

Abstract
Lipid droplets (LDs) are intracellular storage organelles composed of neutral lipids, such as triacylglycerol (TG), surrounded by a phospholipid (PL) monolayer decorated with specific proteins. Herein, we investigate the mechanism of protein association during LD and bilayer membrane expansion. We find that the neutral lipids play a dynamic role in LD expansion by further intercalating with the PL monolayer to create more surface-oriented TG molecules (SURF-TG). This interplay both reduces high surface tension incurred during LD budding or growth and also creates expansion-specific surface features for protein recognition. We then show that the auto-inhibitory (AI) helix of CTP:phosphocholine cytidylyltransferase, a protein known to target expanding monolayers and bilayers, preferentially associates with large packing defects in a sequence-specific manner. Despite the presence of three phenylalanines, the initial binding with bilayers is predominantly mediated by the sole tryptophan due to its preference for membrane interfaces. Subsequent association is dependent on the availability of large, neighboring defects that can accommodate the phenylalanines, which are more probable in the stressed systems. Tryptophan, once fully associated, preferentially interacts with the glycerol moiety of SURF-TG in LDs. The calculation of AI binding free energy, hydrogen bonding and depth analysis, and in-silico mutation experiments support the findings. Hence, SURF-TG can both reduce surface tension and mediate protein association, facilitating class II protein recruitment during LD expansion.
Graphical Abstract
INTRODUCTION
Lipid droplets (LDs) are lipid and energy storage organelles, with cores composed of neutral lipids, such as triacylgycerol (TG) and sterol esters (SE), and bounded by a phospholipid (PL) monolayer.1–4 LDs dynamically recruit proteins to their surfaces throughout their lifecycle in response to cellular needs. For example, when extra energy and/or lipids need to be stored, lipogenesis proteins associate with the LD surface. When energy is required, lipolysis proteins associate.5–6 How LDs dynamically recruit different proteins depending on metabolic status is not fully understood and the focus of this research.
LD proteins are classified into two types depending on their origin. Class I proteins relocate from the ER membrane to LDs via ER-LD contact sites.7–8 One example is glycerol-3-phosphate acyltransferase 4 (GPAT4), an enzyme that catalyzes one of the TG synthesis reactions.9–10 They often contain a hydrophobic hairpin motif, which was proven to be sufficient for LD targeting by itself in the case of GPAT4.7,11 The main driving force of the ER-to-LD targeting has been suggested to be sequence and position-sensitive interactions between hydrophobic residues and the LD PL monolayer.7 This is supported by the finding that LD targeting can be abolished when the order of hydrophobic residues is shuffled. Tryptophan residues in particular were shown to gain significant energetic stabilization by relocating from unfavorable locations in the middle of the ER bilayer to a favorable region just below the glycerol groups in the LD monolayer. Class II proteins, in contrast, target LDs from the cytosol and generally contain one or more amphipathic helices (AHs). For example, the perilipin family exclusively target LDs via their conserved, 11-mer repeat regions.12–13 CTP:phosphocholine cytidylyltransferase (CCT) associates with expanding bilayers and LDs to cover the newly added surface area with phosphatidylcholine (PC), but disassociates once expansion stops and surface tension decreases.14–16 Further information on LD targeting can be found in several reviews.6,17–18
Over the last decade, AH–bilayer interactions have been extensively studied.19 The general mechanism of AH targeting to a bilayer surface is thought to consist of three steps: 1) sensing of packing defects by large hydrophobic residues, 2) peptide folding, and 3) stable association at the surface. Molecular dynamics (MD) simulations have provided insight into the molecular interactions involved in AH—bilayer targeting. All-atom resolution simulations have mainly focused on the first step because the timescale of peptide folding is much longer than most simulation limits (several microseconds). For instance, Voth and co-workers characterized packing defects using solvent-accessible surface area and colocalized those defects with hydrophobic residues upon the binding of an AH.20 Vanni and co-workers characterized packing defects using a Cartesian-based approach and proposed that deep defects are responsible for peptide targeting to bilayers.21 Monje-Galvan and Klauda approached peptide targeting from a thermodynamic context, computing interaction energies and contact frequencies for each amino acid with the membranes of the various PL compositions.22 For two bound conformations, they found that a Ser region or Tyr residue first approach the membrane and anchor the peptide, whereas charged residues, such as Lys, or bulky residues, such as Trp and Phe, contribute to stable association according to the calculated interaction energies.22
The targeting of AHs to LDs is thought to be somewhat similar to that to bilayers. Previous work attributed the general AH targeting preference for LDs over bilayers to LDs’ larger, more persistent packing defects.23 This general packing defect model can describe nonspecific adsorption of AHs to LDs; however, it does not explain differential targeting. For instance, the localization of perilipin family members can depend on the LD neutral lipid composition: Perilipins 1a, 1b, and 5 associate with TG-abundant LDs while perilipins 1c and 4 associate with SE-abundant LDs.24 In contrast, Arf GTPase-Activating Protein 1 has a broad spectrum of association, targeting both bilayers and LDs.25–28 This suggests that not all AHs are equal, and that their differences combined with bilayer/monolayer differences enable organelle-specific protein targeting.
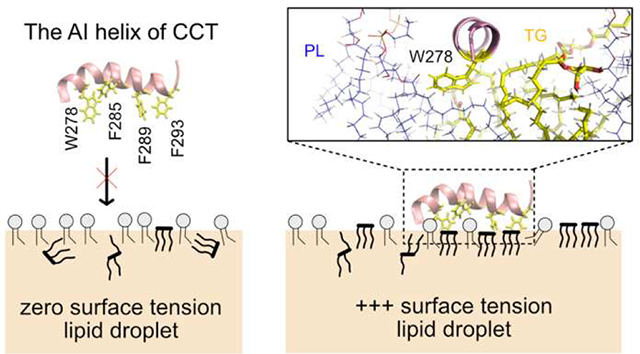
Another example of differential targeting, this time based on the condition of membrane surfaces, is CCT.29–31 CCT catalyzes the rate-limiting step of the PC synthesis. It targets both expanding bilayers and LDs but disassociates when surfaces stop expansion.14–16 Biologically, CCT is up-regulated by PC deficiency in order to catalyze the PC synthesis pathway. Biophysically, the CCT M domain, which is responsible for membrane association, targets PC-deficient and negatively charged membrane surfaces. PC-deficiency is thought to play a role because PC is a cylindrical shape that creates few packing defects, while PC’s precursors such as diacylglycerol (DAG) and phosphatidate (PA) or other lipids such as phosphatidylethanolamine (PE) are packing-discontinuity inducing lipids that facilitate CCT association.32 Negative charges are thought to attract the positively charged M domain (15 basic residues with a net charge of +1).30,33 Domain M of mammalian CCTα consists of three segments:30 The first segment closer to the N-terminus (residues 234–255) is basic and moderately amphipathic. The second segment (residues 256–271) is not conserved and nonessential for the binding of CCTα. The last segment (residues 272–293), the auto inhibitory (AI) helix, is highly amphipathic and contains a number of aromatic residues, including one Trp and three Phe residues. The AI helix is important in CCTα activation as well as membrane association. 34–35 In the cytosol, CCTα is inactive because the AI helix is docked onto the catalytic domain and thus suppresses the reaction. On the other hand, when CCTα binds to a membrane surface, domain M becomes a long, unbroken helix and the AI helix is released from the catalytic domain.36–37
We recently characterized several physical properties of LD monolayers that are different from bilayers.38 Surface-oriented TG molecules (SURF-TG) were found to occupy 5–8% of the LD surface (under zero-surface tension) and to display order in an analogous manner to PLs. The exposed glycerol groups and acyl chains of these SURF-TG molecules in turn create packing defects that are chemically distinct from those found in bilayers. While SURF-TG molecules modulate the area per PL (APL), other TG molecules (considered CORE-TG) interdigitate dynamically with the PL tail region, increasing PL acyl defects and the degree of PL order. The hydration of the LD core was also found to be larger than previously thought, which plays an important role in class I targeting.7,38 The display of TG at the LD surface suggests a plausible hypothesis for differential targeting in which neutral lipids either mediate LD protein interactions directly and/or alter monolayer properties to enable protein specificity. This idea is supported by recent experimental work that measured different binding affinities for a peptide to LDs with different neutral lipid compositions.39 The same order of binding affinities was demonstrated for the bare neutral lipids, implying direct interactions between a peptide and neutral lipids. Interestingly, changing the surface tension of these LDs shifted the binding affinities, however the order of the binding affinities was maintained.
Here we present all-atom MD simulations of LDs under an applied surface tension (i.e., stressed) to study the influence of surface expansion on the physical properties of stressed LDs and SURF-TG’s involvement in CCTα targeting. We demonstrate that under an applied surface tension the LD system expands and more TG molecules are integrated into the monolayer surface. The integration of more SURF-TG thus decreases the applied surface tension, apparent in an inverse correlation between the number of SURF-TG and surface tension once expansion is stopped until equilibrium is reached. Thus, a crucial interplay exists between surface composition and expansion that both enables stage-dependent protein recruitment and stress reduction under LD growth. We follow this with extensive MD simulations (13–14 replicas for each membrane) of the CCTα AI helix to study its association with both unstressed and stressed bilayers and LDs. Within the limited simulation time (1 μs for each replica), we observe both the initial binding event and subsequent residue association are important for peptide targeting. Except for the unstressed bilayer membrane, which has the smallest packing defects, most of the other trajectories have at least one binding event. For the stressed surfaces, other residues can subsequently bind to the membrane surfaces, further leading to targeting; however, finding large defects near the initially inserted Trp/Phe residue was challenging for unstressed surfaces. These factors significantly contribute to the reported binding success to expanding LDs and bilayers. In the stressed bilayers, the initial binding event is mostly mediated by Trp278 (the only Trp in the AI helix) rather than any of the three Phe residues. This is likely due to its preference for membrane interfaces. In the stressed LDs, each Trp/Phe residue has an equal frequency of initial association, but the association site is preferentially TG defects. Once the AI helix is fully associated in the stressed LDs, Trp278 is highly coordinated with the glycerol moiety of SURF-TG via hydrogen bonding. The portion of time frames that have hydrogen bonding between SURF-TG and Trp278 was computed to be 0.25, which is relatively high provided that the LD surface under study contains ~18% SURF-TG. The depth analysis shows that the insertion depth of Trp278 is well aligned with the glycerol moiety of SURF-TG, but not with that of PLs. When Trp278 is mutated to Val278, the high coordination number disappears and the binding success decreases. Finally, the umbrella sampling simulation to compute the free energy of the AI binding to the stressed lipid droplet shows the good agreement with the unbiased simulations. The binding free energy driving from water to the bound state is determined to be ~15.8 kcal/mol. Collectively, our data demonstrate the role of SURF-TG as a stress reducer and peptide targeting mediator, additionally showing the importance of the specific interaction between neutral lipids exposed at the LD surface with the peptide in LD targeting.
METHODS
System setup and simulation details
Four different types of membranes were studied in this work: unstressed bilayers (BI0), stressed bilayers (BIe), unstressed lipid droplets (LD0), and stressed lipid droplets (LDe) where ‘e’ and ‘0’ represent expanding and zero-surface tension, respectively. Every leaflet of the membrane systems was composed of 88 3-palmitoyl-2-oleoyl-D-glycero-l-phosphatidylcholine (POPC), 37 2,3-dioleoyl-D-glycero-l-phosphatidylethanolamine (DOPE), and l0 phosphatidylinositol (SAPI) molecules, which is representative of the ER membrane and LD monolayer surfaces of human cells.40–41 BI0 was prepared with the CHARMM-GUI membrane builder.42–45 After l00 ns of the NPT (constant particle number, pressure, and temperature) simulation, a constant surface tension of 15 mN/m per leaflet (or 30 mN/m for the whole system) was applied in the XY-dimensions for 100 ns. The last structure was used for further simulations of the stressed bilayer (BIe). The LD system was modeled with a tri-layer structure made up of an 8nm-thick bulk TG region in between the two PL monolayers. The initial structure was taken from the end of a 10 μs simulation described in previous work.38 More water was added in the Z-dimension, and the system was equilibrated for 1 μs to create the LD0 system. A constant surface tension of 15 mN/m per leaflet was then applied for 2.3 μs to model an expanding LD. The 300 ns structure from the constant surface tension simulation was taken for the further simulations of the LDe system. Of note, the surface tensions of stable, non-expanding LDs (in vitro) and purified LDs (in vivo) were measured to be 1.63 mN/m and 3.5 mN/m, respectively.39,46 The larger surface tension used herein was selected to capture expansion conditions. Importantly, no membrane instability such as pore formation was detected. TG was modeled with triolein in this work and the topology of triolein (TRIO.rtf) is available in https://github.com/ksy141/TG. All systems were solvated with TIP3P water,47–48 and 0.15M NaCl was added. Details of the system setup are described in Fig. S1.
