I.; Porter J. R.; Ward M. D.; Singh S.; Vithani N.; Meller A.; Mallimadugula U. L.; Kuhn C. E.; Borowsky J. H.; Wiewiora R. P.; et al. SARS-CoV-2 Simulations Go Exascale to Predict Dramatic Spike Opening and Cryptic Pockets across the Proteome. Nat. Chem. 2021, 13, 651. 10.1038/s41557-021-00707-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mycroft-West C. J.; Su D.; Pagani I.; Rudd T. R.; Elli S.; Gandhi N. S.; Guimond S. E.; Miller G. J.; Meneghetti M. C. Z.; Nader H. B.; et al. Heparin Inhibits Cellular Invasion by SARS-CoV-2: Structural Dependence of the Interaction of the Spike S1 Receptor-Binding Domain with Heparin. Thromb. Haemostasis 2020, 120 (12), 1700–1715. 10.1055/s-0040-1721319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milewska A.; Zarebski M.; Nowak P.; Stozek K.; Potempa J.; Pyrc K. Human Coronavirus NL63 Utilizes Heparan Sulfate Proteoglycans for Attachment to Target Cells. J. Virol. 2014, 88 (22), 13221 LP–13230. 10.1128/JVI.02078-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madu I. G.; Chu V. C.; Lee H.; Regan A. D.; Bauman B. E.; Whittaker G. R. Heparan Sulfate Is a Selective Attachment Factor for the Avian Coronavirus Infectious Bronchitis Virus Beaudette (El Heparán Sulfato Es Un Factor de Unión Para La Cepa Beaudette Del Virus de Bronquitis Infecciosa). Avian Dis. 2007, 51 (1), 45–51. 10.1637/0005-2086(2007)051[0045:HSIASA]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Kim S. Y.; Jin W.; Sood A.; Montgomery D. W.; Grant O. C.; Fuster M. M.; Fu L.; Dordick J. S.; Woods R. J.; Zhang F.; Linhardt R. J. Characterization of Heparin and Severe Acute Respiratory Syndrome-Related Coronavirus 2 (SARS-CoV-2) Spike Glycoprotein Binding Interactions. Antiviral Res. 2020, 181, 104873. 10.1016/j.antiviral.2020.104873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen T. M.; Sandoval D. R.; Spliid C. B.; Pihl J.; Perrett H. R.; Painter C. D.; Narayanan A.; Majowicz S. A.; Kwong E. M.; McVicar R. N.; et al. Esko, J. D. SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell 2020, 183 (4), 1043–1057.e15. 10.1016/j.cell.2020.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Chen C. Z.; Swaroop M.; Xu M.; Wang L.; Lee J.; Wang A. Q.; Pradhan M.; Hagen N.; Chen L.; et al. Heparan Sulfate Assists SARS-CoV-2 in Cell Entry and Can Be Targeted by Approved Drugs in Vitro. Cell Discovery 2020, 6 (1), 80. 10.1038/s41421-020-00222-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L.; Chopra P.; Li X.; Bouwman K. M.; Tompkins S. M.; Wolfert M. A.; de Vries R. P.; Boons G.-J. Heparan Sulfate Proteoglycans as Attachment Factor for SARS-CoV-2. ACS Cent. Sci. 2021, 7 (6), 1009–1018. 10.1021/acscentsci.1c00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M.; Zhang T.; Zhang W.; Sun Q.; Li H.; Li J. Elucidating the Interactions Between Heparin/Heparan Sulfate and SARS-CoV-2-Related Proteins—An Important Strategy for Developing Novel Therapeutics for the COVID-19 Pandemic. Front. Mol. Biosci. 2021, 490. 10.3389/fmolb.2020.628551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalra R. S.; Kandimalla R. Engaging the Spikes: Heparan Sulfate Facilitates SARS-CoV-2 Spike Protein Binding to ACE2 and Potentiates Viral Infection. Signal Transduct. Target. Ther. 2021, 6 (1), 39. 10.1038/s41392-021-00470-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan C. A. M.; Li Z.; te Lintelo E.; Bosch B. J.; Haijema B. J.; Rottier P. J. M. Murine Coronavirus with an Extended Host Range Uses Heparan Sulfate as an Entry Receptor. J. Virol. 2005, 79 (22), 14451 LP–14456. 10.1128/JVI.79.22.14451-14456.