

Abstract
Gastrointestinal (GI) malignancies are among the most commonly diagnosed cancers worldwide. Despite the introduction of targeted and immunotherapy agents in the treatment landscape, cytotoxic agents, such as fluoropyrimidines and irinotecan, remain as the cornerstone of chemotherapy for many of these tumors. Pharmacogenetics (PGx) is a rapidly evolving field that accounts for interpatient variability in drug metabolism to predict therapeutic response and toxicity. Given the significant incidence of severe treatment-related adverse events associated with cytotoxic agents, utilizing PGx can allow clinicians to better anticipate drug tolerability while minimizing treatment interruptions or delays. In this review, the PGx profiles of drug-gene pairs with potential impact in GI malignancy therapy – DPYD-5-fluorouracil/capecitabine and UGT1A1-irinotecan – and the available clinical evidence of their roles in reducing severe adverse events are discussed. Considerations for clinical implementation, such as optimal laboratory workflows, electronic health record integration, and stakeholder engagement, as well as provider education, are addressed. Last, exploratory PGx markers in GI malignancy treatment are described. As the PGx knowledge base rapidly evolves, pharmacists will be vital in leveraging their pharmacology knowledge and clinical skills to implement PGx testing in the clinic.
Keywords: gastrointestinal neoplasms, fluorouracil, irinotecan, drug-related side effects, adverse reactions, pharmacogenetics, pharmacogenomics, toxicity, implementation science, pharmacology, fluoropyrimidine
There are over 100 medications known to be impacted by actionable pharmacogenetic (PGx) germline variants.1 The application of precision medicine in the ambulatory environment will allow clinicians to tailor treatment to individual patients based on germline and somatic genetic variants to better predict drug response and risk of toxicity, which is sorely lacking in current practice.2 The US Food and Drug Administration (FDA) has continued to update product labeling for these drugs and recently released an updated table of gene-drug associations with sufficient scientific evidence to guide therapy management.1 To facilitate PGx integration into clinical care, the National Institutes of Health-funded Clinical Pharmacogenetics Implementation Consortium (CPIC) was formed.3 The CPIC publishes peer-reviewed, evidence-based guidelines for specific drug-gene pairs to translate PGx results into practical guidance for informed prescribing decisions.
Gastrointestinal (GI) malignancies include cancers of the colon and rectum, esophagus and stomach, gallbladder, liver, pancreas, appendix, and anus and account for 4.5 million global deaths per year.4 Standard first-line systemic chemotherapy for the majority of patients with GI malignancies often consists of a fluoropyrimidine, such as 5-fluorouracil or capecitabine, in combination with irinotecan, or oxaliplatin, with or without targeted therapy. Although the treatment backbone has remained largely the same for decades, there is heterogeneity in drug response and tolerability with a subset of patients at an inherent risk of developing severe, chemotherapy-related adverse events. These potentially life-threatening toxicities are due to germline variants in the dihydropyrimidine dehydrogenase (DPYD) and uridine diphosphate-glucuronosyltransferase isoform 1A1 (UGT1A1) genes encoding the enzymes responsible for the metabolism of fluoropyrimidines and irinotecan, respectively. In current practice, these germline variants are not determined in individual patients until chemotherapy is initiated and severe toxicity develops.
In this review, the PGx variants impacting response to fluoropyrimidines and irinotecan will be described with evidence detailing the associations of genetic polymorphisms and chemotherapy-induced severe toxicity and safety outcomes from implementing preemptive PGx testing in practice. Factors important for clinical implementation, such as laboratory workflow requirements, integration and interpretation of test results in the electronic health record, and stakeholder engagement with clinical providers and institutional leadership are addressed. As the PGx knowledge base expands, exploratory biomarkers in GI cancer treatment are also discussed. In the era of precision medicine, DPYD testing in patients before the initiation of fluoropyrimidine therapy to mitigate the risk of severe chemotherapy-related adverse events should be considered if optimal clinical and laboratory workflows are in place. Due to the limited availability of genotype-guided dosing guidelines, testing for UGT1A1 polymorphisms can be performed on a case-by-case basis. As essential members of the health care team, pharmacists can play a vital role in leading and participating in PGx implementation efforts. By applying genotype-guided dose adjustments to fluoropyrimidine and irinotecan therapy, it is expected that treatment-related hospitalizations and interruptions in treatment can be prevented while preserving quality of life in patients.
DPYD and Fluoropyrimidines
Metabolism of Fluoropyrimidines
Over the last 40 years, fluoropyrimidines have become among the most widely prescribed anticancer agents with an estimated annual treatment population of two million patients worldwide for solid tumors involving the GI tract, pancreas, and breast.5 In the United States, 5-fluouracil (5-FU) and capecitabine are routinely used in clinical practice. Five-fluouracil (5-FU) is an intravenous fluorine-substituted analogue of uracil that undergoes conversion to fluorodeoxyuridine (FUDR) then fluorodeoxyuridine monophosphate (FdUMP). This active metabolite forms a stable complex with thymidylate synthase (TS) to inhibit the production of deoxythymidine monophosphate (dTMP). A downstream depletion of pyrimidine and deoxyribonucleic acid (DNA) synthesis occurs, resulting in cytotoxicity and apoptosis. Partial incorporation of 5-FU and its metabolites in ribonucleic acid (RNA) have also been shown to contribute to drug metabolism and RNA damage.6,7
Advances in the understanding of the mechanism of action of 5-FU over time have led to changes in drug administration. When leucovorin is given in conjunction with a 5-FU bolus, the folic acid analog forms a ternary complex and stabilizes the binding of FdUMP to TS, extending the drug’s short half-life and enhancing antineo-plastic activity. In addition to bolus dose administration, 5-FU is usually administered as a continuous infusion over 46 hours to improve patient tolerability and drug exposure without compromising clinical efficacy. As an oral prodrug of 5-FU, capecitabine is converted to 5-FU via a three-step enzymatic cascade.6 The amount of 5-FU available to exert its anticancer effect is directly regulated by its catabolism. Dihydropyrimidine dehydrogenase (DPD) is responsible for the initial and rate-limiting step of 5-FU catabolism. Encoded by DPYD, the enzyme converts ~80% of 5-FU in the liver into inactive dihydrofluorouracil (DHFU).6 Figure 1 shows the metabolism pathway of fluoropyrimidines. Patients with inherited metabolic disorders, such as a DPD deficiency, may experience variable systemic clearance of fluoropyrimidines and subsequent drug toxicity.
Figure 1. Fluoropyrimidine metabolism.
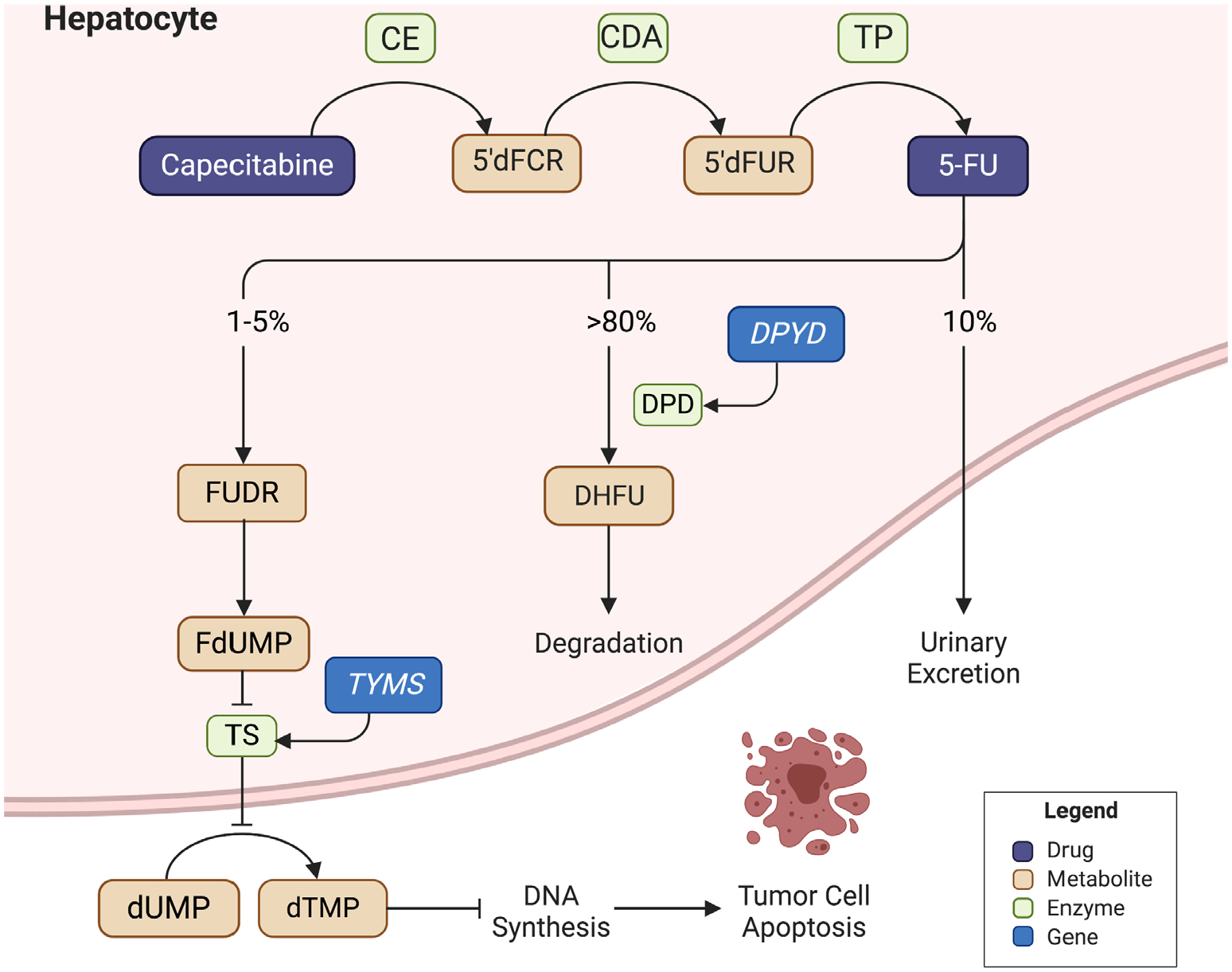
Capecitabine is an oral prodrug that undergoes conversion to 5-fluorouracil via a three-step enzymatic cascade. After metabolism to fluorodeoxyuridine and fluorodeoxyuridine monophosphate, a stable complex with thymidylate synthase is formed to inhibit deoxythymidine monophosphate production. A downstream depletion of deoxyribonucleic acid (DNA) synthesis occurs, leading to cytotoxicity. Catabolism is mediated by dihydrofluorouracil via dihydropyrimidine dehydrogenase. 5′dFCR: 5′-deoxy-5-fluorocytidine; 5-FU = fluorouracil; CDA = cytidine deaminase; CES = carboxylesterase; DHFU = dihydrofluorouracil; DPD = dihydropyrimidine dehydrogenase (encoded by DPYD); dTMP = deoxythymidine monophosphate; dUMP = deoxyuridine monophosphate; FdUMP = fluorodeoxyuridine monophosphate; FUDR = fluorodeoxyuridine; TP = thymidine phosphorylase; TS = thymidylate synthase (encoded by TYMS).
Genetic Variants Associated with DPD Deficiency and Chemotherapy-Related Toxicity
Although treatment with fluoropyrimidines is generally well tolerated, up to 30% of patients may develop severe toxicity in the form of myelosuppression, diarrhea, hand-foot syndrome, or mucositis during early treatment due to its narrow therapeutic index.8 These therapy-related adverse events can be fatal in 1% of treated patients.5 In some cases, fluoropyrimidine toxicity can be traced back to variants in DPYD that alter the protein sequence or mRNA splicing and result in a truncated protein with compromised enzyme activity.9 When DPD is inactive or harbors reduced activity, the rate of 5-FU clearance decreases, leading to the development of severe fluoropyrimidine-related adverse events from prolonged 5-FU exposure.
