

Abstract
Enzymes from secondary metabolic pathways possess broad potential for the selective synthesis of complex bioactive molecules. However, the practical application of these enzymes for organic synthesis is dependent on the development of efficient, economical, operationally-simple and well-characterized systems for preparative scale reactions. We sought to bridge this knowledge gap for the selective biocatalytic synthesis of β-hydroxy-α-amino acids, which are important synthetic building blocks. To achieve this goal, we demonstrated the ability of ObiH, an l-threonine transaldolase, to achieve selective milligram-scale synthesis of a diverse array of nonstandard amino acids (nsAAs) using a scalable whole cell platform. We show how the initial selectivity of the catalyst is high and how the diastereomeric ratio of products decreases at high conversion due to product re-entry into the catalytic cycle. ObiH-catalyzed reactions with a variety of aromatic, aliphatic and heterocyclic aldehydes selectively generated a panel of β-hydroxy-α-amino acids possessing broad functional-group diversity. Furthermore, we demonstrated that ObiH-generated β-hydroxy-α-amino acids could be modified through additional transformations to access important motifs, such as β-chloro-α-amino acids and substituted α-keto acids.
Keywords: Biocatalysis, C─C bond formation, threonine transaldolase, PLP
Introduction
Nature has evolved powerful and effective approaches to selectively generate complex natural products from simple metabolites.[1] Biosynthetic enzymes often catalyze transformations that are challenging to achieve using small molecule methods and can enable efficient access to valuable molecules.[2] In comparison to traditional synthetic transformations, the three-dimensional architecture of enzyme active sites enables exquisite control over the positioning of reactants, often leading to improved chemo-, site- and stereoselectivity profiles. As a result, a wide variety of new biocatalytic methods have been developed in recent years which seek to resolve selectivity challenges in complex molecule synthesis.[3] Functional group interconversions are the principle class of organic transformations for which high quality biocatalytic routes have been successfully developed.[3] In contrast, the production of novel biocatalytic methods for stereocontrolled C─C bond formation remains a promising, but underexplored area, with many exciting new advances underway.[4,5] Such biocatalytic methods possess unique potential for streamlining the construction of complex molecular building blocks, natural products, and pharmaceuticals.[4,6] We envisioned that one such important class of molecules, β-hydroxy-α-amino acids, could be selectively generated in an efficient and highly scalable fashion by designing and employing a biocatalytic reaction platform.
β-Hydroxy-α-amino acids are a class of molecules commonly found in polypeptide natural products and pharmaceuticals (Figure 1A).[1] In particular, these molecules are prominent intermediates in the biosynthesis of cyclic peptide and small molecule antibiotics, such as the compounds hypeptin (1), katanosin B (2), and chloramphenicol (4), as well as other bioactive molecules.[7-9] A variety of organocatalytic methods have been developed to install the β-hydroxyl group, often via the aldol addition of a glycyl Cα-nucleophile into various aldehydes.[10-15] These methods are effective at generating β-hydroxy-α-amino acids, but often require orthogonal, multi-step protecting group strategies to produce the desired product which can increase costs and reduce overall yields. Consequently, researchers have sought to develop efficient biocatalytic approaches to β-hydroxy-α-amino acid synthesis.
Figure 1.

A. Bioactive natural products and pharmaceuticals containing β-hydroxy-α-amino moieties. B. ObiH native reaction in the context of obafluorin biosynthesis. C. Previous substrate scope studies using l-threonine transaldolases (lTTAs). D. This work: highly scalable and efficient generation of diverse β-hydroxy-α-amino acids.