The AI motif of CCTα (275–295 amino acid) was taken from the crystal structure (PDB 4MVC).34 The N- and C-termini were capped with acetylated N-terminus (ACE) and amidated C-terminus (CT2), respectively, to neutralize both ends. The helix was then located 1 nm above the highest atomic position of PL molecules, oriented such that the hydrophobic side of the AI helix faced the membrane surfaces. For each membrane system containing the AI helix, 13–14 replicas of MD simulations (NVT) were conducted with randomly generated initial velocities (each replica was run for 1 μs). A point mutation (W278V) was also prepared to test the importance of this tryptophan via an in silico mutation experiment. The binding behavior of the mutant (W278V) with LDe was compared to that for the wildtype helix.
The MD simulations were performed using the GROMACS (version 2018)49 simulation engine with the CHARMM36m lipid and protein force field.50–51 Simulations were integrated with a 2-fs timestep. Lennard-Jones pair interactions were cut off at 12 Å with a force-switching function between 10 Å and 12 Å. The Particle Mesh Ewald algorithm was used to evaluate long-range electrostatic interactions.52 The LINCS algorithm was used to constrain every bond involving a hydrogen atom.53 Biased simulations (described below) were conducted with the external plugin, PLUMED2.54–55 The pressure was maintained semi-isotropically using the Parrinello-Rahman barostat56 at a pressure of 1.0 bar, a compressibility of 4.5×10−5 bar−1, and a coupling time constant of 5.0 ps. The temperature was maintained using the Nose-Hoover57–58 thermostat at 310 K with a coupling time constant of 1 ps. For NPγT (constant particle number, normal pressure, surface tension, and temperature) simulations, the Berendsen thermostat and barostat were used with the same parameters.59 The umbrella sampling (US) simulations were carried out with NAMD 2.14 to quantify the free energy of CCTα binding to LDe with NAMD 2.14.60 The temperature was maintained using the Langevin thermostat at 310 K with a Langevin coupling coefficient of 1/ps (NVT ensemble) and no coupling for hydrogen atoms.
Molecular images included in this work were rendered using Visual Molecular Dynamics (VMD)61 and PyMOL, and the trajectories were analyzed with MDAnalysis.62 Details of each simulation are described in Table 1.
Table 1.
Description of simulations.
| Simulations | Lipid composition a) | Time | Ensemble | Replicas | Initial struct. |
|---|---|---|---|---|---|
| Bilayer membrane (100 ns structure referred to as BI0) | 176 POPC + 74 DOPE + 20 SAPI | 100 ns | NPT | 1 | CHARMM-GUI |
| Bilayer membrane (100 ns structure referred to as BIe) | 176 POPC + 74 DOPE + 20 SAPI | 100 ns | NPγT | 1 | From above sim. |
| Lipid droplet (1 μs structure referred to as LD0) | 176 POPC + 74 DOPE + 20 SAPI + 429 TRIO | 2 μs | NPT | 1 | From previous simulation38 |
| Lipid droplet (300 ns structure referred to as LDe) | 176 POPC + 74 DOPE + 20 SAPI + 429 TRIO | 2.3 μs | NPγT | 1 | From above sim. |
| BI0 production run | Same as BI0 or BIe | 400 ns | NVT | 1 | BI0 |
| BIe production run | Same as BI0 or BIe | 400 ns | NVT | 1 | BIe |
| LD0 production run | Same as LD0 or LDe | 2 μs | NVT | 1 | LD0 |
| LDe production run | Same as LD0 or LDe | 2.3 μs | NVT | 1 | LDe |
| CCTα folding PMF | N/A | 100 ns b) | NVT | 10 c) | CHARMM-GUI |
| CCTα binding PMF | Same as LD0 or LDe | 162 ns b) | NVT | 36 | LDe |
| BI0 + CCTα | Same as BI0 or BIe | 1 μs b) | NVT | 14 | BI0 |
| BIe + CCTα | Same as BI0 or BIe | 1 μs b) | NVT | 13 | BIe |
| LD0 + CCTα | Same as LD0 or LDe | 1 μs b) | NVT | 14 | LD0 |
| LDe + CCTα | Same as LD0 or LDe | 1 μs b) | NVT | 13 | LDe |
| LDe + W278V | Same as LD0 or LDe | 1 μs b) | NVT | 13 | LDe |
The total number of lipid molecules of each type in a system.
Simulation time length for each replica or window.
The number of windows in the replica-exchange umbrella sampling simulation.
Packing defects
We used the Cartesian-based algorithm to evaluate lipid-packing defects. For the upper leaflet, the lipid atoms (PL and TG) whose Z positions were greater than the threshold (zthr) were selected and projected onto a 1 Å-spacing two-dimensional (XY) grid. The threshold (zthr) was set to the average Z position of the phosphorus atoms of the upper leaflet minus 2 nm, which includes virtually all the lipid atoms of the upper leaflet. If a grid point overlaps with an atom (we define “overlapping” if the distance between the center of the atom and the grid point is less than the atom’s radius, which is taken from the CHARMM36 parameter set,50 plus the half of the diagonal of the grid, √2/2 Å), the Z position of the atom and the atom type are saved in the grid point. If a grid point overlaps with polar atoms (PL head groups or PL glycerol moieties), the grid point is considered no defect. The other grid points were assigned to be PL acyl defects, TG acyl defects, or TG glycerol defects, based on the atom types and Z coordinates saved in the grid point. The atom which has the highest Z position determines the defect type. This algorithm is equivalent to finding a lipid atom that has the highest Z position at each grid point. The same procedure was repeated for the lower leaflet.
For each defect type, the neighboring elementary defects were clustered into one. If the clustered defect contains N elementary defects, it is considered to have a defect size of N Å2. Consistent with other work,20–21,38,63–65 the probability of finding a defect with the size of N Å2 was computed and fit to an exponential decay function: P(N) = ce−N/π, where c is the normalization constant and π is the packing defect constant. If a defect is smaller than 15 Å2 or the probability is lower than 10−4, the data was not included in the fit. The packing defect constant represents how slowly the decay function falls off. The higher the packing defect constant, the higher the probability of finding larger defects.
By using a deeper threshold (zthr) than previous applications of this algorithm,63–64 every grid point overlaps with at least one atom, eliminating so-called ‘deep’ defects. (Previous papers set zthr to 0.1 nm below the sn-2 carbon of the nearest glycerol, while we set it to 2 nm below the average phosphorus level of the either leaflet.) Deep defects have been useful in characterizing the very first step of peptide targeting (sensing of packing defects by large hydrophobic residues).21 However, when a peptide is fully associated with a membrane, almost all grid points where the peptide is located become deep defects. Therefore, the concept of deep defects was eliminated in this work to avoid losing important information about the helix—packing defect interactions. The packing defect analysis script is available in https://github.com/ksy141/SMDAnalysis with an example. The MDAnalysis62 and parallel MDAnalysis66 packages are required.
Helicity PMF
To determine the stability of the AI helix in water, replica-exchange67 umbrella sampling68 (REUS) simulations were carried out to compute the potential of mean force (PMF) of the AI helix as a function of the distance between the carbon alpha atom of the first residue (Ile275) and that of the last residue (Pro295). Harmonic restraints with a force constant of 400 kJ/mol/nm2 were placed in each of the 10 umbrella sampling windows with a 0.2 nm spacing over a range of 2.9 nm to 4.7 nm. An initial structure for each window was obtained from steered MD simulations, in which the same collective variable was biased with a force constant of 500 kJ/mol/nm2 for 20 ns. The exchange was attempted every 1000 steps. The PMF was generated using the weighted histogram analysis method (WHAM) with a bin spacing of 0.02 nm.69–70 The REUS simulations were run for 100 ns, and the trajectories were divided into five blocks, each containing a 20-ns trajectory. The first two blocks were considered equilibration and discarded. The error bars were the standard deviation of the block averages of the rest three blocks.
Binding PMF
The free energy of binding of the AI helix to the LDe surface was estimated using umbrella sampling (US) simulations in the NVT ensemble. The PMF was calculated as a function of the perpendicular distance zCM between the centers of mass (COMs) of the helix and the tri-layer system. 36 windows were prepared with a 0.1 nm spacing over a range of 3.5 nm to 7.0 nm. For the constraints, a harmonic potential with a force constant of 2.5 kcal/mol/Å2 was used in each window. An initial structure for each window was obtained from steered MD simulations, in which the alpha helix content was biased with a force constant of 700 kcal/mol/α2 to maintain the helical structure. Each replica was then equilibrated for 90 ns. The PMF was generated using WHAM with a bin spacing of 0.01 nm. The US simulations were run for 72 ns (with no constraints on the alpha helical content), and the trajectories were divided into six blocks, each containing a 12-ns trajectory. The first 36 ns were discarded for the PMF calculation, and the error bars were obtained from the standard deviation of the last three blocks. For comparison, the normalized probability distribution of P(zCM) and the average location ‹z›P of the phosphorous atoms of the PL molecules in the binding leaflet were obtained from the unbiased MD simulations. The simulation described here was conducted with NAMD 2.14.60
Classification of TG
TG molecules were categorized into two groups: SURF-TG and CORE-TG. The former is surface-oriented with their glycerol moieties exposed to the cytosol and acyl chains extended toward the LD core. The rest of TG molecules are considered CORE-TG. The designation was based on the distance between the TG glycerol moiety and the average position of PL tails (along the Z axis) of the closer leaflet. If the center of mass (COM) of the TG glycerol moiety is located above that of the PL acyl chains of the closer leaflet, it was classified as SURF-TG. Otherwise, it was denoted by CORE-TG.
Binding of AI helix
Binding was defined based on the relative Z position of the Trp/Phe residues of the AI helix (Trp278, Phe285, Phe289, and Phe293) with respect to the phosphorus atoms. Specifically, successful binding was defined if the Z positions of all four residues are lower than the average Z position of the phosphorus atoms of the binding leaflet for more than 100 ns. Complete binding was defined if the average Z position of the four residues was less than the average Z position of the phosphorus atoms by more than 0.5 nm. Once the peptide binds to a membrane surface completely, the Z positions of all the residues remain the same. We used the 14 complete binding trajectories (9 and 5 for BIe and LDe, respectively) for the normalized coordination number analysis.
Molecular groupings to analyze normalized coordination number
To compute the interaction preference of the Trp/Phe residues (Trp278, Phe285, Phe289, and Phe293) with each membrane component, the coordination number was calculated between molecular groups. Each PL molecule was represented by 11 groups: polar head group, phosphate group, glycerol moiety, and four groups for each acyl chain. Similarly, each TG molecule was divided into 13 groups: glycerol moiety and four groups for each acyl chain. Each amino acid was represented by one backbone group and one side chain group. The coordination number between a side chain group of a Trp/Phe residue and each membrane group was computed by , where r0 = 0.4 nm and ri is the distance between the side chain group and the membrane group. The normalized coordination numbers (‖ s ‖) for POPC, DOPE, and SAPI were obtained by dividing the coordination numbers by the numbers of the corresponding PL molecules, which are, 88, 35, and 10 (in one leaflet), respectively. The normalized coordination number (‖ s ‖) for TG was obtained by dividing the number of SURF-TG molecules of the binding leaflet at each time frame.
RESULTS AND DISCUSSION
Physical properties of unstressed and stressed bilayers and LDs
MD simulations of the unstressed bilayer (BI0), stressed bilayer (BIe), unstressed LD (LD0), and stressed LD (LDe) were conducted to characterize the physical and structural properties of unstressed and stressed bilayers and LDs. The stressed bilayer and LD were prepared by applying the surface tension of 15 mN/m per leaflet to the equilibrated bilayer and LD, respectively (Fig. SI and Methods). While the surface tension was maintained at 15 mN/m per leaflet in the NPγT ensemble, different responses to the stress were apparent for the bilayer and LD systems (Figs. 1a and 1b). The X-dimension of the bilayer increases at the beginning and then levels off at ~10 ns, while that of the LD continues to increase with simulation time and takes longer to level off (~1.8 μs). The bilayer response is expected. Under stress the bilayer expands, and more hydrophobic acyl chains are exposed to the cytosol (packing defects), but it can only expand so much. For the LD, on the other hand, stress induces much greater expansion than the bilayer membrane even though the same PL composition was used for both systems (7.9% increase for the bilayer and 38.1% increase for the LD in the X dimension, or equivalently 16.4% and 90.8% increase in the APL). The reason is that more TG molecules are pulled from underneath the PL monolayer (CORE-TG) to create more surface-oriented TG (SURF-TG) in response to the high surface tension. Since the simulation continues to keep the surface tension at the applied 15 mN/m per leaflet, the expansion continues, and more SURF-TG is created. Therefore, the stressed bilayer has increased PL acyl defects compared to the unstressed bilayer, while in the LD there is an increase in both PL acyl defects and TG-related defects (Figs. 1c, Id, S2).
Figure 1:

