

Abstract
Apoptosis is a highly regulated form of cell death that is required for many homeostatic and pathological processes. Recently, alternative cell death pathways have emerged whose regulation is dependent on proteins with canonical functions in apoptosis. Dysregulation of apoptotic signaling frequently underlies the pathogenesis of many cancers, reinforcing the need to develop therapies that initiate alternative cell death processes. This review outlines the convergence points between apoptosis and other death pathways with the purpose of identifying novel strategies for the treatment of apoptosis-refractory cancers. Apoptosis proteins can play key roles in the initiation, regulation, and execution of non-apoptotic death processes that include necroptosis, autophagy, pyroptosis, mPTP-mediated necrosis and ferroptosis. Notably, recent evidence illustrates that dying cells can exhibit biochemical and molecular characteristics of more than one different type of regulated cell death. Thus, this review highlights the amazing complexity and interconnectivity of cell death processes and also raises the idea that a top-to-bottom approach to describing cell death mechanisms may be inadequate for fully understanding the means by which cells die.
Keywords: apoptosis, necroptosis, autophagy, ferroptosis, pyroptosis, mPTP, caspase, BCL-2, cancer
Graphical Abstract

Apoptosis is a form of regulated cell death which is critical for many developmental and physiological processes, however, dysregulation of apoptosis underlies cancer pathogenesis. Recent evidence indicates that many proteins involved in the regulation of apoptosis also have key functions in other regulated cell death pathways, revealing potential novel targets for the treatment of apoptosis-refractory cancers.
Introduction
Over 50 years have passed since Richard Lockshin and Carroll Williams first described the sequential destruction of intersegmental muscles in the silkmoth as a process called “programmed cell death” [1]. This developmentally regulated process of cell elimination was also simultaneously being characterized in vertebrates, resulting in the establishment of the term “apoptosis” by Kerr, Wyllie and Currie to describe the series of morphological events leading to controlled cell removal [2]. Ever since these initial observations, apoptosis has now become the most well understood cell death pathway in the molecular era. Apoptosis is a process in which cells undergo discrete morphological and biochemical steps leading to cell demise in response to developmental or pathological cues [3,4]. Recently there has been a renaissance in the field, such that the number of distinct regulated cell death modalities tally to more than a dozen distinct death pathways [5]. While the core components of the apoptotic pathway have been extensively described in the context of apoptosis [3,4,6], a large body of literature has emerged revealing that apoptotic proteins actively engage with and regulate other forms of cell death. The purpose of this review is to bring to light the extensive crosstalk between proteins well known for their function in apoptosis with emerging nonapoptotic cell death pathways (Figure 1). It is becoming increasingly clear that these pathways are not strictly linear, but form a network of entwined physical and functional interactions. While some of these players simply inhibit one pathway while facilitating activation of another, some apoptosis proteins are directly involved in the activation and amplification of nonapoptotic death signals. In an era in which small molecule inhibitors of apoptosis are in clinical use [7–9], the consequences of pharmacologically modulating apoptosis must be assessed for their influence on these entwined death pathways, and how understanding the crosstalk between apoptotic and nonapoptotic death pathways may reveal potential new therapeutically druggable targets.
Figure 1: Apoptosis regulators as double agents of cell death.
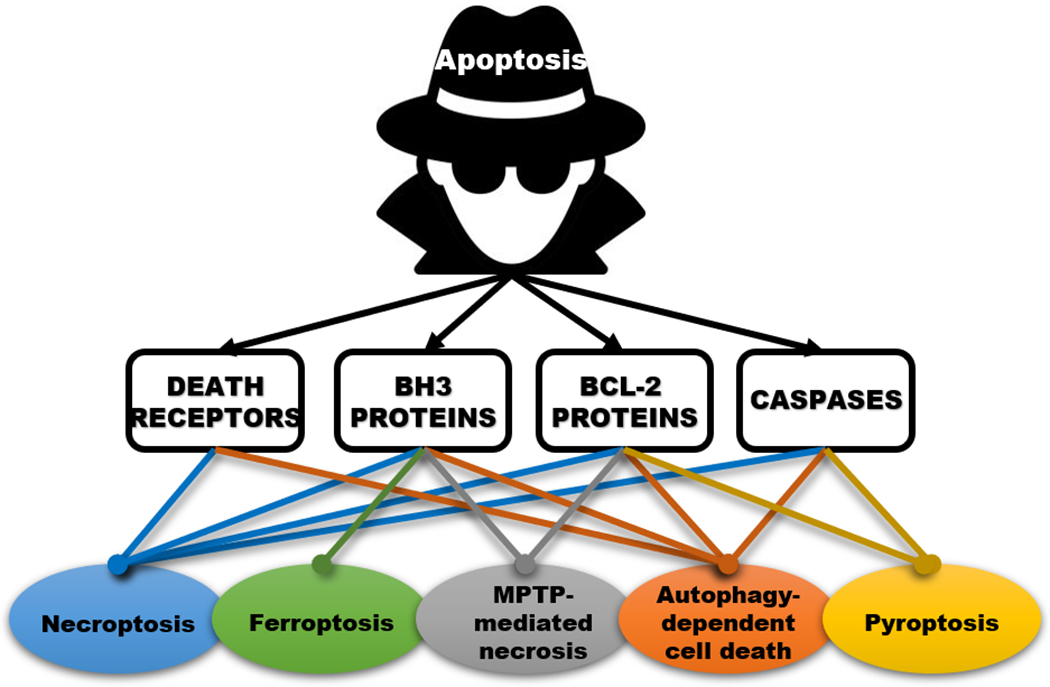
Several classes of proteins with key functions in apoptosis also participate in the execution and/or regulation of other death pathways. Colored lines indicate a documented linkage between each class of apoptotic protein (black) with nonapoptotic death processes.
A brief review of the canonical apoptosis pathway
Apoptotic cell death can be initiated either in response to death receptor (DR) signaling from the plasma membrane or mitochondrial signaling following intracellular stress (Figure 2). DR-mediated apoptosis is initiated upon ligand binding (ie. Fas, tumor necrosis factor (TNF), TNF-related apoptosis-inducing factor (TRAIL)) to their cognate receptor. Ligand binding results in DR clustering and assembly of intracellular signaling complexes through homotypic interactions with cytoplasmic death domain (DD) and death effector domain (DED)-containing proteins [10,11]. Activation of CD95/Fas or TRAIL receptors lead to the formation of the death-inducing signaling complex (DISC), consisting of the adaptor molecule Fas-Associated Death Domain (FADD) and procaspase-8/−10, whereas activated TNF receptors recruit distinct complexes which mediate either cell death or survival (described in more detail in the section “Crosstalk between apoptosis and necroptosis”, below). DISC assembly mediates the recruitment and homo-oligomerization of additional procaspase-8 molecules to form a macromolecular filament [12] that facilitates self-cleavage to yield active caspase-8. From there, caspase-8 possesses dual functions by either directly activating downstream executioner caspases or feeding into the intrinsic (mitochondrial) apoptosis pathway.
Figure 2: Crosstalk between apoptosis and necroptosis pathways.

Signaling from the TNF receptor (TNFR) can induce distinct cell death outcomes through the dual activity of common factors such as caspase-8 and the IAPs. Factors and pathways with known functions in apoptosis or necroptosis are indicated in black or blue shaded boxes/arrows, respectively. Those factors with dual functions (such as caspase-8, cIAP1/2 or XIAP) are highlighted with black and blue boxes. Pharmacological inhibitors of apoptosis or necroptosis are indicated in red. The ubiquitination state of RIPK1 is regulated by cIAP1/2, which keeps the kinase inactive and promotes a survival signal through NFκB. Inhibition of cIAPs by Smac mimetic compounds coupled with TNFR signaling liberates RIPK1 from the receptor complex, allowing it to interact with downstream factors leading to apoptosis (FADD, caspase-8) or necroptosis (RIPK3, MLKL). While caspase-8 and IAPs possess a direct role in the regulation of RIPK1, other apoptosis regulators, including BAX, BAK and several BH3-only proteins, have been shown to influence necroptotic signaling via mechanisms that are not fully understood.
The central regulators of the mitochondrial apoptosis pathway are members of the BCL-2 (B-cell lymphoma-2) family, a collection of pro- and anti-death proteins whose engagement with each other dictates cell fate. The founding member of the family, BCL-2, was identified at a chromosomal breakpoint t(14,18) common in patients with B-cell follicular lymphoma [13], in which the BCL-2 gene was translocated downstream of the immunoglobulin heavy chain locus (IgH) resulting in constitutive expression of the protein. By characterizing the cell cycle profile of BCL-2 expressing cells, David Vaux, Suzanne Cory and Jerry Adams made the seminal observation that BCL-2 was sufficient to mediate survival of the cells after a death signal (interleukin-3 withdrawal), and did not simply promote growth as was previously believed to be solely responsible for tumorigenesis [14]. Since then, the BCL-2 family has expanded to include members that can either promote survival (BCL-2, BCL-xL, MCL-1, A1, BCL-w, and others) or apoptotic cell death (BAX, BAK, BOK) [15]. Additionally, a third branch of the apoptotic BCL-2 protein family tree is the BCL-2-homology 3 (BH3)-only proteins (Bid, Bim, Bad, NOXA, PUMA and others), a class of sensitizer and activator proteins charged with the task of integrating signals from the intracellular environment to modulate interactions between the survival and death-promoting family members [16].
Activation of the intrinsic apoptosis pathway can integrate with extrinsic apoptotic signaling through caspase-8-mediated cleavage of the BH3-only protein, BID. Truncated BID (tBID) directly engages prodeath BCL-2 family members, BAX and BAK, leading to their activation by inducing conformational changes, oligomerization, and mitochondrial outer membrane permeabilization (MOMP) [17]. The release of mitochondrial factors, including cytochrome c and Smac/DIABLO (second mitochondria-derived activator of caspases/direct IAP-binding protein with low pI), leads to the initiation of several downstream consequences including DNA fragmentation, loss of mitochondrial function, inhibition of anti-death proteins, and assembly of the apoptosome complex. Cytochrome c released from mitochondria stimulates the assembly of the apoptosome by complexing with Apaf-1 and procaspase-9, leading to proteolytic activation of caspase-9 and subsequent activation of the executioner caspases-3/7. Regardless of whether directly activated by caspase-8 (extrinsic) or through the apoptosome (intrinsic), activation of the executioner caspases-3/−7 executes downstream events leading to the morphological and biochemical features of apoptosis.
A number of excellent reviews are available for more detailed discussions of the canonical apoptosis pathway [4][3][6][15][18][19].
Targeted approaches for inducing apoptosis in cancers
Therapeutic manipulation of apoptotic pathways has been a long sought out strategy for the treatment of cancers. Most chemotherapeutic and radiotherapeutic interventions interfere with tumor cell proliferation by inducing DNA damage, interfering with the cell cycle or halting DNA replication, the downstream consequence of which leads to apoptotic cell death. However, many cancers contain inactivating mutations or upregulate antiapoptotic proteins that prevent damaged/injured cells from undergoing apoptosis [20]. Recent clinical strategies for activating apoptosis in human cancers have focused on the development of bioactive small molecules, including Smac mimetics/IAP antagonists and inhibitors of antiapoptotic BCL-2 family members (BH3 mimetics). Upon its release from the mitochondria during apoptosis, Smac potentiates an apoptotic signal by directly binding and inactivating endogenous inhibitor of apoptosis proteins (IAPs) [21], a family of negative regulators of intrinsic and extrinsic apoptotic signaling [22]. Although eight family members have been identified, the IAPs predominantly involved in apoptosis inhibition include X-chromosome-linked IAP (XIAP), cellular IAP1 (cIAP1), and cellular IAP2 (cIAP2). Consequently, IAPs are frequently upregulated in human cancers [23], with their expression correlating with chemoresistance and poor prognosis. Monovalent and bivalent small molecule mimetics contain four key amino acids of the Smac N-terminus (Ala-Val-Pro-Ile) that engage the baculoviral IAP repeat (BIR) domains of IAPs. As a result, Smac mimetics functionally disrupt the inhibitory interaction of XIAP with caspases or stimulate the E3 ubiquitin ligase activity of cIAPs leading to a reduction in canonical NF-κB-mediated survival signals and increased noncanonical NF-κB signaling resulting in autocrine TNF secretion [23,24]. Clinical use of Smac mimetics are being explored for the treatment of multiple cancers, both as a single agent or in combination with existing chemotherapeutic agents [23,25]. Although well tolerated in Phase I clinical trials [26], Smac mimetics possessed limited efficacy as a single agent therapeutic in patients with a variety of solid tumors and hematological malignancies, including triple-negative breast cancer, ovarian, primary peritoneal, fallopian tube cancer, and multiple myeloma [27]. Upregulation of circulating inflammatory cytokines was also observed in a Phase I study after Smac mimetic treatment in patients with solid tumors, likely due the effects of cIAP1/2 inhibition activating noncanonical NF-κB signaling [28]. However, IAP inhibition appears to sensitize tumors to standard chemotherapeutic and radiologic interventions [27].