In Nature, the β-hydroxy functional group is often installed on a pre-assembled amino acid through site-selective oxidation by cytochromes P450 or α-ketoglutarate dependent non-heme iron enzymes.[16-19] However, the ability to assemble the side chain while simultaneously setting the stereochemistry of the β-hydroxy group offers a more versatile, complementary approach for the preparative scale synthesis of structurally diverse nsAAs. Researchers have therefore been drawn to the pyridoxal phosphate (PLP)-dependent threonine aldolases (TAs) and serine hydroxymethyl transferases (SHMTs) for synthesis of β-hydroxy-α-amino acids.[20]These enzymes natively catalyze the retro-aldol cleavage of l-threonine (Thr) and l-serine (Ser), respectively, generating glycine for use in primary metabolism.[21] By running these transformations in reverse with very high concentrations of glycine, TAs and SHMTs have been leveraged for the synthesis of β-hydroxy-α-amino acids.[22-27] However, most native TAs catalyze aldol reactions with low stereoselectivity at Cβ, leading to poor diastereoselectivity profiles.[25,28,29] These challenges can be overcome in exceptional cases, either through intensive directed evolution or through the use of diastereoselective crystallization, but these routes are limited to one or a handful of substrates.[28,30]
An alternative and appealing enzymatic strategy for the synthesis of β-hydroxy-α-amino acid draws directly from Nature’s own approach. Rather than contend with the endergonic challenge of reversing the TA reaction, there are a set of mechanistically-related l-threonine transaldolase (LTTA) enzymes that catalyze removal of the Thr sidechain as acetaldehyde and its replacement with a new aldehyde (5) to generate a new β-hydroxy-α-amino acid (Figure 1B, Figure S2). This reaction does not release Gly as an intermediate and is approximately net-thermoneutral. High yields are enabled by coupling to downstream metabolism.[31,32] Notable among LTTA enzymes is ObiH, which catalyzes the diastereoselective synthesis of β-hydroxy-α-amino acid 7 in the biosynthesis of the aminoacyl tRNA synthetase inhibitor obafluorin (Figure 1B, 9).[31-33] Previous efforts to utilize LTTAs for synthetic purposes focused on optimization of analytical scale reactions with electron-deficient aromatic aldehydes (Figure 1C).[33,34] Protein engineering has also been deployed to increase reactivity with p-methylsulfonylbenzaldehyde en route to the antibiotic thiamphenicol.[35] Mirroring Nature’s approach, preparative scale LTTA transformations with this substrate were driven to high yield by coupling to a downstream enzymatic reduction of acetaldehyde.[34] These promising results prompted deeper study to understand the difference in reactivity between TA and LTTA enzymes, which we recently addressed through a detailed structural and mechanistic study of ObiH.[36] Our results showed that the nucleophilic intermediate, a PLP bound glycyl quinonoid, E(QGly) 15, is formed by retro-aldol cleavage of Thr. Further spectroscopic analysis showed this intermediate is kinetically shielded from protonation. Monitoring persistence of the E(QGly) in the presence of small aliphatic aldehydes indicated that ObiH has reactivity toward these electrophiles, which we confirmed via synthesis of β-hydroxyl-l-leucine using purified ObiH.[36] Translating these exciting advances in enzymology to a practical biocatalytic route remains, however, a separate challenge.
For facile adoption by the synthetic community, an LTTA route to β-hydroxy-α-amino acid must be deployed in the most operationally-simple fashion and the limitations of the aldehyde scope established. It has been previously observed that the diastereomeric ratio (dr) of the products of LTTA reactions decrease through an unconfirmed mechanism, confounding the reproducibility of stereochemical outcomes.[37] Furthermore, biocatalytic methods often yield unprotected, hydrophilic products and demonstration of standardized methods for their isolation and downstream manipulation further increase the attractiveness of an enzymatic route. Here, we tackle these challenges and demonstrate how ObiH be utilized as an effective catalyst for the synthesis of structurally diverse β-hydroxy-α-amino acid from inexpensive and readily available starting materials (Figure 1D).
Results and Discussion
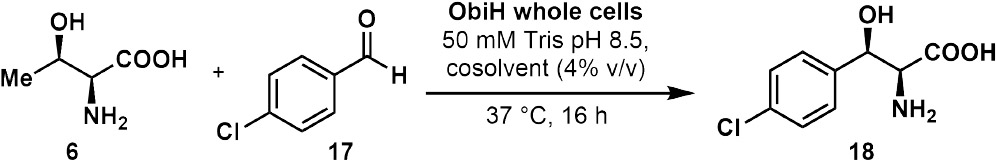
We sought to develop a scalable and operationally-simple method for the selective synthesis of a variety of β-hydroxy-α-amino acids. We reasoned that using a well-studied LTTA as the biocatalyst would provide an ideal starting point for method development. ObiH is a structurally and mechanistically characterized enzyme that has been shown to possess high transaldolase activity with several electron-poor aldehydes.[31,32,36] We first attempted reactions using wet, whole E. coli cells that contained heterologously-expressed N-His-ObiH. Biocatalytic transformations using whole cells have been demonstrated as an economical and efficient route to access valuable molecules without the need for time and resourceintensive enzyme purification procedures.[38-40] Initial optimization reactions with 4-chlorobenzaldehyde (17) revealed that ObiH was catalytically active in the whole cell format (Table 1, entry 1), providing product 18 with 88% conversion, as measured by UPLC-MS. We screened alternative cosolvent compositions at a whole cell concentration of 5 mg/mL (0.5 wt %). In general, there was only a modest impact on reactivity. MeOH and toluene (4% v/v) each had no significant effect on activity when compared to reactions lacking cosolvent (Table 1, entries 1-3), whereas MeCN was detrimental to enzyme activity. Further optimization of wet whole cell loading was performed using MeOH as a cosolvent (Table 1, entries 5-7). Increasing whole cell loadings as high as 4% (20 mg/mL) did not result in appreciable increase in conversions. Anticipating that alternative electrophiles may not react as efficiently with ObiH, we elected to use 1–2% wet whole cells and 4% (v/v) MeOH (Table 1, entry 6) for preparative scale reactions. Cell loadings of this magnitude are sufficiently low for process scale reactions and we attribute this excellent reactivity of ObiH to its high soluble expression in E. coli, boasting >150 mg protein per L culture.
