

Abstract
The development and function of tissues, blood, and the immune system is dependent upon proximity for cellular recognition and communication. However, the detection of cell-to-cell contacts is limited due to a lack of reversible, quantitative probes that can function at these dynamic sites of irregular geometry. Described here is a novel chemo-genetic tool developed for fluorescent detection of protein–protein proximity and cell apposition that utilizes the Fluorogen Activating Protein (FAP) in combination with a Dye Activated by Proximal Anchoring (DAPA). The FAP–DAPA system has two protein components, the HaloTag and FAP, expressed on separate protein targets or in separate cells. The proteins function to bind and activate a compound that has the hexyl chloride (HexCl) ligand connected to malachite green (MG), the FAP fluorogen, via a poly(ethylene glycol) spacer spanning up to 28 nm. The dehalogenase protein, HaloTag, covalently binds the HexCl ligand, locally concentrating the attached MG. If the FAP is within range of the anchored fluorogen, it will bind and activate MG specifically when the bath concentration is too low to saturate the FAP receptor. A new FAP variant was isolated with a 1000-fold reduced KD of ~10–100 nM so that the fluorogen activation reports proximity without artificially enhancing it. The system was characterized using purified FRB and FKBP fusion proteins and showed a doubling of fluorescence upon rapamycin induced complex formation. In cocultured HEK293 cells (HaloTag and FAP-expressing) fluorescence increased at contact sites across a broad range of labeling conditions, more reliably providing contact-specific fluorescence activation with the lower-affinity FAP variant. When combined with suitable targeting and expression constructs, this labeling system may offer significant improvements in on-demand detection of intercellular contacts, potentially applicable in neurological and immunological synapse measurements and other transient, dynamic biological appositions that can be perturbed using other labeling methods that stabilize these interactions.
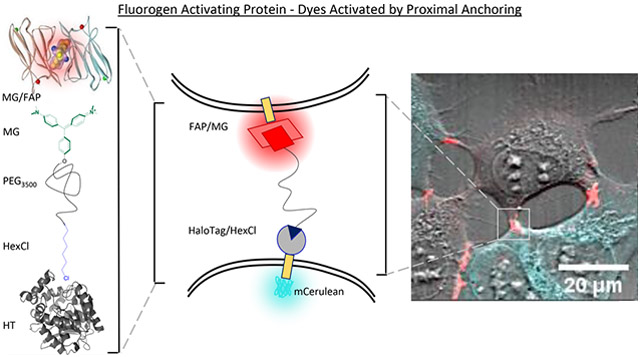
Graphical Abstract
INTRODUCTION
The detection and characterization of sites of cellular apposition are critical for understanding processes such as nerve impulses, the immune response, embryogenesis, and neural development where membrane apposition is required for the transmission of signals between cells. Detection of neural development is particularly challenging due to the complexity of the system and the need to map the connectivity dynamically across length scales from synapses (nm) to cells (mm). Techniques used to observe these systems often utilize data collection at only one of these scales, and at a fixed point in time. For instance, high resolution imaging techniques such as electron microscopy can acquire beautiful images of exact neuronal synapse morphology1,2 but generate enormous data sets when attempting to analyze neuronal connectivity in the brain,3 limiting data collection and analysis to mm3 volumes. Conversely, fluorescence microscopy can image large sections of tissue4 but is constrained by fundamental limits in resolution5 and a general lack of contrast for other cellular structures unless additional fluorophores are employed.
Another approach is to develop fluorescent tools that provide an emergent signal at sites of cellular apposition. These tools must possess all attributes of a good sensor of protein–protein interactions6-8 and also function in tissue and across distances of about 20 nm, the dimension typical at sites of cellular apposition including adherent cell contact sites, neuronal and immunological synapses, and vesicular contact sites.9-11 Therefore, only a few tools have successfully been implemented for the detection of cell contacts.
The most prevalent tool for detection of protein proximity across sites of cellular apposition is the class of split fluorescent proteins applied to detection of neuronal synapses.12 These tools are advantageous because they emit fluorescence in multiple colors13,14 and have been demonstrated to report different aspects of synaptic anatomy and function.15-19 For example, depending on proteins functionalized with the fluorescent protein fragments, reconstitution may reflect the presence14-17 or activity of a neuronal synapse.13 Outside of neuroscience, split fluorescent protein tools have also been applied to the detection of organelle interaction.20 A limitation of contact sensors based on split fluorescent proteins is that they are signal integrators not instantaneous reporters because the complemented proteins are generally not readily dissociated.21,22
The fluorogen activating protein (FAP) system is a fusion protein tool that has enabled very detailed investigations of biological systems.23-30 These FAPs act as fluorescent labels by binding nonfluorescent dyes called fluorogens and activate the dark fluorogen for fluorescence with high brightness and photostability.31 This fluorescence readout upon binding of fluorogen to the FAP can be targeted to various subcellular locations32 and expressed in various model species.23,24,27,33 Additionally, flexibility of the fluorogen to chemical modification has allowed for rational fluorogenic ligand designs to probe various physicochemical properties34-37 and enabled targeted photoablation systems using the same set of fluorogen activating proteins.23,38 The modularity of this system enables functional changes via rational chemical modifications of the fluorogenic ligand which are not possible in other biochemical and genetically encoded tools for monitoring protein and cellular proximity.
To this end, our colleagues have pioneered a FAP based reporter of cell-apposition in the Trans-TEFLA system.38-40 This system involves reversible binding of two fluorogens that are tethered together by a polymeric linker, whereby colocalized fluorescence indicates that the FAP tagged proteins are within range of binding for the tethered fluorophore system. This has been demonstrated in the detection of instances of protein–protein interactions and sites of cell-apposition in live cells. Advantages of this system over FP analogues include temporal control afforded by the addition of an exogenous reagent required for detection. Temporal control allows for sequential detection with multiple spectrally similar fluorophores for “pulse-chase” experiments, reduces interference with other applied reagents, and most significantly, minimizes induced interaction by biochemical tethering across sites of cell apposition until the moment of reagent application. However, in this system, the two fluorogens are independent ligands for their cognate FAP proteins, and equilibrium considerations of the labeling requirements are complex due to competing association and dissociation dynamics. When considering the kinetics, having one end of the molecule covalently anchored and the other end activated selectively by a proximal activator substantially simplifies the validation and optimization space (Figure 1A). Therefore, we designed the FAP–DAPA (Fluorogen Activating Protein–Dye Activated by Proximal Anchoring) system to incorporate the HaloTag/Hexyl chloride ligand as a covalent anchor and targeting component to establish proximity-dependent fluorescence activation from the FAP/Fluorogen system.
Figure 1.