More than 160 different allelic variants in DPYD have been discovered, although most have unclear functional effects on DPD enzyme activity and therefore limited clinical relevance.5 At this time, five DPYD variants known to impact fluoropyrimidine therapy are of primary importance due to their functional consequence: DPYD*2A (rs3918290), DPYD*13 (rs55886062), c.2846A>T (rs67376798), haplotype B3 (rs56038477 and rs75017182), and c.557A>G (rs115232898). The location of DPYD*2A in the intron boundary of exon 14 results in an exon loss, rendering the protein nonfunctional. HapB3 affects pre-mRNA splicing and causes partial production of a nonfunctional protein. DPYD*13, c.2846A>T, and c.557A>G are missense mutations that affect protein function.10 A partial DPD deficiency is present in about 3–5% of individuals of European ancestry, whereas complete deficiency occurs less frequently at a rate of 0.2%.10 The c.557A>G is also of significance given its higher frequency in populations of African ancestry. Approximately 8% of African American individuals have a partial DPD deficiency.5,11 Variants with the most deleterious DPD enzyme activity are DPYD*2A and DPYD*13, whereas the other three variants have been reported to result in a more moderate reduction.10 Table 1 shows the allele frequencies and functional effects of clinically relevant DPYD variants.
Table 1.
Actionable Pharmacogenetic Variants Impacting Response to Fluoropyrimidines
| DPYD variant | Minor allele frequency | |||||
|---|---|---|---|---|---|---|
| Haplotype | Nucleotide Change | rsID | EA | AA | Allele function | Enzyme activity10 |
| HapB3 | c.1236G>A | rs56038477 | 0.024 | 0.003 | Decreased | 25% reduction in DPD activity |
| c. 1129–5923C>G | rs75017182 | |||||
| c.2846A>T | rs67376798 | 0.004 | 0.003 | |||
| c.557A>G | rs115232898 | 0.000 | 0.012 | |||
| *2A | c.1905+1G>A | rs3918290 | 0.008 | 0.003 | None | 50% reduction in DPD activity |
| *13 | c.1679T>G | rs55886062 | 0.001 | 0.000 | None | 50% reduction in DPD activity |
AA = African American; EA = European ancestry; rsID = reference SNP cluster ID; DPYD = dihydropyrimidine dehydrogenase.
The relationship between these variants and fluoropyrimidine-induced severe toxicity has been widely explored and confirmed in the literature (Table 2).8,12–18 In 2013, Terrazzino and colleagues confirmed the clinical validity of variant DPYD*2A and c.2846A>T alleles as risk factors for severe toxicities after fluoropyrimidine use.15 Pooled data showed that individuals with DPYD*2A polymorphisms were likely to experience grade ≥ 3 hematologic toxicity, mucositis, and diarrhea. A strong association was also found among c.2846A>T variant carriers and grade ≥ 3 diarrhea. The meta-analysis concluded that a 5-fold and 8-fold increased risk of overall grade ≥ 3 toxicity is present in *2A and c.2846A>T variant carriers, respectively, compared with wild-type patients.15 A subsequent meta-analysis by Rosmarin and colleagues supported these findings, concluding that although the DPYD*2A and c.2846A>T variants are rare, the risk of associated toxicity is relatively high.16 Significant associations of global toxicity (grade 0–2 vs grade ≥ 3) with capecitabine were found in variant carriers. Evidence of toxicity in DPYD*2A and c.2846A>T carriers with 5-FU bolus (p = 0.0068) and infusional (p = 0.042) monotherapies were also observed.16 A 2015 meta-analysis by Meulendijks and colleagues also found evidence for additional variants, DPYD*13 (c.1679T>G) and haplotype B3 (c.1236G>A), as predictors of fluoropyrimidine-related hematological and gastrointestinal toxicities (p < 0.0001).17
Table 2.
Literature Summary of DPYD Variant Associations with Severe Toxicity
| Study | Study design | Total n patients | Tumor type | Regimen | DPYD variant | Initial dose ↓a | Main findings of grade ≥ 3 toxicity | p value | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Kleinjan et al., 201912 | Retrospective dose escalation, single center, European population | 185 | Solid, various | CAPE-based | c.1236G>A | 20–40% | Dose-reduced variant carriers vs WT atients given standard dose | |||
| c.2846A>T | 20–40% | |||||||||
| Any toxicity | 27.3% vs 37.9% | 0.54 | ||||||||
| Diarrhea | 19.5% vs 18.2% | 1.00 | ||||||||
| Diarrhea-related hospitalization | 11.5% vs 18.2% | 0.62 | ||||||||
| Hematological toxicity | 3.4% vs 9.1% | 0.35 | ||||||||
| HFS (grade ≥ 2) | 20.1% vs 0% | 0.13 | ||||||||
| Toxicity in first cycle of dose-reduced variant carriers | ||||||||||
| c.1236G>A | 2/6 patients | NR | ||||||||
| c.2846A>T | 0/1 patients | NR | ||||||||
| *2A | 0/4 patients | NR | ||||||||
| Henricks et al., 201912 | Matched pair analysis, European population | 1646 | Solid, various | 5-FU- or CAPE-based | *2A | 50% | Dose-reduced *2A carriers vs WT patients given standard dose | |||
| Overall toxicity | 18% vs 23% | 0.57 | ||||||||
| GI toxicity | 10% vs 9% | 0.78 | ||||||||
| Hematological; toxicity | 10% vs 10% | > 0.99 | ||||||||
| HFS | 5% vs 5% | > 0.99 | ||||||||
| Dose-reduced *2A carriers vs *2A carriers given standard dose in historical cohort | ||||||||||
| Overall toxicity | 18% vs 77% | < 0.001 | ||||||||
| GI toxicity | 10% vs 38% | 0.001 | ||||||||
| Hematological toxicity | 10% vs 56% | < 0.001 | ||||||||
| HFS | 5% vs unknown | NR | ||||||||
| Henricks et al., 20188 | Prospective multicenter, European population | 1103 | Solid, various | 5-FU- or CAPE-based | c.1236G>A | 25% | Dose-reduced variant carriers vs WT patients given standard dose | |||
| c.2846A>T | 25% | |||||||||
| *2A | 50% | Overall toxicity | 39% vs 23% | 0.0013 | ||||||
| *13 | 50% | GI toxicity | 20% vs 8% | 0.00089 | ||||||
| Hematological toxicity | 15% vs 6% | 0.0043 | ||||||||
| HFS | 1% vs 4% | 0.41 | ||||||||
| Dose-reduced variant carriers vs variant carriers given standard dose in historical cohort | ||||||||||
| *2A | 31% vs 72% (RR = 2.87, 95% CI = 2.14–3.86) | NR | ||||||||
| *13 | 0% vs 55% (RR = 4.30, 95% CI = 2.10–8.80) | NR | ||||||||
| c.2846A>T | 47% vs 62% (RR = 3.11, 95% CI = 2.25–4.28) | NR | ||||||||
| c.1236G>A | 49% vs 37% (RR = 1.72, 95% CI = 1.22–2.42) | NR | ||||||||
| Deenen et al., 201614 | Prospective multicenter, | 1631 | Solid, various | 5-FU- or CAPE-based | *2A | 50–85% | Dose-reduced *2A carriers vs WT patients given standard dose | |||
| European population | Any toxicity | 23% vs 28% | 0.64 | |||||||
| Diarrhea | 8% vs 6% | 0.68 | ||||||||
| Hematological toxicity | 10% vs 17% | 0.34 | ||||||||
| Hand-foot syndrome | 5% vs 11% | 0.28 | ||||||||
| Dose-reduced *2A carriers vs *2A carriers given standard dose in historical cohort | ||||||||||
| Overall toxicity | 28% vs 73% | < 0.001 | ||||||||
| GI toxicity | 11% vs 56% | 0.001 | ||||||||
| Hematological toxicity | 17% vs 66% | < 0.001 | ||||||||
| Drug-induced death | 0% vs 10% | 0.19 | ||||||||
| Terrazzino et al., 201315 | Meta-analysis (15 studies), European population | 4573 | Solid, various | 5-FU- or CAPE-based |
*2A c.2846A>T |
N/A | *2A variant carriers | |||
| Overall toxicity | (OR = 5.42, 95% | < 0.001 | ||||||||
| Diarrhea | CI = 2.79–10.52) (OR = 5.54, 95% CI = 2.31–13.29) | 0.001 | ||||||||
| Hematological toxicity | (OR = 15.77, 95% CI = 6.36–39.06) | < 0.001 | ||||||||
| Mucositis | (OR = 7.48, 95% CI = 3.03–18.47) | < 0.001 | ||||||||
| c.2846A>T variant carriers | ||||||||||
| Overall toxicity | (OR = 8.18, 95% CI = 2.65–25.25) | < 0.001 | ||||||||
| Diarrhea | (OR = 6.04, 95% CI = 1.77–20.66) | 0.004 | ||||||||
| Rosmarin et al., 201416 | Meta-analysis (17 studies), European, Australian, North American populations | 5782 | Solid, various | 5-FU- or CAPE-based |
*2A c.2846A>T |
N/A | *2A variant carriers | |||
| Overall toxicity | Capecitabine | (OR = 5.51, 95% CI = 1.95–15.51) | 0.0013 | |||||||
| Overall toxicity | Infusional 5-FU | (OR = 6.71, 95% CI = 1.66–27.1) | 0.0075 | |||||||
| Diarrhea | Infusional 5-FU | (OR = 7.71, 95% CI = 1.6–36.9) | 0.011 | |||||||
| Neutropenia | Bolus 5-FU | (OR = 12.90, 95% CI = 3.13–53.3) | 0.0061 | |||||||
| Mucositis | Bolus 5-FU | (OR = 7.15, 95% CI = 1.75–29.1) | 0.0004 | |||||||
| c.2846A>T variant carriers | ||||||||||
| Overall toxicity | Capecitabine | (OR = 9.35, 95% CI = 2.01–13.4) | 0.0043 | |||||||
| Diarrhea | Capecitabine | (OR = 3.14, 95% CI = 0.82–11.9) | 0.093 | |||||||
| HFS | Capecitabine | (OR = 1.31, 95% CI = 0.35–1.96) | 0.69 | |||||||
| Meulendijks et al., 201517 | Meta-analysis (8 studies), European, Australian, North American populations | 7365 | Solid, various | 5-FU- or CAPE-based | *1/*1 c.1236G>A c.2846A>T *2A *13 |
N/A | Overall toxicity in variant carriers vs WT patients | |||
| *2A | (RR = 2.85, 95% CI = 1.75–4.62) | < 0.0001 | ||||||||
| *13 | (RR = 4.40, 95% CI = 2.08–9.30) | < 0.0001 | ||||||||
| c.2846A>T | (RR = 3.02, 95% CI = 2.22–1.10) | < 0.0001 | ||||||||
| c.!236G>A | (RR = 1.59, 95% CI = 1.29–1.97) | < 0.0001 | ||||||||
| GI toxicity in variant carriers vs WT patients | ||||||||||
| *13 | (RR = 5.72, 95% CI = 1.40–23.33) | 0.0158 | ||||||||
| c.2846A>T | (RR = 2.04, 95% CI = 1.49–2.78) | < 0.0001 | ||||||||
| Hematological toxicity in variant carriers vs WT patients | ||||||||||
| *13 | (RR = 9.76, 95% CI = 3.03–31.48) | 0.00014 | ||||||||
| c.2846A>T | (RR = 2.07, 95% CI = 1.17–3.68) | 0.013 | ||||||||
| Saif et al., 201418 | Case report, North American | 1 | Colon | 5-FU-based | c.557A>G | N/A | Hematological toxicity after first cycle in African American patient | |||
| Complete blood count on hospital admission: | ||||||||||
| White blood cell count | 2.4 × 109/L | N/A | ||||||||
| Absolute neutrophil count | 1.2 × 109/L | N/A | ||||||||
| Hemoglobin level | 10.8 g/dl | N/A | ||||||||
| Platelet count | 80 × 109/L | N/A | ||||||||
5-FU = 5-fluorouracil; CAPE = capecitabine; CI = confidence interval; DPD = dihydropyrimidine dehydrogenase; DPYD = dihydropyrimidine dehydrogenase; GI = gastrointestinal; HFS = hand-foot syndrome; N/A = not applicable; NR = not reported; OR: odds ratio; RR: relative risk; WT = wild-type (*1/*1).