Table 1.
Analytical scale optimization of ObiH-catalyzed aldol reactions with 4-chlorobenzaldehyde (17) Conditions: 10 mM aldehyde, 100 mM l-Thr, 50 mM Tris pH 8.5 and 0.5–4% ObiH wet whole cells, with 4% (v/v) co-solvent.. Conversion values are reported as the average of duplicate experiments.
| |||
|---|---|---|---|
| entry | whole cell concentration (%) | cosolvent | percent conversion |
| 1 | 0.5% (5 mg/mL) | MeOH | 88% |
| 2 | 0.5% (5 mg/mL) | no cosolvent | 86% |
| 3 | 0.5% (5 mg/mL) | toluene | 82% |
| 4 | 0.5% (5 mg/mL) | MeCN | 69% |
| 5 | 1.0% (10 mg/mL) | MeOH | 90% |
| 6 | 2.0% (20 mg/mL) | MeOH | 91% |
| 7 | 4.0% (40 mg/mL) | MeOH | 88% |
With these conditions in hand, we were motivated to explore the limits of ObiH-catalyzed reactions by probing the native substrate promiscuity of the catalyst. Previous efforts with an ObiH homolog focused on the analytical scale synthesis of phenylserine derivatives, with a single example of product isolation.[34] To gain a more comprehensive view of ObiH reactivity, we chose to perform analytical and preparative scale reactions and compare the relative yields and observed selectivities. Unprotected β-hydroxy-α-amino acid with halogen functionalities were swiftly generated in high yields on analytical scale, but with low to moderate diastereoselectivity after functionalization with Marfey’s reagent (Figure 2). In comparison, preparative scale reactions with the same substrates delivered products with enhanced diastereoselectivity, albeit with reduced yields. These data suggest that ObiH is subject to a selectivity phenomenon observed with other LTTAs where a precipitous drop in the diastereomeric ratio of the products occurs when reactions are run to high conversions, due to erosion of the stereopurity at the β-position.[37,41] While there is a clear tradeoff between conversion and diastereoselectivity for LTTA-catalyzed reactions, we have demonstrated that a balance can be struck to deliver stereo-enriched products with a moderate sacrifice in isolated yields. Notably, the enantioselectivity of ObiH-catalyzed reactions is high (>99% ee for all products). Phenylserine analogs generated under these conditions include p-chloro-phenylserine (18), p-bromo-phenylserine (19) and m-bromo-phenylserine (20). To unambiguously assign the absolute configuration of ObiH-generated product 18, the compound was recrystallized and characterized by small-molecule X-ray diffraction. This experiment confirmed that the major product of ObiH-catalyzed aldol addition was the (2S, 3R), ‘threo’ isomer, which matched the observed stereochemistry of the native product 9 in obafluorin biosynthesis.[32,42]
Figure 2.

Analytical and preparative-scale synthesis of β-hydroxy-α-amino acids by ObiH. Reactions were performed using 20 mM aldehyde, 100 mM l-Thr, 50 mM Tris pH 8.5 and 1–2% ObiH wet whole cells, with 4% (v/v) MeOH as co-solvent. Preparative scale reactions were incubated at 37 °C for 18 h before quenching with 1 volume equivalent of MeCN, followed by freeze-thaw and centrifugation to remove cell debris. Purification was achieved using a Biotage purification system via reverse-phase chromatography. Yields are reported as isolated product mass after lyophilization. 1H NMR hydration analysis was used to correct yield values for excess water. Analytical scale product yields determined by UPLC-PDA-MS following derivatization with Marfey’s reagent.
Reactions with p-nitrobenzaldehyde on analytical and preparative scale indicated good yields, but low diastereoselectivity, delivering p-nitro-phenylserine (21). This molecule is an intermediate in the synthesis of the antibiotic chloramphenicol (4), as well as a precursor to other nonstandard amino acids, such as p-amino-phenylserine.
We considered a reaction with 4-fluoro-3-nitrobenzaldehyde, (see Figure S1) which would yield an intermediate in the recent total synthesis of vancomycin, a peptide antibiotic.[43] However, the highly electrophilic aldehyde starting material condensed with Thr, precluding effective entry into the active site. A recent directed evolution campaign generated TAs that can catalyze a selective aldol reaction with electron deficient benzaldehydes, and we were therefore less compelled to study this reaction further.[29] Instead, to further probe the limits of the ObiH active site, we sought to challenge the enzyme with sterically bulky aromatic aldehydes to generate p-napth-1-ylserine (22) and biphenylserine (23) (Figure 2). Preparative scale yields for these products were reduced relative to reactions with less bulky benzaldehydes and may be limited by their poor solubility in aqueous conditions. Nevertheless, these reactions show that bulky β-hydroxy amino acids can be isolated with high diastereselectivity, delivering these challenging products in a scalable manner.