Small molecule mimetics have also been developed that inhibit anti-apoptotic BCL-2 family members, with the goal of triggering an apoptotic response in tumor cells. Designed to mimic the BH3 domain conserved among many BCL-2 family members, BH3 mimetics engage the hydrophobic groove of anti-apoptotic BCL-2 proteins with high affinity and specificity, relieving inhibition of pro-apoptotic family members, BAX and BAK, and resulting in a caspase-dependent cell death [29]. The first FDA approved BCL-2 selective inhibitor, venetoclax, is in clinical use for the treatment of chronic lymphocytic leukemia (CLL) [8,30]. More recently, venetoclax was approved in combination with demethylating chemotherapeutic agents for the treatment of acute myeloid leukemia (AML) [31]. However, the recent emergence of venetoclax-refractory tumors have reinforced the need for the development of additional treatment strategies [32–34]. Venetoclax resistance has been observed due to acquired mutations within the BCL-2 protein, by upregulation of other anti-apoptotic family members (such as MCL-1) or by altering other signaling pathways (JNK, autophagy) [32,35]. Thus new strategies for the treatment of apoptosis-refractory cancers are needed to complement existing therapeutics, perhaps through the pharmacological manipulation of alternative, non-apoptotic forms of regulated cell death.
Apoptosis proteins as double agents of cell death
Historically, apoptosis was the only known form of regulated cell death, both in developmental and pathological contexts, with any other modes of cell death described as accidental (necrotic). However, an abundance of evidence has identified several additional forms of regulated cell death, many of which are beginning to be understood in mechanistic detail [36–39]. Quite striking is the fact that many apoptotic proteins play key roles in the regulation of these alternative cell death pathways. Here, this review provides brief descriptions of each of these emerging pathways, and highlights how apoptotic proteins function within those pathways.
Crosstalk between apoptosis and necroptosis.
Over a decade ago, Junying Yuan and colleagues described a novel caspase-independent, death receptor-induced form of inflammatory cell death they termed necroptosis [40]. Since then, the cell death field has experienced a renaissance, during which a detailed mechanism of this regulated necrosis pathway has begun to emerge. Unexpectedly, several key regulators of apoptosis also have important functions in the necroptosis pathway, suggesting a sophisticated mechanism for cross-regulation (Figure 2). The pathway of regulated necroptosis centers around the activity of the kinases Receptor-Interacting Protein Kinase 1 (RIPK1) and RIPK3, the activity of which are regulated in response to TNF-mediated trimerization of TNFR1. While the default outcome of TNFR1 activation leads to survival signaling through NF-κB, TNFR1 can also initiate distinct apoptotic or necroptotic pathways depending on the faction of regulatory components that are engaged with the receptor’s cytoplasmic domain [36]. As one of many components of TNFR1 complex-1, RIPK1 undergoes distinct subsets of posttranslational modifications that dictate whether the complex potentiates a survival signal by activating NF-κB or initiates a death pathway. In their canonical antiapoptotic role, cIAP1/2 prevents TNFR1-mediated apoptosis by constitutively ubiquitinating RIPK1 to inactivate the kinase and facilitate cell survival [41,42]. Loss of these inhibitory signals following a death stimulus allow RIPK1 to dissociate from complex-1 and form complex-2a (ripoptosome) with FADD and procaspase-8, which leads to death by the extrinsic apoptotic pathway [42]. Additionally, active caspase-8 functions as a negative regulator of necroptosis by cleaving RIPK1 within its kinase domain (Asp324), thereby ensuring that the cell proceeds by an apoptotic death pathway [43].
Necroptosis was initially observed only in the absence of caspase activity (either by treatment with caspase inhibitors or genetic deletion) [40]. When caspases are inhibited during TNFR1 activation, RIPK1 is relieved from inhibitory caspase-8 cleavage, allowing it to heterodimerize and activate RIPK3. Together, the RIPK1/RIPK3 necrosome (complex-2b) phosphorylates the pseudokinase, mixed-lineage kinase domain-like protein (MLKL), inducing a conformational change that liberates an N-terminal four-helix bundle (4HB) domain from the molecule that favors MLKL oligomerization and formation of higher order amyloid-like structures [44]. Activated MLKL subsequently translocates to the plasma membrane, leading to membrane permeabilization by a mechanism that has not been fully delineated [45–47]. While many initially questioned the physiological relevance of a death pathway that was apparently observed only after apoptosis inhibition, it has become clear that necroptosis represents a bona fide cell death pathway with clear roles in neurodegenerative disease pathology and neuroinflammation [48,49].
In addition to caspase-8 and cIAPs, other apoptotic regulators have also been shown to modulate the necroptotic death process. The other major IAP family member, XIAP, can also play dual roles in preventing apoptosis or necroptosis. To inhibit apoptosis, XIAP engages specific subsets of effector caspases in the cytoplasm either by direct binding or by ubiquitinating them for proteasomal degradation. Necroptosis inhibition by XIAP occurs downstream of TNFR1 by ubiquitination of RIPK1 in the context of complex-2b [50]. Moreover, BCL-2 family proteins have also been identified as both accomplices and adversaries of the necroptotic death process. Mouse embryonic fibroblasts (MEFs) deficient for both BAX and BAK are resistant to necroptosis induced by TNF, cycloheximide, and zVAD treatment [51]. In a separate study, BAK deficient cells underwent delayed necroptosis in response to IAP inhibition (with Smac mimetic) combined with glucocorticoid treatment [52]. Necroptosis induction also depletes mitochondrial localized MCL-1 in a RIPK3-dependent manner [53], thus leading to mitochondrial dysfunction. Very recently, endogenous MLKL was shown to interact with BCL-2 via a putative BH3 domain, inhibiting RIPK3-dependent MLKL phosphorylation and oligomerization after necroptosis stimulation [54]. In addition, knockout MEFs of the BH3 protein Bmf were mildly resistant to death induced by cadmium treatment, a known inducer of necroptosis [55], suggesting that BH3 proteins may also be involved in RIPK1-mediated death. Another group discovered that the proapoptotic BH3 protein, PUMA is transcriptionally upregulated upon necroptosis initiation by the p65 subunit of NF-κB, which amplifies necroptosis through PUMA-mediated signaling back to RIPK1 and MLKL [56]. Of note, there is mixed evidence as to whether mitochondria are required for necroptotic cell death. While mitophagy-mediated depletion of mitochondria did not alter the kinetics of RIPK3-mediated necroptotic cell death in 3T3 cells [57], other studies have demonstrated that mitochondrially-generated reactive oxygen species (ROS) potentiates the necroptotic signal [58]. Whether or not mitochondria are required for necroptosis, many nonapoptotic functions of BCL-2 proteins occur at non-mitochondrial sites throughout the cell (ie. endoplasmic reticulum, cytoplasm, nucleus) [59], thus opening up the possibility that BCL-2 proteins may exert direct influence on the necroptotic pathway.
Small molecule modulators of the apoptotic pathway have been shown to stimulate necroptotic cell death in apoptosis-deficient cells. Smac mimetics, which were designed to functionally inhibit IAP proteins to stimulate apoptosis, have been shown to cause necroptotic cell death in apoptosis-deficient cells [60–62]. In AML mouse models and patient-derived xenografts, combined pharmacological inhibition of IAPs and caspase-8 induced necroptotic cell death of leukemic cells, resulted in reduced tumor burden and enhanced survival [63], thus paving the way toward selective activation of necroptosis for the treatment of hematological malignancies. Many commonly used anti-cancer agents can also stimulate RIPK1-dependent necroptosis [64], but cancer cells also frequently epigenetically silence RIPK3 leading to tumor resistance [65,66].
The intersection of apoptosis and necroptosis provides an opportunity for the development of therapeutics to selectively target both pathways, yet assessing the prudency of altering the activity of one pathway to alternatively stimulate the other is required. While initiating an inflammatory response may be an effective way to combat cancer cells that have become apoptosis resistant, inflammatory responses would require precise control to avoid extensive pathology due to acute or chronic inflammation [67,68]. Paradoxically, necroptosis-induced inflammatory responses have been implicated in promoting tumor progression and metastasis [69]. For example, high RIPK1 expression in glioblastoma patients correlates with a worse prognosis, likely due to increased NF-κB signaling leading to an upregulation of mdm2 and loss of p53 tumor suppressor activity [70]. Pharmacological inhibition of RIPK1 with necrostatin-1 (a small molecule inhibitor that locks RIPK1 in its inactive conformation [71]) or tissue specific ablation of RIPK3 in mouse endothelial cells reduced metastasis that occurs in response to endothelial necroptosis caused by tumor cells [72]. Thus, necroptosis activation may prove advantageous for the treatment of apoptosis-resistant tumors, but whether engaging an immune response would be beneficial or deleterious has yet to be determined in patients.
Crosstalk between apoptosis and autophagy.
Autophagy is a homeostatic process in which cytoplasmic contents and/or dysfunctional organelles are self-consumed and recycled into components to be utilized by the cell [73]. Most typically thought of as a recycling mechanism in the face of nutrient depletion, autophagy is morphologically characterized by the de novo formation of a double-membrane vesicle containing cytoplasmic contents that fuses with the lysosome [74]. Dual roles for apoptosis proteins in canonical autophagy regulation have been described with proteins engaged at nearly every stage of the apoptotic pathway, including death receptor ligands, BCL-2 family members, BH3 proteins, and caspases, emphasizing the importance of precise regulation between these pathways (Figure 3). Inhibition of the inflammatory factor and death receptor ligand TNF resulted in protection of photoreceptors after retinal detachment, presumably due to a transient increase in autophagy and a corresponding reduction in apoptosis [75]. Anti-apoptotic BCL-2 proteins also bind to the autophagy protein Beclin 1, preventing it from interacting with the VPS34 PI3K complex responsible for autophagy initiation [76–78]. In addition to binding Beclin 1, BCL-2 has also been reported to directly bind to and regulate the lipidation of GABARAP, a protein important for autophagosome formation [79], but this interaction has not been independently confirmed. BCL-xL has been shown to inhibit autophagosome:lysosome fusion and bind Beclin 1:UVRAG to suppress bacterial internalization in a model of Group A Streptococcus infection [80]. Atg12, which when complexed with Atg5 and Atg16 at sites of autophagosome assembly, binds and inhibits anti-apoptotic BCL-2 proteins through a novel BH3 domain [81]. Selective degradation of mitochondria, known as mitophagy, is regulated by BCL-2 through a direct interaction with Parkin [82]. A BH3 containing protein, BNIP3, functions as a mitophagy receptor that recruits autophagosomes by directly binding LC3 [83]. Beclin 1 also engages at the mitochondria with the other major anti-apoptotic BCL-2 family member, MCL-1, leading to inhibition of autophagy initiation [84,85]. Under nutrient rich conditions, siRNA depletion of MCL-1 or Beclin-1 results in stabilization of the other, and their degradation occurred through competitive binding to the USP9X deubiqutinase [84]. A number of BH3 proteins have been shown to regulate the apoptosis/autophagy rheostat, including BIM, BMF, and NOXA, and which serve to either mislocalize autophagy components or are themselves targets of autophagy [86–89].
Figure 3: Crosstalk between apoptosis and autophagy.
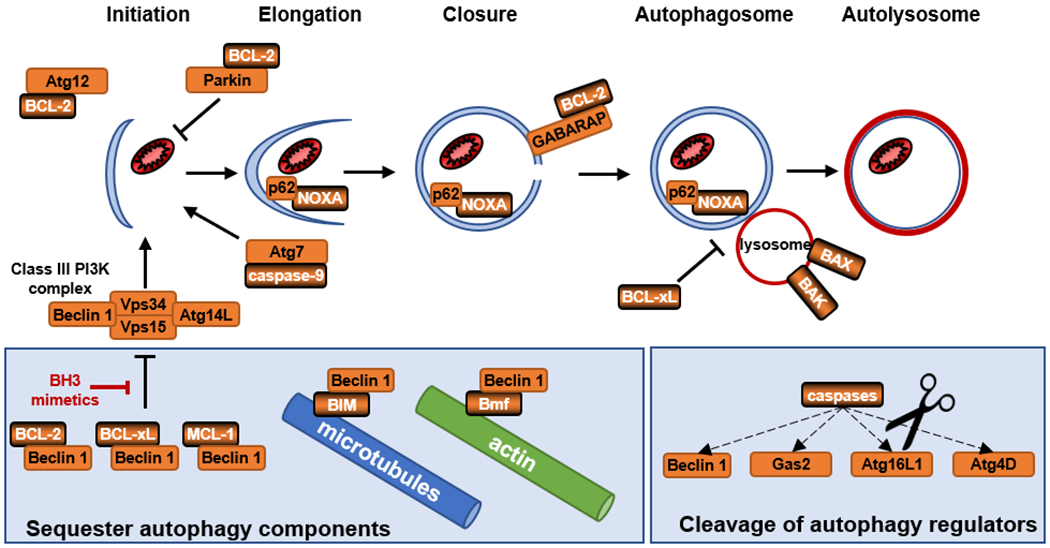
Apoptosis proteins can engage with the autophagy pathway throughout all steps of autophagosome formation, either by sequestering autophagy proteins away from active complex components, by associating with critical autophagy factors or cargo, by caspase-mediated cleavage of autophagy regulators, or by directly influencing autophagosome/lysosome function. Autophagy proteins are indicated in orange, with dual function apoptotic proteins highlighted in orange boxes with black shading.
Cleavage of autophagy proteins by caspases is another mechanism by which apoptosis and canonical autophagy are suggested to be differentially regulated, and may perhaps represent additional therapeutic targets for manipulating autophagy. Caspases have been shown to cleave Beclin 1 and Atg4D to inhibit autophagy and promote apoptosis [90,91]. Interestingly, a Crohn’s-disease associated mutation in Atg16L1 generates a new caspase-3 cleavage site, leading to compromised xenophagy of Yersinia entercolitica [92]. In rice, the glucosidase II β subunit Gas2 is downregulated to initiate autophagy in response to mild ER stress, while severe ER stress results in Gas2 cleavage by caspase-3 to participate in cell death [93]. Further down the apoptotic pathway, caspase-9 forms a complex with Atg7 to promote early events leading to autophagosome formation independently of its protease activity [94]. Remarkably, knockdown of caspase-9 expression in BAX−/− HCT116 cells (BAX being the predominant pro-death protein in this cell type [95]) conferred sensitivity to death after autophagy induction by TRAIL treatment or nutrient deprivation, and caspase-9 knockout MEFs showed reduced autophagosome formation under basal and stimulated conditions, strongly indicating that caspase-9 possesses nonapoptotic functions in regulating autophagy [94].