Toxicity defined per Common Terminology Criteria for Adverse Events (CTCAE) guidelines used by primary study authors.
If preemptive dosage reduction was performed.
Evidence from these meta-analyses has shown the clinical validity of DPYD variants as risk factors for developing fluoropyrimidine-related toxicity, allowing investigators to conduct prospective studies and demonstrate the utility of DPYD genotyping in clinical practice. In 2016, Deenen and colleagues performed a safety analysis of preemptive testing for DPYD*2A variant carriers, concluding that genotype-guided fluoropyrimidine dosing improved toxicity outcomes in individuals with the polymorphism. A similar incidence of severe toxicity was found among dose-reduced variant carriers and wild-type patients given standard dose (23%, p = 0.64) with additional data showing similar systemic 5-FU exposure between the two groups.14 When compared with a historical cohort, the risk of grade ≥ 3 toxicity was significantly reduced in the current dose-reduced variant carrier population compared with variant carriers, the historical cohort that received the standard, full dose (73% vs 28%, p < 0.001).14 It was also noted that toxicity events in the genotype-guided group were short in duration as opposed to the long-lasting and life-threatening toxicity that typically occurs with full dosing. Furthermore, an absolute risk reduction in the incidence of drug-induced death was observed from 10% to 0%.14
In a 2018 multicenter study performed in the Netherlands, Henricks and colleagues further demonstrated the feasibility of prospective genotype-guided dosing.8 Even though fluoropyrimidine-related severe toxicity was found to be higher in variant carriers (39% vs 23%, p = 0.0013), reduced rates of severe toxicity were evident when compared with historical control groups. Dose reductions based on guideline recommendations for common DPYD variants in patients of European ancestry (*2A, *13, c.1129–5923C>G, and c.2846A>T) confirmed a 50% dose reduction was adequate for *2A and *13 carriers, but the 25% performed for c.1129–5923C>G and c.2846A>T variant carriers was likely not enough.8 Since the publication of the study, the CPIC has updated its guidelines on their website to recommend a 50% dose reduction of heterozygous carriers of c.1129–5923C>G and c.2846A>T.10 A study conducted by Kleinjan and colleagues in 2019 supports the practice of DPYD genotype-guided dosing, as initial dose reductions of capecitabine in heterozygous DPYD variant carriers followed by tolerance-based dose escalation did not lead to higher toxicity when compared with wild-type patients (37.9% vs 27.3%, p = 0.54).12 Of the 11 variant carriers, only 6 (54.5%) tolerated dose escalations, achieving a median increase of 8.5% (4–31%). Despite the frequency of the c.557A>G variant in individuals of African descent, there are few studies directly investigating fluoropyrimidine toxicity with the decreased function variant.18 With evidence currently limited to case reports, additional investigation is warranted for c.557A>G testing.
From a resource utilization standpoint, development of drug toxicity is an economic burden to both the patient and health system. A 2016 US study assessing the direct health care costs of common adverse events among patients with metastatic colorectal cancer found that over 90% of the population developed at least one toxicity event, with management strategies costing over a thousand dollars on average.19 Evidence from genotype-guided dose individualization studies has demonstrated that upfront DPYD screening and treatment of severe treatment-related toxicities do not exceed standard of care treatment and management strategies. Cost-minimization analyses from European studies have shown that average total treatment costs were lower in screened patients (€2772–€2599 [US $2830–$3767]) than in non-screened populations (€2650–€2817 [US $2886–$3828]).14,20
Although the results of these studies support the utility of preemptive PGx testing to guide chemotherapy dosing, they demonstrate favorable safety profiles without additional spending from health care payors or institutions in primarily European populations. It should be noted that variants that are frequently cited in the literature are those that are common in individuals of European ancestry (c.2846A>T, *2A, *13, and HapB3) and thus are the ones most often included in cost-effectiveness studies. In the US population, a slightly lower incidence of these four variants would be expected while a higher incidence of variants found in individuals of other races and ethnicities, such as the c.557A>G variant in African Americans, is likely to occur. As a result, cost-effectiveness studies are needed that reflect the frequency of alleles in the US population. A recent analysis from the University of Minnesota evaluated the costs of DPYD and UGT1A1 screening in patients with metastatic colorectal cancer (mCRC) receiving infusional 5-FU and irinotecan (FOLFIRI) with bevacizumab from a US health care system modeling perspective. It was reported that total costs in the genetic testing group were US $25,563 as compared with US $25,515 in the standard of care group, resulting in an incremental cost-effectiveness ratio (ICER) of US $4963 per quality-adjusted life year (QALY) gained. The authors concluded that preemptive screening was cost-effective and significantly lower than typical oncology ICERs of US $50,000–100,000 per QALY.21
Role of DPD Activity Testing and Therapeutic Drug Monitoring
It has been recognized that individuals with normal DPD enzyme activity may still present with elevated plasma concentrations of 5-FU and drug toxicity, indicating that other factors contribute to fluoropyrimidine metabolism.22 In these patients, therapeutic drug monitoring (TDM) may serve as an alternative dosing method to optimize systemic drug exposure and pharmacodynamic responses to improve clinical outcomes. Although pharmacokinetic-guided 5-FU administration protocols using validated TDM algorithms have shown improved treatment efficacy and tolerability, they are not a standard of care in practice due to implementation barriers, which include a long sampling time and costly workflow.22 There have also been varying target areas under the curve levels reported in the literature, with some studies targeting ranges between 18 and 28 mg h/L and others targeting plasma levels at 20–24 mg·h/L or 20–30 mg·h/L.22,23
In certain circumstances, phenotyping tests may be used to screen patients for DPD deficiency if sufficient clinical and laboratory resources are available. The gold standard of DPD phenotyping is an assay that can determine DPD enzyme activity in peripheral blood mononuclear cells (PMBCs), as evidenced by a correlation between activity in PMBCs and DPD activity in the liver.8 Other methods for phenotyping include a measure of baseline dihydrouracil/uracil (UH2/U) ratio, plasma levels of uracil after a uracil test dose, and uracil breath test after a dose of [2–13C]-labeled uracil.8,24 A consensus for an optimal assay in terms of predicting toxicity, sensitivity, and specificity, and cost-effectiveness has not yet been fully established due to the heterogeneity in the analytical methods used among laboratories. The lack of Clinical Laboratory Improvement Amendments (CLIA)-approved availability for enzyme testing has led to slow uptake in clinical practice. According to the Genetic Testing Registry, the only CLIA-approved tests for assessing DPD deficiency are genetic assays for analyzing the entire coding region, deletion/duplication, and/or targeted variants of DPYD.25
Genotype-Guided Prescribing
Despite evidence demonstrating the feasibility and cost-effectiveness of assessing DPD deficiency through DPYD genotyping, much debate still exists regarding its clinical implementation and utility in tailoring fluoropyrimidine therapy.26–28 Nonetheless, regulatory authorities recognize the impact of PGx and have made progress in updating prescribing information for applicable drugs. Multidisciplinary clinical experts have also developed guideline recommendations for PGx integration into patient care and optimal therapeutic decision making.
In 2016, the FDA revised the drug labeling for 5-FU and capecitabine, warning that “patients with certain homozygous or certain compound heterozygous mutations in the DPD gene that result in complete or near complete absence of DPD activity are at increased risk for acute early-onset of toxicity and severe, life-threatening, or fatal adverse reactions.”29,30 It is recommended to withhold or permanently discontinue drug therapy based on clinical assessment of onset, duration, and severity of observed toxicities in these patients. Although the FDA acknowledges DPD deficiency as a risk factor for fluoropyrimidine toxicity and DPYD is listed as a valid biomarker, testing is not required before drug initiation and specific dose recommendations for variant carriers are yet to be published. Similarly, the National Comprehensive Cancer Network (NCCN) Guidelines for colon cancer state that carriers of certain DPYD variants “have a significantly elevated risk for severe, life-threatening toxicity after a standard dose of fluoropyrimidine” but testing is not mandated nor is this statement reflected in guidelines for other tumor types where a fluoropyrimidine is recommended.31
In 2013, the CPIC published genotype-guided guidelines for fluoropyrimidine dosing to help clinicians with the translation of PGx test results into drug treatment decisions.10 A gene activity score (AS) is used to interpret DPYD genetic test results and assign phenotypes and is determined by the function of alleles the patient carries. Each DPYD variant allele is assigned a value according to its enzyme function: 1 for normal function, 0.5 for decreased function, and 0 for no function (or minimal DPD activity). The AS is then calculated as the sum of the two DPYD variants with the lowest variant activity score and corresponds to a phenotype. Patients with an AS of 0 (carriers of two no function variants) or 0.5 (carries of one decreased function variant) are typically classified as poor metabolizers. Those with an AS of 1 (carriers of two decreased function variants or only one no function variant) or 1.5 (carriers of only one decreased function variant) are considered intermediate metabolizers, and those with an AS of 2 are referred to as normal metabolizers.
Clinicians should refer to the DPYD Allele Functionality Table available from the CPIC for the most up to date information when correlating an allele to a function and AS. For example, if a patient’s DPYD PGx test results were reported as DPYD *1/*2A and the table lists *1 allele with a value of 1 and the *2A allele with a value of 0, the sum of these would yield an AS of 1. This patient would then be classified as having an intermediate metabolizer phenotype. Although different scores equate to similar phenotypes, genotype-guided dosing recommendations are dependent on the AS itself. In the case for DPYD poor metabolizers, the CPIC advises against therapy with fluoropyrimidines for patients with an AS of 0 as they may be at the highest risk for severe or fatal drug-related toxicity. However, for individuals with an AS of 0.5, selection of an alternative drug or a strongly reduced dose with TDM can be considered. In intermediate metabolizers with an AS of 1 or 1.5, a 50% dose reduction from the full standard dose is recommended according to a guideline update in November 2018. Before this update, it was recommended for patients with an AS of 1.5 to receive an ambiguous 25–50% dose reduction due to the limited evidence for dosing recommendations in the setting of decreased function variants. Dose escalation remains a consideration in intermediate metabolizers based on clinical judgment and TDM if feasible. With regard to normal metabolizers, an AS of 2 indicates a “normal” risk of fluoropyrimidine toxicity that does not warrant any preemptive dose adjustment.
As of April 2020, the European Medicines Agency (EMA) recommends testing for DPD deficiency before treatment with intravenous 5-FU, capecitabine, or tegafur. Screening can include phenotyping and/or genotyping methods by measuring uracil levels in the plasma or testing for DPYD variants.32 The agency’s therapeutic recommendations are consistent with relevant clinical guidelines where a reduced starting dose should be considered in patients with a partial deficiency and treatment is contraindicated in patients with a known complete DPD deficiency. The EMA also recommends TDM of 5-FU in patients receiving continuous infusions to improve clinical outcomes. At this time, the European Society for Medical Oncology (ESMO) consensus guidelines for the management of patients with mCRC consider DPD testing remain as an option rather than a routine recommendation before fluoropyrimidine therapy.33
Recent prescribing recommendations made available by the Dutch Pharmacogenetics Working Group (DPWG) of the Royal Dutch Association for the Advancement of Pharmacy are similar to CPIC guidelines.5 Notable differences in dosage reductions and phenotypic translations include: (1) recommendations for tegafur, an oral fluoropyrimidine not available in the United States, (2) recommendations for cutaneous routes of 5-FU administration, (3) a 50% reduction in the starting dose in patients with an activity score of 1.5, and (4) and the recommendation to perform phenotyping in patients with an equivalent AS of 0.5 due to the unpredictability of enzymatic activity. According to the multidisciplinary group, it is recommended to determine DPD activity in these patients with an additional phenotyping test then adjust the initial fluoropyrimidine dose based on available data or select an alternative agent.5
Gaps in Evidence Base
Randomized controlled trials are rightly considered the gold standard in applying study results to practice; however, this type of trial design for DPYD research bears ethical concerns (i.e., the risk of drug-induced toxicity in variant carriers given standard doses). Insufficient randomized controlled trials may be a contributing factor for the lack of endorsement from the FDA and national oncology guidelines. Despite newer evidence from studies using historical cohorts, dose escalation trials, and cost analyses support preemptive dosing strategies, the current fluoropyrimidine drug labeling does not reflect the results of these research efforts. Nonetheless, NCCN guidelines for colon cancer recognize the two prospective studies by Henricks and colleagues and Deenen and colleagues, stating that they “have shown DPYD genotyping and fluoropyrimidine dose individualization to be feasible in clinical practice, improve patient safety, and be cost-effective.”8,14 Ongoing investigations will help determine the ideal fluoropyrimidine dose reduction in c.1236G>A and c.2846A>T variant carriers while providing more information on DPYD genotyping in more diverse populations (ClinicalTrials.gov identifier NCT04300361, NCT04194957).