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandon R.; Sharp J. S.; Zhang F.; Pomin V. H.; Ashpole N. M.; Mitra D.; McCandless M. G.; Jin W.; Liu H.; Sharma P. Effective Inhibition of SARS-CoV-2 Entry by Heparin and Enoxaparin Derivatives. J. Virol. 2021, 10.1128/JVI.01987-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbison A. M.; Fogarty C. A.; Phung T. K.; Satheesan A.; Schulz B. L.; Fadda E. Fine-Tuning the Spike: Role of the Nature and Topology of the Glycan Shield in the Structure and Dynamics of SARS-CoV-2 S. Chem. Sci. 2021, 04 (1), 438036. 10.1039/D1SC04832E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paiardi G.; Richter S.; Rusnati M.; Wade R. C.; Oreste P.; Urbinati C. Three-fold mechanism of inhibition of SARS-CoV-2 infection by the interaction of the spike glycoprotein with heparin. arXiv.org 2021, 2103.07722v3. [Google Scholar]
- Schuurs Z. P.; Hammond E.; Elli S.; Rudd T. R.; Mycroft-West C. J.; Lima M. A.; Skidmore M. A.; Karlsson R.; Chen Y.-H.; Bagdonaite I.; et al. Evidence of a Putative Glycosaminoglycan Binding Site on the Glycosylated SARS-CoV-2 Spike Protein N-Terminal Domain. Comput. Struct. Biotechnol. J. 2021, 19, 2806–2818. 10.1016/j.csbj.2021.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan C. A. M.; Haijema B. J.; Schellen P.; Schreur P. W.; te Lintelo E.; Vennema H.; Rottier P. J. M. Cleavage of Group 1 Coronavirus Spike Proteins: How Furin Cleavage Is Traded Off against Heparan Sulfate Binding upon Cell Culture Adaptation. J. Virol. 2008, 82 (12), 6078 LP–6083. 10.1128/JVI.00074-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vankadari N. Structure of Furin Protease Binding to SARS-CoV-2 Spike Glycoprotein and Implications for Potential Targets and Virulence. J. Phys. Chem. Lett. 2020, 11 (16), 6655–6663. 10.1021/acs.jpclett.0c01698. [DOI] [PubMed] [Google Scholar]
- Mohammad A.; Alshawaf E.; Marafie S. K.; Abu-Farha M.; Abubaker J.; Al-Mulla F. Higher Binding Affinity of Furin for SARS-CoV-2 Spike (S) Protein D614G Mutant Could Be Associated with Higher SARS-CoV-2 Infectivity. Int. J. Infect. Dis. 2021, 103, 611–616. 10.1016/j.ijid.2020.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasik D.; Mulchandani A.; Yates M. V. A Heparin-Functionalized Carbon Nanotube-Based Affinity Biosensor for Dengue Virus. Biosens. Bioelectron. 2017, 91, 811–816. 10.1016/j.bios.2017.01.017. [DOI] [PubMed] [Google Scholar]
- Jelinek R.; Kolusheva S. Carbohydrate Biosensors. Chem. Rev. 2004, 104 (12), 5987–6016. 10.1021/cr0300284. [DOI] [PubMed] [Google Scholar]
- Griffith A. R.; Rogers C. J.; Miller G. M.; Abrol R.; Hsieh-Wilson L. C.; Goddard W. A. Predicting Glycosaminoglycan Surface Protein Interactions and Implications for Studying Axonal Growth. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (52), 13697 LP–13702. 10.1073/pnas.1715093115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B. H.; Kim S. H.; Ko Y.; Park J. C.; Ji S.; Gu M. B. The Sensitive Detection of ODAM by Using Sandwich-Type Biosensors with a Cognate Pair of Aptamers for the Early Diagnosis of Periodontal Disease. Biosens. Bioelectron. 2019, 126, 122–128. 10.1016/j.bios.2018.10.040. [DOI] [PubMed] [Google Scholar]
- Kim S. H.; Lee J.; Lee B. H.; Song C.-S.; Gu M. B. Specific Detection of Avian Influenza H5N2 Whole Virus Particles on Lateral Flow Strips Using a Pair of Sandwich-Type Aptamers. Biosens. Bioelectron. 2019, 134, 123–129. 10.1016/j.bios.2019.03.061. [DOI] [PubMed] [Google Scholar]
- Posthuma-Trumpie G. A.; Korf J.; van Amerongen A. Lateral Flow (Immuno)Assay: Its Strengths, Weaknesses, Opportunities and Threats. A Literature Survey. Anal. Bioanal. Chem. 2009, 393 (2), 569–582. 10.1007/s00216-008-2287-2. [DOI] [PubMed] [Google Scholar]