The complex crosstalk between autophagic processes and apoptosis is apparent from observations that autophagy can act as a bystander witness to a cell’s demise (autophagy-associated), an accomplice (autophagy-mediated) or even as the murderer itself (autophagy-dependent) [96,97]. In autophagy-associated cell death, an autophagic response occurs concurrently with a cell death process (usually apoptosis) but does not directly participate in the death pathway. Such responses are typically interpreted as the cell’s effort to maintain essential functions in the face of cellular stress [98]. Autophagy-mediated cell death requires the activation or utilization of autophagic processes/proteins to directly promote a subsequent apoptotic or necroptotic response, such as through the degradation of survival factors or by facilitating the assembly of cell death complexes. Finally, autophagy-dependent cell death (ADCD) requires the establishment of causality, such that cell death is a direct consequence of autophagy and is completely independent of other cell death processes. Accurately characterizing the involvement of autophagy in any death process is impossible simply by assessment of cellular morphology or autophagic flux, as all three autophagy-related death processes frequently display increased autophagic activity. Instead, precise genetic, biochemical, and pharmacological manipulation of essential autophagy processes are required for fully establishing the involvement of autophagy in regulated cell death [5,97].
Physiological ADCD in nonmammalian model organisms has become widely accepted to occur during development [39,99], while experimentally identifying ADCD processes in mammalian cells has been challenging due to higher organismal complexity and the interconnected nature of cell death pathways [100]. ADCD has been observed during midgut regression in Drosophila [101] and germ cell and ventral cord neuron development in C. elegans [102]. Lacking caspases and other apoptotic machinery, the slime mold Dictyostelium discoideum requires ADCD for development of the fruiting body [103]. Although analogous evidence in mammalian cells is highly context dependent, several compelling examples of ADCD have been described. Recently, a novel form of ADCD known as autosis, was described that occurs in response to autophagy-inducing peptides, starvation, or hypoxia/ischemia [104,105]. Autosis requires core components of the autophagy pathway (knockdown of Beclin 1, Atg13 and Atg14 inhibited death), and cell death proceeds via a mechanism independent of apoptosis or necroptosis [104–106]. Interestingly, autosis was also inhibited by cardiac glycosides, suggesting a yet to be characterized role for the Na+,K+-ATPase in mediating this form of ADCD [104]. In MEFs, abolishing apoptosis by genetic knockout of BAX and BAK lead to nonapoptotic cell death after treatment with etoposide or staurosporine [107]. This nonapoptotic cell death was inhibited after treatment with an autophagy inhibitor (3-methyl adenine) or by knockdown of the autophagy genes Atg5 or Beclin 1, suggesting that the death process in BAX/BAK double knockout (DKO) cells was due to ADCD [107]. In the context of mouse development, genetic deletion of Atg5, BAX, and BAK caused early embryonic lethality with an increased occurrence of brain malformations (exencephaly), indicating that ADCD partially compensates for the lack of developmental cell death in apoptosis-deficient mice [108]. Compared with DKO mice, TKO mice also had a significant delay in interdigital web elimination, a hallmark trait of mice lacking pro-apoptotic proteins BAX and BAK, confirming that ADCD plays a role in the embryonic development of DKO mice [108].
Can autophagy pathways be therapeutically manipulated to tip the balance from a survival function toward ADCD in apoptosis-deficient cancers? This remains under investigation as a screen of over 14000 compounds failed to identify any single agent that could enhance autophagic flux past a toxic threshold to induce ADCD in U2OS cells [109]. Furthering our understanding of the detailed mechanisms of ADCD, particularly any differences between ADCD and canonical autophagy, may yield novel strategies for bypassing the survival function of autophagy and specifically stimulate a death activity. For example, blocking autophagosome fusion with the lysosome does not prevent death due to autosis [104], indicating that this death process does not proceed along the canonical downstream autophagy pathway. Despite the developmental requirement for ADCD in the Drosophila midgut, the autophagic death process does not require components of the Atg8 lipid conjugation pathway, Atg7 and Atg3, instead relying on the E1 ubiquitin ligase Uba1 [110]. Anticancer drugs that possess ADCD-inducing activities include the BH3 mimetics obatoclax and gossypol, which disrupt the inhibitory interaction between Beclin 1 and anti-apoptotic BCL-2 proteins allowing Beclin 1 to initiate autophagosome formation as part of the VPS34 PI3K complex (Figure 3) [111].
Given that autophagy processes have been shown in some cases to promote tumor survival, contribute to metastasis, and mediate resistance [112], it might be somewhat counterintuitive to activate an autophagic pathway for the treatment of cancers. Autophagy inhibition is also a therapeutic strategy being tested in cancer patients, with moderate responses observed in cases of glioblastoma, melanoma, and certain types of brain cancer after treatment with the lysosomal inhibitor, chloroquine, in combination with radiation and/or alkylating chemotherapeutics [112]. Thus, selective regulation of ADCD and autophagy-related processes may provide a novel therapeutic approach for cancer therapy.
Crosstalk between apoptosis and pyroptosis.
Another emerging death pathway that has received much attention is the inflammatory process known as pyroptosis. Pyroptosis is a form of cell death most commonly occurring as an innate immune response to pattern recognition receptor (PRR) activation, leading to an inflammatory response [113]. The presence of intracellular toxins, bacterial or viral infections leads to the assembly of a complex of factors collectively known as inflammasomes, which act as a molecular scaffold to mediate the cleavage and activation of the inflammatory caspase-1. Alternatively, sensing of bacterial LPS challenge leads to an inflammasome-independent activation of other inflammatory caspases-4,−5,or −11 [114]. Active inflammatory caspases lead to cleavage of gasdermin D, that when cleaved assembles into pores on the plasma membrane [115]. Plasma membrane poration by gasdermin D leads to cell swelling and lysis, which may mediate the release of the proinflammatory cytokine IL-1β (although gasdermin independent IL1-β release has been described [116]). Although the inflammatory caspases have no known role in the canonical apoptosis pathway, new evidence has brought to light that cleavage of gasdermin family members by apoptotic caspases initiates pyroptosis. The activation of pyroptosis by caspase-3 is dependent on the expression level of endogenous gasdermin E, and gasdermin E knockout mice are protected from lung and intestinal toxicity that results after treatment with cisplatin, a chemotherapeutic believed to induce caspase-3 dependent apoptosis in tumors. Thus, toxicity associated with chemotherapy drugs may be due to a secondary pyroptotic death process in cells expressing gasdermin E [117,118]. Additionally, many cancers epigenetically silence gasdermin E expression as another means to bypass endogenous cell death processes [119–121], suggesting that therapeutic reactivation of pyroptosis may hold promise in the treatment of tumors refractory to conventional chemotherapeutics. Caspase-8 has also been reported to cleave gasdermin D and gasdermin E in response to Yersinia infection [122,123]. Finally, endogenous gasdermin D directly binds BCL-2 through a putative BH3 motif, effectively altering the specificity of its cleavage by the inflammatory caspases to generate a fragment less competent to induce pyroptotic cell death [54].
Crosstalk between apoptosis and mPTP-mediated necrosis.
In response to ischemic injury or neurodegenerative processes, neurons and cardiomyocytes succumb to a form of cell death known as mitochondrial permeability transition pore (mPTP)-mediated necrosis. Distinct from the outer mitochondrial membrane permeabilization induced by apoptosis, the hallmark characteristic of mPTP-mediated necrosis is the formation of inner membrane pores that allow the free passage of ions and solutes up to 1.5 kD [124–126]. Triggered by mitochondrial calcium overload, this breech of inner mitochondrial membrane integrity leads to disastrous consequences for the cell, ultimately sending the cell into a bioenergetic crisis by disrupting the electrochemical proton gradient and ceasing respiration. After decades of intense research, the molecular composition of the mPTP pore/channel was recently confirmed. Previously, only regulatory components were identified including cyclophilin D (CypD), a chaperone that when inhibited by cyclosporin A (CsA) prevents mPTP-mediated necrosis, and the adenine nucleotide translocase (ANT) [125]. Other candidate pore complex regulators included the voltage-dependent anion channels (VDAC), the phosphate carrier (PiC), and the translocator protein (TSPO), but genetic analysis failed to prove essential functions for these components in mPTP-mediated necrosis [125]. A turning point in the field was the revelation that the F1F0 ATP synthase could function as a component of the mPTP pore in one of two ways, by passing solutes either through the border between two ATP synthase monomers or through the c-subunit ring that spans the inner mitochondrial membrane [127,128], but this remains controversial [129]. In two independent studies, highly purified ATP synthase complexes were recently confirmed to possess channel activity consistent with the biophysical characteristics of the mPTP, strongly supporting the identification of the ATP synthase as the sole component of the mPTP pore [130,131]. Further, monomeric complexes were sufficient to mediate mPTP-like channel activity [131], thus distinguishing the channel functions of ATP synthase monomers from the role of ATP synthase dimers in mitochondrial cristae formation [132].
Since proapoptotic BCL-2 family members are famous for forming pores in mitochondrial membranes, it seemed reasonable to assess whether they play a role in mPTP-mediated necrosis. BAX and BAK have been reported to directly interact with and influence the activity of mPTP regulators ANT and VDAC [133,134]. In mice lacking BAX and BAK, mPTP-mediated cell death and associated mitochondrial damage is inhibited in cardiomyocytes after myocardial infarction [135]. Similar results were described in mouse embryonic fibroblasts genetically deleted for BAX and BAK, which are resistant to OMM permeabilization and cell death after pharmacological stimulation of mitochondrial permeability transition, despite ongoing permeabilization of the inner membrane [136]. While necessary for pore formation in the outer membrane after mPTP, BAX and BAK are not directly involved in the CypD-regulated inner membrane permeabilization.
Conversely, antiapoptotic BCL-2 family members can also influence mPTP activity through direct interactions with regulatory components. While initially believed to exist solely on the outer mitochondrial membrane, a role for BCL-xL on the inner membrane as a regulator of the ATP synthase is becoming widely accepted [137,138]. BCL-xL has also been reported to influence ATP/ADP exchange across the outer mitochondrial membrane by way of ANT and VDAC [139,140]. Pharmacological inhibition of BCL-2 and/or BCL-xL may also sensitize cells to mPTP-mediated necrosis [141,142]. The BH3 protein, BID, has also been reported to functionally interact with components of the mPTP to mediate necrosis [143]. Importantly, BCL-2 proteins are known to bind to calcium channels and calcium channel activity at the endoplasmic reticulum [144–146] or mount an apoptotic response to ER stress [147], and therefore may promote ER calcium release that triggers mPTP-mediated necrosis. Although BCL-2 proteins have not been identified as physical components of the mPTP pore, modulation of their activities may prove beneficial for regulating mPTP-mediated necrosis.
Crosstalk between apoptosis and ferroptosis.
Ferroptosis is another emerging cell death pathway whose molecular mechanisms are just beginning to be delineated. The hallmarks of ferroptotic cell death include the oxidation of polyunsaturated fatty acid (PUFA)-containing phospholipids, loss of the ability to repair and detoxify lipid peroxides, and the accumulation of redox-active iron [148]. Morphological features of ferroptosis that have been reported include mitochondrial shrinkage and rounding up of cells with plasma membrane rupture [37,149,150], but these observations remain controversial. A large number of small molecule inducers and inhibitors of ferroptosis have been identified, and recently it was discovered that monounsaturated fatty acids (MUFAs) can protect membranes by blocking lipid ROS accumulation [151]. Ferroptosis inducers have been used to sensitize cancer cells to traditional chemotherapeutic agents or to overcome drug resistance [152,153]. While several proteins have been implicated in mediating ferroptosis, it is clear that this mode of regulated cell death occurs in response to disruptions of normal cellular homeostatic processes, including but not limited to imbalances in iron metabolism and decreases in cellular antioxidants.
While much of the ferroptotic death pathway identified thus far exists independently of the classical apoptotic pathway, initiation of ferroptosis appears to influence the activity or localization of select apoptotic proteins. Notably, the small molecular ferroptosis inducer, erastin, induced caspase-dependent apoptosis in colorectal cancer cell lines [154], perhaps due to mitochondrial translocation of the BH3 protein, BID [155]. HT-22 cells genetically disrupted for BID were resistant to erastin-induced cell death, maintained normal mitochondrial morphology and respiration, and reduced lipid peroxide formation [155]. Crosstalk has also been observed between the tumor suppressor p53 and SLC7A11, a solute carrier receptor necessary for cystine import and subsequent antioxidant response [156]. During the induction of apoptosis, the tumor suppressor p53 becomes stabilized upon integrating cellular stress signals, leading to the transcriptional activation of several apoptotic regulators, including BIM, NOXA and PUMA [157]. P53 can also directly repress SLC7A11 expression, leading to increased sensitivity to erastin, and p53 knockout MEFs were resistant to erastin-induced ferroptosis [156]. Another target of p53-mediated transcriptional regulation, SAT1, was found to induce lipid peroxidation and promote ferroptosis in response to ROS stress [158]. Thus, in addition to regulating apoptosis, p53 appears to coordinate the cellular response to ferroptotic death stimuli by promoting lipid peroxidation and decreasing antioxidant response [159]. Ferroptosis induction also results in upregulation of the BH3 protein, PUMA, independently of p53, but the consequence of PUMA expression is unknown [160,161]. Another p53 family member, ΔNp63α, also inhibits ferroptosis by transcriptionally upregulating genes involved in glutathione metabolism [162]. As the pathways regulating ferroptosis are more precisely delineated, the regulators controlling ferroptosis and apoptosis are likely to become more intertwined.
