

San Miguel et al evaluated the impact of dynamic, rather than single-point, assessment of measurable/minimal residual disease (MRD) by next-generation sequencing at a sensitivity of 10−5 during remission on outcome in over 1400 patients in 2 randomized trials of first-line myeloma therapy. MRD negativity sustained over 12 months is associated with superior progression-free survival over either MRD positivity or nonsustained MRD negativity, regardless of frontline treatment regimen.
Key Points
In patients with transplant-ineligible NDMM, durable MRD negativity is associated with improved PFS.
Daratumumab-based therapies are associated with higher rates and durability of MRD negativity.
Visual Abstract

Abstract
In patients with transplant-ineligible newly diagnosed multiple myeloma (NDMM), daratumumab reduced the risk of disease progression or death by 44% in MAIA (daratumumab/lenalidomide/dexamethasone [D-Rd]) and 58% in ALCYONE (daratumumab/bortezomib/melphalan/prednisone [D-VMP]). Minimal residual disease (MRD) is a sensitive measure of disease and response to therapy. MRD-negativity status and durability were assessed in MAIA and ALCYONE. MRD assessments using next-generation sequencing (10−5) occurred for patients achieving complete response (CR) or better and after at least CR at 12, 18, 24, and 30 months from the first dose. Progression-free survival (PFS) by MRD status and sustained MRD negativity lasting ≥6 and ≥12 months were analyzed in the intent-to-treat population and among patients achieving at least CR. In MAIA (D-Rd, n = 368; lenalidomide and dexamethasone [Rd], n = 369) and ALCYONE (D-VMP, n = 350; bortezomib/melphalan/prednisone [VMP], n = 356), the median duration of follow-up was 36.4 and 40.1 months, respectively. MRD-negative status and sustained MRD negativity lasting ≥6 and ≥12 months were associated with improved PFS, regardless of treatment group. However, daratumumab-based therapy improved rates of MRD negativity lasting ≥6 months (D-Rd, 14.9% vs Rd, 4.3%; D-VMP, 15.7% vs VMP, 4.5%) and ≥12 months (D-Rd, 10.9% vs Rd, 2.4%; D-VMP, 14.0% vs VMP, 2.8%), both of which translated to improved PFS vs control groups. In a pooled analysis, patients who were MRD negative had improved PFS vs patients who were MRD positive. Patients with NDMM who achieved MRD-negative status or sustained MRD negativity had deep remission and improved clinical outcomes. These trials were registered at www.clinicaltrials.gov as #NCT02252172 (MAIA) and #NCT02195479 (ALCYONE).
Introduction
Among patients with newly diagnosed multiple myeloma (NDMM), recent treatment advancements have improved long-term outcomes. However, with these improvements come unique challenges as clinicians evaluate the efficacy of emerging therapies. Specifically, the duration until read-out of clinical trials is long for traditionally used end points such as progression-free survival (PFS) and overall survival (OS), resulting in increased time until novel therapies are translated into clinical practice. Therefore, new disease assessment methods are needed that could serve as surrogate endpoints with more expedient read-out.
Minimal residual disease (MRD) is a sensitive measure of tumor cells in bone marrow that reflects remission status. Many studies have demonstrated that MRD-negative status is indicative of a deep response to therapy that is associated with improved PFS and OS.1-11 Although PFS and OS remain key outcomes in clinical studies, MRD status is being explored as a coprimary end point in clinical trials for multiple myeloma.9 Importantly, however, several aspects of MRD assessment require optimization and standardization, including patient selection, timing of assessment, sensitivity thresholds, frequency of monitoring, and testing methodologies.12 To this end, the International Myeloma Working Group (IMWG) criteria for assessing MRD negativity state are that patients must achieve a complete response or better (≥CR) and MRD-negative status, with a minimum sensitivity of 1 nucleated tumor cell in 100 000 normal cells (a 10‒5 threshold) either by next-generation sequencing or next-generation flow cytometry.13
Daratumumab is a human immunoglobulin Gκ monoclonal antibody targeting CD38 with a direct on-tumor14-17 and immunomodulatory18-20 mechanism of action. Daratumumab is approved across multiple lines of therapy for multiple myeloma21; daratumumab-based regimens consistently improve rates of MRD negativity and long-term outcomes such as PFS and OS relative to standard of care. Two phase 3 clinical studies, MAIA and ALCYONE, have evaluated daratumumab-based regimens for patients with transplant-ineligible NDMM.
In the primary analysis of MAIA with 28.0 months of median follow-up, daratumumab plus lenalidomide and dexamethasone (D-Rd) reduced the risk of disease progression or death by 44% compared with the control group (lenalidomide and dexamethasone [Rd]). Additionally, more D-Rd patients achieved MRD negativity compared with those who received Rd (24% vs 7%; P ≤ .001).10 With longer follow-up of MAIA (36.4 months), D-Rd vs Rd continued to improve clinical outcomes and also demonstrated improved MRD durability lasting ≥6 months (15% vs 4%; P < .0001) and ≥12 months (11% vs 2%; P < .0001).22 In the primary analysis of ALCYONE, daratumumab plus bortezomib, melphalan, and prednisone (D-VMP) reduced the risk of disease progression or death by 50% with 16.5 months of median follow-up compared with the control group (bortezomib, melphalan, and prednisone [VMP]).23 In support of the primary end point, the MRD-negativity rate at that time was also improved for D-VMP vs VMP (22% vs 6%; P < .001). With longer follow-up of ALCYONE (40.1 months), the clinical benefit of D-VMP was maintained; importantly, D-VMP reduced the risk of death by 40% compared with VMP (P = .0003). At the time of this longer follow-up, more patients who received D-VMP vs VMP achieved durable MRD negativity lasting ≥6 months (16% vs 5%; P < .0001) and ≥12 months (14% vs 3%; P < .0001).11 Both MAIA and ALCYONE demonstrated that daratumumab-based regimens improved outcomes compared with standard of care; in addition, they also demonstrated that achievement of MRD negativity was associated with longer PFS, irrespective of trial treatments.