When considering clinical oncologic outcomes, prospective studies have shown that genotype-guided dose reductions do not compromise overall drug exposure and that 5-FU concentrations are similar in variant allele carriers receiving reduced dose fluoropyrimidines compared with wild-type patients receiving fluoropyrimidines at full dose.13,34
UGT1A1 and Irinotecan
Metabolism of Irinotecan
Irinotecan is a semisynthetic camptothecin derivative with antitumor activity against lung, colon, gastric, and gynecological cancers often given in combination with fluoropyrimidine therapy. After intravenous administration, the prodrug enters hepatic cells via passive diffusion then undergoes conversion to its active metabolite, 7-ethyl-10-hydroxycamptothecin (SN-38), via carboxylesterase-mediated hydrolysis (Figure 2). CYP3A4 and CYP3A5 simultaneously mediate the oxidation pathway of irinotecan to form the inactive metabolites APC (7-ethyl-10-[4-N-(5-aminopentanoic acid)-1-piperidino] carbonyloxycamptothecin) and NPC (7-ethyl-10-[4-(1-piperidino)-1-amino] carbonyloxycamptothecin). Irinotecan uptake and transport is facilitated by drug-metabolizing enzymes and transporters, which include ABCB1, MRP1 (ABCC1), MRP2 (ABCC2), and MXR (ABCG2).35
Figure 2. Irinotecan metabolism.
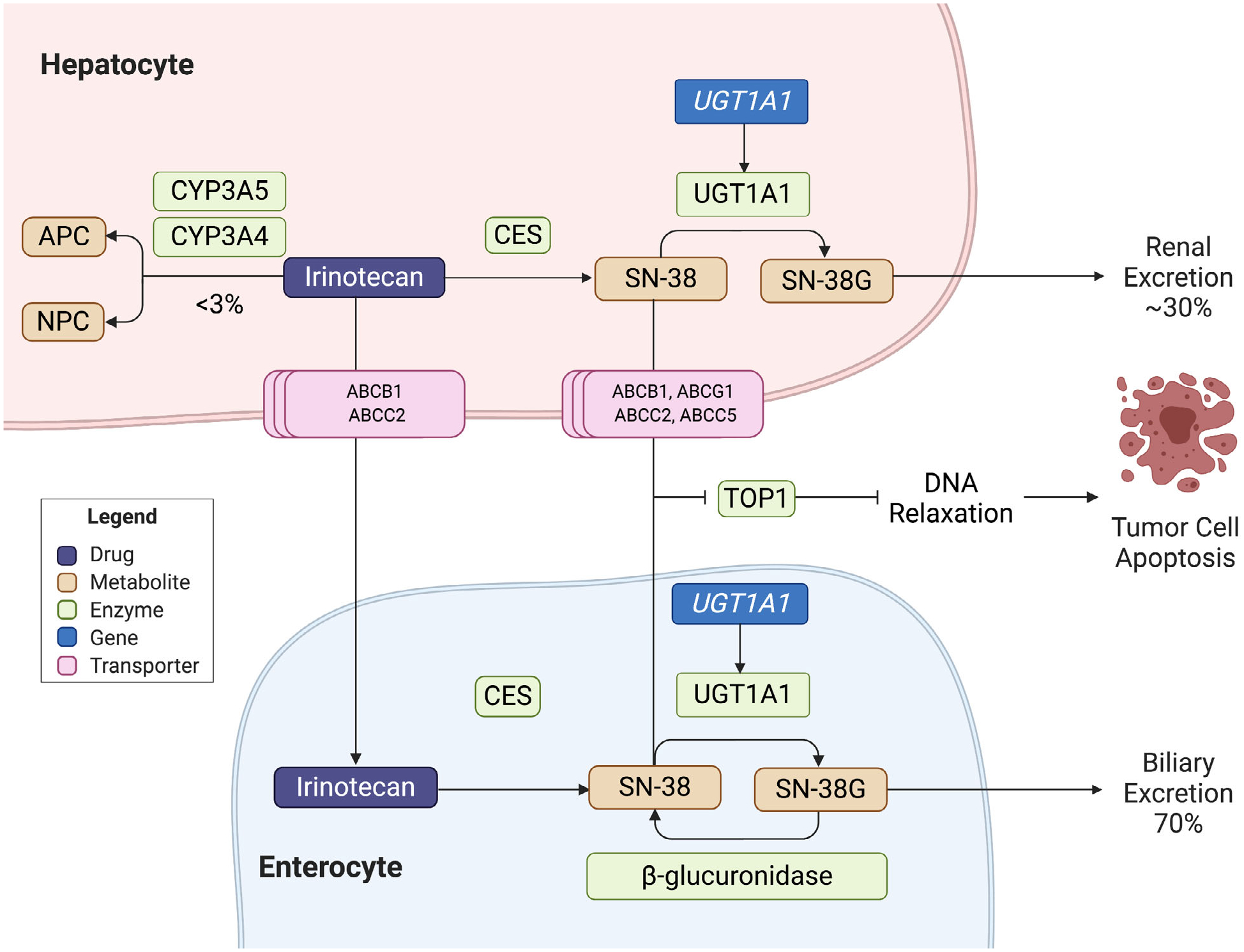
Irinotecan is a prodrug that undergoes conversion to its active metabolite, 7-ethyl-10-hydroxycamptothecin (SN-38), via carboxylesterase (CES)-mediated hydrolysis. CYP3A4/CYP3A5 oxidize SN-38 into inactive APC and NPC. SN-38 targets topoisomerase I to cause apoptosis. Detoxification occurs via uridine diphosphate-glucuronosyltransferase isoform 1A1 (UGT1A1). As the resulting glucuronide, SN-38G, is primarily excreted into bile, bacterial beta-glucuronidases can re-activate the metabolite. CES = carboxylesterases; NPC = 7-ethyl-10-[4-(1-piperidino)-1-amino] carbonyloxycamptothecin; APC = 7-ethyl-10-[4-N-(5-aminopentanoic acid)-1-piperidino] carbonyloxycamptothecin; ABCB1, ABCC2, ABCG2 = ABC dATP-binding cassette transporters; SN-38 = 7-ethyl-10-hydroxycamptothecin; SN-38G = glucuronidated SN-38; TOP1 = DNA Topoisomerase I; UGT1A1 = uridine-diphosphoglucuronate glucuronosyltransferase 1A1 (encoded by UGT1A1).
The SN-38 targets topoisomerase I to exert its cytotoxic effects by preventing DNA re-ligation of single strand breaks, establishing lethal double-stranded breaks that result in irreparable molecular damage and cell apoptosis. Due to the lipophilic nature of SN-38, the metabolite undergoes glucuronidation and detoxification by uridine diphosphate-glucuronosyltransferase isoform 1A1 (UGT1A1) encoded by the UGT1A1 gene in the liver and GI tract. The resulting water-soluble conjugated glucuronide, SN-38G, is primarily excreted via active transport into bile while ~30% undergoes renal elimination.35 Reduced enzymatic activity of UGT1A1 can lead to elevated levels of SN-38 and subsequent unconjugated (indirect) bilirubin. The concentration of SN-38 in its corresponding cellular location typically corresponds to the toxicities observed. For example, higher rates of neutropenia are seen in individuals when increased concentrations of SN-38 are present in the plasma and the reversal of SN-38G back into active SN-38 by bacterial beta-glucuronidases in the intestinal lumen may further contribute to severe diarrhea and mucosal damage.35
Genetic Variants Associated with UGT1A1 and Chemotherapy-Related Toxicity
Genetic polymorphisms in the UGT1A1 gene can result in varying levels of UGT1A1 enzyme activity and severe dose-limiting toxicities in as many as 25% of patients treated with irinotecan.36 Although data for over 135 genetic variants of UGT1A1 is available, the *28 (rs8175347), *6 (rs4148323), *37 (rs8175347), and *80 (rs887829) alleles are commonly associated with reduced enzyme activity, with the two former variants directly related to irinotecan toxicity.36,37 Functional variants with clinical relevance are typically a result of alterations in protein formation or the number of repeat thy-mine-adenine (TA) dinucleotides within the DNA promoter region of the UGT1A1 gene.37
The gene UGT1A1*28 contains seven TA repeats (TA7), differing from the standard six TA repeats in the wild-type allele (TA6), and thus is referred to as an indel polymorphism. This extra repeat decreases the rate of transcription initiation of the UGT1A1 gene, leading to decreased enzyme activity and reduced glucuronidation of bilirubin and irinotecan.36 The *28 variant is also a common cause of Gilbert syndrome (a mild condition of reduced hepatic UGT1A1 activity resulting in indirect hyperbilirubinemia) and its more aggressive childhood subtype, Crigler-Najjar syndrome.36 Individuals with one copy of the *28 allele have a 35% decrease in transcriptional activity, whereas homozygous individuals may experience as much as 70%.36 Eight TA repeats occur in the UGT1A1*37 variant (TA8), leading to reduced promoter activity of UGT1A1 to levels lower than that of the *28 allele. In the *6 variant, an amino acid switch occurs from glycine to argi-nine at position 71 within the protein coding region, producing a missense mutation and reduced UGT1A1 enzyme activity. The *80 variant is reported to have uncertain function by itself, but when its reported with *28 and *37, due to linkage disequilibrium, its presence results in the classification of intermediate or poor metabolizer types.37 The presence of these genetic variants associated with reduced enzyme activity results in reduced glucuronidation and subsequent hyperbilirubinemia, ultimately pre-disposing individuals to irinotecan toxicity.
The gene UGT1A1*28 is the most common variant allele with a frequency of 42–45% in African Americans, 26–31% in individuals of European ancestry, and 9–16% in Asian populations (Table 3).36,37 The UGT1A1*6 variant is common in Asian populations and rarely found in European and African populations. With a frequency of 15–30% in Chinese, Korean, and Japanese populations, the presence of this variant in homozygous individuals (UGT1A1*6/*6) has been reported to serve as a predictor of severe toxicity within this patient population.36 The UGT1A1*37 is found almost exclusively in populations of African origins (2–7%), whereas the *80 variant occurs frequently in both African and European populations (30–45%).37
Table 3.
Actionable Pharmacogenetic Variants Impacting Response to Irinotecan
| UGT1A1 variant | Minor allele frequency | ||||||
|---|---|---|---|---|---|---|---|
| Haplotype | Nucleotide change | rsID | EA | AA | AN | Allele function | Impact on enzyme activity37 |
| *6 | c.211G>A | rs4148323 | 0.008 | 0.004 | 0.146 | Decreased | Decreased UGT1A1 activity |
| *28 | c.-53_-52[8] | rs8175347 | 0.317 | 0.373 | 0.148 | Decreased | Decreased UGT1A1 activity |
| *37 | c.-53_-52TA[9] | rs8175347 | 0.001 | 0.057 | 0.000 | Decreased | Decreased UGT1A1 activity |
| *80 | c.-364C>T | rs887829 | 0.314 | 0.450 | NR | Uncertain | Uncertain UGT1A1 activity |
AA = African American; AN = Asian (East); EA = European ancestry; NR = not reported; rsID = reference SNP cluster ID; UGT1A1 = uridine diphosphate-glucuronosyltransferase 1A1.