Conclusions and future perspectives
In the ever expanding and rapidly changing field of cell death, surprises still loom on the horizon, which will again reshape our thinking of the apoptosis pathway. Although apoptosis is commonly believed to occur independently of immune signaling, two recent paradigm-shifting studies have described a novel cell death mechanism in macrophages in which initiation of an apoptotic pathway leads to a downstream inflammatory response [163,164]. In LPS-primed macrophages, genetic or pharmacological inhibition of anti-apoptotic BCL-2 proteins leads to BAX/BAK-mediated MOMP and release of inhibitors of IAP proteins (such as Smac/DIABLO). The resulting IAP depletion leads to the activation of the caspase-8:RIPK1 complex to promote cleavage and release of the inflammatory cytokine IL-1β [163,164]. Surprisingly, BAX/BAK-mediated IL-1β release is independent of the canonical pyroptosis effectors, gasdermin-D and -E; however the role of the NLRP3 inflammasome in this process is disputed [163,164]. The unexpected implication that apoptosis proteins can mediate inflammatory responses awakens a new appreciation for the complexity and interconnectivity of these death pathways. While this seemingly “inflammatory apoptosis” process was observed in LPS-activated macrophages, it remains to be determined whether apoptotic regulators can engage in additional novel cell death pathways in other cell types or tissues.
It is not coincidental that mammalian cells have evolved mechanisms to coregulate independent death pathways by the same molecular factors, surely with the intention of installing a fail-safe mechanism for ensuring the success of an initiated death program or by amplifying PCD signals. As we learn more about the detailed mechanisms of nonapoptotic cell death pathways, more opportunities for crosstalk with apoptotic regulators will likely emerge as possible targets for therapeutic interventions. Since many tumors find ways of inactivating the apoptotic pathway [18], stimulating an alternative death pathway that initiates a localized inflammatory response may more effectively arm the body to combat cancer. Identifying common regulatory factors that participate in multiple death pathways, such as caspase-8 functioning in apoptosis, necroptosis, and pyroptosis, are likely to facilitate the development and implementation of novel strategies that alter their response in favor of progressing down other death pathways.
Acknowledgements:
This work was supported by The Hartwell Foundation and the National Institutes of Health F32 GM095253 and RO1 NS037402. Thanks to Marie Hardwick, Jason Huska, Madhura Kulkarni, Elizabeth Jonas, Junying Yuan, Holly Tang and Hogan Tang for critical reading of the manuscript.
Abbreviations:
- ADCD
autophagy-dependent cell death
- AML
acute myeloid leukemia
- ANT
adenine nucleotide translocator
- ATP
adenosine triphosphate
- BCL-2
B-cell lymphoma-2
- BH3
BCL-2 homology-3
- BIR
baculoviral IAP repeat
- cIAP
cellular inhibitor of apoptosis protein
- CLL
chronic lymphocytic leukemia
- CsA
cyclosporin A
- CypD
cyclophilin D
- DD
death domain
- DED
death effector domain
- DISC
death-inducing signaling complex
- DKO
double knock out
- DR
death receptor
- ER
endoplasmic reticulum
- FADD
Fas-associated death domain
- IAP
inhibitor of apoptosis protein
- IgH
immunoglobulin H
- MEF
mouse embryonic fibroblasts
- MLKL
mixed-lineage kinase-like
- MOMP
mitochondrial outer membrane permeabilization
- mPTP
mitochondrial permeability transition pore
- MUFA
monounsaturated fatty acid
- PI3K
phosphatidylinositide 3-kinase
- PiC
phosphate carrier
- PRR
pattern recognition receptor
- PUFA
polyunsaturated fatty acids
- RIPK
receptor-interacting serine/threonine kinase
- ROS
reactive oxygen species
- Smac/DIABLO
second mitochondria-derived activator of caspases/direct IAP-binding protein with low Pi
- TNF
tumor necrosis factor
- TNFR
tumor necrosis factor receptor
- TRAIL
TNF-related apoptosis-inducing factor
- TSPO
translocator protein
- VDAC
voltage-dependent anion channel
- XIAP
X-chromosome-linked IAP
Footnotes
Conflicts of Interest: none
References:
- 1.Lockshin RA & Williams CM (1965) PROGRAMMED CELL DEATH--I. CYTOLOGY OF DEGENERATION IN THE INTERSEGMENTAL MUSCLES OF THE PERNYI SILKMOTH. J. Insect Physiol 11, 123–33. [DOI] [PubMed] [Google Scholar]
- 2.Kerr JF., Wyllie A & Currie A (1972) Apoptosis: a Basic Biological Phenomenon With Wide- Ranging Implications in Tissue Kinetics. Br J Cancer 26, 239–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol. Pathol 35, 495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taylor RC, Cullen SP & Martin SJ (2008) Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol 9, 231–241. [DOI] [PubMed] [Google Scholar]
- 5.Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, Annicchiarico-Petruzzelli M, Antonov AV., Arama E, Baehrecke EH, Barlev NA, Bazan NG, Bernassola F, Bertrand MJM, Bianchi K, Blagosklonny MV., Blomgren K, Borner C, Boya P, Brenner C, Campanella M, Candi E, Carmona-Gutierrez D, Cecconi F, Chan FK-M, Chandel NS, Cheng EH, Chipuk JE, Cidlowski JA, Ciechanover A, Cohen GM, Conrad M, Cubillos-Ruiz JR, Czabotar PE, D’Angiolella V, Dawson TM, Dawson VL, De Laurenzi V, De Maria R, Debatin K-M, DeBerardinis RJ, Deshmukh M, Di Daniele N, Di Virgilio F, Dixit VM, Dixon SJ, Duckett CS, Dynlacht BD, El-Deiry WS, Elrod JW, Fimia GM, Fulda S, García-Sáez AJ, Garg AD, Garrido C, Gavathiotis E, Golstein P, Gottlieb E, Green DR, Greene LA, Gronemeyer H, Gross A, Hajnoczky G, Hardwick JM, Harris IS, Hengartner MO, Hetz C, Ichijo H, Jäättelä M, Joseph B, Jost PJ, Juin PP, Kaiser WJ, Karin M, Kaufmann T, Kepp O, Kimchi A, Kitsis RN, Klionsky DJ, Knight RA, Kumar S, Lee SW, Lemasters JJ, Levine B, Linkermann A, Lipton SA, Lockshin RA, López-Otín C, Lowe SW, Luedde T, Lugli E, MacFarlane M, Madeo F, Malewicz M, Malorni W, Manic G, Marine J-C, Martin SJ, Martinou J-C, Medema JP, Mehlen P, Meier P, Melino S, Miao EA, Molkentin JD, Moll UM, Muñoz-Pinedo C, Nagata S, Nuñez G, Oberst A, Oren M, Overholtzer M, Pagano M, Panaretakis T, Pasparakis M, Penninger JM, Pereira DM, Pervaiz S, Peter ME, Piacentini M, Pinton P, Prehn JHM, Puthalakath H, Rabinovich GA, Rehm M, Rizzuto R, Rodrigues CMP, Rubinsztein DC, Rudel T, Ryan KM, Sayan E, Scorrano L, Shao F, Shi Y, Silke J, Simon H-U, Sistigu A, Stockwell BR, Strasser A, Szabadkai G, Tait SWG, Tang D, Tavernarakis N, Thorburn A, Tsujimoto Y, Turk B, Vanden Berghe T, Vandenabeele P, Vander Heiden MG, Villunger A, Virgin HW, Vousden KH, Vucic D, Wagner EF, Walczak H, Wallach D, Wang Y, Wells JA, Wood W, Yuan J, Zakeri Z, Zhivotovsky B, Zitvogel L, Melino G & Kroemer G (2018) Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 25, 486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Youle RJ & Strasser A (2008) The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol 9, 47–59. [DOI] [PubMed] [Google Scholar]
- 7.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O’Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW & Rosenberg SH (2005) An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435, 677–681. [DOI] [PubMed] [Google Scholar]
- 8.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DCS, Hymowitz SG, Jin S, Khaw SL, Kovar PJ, Lam LT, Lee J, Maecker HL, Marsh KC, Mason KD, Mitten MJ, Nimmer PM, Oleksijew A, Park CH, Park C-M, Phillips DC, Roberts AW, Sampath D, Seymour JF, Smith ML, Sullivan GM, Tahir SK, Tse C, Wendt MD, Xiao Y, Xue JC, Zhang H, Humerickhouse R a, Rosenberg SH & Elmore SW (2013) ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med 19, 202–8. [DOI] [PubMed] [Google Scholar]
- 9.Sarosiek KA & Letai A (2016) Directly targeting the mitochondrial pathway of apoptosis for cancer therapy using BH3 mimetics - recent successes, current challenges and future promise. FEBS J. 283, 3523–3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lavrik I, Golks A & Krammer PH (2005) Death receptor signaling. J. Cell Sci 118, 265–7. [DOI] [PubMed] [Google Scholar]
- 11.Fulda S (2015) Targeting extrinsic apoptosis in cancer: Challenges and opportunities. Semin. Cell Dev. Biol 39, 20–25. [DOI] [PubMed] [Google Scholar]
- 12.Tummers B & Green DR (2017) Caspase-8: regulating life and death. Immunol. Rev 277, 76–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsujimoto Y, Cossman J, Jaffe E & Croce CM (1985) Involvement of the bcl-2 gene in human follicular lymphoma. Science 228, 1440–3. [DOI] [PubMed] [Google Scholar]
- 14.Vaux DL, Cory S & Adams JM (1988) Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 335, 440–442. [DOI] [PubMed] [Google Scholar]
- 15.Huska JD, Lamb HM & Hardwick JM (2019) Overview of BCL-2 Family Proteins and Therapeutic Potentials. In Methods in molecular biology (Clifton, N.J.) pp. 1–21. [DOI] [PubMed] [Google Scholar]
- 16.Glab JA, Mbogo GW & Puthalakath H (2017) BH3-Only Proteins in Health and Disease. In International review of cell and molecular biology pp. 163–196. [DOI] [PubMed]
- 17.Li H, Zhu H, Xu CJ & Yuan J (1998) Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94, 491–501. [DOI] [PubMed] [Google Scholar]
- 18.Letai A (2017) Apoptosis and Cancer. Annu. Rev. Cancer Biol 1, 275–294. [Google Scholar]
- 19.Fulda S & Debatin K-M (2006) Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 25, 4798–4811. [DOI] [PubMed] [Google Scholar]
- 20.Dimri M & Dimri GP (2016) Senescence, apoptosis, and cancer. In The Molecular Basis of Human Cancer pp. 183–196. Springer; New York. [Google Scholar]
- 21.Du C, Fang M, Li Y, Li L & Wang X (2000) Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 102, 33–42. [DOI] [PubMed] [Google Scholar]
- 22.Silke J & Vince J (2017) IAPs and cell death. In Current Topics in Microbiology and Immunology pp. 95–117. Springer Verlag. [DOI] [PubMed] [Google Scholar]
- 23.Fulda S (2017) Smac Mimetics to Therapeutically Target IAP Proteins in Cancer. In International Review of Cell and Molecular Biology pp. 157–169. Elsevier Inc. [DOI] [PubMed] [Google Scholar]
- 24.Derakhshan A, Chen Z & Van Waes C (2017) Therapeutic small molecules target inhibitor of apoptosis proteins in cancers with deregulation of extrinsic and intrinsic cell death pathways. Clin. Cancer Res 23, 1379–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu H, Li Y, Liu Y & Han B (2019) Bivalent SMAC Mimetics for Treating Cancer by Antagonizing Inhibitor of Apoptosis Proteins. ChemMedChem 14, 1951–1962. [DOI] [PubMed] [Google Scholar]