Bilayer and LD under the constant surface tension. The X dimension Lx (black) and surface tension per leaflet γ (blue) for the bilayer (a) and LD (b) in the NPγT ensemble (γ = 15 mN/m per leaflet). The surface tension per leaflet (blue) was smoothed using a 2,000-point running average for the bilayer and a 6,000-point running average for the LD. The 100 ns and 300 ns structures of the NPγT simulations for the bilayer (c) and LD (d), respectively, which are referred to as BIe and LDe in this study. The light and dark blue indicate polar groups (head groups and glycerol moieties) and acyl chains of PLs, respectively. The green and yellow indicate glycerol moieties and acyl chains of TG, respectively. PL acyl defects are shown in red and the other defects are not shown for visual clarity. The snapshots of unstressed bilayer (BI0) and LD (LD0) are shown in Fig. S2.
To compare the surface, molecular, and physical properties of the four membranes under a fixed level of expansion, NVT simulations were conducted (production run). The packing defect constants (π) for four surfaces were evaluated (Fig. 2). From BI0 to BIe, the packing defect constant for PL acyl defects significantly increased (Fig. 2), consistent with the snapshots (Fig. 1c and S2). The packing defect constant for PL acyl defects also increased from LD0 to LDe, making them nearly equivalent to those in BIe. However, the increase from LD0 to LDe is not as large as the increase from BI0 to BIe, partially because LD0 has more PL acyl defects than BI0 due to the interdigitation with CORE-TG, as previously described.38 The increase is also less significant because the increased surface area in LDe includes increased TG acyl and glycerol defects (Fig. 2). Overall, the LD surfaces are more enriched with chemically distinct defects and therefore will more easily recruit certain proteins, as demonstrated in the previous work.23
Figure 2:

Packing defect constants (π) of PL acyl, TG acyl, and TG glycerol defects for BI0 (orange), BIe (red), LD0 (light blue), and LDe (dark blue).
In the NVT simulation of LDe, the surface tension relaxes to 10.0 ± 0.6 mN/m from 15 mN/m in ~500 ns (Fig. 3). In contrast, the surface tension of BIe under the NVT ensemble remained 14.4 ± 0.3 mN/m. We suspected that the decrease in the surface tension could be due to increasing numbers of SURF-TG. We therefore define the SURF-TG ratio (rSURF–TG) with respect to PL as the number of SURF–TG (NSURF–TG) divided by the sum of the number of SURF–TG (NSURF–TG) and the number of PL (NPL): rSURF–TG = NSURF–TG/(NSURF–TG + Npl(. Consistent with our expectations, the amount of SURF–TG and surface tension are inversely correlated (Fig. 3), demonstrating SURF-TG does effectively reduce high surface tension. The system equilibrates when the induced surface tension from SURF-TG being a less effective surfactant than PLs balances the applied surface tension. Of note, we showed that SURF-TG can occupy 5–8% of the surface under zero-surface tension in our previous work.38 Under stress, the percentage of the SURF-TG in LDe increases to ~20% (Fig. 3). We expect the role of SURF-TG as a stress reducer is critical when LDs are budding or growing under the limited supply of PLs.
Figure 3:

NVT simulation of the stressed LD (LDe). The SURF-TG ratio (black), rSURF–TG = Nsurf–tg/(NSUrf–tg + NPL), is inversely correlated with the surface tension per leaflet (blue). The surface tension per leaflet was block-averaged every 20 ns.
We then calculated the PMF of PL and TG with respect to the Z position by histogramming the Z position of C2 atoms for PLs and TGs (Fig. 4). In our previous work, we discussed how transitioning from CORE-TG to SURF-TG is a slow process such that SURF-TG only equilibrates after 2 μs of simulation. Consistent with this, we argue the high transition barrier (~2.6 kcal/mol) from CORE-TG to SURF-TG in the unstressed LD has eluded the observation of SURF-TG in the previous shorter all-atom simulations.23,71 SURF-TG is increasingly stabilized in the stressed system, making the transition barrier from SURF-TG to CORE-TG even higher in LDe. This implies SURF-TG stays longer at the LD surface in the stressed LD. The location of the local minimum of SURF-TG (indicated with dashed lines in Fig. 4) also shifts from −0.42 nm in LD0 to −0.31 nm in LDe. Thus, the depth of SURF-TG is more aligned with that of PLs in the stressed LD. We will later discuss how the insertion depth of Trp278 of CCTα aligns with SURF-TG’s glycerol moiety depth, and they interact via hydrogen bonding. Finally, as one may expect, pulling PL from the averaged position resembles a harmonic potential (for the regions sampled) and is much more resistant than TG.
Figure 4:

Transition PMFs for PLs (continuous lines) and TG molecules (lines with squares) in the unstressed LD (gray) and stressed LD (black) from unbiased NVT simulations. The average Z position of PL’s C2 atom of either leaflet was set to zero and the positive Z is toward water. The vertical dashed lines indicate the local minima for the TG PMFs.
In order to probe the properties of PLs and SURF-TGs under the stress, the order parameters of PL and SURF-TG acyl chains were calculated (Fig. 5). There is a significant decrease in the PL’s order parameters under surface tension for both bilayers and LDs. This is because the expansion in XY-dimensions causes the low density at each Z position (Fig. S3). Therefore, the acyl chains have more space for fluctuations, resulting in a reduction in their order. The order parameters of PLs in the unstressed LD (light blue) are slightly higher than those in the unstressed bilayer (orange), especially at the tail region, which is consistent with previous work.38,71 This is because CORE-TG interdigitates with the PL acyl chains, resulting in a higher density in the lower tail region (−2 nm ~ −1.4 nm in Fig. S3). SURF-TG tracks DOPE’S order parameters quite well for both stressed and unstressed LD, demonstrating its ability to act as a membrane component.
Figure 5:

Order parameters of POPC (top) and DOPE (bottom) from BI0 (orange), BIe (red), LD0 (light blue), and LDe (dark blue). Gray and black squares indicate the order parameters of SURF-TG from LD0 and LDe, respectively.
CCTα and the AI helicity
We studied association of the CCTα AI helix (residues 275–295) to four the membrane systems (BI0, BIe, LD0, and LDe) using the extensive MD simulations. The AI helix was taken from the resolved structure of CCTα (PDB 4MVC) (Fig. 6a).34 The AI motif is amphipathic and contains four Trp/Phe residues (Trp278, Phe285, Phe289, and Phe293), which play a key role in protein association with the membrane surfaces (Fig. 6b and 6c). Previous studies investigated the LD targeting behavior of CCTα P2, a longer peptide extending toward the N-terminus (residues 267–294) of the AI helix.23 The P2 peptide targets expanding LD surfaces, but when the large hydrophobic residues (Ile, Leu, Phe, and Trp), including the four Trp/Phe residues (Trp278, Phe285, Phe289, Phe293), are mutated to Val, LD targeting is abolished. In this work, we also conducted the mutant experiment in silico with a single mutation, W278V, which will be discussed later.
Figure 6:

CCTα and the AI helix, (a) The resolved CCTα structure (PDB code: 4MVC). CCTα is a dimeric protein, each monomer represented with a different color. The pink segment represented as a ribbon is the AI helix (residues 275–295) used in this study. (b) The AI helix in pink and four Trp/Phe residues in yellow. (c) The helical wheel of the AI helix computed using Heliquest (https://heliquest.ipmc.cnrs.fr/).74 (d) The helical PMF (left) along the end-to-end distance, d, with the error bars (thin lines). The last structures of the first and the last window (right) with the carbon alpha atom of the first residue (red) and the last residue (purple).
Previous circular dichroism (CD) analysis of the peptides, CCTα PEPC22 (residues 267–288) and domain M (residues 237–293), demonstrated that these peptides are helical in the presence of anionic lipid vesicles and unstructured in water.72–73 On the other hand, the AI motif remains α-helical in the solution form of CCTα because it is docked onto the catalytic domain. When binding to a membrane, the M domain of CCTα becomes a long α-helix.34–35 Therefore, we hypothesized that the AI motif could remain helical over the course of CCTα membrane association. To test this hypothesis, we conducted a REUS simulation of the AI helix in an aqueous solution to obtain the PMF using the end-to-end distance as a collective variable. The biased simulations suggest that the helical structure of the peptide is stable in water within the accuracy of the force field (Fig. 6d). This motivates us to employ the helical structure as an initial structure of the AI motif in our membrane binding simulations.
CCTα Al-membrane binding
We first placed the AI helix 1 nm above the highest Z position of PLs in the unstressed and stressed bilayers and LDs. The AI helix was oriented such that the hydrophobic side faces the membrane surface. In order to obtain sufficient statistics, we conducted 13–14 replicas for each system, with each trajectory was run for 1 μs. We define the binding of the AI helix to a membrane using the Z position of the average phosphorus atoms of the binding leaflet and the Z positions of the four Trp/Phe residues (Trp278, Phe285, Phe289, and Phe293). When the four residues remain below the average phosphorus level (referring to the upper leaflet) for more than 100 ns, the AI helix was considered to have successfully associated with the membrane. This definition is equivalent to the previous paper23 except the previous paper used only one large hydrophobic residue while we used a stronger condition of all four Trp/Phe residues. The difference was chosen because the previous work used the random coil as an initial structure and studied the very first association of the binding, while we used the helical structure and therefore can better sample the phase space in which the peptide can fully associate. For the mutant (W278V) trajectories, two trajectories were excluded from the successful binding category that satisfy the above condition but have high fluctuations in the Val278 depth profile in the last 100 ns. Val278 effectively disassociates from the membrane surface, which did not occur in the natural (unmutated) simulations.
The binding success was simply calculated as the occurrence by dividing the number of trajectories that have successful association within our simulation length by the number of total trajectories (13–14 replicas). Consistent with the experimental observation,14–16 the AI helix shows the high binding success with the stressed bilayer and the stressed LD (~90%) in the timescale of microseconds (Fig. 7). The single mutation (W278V) reduces the binding success with the stressed LD to ~54%. Further analysis and simulations would be required to confirm the previous predictions that the AI helix should target LD0 over BI0 due to the bigger, more persistent packing defects present at the LD surface.23 However, analyzing the depth profile of each Trp/Phe residue does support this argument. For 4 out of 14 BI0 trajectories, no association of any Trp/Phe residues was observed. On the other hand, 13 out of 14 LD0 trajectories have at least one initial association.
Figure 7:

Binding success (occurrence) of the AI helix (wildtype in black, mutant in crosshatch) to four different surfaces. For each membrane, the simulations were replicated 13–14 times.
Comparing association with BI0 versus BIe, the lack of initial association of any Trp/Phe residues in 4 out of 14 BI0 trajectories is likely due to the limited packing defects in the BI0 surface. In contrast, all the BIe trajectories include association with at least one of the Trp/Phe residues. We also observed the active role of Trp in peptide targeting in BIe using the depth profile analysis. Although the AI helix has three Phe and only one Trp, it was Trp278 that first associated in 8 out of 13 BIe trajectories. We expect this is because of Trp’s preference for membrane interfaces.7,75 In order to have successful binding, subsequent association of the other large hydrophobic residues should follow. We focused on two bilayer simulations, one with unsuccessful association to BI0 (Fig. 8) and another with successful association to BIe (Fig. 9). For both simulations, Trp278 was first inserted and the peptide stayed at the membrane surface. In the unsuccessful binding (Fig. 8), Trp278 is located at the average phosphorus level of the upper leaflet (a depth of 0 represents the average phosphorus level). However, it could not further embed, nor could the other Phe residues embed due to a lack of large, neighboring packing defects. In contrast, in the successful simulation (Fig. 9), Trp278 was first inserted at 25 ns, followed by Phe285 at 70 ns. At 75 ns, a large, neighboring defect emerged near the defect that eventually accommodated the other Phe residues (Phe289 and Phe293) resulting in full association.
Figure 8:

Unsuccessful binding of the AI helix in BI0. The depth profile is shown in top left. The average Z position of the phosphorus atoms of the upper leaflet was set to 0. The positive Z is toward water and the negative Z toward the membrane center. Snapshots with simulation times are shown. The light and dark blue indicate polar groups and acyl chains of PLs, respectively. The PL acyl defects (red) are colocalized. The AI helix is shown in pink. Only the part of the system is shown in this figure. The actual membrane system is larger.
Figure 9:

Successful binding of the AI helix in BIe. The same color code was used as in Figure 8.
Focusing on the LDe simulations demonstrates that TG defects play a significant role in peptide targeting. In 9 out of 13 LDe trajectories, the initial binding happened at a TG defect, suggesting that SURF-TG facilitate the initial association of Trp/Phe residues. In contrast to the BIe simulations, the four Trp/Phe residues have an equal frequency of initiating binding to LDe. Specifically, the number of trajectories in which Trp278, Phe285, Phe289, and Phe293 initially associated was 4, 3, 3, and 3, respectively. We expect this is because of Phe’s stabilization at the LD interface due to interactions with SURF-TG. Our previous work supports this argument showing greater stabilization of Phe during permeation into a relaxed LD compared to that into a relaxed bilayer.7 We expect the stabilization will be even greater in LDe because of the increased amount of SURF-TG compared to LD0. Finally, one successful binding trajectory to LDe is shown in Fig. 10. Trp278 initially associates with the TG acyl defects at 27 ns and other Phe residues target the PL acyl defects, emerging near Trp278 at 95 ns. Once the peptide is fully associated, the peptide preferentially interacts with the hydrophobic surface of the membrane via PL and TG acyl defects as shown in Fig. 10 (snapshot from 857 ns).
Figure 10:

Successful binding of the AI helix in LDe. The same color code for PL and protein was used as in Figure 8. Additionally, the glycerol moieties and acyl chains of TG are shown in green and yellow, respectively. Defects are not shown for visual clarity.
To quantify the frequency of interactions between each Trp/Phe residue and each membrane group once the peptide is fully associated, we selected and analyzed the trajectories that have complete binding. We define the complete binding if the COM of the four Trp/Phe residues are 0.5 nm below that of the phosphorus atoms of the binding leaflet. This is an even stronger condition than that used to define the successful binding above. In these trajectories, the Z position of each residue remains almost the same following full association. Nine BIe trajectories and five LDe trajectories satisfied this condition. For these simulations, we first reduced the resolution of the all-atom trajectories to collective coordinates as described in the Methods and shown in Figs. 11a and 11b. This was done to reduce the amount of redundant information and increase the efficiency of the coordination analysis. In each trajectory, the coordination number between each Trp/Phe residue and each membrane group was computed (averaged over the frames that have complete binding). The normalized coordination number (|| s ||) was obtained by dividing the coordination number by the number of molecules of each type at the binding surface (see the Methods). We then averaged the normalized coordination number over the trajectories that have complete binding for each system (Nine trajectories for BIe, five trajectories for LDe, and three trajectories for W278V-LDe). The error bars (Fig. S4) revealed that the peptide-SAPI interactions were not statistically reliable. This is not a surprise given that each trajectory samples different phase space of peptide targeting, and SAPI is the minor membrane component with only 10 molecules per leaflet. Thus, SAPI was excluded from this analysis. In general, the Trp/Phe hydrophobic residues have higher preference for membrane molecules with two or more unsaturated double bonds (DOPE, and TG) over POPC, which has only one double bond in the acyl chains (Figs. 11 and S4). Interestingly, the high contact between the glycerol moiety (GL) of TG and Trp278 was found in LDe simulations (Fig. 11c and S4). This is explained by hydrogen bonding between Trp278 and the GL of SURF-TG, which can be less than 2 Å in the trajectories, indicative of a stable hydrogen bond (Fig. 11d). In contrast, the phenylalanine residues which lack the hydrogen bonding ability do not show a clear preference for TG’s GL group (Fig. S4).
Figure 11:

Molecular groupings (MG) of all-atom trajectories to analyze the normalized coordination number. (a) PL and TG, represented with 11 and 13 molecular groups, respectively. (b) The all-atom (left) snapshot of the first timeframe and the corresponding collective coordinates (right). Each amino acid consists of one backbone group and one side chain group. Water and ions are not shown for visual clarity. (c) The normalized coordination number (|| s ||) between Trp278 and each membrane group, averaged over the trajectories that have the complete binding for the stressed LD. The filled circles, unfilled circles, and triangles represent sn-1, sn-2, and sn-3 chains, respectively, (d) The snapshot showing the hydrogen bond between Trp278 (represented as sticks) and the glycerol moiety of TG. The same color code was used as in Fig. 10.
This interaction is also apparent in the fraction of frames that have the hydrogen bond between Trp278 and the oxygen atoms of SURF-TG, which is 0.244 ± 0.125 from the frames that have the complete binding in the LDe trajectories. The hydrogen bond was defined with the donor-acceptor distance cutoff of 0.4 nm and the angle cutoff of 150°. Thus, Trp278 forms a hydrogen bonding with SURF-TG in approximately one out of four frames, which is relatively high number considering the lipid composition of the stressed LD surface is only ~15–18% of SURF-TG. Depth analysis further supports Trp278’s preferential interaction with SURF-TG over the PL glycerol group. The preferred insertion depth of Trp278 (−3.7 Å averaged from bound simulations where the depth of 0 represents the average Z position of the PL C2 atom of the binding leaflet) is well aligned with the Z positions of the six SURF-TG oxygen atoms (centered at −2.9 Å), but far from that of the four PL glycerol oxygen atoms (centered at +1.3 Å).
The computed binding free energy of the AI helix (Fig. 12) also supports our findings. To ensure the findings from our unbiased simulations are statistically informative, we calculated the free energy profile (PMF) of the helix association with the LDe surface with US simulations. The distance zCM between the COMs of the helix and the tri-layer system was used as the collective variable. The free energy minimum is located at zCM ≈ 4.35 nm, which is below the average location of the phosphorous atoms of the PL molecules in the binding leaflet ‹z›P ≈ 4.48 nm (green dashed line) and consistent with hydrogen bonding between Trp278 and SURF-TG. The probability distribution P(zCM) of the helix from the unbiased simulations in which the AI helix is bound (shown by red bar graph) aligns well with the free energy profile. The slight shift to toward the LD center in this distribution is expected given that the simulations in which association is weak or absent are not included. The alignment along with the barrierless PMF and large binding free energy (−15.8 kcal/mol) support the validity of the data gathered from the unbiased simulations and the observed rapid association with the LDe surface.
Figure 12:

PMF (blue) of the AI helix binding to the LDe surface, obtained with the umbrella sampling simulations. The error bars (blue dashed lines) were obtained from the standard deviation of the last three blocks. The normalized probability distribution of the AI helix P(zCM) (red bars) and the average location of the PL phosphorous atoms ‹z›P (green dashed line) were obtained from the unbiased MD simulations.
Furthermore, a point mutation (W278V) was prepared to confirm the importance of Trp278 via an in-silico mutation experiment. In the mutated trajectories (W278V), three LDe trajectories satisfied the complete binding condition in which the COM of the four Val/Phe residues were 0.5 nm below that of the phosphorus atoms of the binding leaflet. Consistent with our expectations, Val278 does not show a preference for the GL of SURF-TG due to the lack of the hydrogen bonding ability (Fig. 13). Taken together, this strongly supports the frequent interaction with the GL of SURF-TG being specific to tryptophan due to its combined features of being a large aromatic residue with the ability to hydrogen bond.
Figure 13:

Normalized coordination number between Val278 and each membrane group, averaged over the mutant trajectories (W278V) that have complete binding with the stressed LD. The filled circles, unfilled circles, and triangles represent sn-1, sn-2, and sn-3 chains, respectively.
Finally, we characterized the nature of the defect-residue interactions by calculating the area of each defect type under Trp278 and Phe285 when they are fully associated (i.e. when the residue COM is 5 Å below the average phosphorus level of the binding leaflet). Figure 14 shows the normalized joint probability distribution P(aPL,aTG) of each residue associating with PL and TG defects (aPL and aTG, respectively). The heat map adopts a lower triangular form, consistent with maximum areas of 18 Å2 and 14 Å2 for Trp and Phe, respectively. Lower cumulative values are due to overlap with PL head groups. Consistent with lipid packing defects, which are much larger than the dimensions of the residues and characterized by compositional heterogeneity, the residues often interact with a mixture of PLs and TGs. In fact, the probability that the residues reside on a mixture of PL and TG elementary defects, simply the integral of P(aPL ≠ 0,aTG ≠ 0), turns out to be 74.5% for Trp278 and 60.1% for Phe285. Therefore, when Trp278 is completely bound to the stressed LD, its interaction with SURF-TG does not necessitate its localization to a homogeneous packing defect.
Figure 14:

Normalized joint probability distribution of the defect areas, aPL and aTG, that Trp278 (a) or Phe285 (b) associates with, given that each residue is completely associated.
CONCLUSIONS
LDs store neutral lipids (e.g. TG) surrounded by a PL monolayer. We recently demonstrated that TGs interdigitate with the PL monolayer as surface-oriented (SURF-TG) molecules creating chemically distinct packing defects from those found in PL bilayers.38 In this work, we further characterize the physical and surface properties of stressed LDs under surface tension, which is expected to increase during various LD lifecycle stages such as budding and growth. Although SURF-TG molecules are worse surfactants than PLs, and increase surface tension under non-expansion conditions, they can decrease applied high surface tension during expansion. As shown herein the SURF-TG percentage increases from 5–8% under zero-surface tension conditions to ~18% at ~10mN/m. Consistently, once expansion is stopped, the inverse correlation between the percentage of SURF-TG with the surface tension demonstrates that SURF-TG can effectively reduce high surface tension. Thus, the interplay between SURF-TG and surface tension will facilitate growth, such as during the initial stages of LD formation, by mediating stress alongside the association of proteins like lipid droplet assembly factor 1.76 Similarly, growth and shrinkage will alter the LD surface, enabling stage-dependent protein recruitment.
We also investigate how SURF-TG is involved in AH targeting in both stressed and relaxed LDs. We chose the AI helix of CCTα as a test peptide, motivated by its conditional binding to the expanding (stressed) monolayer and bilayer surfaces. In water, the AI helix is helical due to its association with the catalytic domain of CCTα.34–35 Therefore, we used the AI helix as our initial structure. We further support the validity of this choice by showing that the helix is also stable in water with enhanced free energy simulations. The advantage of being able to use the helix as the initial peptide structure is that it enables sampling of the membrane-associated phase space and bypasses the folding step common for other AH helices. This allows us to discuss residue—membrane interactions in the fully associated state, which will be valuable when considering disassociation as well. The computational cost of folding an AH helix during membrane association is what limited most previous MD simulations to the very first step of peptide targeting (sensing packing defects by hydrophobic residues).20–21,23
Comparing trajectories for stressed and relaxed bilayers (BIe and Bl0, respectively) and LDs (LDe and LD0, respectively) reveals that the initial association is attempted more frequently for the stressed systems, and least frequently for the unstressed bilayer. Full association is dependent on the presence of large defects near the initially inserted residue, which is far less probable in the unstressed surfaces. Thus, the increase in size and prevalence of packing defects in expanding bilayer and monolayer surfaces is what drives conditional CCTα association during expansion. Our bilayer results further reveal that Trp is more “active” than Phe in the initial targeting. The number of trajectories in which Trp first associates with a surface is greater than those in which Phe first associates, despite the fact that there are three Phe residues to only one Trp. Additionally, the inserted Trp effectively holds the AI helix localized at the interface while the other large hydrophobic residues are looking for the neighboring defects. In contrast, when Phe first associates, it is sometimes kicked out of the membrane surface. The active role of Trp in peptide targeting is likely due to its preference for membrane interfaces.7,75 It is interesting to note that Trp278 is also located at the bottom of the helix, closest to where CCTα would interface with the membrane. In contrast, the specificity of Trp in the initial targeting is reduced in the stressed LD, potentially because of Phe’s stabilization at the LD surface. The frequency that Trp or Phe residues initially associated was comparable, but both preferentially targeted TG defects over PL defects in our stressed LD simulations.
Once the AI helix is fully associated with the stressed LD, the interaction between the glycerol moiety of SURF-TG and Trp278 was notably high, and the interaction frequency was every out of four frames on average. This specific interaction is driven by hydrogen bonding, which is lacking for the other Phe residues (Phe285, Phe289, and Phe293). The preference of Trp278 for the glycerol moiety of SURF-TG over that of PL is further supported by the alignment of SURF-TG and the insertion depth of Trp278. Additionally, the single mutation (W278V) in the helix reduces the binding success from ~90% to ~54%. There is no preferential interaction between the glycerol moiety of SURF-TG and Val278. Finally, the US simulations confirmed the unbiased MD results, demonstrating the free energy difference between the unbound and bound states is 15.8 kcal/mol. Collectively, this suggests that tryptophan residues play a special role in membrane and LD association due to the combination of their bulky aromatic group and ability to hydrogen bond. We anticipate this hydrophilic interaction at the hydrophobic interface can increase binding affinities. For instance, the LD targeting domain of comparative gene identification-58 (CGI-58 or ABHD5), an activator of adipose triglyceride lipase (ATGL), is enriched with Trp and mutating those residues to Ala abolishes LD targeting.77–78 Also, during lipolysis, CCT1 has the weakest binding affinity with LD surfaces and CGI-58 has one of the strongest among the proteins tested.79 The possible explanation could be the number of Trp residues and the interaction between Trp and SURF-TG. The whole membrane binding domain of rat CCTα, which ranges from residue 234 to 293, has only one Trp residue, whereas CGI-58 has three.
Taken together, our results provide new perspectives on the mechanisms behind preferential AH targeting to expanding bilayers and LD monolayers, the interplay between SURF-TG molecules and surface tension, and the specific role of SURF-TG molecules in LD protein association.
Supplementary Material
ACKNOWLEDGEMENTS
S.K. thanks Dr. Rosemary Cornell for useful discussions on CCTα. We thank Dr. Jeeyun Chung, Dr. Rich Pastor, Dr. Robert V. Farese, Jr., Dr. Tobias C. Walther, and Dr. Gregory A. Voth for general discussions. This research was supported by the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) through grant 5R01GM097194. S.K. was supported by the National Institute of General Medical Sciences of the National Institutes of Health under NIH award number R01-GM063796 (G. A. Voth, Principal Investigator). The MD simulations were performed on the high-performance GPU cluster (GM4) at the University of Chicago Research Computing Center, supported by NSF grant DMR-1828629 and the Stampede2 supercomputer at the Texas Advanced Computing Center (TACC) through allocation MCB200018 with resources provided by the Extreme Science and Engineering Discovery Environment (XSEDE) supported by NSF grant ACI-1548562.
Footnotes
SUPPORTING INFORMATION
Figures S1–S4 and the physical properties of the more expanded LD structure (Figures S5–S9) are included in Supporting Information.
REFERENCES
- 1.Wilfling F; Haas JT; Walther TC; R. V. F. Jr, Lipid droplet biogenesis. Current Opinion in Cell Biology 2014, 29, 39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walther TC; Chung J; Farese RV, Lipid Droplet Biogenesis. Annual Review of Cell and Developmental Biology 2017, 33 (1), 491–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Farese RV; Walther TC, Lipid Droplets Finally Get a Little R-E-S-P-E-C-T. Cell 2009, 139 (5), 855–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olzmann JA; Carvalho P, Dynamics and functions of lipid droplets. Nature Reviews Molecular Cell Biology 2019, 20 (3), 137–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ducharme NA; Bickel PE, Minireview: Lipid Droplets in Lipogenesis and Lipolysis. Endocrinology 2008, 149 (3), 942–949. [DOI] [PubMed] [Google Scholar]
- 6.Kory N; Farese RV; Walther TC, Targeting Fat: Mechanisms of Protein Localization to Lipid Droplets. Trends in Cell Biology 2016, 26 (7), 535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olarte MJ; Kim S; Sharp ME; Swanson JMJ; Farese RV Jr.; Walther TC, Determinants of Endoplasmic Reticulum-to-Lipid Droplet Protein Targeting. Dev Cell 2020, 54 (4), 471–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caillon L; Nieto V; Gehan P; Omrane M; Rodriguez N; Monticelli L; Thiam AR, Triacylglycerols sequester monotopic membrane proteins to lipid droplets. Nature Communications 2020, 11 (1), 3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilfling F; Wang H; Haas T, Joel; Krahmer N; Gould J, Travis; Uchida A; Cheng J-X; Graham M; Christiano R; Fröhlich F, et al. , Triacylglycerol Synthesis Enzymes Mediate Lipid Droplet Growth by Relocalizing from the ER to Lipid Droplets. Developmental Cell 2013, 24 (4), 384–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang H; Airola MV; Reue K, How lipid droplets “TAG” along: Glycerolipid synthetic enzymes and lipid storage. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids 2017, 1862 (10), 1131–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang H; Becuwe M; Housden BE; Chitraju C; Porras AJ; Graham MM; Liu XN; Thiam AR; Savage DB; Agarwal AK, et al. , Seipin is required for converting nascent to mature lipid droplets. eLife 2016, 5, e16582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Copic A; Antoine-Bally S; Gimenez-Andres M; La Torre Garay C; Antonny B; Manni MM; Pagnotta S; Guihot J; Jackson CL, A giant amphipathic helix from a perilipin that is adapted for coating lipid droplets. Nat Commun 2018, 9 (1), 1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rowe ER; Mimmack ML; Barbosa AD; Haider A; Isaac I; Ouberai MM; Thiam AR; Patel S; Saudek V; Siniossoglou S, et al. , Conserved Amphipathic Helices Mediate Lipid Droplet Targeting of Perilipins 1–3. Journal of Biological Chemistry 2016, 291 (13), 6664–6678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yue L; McPhee MJ; Gonzalez K; Charman M; Lee J; Thompson J; Winkler DFH; Cornell RB; Pelech S; Ridgway ND, Differential dephosphorylation of CTP:phosphocholine cytidylyltransferase upon translocation to nuclear membranes and lipid droplets. Mol Biol Cell 2020, 31 (10), 1047–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krahmer N; Guo Y; Wilfling F; Hilger M; Lingrell S; Heger K; Newman W, Heather; Schmidt-Supprian M; Vance E, Dennis; Mann M, et al. , Phosphatidylcholine Synthesis for Lipid Droplet Expansion Is Mediated by Localized Activation of CTP:Phosphocholine Cytidylyltransferase. Cell Metabolism 2011, 14 (4), 504–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Northwood IC; Tong AH; Crawford B; Drobnies AE; Cornell RB, Shuttling of CTP:Phosphocholine cytidylyltransferase between the nucleus and endoplasmic reticulum accompanies the wave of phosphatidylcholine synthesis during the G(0) --> G(1) transition. J Biol Chem 1999, 274 (37), 26240–8. [DOI] [PubMed] [Google Scholar]
- 17.Dhiman R; Caesar S; Thiam AR; Schrul B, Mechanisms of protein targeting to lipid droplets: A unified cell BI0logical and BI0physical perspective. Seminars in Cell & Developmental Biology 2020. [DOI] [PubMed] [Google Scholar]
- 18.Bersuker K; Olzmann JA, Establishing the lipid droplet proteome: Mechanisms of lipid droplet protein targeting and degradation. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids 2017, 1862 (10), 1166–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gimenez-Andres M; Copic A; Antonny B, The Many Faces of Amphipathic Helices. Biomolecules 2018, 8 (3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cui H; Lyman E; Voth GA, Mechanism of Membrane Curvature Sensing by Amphipathic Helix Containing Proteins. Biophysical Journal 2011, 100 (5), 1271–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vanni S; Vamparys L; Gautier R; Drin G; Etchebest C; Fuchs FJ, Patrick; Antonny, B., Amphipathic Lipid Packing Sensor Motifs: Probing Bilayer Defects with Hydrophobic Residues. Biophysical Journal 2013, 104 (3), 575–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Monje-Galvan V; Klauda JB, Preferred Binding Mechanism of Osh4’s Amphipathic Lipid-Packing Sensor Motif, Insights from Molecular Dynamics. J Phys Chem B 2018, 122 (42), 9713–9723. [DOI] [PubMed] [Google Scholar]