In addition to the variety of phenylserine analogs that were produced using this method, we sought to generate β-hydroxy-α-amino acids which could be used directly in the synthesis of natural products and pharmaceuticals. For example, β-hydroxytyrosine is an nsAA found in several cyclic peptide natural products, including the antibiotic hypeptin (Figure 1, 1).[7] We initially attempted to directly generate β-hydroxytyrosine via an ObiH-catalyzed aldol reaction with 4-hydroxybenzaldehyde, but observed no product formation. This lack of reactivity has been mirrored in other TA systems, but no strategy to access these products has been put forth.[29] We reasoned that, because the pKa of the phenol is low relative to the pH of the reaction, the predominant form in solution is the highly electron rich phenolate. Consequently, this molecule is an intrinsically inert substrate for biocatalytic aldol reactions. We anticipated that protection of the phenolic group on this substrate with an electron-withdrawing substituent would prevent ionization and increase the electrophilicity of the benzaldehyde, enabling a productive catalytic reaction. This hypothesis was confirmed in reactions with pivaloyl-protected 4-hydroxybenzaldehyde, generating the protected β-hydroxytyrosine 24 in 25% yield. This protection strategy was also successful with a trifluoromethylsulfonyl protected aldehyde, delivering the triflated β-hydroxytyrosine 25 in 22% yield. Through this simple substrate modification procedure, we generated a pair of aldehydes that could undergo productive catalysis with ObiH, enabling efficient and selective access to protected β-hydroxytyrosine. Furthermore, the protection as a triflate provides a useful functional handle compatible for downstream diversification through cross-coupling reactions.[44]
Heterocyclic amino acids possess broad potential to be used as synthetic building blocks in the production of pharmaceuticals.[45] Notably, access to heterocyclic β-hydroxy-α-amino acid has also been a challenge for TA catalysis and we therefore tested ObiH for reactivity with heterocyclic aldehydes.[46] These transformations proceeded smoothly, delivering pyridine-2-yl-serine (27) in good yield (59%) and with moderate diastereoselectivity (10:1 dr), as well as 3-thienylserine (26) in diminished yield (23%). We further envisioned that ObiH could be used to generate β-hydroxy-tryptophan through reactions with indole-3-carboxaldehyde (Figure S1). However, no amino acid product was observed in these reactions, potentially due to the electron-rich nature of the aldehyde. Reactions with imidazole-4-carboxyaldehyde gave rise to a prominent peak in the UPLC-MS indicative of β-hydroxy-histidine (28). However, the highly polar nature of this amino acid hindered its isolation. Nevertheless, Marfey’s analysis indicates that ObiH can generate 28 in 27% yield based on UPLC analysis with 9:1 diastereoselectivity (Figure 2).
Following successful preparative-scale synthesis of diverse aromatic and heterocyclic amino acids using ObiH, we were inspired to explore the abilities of the enzyme to catalyze reactions with aliphatic aldehydes. Preparative scale reactions with branched-chain aliphatic aldehydes proceeded smoothly, with excellent diastereomeric ratios (>20:1). As with all ObiH reactions performed in this study, we again observed reduced diastereoselectivity in higher yielding analytical scale reactions. Nevertheless, we isolated β-hydroxy-cyclopentyl product 29 in 46% yield. Productive catalysis was also observed with isobutyraldehyde, delivering β-hydroxyleucine (31, 42% yield) and we obtained β-hydroxyhomoleucine (32) in 55% yield. The straight-chain hexanyl product 30 was also generated with high diastereoselectivity (>20:1), albeit with a reduced yield of 22%, indicating that ObiH can undergo productive catalysis with sterically bulky, unstrained aldehydes. However, conversion was not observed in reactions with larger straight-chain aldehyde dodecanal, suggesting that there are practical limitations in substrate size for ObiH-catalyzed reactions. The success of ObiH in generating a variety of aliphatic β-hydroxy-α-amino acid is a significant advance, as biocatalytic access to these motifs has generally been limited to a few examples of threonine aldolases with low to moderate diastereoselectivities.[47,48] While biosynthetic strategies which involve late stage selective hydroxylation have found success in generating aliphatic β-hydroxy-α-amino acid, this approach is highly substrate specific and precludes a generalized route to simultaneously install a diverse variety of side chains and the β-hydroxyl group. [18,19] ObiH-catalyzed aldol reactions therefore offer a streamlined enzymatic route to access diverse β-hydroxy-α-amino acid.