- 26.Beug ST, Conrad DP, Alain T, Korneluk RG & Lacasse EC (2015) Combinatorial cancer immunotherapy strategies with proapoptotic small-molecule IAP antagonists. Int. J. Dev. Biol 59, 141–147. [DOI] [PubMed] [Google Scholar]
- 27.Cong H, Xu L, Wu Y, Qu Z, Bian T, Zhang W, Xing C & Zhuang C (2019) Inhibitor of Apoptosis Protein (IAP) Antagonists in Anticancer Agent Discovery: Current Status and Perspectives. J. Med. Chem 62, 5750–5772. [DOI] [PubMed] [Google Scholar]
- 28.Infante JR, Dees EC, Olszanski AJ, Dhuria S V., Sen S, Cameron S & Cohen RB (2014) Phase i dose-escalation study of LCL161, an oral inhibitorof apoptosis proteins inhibitor, in patients with advanced solid tumors. J. Clin. Oncol 32, 3103–3110. [DOI] [PubMed] [Google Scholar]
- 29.Garner TP, Lopez A, Reyna DE, Spitz AZ & Gavathiotis E (2017) Progress in targeting the BCL-2 family of proteins. Curr. Opin. Chem. Biol 39, 133–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Juárez-Salcedo LM, Desai V & Dalia S (2019) Venetoclax: evidence to date and clinical potential. Drugs Context 8, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Richard-Carpentier G & DiNardo CD (2019) Venetoclax for the treatment of newly diagnosed acute myeloid leukemia in patients who are ineligible for intensive chemotherapy. Ther. Adv. Hematol 10, 204062071988282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tahir SK, Smith ML, Hessler P, Rapp LR, Idler KB, Park CH, Leverson JD & Lam LT (2017) Potential mechanisms of resistance to venetoclax and strategies to circumvent it. BMC Cancer 17, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tausch E, Close W, Dolnik A, Bloehdorn J, Chyla B, Bullinger L, Döhner H, Mertens D & Stilgenbauer S (2019) Venetoclax resistance and acquired BCL2 mutations in chronic lymphocytic leukemia. Haematologica 104, e434–e437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blombery P, Birkinshaw RW, Nguyen T, Gong J nan, Thompson ER, Xu Z, Westerman DA, Czabotar PE, Dickinson M, Huang DCS, Seymour JF & Roberts AW (2019) Characterization of a novel venetoclax resistance mutation (BCL2 Phe104Ile) observed in follicular lymphoma. Br. J. Haematol [DOI] [PubMed] [Google Scholar]
- 35.Bodo J, Zhao X, Durkin L, Souers AJ, Phillips DC, Smith MR & Hsi ED (2016) Acquired resistance to venetoclax (ABT-199) in t(14;18) positive lymphoma cells. Oncotarget 7, 70000–70010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shan B, Pan H, Najafov A & Yuan J (2018) Necroptosis in development and diseases. Genes Dev. 32, 327–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK, Kagan VE, Noel K, Jiang X, Linkermann A, Murphy ME, Overholtzer M, Oyagi A, Pagnussat GC, Park J, Ran Q, Rosenfeld CS, Salnikow K, Tang D, Torti FM, Torti S V, Toyokuni S, Woerpel KA & Zhang DD (2017) Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 171, 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Man SM, Karki R & Kanneganti T-D (2017) Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev 277, 61–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bialik S, Dasari SK & Kimchi A (2018) Autophagy-dependent cell death – where, how and why a cell eats itself to death. J Cell Sci 131, jcs215152. [DOI] [PubMed] [Google Scholar]
- 40.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA & Yuan J (2005) Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol 1, 112–119. [DOI] [PubMed] [Google Scholar]
- 41.de Almagro MC, Goncharov T, Newton K & Vucic D (2015) Cellular IAP proteins and LUBAC differentially regulate necrosome-associated RIP1 ubiquitination. Cell Death Dis. 6, e1800–e1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feltham R, Vince JE & Lawlor KE (2017) Caspase-8: not so silently deadly. Clin. Transl. Immunol 6, e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin Y, Devin A, Rodriguez Y & Liu ZG (1999) Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 13, 2514–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu S, Liu H, Johnston A, Hanna-Addams S, Reynoso E, Xiang Y & Wang Z (2017) MLKL forms disulfide bond-dependent amyloid-like polymers to induce necroptosis. Proc. Natl. Acad. Sci. U. S. A 114, E7450–E7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang J, Yang Y, He W & Sun L (2016) Necrosome core machinery: MLKL. Cell. Mol. Life Sci 73, 2153–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hildebrand JM, Tanzer MC, Lucet IS, Young SN, Spall SK, Sharma P, Pierotti C, Garnier JM, Dobson RCJ, Webb AI, Tripaydonis A, Babon JJ, Mulcair MD, Scanlon MJ, Alexander WS, Wilks AF, Czabotar PE, Lessene G, Murphy JM & Silke J (2014) Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc. Natl. Acad. Sci. U. S. A 111, 15072–15077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Petrie EJ, Hildebrand JM & Murphy JM (2017) Insane in the membrane: a structural perspective of MLKL function in necroptosis. Immunol. Cell Biol 95, 152–159. [DOI] [PubMed] [Google Scholar]
- 48.Yuan J, Amin P & Ofengeim D (2018) Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heckmann BL, Tummers B & Green DR (2018) Crashing the computer: apoptosis vs. necroptosis in neuroinflammation. Cell Death Differ., 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yabal M, Müller N, Adler H, Knies N, Groß CJ, Damgaard RB, Kanegane H, Ringelhan M, Kaufmann T, Heikenwälder M, Strasser A, Groß O, Ruland J, Peschel C, Gyrd-Hansen M & Jost PJ (2014) XIAP Restricts TNF- and RIP3-Dependent Cell Death and Inflammasome Activation. Cell Rep. 7, 1796–1808. [DOI] [PubMed] [Google Scholar]
- 51.Irrinki KM, Mallilankaraman K, Thapa RJ, Chandramoorthy HC, Smith FJ, Jog NR, Gandhirajan RK, Kelsen SG, Houser SR, May MJ, Balachandran S & Madesh M (2011) Requirement of FADD, NEMO and BAX/BAK for Aberrant Mitochondrial Function in TNF{alpha}-Induced Necrosis. Mol. Cell. Biol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rohde K, Kleinesudeik L, Roesler S, Löwe O, Heidler J, Schröder K, Wittig I, Dröse S & Fulda S (2017) A Bak-dependent mitochondrial amplification step contributes to Smac mimetic/glucocorticoid-induced necroptosis. Cell Death Differ. 24, 83–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Karch J, Kanisicak O, Brody MJ, Sargent MA, Michael DM & Molkentin JD (2015) Necroptosis Interfaces with MOMP and the MPTP in Mediating Cell Death. PLoS One 10, e0130520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shi CS & Kehrl JH (2019) Bcl-2 regulates pyroptosis and necroptosis by targeting BH3-like domains in GSDMD and MLKL. Cell Death Discov. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tischner D, Manzl C, Soratroi C, Villunger A & Krumschnabel G (2012) Necrosis-like death can engage multiple pro-apoptotic Bcl-2 protein family members. Apoptosis 17, 1197–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen D, Tong J, Yang L, Wei L, Stolz DB, Yu J, Zhang J & Zhang L (2018) PUMA amplifies necroptosis signaling by activating cytosolic DNA sensors. Proc. Natl. Acad. Sci 115, 3930–3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tait SWG, Oberst A, Quarato G, Milasta S, Haller M, Wang R, Karvela M, Ichim G, Yatim N, Albert ML, Kidd G, Wakefield R, Frase S, Krautwald S, Linkermann A & Green DR (2013) Widespread Mitochondrial Depletion via Mitophagy Does Not Compromise Necroptosis. Cell Rep. 5, 878–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marshall KD & Baines CP (2014) Necroptosis: Is there a role for mitochondria? Front. Physiol 5 AUG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gross A & Katz SG (2017) Non-apoptotic functions of BCL-2 family proteins. Cell Death Differ. 24, 1348–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Safferthal C, Rohde K & Fulda S (2017) Therapeutic targeting of necroptosis by Smac mimetic bypasses apoptosis resistance in acute myeloid leukemia cells. Oncogene 36, 1487–1502. [DOI] [PubMed] [Google Scholar]
- 61.Cekay MJ, Roesler S, Frank T, Knuth A-K, Eckhardt I & Fulda S (2017) Smac mimetics and type II interferon synergistically induce necroptosis in various cancer cell lines. Cancer Lett. 410, 228–237. [DOI] [PubMed] [Google Scholar]
- 62.McComb S, Aguadé-Gorgorió J, Harder L, Marovca B, Cario G, Eckert C, Schrappe M, Stanulla M, von Stackelberg A, Bourquin J-P & Bornhauser BC (2016) Activation of concurrent apoptosis and necroptosis by SMAC mimetics for the treatment of refractory and relapsed ALL. Sci. Transl. Med 8, 339ra70. [DOI] [PubMed] [Google Scholar]
- 63.Brumatti G, Ma C, Lalaoui N, Nguyen NY, Navarro M, Tanzer MC, Richmond J, Ghisi M, Salmon JM, Silke N, Pomilio G, Glaser SP, De Valle E, Gugasyan R, Gurthridge MA, Condon SM, Johnstone RW, Lock R, Salvesen G, Wei A, Vaux DL, Ekert PG & Silke J (2016) The caspase-8 inhibitor emricasan combines with the SMAC mimetic birinapant to induce necroptosis and treat acute myeloid leukemia. Sci. Transl. Med 8. [DOI] [PubMed] [Google Scholar]
- 64.Chen D, Yu J & Zhang L (2016) Necroptosis: an alternative cell death program defending against cancer. Biochim. Biophys. Acta 1865, 228–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Koo G-B, Morgan MJ, Lee D-G, Kim W-J, Yoon J-H, Koo JS, Kim S Il, Kim SJ, Son MK, Hong SS, Levy JMM, Pollyea DA, Jordan CT, Yan P, Frankhouser D, Nicolet D, Maharry K, Marcucci G, Choi KS, Cho H, Thorburn A & Kim Y-S (2015) Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. 25, 707–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Su Z, Yang Z, Xie L, DeWitt JP & Chen Y (2016) Cancer therapy in the necroptosis era. Cell Death Differ. 23, 748–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Silva MT (2010) Secondary necrosis: The natural outcome of the complete apoptotic program. FEBS Lett. 584, 4491–4499. [DOI] [PubMed] [Google Scholar]
- 68.Mezzatesta C & Bornhauser BC (2019) Exploiting necroptosis for therapy of acute lymphoblastic leukemia. Front. Cell Dev. Biol 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gong Y, Fan Z, Luo G, Yang C, Huang Q, Fan K, Cheng H, Jin K, Ni Q, Yu X & Liu C (2019) The role of necroptosis in cancer biology and therapy. Mol. Cancer 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Park S, Hatanpaa KJ, Xie Y, Mickey BE, Madden CJ, Raisanen JM, Ramnarain DB, Xiao G, Saha D, Boothman DA, Zhao D, Bachoo RM, Pieper RO & Habib AA (2009) The receptor interacting protein 1 inhibits p53 induction through NF-KB activation and confers a worse prognosis in glioblastoma. Cancer Res. 69, 2809–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xie T, Peng W, Liu Y, Yan C, Maki J, Degterev A, Yuan J & Shi Y (2013) Structural basis of RIP1 inhibition by necrostatins. Structure 21, 493–499. [DOI] [PubMed] [Google Scholar]