- 23.Prévost Coline; E. Sharp M; Kory Nora; Lin Qingqing; A. Voth G; V. Farese R; C. Walther T, Mechanism and Determinants of Amphipathic Helix-Containing Protein Targeting to Lipid Droplets. Developmental Cell 2018, 44 (1), 73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsieh K; Lee YK; Londos C; Raaka BM; Dalen KT; Kimmel AR, Perilipin family members preferentially sequester to either triacylglycerol-specific or cholesteryl-ester-specific intracellular lipid storage droplets. Journal of Cell Science 2012, 125 (17), 4067–4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Antonny B; Huber I; Paris S; Chabre M; Cassel D, Activation of ADP-ribosylation Factor 1 GTPase-Activating Protein by Phosphatidylcholine-derived Diacylglycerols. Journal of Biological Chemistry 1997, 272 (49), 30848–30851. [DOI] [PubMed] [Google Scholar]
- 26.Bigay J; Gounon P; Robineau S; Antonny B, Lipid packing sensed by ArfGAP1 couples COPI coat disassembly to membrane bilayer curvature. Nature 2003, 426 (6966), 563–566. [DOI] [PubMed] [Google Scholar]
- 27.Wilfling F; Thiam AR; Olarte M-J; Wang J; Beck R; Gould TJ; Allgeyer ES; Pincet F; Bewersdorf J; Farese RV, et al. , Arf1/COPI machinery acts directly on lipid droplets and enables their connection to the ER for protein targeting. eLife 2014, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bersuker K; Peterson CWH; To M; Sahl SJ; Savikhin V; Grossman EA; Nomura DK; Olzmann JA, A Proximity Labeling Strategy Provides Insights into the Composition and Dynamics of Lipid Droplet Proteomes. Developmental Cell 2018, 44 (1), 97–112.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cornell RB; Ridgway ND, CTP:phosphocholine cytidylyltransferase: Function, regulation, and structure of an amphitropic enzyme required for membrane biogenesis. Prog Lipid Res 2015, 59, 147–71. [DOI] [PubMed] [Google Scholar]
- 30.Cornell RB, Membrane lipid compositional sensing by the inducible amphipathic helix of CCT. Biochim Biophys Acta 2016, 1861 (8 Pt B), 847–861. [DOI] [PubMed] [Google Scholar]
- 31.Cornell RB, Membrane Lipids Assist Catalysis by CTP: Phosphocholine Cytidylyltransferase. Journal of Molecular Biology 2020, 432 (18), 5023–5042. [DOI] [PubMed] [Google Scholar]
- 32.Cornell RB, Regulation of CTP:phosphocholine cytidylyltransferase by lipids. 2. Surface curvature, acyl chain length, and lipid-phase dependence for activation. Biochemistry 1991, 30 (24), 5881–8. [DOI] [PubMed] [Google Scholar]
- 33.Cornell RB, Regulation of CTP:phosphocholine cytidylyltransferase by lipids. 1. Negative surface charge dependence for activation. Biochemistry 1991, 30 (24), 5873–80. [DOI] [PubMed] [Google Scholar]
- 34.Lee J; Taneva SG; Holland BW; Tieleman DP; Cornell RB, Structural basis for autoinhibition of CTP:phosphocholine cytidylyltransferase (CCT), the regulatory enzyme in phosphatidylcholine synthesis, by its membrane-binding amphipathic helix. J BI0l Chem 2014, 289 (3), 1742–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramezanpour M; Lee J; Taneva SG; Tieleman DP; Cornell RB, An auto-inhibitory helix in CTP:phosphocholine cytidylyltransferase hijacks the catalytic residue and constrains a pliable, domain-bridging helix pair. J Biol Chem 2018, 293 (18), 7070–7084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cornell RB; Northwood IC, Regulation of CTP:phosphocholine cytidylyltransferase by amphitropism and relocalization. Trends in Biochemical Sciences 2000, 25 (9), 441–447. [DOI] [PubMed] [Google Scholar]
- 37.Davies SM; Epand RM; Kraayenhof R; Cornell RB, Regulation of CTP: phosphocholine cytidylyltransferase activity by the physical properties of lipid membranes: an important role for stored curvature strain energy. Biochemistry 2001, 40 (35), 10522–31. [DOI] [PubMed] [Google Scholar]
- 38.Kim S; Swanson JMJ, The Surface and Hydration Properties of Lipid Droplets. Biophys J 2020, I19 (10), 1958–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chorlay A; Thiam AR, Neutral lipids regulate amphipathic helix affinity for model lipid droplets. J Cell Biol 2020, 219 (4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang Y; Lee M; Fairn GD, Phospholipid subcellular localization and dynamics. J Biol Chem 2018, 293 (17), 6230–6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Meer G; Voelker DR; Feigenson GW, Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 2008, 9 (2), 112–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jo S; Kim T; Iyer VG; Im W, CHARMM-GUI: A web-based graphical user interface for CHARMM. Journal of Computational Chemistry 2008, 29 (11), 1859–1865. [DOI] [PubMed] [Google Scholar]
- 43.Jo S; Lim JB; Klauda JB; Im W, CHARMM-GUI Membrane Builder for Mixed Bilayers and Its Application to Yeast Membranes. Biophysical Journal 2009, 97 (1), 50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu EL; Cheng X; Jo S; Rui H; Song KC; Dávila-Contreras EM; Qi Y; Lee J; Monje-Galvan V; Venable RM, et al. , CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. Journal of Computational Chemistry 2014, 35 (27), 1997–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee J; Cheng X; Swails JM; Yeom MS; Eastman PK; Lemkul JA; Wei S; Buckner J; Jeong JC; Qi Y, et al. , CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J Chem Theory Comput 2016, 12 (1), 405–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ben M’barek K; Ajjaji D; Chorlay A; Vanni S; Foret L; Thiam AR, ER Membrane Phospholipids and Surface Tension Control Cellular Lipid Droplet Formation. Dev Cell 2017, 41 (6), 591–604 e7. [DOI] [PubMed] [Google Scholar]
- 47.Jorgensen WL, Quantum and statistical mechanical studies of liquids. 10. Transferable intermolecular potential functions for water, alcohols, and ethers. Application to liquid water. Journal of the American Chemical Society 1981, 103 (2), 335–340. [Google Scholar]
- 48.Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML, Comparison of simple potential functions for simulating liquid water. The Journal of Chemical Physics 1983, 79 (2), 926–935. [Google Scholar]
- 49.Van Der Spoel D; Lindahl E; Hess B; Groenhof G; Mark AE; Berendsen HJC, GROMACS: Fast, flexible, and free. Journal of Computational Chemistry 2005, 26 (16), 1701–1718. [DOI] [PubMed] [Google Scholar]
- 50.Klauda JB; Venablee RM; Freites JA; O’Connor JW; Tobias DJ; Mondragon-Ramirez C; Vorobyov I; Mackerell AD; Pastor RW, Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. The Journal of Physical Chemistry B 2010, 114 (23), 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Best RB; Zhu X; Shim J; Lopes PEM; Mittal J; Feig M; Mackerell AD, Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone and ϕ,ψ Side-Chain χ1 and χ2 Dihedral Angles. Journal of Chemical Theory and Computation 2012, 8 (9), 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Essmann U; Perera L; Berkowitz ML; Darden T; Lee H; Pedersen LG, A smooth particle mesh Ewald method. The Journal of Chemical Physics 1995, 103 (19), 8577–8593. [Google Scholar]
- 53.Hess B, P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. Journal of Chemical Theory and Computation 2008, 4 (1), 116–122. [DOI] [PubMed] [Google Scholar]
- 54.Tribello GA; Bonomi M; Branduardi D; Camilloni C; Bussi G, PLUMED 2: New feathers for an old bird. Computer Physics Communications 2014, 185 (2), 604–613. [Google Scholar]
- 55.Bussi G; Tribello GA, Analyzing and Biasing Simulations with PLUMED. In Methods in Molecular Biology, Springer; New York: 2019; pp 529–578. [DOI] [PubMed] [Google Scholar]
- 56.Parrinello M; Rahman A, Polymorphic transitions in single crystals: A new molecular dynamics method. Journal of Applied Physics 1981, 52 (12), 7182–7190. [Google Scholar]
- 57.Nosé S, A unified formulation of the constant temperature molecular dynamics methods. The Journal of Chemical Physics 1984, 81 (1), 511–519. [Google Scholar]
- 58.Hoover WG, Canonical dynamics: Equilibrium phase-space distributions. Physical Review A 1985, 31 (3), 1695–1697. [DOI] [PubMed] [Google Scholar]
- 59.Berendsen HJC; Postma JPM; Van Gunsteren WF; Dinola A; Haak JR, Molecular dynamics with coupling to an external bath. The Journal of Chemical Physics 1984, 81 (8), 3684–3690. [Google Scholar]
- 60.Phillips JC; Hardy DJ; Maia JDC; Stone JE; Ribeiro JV; Bernardi RC; Buch R; Fiorin G; Henin J; Jiang W, et al. , Scalable molecular dynamics on CPU and GPU architectures with NAMD. J Chem Phys 2020, 153 (4), 044130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Humphrey W; Dalke A; Schulten K, VMD: Visual molecular dynamics. Journal of Molecular Graphics 1996, 14 (1), 33–38. [DOI] [PubMed] [Google Scholar]
- 62.Michaud-Agrawal N; Denning EJ; Woolf TB; Beckstein O, MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. Journal of Computational Chemistry 2011, 32 (10), 2319–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gautier R; Bacle A; Tiberti ML; Fuchs PF; Vanni S; Antonny B, PackMem: A Versatile Tool to Compute and Visualize Interfacial Packing Defects in (ipid Bilayers. Biophysical Journal 2018, 115 (3), 436–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vamparys L; Gautier R; Vanni S; Bennett WFD; Tieleman DP; Antonny B; Etchebest C; Fuchs FJ, Patrick, Conical (ipids in Flat Bilayers Induce Packing Defects Similar to that Induced by Positive Curvature. Biophysical Journal 2013, 104 (3), 585–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bacle A; Gautier R; Jackson CL; Fuchs PFJ; Vanni S, Interdigitation between Triglycerides and (ipids Modulates Surface Properties of (ipid Droplets. Biophys J 2017, 112 (7), 1417–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fan S; Linke M; Paraskevakos I; Gowers R; Gecht M; Beckstein O In PMDA - Parallel Molecular Dynamics Analysis, Proceedings of the 18th Python in Science Conference, 2019–01-01; SciPy: 2019. [Google Scholar]
- 67.Sugita Y; Okamoto Y, Replica-exchange molecular dynamics method for protein folding. Chemical Physics Letters 1999, 314 (1–2), 141–151. [Google Scholar]
- 68.Torrie GM; Valleau JP, Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. Journal of Computational Physics 1977, 23 (2), 187–199. [Google Scholar]
- 69.Grossfied A, version 2.0.9. WHAM: an implementation of the weighted histogram analysis method. [Google Scholar]
- 70.Kumar S; Rosenberg JM; Bouzida D; Swendsen RH; Kollman PA, THE weighted histogram analysis method for free-energy calculations on Biomolecules. I. The method. Journal of Computational Chemistry 1992, 13 (8), 1011–1021. [Google Scholar]
- 71.Gordon SM; Pourmousa M; Sampson M; Sviridov D; Islam R; Perrin BS Jr.; Kemeh G; Pastor RW; Remaley AT, Identification of a novel lipid binding motif in apolipoprotein B by the analysis of hydrophobic cluster domains. Biochim Biophys Acta Biomembr 2017, 1859 (2), 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dunne SJ; Cornell RB; Johnson JE; Glover NR; Tracey AS, Structure of the membrane binding domain of CTP:phosphocholine cytidylyltransferase. Biochemistry 1996, 35 (37), 11975–84. [DOI] [PubMed] [Google Scholar]
- 73.Taneva S; Johnson JE; Cornell RB, Lipid-induced conformational switch in the membrane binding domain of CTP:phosphocholine cytidylyltransferase: a circular dichroism study. Biochemistry 2003, 42 (40), 11768–76. [DOI] [PubMed] [Google Scholar]
- 74.Gautier R; Douguet D; Antonny B; Drin G, HEUIQUEST: a web server to screen sequences with specific -helical properties. Bioinformatics 2008, 24 (18), 2101–2102. [DOI] [PubMed] [Google Scholar]
- 75.Yau W-M; Wimley WC; Gawrisch K; White SH, The Preference of Tryptophan for Membrane Interfaces. Biochemistry 1998, 37 (42), 14713–14718. [DOI] [PubMed] [Google Scholar]
- 76.Chung J; Wu X; Lambert TJ; Lai ZW; Walther TC; Farese RV Jr., LDAF1 and Seipin Form a Lipid Droplet Assembly Complex. Dev Cell 2019, 51 (5), 551–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gruber A; Cornaciu I; Lass A; Schweiger M; Poeschl M; Eder C; Kumari M; Schoiswohl G; Wolinski H; Kohlwein SD, et al. , The N-terminal Region of Comparative Gene Identification-58 (CGI-58) Is Important for Lipid Droplet Binding and Activation of Adipose Triglyceride Lipase. Journal of Biological Chemistry 2010, 285 (16), 12289–12298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Boeszoermenyi A; Nagy HM; Arthanari H; Fillip CJ; Lindermuth H; Luna RE; Wagner G; Zechner R; Zangger K; Oberer M, Structure of a CGI-58 Motif Provides the Molecular Basis of Lipid Droplet Anchoring. Journal of Biological Chemistry 2015, 290 (44), 26361–26372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kory N; Thiam A-R; Farese V, Robert Walther, C., Tobias, Protein Crowding Is a Determinant of Lipid Droplet Protein Composition. Developmental Cell 2015, 34 (3), 351–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.