We last sought to challenge ObiH with a large, fluorescent aldehyde derived from the valuable probe molecule BODIPY.[49] An initial whole cell ObiH reaction was performed on milligram scale to generate BODIPY-containing β-hydroxy-α-amino acid (Figure 3). Aldehyde 33 underwent productive catalysis with ObiH, generating product 34 in poor yield (~1%). We improved the yields by using purified ObiH as catalyst and by the addition of 10% DMSO as co-solvent to increase the solubility of 33. These changes improved the yield to 7% with high diastereoselectivity (>20:1). While protein engineering would be needed to further increase the reactivity of ObiH with this aldehyde, such efforts are straightforward once initial activity is demonstrated, which we provide here. Fluorescence spectra showed that product 34 exhibits characteristic fluorescence after excitation at 501 nm and emission at 517 nm (Figure 3). We anticipate that this amino acid could be a valuable tool in chemical biology applications. This reaction further demonstrates the potential of ObiH to react with a range of aldehydes to generate functional amino acid products. Despite some limitations in its native reactivity, we used ObiH to generate numerous benzylic, heterocyclic and aliphatic β-hydroxy amino acids on preparative scale. These efforts demonstrate the native promiscuity of ObiH toward a variety of inexpensive aldehydes, selectively producing important synthetic precursors in one step. Additionally, this biocatalytic approach enables the use of Thr as an inexpensive substrate without any pre-functionalization, which is a considerable advantage over previous synthetic approaches. However, observation of an inverse correlation between yield and diastereoselectivities in these transformations spurred further mechanistic questions about this common phenomenon in LTTAs.
Figure 3.

A. ObiH-catalyzed synthesis of BODIPY-containing β-hydroxy-α-amino acid 34. Reactions were performed using 20 mM aldehyde, 125 mM L-Thr, 50 mM Tris pH 8.5 and 40 μM purified ObiH with 10% (v/v) DMSO as co-solvent. Reactions were incubated at 37 °C for 20 h before quenching with 1 volume equivalent of MeCN and centrifugation to remove protein debris. Purification was achieved using a Biotage purification system via reverse-phase chromatography. Yields are reported as isolated product mass after lyophilization. 1H NMR hydration analysis was used to correct yield values for excess water. B. Absorbance and emission spectra of 20 μM amino acid 34 dissolved in MeOH. C. 20 μM of amino acid 34 in MeOH under UV light.
To characterize catalyst behavior throughout the reaction, we measured a time course of the ObiH reaction with 4-chlorobenzaldehyde. This reaction yielded two well-resolved diastereomers, enabling direct comparison (via UPLC-MS) of the observed yield of product 18 and diastereomeric ratios for each timepoint. At early timepoints, the diastereomeric excess was high, indicating that ObiH exhibits superb diastereoselectivity. Analysis of later time points showed a notable reduction in the diasteromeric excess of product 18 as yield increased beyond 50% (Figure 4A). Based on our previous studies of the mechanism of ObiH,[36] we hypothesized that this drop in the diastereomeric excess is caused by the reversibility of the transaldolase reaction and product reentry into the catalytic cycle.
Figure 4.

A. Plot of diastereoselectivity of ObiH-catalyzed reaction with 4-chlorobenzaldehyde versus yield at varying timepoints. B. UV-visible spectrum following titration of ObiH with 4-chloro-phenylserine (18) demonstrating glycyl quinonoid (15) formation (λmax = 494 nm). C. ObiH-catalyzed racemization of diastereomerically-enriched 4-chloro-phenylserine (18). D. ObiH active site model with 4-chloro-phenylserine bound to PLP as the external aldimine (EAex). The active site is at the dimer interface and individual monomers are colored in light and dark grey. Active site residues are shown as sticks. The erythro isomer is colored salmon and the threo isomer is colored cyan. Hydrogen bonds are shown as black dashes.
To demonstrate that product can reenter the catalytic cycle and generate glycyl-quinonoid intermediate 15, we performed a UV-vis experiment (Figure 4B) in which ObiH was mixed with p-chloro-phenylserine (18). Following the addition of 5 mM 18, we observed a shift from the internal aldimine:PLP signal (405 nm) to a new peak at 494 nm, corresponding to stabilized glycyl-quinonoid intermediate 15.[36] This same intermediate was also observed upon titration of ObiH with native transaldolase substrate Thr, suggesting that the 4-chloro-phenylserine product can indeed reenter the catalytic cycle through retro-aldol cleavage.[36] For direct evidence of product reentry leading to diastereomeric erosion, we monitored an ObiH reaction with p-chloro-phenylserine under turnover conditions in the presence of 4-chlorobenzaldehyde (17) and observed a slow decrease in diastereomeric excess over time, beginning with a de value of 82% and ending with a de of 64% after 18 h (Figure 4C). These experiments confirm that ObiH product 18 can indeed reenter the catalytic cycle to produce an on-cycle quinonoid intermediate and that this behavior leads directly to reduced d.e. for ObiH products. These data are consistent with the observation that ObiH reactions which exhibit high conversion also exhibit reduced d.e. for all substrates tested.
Based on these observations, we rationalized that product reentry leads to scrambling at the β-position through iterative cycles of retro-aldol product cleavage, followed by the forward aldol reaction with 4-chlorobenzaldehyde. Because the major isomer is at a higher concentration at later time points, it will preferentially re-enter the active site and be broken down, only to be re-formed as a mixture of threo and erythro isomers. In this way, the kinetically disfavored erythro isomer slowly accumulates over the course of the reaction and the diastereomeric ratio shifts towards an equilibrium ratio. Notably, the stereochemistry at Cα is maintained within the limits of detection during all the experiments described here.