- 72.Strilic B, Yang L, Albarrán-Juárez J, Wachsmuth L, Han K, Müller UC, Pasparakis M & Offermanns S (2016) Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature 536, 215–218. [DOI] [PubMed] [Google Scholar]
- 73.Yin Z, Pascual C & Klionsky D (2016) Autophagy: machinery and regulation. Microb. Cell 3, 588–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Morishita H & Mizushima N (2019) Diverse Cellular Roles of Autophagy. Annu. Rev. Cell Dev. Biol 35, 453–475. [DOI] [PubMed] [Google Scholar]
- 75.Xie J, Zhu R, Peng Y, Gao W, Du J, Zhao L, Chi Y & Yang L (2017) Tumor necrosis factor-alpha regulates photoreceptor cell autophagy after retinal detachment. Sci. Rep 7, 17108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B & Levine B (1998) Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J. Virol 72, 8586–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Oberstein A, Jeffrey PD & Shi Y (2007) Crystal Structure of the Bcl-X L -Beclin 1 Peptide Complex. J. Biol. Chem 282, 13123–13132. [DOI] [PubMed] [Google Scholar]
- 78.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD & Levine B (2005) Bcl-2 Antiapoptotic Proteins Inhibit Beclin 1-Dependent Autophagy. Cell 122, 927–939. [DOI] [PubMed] [Google Scholar]
- 79.Ma P, Schwarten M, Schneider L, Boeske A, Henke N, Lisak D, Weber S, Mohrlüder J, Stoldt M, Strodel B, Methner A, Hoffmann S, Weiergräber OH & Willbold D (2013) Interaction of Bcl-2 with the Autophagy-related GABA A Receptor-associated Protein (GABARAP). J. Biol. Chem 288, 37204–37215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nakajima S, Aikawa C, Nozawa T, Minowa-Nozawa A, Toh H & Nakagawa I (2017) Bcl-xL Affects Group A Streptococcus-Induced Autophagy Directly, by Inhibiting Fusion between Autophagosomes and Lysosomes, and Indirectly, by Inhibiting Bacterial Internalization via Interaction with Beclin 1-UVRAG. PLoS One 12, e0170138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rubinstein AD, Eisenstein M, Ber Y, Bialik S & Kimchi A (2011) The autophagy protein atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol. Cell 44, 698–709. [DOI] [PubMed] [Google Scholar]
- 82.Hollville E, Carroll RG, Cullen SP & Martin SJ (2014) Bcl-2 Family Proteins Participate in Mitochondrial Quality Control by Regulating Parkin/PINK1-Dependent Mitophagy. Mol. Cell 55, 451–466. [DOI] [PubMed] [Google Scholar]
- 83.Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S & Gustafsson ÅB (2012) Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J. Biol. Chem 287, 19094–19104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Elgendy M, Ciro M, Abdel-Aziz AK, Belmonte G, Zuffo RD, Mercurio C, Miracco C, Lanfrancone L, Foiani M & Minucci S (2014) Beclin 1 restrains tumorigenesis through Mcl-1 destabilization in an autophagy-independent reciprocal manner. Nat. Commun 5, 5637. [DOI] [PubMed] [Google Scholar]
- 85.Germain M, Nguyen AP, Le Grand JN, Arbour N, Vanderluit JL, Park DS, Opferman JT & Slack RS (2011) MCL-1 is a stress sensor that regulates autophagy in a developmentally regulated manner. EMBO J. 30, 395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dai Y & Grant S (2015) BCL2L11/Bim as a dual-agent regulating autophagy and apoptosis in drug resistance. Autophagy 11, 416–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Luo S, Garcia-Arencibia M, Zhao R, Puri C, Toh PPC, Sadiq O & Rubinsztein DC (2012) Bim Inhibits Autophagy by Recruiting Beclin 1 to Microtubules. Mol. Cell 47, 359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Delgado M & Tesfaigzi Y (2013) BH3-only proteins, Bmf and Bim, in autophagy. Cell Cycle 12, 3453–3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang J, Cui D, Gu S, Chen X, Bi Y, Xiong X & Zhao Y (2018) Autophagy regulates apoptosis by targeting NOXA for degradation. Biochim. Biophys. Acta - Mol. Cell Res 1865, 1105–1113. [DOI] [PubMed] [Google Scholar]
- 90.Wirawan E, Vande Walle L, Kersse K, Cornelis S, Claerhout S, Vanoverberghe I, Roelandt R, De Rycke R, Verspurten J, Declercq W, Agostinis P, Vanden Berghe T, Lippens S & Vandenabeele P (2010) Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 1, e18–e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Betin VMS & Lane JD (2009) Caspase cleavage of Atg4D stimulates GABARAP-L1 processing and triggers mitochondrial targeting and apoptosis. J. Cell Sci 122, 2554–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Murthy A, Li Y, Peng I, Reichelt M, Katakam AK, Noubade R, Roose-Girma M, DeVoss J, Diehl L, Graham RR & van Lookeren Campagne M (2014) A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature 506, 456–462. [DOI] [PubMed] [Google Scholar]
- 93.Cui J, Chen B, Wang H, Han Y, Chen X & Zhang W (2016) Glucosidase II β-subunit, a novel substrate for caspase-3-like activity in rice, plays as a molecular switch between autophagy and programmed cell death. Sci. Rep 6, 31764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Han J, Hou W, Goldstein LA, Stolz DB, Watkins SC & Rabinowich H (2014) A Complex between Atg7 and Caspase-9. J. Biol. Chem 289, 6485–6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang C & Youle RJ (2012) Predominant requirement of Bax for apoptosis in HCT116 cells is determined by Mcl-1’s inhibitory effect on Bak. Oncogene 31, 3177–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Denton D & Kumar S (2018) Autophagy-dependent cell death. Cell Death Differ., 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Klionsky DJ, Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Arozena AA, Adachi H, Adams CM, Adams PD, Adeli K, Adhihetty PJ, Adler SG, Agam G, Agarwal R, Aghi MK, Agnello M, Agostinis P, Aguilar P V., Aguirre-Ghiso J, Aguirre-Ghiso J, Airoldi EM, Airoldi EM, Ait-Si-Ali S, Akematsu T, Akporiaye ET, Al-Rubeai M, Albaiceta GM, Albanese C, Albani D, Albert ML, Aldudo J, Algül H, Alirezaei M, Alloza I, Alloza I, Almasan A, Almonte-Beceril M, Alnemri ES, Alonso C, Altan-Bonnet N, Altieri DC, Alvarez S, Alvarez-Erviti L, Alves S, Amadoro G, Amano A, Amantini C, Ambrosio S, Amelio I, Amer AO, Amessou M, Amon A, An Z, Anania FA, Andersen SU, Andley UP, Andreadi CK, Andrieu-Abadie N, Anel A, Ann DK, Anoopkumar-Dukie S, Antonioli M, Antonioli M, Aoki H, Apostolova N, Aquila S, Aquilano K, Araki K, Arama E, Aranda A, Araya J, Arcaro A, Arias E, Arimoto H, Ariosa AR, Armstrong JL, Arnould T, Arsov I, Asanuma K, Askanas V, Asselin E, Atarashi R, Atherton SS, Atkin JD, Attardi LD, Auberger P, Auburger G, Aurelian L, Autelli R, Avagliano L, Avagliano L, Avantaggiati ML, Avrahami L, Awale S, Azad N, Bachetti T, Backer JM, Bae DH, Bae JS, Bae ON, Bae SH, Baehrecke EH, Baek SH, Baghdiguian S, Bagniewska-Zadworna A, Bai H, Bai J, Bai XY, Bailly Y, Balaji KN, Balduini W, Ballabio A, Balzan R, Banerjee R, Bánhegyi G, Bao H, Barbeau B, Barrachina MD, Barreiro E, Bartel B, Bartolomé A, Bassham DC, Bassi MT, Bast RC, Basu A, Batista MT, Batoko H, Battino M, Bauckman K, Baumgarner BL, Bayer KU, Beale R, Beaulieu JF, Beck GR, Beck GR, Becker C, Beckham JD, Bédard PA, Bednarski PJ, Begley TJ, Behl C, Behrends C, Behrens GMN, Behrns KE, Bejarano E, Belaid A, Belleudi F, Bénard G, Berchem G, Bergamaschi D, Bergami M, Berkhout B, Berliocchi L, Bernard A, Bernard M, Bernassola F, Bertolotti A, Bess AS, Besteiro S, Bettuzzi S, Bhalla S, Bhattacharyya S, Bhutia SK, Biagosch C, Bianchi MW, Bianchi MW, Bianchi MW, Biard-Piechaczyk M, Billes V, Bincoletto C, Bingol B, Bird SW, Bitoun M, Bjedov I, Blackstone C, Blanc L, Blanco GA, Blomhoff HK, Boada-Romero E, Böckler S, Boes M, Boesze-Battaglia K, Boise LH, Boise LH, Bolino A, Boman A, Bonaldo P, Bordi M, Bordi M, Bosch J, Botana LM, Botti J, Bou G, Bouché M, Bouchecareilh M, Boucher MJ, Boulton ME, Bouret SG, Boya P, Boyer-Guittaut M, Bozhkov P V., Brady N, Braga VMM, Brancolini C, Braus GH, Bravo-San-Pedro JM, Bravosan-Pedro JM, Bravosan-Pedro JM, Bravosan-Pedro JM, Brennan LA, Bresnick EH, Brest P, Bridges D, Bringer MA, Brini M, Brito GC, Brodin B, Brookes PS, Brown EJ, Brown K, Broxmeyer HE, Bruhat A, Bruhat A, Brum PC, Brumell JH, Brunetti-Pierri N, Brunetti-Pierri N, Bryson-Richardson RJ, Buch S, Buchan AM, Budak H, Bulavin D V., Bulavin D V., Bulavin D V., Bultman SJ, Bultynck G, Bumbasirevic V, Burelle Y, Burke RE, Burke RE, Burmeister M, Bütikofer P, Caberlotto L, Cadwell K, Cahova M, Cai D, Cai J, Cai Q, Calatayud S, Camougrand N, Campanella M, Campbell GR, Campbell M, Campello S, Campello S, Candau R, Caniggia I, Cantoni L, Cao L, Caplan AB, Caraglia M, Cardinali C, Cardoso SM, Carew JS, Carleton LA, Carlin CR, Carloni S, Carlsson SR, Carmona-Gutierrez D, Carneiro LAM, Carnevali O, Carra S, Carrier A, Carroll B, Casas C, Casas J, Cassinelli G, Castets P, Castro-Obregon S, Cavallini G, Ceccherini I, Cecconi F, Cecconi F, Cecconi F, Cederbaum AI, Ceña V, Ceña V, Cenci S, Cenci S, Cerella C, Cervia D, Cetrullo S, Chaachouay H, Chae HJ, Chagin AS, Chai CY, Chai CY, Chakrabarti G, Chamilos G, Chan EYW, Chan MTV, Chandra D, Chandra P, Chang CP, Chang RCC, Chang TY, Chatham JC, Chatterjee S, Chauhan S, Che Y, Cheetham ME, Cheluvappa R, Chen CJ, Chen G, Chen G, Chen GC, Chen G, Chen H, Chen JW, Chen JK, Chen JK, Chen M, Chen M, Chen P, Chen Q, Chen Q, Chen S Der, Chen S, Chen SSL, Chen W, Chen WJ, Chen WQ, Chen W, Chen X, Chen YH, Chen YG, Chen Y, Chen Y, Chen Y, Chen Y, Chen YJ, Chen YQ, Chen Y, Chen Z, Chen Z, Cheng A, Cheng CHK, Cheng H, Cheong H, Cherry S, Chesney J, Cheung CHA, Chevet E, Chi HC, Chi SG, Chiacchiera F, Chiang HL, Chiarelli R, Chiariello M, Chiariello M, Chiariello M, Chieppa M, Chin LS, Chiong M, Chiu GNC, Cho DH, Cho SG, Cho WC, Cho YY, Cho YS, Choi AMK, Choi EJ, Choi EK, Choi EK, Choi EK, Choi EK, Choi J, Choi ME, Choi S Il, Choi S Il, Chou TF, Chouaib S, Choubey D, Choubey V, Chow KC, Chowdhury K, Chu CT, Chuang TH, Chun T, Chung H, Chung T, Chung YL, Chwae YJ, Cianfanelli V, Ciarcia R, Ciechomska IA, Ciriolo MR, Cirone M, Claerhout S, Claerhout S, Clague MJ, Clària J, Clarke PGH, Clarke R, Clementi E, Clementi E, Cleyrat C, Cnop M, Coccia EM, Cocco T, Codogno P, Coers J, Cohen EEW, Colecchia