Here we provide an evaluation of sustained MRD negativity in patients with transplant-ineligible NDMM; although the benefit of achieving MRD negativity has been well established, this study is the first to assess the prognostic value of sustained MRD negativity lasting ≥6 or ≥12 months in NDMM. Specifically, we present an analysis of the association of MRD durability with PFS using data from the phase 3 MAIA and ALCYONE studies after 36.4 and 40.1 months of median follow-up, respectively. These data support the use of MRD durability as a predictive and prognostic tool in NDMM and provide context for the length of MRD durability that is clinically meaningful.
Methods
Trial design and oversight
The study designs of the phase 3 randomized, open-label, multicenter MAIA (clinicaltrials.gov identifier: #NCT02252172)10 and ALCYONE (#NCT02195479)23 studies have been published previously with the primary end point analyses for each study. Briefly, MAIA and ALCYONE evaluated daratumumab plus Rd or VMP, respectively, in patients with transplant-ineligible NDMM. In both studies, patients had documented measurable disease according to IMWG criteria24 and were ineligible for high-dose chemotherapy or stem cell transplantation because of age (≥65 years) or unacceptable coexisting conditions. All patients provided written informed consent, and the studies were approved by independent ethics committees/institutional review boards and conducted in accordance with the Declaration of Helsinki and current International Conference on Harmonization Good Clinical Practice guidelines.
Randomization and study treatment
In each study, patients were randomized (1:1) to each treatment group based on stratification factors (International Staging System [ISS] disease stage [I vs II vs III, with higher stages indicating a poorer prognosis], geographic region [North America vs other for MAIA; Europe vs other for ALCYONE], and age [<75 vs ≥75 years]).10,23 In MAIA, all patients received lenalidomide (25 mg orally on days 1-21) and dexamethasone (40 mg weekly) during each 28-day cycle. Patients in the D-Rd group received daratumumab (16 mg/kg) weekly for cycles 1 and 2, every other week for cycles 3 to 6, and every 4 weeks thereafter. Study treatment continued until progressive disease or unacceptable toxicity. In ALCYONE, all patients received up to nine 42-day cycles of bortezomib (1.3 mg/m2 subcutaneously twice weekly during weeks 1, 2, 4, and 5 of cycle 1 and once weekly during weeks 1, 2, 4, and 5 of cycles 2-9), melphalan (9 mg/m2 orally on days 1-4 of each cycle), and prednisone (60 mg/m2 orally on days 1-4 of each cycle). In the D-VMP group, patients received daratumumab (16 mg/kg intravenously) weekly in cycle 1, every 3 weeks in cycles 2 to 9, and every 4 weeks thereafter until disease progression or unacceptable toxicity. For each study, pre- and postinfusion medications and dose modifications have been previously described.10,23
End points and assessments
For MAIA and ALCYONE, the primary end point was PFS and was reported previously.10,23 Response assessments and disease assessments were conducted using a central laboratory and a validated computer algorithm according to IMWG criteria.13,25,26 MRD assessments were to occur for all patients who achieved ≥CR. For patients who achieved ≥CR, additional MRD assessments occurred at 12, 18, 24, and 30 months after the first dose. MRD was assessed from bone marrow aspirates and evaluated with next-generation sequencing using the clonoSEQ assay (v.2.0; Adaptive Biotechnologies, Seattle, WA),27 according to IMWG criteria.13 MRD-negativity rate was defined as the proportion of patients who achieved ≥CR with negative MRD test results at any time during treatment. A minimum cell input equivalent to the given sensitivity threshold was required to determine MRD negativity (eg, MRD at 10−5 required that ≥100 000 cells were evaluated). A patient was considered MRD positive if MRD negativity was not achieved, if a test was inconclusive or missing, or if they did not reach a best response of ≥CR. Sustained MRD negativity, which was evaluated in the intent-to-treat (ITT) population, was defined as the maintenance of MRD negativity in bone marrow confirmed ≥6 or ≥12 months apart.
Statistical analyses
Methods supporting sample size determination and protocol-specified statistical analyses have been previously described.10,23 Post hoc analyses of PFS by MRD status and/or response category were evaluated, and a 2-sided P value is presented. PFS was compared between groups based on a log-rank test, and hazard ratios (HRs) and 95% confidence intervals (CIs) were estimated with a Cox regression model. Analysis with time-varying covariates were used to evaluate the correlation between PFS and response with MRD status. A univariate model was tested with MRD negativity at multiple time points as the sole time-varying covariate. All patients were considered MRD positive at baseline. A multivariate model with the following factors as covariates was also performed to determine whether the correlation was affected by any of these baseline factors: age (as reported in the case report form), ISS disease stage (I, II, III), baseline renal function (>60 mL/min, ≤60 mL/min), and cytogenetic risk (high, standard; risk was determined by fluorescence in situ hybridization or karyotype testing with high risk denoted by a positive test for any of the del17p, t[14;16], or t[4;14] molecular abnormalities). If values in baseline renal function or cytogenetic risk were missing, those patients were excluded from the multivariate model.
Results
Patients
In total, 737 patients in MAIA (D-Rd, n = 368; Rd, n = 369) and 706 patients in ALCYONE (D-VMP, n = 350; VMP, n = 356) were randomized to the daratumumab and control groups (supplemental Figure 1, available on the Blood Web site). Baseline characteristics were previously published.10,23 The median duration of follow-up was 36.4 (range, 0.0-49.9) months in MAIA and 40.1 (range, 0.0-52.1) months in ALCYONE. The majority (D-Rd/Rd: 93.1%; D-VMP/VMP: 91.9%) of patients eligible for MRD assessments (≥CR) provided a sample for MRD testing with successful calibration. A few eligible patients in each treatment group did not have a sample for MRD testing (D-VMP, 4.4% [7/160]; VMP, 7.8% [7/90]; D-Rd, 1.6% [3/182]; Rd, 5.0% [5/100]; supplemental Table 1).
MRD negativity and durability
In both MAIA and ALCYONE, daratumumab-based therapy led to improved rates of MRD negativity (10−5 threshold) compared with the standard of care in both the ITT populations (D-Rd, 28.8% vs Rd, 9.2%; P < .0001; D-VMP, 26.9% vs VMP, 7.0%; P < .0001) and among patients who achieved ≥CR (D-Rd, 58.2% vs Rd, 34.0%; P = .0001; D-VMP, 58.8% vs VMP, 27.8%; P < .0001; Table 1). MRD-negativity rates at the more stringent threshold of 10‒6 were also improved for the daratumumab treatment groups vs standard of care in the ITT populations of each study (D-Rd, 9.2% vs Rd, 3.3%; P = .0007; D-VMP, 9.1% vs VMP, 0.8%; P < .0001).