To probe the structural basis for the diastereoselectivity of ObiH, we turned to the recently described structure at 1.66 Å (PDB ID: 7K34).[36] This structure is of the internal aldimine state of the enzyme and, despite extensive efforts, we were unable to determine a substrate-bound structure under these conditions. We previously turned to molecular dynamics, which provided a plausible structure of ObiH with Thr bound as the external aldimine.[36] Here, we considered how a β-arene would fit into the active site and found that it was a highly constricted environment. Only a single staggered rotamer could be formed without a significant steric clash with protein backbone (Figure 4D). While it is possible that some extensive conformational rearrangement occurs upon substrate binding, these areas of contact were stable under the conditions of our previous molecular dynamics simulations. We therefore hypothesize that the bulky substrates described here bind in a single, common orientation extending towards Trp68, which is found on a highly flexible loop. In this model the preferred, on-pathway, mode of binding delivers the si-face of the electrophile to the E(QGly) nucleophile and aldol addition gives rise to the threo product.
We also sought to further showcase the synthetic utility of ObiH-generated products through downstream functionalization reactions to access important chemical building blocks. For example, β-chloroleucine is a nsAA found in the aerugenosins (Figure 5B, see 39 and 40), a class of peptide natural products which act as protease inhibitors.[50] Following ObiH-catalyzed generation of threo-β-hydroxyleucine (36), we subjected the purified material to conditions for β-chlorination. Amino acid 36 was dissolved in neat thionyl chloride (10 equiv) and heated to 60 °C overnight. Following a quench to hydrolyze the resulting acid chloride material, erythro-β-chloroleucine (37) was isolated in a 42% yield and high diastereoselectivity. This synthesis leverages the selectivity of ObiH in asymmetric C-C bond formation, enabling diastereoselective access to halogenated amino acids.
Figure 5.

Downstream chemical modifications of ObiH-generated β-hydroxyleucine.
Finally, we aimed to demonstrate the utility of ObiH derived β-hydroxy-α-amino acids as precursors to aliphatic α-ketoacids (Figure 5A). The phenylserine dehydratase from Ralstonia pickettii was previously characterized as a soluble and functional enzyme under heterologous expression conditions in E. coli.[51] Here, we demonstrate the scope of dehydratase activity can be extended beyond aromatic amino acids by developing a one-pot, telescoped sequence for generating α-keto acid 38 directly from Thr (6) and isobuyraldehyde (36).[51] α-keto acid 38 was formed in 37% yield through this sequence. Such α-keto acid products are highly desirable and have found use as intermediates in biocatalytic cascade reactions, acylating agents in organic synthesis and as precursors for the synthesis of biofuels and pharmaceuticals.[52-57] Through these functionalization reactions, we have demonstrated the diverse products which can be accessed through ObiH-catalyzed transformations.
Conclusions
Here we have demonstrated the synthetic utility of ObiH for the diastereoselective production of diverse and valuable β-hydroxy-α-amino acids from commercially available starting materials. These molecules represent an important starting point for the synthesis of natural products and pharmaceuticals and can be probes for biological systems. Recognizing the value of a transformation that directly produces β-hydroxy-α-amino acids, we sought to efficiently generate a variety of these materials through preparative-scale reactions using whole E. coli cells. We have characterized the native reactivity of ObiH toward a panel of aldehydes, probing the abilities and limitations of ObiH-catalyzed aldolase reactions. This substrate scope analysis was supported by detailed characterization of ObiH diastereoselectivity and underpinning mechanistic implications of observed selectivity trends. Through these efforts, we have developed an efficient route to accessing important organic building blocks, including downstream functionalization reactions to directly synthesize β-halogenated amino acids and α-keto acids. Based on the simplicity and versatility of this reaction platform, we advance ObiH as a useful biocatalyst for selective C─C bond formation and we anticipate that this enzyme will serve as a new and effective implement in the organic chemist’s toolbox.
Supplementary Material
Acknowledgements
This work was supported by the Office of the Vice Chancellor for Research and Graduate Education at the University of Wisconsin-Madison with funding from the Wisconsin Alumni Research Foundation and the NIH DP2-GM137417 to A.R.B.; Morgridge Institute for Research – Metabolism Theme Fellowship to P.K.; The NMR spectrometers were supported by the Bender Fund. The purchase of the Bruker D8 VENTURE Photon III X-ray diffractometer was partially funded by NSF Award #CHE-1919350 to the UW–Madison Department of Chemistry.
We acknowledge the invaluable support, assistance and advice from our colleagues in the Buller group.