D, Colecchia D, Colecchia D, Coletto L, Coll NS, Colucci-Guyon E, Comincini S, Condello M, Cook KL, Coombs GH, Cooper CD, Cooper JM, Coppens I, Corasaniti MT, Corazzari M, Corazzari M, Corbalan R, Corcelle-Termeau E, Cordero MD, Corral-Ramos C, Corti O, Corti O, Cossarizza A, Costelli P, Costes S, Cotman SL, Coto-Montes A, Cottet S, Cottet S, Couve E, Covey LR, Cowart LA, Cox JS, Coxon FP, Coyne CB, Cragg MS, Craven RJ, Crepaldi T, Crespo JL, Criollo A, Crippa V, Cruz MT, Cuervo AM, Cuezva JM, Cui T, Cutillas PR, Czaja MJ, Czyzyk-Krzeska MF, Dagda RK, Dahmen U, Dai C, Dai W, Dai Y, Dalby KN, Valle LD, Dalmasso G, D’amelio M, Damme M, Darfeuille-Michaud A, Dargemont C, Darley-Usmar VM, Dasarathy S, Dasgupta B, Dash S, Dass CR, Davey HM, Davids LM, Dávila D, Davis RJ, Dawson TM, Dawson TM, Dawson VL, Daza P, de Belleroche J, de Figueiredo P, de Figueiredo P, de Figueiredo RCBQ, de la Fuente J, De Martino L, De Matteis A, De Meyer GRY, De Milito A, De Santi M, de Souza W, De Tata V, De Zio D, Debnath J, Dechant R, Decuypere JP, Decuypere JP, Deegan S, Dehay B, Del Bello B, Del Re DP, Delage-Mourroux R, Delbridge LMD, Deldicque L, Delorme-Axford E, Deng Y, Dengjel J, Denizot M, Dent P, Der CJ, Deretic V, Derrien B, Deutsch E, Devarenne TP, Devenish RJ, Di Bartolomeo S, Di Daniele N, Di Domenico F, Di Nardo A, Di Paola S, Di Pietro A, Di Renzo L, Di Antonio A, Díaz-Araya G, Díaz-Laviada I, Diaz-Meco MT, Diaz-Nido J, Dickey CA, Dickson RC, Diederich M, Digard P, Dikic I, Dinesh-Kumar SP, Ding C, Ding WX, Ding Z, Dini L, Distler JHW, Diwan A, Djavaheri-Mergny M, Dmytruk K, Dobson RCJ, Doetsch V, Dokladny K, Dokudovskaya S, Donadelli M, Dong XC, Dong X, Dong Z, Donohue TM, Donohue-jr TM, Doran KS, D’orazi G, Dorn GW, Dosenko V, Dridi S, Drucker L, Du J, Du LL, Du L, du Toit A, Dua P, Duan L, Duann P, Dubey VK, Duchen MR, Duchosal MA, Duez H, Dugail I, Dumit VI, Duncan MC, Dunlop EA, Dunn WA, Dupont N, Dupuis L, Dupuis L, Durán R V., Durcan TM, Duvezin-Caubet S, Duvvuri U, Eapen V, Ebrahimi-Fakhari D, Echard A, Eckhart L, Edelstein CL, Edinger AL, Eichinger L, Eisenberg T, Eisenberg-Lerner A, Eissa NT, El-Deiry WS, El-Khoury V, Elazar Z, Eldar-Finkelman H, Elliott CJH, Emanuele E, Emmenegger U, Engedal N, Engelbrecht AM, Engelender S, Enserink JM, Erdmann R, Erenpreisa J, Eri R, Eriksen JL, Erman A, Escalante R, Eskelinen EL, Espert L, Esteban-Martínez L, Evans TJ, Fabri M, Fabrias G, Fabrizi C, Facchiano A, Færgeman NJ, Faggioni A, Fairlie WD, Fairlie WD, Fairlie WD, Fan C, Fan D, Fan J, Fang S, Fanto M, Fanzani A, Farkas T, Faure M, Favier FB, Favier FB, Fearnhead H, Federici M, Fei E, Felizardo TC, Feng H, Feng Y, Feng Y, Feng Y, Ferguson TA, Fernández ÁF, Fernandez-Barrena MG, Fernandez-Checa JC, Fernandez-Checa JC, Fernández-López A, Fernandez-Zapico ME, Feron O, Ferraro E, Ferreira-Halder CV, Fesus L, Feuer R, Fiesel FC, Filippi-Chiela EC, Filomeni G, Filomeni G, Fimia GM, Fimia GM, Fingert JH, Fingert JH, Finkbeiner S, Finkbeiner S, Finkel T, Fiorito F, Fiorito F, Fisher PB, Flajolet M, Flamigni F, Florey O, Florio S, Floto RA, Folini M, Follo C, Fon EA, Fornai F, Fornai F, Fortunato F, Fraldi A, Franco R, Francois A, Francois A, François A, Frankel LB, Fraser IDC, Frey N, Freyssenet DG, Frezza C, Friedman SL, Frigo DE, Frigo DE, Fu D, Fuentes JM, Fueyo J, Fujitani Y, Fujiwara Y, Fujiya M, Fukuda M, Fulda S, Fusco C, Gabryel B, Gaestel M, Gailly P, Gajewska M, Galadari S, Galadari S, Galili G, Galindo I, Galindo MF, Galliciotti G, Galluzzi L, Galluzzi L, Galluzzi L, Galluzzi L, Galluzzi L, Galy V, Gammoh N, Gandy S, Gandy S, Ganesan AK, Ganesan S, Ganley IG, Gannagé M, Gao FB, Gao F, Gao JX, Nannig LG, Véscovi EG, Garcia-Macía M, Garcia-Ruiz C, Garg AD, Garg PK, Gargini R, Gassen NC, Gatica D, Gatica D, Gatti E, Gatti E, Gatti E, Gavard J, Gavathiotis E, Ge L, Ge P, Ge S, Gean PW, Gelmetti V, Genazzani AA, Geng J, Genschik P, Gerner L, Gestwicki JE, Gewirtz DA, Ghavami S, Ghigo E, Ghosh D, Giammarioli AM, Giammarioli AM, Giampieri F, Giampietri C, Giatromanolaki A, Gibbings DJ, Gibellini L, Gibson SB, Ginet V, Giordano A, Giordano A, Giorgini F, Giovannetti E, Giovannetti E, Girardin SE, Gispert S, Giuliano S, Giuliano S, Gladson CL, Glavic A, Gleave M, Godefroy N, Gogal RM, Gokulan K, Goldman GH, Goletti D, Goligorsky MS, Gomes AV., Gomes LC, Gomez H, Gomez-Manzano C, Gómez-Sánchez R, Gonçalves DAP, Goncu E, Gong Q, Gongora C, Gonzalez CB, Gonzalez-Alegre P, Gonzalez-Cabo P, Gonzalez-Cabo P, González-Polo RA, Goping IS, Gorbea C, Gorbunov N V., Goring DR, Gorman AM, Gorski SM, Gorski SM, Goruppi S, Goto-Yamada S, Gotor C, Gottlieb RA, Gozes I, Gozuacik D, Graba Y, Graef M, Granato GE, Grant GD, Grant S, Gravina GL, Green DR, Greenhough A, Greenwood MT, Grimaldi B, Gros F, Grose C, Groulx JF, Gruber F, Grumati P, Grumati P, Grune T, Guan JL, Guan KL, Guerra B, Guillen C, Guillen C, Gulshan K, Gunst J, Guo C, Guo L, Guo M, Guo W, Guo XG, Gust AA, Gustafsson ÅB, Gutierrez E, Gutierrez MG, Gwak HS, Haas A, Haber JE, Hadano S, Hagedorn M, Hahn DR, Halayko AJ, Hamacher-Brady A, Hamada K, Hamai A, Hamann A, Hamasaki M, Hamer I, Hamid Q, Hammond EM, Han F, Han W, Handa JT, Hanover JA, Hansen M, Harada M, Harhaji-Trajkovic L, Harper JW, Harrath AH, Harris AL, Harris J, Hasler U, Hasselblatt P, Hasui K, Hawley RG, Hawley TS, He C, He CY, He F, He G, He RR, He XH, He YW, He YY, Heath JK, Hébert MJ, Heinzen RA, Helgason GV, Hensel M, Henske EP, Her C, Herman PK, Hernández A, Hernandez C, Hernández-Tiedra S, Hetz C, Hetz C, Hiesinger PR, Higaki K, Hilfiker S, Hill BG, Hill JA, Hill WD, Hill WD, Hill WD, Hill WD, Hino K, Hofius D, Hofman P, Höglinger GU, Höglinger GU, Höhfeld J, Holz MK, Holz MK, Hong Y, Hood DA, Hoozemans JJM, Hoppe T, Hsu C, Hsu CY, Hsu LC, Hu D, Hu G, Hu HM, Hu H, Hu MC, Hu YC, Hu ZW, Hua F, Hua Y, Huang C, Huang C, Huang HL, Huang KH, Huang KY, Huang S, Huang S, Huang WP, Huang YR, Huang Y, Huang Y, Huber TB, Huber TB, Huber TB, Huebbe P, Huh WK, Hulmi JJ, Hulmi JJ, Hur GM, Hurley JH, Husak Z, Hussain SNA, Hussain SNA, Hussain S, Hwang JJ, Hwang S, Hwang TIS, Ichihara A, Imai Y, Imbriano C, Inomata M, Into T, Iovane V, Iovanna JL, Iozzo R V., Ip NY, Irazoqui JE, Iribarren P, Isaka Y, Isakovic AJ, Ischiropoulos H, Ischiropoulos H, Isenberg JS, Ishaq M, Ishida H, Ishii I, Ishmael JE, Isidoro C, Isobe KI, Isono E, Issazadeh-Navikas S, Itahana K, Itakura E, Ivanov AI, Iyer AK V., Izquierdo JM, Izumi Y, Izzo V, Izzo V, Izzo V, Izzo V, Jäättelä M, Jaber N, Jackson DJ, Jackson WT, Jacob TG, Jacques TS, Jagannath C, Jain A, Jain A, Jana NR, Jang BK, Jani A, Jani A, Janji B, Jannig PR, Jansson PJ, Jean S, Jendrach M, Jeon JH, Jessen N, Jeung EB, Jia K, Jia L, Jiang H, Jiang H, Jiang L, Jiang T, Jiang X, Jiang X, Jiang X, Jiang Y, Jiang Y, Jiang Y, Jiang Y, Jiménez A, Jin C, Jin H, Jin L, Jin M, Jin M, Jin S, Jinwal UK, Jo EK, Johansen T, Johnson DE, Johnson GVW, Johnson JD, Jonasch E, Jones C, Joosten LAB, Jordan J, Joseph AM, Joseph B, Joubert AM, Ju D, Ju J, Juan HF, Juenemann K, Juhász G, Jung HS, Jung JU, Jung YK, Jungbluth H, Jungbluth H, Jungbluth H, Justice MJ, Justice MJ, Jutten B, Kaakoush NO, Kaarniranta K, Kaasik A, Kabuta T, Kaeffer B, Kågedal K, Kahana A, Kajimura S, Kakhlon O, Kalia M, Kalvakolanu D V., Kamada Y, Kambas K, Kaminskyy VO, Kampinga HH, Kandouz M, Kang C, Kang C, Kang R, Kang TC, Kanki T, Kanneganti TD, Kanno H, Kanthasamy AG, Kantorow M, Kaparakis-Liaskos M, Kapuy O, Karantza V, Karim MR, Karmakar P, Kaser A, Kaushik S, Kawula T, Kaynar AM, Kaynar AM, Ke PY, Ke ZJ, Kehrl JH, Keller KE, Kemper JK, Kenworthy AK, Kepp O, Kern A, Kesari S, Kessel D, Ketteler R, Kettelhut I do C, Khambu B, Khan MM, Khandelwal VKM, Khare S, Kiang JG, Kiger AA, Kihara A, Kim AL, Kim CH, Kim DR, Kim DH, Kim EK, Kim EK, Kim HY, Kim HR, Kim JS, Kim JH, Kim JH, Kim JC, Kim JH, Kim JH, Kim KW, Kim MD, Kim MM, Kim PK, Kim SW, Kim SY, Kim YS, Kim Y, Kimchi A, Kimmelman AC, Kimura T, King JS, Kirkegaard K, Kirkin V, Kirshenbaum LA, Kishi S, Kitajima Y, Kitamoto K, Kitaoka Y, Kitazato K, Kley RA, Klimecki WT, Klinkenberg M, Klucken J, Knævelsrud H, Knecht E, Knuppertz L, Ko JL, Kobayashi S, Koch JC, Koechlin-Ramonatxo C, Koenig U, Koh YH, Köhler K, Kohlwein SD, Koike M, Komatsu M, Kominami E, Kong D, Kong HJ, Konstantakou EG, Kopp BT, Korcsmaros T, Korhonen L, Korolchuk VI, Koshkina N V., Kou Y, Koukourakis MI, Koumenis C, Kovács AL, Kovács T, Kovacs WJ, Koya D, Kraft C, Krainc D, Kramer H, Kravic-Stevovic T, Krek W, Kretz-Remy C, Kretz-Remy C, Kretz-Remy C, Krick R, Krishnamurthy M, Kriston-Vizi J, Kroemer G, Kroemer G, Kroemer G, Kroemer G, Kruer MC, Kruger R, Ktistakis NT, Kuchitsu K, Kuhn C, Kumar AP, Kumar A, Kumar A, Kumar D, Kumar D, Kumar R, Kumar S, Kundu M, Kung HJ, Kung HJ, Kuno A, Kuo SH, Kuret J, Kurz T, Kwok T, Kwok T, Kwon TK, Kwon YT, Kyrmizi I, La Spada AR, La Spada AR, Lafont F, Lahm T, Lakkaraju A, Lam T, Lamark T, Lancel S, Landowski TH, Lane DJR, Lane JD, Lanzi C, Lapaquette P, Lapierre LR, Laporte J, Laukkarinen J, Laurie GW, Lavandero S, Lavandero S, Lavie L, Lavoie MJ, Law BYK, Law HKW, Law KB, Layfield R, Lazo PA, Lazo PA, Le Cam L, Le Cam L, Le Cam L, Le Roch KG, Le Stunff H, Le Stunff H, Leardkamolkarn V, Lecuit M, Lee BH, Lee CH, Lee EF, Lee EF, Lee EF, Lee GM, Lee HJ, Lee H, Lee JK, Lee J, Lee JH, Lee JH, Lee JH, Lee M, Lee MS, Lee PJ, Lee SW, Lee SJ, Lee SJ, Lee SY, Lee SH, Lee SS, Lee SS, Lee SJ, Lee S, Lee YR, Lee YJ, Lee YH, Leeuwenburgh C, Lefort S, Legouis R, Lei J, Lei QY, Leib DA, Leibowitz G, Lekli I, Lemaire SD, Lemasters JJ, Lemberg MK, Lemoine A, Leng S, Lenz G, Lenzi P, Lerman LO, Barbato DL, Leu JIJ, Leung HY, Leung HY, Levine B, Levine B, Lewis PA, Lewis PA, Lezoualch F, Li C, Li F, Li FJ, Li J, Li K, Li L, Li M, Li M, Li Q, Li R, Li S, Li W, Li W, Li X, Li Y, Lian J, Liang C, Liang Q, Liao Y, Liberal J, Liberski PP, Lie P, Lieberman AP, Lim HJ, Lim KL, Lim KL, Lim K, Lima RT, Lima RT, Lima RT, Lin CS, Lin CS, Lin CF, Lin F, Lin F, Lin FC, Lin K, Lin KH, Lin PH, Lin T, Lin WW, Lin YS, Lin Y, Linden R, Lindholm D, Lindqvist LM, Lingor P, Linkermann A, Liotta LA, Lipinski MM, Lira VA, Lisanti MP, Liton PB, Liu B, Liu C, Liu CF, Liu F, Liu HJ, Liu J, Liu JJ, Liu JL, Liu K, Liu L, Liu L, Liu Q, Liu RY, Liu S, Liu S, Liu W, Liu X De, Liu X, Liu XH, Liu X, Liu X, Liu X, Liu X, Liu X, Liu Y, Liu, Liu Z, Liu Z, Liuzzi JP, Lizard G, Ljujic M, Lodhi IJ, Logue SE, Lokeshwar BL, Long