- Mak W. C.; Beni V.; Turner A. P. F. Lateral-Flow Technology: From Visual to Instrumental. TrAC, Trends Anal. Chem. 2016, 79, 297–305. 10.1016/j.trac.2015.10.017. [DOI] [Google Scholar]
- Ahmad Raston N. H.; Nguyen V.-T.; Gu M. B. A New Lateral Flow Strip Assay (LFSA) Using a Pair of Aptamers for the Detection of Vaspin. Biosens. Bioelectron. 2017, 93, 21–25. 10.1016/j.bios.2016.11.061. [DOI] [PubMed] [Google Scholar]
- Li J.; Jing L.; Song Y.; Zhang J.; Chen Q.; Wang B.; Xia X.; Han Q. Rapid Detection of Rongalite via a Sandwich Lateral Flow Strip Assay Using a Pair of Aptamers. Nanoscale Res. Lett. 2018, 13 (1), 296. 10.1186/s11671-018-2709-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koczula K. M.; Gallotta A. Lateral Flow Assays. Essays Biochem. 2016, 60 (1), 111–120. 10.1042/EBC20150012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanack K.; Messerschmidt K.; Listek M. Adv. Exp. Med. Biol. 2016, 917, 11–22. 10.1007/978-3-319-32805-8_2. [DOI] [PubMed] [Google Scholar]
- Mercer T. R.; Salit M. Testing at Scale during the COVID-19 Pandemic. Nat. Rev. Genet. 2021, 22, 415. 10.1038/s41576-021-00360-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mina M. J.; Peto T. E.; García-Fiñana M.; Semple M. G.; Buchan I. E. Clarifying the Evidence on SARS-CoV-2 Antigen Rapid Tests in Public Health Responses to COVID-19. Lancet 2021, 397 (10283), 1425–1427. 10.1016/S0140-6736(21)00425-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crozier A.; Rajan S.; Buchan I.; McKee M. Put to the Test: Use of Rapid Testing Technologies for Covid-19. BMJ. 2021, 372, n208. 10.1136/bmj.n208. [DOI] [PubMed] [Google Scholar]
- Bermejo-Jambrina M.; Eder J.; Kaptein T. M; Hamme J. L; Helgers L. C; Vlaming K. E; Brouwer P. J M; Nuenen A. C; Spaargaren M.; Bree G. J; Nijmeijer B. M; Kootstra N. A; Gils M. J; Sanders R. W; Geijtenbeek T. B H Infection and Transmission of SARS-CoV-2 Depend on Heparan Sulfate Proteoglycans. EMBO J. 2021, 40 (20), 2020.08.18.255810. 10.15252/embj.2020106765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittum J. E.; Sankaranarayanan N. V.; O’Hara C. P.; Desai U. R. On the Selectivity of Heparan Sulfate Recognition by SARS-CoV-2 Spike Glycoprotein. ACS Med. Chem. Lett. 2021, 12, 1710. 10.1021/acsmedchemlett.1c00343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao W.; Ma B.; Li Z.; Wang X.; Gao X.; Li Y.; Qin B.; Shang S.; Cui S.; Tan Z. Binding of the SARS-CoV-2 Spike Protein to Glycans. Sci. Bull. 2021, 66 (12), 1205–1214. 10.1016/j.scib.2021.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esko J. D.; Lindahl U. Molecular Diversity of Heparan Sulfate. J. Clin. Invest. 2001, 108 (2), 169–173. 10.1172/JCI13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D.; Esko J. D. Demystifying Heparan Sulfate–Protein Interactions. Annu. Rev. Biochem. 2014, 83 (1), 129–157. 10.1146/annurev-biochem-060713-035314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue J.; Jin W.; Yang H.; Faulkner J.; Song X.; Qiu H.; Teng M.; Azadi P.; Zhang F.; Linhardt R. J.; Wang L. Heparan Sulfate Facilitates Spike Protein-Mediated SARS-CoV-2 Host Cell Invasion and Contributes to Increased Infection of SARS-CoV-2 G614 Mutant and in Lung Cancer. Front. Mol. Biosci. 2021, 10.3389/fmolb.2021.649575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chautard E.; Fatoux-Ardore M.; Ballut L.; Thierry-Mieg N.; Ricard-Blum S. MatrixDB, the Extracellular Matrix Interaction Database. Nucleic Acids Res. 2011, 39 (Database), D235–D240. 