Graphical outline of FAP–DAPA system and reagents utilized. (A) Interactions of FAP and HaloTag receptors with DAPA dye showing how (a) the distribution of receptors bound is dictated by differential in KD(FAP/fluorogen) and kon(HT/HexCl), and concentration of DAPA. At sub KD concentrations of DAPA, the DAPA HexCl ligand will first bind to the HaloTag (b, c), and then DAPA Malachite Green (MG) ligand will bind to FAP if the distance D is less than ~20 nm (c). (B) Structure of dimerized L5**-MG complex (PDB: 4K3H) with MG highlighted in green. (C) Structure of the HaloTag protein (PDB: 5UXZ) with estimated HexCl ligand extension. (D) Synthesis of DAPA dye, MG-PEG3500-HexCl using a fluorogenic alkyne for Cu-“click” cycloaddition to the azide terminus and MG-NHS ester for reaction with amino-terminal end of the NH2–PEG3500-N3 polymer.
The HaloTag system is derived from a bacterial dehalogenase enzyme that was modified to covalently bind linear haloalkanes. The rapid kinetics and irreversible covalent bond formation make the HaloTag system ideal for pairing with the FAP/Fluorogen system, which also has rapid association rates and low equilibrium binding concentrations. Additionally, the HaloTag system has been demonstrated in a variety of relevant systems and species. Most notably, the HaloTag/Hexyl chloride interaction was used in the DART (Drugs Acutely Restricted by Tethering) system to target chlorohexyl modified drugs to specific neurons using the same principle of tether-mediated increase in local concentration41 to achieve selective action of drugs to receptors only on HaloTag expressing cells.
Our anchored fluorogen system is expected to work as shown in Figure 1A and has three intertwined principles dictating whether a reporter fluorogen will show specific fluorescence at contact sites: DAPA tether length, FAP/HaloTag ligand binding affinity, and concentration of DAPA dye. First, the tether must be long enough for both ligands to easily bind across typical intertarget spacing. Like compressing or overstretching a spring increases potential energy, compressing a polymer below its equilibrium radius (eq 1) or stretching the polymer to its full contour length (eq 2) also pulls the system out of equilibrium, with an associated energetic penalty.
| (1) |
| (2) |
For poly(ethylene glycol) (PEG) systems, the lower limit for tethered ligand binding is predicted to be near the Flory radius (eq 1), which describes the equilibrium size of a globular polymer in terms of monomer number (N, N = 80) and monomer length (a, a = 0.35 nm).42,43 The upper limit of tethered ligand binding must be below the contour length for the polymer at 28 nm (eq 2)44-47 but has also been shown to depend on the binding affinity for the receptor.44,47 The predicted maximal binding distance (Rmax) is 16–17 nm for PEG3500 for all discussed FAPs (10 pM to 100 nM).42,44,47 Between Rmin and Rmax, the HaloTag–DAPA complex will exhibit an increase in local concentration of the fluorogen ligand (1 molecule/30 nm3 = ~30 μM local concentration), essentially guaranteeing that the ligand will bind any receptor that enters the region. However, conditional activation is still dependent on proper DAPA concentration; if the concentration of the DAPA reagent is far above the KD of the FAP/fluorogen reporter, then the system will show nonspecific fluorescence at every site expressing FAP. Conversely, if the concentration of the DAPA reagent is too low, then slow labeling of the HaloTag anchor could compromise measurement of true dynamics in the system. Therefore, we expect the system to show coincidence specificity at ranges appropriate to detect cell contact sites, but only if applied DAPA concentration falls between the effective binding concentrations of the anchor and activator proteins.
In this paper, we describe the synthesis of the DAPA and validation of FAP/DAPA and HaloTag/DAPA interactions in vitro and in cell culture. In order to widen the concentration range of coincidence specificity, a lower affinity FAP was identified via site directed mutagenesis of the existing FAP, dL5**. The selected variant, dL5(E52K), abbreviated dK, possesses a much faster half-life of dissociation and similar fluorescence quantum yield. Use of the dK protein in vitro in a homogeneous (no-wash) rapamycin chemical inducer of dimerization (CID) assay demonstrated selective activation of the FAP-fluorogen signal upon dimerization. Selective identification of sites of cellular apposition was investigated in HEK293 cells using both the low and high affinity FAP. Together, these results demonstrate that biochemical tuning of the relative affinities of the anchoring and activating moieties produces high specificity labeling of cell-contact sites with minimal off-target background signal and opens a broad reagent working range for selective labeling of these sites, potentially suitable for more complex specimens where precise control of reagent concentrations is impossible such as organoids, tissues, and live animals.
RESULTS AND DISCUSSION
Synthesis and Validation of the Proximity Sensing Reagent.
DAPA, MG-PEG3500-HexCl, was synthesized from a commercially available heterobifunctionalized PEG polymer with alkyne and amine functional ends (Figure 1D). A coumarin alkyne derivative of the standard hexyl chloride HaloTag ligand was prepared and utilized for ease of characterization of the macromolecular conjugate (Figure 1D, Supporting Information).48,49 After the copper catalyzed azide–alkyne cyclocondensation reaction and size exclusion chromatography, Malachite Green–ethylenediamine–succinate28,40 was conjugated to the PEG amino terminus and purified by size exclusion and silica gel chromatography to isolate the pure bifunctional reagent.
Binding of the individual moieties of MG-PEG3500-hexCl to dL5** or the HaloTag was evaluated using purified proteins. Binding affinities, assessed by the ratio of rates of association and dissociation, demonstrated tight binding affinities in the midpicomolar range when DAPA was complexed with dL5** (Table 1). Brightness of the complex was somewhat lower than other MG derivatives with a quantum yield of fluorescence of 0.11 for dL5** (Table 1, Supporting Information Figure 10). The hexyl chloride moiety was also demonstrated to irreversibly bind to a purified HaloTag (HT) protein fusion as indicated by increased molecular weight of the protein/DAPA complex using denaturing SDS-PAGE (Supporting Information Figure 4).
Table 1.
Table of dye properties and activation characteristics with the two FAPs, dL5** (dL5(E52D) and dK (dL5(E52K))
| quantum yield, Φfa |
dissociation constant, KDb (nM) |
on rate, kon (×106 M−1 s−1) |
off rate, koff (×10−4 s−1) |
||||||
|---|---|---|---|---|---|---|---|---|---|
| dye | dL5** | dK | dL5** | dK | dL5** | dK | dL5** | dK | cell permeant |
| DAPA | 0.11 | 0.05 | 0.031 ± 0.002 | 13.7 ± 0.5 | 3.2 ± 0.2 | 0.39 ± 0.03 | 1.23 ± 0.06 | 53 ± 2 | no |
| MG-2p | 0.20c | 0.10 | 0.0197 ± 0.0005 | 4.9 ± 0.2 | 2.82 ± 0.02 | 0.71 ± 0.05 | 0.556 ± 0.028 | 34 ± 1 | no |
| MG-BTau | 0.19c | 0.11 | N.A. | 23.5 ± 0.9 | 1.37 ± 0.03 | 0.30 ± 0.02 | N.A. | 70 ± 2 | no |
| MG-nBu | 0.20d | 0.09 | N.A. | 0.68 ± 0.03 | 7.5 ± 0.8 | 3.0 ± 0.2 | N.A. | 20.0 ± 0.9 | yes |
Quantum yields were determined relative to MG-2p/dL5 for samples at 2, 1, 0.75, and 0.5 μM dye with 10× FAP using equation Φunk = Φstd(slopeunk/slopestd), slopes (integrated fluorescence versus concentration).