Variability in UGT1A1 activity was first discovered in 1998 by Ratain and colleagues, who later went on to lead a phase I trial in 2002 that correlated the presence of genetic variants to evident levels of toxicity.38,39 Since then, the development of severe side effects after treatment with irinotecan has been extensively studied with the *28 and *6 alleles (Table 4).40–48 A 2007 meta-analysis by Hoskins et al.40 evaluated the association between UGT1A1*28 and irinotecan-related toxicity, finding that the risk of severe neutropenia was dependent on the dose of irinotecan administered in homozygous individuals. The authors advised genotyping for the *28 allele in patients receiving irinotecan at doses of 250 mg/m2 or higher to mitigate the increased risk of drug-induced hematological toxicity. A subsequent 2010 meta-analysis by Hu and colleagues reported that the genotype was also associated with an increased risk of neutropenia at medium doses of 150–250 mg/m2 (relative risk [RR] = 2.0, p < 0.01) as well as low doses (< 150 mg/m2; RR = 2.4, p < 0.01).41 Although there has been mixed data regarding the development of severe diarrhea and the *28 allele, a 2017 meta-analysis by Liu and colleagues evaluating 58 studies in patients with GI and lung cancers determined that patients with heterozygous or homozygous genotypes had a greater prevalence of diarrhea when compared with wild-type patients (odds ratio [OR] = 2.18, p < 0.001).45
Table 4.
Literature Summary of UGT1A1 Variant Associations with Severe Toxicity
| Study | Study design | Total n patients | Tumor type | Regimen | UGT1A1 variant | Irinotecan dose | Main findings of grade > 3 toxicity | p value | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Páez et al., 201946 | Prospective randomized multicenter, European population | 82 | mCRC | FOLFIRI-based | *28 | Experimental (geno typing): *1/* 1–300 mg/m2 *1/*28–260 mg/m2 Control (no genotyping): 180 mg/m2 |
Toxicity in experimental vs control groups | |||
| Neutropenia | 15% vs 20.5% | NS | ||||||||
| Diarrhea | 2.5% vs 7.7% | NS | ||||||||
| Liu et al., 201745 | Meta-analysis (58 studies), Asian, white, mixed populations | 4898 | mCRC, SCLC, mNSCLC | Irinotecan-based | *28 | Multiple dosing protocols | Toxicity in *28/*28 and *1/*28 vs WT individuals | |||
| Neutropenia | (OR = 2.15, 95% CI = 1.71–2.70) | < 0.001 | ||||||||
| Diarrhea | (OR = 2.18, 95% CI = 1.68–2.83) | < 0.001 | ||||||||
| Toffoli et al., 201744 | Prospective dose escalation multicenter, European, North American populations | 48 | mCRC | FOLFIRI plus BEV | *28 | Initial to maximum tolerated: *1/*1, *1/*28: 260–370 mg/m2 |
Toxicity according to dose in *1/*28 vs WT individuals | |||
| Neutropenia | 260 mg/m | 1/10 vs 0/10 patients | NR | |||||||
| 310 mg/m2 | 2/10 vs 0/10 patients | NR | ||||||||
| Neutropenic sepsis | 370 mg/m2 | 0/3 vs 1/5 patients | NR | |||||||
| Diarrhea | 260 mg/m2 | 0/10 vs 1/10 patients | NR | |||||||
| 310 mg/m2 | 1/10 vs 2/10 patients | NR | ||||||||
| 370 mg/m2 | 1/3 vs 0/4 patients | NR | ||||||||
| Arrhythmia | 260 mg/m2 | 1/10 vs 0/10 patients | NR | |||||||
| Mucositis | 310 mg/m2 | 0/10 vs 1/10 patients | NR | |||||||
| N/V | 370 mg/m2 | 0/3 vs 1/4 patients | NR | |||||||
| Marcuello et al., 201143 | Prospective dose escalation, European population | 98 | mCRC | FOLFIRI | *28 | Initial to maximum tolerated: *1/*28: 110–390 mg/m2 *28/*28: 90–150 mg/m2 |
Toxicity according to dose in *1/*28 vs WT individuals | |||
| Neutropenia | 300 mg/m | 1/6 vs 0/3 patients | NR | |||||||
| 390 mg/m2 | 1/2 vs 0/6 patients | NR | ||||||||
| Diarrhea | 220 mg/m2 | 0/3 vs 1/6 patients | NR | |||||||
| 340 mg/m2 | 1/6 vs 0/6 patients | NR | ||||||||
| Asthenia | 260 mg/m2 | 0/3 vs 1/12 patients | NR | |||||||
| 390 mg/m2 | 1/2 vs 1/6 patients | NR | ||||||||
| Toxicity according to dose in *28/*28 individuals | ||||||||||
| Neutropenia | 150 mg/m | 1/5 patients | NR | |||||||
| Diarrhea | 90 mg/m2 | 1/6 patients | NR | |||||||
| Asthenia | 150 mg/m2 | 1/5 patients | NR | |||||||
| Toffoli et al., 201042 | Prospective dose escalation multicenter, European population | 59 | mCRC | FOLFIRI | *28 | Initial to maximum tolerated: *1/*1, *1/*28: 215–370 mg/m2 |
Toxicity according to dose in *1/*28 vs WT individuals | |||
| Neutropenia | 370 mg/m2 | 1/4 vs 0/10 patients | NR | |||||||
| Diarrhea | 215 mg/m2 | 1/6 vs 0/4 patients | NR | |||||||
| 260 mg/m2 | 0/4 vs 1/12 patients | NR | ||||||||
| Asthenia | 370 mg/m2 | 1/4 vs 0/10 patients | NR | |||||||
| Stomatitis | 310 mg/m2 | 0/10 vs 1/6 patients | NR | |||||||
| Hu et al., 201041 | Meta-analyses (9 studies), European, North American, Asian populations | 1998 | Solid, various | Irinotecan-based | *28 | High: ≥ 250 mg/m2
Medium: 150–250 mg/mg2 Low: < 150 mg/m2 |
Neutropenia in *28/*28 vs. *1/*28 and WT individuals | |||
| High dose | (RR = 7.22, 95% CI = 3.10–16.78) | 0.003 | ||||||||
| Medium dose | (RR = 2.00, 95% CI = 1.62–2.47) | < 0.001 | ||||||||
| Low dose | (RR = 2.43, 95% CI = 1.34–4.39) | < 0.001 | ||||||||
| Neutropenia in *1/*28 vs WT individuals | ||||||||||
| High dose | (RR = 2.65, 95% CI = 0.70–9.94) | 0.149 | ||||||||
| Medium dose | (RR = 1.29, 95% CI = 1.04–1.62) | 0.023 | ||||||||
| Low dose | (RR = 2.94, 95% CI = 1.36–6.35) | 0.006 | ||||||||
| Hoskins et al., 200740 | Meta-analyses (9 studies), European, North American populations | 821 | Solid, various | Irinotecan-based | *28 | High: > 250 mg/m2
Medium: 150–250 mg/mg2 Low: < 150 mg/m2 |
Hematological toxicity in *28/*28 vs *1/*28 and WT individuals | |||
| High dose | (OR = 27.8, 95% CI = 4.0–195) | 0.005 | ||||||||
| Medium dose | (OR = 3.22, 95% CI = 1.52–6.81) | 0.008 | ||||||||
| Low dose | (OR = 1.80, 95% CI = 0.37–8.84) | 0.41 | ||||||||
| Diarrhea | 10.5% vs 5.1% vs 5.6% | 0.648 | ||||||||
| Zhang et al., 201748 | Meta-analyses (12 studies), Asian population | 1140 | Solid, various | Irinotecan-based | *6 | High: ≥ 150 mg/m2
Low: < 150 mg/m2 |
Neutropenia in *28/*28 vs. *1/*28 and WT individuals | |||
| High dose | (OR = 1.97, 95% CI = 1.47–2.67) | < 0.001 | ||||||||
| Low dose | (OR = 2.66, 95% CI = 1.10–6.45) | 0.03 | ||||||||
| Neutropenia in *28/*28 vs. WT individuals | ||||||||||
| High dose | (OR = 2.89, 95% CI = 1.69–4.94) | < 0.001 | ||||||||
| Low dose | (OR = 3.17, 95% CI = 1.11–9.04) | 0.19 | ||||||||
| Neutropenia in *1/*28 vs. WT individuals | ||||||||||
| High dose | (OR = 1.65, 95% CI = 1.15–2.35) | 0.003 | ||||||||
| Low dose | (OR = 2.36, 95% CI = 1.28–4.35) | 0.009 | ||||||||
| Cheng et al., 201447 | Meta-analyses (11 studies), Asian population | 1141 | Solid, various | Irinotecan-based | *6 | High/medium: ≥ 150 mg/m2a
Low: < 150 mg/m2a |
Toxicity in *28/*28 vs. WT individuals | |||
| Neutropenia | (OR = 4.44, 95% CI = 2.42–8.14) | < 0.001 | ||||||||
| Diarrhea | (OR = 3.51, 95% CI = 1.41–8.73’) | 0.007 | ||||||||
| Toxicity in *1/*28 vs. WT individuals | ||||||||||
| Neutropenia | (OR = 1.98, 95% CI = 1.45–2.71) | < 0.001 | ||||||||
| Diarrhea | (OR = 1.44, 95% CI = 0.84–2.49) | 0.186 | ||||||||
| Toxicity in *1/*28 vs. WT individuals | ||||||||||
| High dose | (OR = 1.65, 95% CI = 1.15–2.35) | 0.003 | ||||||||
| Low dose | (OR = 2.36, 95% CI = 1.28–4.35) | 0.009 | ||||||||
BEV = bevacizumab; CI = confidence interval; FOLFIRI = 5-fluorouracil and irinotecan; mCRC = metastatic colorectal cancer; mNSCLC = metastatic non-small cell lung cancer; N/V = nausea/vomiting; NR = not reported; NS = not significant; OR = odds ratio; RR = relative risk; SCLC = small cell lung cancer; UGT1A1 = uridine diphosphate-glucuronosyltransferase 1A1; WT = wild-type (*1/*1).
Toxicity defined per Common Terminology Criteria for Adverse Events (CTCAE) guidelines used by primary study authors.
Dose comparisons not analyzed in overall findings.
A number of genotype-guided irinotecan dose escalation studies have been conducted over the past decade. In a 2010 study by Toffoli and colleagues, it was demonstrated that higher doses of irinotecan in UGT1A1 wild-type (370 mg/m2 in *1/*1 genotype) and heterozygous individuals (310 mg/m2 in*1/*28 genotype) could safely be administered with infusional 5-FU every two weeks (FOLFIRI regimen) for mCRC compared with the standard dose of irinotecan 180 mg/m2.42 In 2017, Toffoli and colleagues evaluated irinotecan doses in patients treated with FOLFIRI plus bevacizumab, finding that slightly lower doses of 310 and 260 mg/m2 were tolerated in wild-type and heterozygous patients, respectively, although these were still higher than the standard dose.44 When considering patients with a homozygous genotype (*28/*28), a prospective dose-finding study by Marcuello and colleagues initiated these individuals on a biweekly dose of irinotecan 90 mg/m2 for a maximally tolerated dose of 130 mg/m2, which is an ~30% reduction in the standard dose.43 The authors also noted a poor overall tumor response rate of 13% in homozygous individuals, compared with rates of 60% in wild-type and 39% in heterozygous patients (p = 0.049), although these findings were primarily exploratory.43 The findings of these early-phase trials affirmed that higher than standard doses can be safely administered to UGT1A1*1/*1 and *1/*28 patients with colorectal cancer receiving FOLFIRI, leading to a recent multicenter randomized phase II trial by P aez and colleagues,46 which found that wild-type patients treated with a 300 mg/m2 dose of irinotecan and heterozygous patients treated with a 260 mg/m2 dose compared with those treated with standard dose yielded higher overall tumor response rates (67.5% vs 43.6%, p = 0.001). Significant differences in neutropenia, diarrhea, or asthenia were not evident between the groups.