10.1093/nar/gkq830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chautard E.; Ballut L.; Thierry-Mieg N.; Ricard-Blum S. MatrixDB, a Database Focused on Extracellular Protein–Protein and Protein–Carbohydrate Interactions. Bioinformatics 2009, 25 (5), 690–691. 10.1093/bioinformatics/btp025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Launay G.; Salza R.; Multedo D.; Thierry-Mieg N.; Ricard-Blum S. MatrixDB, the Extracellular Matrix Interaction Database: Updated Content, a New Navigator and Expanded Functionalities. Nucleic Acids Res. 2015, 43 (D1), D321–D327. 10.1093/nar/gku1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerc O.; Deniaud M.; Vallet S. D.; Naba A.; Rivet A.; Perez S.; Thierry-Mieg N.; Ricard-Blum S. MatrixDB: Integration of New Data with a Focus on Glycosaminoglycan Interactions. Nucleic Acids Res. 2019, 47 (D1), D376–D381. 10.1093/nar/gky1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.-J.; Lee J.; Patel D. S.; Ma H.; Lee H. S.; Jo S.; Im W. Glycan Reader Is Improved to Recognize Most Sugar Types and Chemical Modifications in the Protein Data Bank. Bioinformatics 2017, 33 (19), 3051–3057. 10.1093/bioinformatics/btx358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S.; Song K. C.; Desaire H.; MacKerell A. D. Jr.; Im W. Glycan Reader: Automated Sugar Identification and Simulation Preparation for Carbohydrates and Glycoproteins. J. Comput. Chem. 2011, 32 (14), 3135–3141. 10.1002/jcc.21886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guvench O.; Hatcher E.; Venable R. M.; Pastor R. W.; MacKerell A. D. CHARMM Additive All-Atom Force Field for Glycosidic Linkages between Hexopyranoses. J. Chem. Theory Comput. 2009, 5 (9), 2353–2370. 10.1021/ct900242e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S.; Kim T.; Iyer V. G.; Im W. CHARMM-GUI: A Web-Based Graphical User Interface for CHARMM. J. Comput. Chem. 2008, 29 (11), 1859–1865. 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- Cladera J.; Martin I.; O’Shea P. The Fusion Domain of HIV Gp41 Interacts Specifically with Heparan Sulfate on the T-Lymphocyte Cell Surface. EMBO J. 2001, 20 (1–2), 19–26. 10.1093/emboj/20.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell B. J.; Lortat-Jacob H. Human Immunodeficiency Virus and Heparan Sulfate: From Attachment to Entry Inhibition. Front. Immunol. 2013, 10.3389/fimmu.2013.00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrop H. A.; Rider C. C. Heparin and Its Derivatives Bind to HIV-1 Recombinant Envelope Glycoproteins, Rather than to Recombinant HIV-1 Receptor, CD4. Glycobiology 1998, 8 (2), 131–137. 10.1093/glycob/8.2.131. [DOI] [PubMed] [Google Scholar]
- Hansen J.; Baum A.; Pascal K. E.; Russo V.; Giordano S.; Wloga E.; Fulton B. O.; Yan Y.; Koon K.; Patel K.; et al. Studies in Humanized Mice and Convalescent Humans Yield a SARS-CoV-2 Antibody Cocktail. Science (Washington, DC, U. S.) 2020, 369 (6506), 1010–1014. 10.1126/science.abd0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R.; Zhang Y.; Li Y.; Xia L.; Guo Y.; Zhou Q. Structural Basis for the Recognition of SARS-CoV-2 by Full-Length Human ACE2. Science (Washington, DC, U. S.) 2020, 367 (6485), 1444–1448. 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan J.; Ge J.; Yu J.; Shan S.; Zhou H.; Fan S.; Zhang Q.; Shi X.; Wang Q.; Zhang L.; Wang X. Structure of the SARS-CoV-2 Spike Receptor-Binding Domain Bound to the ACE2 Receptor. Nature 2020, 581 (7807), 215–220. 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- Chi X.; Yan R.; Zhang J.; Zhang G.; Zhang Y.; Hao M.; Zhang Z.; Fan P.; Dong Y.; Yang Y.; et al. Neutralizing Human Antibody Binds to the N-Terminal Domain of the Spike Protein of SARS-CoV-2. Science (Washington, DC, U. S.) 2020, 369 (6504), 650–655. 10.1126/science.abc6952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrake A.; Rupley J. A. Environment and Exposure to Solvent of Protein Atoms. Lysozyme and Insulin. J. Mol. Biol. 1973, 79 (2), 351–371. 10.1016/0022-2836(73)90011-9. [DOI] [PubMed] [Google Scholar]