Table 1.
Rates of sustained MRD-negativity status in transplant-ineligible NDMM
| MRD negativity (10‒5) | MAIA | ALCYONE* | ||||
|---|---|---|---|---|---|---|
| D-Rd | Rd | P † | D-VMP | VMP | P † | |
| Intention-to-treat | n = 368 | n = 369 | n = 350 | n = 356 | ||
| MRD-negative status, n (%) | 106 (28.8) | 34 (9.2) | <.0001 | 94 (26.9) | 25 (7.0) | <.0001 |
| ≥6 mo sustained | 55 (14.9) | 16 (4.3) | <.0001 | 55 (15.7) | 16 (4.5) | <.0001 |
| ≥12 mo sustained | 40 (10.9) | 9 (2.4) | <.0001 | 49 (14.0) | 10 (2.8) | <.0001 |
| Complete response or better | n = 182 | n = 100 | n = 160 | n = 90 | ||
| MRD-negative status, n (%) | 106 (58.2) | 34 (34.0) | .0001 | 94 (58.8) | 25 (27.8) | <.0001 |
| ≥6 mo sustained | 55 (30.2) | 16 (16.0) | .0097 | 55 (34.4) | 16 (17.8) | .0055 |
| ≥12 mo sustained | 40 (22.0) | 9 (9.0) | .0053 | 49 (30.6) | 10 (11.1) | .0006 |
MRD data on durability from the ITT population of ALCYONE were reported previously.11
P value was calculated using Fisher's exact test.
MRD durability was assessed among patients achieving ≥2 MRD-negative results lasting ≥6 or ≥12 months with no MRD positive result in between. In each study, daratumumab was associated with higher rates of sustained MRD negativity in the ITT population lasting ≥6 months (MAIA: D-Rd, 14.9% vs Rd, 4.3%; P < .0001; ALCYONE: D-VMP, 15.7% vs VMP, 4.5%; P < .0001) and ≥12 months (D-Rd, 10.9% vs Rd, 2.4%; P < .0001; D-VMP, 14.0% vs VMP, 2.8%; P < .0001; Table 1). Similar observations occurred among patients who achieved ≥CR; daratumumab-based therapies were associated with improved MRD durability lasting ≥6 months (MAIA: D-Rd, 30.2% vs 16.0%; P = .0097; ALCYONE: D-VMP, 34.4% vs VMP, 17.8%; P = .0055) and ≥12 months (D-Rd, 22.0% vs Rd, 9.0%; P = .0053; D-VMP, 30.6% vs VMP, 11.1%; P = .0006; Table 1). Among patients who achieved MRD durability lasting ≥6 months but not ≥12 months, the main reason was because of unavailable data to demonstrate 12 months of durability (MAIA: 18 of 22; ALCYONE; 7 of 12), with only 4 patients in MAIA and 5 patients in ALCYONE having an MRD assessment at 12 months that was indeterminate or that turned MRD positive.
Baseline demographic and disease characteristics by MRD durability (MRD negativity lasting ≥6 months, not lasting ≥6 months, lasting ≥12 months, or not lasting ≥12 months) among patients in MAIA and ALCYONE are summarized in supplemental Tables 2 and 3. In general, baseline characteristics were comparable among patients who achieved sustained MRD negativity ≥12 months vs those who did not achieve ≥12-month MRD negativity within each study. Most characteristics reflected a comparable percentage of patients between treatment arms, but it should be noted that few patients (≤10) in the control arm of each study achieved sustained MRD negativity lasting ≥12 months. Among the small number of patients in the control arms who did achieve sustained MRD negativity ≥12 months, the majority were categorized as ISS stage I or II and had standard cytogenetic risk. In MAIA and ALCYONE, the proportion of patients with standard vs high cytogenetic risk was generally similar for those who achieved sustained MRD negativity compared with the ITT population (supplemental Tables 2 and 3), although the number of patients in the high cytogenetic risk subgroups was small.
PFS and MRD negativity
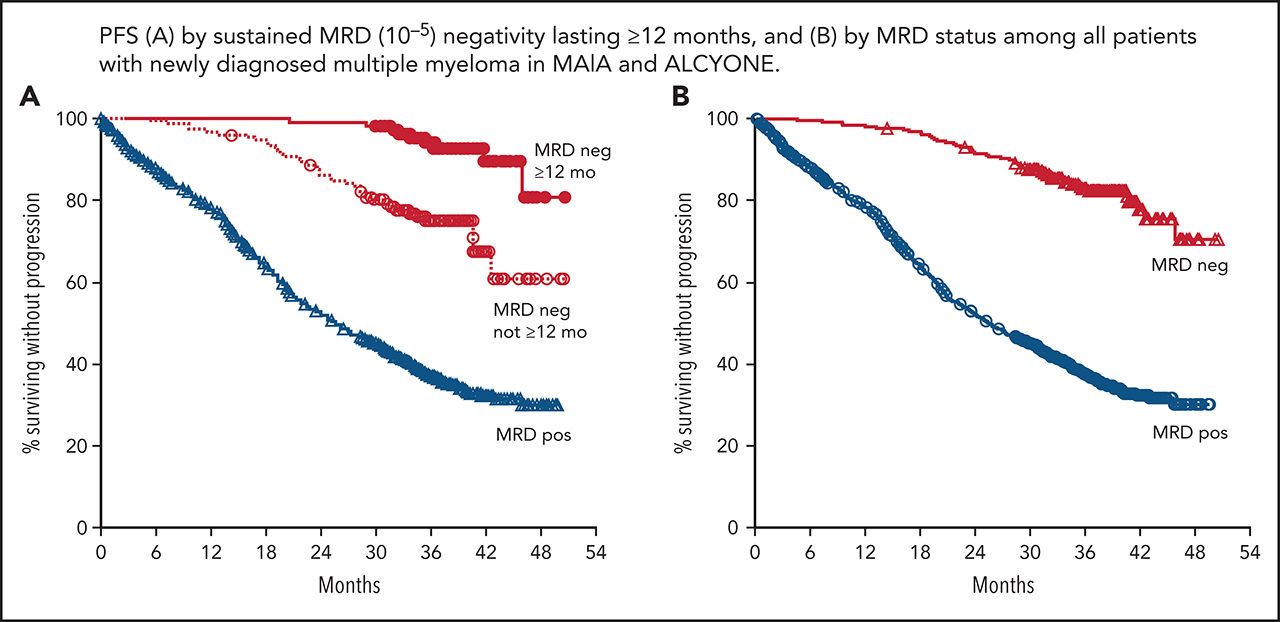
In the ITT populations of MAIA and ALCYONE, MRD-negative patients had improved PFS compared with MRD-positive patients (MAIA: HR, 0.15 [95% CI, 0.09-0.26]; P < .0001; ALCYONE: HR, 0.21 [95% CI, 0.15-0.30]; P < .0001; Figure 1; supplemental Figure 2). Consistent with these findings, PFS was also improved for patients who achieved sustained MRD negativity lasting ≥6 months (supplemental Figure 3A-C) or ≥12 months (Figure 2); similar analyses by treatment group demonstrated that the association of improved PFS with sustained MRD negativity was maintained regardless of treatment arm (supplemental Figures 3D-E and 4). Furthermore, a combined analysis of patients from MAIA and ALCYONE who received daratumumab-containing regimens (D-Rd and D-VMP, n = 718) or standard of care (Rd and VMP, n = 725) also demonstrated the clinical benefit of MRD negativity. PFS was prolonged in patients with sustained MRD durability lasting ≥6 months (supplemental Figure 5A) or ≥12 months (supplemental Figure 5B) compared with patients who did not achieve sustained MRD negativity or patients who were MRD positive. These data are supported by a Cox proportional hazards model showing that MRD negativity lasting ≥6 or ≥12 months are each associated with improved PFS in both univariate and multivariate analyses (Table 2).