KD = koff/kon. Errors are reported as the standard deviation for N = 4 replicates.
Yan et al.50
Perkins et al.26 N.A. data were not acquired.
Development of a Low Affinity FAP for Proximity Detection.
The DAPA sensing reagent must be present at concentrations below the KD of the FAP reporter, to ensure generally low occupancy of the FAP unless it is in proximity of the anchoring protein. For the current FAP, dL5**, the in vitro KD is on the order of 20 pM, with slightly weaker empirical cellular labeling values.50 In order to meet conditions for proximity-specific fluorescence activation under reliable labeling conditions, DAPA concentrations should interpose FAP and HaloTag saturation binding ranges, and a lower affinity variant of the FAP with comparable brightness is needed to extend the working concentration range of the FAP–DAPA system.
We prepared a library of variants to identify potential FAPs with low affinity and high brightness. For ease of evaluation, modifications were performed on the monomeric version of dL5**, mL5(L91S E52D). Previous work had demonstrated strong dependence of FAP binding affinity on the identity of residue 52, a residue located near the back of the binding pocket (Figure 2A).31,34 Therefore, residue 52 was mutated to the 20 possible amino acids via homologous recombination in yeast. Each mL5 mutant was evaluated for fluorescence activation and surface effective KD displayed on the surface of yeast. Each mutant was then secreted and characterized by biochemical and fluorescence methods to determine binding energetics including Isothermal Titration Calorimetry (ITC), dissociation rate (fluorescence kinetics), and fluorescence brightness (Fluorescence Correlation Spectroscopy (FCS); Figure 2B).
Figure 2.

Site-directed saturation mutagenesis to identify low-affinity L5 FAP mutants. (A) Crystal structure (PDB: 4K3H) of two L5* monomers binding Malachite Green (green) with top and side views. Residue 52 (red, E52D) is highlighted showing how direct participation in the MG binding pocket is unlikely since the residue is distal to the binding pocket and opposite the expected entry point of the dye. (B) Properties of monomeric L5*(E52X) variants: Isothermal calorimetry gave enthalpy values via titration of 4 μM protein with MG-2p. Rates were determined directly via fluorescence measurement (dissociation N = 4, association at four concentrations between 100 and 300 nM with N = 4). E, K dissociation rates were determined using equation kon = koff/KD. The KD was calculated based on fluorescence equilibrium titration (at 5 nM concentration of the L5 monomer protein) and is depicted as the size of the plotted point (nM, log scaling). Molecular brightness was measured by fluorescence correlation spectroscopy and is depicted as a color gradient (red is brighter; Supporting Information Table 3).
The results confirm that residue 52 at the back of the binding pocket was highly involved in binding affinity and brightness. Mutants possessing β-hydroxyl or β-carboxy groups (E52A, E52D, E52S, and E52T) showed similar properties of tight binding, slow dissociation, and high molecular brightness (Figure 2B). Larger side chain molecular weight correlated to decreases in binding enthalpies (r(14) = −0.628, p < 0.01) and a shortening half-life of dissociation (r(14) = −0.564, p < 0.025). The brightness of the complex was also seen to decrease slightly with increasing rate of dissociation (r(15) = −0.586, p < 0.025) and loosening KD (r(15) = −0.405, p < 0.1; Supporting Information Tables 3, 4). Of these mutants, mL5(E52K), with a bulky and cationic residue, shows by far the weakest KD and fastest off rate, though with reduced fluorescence brightness.
To further examine the E52K variant, homodimer dL5(L91S E52K) (dK) with the E52K mutation in both monomers was prepared. When compared to parent dL5**, dK showed a ~250–500-fold looser KD while maintaining half of the molecular brightness (Table 1). It is hypothesized that fluorogen-induced dimerization is suppressed in this variant due to the positive bulky residue causing a shift in the back of the fluorogen binding pocket (Figure 2A). A more negative enthalpy for this mL5 variant, which includes both ligand-binding and interdomain interactions, suggests that while the pocket may be more stable, it is oriented such that activation energy is lower and dissociation is more facile to achieve a moderate overall KD. Due to these energetic and kinetic differences, the dL5(E52K) variant is an effective fluorogen activator and low-affinity, rapid-dissociating fluorogen activating protein variant that was selected for investigation in the FAP–DAPA system (Supporting Information Figures 8, 9).
Testing of FAP–DAPA System for Detection of Protein–Protein Interactions.
In order to test the efficacy of the new lowered-affinity dK variant for detection of protein–protein interactions, the FKBP/FRB/rapamycin chemical inducer of the dimerization (CID) system was used as an in vitro model of inducible proximity.6,51 All permutations of N- and C-terminal HaloTag (HT) and dK fusions of FKBP and FRB were cloned into pET21 vectors, and recombinant proteins were synthesized and purified from E. coli (Rosettagami 2 (DE3) used to enhance disulfide bond formation, Supporting Information Figure 3). All pairs were tested for rapamycin induced fluorescence from the FAP–DAPA system.
Selected fusions FRB-HT/dK-FKBP and dK-FRB/FKBP-HT (Figure 3B, D) show signal increase over background FAP binding (Figure 3C, E). This change corresponds to a 50% (p < 0.01, 2-sided, unpaired t test) or 100% (p < 0.05, 2-sided, unpaired t test) increase in FAP–DAPA signal for rapamycin induced dimerization of FRB-HT/dK-FKBP or dK-FRB/FKBP-HT, respectively. This indicates that the DAPA reagent is long enough to bind to both receptors. Additionally, the increase in fluorescence of FAP/fluorogen upon CID shows that the FAP–DAPA signal is dependent on the proximity of the FAP receptor to the HaloTag anchor. This positive result was somewhat muted in other configurations of the fusions (Supporting Information Figure 6). It is suspected that orientation dependence of the FKBP/FRB fusions, and subsequent differing inter-receptor spacing (Supporting Information Figure 5), plays a major role. The relationship between complementation efficiency and linker length or protein fusion orientation has been widely observed in the FBKP/FRB system and other systems.52,53
Figure 3.