Several studies within Asian populations have demonstrated that UGT1A1*6 can be used as a predictor of irinotecan-induced toxicity. Significant rates of severe neutropenia have been observed in the variant carriers compared with wild-type patients. A 2014, a meta-analysis by Cheng and colleagues evaluating associations between the variant and severe toxicity in Asian patients, reported the *6 polymorphisms could be used as potential biomarkers, as both heterozygous patients (OR = 1.98, p < 0.001) and homozygous patients (OR = 4.44, p < 0.001) had an increased risk of severe neutropenia, whereas severe diarrhea was only of significance in homozygous individuals (OR = 3.51, p = 0.007).47 In 2017, a meta-analysis by Zhang and colleagues48 assessed the association between the *6 allele and toxicity in Chinese, Japanese, Korean, and Thai populations, confirming that variant carriers were at an increased risk of irinotecan-induced neutropenia (p < 0.001). The authors also stated higher rates of irinotecan-induced grade 3–4 neutropenia were seen in heterozygous patients with lung (p = 0.019) and other cancers (excluding colorectal, gastric, and small cell lung; p = 0.001) while noting significant associations among homozygous individuals with colorectal cancer (p = 0.014), gastric cancer (p = 0.009), and other tumor types (excluding lung; p = 0.036).48 Higher rates of severe neutropenia also correlated to geographic region, as significant associations were seen among Chinese (OR = 1.73, p = 0.004) and Japanese (OR = 4.03, p < 0.001) populations.48 The authors concluded that further well-designed studies with the inclusion of more ethnic groups are needed to validate the currently established risks.
Genotype-Guided Prescribing
Given the prospective and retrospective evidence of irinotecan-induced toxicity based on UGT1A1 genotype, the FDA revised its drug labeling for irinotecan, acknowledging the increased risk of hematologic toxicity in *28 allele carriers.39,49–51 To counteract the increased risk of neutropenia, a reduction of irinotecan by at least one dose level (~20–40% reduction in the starting dose) is recommended for UGT1A1*28 homozygous individuals. For liposomal formulations of irinotecan, the recommended starting dose is 50 mg/m2. Subsequent dose modifications for both drug preparations can be considered on an individual patient basis. Therapeutic guidelines from the DPWG recommend an initial dose reduction of 30% for poor metabolizers with subsequent dose escalation guided by patient tolerance and neutrophil counts.52 At this time, neither the FDA nor DPWG recommend dose modifications for intermediate metabolizers (i.e., *1/*28) receiving treatment with irinotecan or therapeutic adjustments based on other UGT1A1 variant alleles.
Gaps in Evidence Base
An analysis by Gold and colleagues,53 showed that preemptive UGT1A1*28 PGx testing in patients with mCRC cost less and yielded slightly improved quality-adjusted life expectancy. In this modeling study, if a 25% dose reduction was performed in homozygous individuals (11% of the study population), it was estimated that 84.5 cases of severe neutropenia would have been avoided per 10,000 patients, saving US $2.7 million in treatment costs.53 Whereas the study showed that preemptive testing reduced costs with an estimated average saving of US $272 per patient, the authors emphasized that further studies are needed to evaluate the efficacy of reduced dose irinotecan in homozygous individuals to prevent compromising tumor outcomes.
Whereas the risk of irinotecan-associated adverse events is greater in patients with UGT1A1 variants due to increased systemic exposure to irinotecan and SN-38, previous genotype-guided dosing strategies were conducted during a time when higher doses (> 180 mg/m2) were commonly studied. Although many of these approaches aimed to demonstrate that increasing irinotecan dose by genotype confers improved response and/or survival compared with the standard dose, the prescribing of these increased doses has not been widely utilized in practice, thus limiting the relevance of genotype-based toxicity results. Implementation of UGT1A1 genotyping has been slow partly due to this lack of consensus in correlating optimal dosage adjustments with doses used in current clinical practice. Given that applicable genotype-adjusted irinotecan doses may improve tumor response as recently evidenced by Catenacci and colleagues, it is anticipated that the results from additional and ongoing studies (ClinicalTrials.gov identifier NCT02138617, NCT01643499, and NCT01639326) will help accelerate UGT1A1 testing uptake into routine practice.54
Implementation of Pharmacogenetics into Clinical Practice
The accumulation of evidence linking genotypes with drug response and toxicity as well as the availability of evidence-based consensus guidelines and interdisciplinary stakeholder support are driving the implementation of PGx testing into practice. As an important member of the health care team, pharmacists can work with other health system leaders, such as physicians, laboratory professionals, and genetic counselors to develop protocols to implement PGx testing. In addition, pharmacists are well suited to operationalize efforts, including the ordering of PGx tests and the reporting and interpretation of test results, to guide optimal drug selection and dosing (Figure 3).55
Figure 3. Considerations for clinical implementation of DPYD and UGT1A1 genotyping.
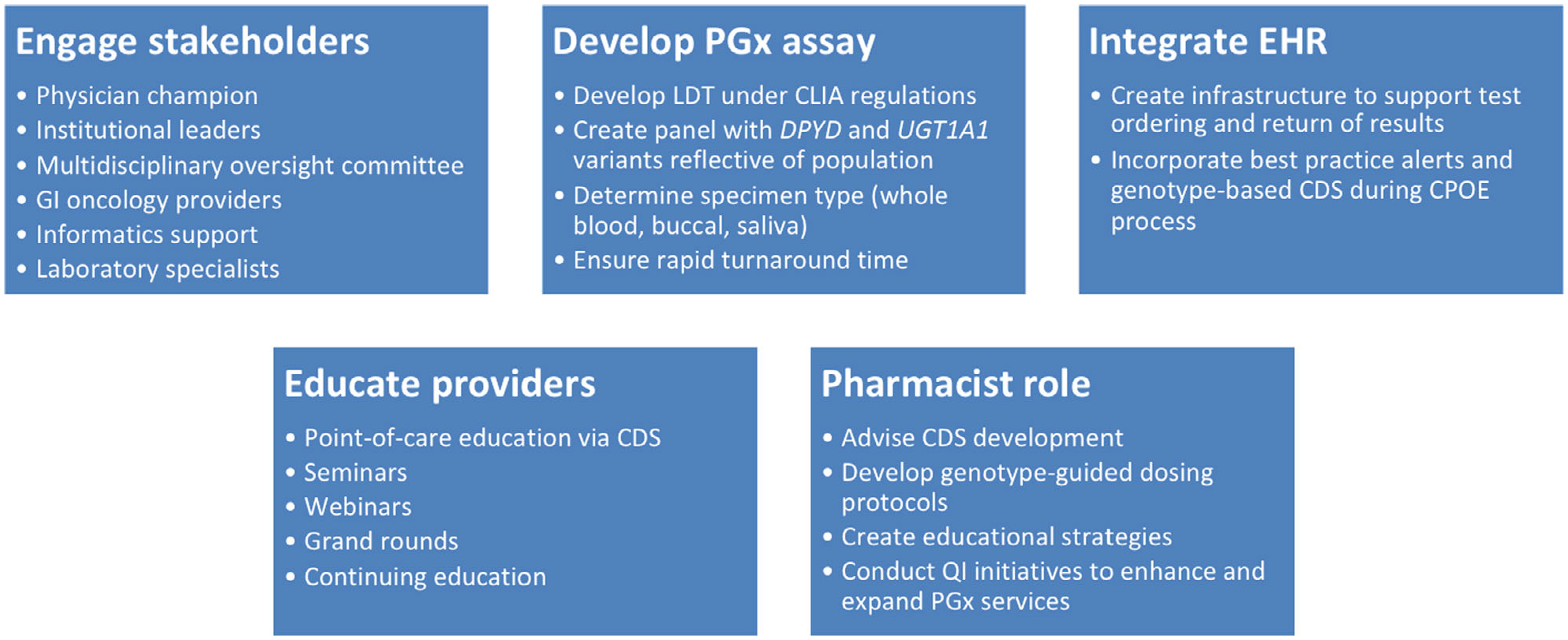
CDS = clinical decision support; CLIA = Clinical Laboratory Improvement Amendments; CPOE = computerized physician order entry; GI = gastrointestinal; LDT = laboratory developed test; PGx = pharmacogenetic; QI = quality improvement.
Assay Availability
Seamless integration of PGx into patient care involves appropriate oversight of genetic testing within the laboratory workspace. Tests can be performed using send-out commercial test kits (if available) or through in-house laboratory developed tests that have met validation and accreditation standards per CLIA regulations set by the Center for Medicaid and Medicare Services. The practice of routine PGx testing is often challenged by laboratory turnaround times (TATs) of results. TATs can vary from days to weeks depending on the test (i.e., single-gene vs multigene panel) and testing technology (i.e., genotyping vs sequencing).
Given that longer TATs are a barrier to the implementation of these tests in clinical practice, studies evaluating the clinical utility of PGx testing have reported that implementation is more practical when PGx results are returned within an acceptable time frame. The study protocol by Henricks and colleagues required a TAT of 7 days at most for treating physicians receiving preemptive genotyping results performed at in-house laboratories.8 A quality improvement initiative by Kasi and colleagues,56 showed that point-of-care send-out panels of PGx results were returned within 3 to 5 days (mean = 3.19 ± 1.69 days). DPYD and UGT1A1 assays should aim to provide results within 3 to 7 days to account for the variability in obtaining chemotherapy prior authorizations from health insurance plans and other clinical workflow logistics. Results should also contain standardized and easily interpretable information to ensure its utility among providers at the time of prescribing.
Integrating Pharmacogenetic Results into the Electronic Health Record
The ordering and storage of PGx test results into the electronic health record (EHR) to assist clinicians in clinical decision making further drives test utility. Electronic health record (EHR) terminologies and standards, such as Health Level Seven (HL7), Logical Observation Identifiers Names and Codes (LOINC), Systematized Nomenclature of Medicine (SNOMED), and Fast Healthcare Interoperability Resources (FHIR), support the discrete transfer of PGx results from the laboratory to the EHR. When paired with individual patient data, appropriate clinical decision support (CDS) can overcome the longstanding barrier of applying PGx test results to patient care. EHR integration of PGx has largely been developed by health care systems themselves, with past experiences noting that CDS elements of user interface design should include simple drug dose recommendations with adverse event implications, the significance and priority levels of applicable recommendations, and references to literature supporting the recommendations.57,58 More than 100 drugs contain genomic information in their FDA-approved product labeling and 24 clinical guidelines for 19 genes with therapeutic recommendations for over 50 drugs are available from the CPIC. As additional guidelines and clinically relevant drug-gene associations are discovered, incorporating adaptable CDS within the EHR will enable clinicians to manage and utilize new PGx evidence to the patient’s benefit.
Stakeholder Engagement
Clinical providers are a key stakeholder group that can propel the successful adoption of preemptive PGx testing strategies. Identifying a physician champion is critical in implementation to advocate for significant drug-gene pairs and the dissemination of evidence among prescribers.59 To increase awareness and garner further support, assessment of provider perceptions toward PGx dosing strategies and preparedness to use test results in practice can aid in identifying barriers and facilitating successful implementation.60 Cultivating support from institutional leadership and the formation of a multidisciplinary oversight committee are also essential in obtaining participation from all end users of clinical PGx services.
Provider Education
Providing a baseline understanding of PGx through clinician education is necessary to support test utility while addressing potential deficits in knowledge. Point-of-care education via appropriately designed CDS alerts is a favorable teaching method in disseminating new information, especially when linked to clinical guidelines and primary literature.57 Ongoing educational programs using evidence-based guidelines and data must be implemented to keep content current, accurate, and relevant in the context of clinical care. Modifying provider behavior is a multifaceted approach, but offering educational resources is vital in the successful implementation of clinical PGx.
Role of Pharmacists
Pharmacists are the medication experts and have long been tailoring medications based on patient-specific characteristics, such as kidney and liver function; incorporation of genetic information in therapeutic decision making falls within the domain of their pharmacy training. The profession has prepared pharmacists to apply PGx in practice by developing required didactic and experiential course offerings within pharmacy curricula for students. Advanced PGx training opportunities are now available with the establishment of residencies and fellowships, certificate programs, and continuing education courses. The need for clinical pharmacist input in research and implementation efforts has also been recognized by professional societies and other health care providers, allowing pharmacists to further demonstrate their value in the health system.61,62 Pre- and post-implementations efforts led by pharmacists in a pilot project at the University of Florida assessing genotype-guided antiplatelet therapy found that successful implementation required expertise in pharmacy informatics (for CDS development in the EHR), medication safety, medication-use policies and processes, and educational strategy development.61 With a deep-rooted background in pharmacology and medication management, pharmacists can build upon their clinical services and help execute PGx efforts as the medical community works toward fully embracing its clinical implementation.