- Yuan M.; Wu N. C.; Zhu X.; Lee C.-C. D.; So R. T. Y.; Lv H.; Mok C. K. P.; Wilson I. A. A Highly Conserved Cryptic Epitope in the Receptor Binding Domains of SARS-CoV-2 and SARS-CoV. Science (80-.). 2020, 368 (6491), 630 LP–633. 10.1126/science.abb7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes C. O.; West A. P. Jr.; Huey-Tubman K. E.; Hoffmann M. A. G.; Sharaf N. G.; Hoffman P. R.; Koranda N.; Gristick H. B.; Gaebler C.; Muecksch F.; et al. Structures of Human Antibodies Bound to SARS-CoV-2 Spike Reveal Common Epitopes and Recurrent Features of Antibodies. Cell 2020, 182 (4), 828–842.e16. 10.1016/j.cell.2020.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M.; Liu H.; Wu N. C.; Wilson I. A. Recognition of the SARS-CoV-2 Receptor Binding Domain by Neutralizing Antibodies. Biochem. Biophys. Res. Commun. 2021, 538, 192–203. 10.1016/j.bbrc.2020.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurrus E.; Engel D.; Star K.; Monson K.; Brandi J.; Felberg L. E.; Brookes D. H.; Wilson L.; Chen J.; Liles K.; et al. Improvements to the APBS Biomolecular Solvation Software Suite. Protein Sci. 2018, 27 (1), 112–128. 10.1002/pro.3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker N. A.; Sept D.; Joseph S.; Holst M. J.; McCammon J. A. Electrostatics of Nanosystems: Application to Microtubules and the Ribosome. Proc. Natl. Acad. Sci. U. S. A. 2001, 98 (18), 10037 LP–10041. 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst M.; Saied F. Multigrid Solution of the Poisson—Boltzmann Equation. J. Comput. Chem. 1993, 14 (1), 105–113. 10.1002/jcc.540140114. [DOI] [Google Scholar]
- Holst M. J.; Saied F. Numerical Solution of the Nonlinear Poisson–Boltzmann Equation: Developing More Robust and Efficient Methods. J. Comput. Chem. 1995, 16 (3), 337–364. 10.1002/jcc.540160308. [DOI] [Google Scholar]
- Gasbarri M.; V’kovski P.; Torriani G.; Thiel V.; Stellacci F.; Tapparel C.; Cagno V. SARS-CoV-2 Inhibition by Sulfonated Compounds. Microorganisms 2020, 8, 1894. 10.3390/microorganisms8121894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi N. S.; Mancera R. L. Free Energy Calculations of Glycosaminoglycan–Protein Interactions. Glycobiology 2009, 19 (10), 1103–1115. 10.1093/glycob/cwp101. [DOI] [PubMed] [Google Scholar]
- Sarkar A.; Yu W.; Desai U. R.; MacKerell A. D.; Mosier P. D. Estimating Glycosaminoglycan–Protein Interaction Affinity: Water Dominates the Specific Antithrombin–Heparin Interaction. Glycobiology 2016, 26 (10), 1041–1047. 10.1093/glycob/cww073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer J.; Sager C. P.; Ernst B. Hydroxyl Groups in Synthetic and Natural-Product-Derived Therapeutics: A Perspective on a Common Functional Group. J. Med. Chem. 2019, 62 (20), 8915–8930. 10.1021/acs.jmedchem.9b00179. [DOI] [PubMed] [Google Scholar]
- Přistoupil T. I.; Kramlová M.; Štěrbíková J. On the Mechanism of Adsorption of Proteins to Nitrocellulose in Membrane Chromatography. J. Chromatogr. A 1969, 42, 367–375. 10.1016/S0021-9673(01)80636-1. [DOI] [PubMed] [Google Scholar]