FAP and HaloTag fusions to FRB and FKBP, induced to dimerize with rapamycin, show increased FAP–DAPA fluorescence, indicating proximity dependent activation. (A) Illustration of FRB-HT and FKBP-dK interacting with rapamycin created in silico from solved crystal structures (PDB: 4FAP, 5UXZ, and 4K3G). (B, D) The cartoons of selected fusions for this study showing pairs where the HT/FAP are on cis or trans faces of the FRB/rapa/FKBP trimer. (C, E) Fluorescence of rapamycin induced dimerization of FKBP and FRB fusions of dK and HT were measured at 100 nM dK fusion, 200 nM HT fusion, 250 nM rapamycin, and 10 nM DAPA. Addition of rapamycin to solutions containing FRB/FKBP fusions show significant increase in FAP–DAPA signal when compared to both proteins in absence of rapamycin or dK fusions alone (unpaired, two sided, t test, *p < 0.05; ****p ≤ 0.0001, ***0.0001 < p ≤ 0.001, **0.0001 < p ≤ 0.01, *p ≤ 0.05).
The results of this experiment suggest that one can use the FAP–DAPA system to monitor CID or protein interactions for suitably oriented systems. Future chemo-biophysical studies with altered ligand–receptor spacing or varied polymer length between the tether and the fluorogen will investigate relationships between linker geometry, receptor spacing, and signal generation, opening the possibility that a set of DAPA dyes, potentially binding a common FAP in multiple colors for example using MG/MHN fluorogens binding to dK FAP (Supporting Information Figure 7), could be used as a molecular ruler, with fluorescence activation depending sensitively on the linker length for a fixed interreceptor distance.
Detection of Cell Membrane Proximity in Cultured Mammalian Cells.
In order to establish the selective detection of cell proximity at the interface of HaloTag and FAP expressing cell lines, stable HEK293 cell lines were created expressing HaloTag or FAP constructs and used to test DAPA properties, including cell permeability, independent and simultaneous binding to the respective protein tags, and selective labeling of intercellular contact sites.
To robustly measure cell contact sites, the DAPA dye should remain cell-excluded to avoid labeling within secretory pathways. Cell permeability of the DAPA reagent was tested in cell culture relative to the highly cell-permeant MG derivative MG-nBu.26 In cell culture, both dyes at saturating concentrations labeled membrane expressed dL5** (dL5**-TM, Addgene: 73206), but only the positive control, MG-nBu, labeled the nuclear FAP (NLS-dL5**, Addgene: 73205; Figure 4A, Supporting Information Figure 11). DAPA and MG-nBu also suitably label dK-TM expressing cells (Supporting Information Figure 12).
Figure 4.

Cell permeability testing for the DAPA. (A) Microscopy analysis: HEK293 cells stably expressing either transmembrane anchored (dL5**-TM, Addgene: 73206) or nuclear localized (NLS-dL5**, Addgene: 73205) FAP were labeled with 500 nM DAPA or MG-nBu, a cell permeant dye.26 Cells were incubated with dyes for 15 min, and then imaged (Zeiss 880 LSM, 40× oil, λex/em: 633/(641–695 nm)). Lookup tables are matched, and laser powers are 0.4% and 6% for the dL5**-TM and the NLS-dL5** cells, respectively. The images show that the DAPA labels surface FAP but cannot label intracellular FAP due to cell impermeability. (B) Flow cytometry analysis: HEK293 cells expressing NLS-dL5** or dL5**-TM were labeled with 100 nM dye. Percentiles corresponding to the median fluorescence of the expressing population (TM, 100% expressing, 50%ile; NLS, 50% expressing, 75%ile) were determined and normalized to the MG-nBu signal, indicating low cell permeability of the DAPA (5% of nBu signal) and transmembrane labeling signal reduced compared with Mg-nBu due to inability to label internal fraction of dL5**-TM (as in the images) and lower QY.
Low cell permeability of the DAPA was also confirmed using flow cytometry. Labeling of NLS-dL5** with DAPA shows only 5% of the signal seen when labeled with MG-nBu (Figure 4B). However, DAPA labeling of the dL5**-TM shows 40% of the MG-nBu fluorescence. The difference in DAPA versus MG-nBu surface labeling fluorescence can likely be attributed to two factors: differences in quantum yield of fluorescence (Table 1) and labeling of dL5**-TM localized in intracellular secretory compartments by MG-nBu as seen in Figure 4A. Overall, this confirms that the DAPA is effectively cell impermeant, which is important for specific labeling at sites of cellular apposition and intercellular interaction. This result with a linear PEG linker is different from the branched polymer p(OEOMA) malachite green conjugates that were previously reported to show high cell permeability.36 For future intracellular application of this system, for example, measurements of organelle contacts or other specific protein interactions, the structure of the intervening polymer will be investigated.
DAPA binding to the HaloTag protein was tested utilizing HEK293 cells expressing the HaloTag (HT) fused to a transmembrane domain (TM) for extracellular expression and an mCerulean3 (mCer3) fluorescent protein54 incorporated at the intracellular C-terminus for positive identification of the transfected cells (HT-TM-mCer3 plasmid, Addgene: 145767; Figure 5). Validation of the cell line confirmed proper surface labeling of the Cy3-hexCl derivative on the surface of the HT-TM-mCer3 expressing cells (Supporting Information Figure 13). To test labeling of the DAPA reagent, HT-TM-mCer3 cells were labeled with DAPA or the cell-excluded MG MG-BTau and extensively washed to remove all DAPA dye unbound by HT-TM-mCer3. Subsequent labeling with saturating concentrations of purified dL5** shows selective FAP fluorescence on the surface of mCer3 positive cells, indicating specificity of the DAPA for labeling HaloTag expressing cells and not WT cells. Additionally, the MG-BTau control does not show any labeling of the HaloTag expressing cells, which confirms that binding of the DAPA is dependent on the HexCl/HaloTag interaction. Finally, this experiment shows that the DAPA is capable of binding both FAP and HaloTag proteins simultaneously in cell culture, indicating that the system will report protein–protein interactions in live cells and protein proximity across two cells provided the DAPA linker spans the distance between the HaloTag and FAP.
Figure 5.

Specific binding of DAPA to HaloTag receptors. HEK 293 cells expressing surface HaloTag (HT-TM-mCerulean3, cyan; Addgene: 145767) were labeled with 500 nM DAPA or MG-BTau anchor-less control, and unbound dye was washed away (6×). Binding of DAPA via hexCl ligand was confirmed by the addition of 1 μM FAP, dL5**, which binds to any captured MG and should show a red fluorescence signal on HaloTag expressing cells. Cells were imaged using the Zeiss 880 LSM using 633 and 405 nm excitation with laser powers of 55 and 25.7% respectively and are depicted with 20 μm scale bars. DAPA specifically labeled mCer3 positive cells, indicating specificity of the DAPA for labeling HT expressing cells and that the anchored fluorogen is readily activated upon interaction with the FAP.
Cell Contact Assay.