Future Directions
Current Limitations to Testing
As PGx testing gains traction as a new clinical standard, test costs and reimbursement consistently remain as barriers to implementation. Given that the cost of testing largely depends on the genotype panel and health insurance coverage, controversy remains as to if and when a test should be ordered and reimbursed by the payor.63 Although PGx testing is a once-in-a-lifetime test and genotype-guided dosing strategies have shown to be less of an overall economic burden when compared with drug-related adverse event management costs, payors are still resistant to reimburse testing. Moreover, a study assessing PGx testing among private insurers found that test coverage policies were not readily accessible on company websites and that reimbursement largely varied according to the listed gene-drug pair, with only about 40% of known pairs covered in the policies.64 Recently, however, the Centers for Medicare and Medicaid Services (CMS) have posted a Local Coverage Determination (LCD) to cover PGx testing, including panel testing, to be effective in the summer of 2020.65
Uncertainty in health insurer coverage can cause providers to wholly refrain from ordering PGx tests to prevent delays in patient care. Moreover, providers are increasingly aware of the development of potential health disparities if patients opt to undergo testing using out-of-pocket expenses. From the patient perspective, while many acknowledge the value of PGx testing, most would only undergo testing if completely reimbursed from payors.66 Because PGx results have anticipated lifetime benefits, which will likely yield greater cost-savings in the future, insurers can help manifest PGx testing into practice by supporting test reimbursement. It is expected that pharmacoeconomic evaluations that demonstrate the benefit of PGx testing to prevent expensive hospitalizations related to drug toxicities will eventually convince payors to cover PGx tests.
Ongoing Collection of Clinical Utility Data
A growing amount of pharmacokinetic and retrospective data show promising results for preemptive DPYD and UGT1A1 testing with recent prospective data demonstrating their clinical validity in known variant carriers. Although there is literature supporting the clinical utility of DPYD and UGT1A1 testing, ongoing collection of these data will help drive policymakers and clinical providers translate this knowledge into routine clinical care. This type of evidence includes information from cost-effectiveness studies, implementation studies, and the number of patients needed to genotype to prevent one chemotherapy-related adverse event. Moreover, it is also important to recognize ongoing health disparities to implement programs to advance health equity among different ancestry groups. Given the greater prevalence of certain variants in different ancestry groups, such as the DPYD c.557A>G variant in the African ancestry population and UGT1A1*6 in the Asian population, further studies are needed to evaluate how these variants impact drug response in these diverse populations.67
Long-term data regarding tumor outcomes, such as progression-free survival (PFS) and overall survival (OS) are also of high interest as the limited data about the impact of PGx variants on survival has understandably limited the enthusiasm for preemptive PGx testing. There is also a concern that patients who carry a variant allele may never develop severe toxicity, and these individuals may end up being underdosed. For many of these reasons, it may be worth analyzing quality of life as an end point in clinical trials, particularly in the palliative (non-curative) setting. Favorable oncologic outcomes from prospective studies would certainly allow for wider acceptance of PGx testing in the health care community.
Additional Factors for Adverse Drug Reactions
When determining the optimal dose for any drug, utilizing genetic information is one piece of the puzzle among a variety of other patient-specific characteristics. These include physiological considerations (i.e., body weight and organ function) and environmental factors, such as concomitant medications, lifestyle habits, and smoking status. Patient or family history of intolerance of similar chemotherapy agents may also prompt prescribers to perform initial dose reductions as a precautionary measure. Because pharmacists are well versed in using laboratory parameters to optimize drug dosing, PGx test results should ultimately be treated as another pharmacokinetic/pharmacodynamic value during clinical assessment.
Exploratory Biomarkers
Many exploratory PGx markers in GI cancer treatment have emerged alongside the growing DPYD and UGT1A1 evidence base. These markers include variants in the thymidylate synthase (TYMS) and 5,10-methylenetetrahydrofolate reductase (MTHFR) genes for predicting toxicity with fluoropyrimidines and CYP3A4 with irinotecan. Genetic polymorphisms in TYMS are associated with drug resistance and lower survival, however, data predicting drug toxicity are not as robust as that of DPYD.68 Variants in MTHFR, a vital enzyme in intracellular folate metabolism and DNA synthesis, are associated with decreased enzyme activity, indirectly increasing the cytotoxic effects of fluoropyrimidines.68 A recent study by Pellicer and colleagues reported that MTHFR rs1801133 is significantly associated with the delayed administration of chemotherapy due to toxicity.69 Although the results of this study reportedly revived an interest of exploring MTHFR’s role in predicting fluoropyrimidine toxicity, validated evidence is not available to support testing of these markers in clinical practice at this time. Additionally, genetic variations of CYP3A4 (i.e., CYP3A4*2, CYP3A4*10, and CYP3A4*17) may play a role in the oxidation of SN-38 to form inactive metabolites and contribute to irinotecan toxicity but significant correlations have not yet been observed between these genotypes, total drug clearance, and symptom frequency.70 Currently, there are no accepted guidelines for managing patients with the aforementioned PGx variants until more evidence demonstrates their clinical relevance to therapy.
Conclusion
In the era of precision medicine, DPYD testing in patients before the initiation of fluoropyrimidine therapy to mitigate the risk of severe chemotherapy-related adverse events should be considered if optimal clinical and laboratory workflows are in place. In situations where individual DPYD pharmacogenetic testing may not be feasible, including the gene on a panel of matched germline genotyping alongside somatic tumor genetic testing in patients with tumor types treated with fluoropyrimidine agents could also be considered, as recently proposed by Hertz and Sahai.71 As more robust data from well-powered randomized controlled clinical trials become available through ongoing and future trials, it is anticipated that these results will be incorporated into clinical guideline recommendations and help drive reimbursement from payors. With the appropriate resources and support, pharmacists will be vital in leveraging their pharmacology knowledge and clinical skills to implement PGx testing in the clinic.
Acknowledgments
Figures 1 and 2 created with BioRender.com.
Footnotes
Conflict of Interest: The authors declare no conflicts of interest.
References
- 1.Food and Drug Administration. Table of Pharmacogenomic Biomarkers in Drug Labeling [Internet], 2020. Available from https://www.fda.gov/drugs/science-and-research-drugs/table-pharmacogenomic-biomarkers-drug-labeling. Accessed April 9, 2020.
- 2.McDermott U, Downing JR, Stratton MR. Genomics and the continuum of cancer care. N Engl J Med 2011;364(4):340–50. [DOI] [PubMed] [Google Scholar]
- 3.Relling MV, Klein TE. CPIC: clinical pharmacogenetics implementation consortium of the pharmacogenomics research network. Clin Pharmacol Ther 2011;89(3):464–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020;70(1):7–30. [DOI] [PubMed] [Google Scholar]
- 5.Lunenburg CATC, van der Wouden CH, Nijenhuis M, Crommentuijn-van Rhenen MH, de Boer-Veger NJ, Buunk AM, Houwink EJF, Mulder H, Rongen GA, van Schaik RHN, van der Weide J, Wilffert B, Deneer VHM, Swen JJ, Guchelaar H-J. Dutch Pharmacogenetics Working Group (DPWG) guideline for the gene-drug interaction of DPYD and fluoropyrimidines. Eur J Hum Genet 2020; 28(4): 508–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thorn CF, Marsh S, Carrillo MW, McLeod HL, Klein TE, Altman RB. PharmGKB summary: fluoropyrimidine pathways. Pharmacogenet Genomics 2011;21(4):237–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang N, Yin Y, Xu S-J, Chen W-S. 5-Fluorouracil: mechanisms of resistance and reversal strategies. Molecules 2008;13 (8):1551–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Henricks LM, Lunenburg CATC, de Man FM, et al. DPYD genotype-guided dose individualisation of fluoropyrimidine therapy in patients with cancer: a prospective safety analysis. Lancet Oncol 2018;19(11):1459–67. [DOI] [PubMed] [Google Scholar]
- 9.Offer SM, Wegner NJ, Fossum C, Wang K, Diasio RB. Phenotypic profiling of DPYD variations relevant to 5-fluorouracil sensitivity using real-time cellular analysis and in vitro measurement of enzyme activity. Cancer Res 2013;73(6):1958–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amstutz U, Henricks LM, Offer SM, et al. Clinical pharmacogenetics implementation consortium (CPIC) guideline for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing: 2017 update. Clin Pharmacol Ther 2018;103 (2):210–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Offer SM, Lee AM, Mattison LK, Fossum C, Wegner NJ, Diasio RB. A DPYD variant (Y186C) in individuals of african ancestry is associated with reduced DPD enzyme activity. Clin Pharmacol Ther 2013;94(1):158–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kleinjan JP, Brinkman I, Bakema R, van Zanden JJ, van Rooijen JM. Tolerance-based capecitabine dose escalation after DPYD genotype-guided dosing in heterozygote DPYD variant carriers: a single-center observational study. Anticancer Drugs 2019;30(4):410–5. [DOI] [PubMed] [Google Scholar]
- 13.Henricks LM, van Merendonk LN, Meulendijks D, et al. Effectiveness and safety of reduced-dose fluoropyrimidine therapy in patients carrying the DPYD*2A variant: a matched pair analysis. Int J Cancer 2019;144(9):2347–54. [DOI] [PubMed] [Google Scholar]
- 14.Deenen MJ, Meulendijks D, Cats A, et al. Upfront genotyping of DPYD*2A to individualize fluoropyrimidine therapy: a safety and cost analysis. J Clin Oncol 2016;34(3):227–34. [DOI] [PubMed] [Google Scholar]
- 15.Terrazzino S, Cargnin S, Del Re M, Danesi R, Canonico PL, Genazzani AA. DPYD IVS14+1G>A and 2846A>T genotyping for the prediction of severe fluoropyrimidine-related toxicity: a meta-analysis. Pharmacogenomics 2013;14(11):1255–72. [DOI] [PubMed] [Google Scholar]
- 16.Rosmarin D, Palles C, Church D, et al. Genetic markers of toxicity from capecitabine and other fluorouracil-based regimens: investigation in the QUASAR2 study, systematic review, and meta-analysis. J Clin Oncol 2014;32(10):1031–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meulendijks D, Henricks LM, Sonke GS, et al. Clinical relevance of DPYD variants c.1679T>G, c.1236G>A/HapB3, and c.1601G>A as predictors of severe fluoropyrimidine-associated toxicity: a systematic review and meta-analysis of individual patient data. Lancet Oncol 2015;16(16):1639–50. [DOI] [PubMed] [Google Scholar]
- 18.Saif MW, Lee AM, Offer SM, McConnell K, Relias V, Diasio RB. A DPYD variant (Y186C) specific to individuals of African descent in a patient with life-threatening 5-FU toxic effects: potential for an individualized medicine approach. Mayo Clin Proc 2014;89(1):131–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Latremouille-Viau D, Chang J, Guerin A, et al. The economic burden of common adverse events associated with metastatic colorectal cancer treatment in the United States. J Med Econ 2017;20(1):54–62. [DOI] [PubMed] [Google Scholar]
- 20.Henricks LM, Lunenburg CATC, de Man FM, et al. A cost analysis of upfront DPYD genotype-guided dose individualisation in fluoropyrimidine-based anticancer therapy. Eur J Cancer 2019;107:60–7. [DOI] [PubMed] [Google Scholar]
- 21.Rivers Z, Stenehjem DD, Jacobson P, Lou E, Nelson A, Kuntz KM. A cost-effectiveness analysis of pretreatment DPYD and UGT1A1 screening in patients with metastatic colorectal cancer (mCRC) treated with FOLFIRI+bevacizumab (FOLFIRI+-Bev). JCO 2020;38(4_suppl):168. [Google Scholar]
- 22.Lee JJ, Beumer JH, Chu E. Therapeutic drug monitoring of 5-fluorouracil. Cancer Chemother Pharmacol 2016;78(3):447–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morawska K, Goirand F, Marceau L, et al. 5-FU therapeutic drug monitoring as a valuable option to reduce toxicity in patients with gastrointestinal cancer. Oncotarget 2018;9 (14):11559–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Staveren MC, Guchelaar HJ, van Kuilenburg ABP, Gelderblom H, Maring JG. Evaluation of predictive tests for screening for dihydropyrimidine dehydrogenase deficiency. Pharmacogenomics J 2013;13(5):389–95. [DOI] [PubMed] [Google Scholar]
- 25.Genetic Testing Registry [Internet]. Available from https://www.ncbi.nlm.nih.gov/gtr/all/tests/?term=dpd&filter=certification:clia. Accessed July 22, 2020.