Figure 1.

PFS based on MRD status (10−5) in MAIA and ALCYONE. Kaplan-Meier estimates of PFS by MRD status among patients in the ITT populations. MRD was assessed at a threshold of 1 tumor cell per 105 white blood cells. Red lines show MRD-negative patient populations and blue lines show MRD-positive patient populations (D-Rd/Rd shown for MAIA [A]; D-VMP/VMP for ALCYONE [B]).
Figure 2.

PFS based on sustained MRD negativity (10−5; ≥12 months) in MAIA, ALCYONE, and in both studies pooled. Kaplan-Meier estimates of PFS by sustained MRD negativity lasting ≥12 months among patients in the ITT populations. MRD status was assessed at a threshold of 1 tumor cell per 105 white blood cells. Red lines show MRD-negative patient populations and blue lines show MRD-positive patient populations (D-Rd/Rd shown for MAIA [A]; D-VMP/VMP for ALCYONE [B]; and D-Rd/Rd/D-VMP/VMP for all studies combined [C]).
Table 2.
Cox proportional hazards model for PFS with MRD durability status as a covariate
| Variable | MRD negativity sustained vs not sustained for ≥6 mo | MRD negativity sustained vs not sustained for ≥12 mo | ||
|---|---|---|---|---|
| HR (95% CI) | P | HR (95% CI) | P | |
| Univariate analysis | ||||
| Sustained MRD negativity (10−5) | 0.10 (0.06-0.18) | <.0001 | 0.09 (0.05-0.17) | <.0001 |
| Multivariate analysis 1 | ||||
| Sustained MRD negativity (10−5) | 0.12 (0.07-0.20) | <.0001 | 0.11 (0.05-0.21) | <.0001 |
| Treatment (daratumumab-containing regimen vs SoC) | 0.55 (0.48-0.64) | <.0001 | 0.55 (0.47-0.64) | <.0001 |
| Multivariate analysis 2 | ||||
| Sustained MRD negativity (10−5) | 0.11 (0.07-0.19) | <.0001 | 0.10 (0.05-0.20) | <.0001 |
| Treatment (daratumumab-containing regimen vs SoC) | 0.54 (0.47-0.63) | <.0001 | 0.54 (0.47-0.63) | <.0001 |
| Age (<75 vs ≥75 y) | 0.99 (0.85-1.15) | .8471 | 1.01 (0.87-1.17) | .9171 |
| ISS disease stage (I vs III) | 0.46 (0.38-0.57) | <.0001 | 0.48 (0.39-0.59) | <.0001 |
| ISS disease stage (II vs III) | 0.82 (0.70-0.96) | .0166 | 0.82 (0.70-0.97) | .0178 |
| Region (other vs NA/EU) | 0.72 (0.62-0.83) | <.0001 | 0.72 (0.62-0.84) | <.0001 |
NA/EU, North America/Europe; SoC, standard of care.
Data are for a univariate and multivariate analysis of combined data from the MAIA and ALCYONE studies among patients who did and did not achieve sustained MRD negativity lasting ≥6 months or patients who did and did not achieve sustained MRD negativity lasting ≥12 months. The following variables were evaluated: sustained MRD negativity, treatment group, age, ISS disease stage, and region.
In MAIA and ALCYONE, the median time to subsequent anticancer therapy (TTSAT) for patients who achieved MRD negativity was not reached among the daratumumab-treated groups or in the control arm of MAIA but was 44.4 months in the control arm of ALCYONE (Table 3). Among patients who were MRD positive, daratumumab therapy was associated with longer median TTSAT (MAIA: D-Rd, not reached vs Rd, 34.8 months; HR, 0.58 [95% CI, 0.44-0.75]; P < .0001; ALCYONE: D-VMP, 43.8 months vs VMP, 24.9 months; HR, 0.53 [95% CI, 0.42-0.68]; P < .0001; Table 3). For patients who were MRD negative at any time before initiating subsequent anticancer therapy, the risk of disease progression or death on the next subsequent line of therapy (PFS2) was not different for patients who received daratumumab-containing regimens or standard of care (supplemental Table 4); however, it should be noted that there were relatively few PFS2 events. Among patients who were MRD positive before subsequent anticancer therapy, PFS2 was not different for D-Rd vs Rd therapy; however, PFS2 was improved for patients who received D-VMP vs VMP (HR, 0.64 [95% CI, 0.49-0.83]; P = .0008; supplemental Table 4).