Finally, the FAP–DAPA system was evaluated as a sensor for membrane apposition using both differential DAPA labeling concentrations and FAP binding affinities. Surface FAP expressing cells (dL5**-TM or dK-TM, Addgene: 145766) were coplated with the HaloTag surface expressing cells (HT-TM-mCer3). The cells were then labeled with DAPA dye and imaged for red fluorescence excited at 633 nm. Brighter red fluorescence was observed at the interface between FAP and HaloTag expressing cells (mCer3 imaged with 405 nm excitation; Figure 6A,B), with clear selectivity for intercellular contact sites in the dK-TM experiments, and at the low DAPA range for the dL5**-TM experiments.
Figure 6.

The dK FAP improves cell contact labeling selectivity. (A, B) HEK293 cells expressing HT-TM-mCerulean3 (cyan) and either dK-TM (A, red) or dL5**-TM (B, red) were coplated on a poly-l-lysine coated dish. The live cells were labeled with DAPA dye at 0.5 to 100 nM in OptiMEM media and imaged immediately without removal of the dye. All imaging was collected using a Zeiss 880 LSM with mCerulean observed with 405 nm excitation, 25% laser power, and FAP observed with 633 nm excitation. The laser powers for the 633 nm channel are (A) 55%, 25%, 25%, 0.4% and (B) 0.4% for all. The scale bars are 20 μm. (C, D) Quantitation of cell contact contrast: In two to five images per labeling condition, brightness of cell contact sites was measured from the perimeter of FAP expressing cells using a 6.37 pixel2 circular ROI. ROIs were separated into contact and noncontact sites based on the presence or absence of proximal mCer3 signal. The brightness of each sample is normalized to the mean noncontact fluorescence and plotted as fold change over mean noncontact fluorescence for each DAPA concentration. As concentration increases from 0.5 nM to 100 nM, the difference between the contact and noncontact zone becomes less significant for dL5** (t test, two sided) but is maintained for dK. Both images and extracted data indicate differential contact site specificity observed in dK versus dL5** and show loss of specificity with increasing DAPA concentration in dL5** panels.
To quantitatively assess the quality of cell-contact site labeling, brightness in the 633 nm channel at contact sites and noncontact sites was measured manually with small ROIs of consistent size in two to five images per labeling condition (Figure 6C,D, Supplemental Tables 5 and 6). Both dL5**-TM or dK-TM expressing cells showed significant differences between contact-site and noncontact site signals at all concentrations. The dK samples, overall, showed more significant differences between contact site and noncontact site fluorescence (two-sided t test, p < 0.002). The best performance was seen by the 10 nM DAPA dye labeled dK sample: a 40-fold signal increase at the contact site versus the noncontact site was observed. The dL5** populations displayed selective labeling of contact sites at concentrations of DAPA dye between 0.5 nM and 10 nM (p < 0.01, ratios > 3.8). Upon an increase in dye concentration to 100 nM, the ratio between the contact and noncontact signal decreases and the difference in contact versus noncontact population fluorescence readings are barely significantly different (p < 0.1, ratio = 1.9), largely due to the significant labeling of the dL5**-TM cell surface by the dye irrespective of the proximity of a HaloTag cell.
The dK-TM cells labeled with the DAPA reagent show a clear signal at the contact sites, with little or no detectable background labeling of noncontact site receptors on the FAP cell indicating specificity. This difference in contact site labeling specificity at this concentration range is expected due to the ~500-fold difference in FAP/DAPA KD between dL5** and dK (Table 1). The selectivity by local activation at cell-contact sites in this labeling system provides a robust and selective tool for quantitation of intercellular proximity. The reduction of the noncontact site fluorescence and the increased working range for the DAPA ligand with the reduced affinity dK FAP provides for a dramatic simplification of the workflow for labeling, detection, and analysis of intercellular contacts, in living cells.
To test the ability of the FAP–DAPA system to function as a reporter of cellular apposition in real-time, time-lapse imaging was performed using cocultured dK-TM and HT-TM-mCer3 cells over a period of about 1 h (Supporting Information Video 1, Supporting Information Figure 14, 15). Typical cell dynamics were observed in this experiment. Dynamic contacts were visible, and examples of multiple events of contact pruning (Supporting Information Figure 15A,B) and widening of sites of cellular apposition (Supporting Information Figure 15C) were documented. This suggests that the FAP–DAPA system utilizing the low affinity FAP, dK, is suitably noninvasive and reversible to detect sites of cellular apposition in cell culture and allow for real-time observation of transient systems such as the observed dynamic contacts on the minutes to hours time scale.
CONCLUSION
This study describes the development of a robust proximity detection system, FAP–DAPA. The properties of the dK-FAP (E52K)—decreased affinity of the FAP/Fluorogen complex, slower binding, quicker dissociation, and yet a similar quantum yield (Figure 2, Table 1)—makes the activator component a transient binder, able to report on proximity while minimizing alterations of the dynamics of the biological system. In cultured cells, the specificity of labeling at cell-contact sites in live-cell microscopy also establishes that this labeling system is coincidence specific and high contrast.
Additionally, we have demonstrated the utility of the FAP–DAPA system as a robust live cell imaging tool for imaging sites of cellular apposition. It is both suitable to detecting transient sites of cell contacts and also robust enough to detect these interactions over long periods of time, even 6 h after labeling HaloTag expressing cells in cell culture (Supporting Information Figure 16). There are two advantages notable in this covalent labeling and proximal activation approach: first, the persistence of the HexCl ligand bound to the HaloTag should enable robust long-term imaging studies, or imaging studies in complex specimens where the concentration of a ligand cannot be carefully and homogeneously controlled; second, the covalent label may be suitable for methods analyzing specimens after fixation and labeling procedures that preserve the function of the dK FAP. This is an improvement upon existing systems using two reversible ligands as reporters which are not suited to long-term studies or complex specimens due to the opportunity for reagent dissociation, fluorogen labeling background, or concentration heterogeneity that may alter labeling efficiency throughout a specimen.38-40
In the rapamycin induced dimerization studies, we saw dependence of signal output on the orientation of the proteins and interestingly obtained the largest signal increases for larger inter-receptor spacing. This is highly contrary to most existing reporter systems. For example, split fluorescent proteins and FRET systems are highly orientation dependent and primarily detect protein–protein interactions at very close range.55 The tethered fluorogen system demonstrated here fills the gap left by these tools for detection of longer-range protein–protein interactions, for example, in large, dynamic, multiprotein complexes, and interprotein recruitment studies. Studies further elucidating geometric requirements for signal activation could be achieved via modulation of either receptor spacing or tether linker length or dynamics and should be powerful and predictable when paired with the development of quantitative modeling occurring in this field.46 This direction is particularly interesting as it could enable a library of tools for investigation of various systems and mapping of inter-receptor distances.