- 26.Ciccolini J, Del Re M, Danesi R, Milano G, Schellens JHM, Raymond E. Predicting fluoropyrimidine-related toxicity: turning wish to will, the PAMM-EORTC position. Ann Oncol 2018;29(9):1893–4. [DOI] [PubMed] [Google Scholar]
- 27.Lunenburg CATC, Henricks LM, Guchelaar H-J, et al. Prospective DPYD genotyping to reduce the risk of fluoropyrimidine-induced severe toxicity: Ready for prime time. Eur J Cancer 2016;54:40–8. [DOI] [PubMed] [Google Scholar]
- 28.Milano G DPD testing must remain a recommended option, but not a recommended routine test. Ann Oncol 2017;28 (6):1399. [DOI] [PubMed] [Google Scholar]
- 29.Xeloda (capecitabine) [package insert] [Internet]. San Francisco, CA: Genentech, Inc., 2001. Available from https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=a1de8bba-3b1d-4c9d-ab8a-32d2c05e67c8. Accessed April 9, 2020. [Google Scholar]
- 30.Fluorouracil [package insert] [Internet]. Spectrum Pharmaceuticals, Inc., 1956. Available from https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/012209s040lbl.pdf. Accessed April 9, 2020. [Google Scholar]
- 31.NCCN Clinical Practice Guidelines in Oncology: Colon Cancer. Version 2.2020 [Internet]. National Comprehensive Cancer Network, 2020. Available from www.nccn.org/professionals/physician_gls/pdf/colon.pdf. Accessed April 22, 2020. [Google Scholar]
- 32.EMA recommendations on DPD testing prior to treatment with fluorouracil, capecitabine, tegafur and flucytosine [Internet]. European Medicines Agency, 2020. Available from https://www.ema.europa.eu/en/news/ema-recommendations-dpd-testing-prior-treatment-fluorouracil-capecitabine-tegafur-flucytosine. Accessed July 22, 2020. [Google Scholar]
- 33.Van Cutsem E, Cervantes A, Adam R, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol 2016;27(8):1386–422. [DOI] [PubMed] [Google Scholar]
- 34.Deenen MJ, Tol J, Burylo AM, et al. Relationship between single nucleotide polymorphisms and haplotypes in DPYD and toxicity and efficacy of capecitabine in advanced colorectal cancer. Clin Cancer Res 2011;17(10):3455–68. [DOI] [PubMed] [Google Scholar]
- 35.Whirl-Carrillo M, McDonagh EM, Hebert JM, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther 2012;92(4):414–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dean L Irinotecan therapy and UGT1A1 genotype. In: Pratt VM, McLeod HL, Rubinstein WS, Scott SA, Dean LC, Kattman BL, Malheiro AJ, eds. Medical genetics summaries [Internet]. Bethesda (MD): National Center for Biotechnology Information, 2012. Available from http://www.ncbi.nlm.nih.gov/books/NBK294473/. Accessed April 9, 2020. [PubMed] [Google Scholar]
- 37.Gammal RS, Court MH, Haidar CE, et al. Clinical pharmacogenetics implementation consortium. clinical pharmacogenetics implementation consortium (CPIC) guideline for UGT1A1 and atazanavir prescribing. Clin Pharmacol Ther 2016;99(4): 363–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iyer L, King CD, Whitington PF, et al. Genetic predisposition to the metabolism of irinotecan (CPT-11). Role of uridine diphosphate glucuronosyltransferase isoform 1A1 in the glucuronidation of its active metabolite (SN-38) in human liver microsomes. J Clin Invest 1998;101(4):847–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Innocenti F, Undevia SD, Iyer L, et al. Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. JCO 2004;22(8):1382–8. [DOI] [PubMed] [Google Scholar]
- 40.Hoskins JM, Goldberg RM, Qu P, Ibrahim JG, McLeod HL. UGT1A1*28 genotype and irinotecan-induced neutropenia: dose matters. J Natl Cancer Inst 2007;99(17):1290–5. [DOI] [PubMed] [Google Scholar]
- 41.Hu Z-Y, Yu Q, Pei Q, Guo C. Dose-dependent association between UGT1A1*28 genotype and irinotecan-induced neutropenia: low doses also increase risk. Clin Cancer Res 2010;16(15):3832–42. [DOI] [PubMed] [Google Scholar]
- 42.Toffoli G, Cecchin E, Gasparini G, et al. Genotype-driven phase I study of irinotecan administered in combination with fluorouracil/leucovorin in patients with metastatic colorectal cancer. J Clin Oncol 2010;28(5):866–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marcuello E, Páez D, Paré L, Salazar J, Sebio A, del Rio E, Baiget M. A genotype-directed phase I-IV dose-finding study of irinotecan in combination with fluorouracil/leucovorin as first-line treatment in advanced colorectal cancer. Br J Cancer 2011;105(1):53–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Toffoli G, Sharma MR, Marangon E, et al. Genotype-guided dosing study of FOLFIRI plus bevacizumab in patients with metastatic colorectal cancer. Clin Cancer Res 2017;23(4):918–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu X-H, Lu J, Duan W, et al. Predictive value of UGT1A1*28 polymorphism in irinotecan-based chemotherapy. J Cancer 2017;8(4):691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Páez D, Tobeña M, Fernández-Plana J, et al. Pharmacogenetic clinical randomised phase II trial to evaluate the efficacy and safety of FOLFIRI with high-dose irinotecan (HD-FOLFIRI) in metastatic colorectal cancer patients according to their UGT1A 1 genotype. Br J Cancer 2019;120(2):190–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheng L, Li M, Hu J, et al. UGT1A1*6 polymorphisms are correlated with irinotecan-induced toxicity: a system review and meta-analysis in Asians. Cancer Chemother Pharmacol 2014;73(3):551–60. [DOI] [PubMed] [Google Scholar]
- 48.Zhang X, Yin J-F, Zhang J, Kong S-J, Zhang H-Y, Chen X-M. UGT1A1*6 polymorphisms are correlated with irinotecan-induced neutropenia: a systematic review and meta-analysis. Cancer Chemother Pharmacol 2017;80(1):135–49. [DOI] [PubMed] [Google Scholar]
- 49.Camptosar (irinotecan) [package insert] [Internet]. New York, NY: Pfizer Laboratories Div Pfizer Inc, 1996. Available from https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=e518dfc6-7e93-4fee-a66c-51e1ab71c056. Accessed April 9, 2020. [Google Scholar]
- 50.McLeod HL, Parodi L, Sargent DJ, Marsh S, Green E, Abreu P, Cisar LA, Goldberg RM. UGT1A1*28, toxicity and outcome in advanced colorectal cancer: results from trial N9741. JCO 2006;24(18_suppl):3520. [Google Scholar]
- 51.Cecchin E, Innocenti F, D’Andrea M, et al. Predictive role of the UGT1A1, UGT1A7, and UGT1A9 genetic variants and their haplotypes on the outcome of metastatic colorectal cancer patients treated with fluorouracil, leucovorin, and irinotecan. J Clin Oncol 2009;27(15):2457–65. [DOI] [PubMed] [Google Scholar]
- 52.Swen JJ, Nijenhuis M, de Boer A, et al. Pharmacogenetics: from bench to byte – an update of guidelines. Clin Pharmacol Ther 2011;89(5):662–73. [DOI] [PubMed] [Google Scholar]
- 53.Gold HT, Hall MJ, Blinder V, Schackman BR. Cost effectiveness of pharmacogenetic testing for uridine diphosphate glucuronosyltransferase 1A1 before irinotecan administration for metastatic colorectal cancer. Cancer 2009;115(17):3858–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Catenacci DVT, Chase L, Lomnicki S, et al. Evaluation of the association of perioperative UGT1A1 genotype-dosed gFOLFIRINOX with margin-negative resection rates and pathologic response grades among patients with locally advanced gastroesophageal adenocarcinoma: a phase 2 clinical trial. JAMA Netw Open 2020;3(2):e1921290. [DOI] [PubMed] [Google Scholar]
- 55.American Society of Health-System Pharmacists. ASHP statement on the pharmacist’s role in clinical pharmacogenomics. Am J Health Syst Pharm 2015;72(7):579–81. [DOI] [PubMed] [Google Scholar]
- 56.Kasi PM, Koep T, Schnettler E, et al. Feasibility of integrating panel-based pharmacogenomics testing for chemotherapy and supportive care in patients with colorectal cancer. Technol Cancer Res Treat 2019;18:1533033819873924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hicks JK, Dunnenberger HM, Gumpper KF, Haidar CE, Hoffman JM. Integrating pharmacogenomics into electronic health records with clinical decision support. Am J Health Syst Pharm 2016;73(23):1967–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Khelifi M, Tarczy-Hornoch P, Devine EB, Pratt W. Design recommendations for pharmacogenomics clinical decision support systems. AMIA Jt Summits Transl Sci Proc 2017;2017:237–46. [PMC free article] [PubMed] [Google Scholar]
- 59.Arwood MJ, Chumnumwat S, Cavallari LH, Nutescu EA, Duarte JD. Implementing pharmacogenomics at your institution: establishment and overcoming implementation challenges. Clin Transl Sci 2016;9(5):233–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Owusu Obeng A, Fei K, Levy KD, Elsey AR, Pollin TI, Ramirez AH, Weitzel KW, Horowitz CR. Physician-reported benefits and barriers to clinical implementation of genomic medicine: a multi-site IGNITE-network survey. J Pers Med 2018;8(3):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Owusu-Obeng A, Weitzel KW, Hatton RC, et al. Emerging roles for pharmacists in clinical implementation of pharmacogenomics. Pharmacotherapy 2014;34(10):1102–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hicks JK, Aquilante CL, Dunnenberger HM, et al. Precision pharmacotherapy: integrating pharmacogenomics into clinical pharmacy practice. J Am Coll Clin Pharm 2019;2(3):303–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Haga SB, Moaddeb J. Comparison of delivery strategies for pharmacogenetic testing services. Pharmacogenet Genomics 2014;24(3):139–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Park SK, Thigpen J, Lee IJ. Coverage of pharmacogenetic tests by private health insurance companies. J Am Pharm Assoc (2003) 2020;60:352–6.e3. [DOI] [PubMed] [Google Scholar]
- 65.Huddart R LCD announces coverage for PGx testing under medicare [Internet]. The PharmGKB Blog, 2020. Available from https://pharmgkb.blogspot.com/2020/06/lcd-announces-coverage-for-pgx-testing.html. Accessed July 31, 2020.
- 66.Bielinski SJ, St Sauver JL, Olson JE, et al. Are patients willing to incur out-of-pocket costs for pharmacogenomic testing? Pharmacogenomics J 2017;17(1):1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang H, De T, Zhong Y, Perera MA. The advantages and challenges of diversity in pharmacogenomics: can minority populations bring us closer to implementation? Clin Pharmacol Ther 2019;106(2):338–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.De Matti E, Roncato R, Dalle Fratte C, Ecca F, Toffoli G, Cecchin E. The use of pharmacogenetics to increase the safety of colorectal cancer patients treatedwith fluoropyrimidines. CDR; 2019. Available from https://cdrjournal.com/article/view/2994. Accessed April 9, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pellicer M, García-González X, García MI, et al. Identification of new SNPs associated with severe toxicity to capecitabine. Pharmacol Res 2017;120:133–7. [DOI] [PubMed] [Google Scholar]
- 70.Panczyk M Pharmacogenetics research on chemotherapy resistance in colorectal cancer over the last 20 years. World J Gastroenterol 2014;20(29):9775–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hertz DL, Sahai V. Including DPYD on cancer genetic panels to prevent fatal fluoropyrimidine toxicity. J Natl Compr Canc Netw 2020;18(4):372–4. [DOI] [PubMed] [Google Scholar]