- Sarrazin S.; Lamanna W. C.; Esko J. D. Heparan Sulfate Proteoglycans. Cold Spring Harbor Perspect. Biol. 2011, 3 (7), a004952–a004952. 10.1101/cshperspect.a004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross G. M. S.; Filippini D.; Nielen M. W. F.; Salentijn G. I. Unraveling the Hook Effect: A Comprehensive Study of High Antigen Concentration Effects in Sandwich Lateral Flow Immunoassays. Anal. Chem. 2020, 92 (23), 15587–15595. 10.1021/acs.analchem.0c03740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceron J.; Lamy E.; Martinez-Subiela S.; Lopez-Jornet P.; Capela-Silva F.; Eckersall P.; Tvarijonaviciute A. Use of Saliva for Diagnosis and Monitoring the SARS-CoV-2: A General Perspective. J. Clin. Med. 2020, 9 (5), 1491. 10.3390/jcm9051491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie A. L.; Fournier J.; Casanovas-Massana A.; Campbell M.; Tokuyama M.; Vijayakumar P.; Warren J. L.; Geng B.; Muenker M. C.; Moore A. J.; Vogels C. B. F.; et al. Saliva or Nasopharyngeal Swab Specimens for Detection of SARS-CoV-2. N. Engl. J. Med. 2020, 383 (13), 1283–1286. 10.1056/NEJMc2016359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D.; Wu C.. Saliva Glycoproteins Bind to Spike Protein of SARS-CoV-2. Preprints, 2020. 10.20944/preprints202005.0192.v1. [DOI]
- Corum J.; Zimmer C.. Coronavirus Variants and Mutations. https://www.nytimes.com/interactive/2021/health/coronavirus-variant-tracker.html (accessed May 25, 2021).
- World Health Organization . Tracking SARS-CoV-2 variants. https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed Aug 8, 2021).
- McCallum M.; Walls A. C.; Sprouse K. R.; Bowen J. E.; Rosen L.; Dang H. V; DeMarco A.; Franko N.; Tilles S. W.; Logue J.; et al. Molecular Basis of Immune Evasion by the Delta and Kappa SARS-CoV-2 Variants. Science 2021, 0, eabl8506. 10.1126/science.abl8506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobeil S. M.-C.; Janowska K.; McDowell S.; Mansouri K.; Parks R.; Stalls V.; Kopp M. F.; Manne K.; Li D.; Wiehe K.; Saunders K. O.; Edwards R. J.; Korber B.; Haynes B. F.; Henderson R.; Acharya P. Effect of Natural Mutations of SARS-CoV-2 on Spike Structure, Conformation, and Antigenicity. Science 2021, 373 (6555), abi6226. 10.1126/science.abi6226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiliaev N.; Lukash T.; Palchevska O.; Crossman D. K.; Green T. J.; Crowley M. R.; Frolova E. I.; Frolov I. Natural and Recombinant SARS-CoV-2 Isolates Rapidly Evolve In Vitro to Higher Infectivity through More Efficient Binding to Heparan Sulfate and Reduced S1/S2 Cleavage. J. Virol. 2021, 95 (21), 2021.06.28.450274. 10.1128/JVI.01357-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parolo C.; Sena-Torralba A.; Bergua J. F.; Calucho E.; Fuentes-Chust C.; Hu L.; Rivas L.; Álvarez-Diduk R.; Nguyen E. P.; Cinti S.; et al. A. Tutorial: Design and Fabrication of Nanoparticle-Based Lateral-Flow Immunoassays. Nat. Protoc. 2020, 15 (12), 3788–3816. 10.1038/s41596-020-0357-x. [DOI] [PubMed] [Google Scholar]
- Yetisen A. K.; Akram M. S.; Lowe C. R. Paper-Based Microfluidic Point-of-Care Diagnostic Devices. Lab Chip 2013, 13 (12), 2210. 10.1039/c3lc50169h. [DOI] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30 (16), 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31 (2), 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa F.; Varoquaux G.; Gramfort A.; Michel V.; Thirion B.; Grisel O.; Blondel M.; Prettenhofer P.; Weiss R.; Dubourg V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12 (85), 2825–2830. [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14 (1), 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Walls A. C.; Park Y.-J.; Tortorici M. A.; Wall A.; McGuire A. T.; Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181 (2), 281–292.e6. 