New tools are needed for studying the dynamics of cell contacts and the FAP–DAPA system has the properties required for a robust and selective tool. The function of both the HaloTag and dL5 related FAP proteins within living cells,32 coupled with the potential to develop macromolecular DAPA ligands with high cell-permeability36 supports the possibility that this approach could be used within live cells to measure transient protein or organelle proximity with high specificity and contrast. Of special interest is the ability to detect neuronal or immunological synapses enabling high throughput and high-contrast mapping of connectivity in higher order systems.
Finally, the availability of several FAP-fluorogen systems with a range of colors and affinities28,34,37,56,57 and other properties58-61 opens the possibility of simultaneous probing of several interactions by anchoring several fluorogens for activation by their cognate FAP only when it “arrives on the scene.” A multiplexed FAP–DAPA labeling approach could be used to map synaptic connectivity between several neural cell types, or to identify multiple proteins assembling into a protein complex in the cell. A number of new directions are opened by this highly fluorogenic, proximity labeling, and activation method.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by funding from the NIH Brain Initiative (R21NS092019 and RF1MH114103), NIH Technology Centers for Networks and Pathways (U54GM103529), and NIH R01EB017268 and a grant from the Kauffman Foundation. Funding for NMR instrumentation at Carnegie Mellon University was partially supported by the National Science Foundation (CHE- 0130903 and CHE-1039870). We thank H. Teng for help with microscopy, Y. Creeger for help with flow cytometry, G. Whithers and R. Gil for help with NMR, M. Bier and L. Plath for help with ESI and MALDI-TOF, and A. Linstedt for use of the Chemidoc gel imager. We also thank M. Naganbabu and K. Noonan and their lab for helpful discussions regarding hexCl-coumarin-alkyne synthesis. NIH TCNP grant U54GM103529 (Y.W., C.T., B.S., M.B.), NIH (5R01EB017268; B.S., M.B., Z.Y.); NIH RF1MH114103 (M.C., Z.Y., M.B); NIH R21NS092019 (M.C., Y.W.).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.0c00419.
Detailed methods, descriptions for all materials and measurements, comprehensive synthetic details and analytical results for all prepared molecules, descriptions of plasmid DNA prepared, and associated Addgene ID for deposited plasmids; extended biochemical data for the various constructs characterized and reported in summary in the main figures; additional imaging and cytometry data supporting the function of prepared plasmids, image data showing a variety of cell-contact experiments, and observations of varied contact behavior using the FAP–DAPA system included as annotated still images (PDF)
Time-lapse video showing dynamic cell contacts visualized over an hour in confocal microscopy (MP4)
The authors declare the following competing financial interest(s): M.P.B. is a Founder and Chief Technology Officer of Sharp Edge Labs, a company that is commercially utilizing the Fluorogen Activating Protein technology.
Contributor Information
M. Alexandra Carpenter, Carnegie Mellon University, Department of Chemistry, Pittsburgh, Pennsylvania 15213, United States.
Yi Wang, Carnegie Mellon University, Department of Biological Sciences, Pittsburgh, Pennsylvania 15213, United States.
Cheryl A. Telmer, Carnegie Mellon University, Department of Biological Sciences and Molecular Biosensor and Imaging Center, Pittsburgh, Pennsylvania 15213, United States
Brigitte F. Schmidt, Carnegie Mellon University, Molecular Biosensor and Imaging Center, Pittsburgh, Pennsylvania 15213, United States
Zhipeng Yang, Carnegie Mellon University, Department of Biological Sciences, Pittsburgh, Pennsylvania 15213, United States.
Marcel P. Bruchez, Carnegie Mellon University, Department of Chemistry, Department of Biological Sciences, and Molecular Biosensor and Imaging Center, Pittsburgh, Pennsylvania 15213, United States.
REFERENCES
- (1).Rees RP, Bunge MB, and Bunge RP (1976) Morphological Changes in the Neuritic Growth Cone and Target Neuron during Synaptic Junction Development in Culture. J. Cell Biol 68 (2), 240–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Burette A, Collman F, Micheva KD, Smith SJ, and Weinberg RJ (2015) Knowing a Synapse When You See One. Front. Neuroanat 9, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Briggman KL, and Bock DD (2012) Volume Electron Microscopy for Neuronal Circuit Reconstruction. Curr. Opin. Neurobiol 22, 154–161. [DOI] [PubMed] [Google Scholar]
- (4).Chung K, Wallace J, Kim S-Y, Kalyanasundaram S, Andalman AS, Davidson TJ, Mirzabekov JJ, Zalocusky KA, Mattis J, Denisin AK, et al. (2013) Structural and Molecular Interrogation of Intact Biological Systems. Nature 497 (7449), 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Abbe E VII (1881) -On the Estimation of Aperture in the Microscope. J. R. Microsc. Soc 1 (3), 388–423. [Google Scholar]
- (6).Gautier A, Nakata E, Lukinavičius G, Tan KT, and Johnsson K (2009) Selective Cross-Linking of Interacting Proteins Using Self-Labeling Tags. J. Am. Chem. Soc 131 (49), 17954–17962. [DOI] [PubMed] [Google Scholar]
- (7).Morell M, Ventura S, and Avilés FX (2009) Protein Complementation Assays: Approaches for the in Vivo Analysis of Protein Interactions. FEBS Lett. 583 (11), 1684–1691. [DOI] [PubMed] [Google Scholar]
- (8).Ngounou Wetie AG, Sokolowska I, Woods AG, Roy U, Loo JA, and Darie CC (2013) Investigation of Stable and Transient Protein-Protein Interactions: Past, Present, and Future. Proteomics 13, 538–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Rees RP, Bunge MB, and Bunge RP (1976) Morphological Changes in the Neuritic Growth Cone and Target Neuron during Synaptic Junction Development in Culture. J. Cell Biol 68 (2), 240–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Dustin ML (2014) The Immunological Synapse. Cancer Immunol. Res 2 (11), 1023–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Gatta AT, and Levine TP (2017) Piecing Together the Patchwork of Contact Sites. Trends Cell Biol. 27 (3), 214–229. [DOI] [PubMed] [Google Scholar]