Table 3.
Time to next therapy by MRD status and durability
| TTSAT by MRD status | MAIA | ALCYONE | ||
|---|---|---|---|---|
| D-Rd (n = 368; ITT) | Rd (n = 369; ITT) | D-VMP (n = 350; ITT) | VMP (n = 356; ITT) | |
| MRD negative (10‒5) at ≥1 time point, n (%) * | 106 (28.8%) | 34 (9.2%) | 94 (26.9%) | 25 (7.0%) |
| Number of events (%); number censored (%)† | 5 (4.7%); 101 (95.3%) | 2 (5.9%); 32 (94.1%) | 13 (13.8%); 81 (86.2%) | 9 (36.0%); 16 (64.0%) |
| Median (95% CI), mo | NR (42.5-NE) | NR (NE-NE) | NR (46.4-NE) | 44.4 (36.5-NE) |
| HR (95% CI), P value | 0.54 (0.10-2.95); P = .4661‡ | 0.38 (0.16-0.88); P = .0197‡ | ||
| 36-mo TTSAT rate, % (95% CI) | 96.9 (90.6-99.0) | 90.5 (64.4-97.8) | 88.7 (80.1-93.8) | 75.3 (53.0-88.1) |
| MRD positive, n (%) * | 262 (71.2%) | 335 (90.8%) | 256 (73.1%) | 331 (93.0%) |
| Number of events (%); number censored (%)† | 82 (31.3%); 180 (68.7%) | 152 (45.4%); 183 (54.6%) | 110 (43.0%); 146 (57.0%) | 203 (61.3%); 128 (38.7%) |
| Median (95% CI), mo | NR (NE-NE) | 34.8 (29.2-NE) | 43.8 (35.3-NE) | 24.9 (21.9-27.3) |
| HR (95% CI), P value | 0.58 (0.44-0.75); P<.0001‡ | 0.53 (0.42-0.68); P<.0001‡ | ||
| 36-mo TTSAT rate, % (95% CI) | 65.4 (58.7-71.2) | 48.7 (42.6-54.5) | 54.9 (48.1-61.2) | 33.2 (27.7-38.8) |
| Achieved and remained MRD negative (10‒5) for ≥ 6 mo, n (%) * | 55 (14.9%) | 16 (4.3%) | 55 (15.7%) | 16 (4.5%) |
| Number of events (%); number censored (%)† | 2 (3.6%); 53 (96.4%) | 0 (0%); 16 (100.0%) | 5 (9.1%); 50 (90.9%) | 3 (18.8%); 13 (81.3%) |
| Median (95% CI), mo | NR (NE-NE) | NR (NE-NE) | NR (46.4-NE) | NR (44.4-NE) |
| HR (95% CI), P value | NR (0-NE); P = .4674‡ | 0.53 (0.13-2.22); P = .3746‡ | ||
| 36-mo TTSAT rate, % (95% CI) | 96.1 (85.2-99.0) | 100.0 (100.0-100.0) | 96.3 (85.9-99.1) | 93.8 (63.2-99.1) |
| MRD negativity (10−5) not lasting ≥6 mo, n (%) * | 51 (13.9%) | 18 (4.9%) | 39 (11.1%) | 9 (2.5%) |
| Number of events (%); number censored (%)† | 3 (5.9%); 48 (94.1%) | 2 (11.1%); 16 (88.9%) | 8 (20.5%); 31 (79.5%) | 6 (66.7%); 3 (33.3%) |
| Median (95% CI), mo | NR (42.48-NE) | NR (34.66-NE) | NR (NE-NE) | 32.6 (14.1-NE) |
| HR (95% CI), P value | 0.30 (0.4-2.17); P = .2069‡ | 0.28 (0.10-0.80); P = .0113‡ | ||
| 36-mo TTSAT rate, % (95% CI) | 98.0 (86.6-99.7) | 78.7 (31.8-95.1) | 77.2 (59.3-87.9) | 38.9 (9.3-68.7) |
| Achieved and remained MRD negative (10−5) for ≥12 mo, n (%) * | 40 (10.9%) | 9 (2.4%) | 49 (14.0%) | 10 (2.8%) |
| Number of events (%); number censored (%)† | 2 (5.0%); 38 (95.0%) | 0 (0%); 9 (100.0%) | 4 (8.2%); 45 (91.8%) | 1 (10.0%); 9 (90.0%) |
| Median (95% CI), mo | NR (NE-NE) | NR (NE-NE) | NR (46.4-NE) | NR (44.4-NE) |
| HR (95% CI), P value | NR (0-NE); P = .4975‡ | 0.99 (0.11-8.86); P = .9897‡ | ||
| 36-mo TTSAT rate, % (95% CI) | 94.6 (80.1-98.6) | 100.0 (100.0-100.0) | 95.8 (84.2-98.9) | 100.0 (100.0-100.0) |
| MRD negativity (10−5) not lasting ≥12 mo, n (%) * | 66 (17.9%) | 25 (6.8%) | 45 (12.9%) | 15 (4.2%) |
| Number of events (%); number censored (%)† | 3 (4.5%); 63 (95.5%) | 2 (8.0%); 23 (92.0%) | 9 (20.0%); 36 (80.0%) | 8 (53.3%); 7 (46.7%) |
| Median (95% CI), mo | NR (42.48-NE) | NR (34.66-NE) | NR (44.2-NE) | 37.0 (27.6-NE) |
| HR (95% CI), P value | 0.30 (0.04-2.17); P = .2082‡ | 0.36 (0.14-0.95); P = .0305‡ | ||
| 36-mo TTSAT rate, % (95% CI) | 98.5 (89.6-99.8) | 85.2 (47.6-96.6) | 80.5 (64.7-89.8) | 57.8 (29.0-78.4) |
NE, not evaluable; NR, not reached.
Percentages calculated using the total number of patients in each column heading (ITT population) as the denominator.
Percentages calculated using the number of patients in each column from the row immediately above number of events (%); number censored (%).
HR and 95% CI from a Cox proportional hazards model with treatment group as the sole explanatory variable. An HR < 1 indicates an advantage for D-Rd or D-VMP. P value is based on the log-rank test.