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangaru S.; Ozorowski G.; Turner H. L.; Antanasijevic A.; Huang D.; Wang X.; Torres J. L.; Diedrich J. K.; Tian J.-H.; Portnoff A. D.; et al. Structural Analysis of Full-Length SARS-CoV-2 Spike Protein from an Advanced Vaccine Candidate. Science (80-.). 2020, 370 (6520), 1089 LP–1094. 10.1126/science.abe1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson M. H. M.; Søndergaard C. R.; Rostkowski M.; Jensen J. H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical PKa Predictions. J. Chem. Theory Comput. 2011, 7 (2), 525–537. 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- Søndergaard C. R.; Olsson M. H. M.; Rostkowski M.; Jensen J. H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of PKa Values. J. Chem. Theory Comput. 2011, 7 (7), 2284–2295. 10.1021/ct200133y. [DOI] [PubMed] [Google Scholar]
- Madhavi Sastry G.; Adzhigirey M.; Day T.; Annabhimoju R.; Sherman W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput.-Aided Mol. Des. 2013, 27 (3), 221–234. 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kalé L.; Schulten K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26 (16), 1781–1802. 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J. C.; Hardy D. J.; Maia J. D. C.; Stone J. E.; Ribeiro J. V.; Bernardi R. C.; Buch R.; Fiorin G.; Hénin J.; Jiang W.; et al. Scalable Molecular Dynamics on CPU and GPU Architectures with NAMD. J. Chem. Phys. 2020, 153 (4), 44130. 10.1063/5.0014475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant O. C.; Montgomery D.; Ito K.; Woods R. J. Analysis of the SARS-CoV-2 Spike Protein Glycan Shield Reveals Implications for Immune Recognition. Sci. Rep. 2020, 10 (1), 14991. 10.1038/s41598-020-71748-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farid R.; Day T.; Friesner R. A.; Pearlstein R. A. New Insights about HERG Blockade Obtained from Protein Modeling, Potential Energy Mapping, and Docking Studies. Bioorg. Med. Chem. 2006, 14 (9), 3160–3173. 10.1016/j.bmc.2005.12.032. [DOI] [PubMed] [Google Scholar]
- Sherman W.; Beard H. S.; Farid R. Use of an Induced Fit Receptor Structure in Virtual Screening. Chem. Biol. Drug Des. 2006, 67 (1), 83–84. 10.1111/j.1747-0285.2005.00327.x. [DOI] [PubMed] [Google Scholar]
- Sherman W.; Day T.; Jacobson M. P.; Friesner R. A.; Farid R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2006, 49 (2), 534–553. 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- van Meer G.; Voelker D. R.; Feigenson G. W. Membrane Lipids: Where They Are and How They Behave. Nat. Rev. Mol. Cell Biol. 2008, 9 (2), 112–124. 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79 (2), 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Huang J.; Rauscher S.; Nawrocki G.; Ran T.; Feig M.; de Groot B. L.; Grubmüller H.; MacKerell A. D. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14 (1), 71–73. 10.1038/nmeth.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; MacKerell A. D. CHARMM36 All-Atom Additive Protein Force Field: Validation Based on Comparison to NMR Data. J. Comput. Chem. 2013, 34 (25), 2135–2145. 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaro R. E.; Mulholland A. J. Biomolecular Simulations in the Time of COVID-19, and After. Comput. Sci. Eng. 2020, 22 (6), 30–36. 10.1109/MCSE.2020.3024155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland A. J.; Amaro R. E. COVID19 - Computational Chemists Meet the Moment. J. Chem. Inf. Model. 2020, 60 (12), 5724–5726. 10.1021/acs.jcim.0c01395. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.