- (12).Lee H, Oh WC, Seong J, and Kim J (2016) Advanced Fluorescence Protein-Based Synapse-Detectors. Front. Synaptic Neurosci 8, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Macpherson LJ, Zaharieva EE, Kearney PJ, Alpert MH, Lin T-Y, Turan Z, Lee C-H, and Gallio M (2015) Dynamic Labelling of Neural Connections in Multiple Colours by Trans-Synaptic Fluorescence Complementation. Nat. Commun 6 (1), 10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Choi JH, Sim SE, Kim J. il, Choi DII, Oh J, Ye S, Lee J, Kim TH, Ko HG, Lim CS, et al. (2018) Interregional Synaptic Maps among Engram Cells Underlie Memory Formation. Science (Washington, DC, U. S.) 360 (6387), 430–435. [DOI] [PubMed] [Google Scholar]
- (15).Tsetsenis T, Boucard AA, Arac D, Brunger AT, and Sudhof TC (2014) Direct Visualization of Trans-Synaptic Neurexin-Neuroligin Interactions during Synapse Formation. J. Neurosci 34 (45), 15083–15096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Kim J, Zhao T, Petralia RS, Yu Y, Peng H, Myers E, and Magee JC (2012) MGRASP Enables Mapping Mammalian Synaptic Connectivity with Light Microscopy. Nat. Methods 9 (1), 96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Feinberg EH, VanHoven MK, Bendesky A, Wang G, Fetter RD, Shen K, and Bargmann CI (2008) GFP Reconstitution Across Synaptic Partners (GRASP) Defines Cell Contacts and Synapses in Living Nervous Systems. Neuron 57 (3), 353–363. [DOI] [PubMed] [Google Scholar]
- (18).Feng L, Zhao T, and Kim J (2012) Improved Synapse Detection for MGRASP-Assisted Brain Connectivity Mapping. Bioinformatics 28 (12), i25–i31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Kinoshita N, Huang AJY, McHugh TJ, Suzuki SC, Masai I, Kim IH, Soderling SH, Miyawaki A, and Shimogori T (2019) Genetically Encoded Fluorescent Indicator GRAPHIC Delineates Intercellular Connections. iScience 15, 28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Harmon M, Larkman P, Hardingham G, Jackson M, and Skehel P (2017) A Bi-Fluorescence Complementation System to Detect Associations between the Endoplasmic Reticulum and Mitochondria. Sci. Rep 7 (1), 17467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Xing S, Wallmeroth N, Berendzen KW, and Grefen C (2016) Techniques for the Analysis of Protein-Protein Interactions in Vivo. Plant Physiol. 171 (2), 727–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Eisenberg-Bord M, Shai N, Schuldiner M, and Bohnert M (2016) A Tether Is a Tether Is a Tether: Tethering at Membrane Contact Sites. Dev. Cell 39 (4), 395–409. [DOI] [PubMed] [Google Scholar]
- (23).He J, Wang Y, Missinato MA, Onuoha E, Perkins LA, Watkins SC, St Croix CM, Tsang M, and Bruchez MP (2016) A Near-Infrared Genetically Targetable and Activatable Photosensitizer. Nat. Methods 13 (3), 263–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Bulgari D, Deitcher DL, Schmidt BF, Carpenter MA, Szent-Gyorgyi C, Bruchez MP, and Levitan ES (2019) Activity-Evoked and Spontaneous Opening of Synaptic Fusion Pores. Proc. Natl. Acad. Sci. U. S. A 116 (34), 17039–17044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Qian W, Kumar N, Roginskaya V, Fouquerel E, Opresko PL, Shiva S, Watkins SC, Kolodieznyi D, Bruchez MP, and Van Houten B (2019) Chemoptogenetic Damage to Mitochondria Causes Rapid Telomere Dysfunction. Proc. Natl. Acad. Sci. U. S. A 116 (37), 18435–18444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Perkins LA, Fisher GW, Naganbabu M, Schmidt BF, Mun F, and Bruchez MP (2018) High-Content Surface and Total Expression SiRNA Kinase Library Screen with VX-809 Treatment Reveals Kinase Targets That Enhance F508del-CFTR Rescue. Mol. Pharmaceutics 15 (3), 759–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kuljis DA, Park E, Telmer CA, Lee J, Ackerman DS, Bruchez MP, and Barth AL (2019) Fluorescence-Based Quantitative Synapse Analysis for Cell-Type Specific Connectomics. eneuro 6, ENEURO.0193-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Szent-Gyorgyi C, Schmidt BF, Creeger Y, Fisher GW, Zakel KL, Adler S, Fitzpatrick JAJ, Woolford CA, Yan Q, Vasilev KV, Berget PB, Bruchez MP, Jarvik JW, and Waggoner A (2008) Fluorogen-Activating Single-Chain Antibodies for Imaging Cell Surface Proteins. Nat. Biotechnol 26 (2), 235–240. [DOI] [PubMed] [Google Scholar]
- (29).Kuljis DA, Park E, Telmer CA, Lee J, Ackerman DS, Bruchez MP, and Barth AL (2019) Fluorescence-Based Quantitative Synapse Analysis for Cell-Type Specific Connectomics. eNeuro 6, ENEURO.0193-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Fouquerel E, Barnes RP, Uttam S, Watkins SC, Bruchez MP, and Opresko PL (2019) Targeted and Persistent 8-Oxoguanine Base Damage at Telomeres Promotes Telomere Loss and Crisis. Mol. Cell 75 (1), 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Szent-Gyorgyi C, Stanfield RL, Andreko S, Dempsey A, Ahmed M, Capek S, Waggoner AS, Wilson IA, and Bruchez MP (2013) Malachite Green Mediates Homodimerization of Antibody VL Domains to Form a Fluorescent Ternary Complex with Singular Symmetric Interfaces. J. Mol. Biol 425 (22), 4595–4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Telmer CA, Verma R, Teng H, Andreko S, Law L, and Bruchez MP (2015) Rapid, Specific, No-Wash, Far-Red Fluorogen Activation in Subcellular Compartments by Targeted Fluorogen Activating Proteins. ACS Chem. Biol 10 (5), 1239–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Pratt CP, and Bruchez AMP Studies of BK Channel Trafficking and Synaptic Vesicle Recycling with Fluorogen-Activating Peptides Applications of FAPs for Neurobiological Imaging. Thesis, Carnegie Mellon University, 2017. DOI: 10.1184/R1/6723275.v1. [DOI] [Google Scholar]
- (34).Szent-Gyorgyi C, Schmidt BF, Fitzpatrick JAJ, and Bruchez MP (2010) Fluorogenic Dendrons with Multiple Donor Chromophores as Bright Genetically Targeted and Activated Probes. J. Am. Chem. Soc 132 (32), 11103–11109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Perkins LA, Yan Q, Schmidt BF, Kolodieznyi D, Saurabh S, Larsen MB, Watkins SC, Kremer L, and Bruchez MP (2018) Genetically Targeted Ratiometric and Activated PH Indicator Complexes (TRApHIC) for Receptor Trafficking. Biochemistry 57 (5), 861–871. [DOI] [PubMed] [Google Scholar]