Although some variation occurred by treatment group, in general, estimated 36-month TTSAT rates were highest for patients with sustained MRD negativity lasting ≥6 months (MAIA: D-Rd, 96.1%, vs Rd, 100.0%; ALCYONE: D-VMP, 96.3% vs VMP, 93.8%; Table 3) and ≥12 months (MAIA: 94.6% vs 100.0%; ALCYONE: 95.8% vs 100.0%; Table 3) compared with patients who did not have MRD negativity lasting ≥6 months (MAIA: 98.0% vs 78.7%; ALCYONE: 77.2% vs 38.9%) and ≥12 months (MAIA: 98.5% vs 85.2%; ALCYONE: 80.5% vs 57.8%). Patients who were MRD positive had the shortest median time to next therapy (Table 3). In addition, estimated 36-month PFS2 rates were higher for MRD-negative patients compared with MRD-positive patients (supplemental Table 4).
Combined analysis of PFS by MRD negativity
In a combined analysis of patients from MAIA and ALCYONE, based on patients who achieved MRD negativity (n = 259) compared with patients who were MRD positive (n = 1184), patients who were MRD negative had improved PFS compared with patients who were MRD positive (HR, 0.19 [95% CI, 0.14-0.26]; P < .0001; Figure 3A). This trend was maintained irrespective of therapy regimen (Figure 3B). Among patients achieving MRD negativity, daratumumab-containing regimens improved PFS compared with standard of care (HR, 0.51 [95% CI, 0.28-0.92]; P < .0253; Figure 3B). In support of the observation that patients in the deepest response level had improved PFS, a Cox regression model with time-varying covariates showed that MRD negativity had an effect on PFS in both univariate and multivariate analyses (supplemental Table 5).
Figure 3.

PFS by MRD status (10−5) among all patients in MAIA and ALCYONE and in the pooled daratumumab-based combination groups vs control groups. Kaplan-Meier estimates of PFS based on MRD negativity in the ITT populations. MRD negativity was assessed at a threshold of 1 tumor cell per 105 white blood cells. (A) The red line shows patients who achieved MRD negativity at any time since randomization; the blue line shows patients who were MRD positive. (B) Red lines show regimens containing daratumumab (D-Rd and D-VMP); blue lines show standard of care regimens (Rd and VMP). A total of 5 patients who achieved a best response of VGPR were also MRD negative (all from the D-VMP arm of ALCYONE). Dara, daratumumab; VGPR, very good partial response.
Discussion
This analysis from two phase 3 studies of daratumumab plus standard-of-care regimens for the treatment of transplant-ineligible NDMM provides evidence that MRD negativity is associated longer PFS and that this benefit is improved for patients who reach durable MRD negativity. Although these data demonstrate the prognostic value of MRD durability lasting ≥12 months, which aligns with IMWG criteria,13 these data also support the clinical relevance and potential prognostic value of assessing MRD durability of shorter duration (ie, MRD negativity lasting ≥6 months). MRD negativity and durability were associated with improved PFS regardless of treatment regimen; however, daratumumab-based therapies drove more patients to achieve MRD-negative status and maintain MRD negativity for ≥6 and ≥12 months. It is possible that daratumumab-based therapies may induce longer periods of MRD negativity and deeper response; however, it is also possible that the continuous exposure to daratumumab alone or in combination with lenalidomide may have contributed to the longer periods of MRD negativity and deeper responses.
Our results are consistent with previous publications showing that MRD negativity is associated with improved PFS and OS for multiple myeloma,1-11 including results from 2 meta-analyses of patients primarily with NDMM.6,9 One analysis included 14 clinical studies and found MRD negativity to be correlated with improved PFS (HR, 0.41 [95% CI, 0.36-0.48]; P < .001) and OS (HR, 0.57 [95% CI, 0.46-0.71]; P < .001).6 Another meta-analysis evaluated 6 NDMM studies, including data from the primary analysis of ALCYONE, which are reported here; in that analysis, a correlation between MRD negativity and PFS was demonstrated by a weighted regression analysis.9 MRD negativity and durability were also measured in the FORTE study in patients with transplant-eligible NDMM.28 The treatment group with the highest rate of sustained MRD negativity (carfilzomib plus lenalidomide/dexamethasone with transplantation) also had improved PFS compared with the other study treatments (carfilzomib plus cyclophosphamide/dexamethasone plus transplantation or carfilzomib plus lenalidomide/dexamethasone without transplantation).28 Collectively, these studies are consistent with our present analysis; however, these studies were based on different MRD assessment methodologies, sensitivity thresholds, and together included diverse patient populations. In our study, we explore the correlation of MRD negativity with long-term outcomes including PFS and PFS2 using consistent assessment techniques, sensitivity thresholds, and similar patient populations. At the clinical cutoff date for these analyses, OS data were immature for MAIA, limiting the analysis of MRD status and durability as a surrogate end point for survival.
The current analysis demonstrated that patients who were MRD negative vs MRD positive had longer times to subsequent anticancer therapy and improved PFS2. Moreover, patients with sustained MRD negativity lasting either ≥6 or ≥12 months had the longest time to subsequent therapy. Although these data support the association of MRD negativity and durability with improved long-term outcomes, the impact on PFS2 requires longer follow-up because of the small number of events. Additionally, in ALCYONE, daratumumab therapy was associated with longer time to subsequent anticancer therapy not only for MRD-negative patients but also among MRD-positive patients, and daratumumab led to improved PFS2 among MRD-positive patients. Interestingly, this observation demonstrates a clinical benefit of daratumumab even among patients who do not reach MRD negativity.
A strength of this study is its focus on patients with transplant-ineligible NDMM with similar baseline demographic and disease characteristics who were prospectively enrolled in one of two phase 3 clinical studies. These patients benefitted from undergoing consistent MRD assessment methodologies at the same sensitivity threshold, underscoring the robustness of the dataset. Moreover, we also present strong evidence from a pooled analysis of patients from MAIA and ALCYONE, showing that patients who achieved MRD negativity had improved PFS compared with patients who were MRD positive. Although the proportion of patients with transplant-ineligible NDMM who achieved MRD negativity is a relatively small proportion of the study population, patients who did achieve deep response had improved outcomes. These data are supported by other studies that demonstrate PFS and OS were prolonged in MRD-negative patients with NDMM1-3,5,7,8 and in a previous report that achievement of CR in the absence of MRD negativity was not associated with prolonged PFS or OS.5 Taken together with data from the current study, this evidence suggests that focusing only on hematologic response (CR) without consideration of MRD status limits the prognostic impact for clinical outcomes.