References
- [1].Blaskovich MAT, J. Med. Chem 2016, 59, 10807–10836. [DOI] [PubMed] [Google Scholar]
- [2].Lin C-I, McCarty RM, Liu H, Angew. Chemie Int. Ed 2017, 56, 3446–3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hönig M, Sondermann P, Turner NJ, Carreira EM, Angew. Chemie Int. Ed 2017, 56, 8942–8973. [DOI] [PubMed] [Google Scholar]
- [4].Zetzsche LE, Narayan ARH, Nat. Rev. Chem 2020, 4, 334–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Watkins-Dulaney E, Straathof S, Arnold F, ChemBioChem 2021, 22, 5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Huffman MA, Fryszkowska A, Alvizo O, Borra-Garske M, Campos KR, Canada KA, Devine PN, Duan D, Forstater JH, Grosser ST, Halsey HM, Hughes GJ, Jo J, Joyce LA, Kolev JN, Liang J, Maloney KM, Mann BF, Marshall NM, McLaughlin M, Moore JC, Murphy GS, Nawrat CC, Nazor J, Novick S, Patel NR, Rodriguez-Granillo A, Robaire SA, Sherer EC, Truppo MD, Whittaker AM, Verma D, Xiao L, Xu Y, Yang H, Science 2019, 366, 1255–1259. [DOI] [PubMed] [Google Scholar]
- [7].Crüsemann M, Wirtz DA, Ludwig KC, Arts M, Marx CE, Krannich S, Barac P, Kehraus S, Josten M, Henrichfreise B, Müller A, König GM, Peoples AJ, Nitti A, Spoering AL, Ling LL, Lewis K, Schneider T, Angew. Chemie Int. Ed 2021, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Maki H, Miura K, Yamano Y, Antimicrob. Agents Chemother 2001, 45, 1823–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Corbett MD, Chipko BR, Antimicrob. Agents Chemother 1978, 13, 193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ooi T, Taniguchi M, Kameda M, Maruoka K, Angew. Chemie Int. Ed 2002, 41, 4542–4544. [DOI] [PubMed] [Google Scholar]
- [11].Singjunla Y, Baudoux J, Rouden J, Org. Lett 2013, 15, 5770–5773. [DOI] [PubMed] [Google Scholar]
- [12].Thayumanavan R, Tanaka F, Barbas CF, Org. Lett 2004, 6, 3541–3544. [DOI] [PubMed] [Google Scholar]
- [13].Mettath S, Srikanth GSC, Dangerfield BS, Castle SL, J. Org. Chem 2004, 6489–6492. [DOI] [PubMed] [Google Scholar]
- [14].Vera S, Vázquez A, Rodriguez R, Del Pozo S, Urruzuno I, de Cózar A, Mielgo A, Palomo C, J. Org. Chem 2021, 86, 7757–7772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Evans DA, Janey JM, Magomedov N, Tedrow JS, Angew. Chemie Int. Ed 2001, 1936–1940. [PubMed] [Google Scholar]
- [16].Cryle MJ, Meinhart A, Schlichting I, J. Biol. Chem 2010, 285, 24562–24574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chen H, Thomas MG, O’Connor SE, Hubbard BK, Burkart MD, Walsh CT, Biochemistry 2001, 40, 11651–11659. [DOI] [PubMed] [Google Scholar]
- [18].Renata H, Shimizu E, Zwick CR, Tetrahedron 2021, 90, 132190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zwick CR, Sosa MB, Renata H, J. Am. Chem. Soc 2021, 143, 1673–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kimura T, Vassilev VP, Shen G-J, Wong C-H, J. Am. Chem. Soc 1997, 119, 11734–11742. [Google Scholar]
- [21].Di Salvo ML, Remesh SG, Vivoli M, Ghatge MS, Paiardini A, D’Aguanno S, Safo MK, Contestabile R, FEBS J. 2014, 281, 129–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hernandez K, Zelen I, Petrillo G, Usón I, Wandtke CM, Bujons J, Joglar J, Parella T, Clapés P, Angew. Chemie Int. Ed 2015, 54, 3013–3017. [DOI] [PubMed] [Google Scholar]
- [23].Gutierrez ML, Garrabou X, Agosta E, Servi S, Parella T, Joglar J, Clapés P, Chem. Eur. J 2008, 14, 4647–4656. [DOI] [PubMed] [Google Scholar]
- [24].Fesko K, Front. Bioeng. Biotechnol 2019, 7, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Fesko K, Appl. Microbiol. Biotechnol 2016, 100, 2579–2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Fesko K, Uhl M, Steinreiber J, Gruber K, Griengl H, Angew. Chemie Int. Ed 2010, 49, 121–124. [DOI] [PubMed] [Google Scholar]
- [27].Steinreiber J, Schürmann M, Wolberg M, Van Assema F, Reisinger C, Fesko K, Mink D, Griengl H, Angew. Chemie Int. Ed 2007, 46, 1624–1626. [DOI] [PubMed] [Google Scholar]