- (36).Magenau AJD, Saurabh S, Andreko SK, Telmer C. a., Schmidt BF, Waggoner AS, and Bruchez MP (2015) Genetically Targeted Fluorogenic Macromolecules for Subcellular Imaging and Cellular Perturbation. Biomaterials 66, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Pratt CP, He J, Wang Y, Barth AL, and Bruchez MP (2015) Fluorogenic Green-inside Red-Outside (GIRO) Labeling Approach Reveals Adenylyl Cyclase-Dependent Control of BKα Surface Expression. Bioconjugate Chem. 26 (9), 1963–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Ackerman DS, Altun B, Kolodieznyi D, Bruchez MP, Tsourkas A, and Jarvik JW (2019) Antibody-Linked Fluorogen-Activating Proteins for Antigen Detection and Cell Ablation. Bioconjugate Chem. 30 (1), 63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Vasilev KV, Gallo E, Shank N, and Jarvik JW (2016) Novel Biosensor of Membrane Protein Proximity Based on Fluorogen Activated Proteins. Comb. Chem. High Throughput Screening 19 (5), 392–399. [DOI] [PubMed] [Google Scholar]
- (40).Ackerman DS, Vasilev KV, Schmidt BF, Cohen LB, and Jarvik JW (2017) Tethered Fluorogen Assay to Visualize Membrane Apposition in Living Cells. Bioconjugate Chem. 28 (5), 1356–1362. [DOI] [PubMed] [Google Scholar]
- (41).Shields BC, Kahuno E, Kim C, Apostolides PF, Brown J, Lindo S, Mensh BD, Dudman JT, Lavis LD, and Tadross MR (2017) Deconstructing Behavioral Neuropharmacology with Cellular Specificity. Science (Washington, DC, U. S.) 356 (6333), eaaj2161. [DOI] [PubMed] [Google Scholar]
- (42).Moreira AG, Jeppesen C, Tanaka F, and Marques CM (2003) Irreversible vs. Reversible Bridging: When Is Kinetics Relevant for Adhesion? Europhys. Lett 62 (6), 876–882. [Google Scholar]
- (43).Wong JY, and Kuhl TL (2008) Dynamics of Membrane Adhesion: The Role of Polyethylene Glycol Spacers, Ligand-Receptor Bond Strength, and Rupture Pathway. Langmuir 24 (4), 1225–1231. [DOI] [PubMed] [Google Scholar]
- (44).Moore NW, and Kuhl TL (2006) The Role of Flexible Tethers in Multiple Ligand-Receptor Bond Formation between Curved Surfaces. Biophys. J 91 (5), 1675–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Jeppesen C (2001) Impact of Polymer Tether Length on Multiple Ligand-Receptor Bond Formation. Science (Washington, DC, U. S.) 293 (5529), 465–468. [DOI] [PubMed] [Google Scholar]
- (46).Bell S, and Terentjev EM (2017) Kinetics of Tethered Ligands Binding to a Surface Receptor. Macromolecules 50 (21), 8810–8815. [Google Scholar]
- (47).Jeppesen C, Israelachvili JN, Mullah N, Zalipsky S, Wong JY, Kuhl TL, Israelachvili JN, Mullah N, Zalipsky S, and Marques CM (2001) Impact of Polymer Tether Length on Multiple Ligand-Receptor Bond Formation. Science (Washington, DC, U. S.) 293 (2001), 465–468. [DOI] [PubMed] [Google Scholar]
- (48).Zhou Z, and Fahrni CJ (2004) A Fluorogenic Probe for the Copper (I)-Catalyzed Azide-Alkyne Ligation Reaction. J. Am. Chem. Soc 126 (I), 8862–8863. [DOI] [PubMed] [Google Scholar]
- (49).Wright AT, Song JD, and Cravatt BF (2009) A Suite of Activity-Based Probes for Human Cytochrome P450 Enzymes. J. Am. Chem. Soc 131 (30), 10692–10700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Yan Q, Schmidt BF, Perkins LA, Naganbabu M, Saurabh S, Andreko SK, and Bruchez MP (2015) Near-Instant Surface-Selective Fluorogenic Protein Quantification Using Sulfonated Triarylmethane Dyes and Fluorogen Activating Proteins. Org. Biomol. Chem 13 (7), 2078–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Choi J, Chen J, Schreiber SL, and Clardy J (1996) Structure of the FKBP12-Rapamycin Complex Interacting with the Binding Domain of Human FRAP. Science 273 (5272), 239–242. [DOI] [PubMed] [Google Scholar]
- (52).Paulmurugan R, and Gambhir SS (2007) Combinatorial Library Screening for Developing an Improved Split-Firefly Luciferase Fragment-Assisted Complementation System for Studying Protein-Protein Interactions. Anal. Chem 79 (6), 2346–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Chrétien A-È, Gagnon-Arsenault I, Dubé AK, Barbeau X, Després PC, Lamothe C, Dion-Côté A-M, Lagüe P, and Landry CR (2018) Extended Linkers Improve the Detection of Protein-Protein Interactions (PPIs) by Dihydrofolate Reductase Protein-Fragment Complementation Assay (DHFR PCA) in Living Cells. Mol. Cell. Proteomics 17 (2), 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Markwardt ML, Kremers GJ, Kraft CA, Ray K, Cranfill PJC, Wilson KA, Day RN, Wachter RM, Davidson MW, and Rizzo MA (2011) An Improved Cerulean Fluorescent Protein with Enhanced Brightness and Reduced Reversible Photoswitching. PLoS One 6 (3), e17896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Piston DW, and Kremers GJ (2007) Fluorescent Protein FRET: The Good, the Bad and the Ugly. Trends Biochem. Sci 32 (9), 407–414. [DOI] [PubMed] [Google Scholar]
- (56).Yushchenko DA, Zhang M, Yan Q, Waggoner AS, and Bruchez MP (2012) Genetically Targetable and Color-Switching Fluorescent Probe. ChemBioChem 13 (11), 1564–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Saunders MJ, Block E, Sorkin A, Waggoner AS, and Bruchez MP (2014) A Bifunctional Converter: Fluorescein Quenching ScFv/Fluorogen Activating Protein for Photostability and Improved Signal to Noise in Fluorescence Experiments. Bioconjugate Chem. 25 (8), 1556–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Zhang M, Chakraborty SK, Sampath P, Rojas JJ, Hou W, Saurabh S, Thorne SH, Bruchez MP, and Waggoner AS (2015) Fluoromodule-Based Reporter/Probes Designed for in Vivo Fluorescence Imaging. J. Clin. Invest 125 (10), 3915–3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Xu S, and Hu HY (2018) Fluorogen-Activating Proteins: Beyond Classical Fluorescent Proteins. Acta Pharm. Sin. B 8 (3), 339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Holleran J, Brown D, Fuhrman MH, Adler SA, Fisher GW, and Jarvik JW (2010) Fluorogen-Activating Proteins as Biosensors of Cell-Surface Proteins in Living Cells. Cytometry, Part A 77A (8), 776–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Pow CL, Marks SA, Jesper LD, Silva GL, Shank NI, Jones EW, Burnette JM, Berget PB, and Armitage BA (2008) A Rainbow of Fluoromodules: A Promiscuous ScFv Protein Binds to and Activates a Diverse Set of Fluorogenic Cyanine Dyes. J. Am. Chem. Soc 130 (38), 12620–12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.