These data together with observations from the current analysis indicate durable MRD negativity lasting ≥6 or ≥12 months may represent yet a deeper level of response with a higher prognostic value, suggesting MRD negativity may be a more robust evaluation of disease control if sustained over time. The present study supports this view by demonstrating improved PFS with sustained MRD negativity.
Supplementary Material
The online version of this article contains a data supplement.
{kind=link}
Acknowledgments
The authors thank the patients, volunteers, coinvestigators, staff members at the trial sites, and clinical site coordinators who participated in the MAIA and ALCYONE studies; representatives of the sponsor who were involved in data collection and analysis; Clarissa M. Uhlar, of Janssen Research & Development, for contributions to study design; and Charlotte Majerczyk, of Cello Health Communications/MedErgy, for medical writing and assistance, which was funded by the sponsor.
The studies were supported by Janssen Research & Development, LLC.
Footnotes
The data sharing policy of Janssen Pharmaceutical Companies of Johnson & Johnson is available at https://www.janssen.com/clinical-trials/transparency. As noted on this site, requests for access to the study data can be submitted through the Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.
The current affiliation for J.U. is US Medical Affairs, Hematology, Genmab US, Inc, Plainsboro, NJ.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: J.S.-M., H.A.-L., B.P., S.K., M.A.D., T.F., M.-V.M., C.T., A.J., S.Z.U., G.C., M.C., and H.Q. contributed to study design, data acquisition, and contributed to data analysis or interpretation; J.U., P.R., H.P., Mia Qi, S.S., J.W., M.K., N.D., C.H., R.V.R., A.K., R.K., and Ming Qi contributed to study design and contributed to data analysis or interpretation; and all authors reviewed the manuscript, approved the final version, decided to publish this report, and vouch for data accuracy and completeness.
Conflict-of-interest disclosure: J.S.-M. served as a consultant and sat on an advisory board for Amgen, Bristol Myers Squibb, Celgene, Janssen, MSD, Novartis, Takeda, Roche, Sanofi, GSK, AbbVie, and Karyopharm. H.A.-L. received honoraria from and served on a speaker’s bureau for Celgene, Amgen, Bristol Myers Squibb, Sanofi, and Janssen and received research funding from Celgene and Janssen. B.P. served as a consultant for and received honoraria from Amgen, Bristol Myers Squibb/Celgene, Janssen, and Takeda and received research support from Bristol Myers Squibb/Celgene, Sanofi, and Roche. S.K. received research funding from, served as a consultant, and sat on an advisory board for Celgene, Takeda, Janssen, AbbVie, Adaptive, KITE, and Medimmune/AstraZeneca; received research funding from Merck, Novartis, Roche, and Sanofi; and was an IRC member for Oncopeptides. M.A.D. received honoraria from Amgen, Takeda, Bristol Myers Squibb, Janssen, Celgene, and Beigene. T.F. served on a speaker’s bureau for Janssen, Bristol Myers Squibb, and Takeda and served on advisory boards for Janssen, Bristol Myers Squibb, Takeda, Roche, Amgen, Karyopharm, Sanofi, and Oncopeptides. M.-V.M. received honoraria from and served on an advisory board for Janssen, Celgene, Amgen, Takeda, AbbVie, GSK, Adaptive, Roche, Genentech, Pfizer, and Regeneron. A.J. received honoraria from and served as a consultant or sat on an advisory board for AbbVie, Amgen, Celgene/Bristol Myers Squibb, GSK, Janssen, and Karyopharm. S.Z.U. received research support personal fees from Amgen, Celgene, Sanofi, Seattle Genetics, Janssen, Takeda, SkylineDX, and Merck; received personal fees from AbbVie and MundiPharma; and received research support from Bristol Myers Squibb and Pharmacyclics. G.C. received honoraria from Amgen, Bristol Myers Squibb, Celgene, Janssen, Takeda, Roche, and Sanofi and received research support from Celgene, Janssen, and Takeda. M.C. received honoraria from and served on a speaker’s bureau for Janssen, Celgene/Bristol Myers Squibb, Sanofi, Takeda, Amgen, MundiPharma, AbbVie, Adaptive, and GSK and served on a speakers bureau for Janssen and Celgene/Bristol Myers Squibb. H.Q. received research funding from Amgen, Sanofi, Celgene, Karyopharm, and GSK and served on steering committees or advisory boards for Amgen, Celgene, Karyopharm, GSK, Janssen-Cilag, and Sanofi. J.U. was an employee of Janssen at the time of study and may hold stock. P.R., H.P., Mia Qi, S.S., J.W., M.K., N.D., C.H., R.V.R., A.K., R.K., and Ming Qi are employees of Janssen and may hold stock. N.J.B. received honoraria from and served as a consultant for AbbVie, Amgen, GSK, Janssen, and Karyopharm; received research funding, honoraria, and served as a consultant for Celgene/Bristol Myers Squibb; and received honoraria from Takeda. All other authors declare no competing financial interests.
Correspondence: Jesus San-Miguel, Clínica Universidad de Navarra, Centro de Investigación Médica Aplicada (CIMA), Instituto de Investigación Sanitaria de Navarra (IDISNA), CIBER-ONC number CB16/12/00369, Av de Pío XII, 55, 31008 Pamplona, Spain; e-mail: sanmiguel@unav.es.