- [28].Chen Q, Chen X, Feng J, Wu Q, Zhu D, Ma Y, ACS Catal. 2019, 9, 4462–4469. [Google Scholar]
- [29].Zheng W, Yu H, Fang S, Chen K, Wang Z, Cheng X, Xu G, Yang L, Wu J, ACS Catal. 2021, 11, 3198–3205. [Google Scholar]
- [30].Goldberg SL, Goswami A, Guo Z, Chan Y, Lo ET, Lee A, Chi Truc V, Natalie KJ, Hang C, Rossano LT, Schmidt MA, Org. Process Res. Dev 2015, 19, 1308–1316. [Google Scholar]
- [31].Schaffer JE, Reck MR, Prasad NK, Wencewicz TA, Nat. Chem. Biol 2017, 13, 737–744. [DOI] [PubMed] [Google Scholar]
- [32].Scott TA, Heine D, Qin Z, Wilkinson B, Nat. Commun 2017, 8, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kreitler DF, Gemmell EM, Schaffer JE, Wencewicz TA, Gulick AM, Nat. Commun 2019, 10, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Xu L, Wang LC, Su BM, Xu XQ, Lin J, Bioresour. Technol 2020, 310, 123439. [DOI] [PubMed] [Google Scholar]
- [35].Dückers N, Baer K, Simon S, Gröger H, Hummel W, Appl. Microbiol. Biotechnol 2010, 88, 409–424. [DOI] [PubMed] [Google Scholar]
- [36].Kumar P, Meza A, Ellis JM, Carlson GA, Bingman CA, Buller AR, ACS Chem. Biol 2021, 16, 86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wang S, Deng H, Appl. Microbiol. Biotechnol 2021, 2, 3507–3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wachtmeister J, Rother D, Curr. Opin. Biotechnol 2016, 42, 169–177. [DOI] [PubMed] [Google Scholar]
- [39].Baker Dockrey SA, Doyon TJ, Perkins JC, Narayan ARH, Chem. Biol. Drug Des 2019, 93, 1207–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Garzón-Posse F, Becerra-Figueroa L, Hernández-Arias J, Gamba-Sánchez D, Molecules 2018, 23, 1265–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Xu L, Wang LC, Xu XQ, Lin J, Catal. Sci. Technol 2019, 9, 5943–5952. [Google Scholar]
- [42].Schaffer JE, Reck MR, Prasad NK, Wencewicz TA, Nat. Chem. Biol 2017, 13, 737–744. [DOI] [PubMed] [Google Scholar]
- [43].Moore MJ, Qu S, Tan C, Cai Y, Mogi Y, Keith DJ, Boger DL, J. Am. Chem. Soc 2020, 142, 16039–16050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Willemse T, Schepens W, Van Vlijmen HWT, Maes BUW, Ballet S, Catalysts 2017, 7, 74–106. [Google Scholar]
- [45].Wang M, Rakesh KP, Leng J, Fang WY, Ravindar L, Channe Gowda D, Qin HL, Bioorg. Chem 2018, 76, 113–129. [DOI] [PubMed] [Google Scholar]
- [46].Beaudoin SF, Hanna MP, Ghiviriga I, Stewart JD, Enzyme Microb. Technol 2018, 119, 1–9. [DOI] [PubMed] [Google Scholar]
- [47].Steinreiber J, Fesko K, Mayer C, Reisinger C, Schürmann M, Griengl H, Tetrahedron 2007, 63, 8088–8093. [Google Scholar]
- [48].Blesl J, Trobe M, Anderl F, Breinbauer R, Strohmeier GA, Fesko K, ChemCatChem 2018, 10, 3453–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Lu H, Shen Z, Front. Chem 2020, 8, 290–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hanessian S, Wang X, Ersmark K, Del Valle JR, Org. Lett 2009, 11, 4232–4235. [DOI] [PubMed] [Google Scholar]
- [51].Heiwa O, Shinji N, Haruo M, Biotechnol. Biochem 2002, 66, 2755–2758. [Google Scholar]
- [52].Xue YP, Cao CH, Zheng YG, Chem. Soc. Rev 2018, 47, 1516–1561. [DOI] [PubMed] [Google Scholar]
- [53].Penteado F, Lopes EF, Alves D, Perin G, Jacob RG, Lenardão EJ, Chem. Rev 2019, 119, 7113–7278. [DOI] [PubMed] [Google Scholar]
- [54].Atsumi S, Hanai T, Liao JC, Nature 2008, 451, 86–89. [DOI] [PubMed] [Google Scholar]
- [55].Marín-Valls R, Hernández K, Bolte M, Parella T, Joglar J, Bujons J, Clapés P, J. Am. Chem. Soc 2020, 142, 19754–19762. [DOI] [PubMed] [Google Scholar]
- [56].Marín-Valls R, Hernández K, Bolte M, Joglar J, Bujons J, Clapés P, ACS Catal. 2019, 9, 7568–7577. [Google Scholar]
- [57].Hernández K, Joglar J, Bujons J, Parella T, Clapés P, Angew. Chemie Int. Ed 2018, 57, 3583–3587. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.