REFERENCES
- 1.Attal M, Lauwers-Cances V, Hulin C, et al. ; IFM 2009 Study . Lenalidomide, bortezomib, and dexamethasone with transplantation for myeloma. N Engl J Med. 2017;376(14): 1311-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paiva B, Vidriales MB, Cerveró J, et al. ; GEM (Grupo Español de MM)/PETHEMA (Programa para el Estudio de la Terapéutica en Hemopatías Malignas) Cooperative Study Groups . Multiparameter flow cytometric remission is the most relevant prognostic factor for multiple myeloma patients who undergo autologous stem cell transplantation. Blood. 2008;112(10):4017-4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rawstron AC, Child JA, de Tute RM, et al. Minimal residual disease assessed by multiparameter flow cytometry in multiple myeloma: impact on outcome in the Medical Research Council Myeloma IX Study. J Clin Oncol. 2013;31(20):2540-2547. [DOI] [PubMed] [Google Scholar]
- 4.Martinez-Lopez J, Lahuerta JJ, Pepin F, et al. Prognostic value of deep sequencing method for minimal residual disease detection in multiple myeloma. Blood. 2014;123(20):3073-3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lahuerta JJ, Paiva B, Vidriales MB, et al. ; GEM (Grupo Español de Mieloma)/PETHEMA (Programa para el Estudio de la Terapéutica en Hemopatías Malignas) Cooperative Study Group . Depth of response in multiple myeloma: a pooled analysis of three PETHEMA/GEM clinical trials. J Clin Oncol. 2017;35(25):2900-2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Munshi NC, Avet-Loiseau H, Rawstron AC, et al. Association of minimal residual disease with superior survival outcomes in patients with multiple myeloma: a meta-analysis. JAMA Oncol. 2017;3(1):28-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Landgren O, Devlin S, Boulad M, Mailankody S. Role of MRD status in relation to clinical outcomes in newly diagnosed multiple myeloma patients: a meta-analysis. Bone Marrow Transplant. 2016;51(12):1565-1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perrot A, Lauwers-Cances V, Corre J, et al. Minimal residual disease negativity using deep sequencing is a major prognostic factor in multiple myeloma. Blood. 2018;132(23):2456-2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Avet-Loiseau H, Ludwig H, Landgren O, et al. Minimal residual disease status as a surrogate endpoint for progression-free survival in newly diagnosed multiple myeloma studies: a meta-analysis. Clin Lymphoma Myeloma Leuk. 2020;20(1):e30-e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Facon T, Kumar S, Plesner T, et al. ; MAIA Trial Investigators . Daratumumab plus lenalidomide and dexamethasone for untreated myeloma. N Engl J Med. 2019; 380(22):2104-2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mateos MV, Cavo M, Blade J, et al. Overall survival with daratumumab, bortezomib, melphalan, and prednisone in newly diagnosed multiple myeloma (ALCYONE): a randomised, open-label, phase 3 trial. Lancet. 2020;395(10218):132-141. [DOI] [PubMed] [Google Scholar]
- 12.Kostopoulos IV, Ntanasis-Stathopoulos I, Gavriatopoulou M, Tsitsilonis OE, Terpos E. Minimal residual disease in multiple myeloma: current landscape and future applications with immunotherapeutic approaches. Front Oncol. 2020;10:860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar S, Paiva B, Anderson KC, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016;17(8):e328-e346. [DOI] [PubMed] [Google Scholar]
- 14.de Weers M, Tai YT, van der Veer MS, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J Immunol. 2011;186(3):1840-1848. [DOI] [PubMed] [Google Scholar]
- 15.Lammerts van Bueren J, Jakobs D, Kaldenhoven N, et al. Direct in vitro comparison of daratumumab with surrogate analogs of CD38 antibodies MOR03087, SAR650984 and Ab79. Blood. 2014; 124(21):3474. [Google Scholar]
- 16.Overdijk MB, Verploegen S, Bögels M, et al. Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. MAbs. 2015;7(2):311-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Overdijk MB, Jansen JH, Nederend M, et al. The therapeutic CD38 monoclonal antibody daratumumab induces programmed cell death via Fcgamma receptor-mediated cross-linking. J Immunol. 2016;197(3):807-813. [DOI] [PubMed] [Google Scholar]
- 18.Krejcik J, Casneuf T, Nijhof IS, et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood. 2016;128(3):384-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adams HC III, Stevenaert F, Krejcik J, et al. High-parameter mass cytometry evaluation of relapsed/refractory multiple myeloma patients treated with daratumumab demonstrates immune modulation as a novel mechanism of action. Cytometry A. 2019;95(3):279-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Casneuf T, Adams HC III, van de Donk N, et al. Deep immune profiling of patients treated with lenalidomide and dexamethasone with or without daratumumab. Leukemia. 2020;35(2):573-584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Darzalex (daratumumab) . Package insert. Janssen Biotech; 2020. [Google Scholar]
- 22.Bahlis N, Facon T, Usmani SZ, et al. Daratumumab plus lenalidomide and dexamethasone (D-Rd) versus lenalidomide and dexamethasone (Rd) in patients with newly diagnosed multiple myeloma (NDMM) ineligible for transplant: updated analysis of MAIA. In: Proceedings from the 61st American Society of Hematology Annual Meeting & Exposition; 7-10 December 2019; Orlando, FL. [Google Scholar]
- 23.Mateos MV, Dimopoulos MA, Cavo M, et al. ; ALCYONE Trial Investigators . Daratumumab plus bortezomib, melphalan, and prednisone for untreated myeloma. N Engl J Med. 2018;378(6):518-528. [DOI] [PubMed] [Google Scholar]
- 24.Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538-e548. [DOI] [PubMed] [Google Scholar]
- 25.Durie BGM, Harousseau JL, Miguel JS, et al. ; International Myeloma Working Group . International uniform response criteria for multiple myeloma [corrections published in Leukemia. 2006;20:2220 and Leukemia. 2007;21:1134]. Leukemia. 2006;20(9):1467-1473. [DOI] [PubMed] [Google Scholar]
- 26.Rajkumar SV, Harousseau JL, Durie B, et al. ; International Myeloma Workshop Consensus Panel 1 . Consensus recommendations for the uniform reporting of clinical trials: report of the International Myeloma Workshop Consensus Panel 1. Blood. 2011;117(18):4691-4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adaptive Biotechnologies Corporation . clonoSEQ® Assay Technical Information. New York, NY: Adaptive Biotechnologies Corporation; 2018. [Google Scholar]
- 28.Gay F, Musto P, Rota Scalabrini D, et al. Survival analysis of newly diagnosed transplant-eligible multiple myeloma patients in the randomized FORTE trial [abstract]. Blood. 2020;136(suppl 1):35-37. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.