YC, Lonial S, Loos B, López-Otín C, López-Vicario C, Lorente M, Lorenzi PL, Lorenzi PL, Lõrincz P, Los M, Lotze MT, Lovat PE, Lu B, Lu B, Lu J, Lu Q, Lu SM, Lu S, Lu Y, Luciano F, Luckhart S, Lucocq JM, Ludovico P, Ludovico P, Lugea A, Lukacs NW, Lum JJ, Lum JJ, Lum JJ, Lund AH, Luo H, Luo J, Luo S, Luparello C, Lyons T, Ma J, Ma Y, Ma Y, Ma Z, Machado J, Machado-Santelli GM, Macian F, MacIntosh GC, MacKeigan JP, Macleod KF, MacMicking JD, MacMillan-Crow LA, Madeo F, Madesh M, Madesh M, Madrigal-Matute J, Maeda A, Maeda T, Maegawa G, Maellaro E, Maes H, Magariños M, Maiese K, Maiti TK, Maiuri L, Maiuri MC, Maki CG, Malli R, Malorni W, Malorni W, Maloyan A, Mami-Chouaib F, Man N, Man N, Mancias JD, Mandelkow EM, Mandell MA, Manfredi AA, Manié SN, Manzoni C, Manzoni C, Mao K, Mao K, Mao Z, Mao ZW, Marambaud P, Marconi AM, Marelja Z, Marfe G, Margeta M, Margittai E, Mari M, Mariani FV., Marin C, Marinelli S, Mariño G, Markovic I, Marquez R, Martelli AM, Martens S, Martin KR, Martin SJ, Martin S, Martin-Acebes MA, Martín-Sanz P, Martinand-Mari C, Martinet W, Martinez J, Martinez-Lopez N, Martinez-Outschoorn U, Martínez-Velázquez M, Martinez-Vicente M, Martins WK, Martins WK, Mashima H, Mastrianni JA, Matarese G, Matarese G, Matarrese P, Mateo R, Matoba S, Matsumoto N, Matsushita T, Matsuura A, Matsuzawa T, Mattson MP, Matus S, Matus S, Matus S, Maugeri N, Mauvezin C, Mayer A, Maysinger D, Mazzolini GD, McBrayer MK, McCall K, McCormick C, McInerney GM, McIver SC, McKenna S, McMahon JJ, McNeish IA, Mechta-Grigoriou F, Medema JP, Medina DL, Megyeri K, Mehrpour M, Mehta JL, Mei Y, Meier UC, Meijer AJ, Meléndez A, Melino G, Melino G, Melino S, de Melo EJT, Mena MA, Meneghini MD, Menendez JA, Menezes R, Menezes R, Meng L, Meng LH, Meng S, Menghini R, Menko AS, Menna-Barreto RFS, Menon MB, Meraz-Ríos MA, Merla G, Merlini L, Merlot AM, Meryk A, Meschini S, Meyer JN, Mi MT, Miao CY, Micale L, Michaeli S, Michiels C, Migliaccio AR, Mihailidou AS, Mihailidou AS, Mijaljica D, Mikoshiba K, Milan E, Milan E, Miller-Fleming L, Mills GB, Mills IG, Mills IG, Mills IG, Minakaki G, Minassian BA, Ming XF, Minibayeva F, Minina EA, Mintern JD, Minucci S, Miranda-Vizuete A, Mitchell CH, Miyamoto S, Miyazawa K, Mizushima N, Mnich K, Mograbi B, Mohseni S, Moita LF, Molinari M, Molinari M, Molinari M, Møller AB, Mollereau B, Mollinedo F, Mongillo M, Monick MM, Montagnaro S, Montell C, Montell C, Moore DJ, Moore MN, Mora-Rodriguez R, Moreira PI, Morel E, Morelli MB, Moreno S, Morgan MJ, Moris A, Moriyasu Y, Morrison JL, Morrison LA, Morselli E, Moscat J, Moseley PL, Mostowy S, Motori E, Mottet D, Mottram JC, Moussa CEH, Mpakou VE, Mukhtar H, Levy JMM, Muller S, Muñoz-Moreno R, Muñoz-Pinedo C, Münz C, Murphy ME, Murray JT, Murthy A, Mysorekar IU, Nabi IR, Nabissi M, Nader GA, Nagahara Y, Nagai Y, Nagata K, Nagelkerke A, Nagy P, Naidu SR, Nair S, Nakano H, Nakatogawa H, Nanjundan M, Napolitano G, Naqvi NI, Nardacci R, Narendra DP, Narita M, Nascimbeni AC, Natarajan R, Navegantes LC, Nawrocki ST, Nazarko TY, Nazarko VY, Neill T, Neri LM, Netea MG, Netea-Maier RT, Neves BM, Ney PA, Nezis IP, Nguyen HTT, Nguyen HP, Nicot AS, Nilsen H, Nilsen H, Nilsson P, Nilsson P, Nishimura M, Nishino I, Niso-Santano M, Niu H, Nixon RA, Njar VCO, Noda T, Noegel AA, Nolte EM, Norberg E, Norga KK, Noureini SK, Notomi S, Notterpek L, Nowikovsky K, Nukina N, Nürnberger T, O’donnell VB, O’donovan T, O’dwyer PJ, Oehme I, Oeste CL, Ogawa M, Ogretmen B, Ogura Y, Oh YJ, Ohmuraya M, Ohshima T, Ojha R, Okamoto K, Okazaki T, Oliver FJ, Ollinger K, Olsson S, Orban DP, Orban DP, Ordonez P, Orhon I, Orosz L, O’rourke EJ, Orozco H, Orozco H, Ortega AL, Ortona E, Osellame LD, Oshima J, Oshima S, Osiewacz HD, Otomo T, Otsu K, Ou JHJ, Outeiro TF, Ouyang DY, Ouyang H, Overholtzer M, Ozbun MA, Ozdinler PH, Ozpolat B, Pacelli C, Paganetti P, Page G, Pages G, Pagnini U, Pajak B, Pajak B, Pak SC, Pakos-Zebrucka K, Pakpour N, Palková Z, Palladino F, Pallauf K, Pallet N, Palmieri M, Paludan SR, Palumbo C, Palumbo S, Pampliega O, Pan H, Pan W, Panaretakis T, Pandey A, Pandey A, Pantazopoulou A, Papackova Z, Papademetrio DL, Papassideri I, Papini A, Parajuli N, Pardo J, Parekh V V., Parenti G, Park JI, Park J, Park OK, Parker R, Parlato R, Parlato R, Parys JB, Parzych KR, Parzych KR, Pasquet JM, Pasquier B, Pasumarthi KBS, Patschan D, Patterson C, Pattingre S, Pattingre S, Pattison S, Pause A, Pavenstädt H, Pavone F, Pedrozo Z, Peña FJ, Peñalva MA, Pende M, Peng J, Penna F, Penninger JM, Pensalfini A, Pepe S, Pereira GJS, Pereira PC, de la Cruz VP, Pérez-Pérez ME, Pérez-Rodríguez D, Pérez-Sala D, Perier C, Perl A, Perlmutter DH, Perrotta I, Pervaiz S, Pervaiz S, Pervaiz S, Pesonen M, Pessin JE, Peters GJ, Petersen M, Petrache I, Petrof BJ, Petrovski G, Petrovski G, Petrovski G, Phang JM, Piacentini M, Pierdominici M, Pierre P, Pierre P, Pierre P, Pierre P, Pierrefite-Carle V, Pietrocola F, Pietrocola F, Pietrocola F, Pietrocola F, Pimentel-Muiños FX, Pinar M, Pineda B, Pinkas-Kramarski R, Pinti M, Pinton P, Piperdi B, Piret JM, Platanias LC, Platanias LC, Platta HW, Plowey ED, Pöggeler S, Poirot M, Polčic P, Poletti A, Poon AH, Popelka H, Popova B, Poprawa I, Poulose SM, Poulton J, Powers SK, Powers T, Pozuelo-Rubio M, Prak K, Prange R, Prescott M, Priault M, Prince S, Proia RL, Proikas-Cezanne T, Prokisch H, Promponas VJ, Przyklenk K, Puertollano R, Pugazhenthi S, Puglielli L, Pujol A, Pujol A, Pujol A, Puyal J, Puyal J, Pyeon D, Qi X, Qian W Bin, Qin ZH, Qiu Y, Qu Z, Quadrilatero J, Quinn F, Raben N, Rabinowich H, Radogna F, Ragusa MJ, Rahmani M, Raina K, Ramanadham S, Ramesh R, Rami A, Randall-Demllo S, Randow F, Randow F, Rao H, Rao VA, Rasmussen BB, Rasse TM, Ratovitski EA, Rautou PE, Rautou PE, Rautou PE, Rautou PE, Ray SK, Razani B, Razani B, Reed BH, Reggiori F, Rehm M, Reichert AS, Rein T, Reiner DJ, Reits E, Ren J, Ren X, Renna M, Reusch JEB, Reusch JEB, Revuelta JL, Reyes L, Rezaie AR, Richards RI, Richardson DR, Richetta C, Riehle MA, Rihn BH, Rikihisa Y, Riley BE, Rimbach G, Rippo MR, Ritis K, Rizzi F, Rizzo E, Roach PJ, Robbins J, Roberge M, Roca G, Roccheri MC, Rocha S, Rodrigues CMP, Rodríguez CI, de Cordoba SR, Rodriguez-Muela N, Roelofs J, Rogov VV., Rohn TT, Rohrer B, Romanelli D, Romani L, Romano PS, Roncero MIG, Rosa JL, Rosello A, Rosen KV., Rosen KV., Rosenstiel P, Rost-Roszkowska M, Roth KA, Roué G, Rouis M, Rouschop KM, Ruan DT, Ruano D, Rubinsztein DC, Rucker EB, Rudich A, Rudolf E, Rudolf R, Ruegg MA, Ruiz-Roldan C, Ruparelia AA, Rusmini P, Russ DW, Russo GL, Russo G, Russo R, Rusten TE, Rusten TE, Ryabovol V, Ryan KM, Ryter SW, Sabatini DM, Sacher M, Sacher M, Sachse C, Sack MN, Sadoshima J, Saftig P, Sagi-Eisenberg R, Sahni S, Saikumar P, Saito T, Saitoh T, Sakakura K, Sakoh-Nakatogawa M, Sakuraba Y, Salazar-Roa M, Salomoni P, Saluja AK, Salvaterra PM, Salvioli R, Samali A, Sanchez AMJ, Sánchez-Alcázar JA, Sanchez-Prieto R, Sandri M, Sanjuan MA, Santaguida S, Santambrogio L, Santoni G, Dos Santos CN, Dos Santos CN, Saran S, Sardiello M, Sargent G, Sarkar P, Sarkar S, Sarrias MR, Sarwal MM, Sasakawa C, Sasaki M, Sass M, Sato K, Sato M, Satriano J, Savaraj N, Saveljeva S, Schaefer L, Schaible UE, Scharl M, Schatzl HM, Schekman R, Scheper W, Scheper W, Scheper W, Schiavi A, Schiavi A, Schipper HM, Schipper HM, Schmeisser H, Schmidt J, Schmitz I, Schmitz I, Schneider BE, Schneider EM, Schneider JL, Schon EA, Schönenberger MJ, Schönthal AH, Schorderet DF, Schorderet DF, Schröder B, Schuck S, Schulze RJ, Schwarten M, Schwarz TL, Sciarretta S, Sciarretta S, Sciarretta S, Scotto K, Scovassi AI, Screaton RA, Screaton RA, Screen M, Seca H, Seca H, Seca H, Sedej S, Segatori L, Segatori L, Segev N, Seglen PO, Seguí-Simarro JM, Segura-Aguilar J, Seiliez I, Seki E, Sell C, Semenkovich CF, Semenza GL, Sen U, Serra AL, Serrano-Puebla A, Sesaki H, Setoguchi T, Settembre C, Shacka JJ, Shajahan-Haq AN, Shapiro IM, Sharma S, She H, Shen CKJ, Shen CC, Shen HM, Shen S, Shen W, Sheng R, Sheng X, Sheng ZH, Shepherd TG, Shi J, Shi J, Shi Q, Shi Q, Shi Y, Shibutani S, Shibuya K, Shidoji Y, Shieh JJ, Shih CM, Shimada Y, Shimizu S, Shin DW, Shinohara ML, Shintani M, Shintani T, Shioi T, Shirabe K, Shiri-Sverdlov R, Shirihai O, Shore GC, Shu CW, Shukla D, Sibirny AA, Sibirny AA, Sica V, Sica V, Sica V, Sica V, Sigurdson CJ, Sigurdsson EM, Sijwali PS, Sikorska B, Silveira WA, Silvente-Poirot S, Silverman GA, Simak J, Simmet T, Simon AK, Simon HU, Simone C, Simons M, Simonsen A, Singh R, Singh S V., Singh SK, Sinha D, Sinha S, Sinicrope FA, Sirko A, Sirohi K, Sishi BJN, Sittler A, Siu PM, Sivridis E, Skwarska A, Slack R, Slaninová I, Slavov N, Smaili SS, Smalley KSM, Smith DR, Soenen SJ, Soleimanpour SA, Solhaug A, Somasundaram K, Son JH, Sonawane A, Song C, Song F, Song HK, Song JX, Song W, Soo KY, Sood AK, Sood AK, Soong TW, Soontornniyomkij V, Sorice M, Sotgia F, Soto-Pantoja DR, Sotthibundhu A, Sousa MJ, Spaink HP, Span PN, Spang A, Sparks JD, Speck PG, Spector SA, Spies CD, Springer W, Clair DS, Stacchiotti A, Staels B, Stang MT, Starczynowski DT, Starokadomskyy P, Steegborn C, Steele JW, Stefanis L, Steffan J, Stellrecht CM, Stenmark H, Stepkowski TM, Stern ST, Stevens C, Stockwell BR, Stockwell BR, Stoka V, Storchova Z, Stork B, Stratoulias V, Stravopodis DJ, Stravopodis DJ, Strnad P, Strohecker AM, Ström AL, Stromhaug P, Stulik J, Su YX, Su Z, Subauste CS, Subramaniam S, Sue CM, Suh SW, Sui X, Sukseree S, Sulzer D, Sun FL, Sun J, Sun J, Sun SY, Sun Y, Sun Y, Sun Y, Sundaramoorthy V, Sung J, Suzuki H, Suzuki K, Suzuki N, Suzuki T, Suzuki YJ, Swanson MS, Swanton C, Swärd K, Swarup G, Sweeney ST, Sylvester PW, Szatmari Z, Szegezdi E, Szlosarek PW, Taegtmeyer H, Tafani M, Taillebourg E, Tait SWG, Takacs-Vellai K, Takahashi Y, Takáts S, Takemura G, Takigawa N, Talbot NJ, Tamagno E, Tamburini J, Tan CP, Tan L, Tan ML, Tan ML, Tan M, Tan YJ, Tan YJ, Tanaka K, Tanaka M, Tang D, Tang D, Tang G, Tanida I, Tanji K, Tannous BA, Tapia JA, Tasset-Cuevas I, Tatar M, Tavassoly I, Tavernarakis N, Tavernarakis N, Tavernarakis N, Taylor A, Taylor GS, Taylor GA, Taylor GA, Taylor GA, Taylor GA, Taylor JP, Taylor MJ, Tchetina E V., Tee AR, Teixeira-Clerc F, Teixeira-Clerc F, Telang S, Tencomnao T, Teng BB, Teng RJ, Terro F, Tettamanti G, Theiss AL, Theron AE, Thomas KJ, Thomé MP, Thomes PG, Thorburn A, Thorner J, Thum T, Thumm M, Thurston TLM, Tian L, Till A, Till A, Ting JPY, Ting JPY, Titorenko VI, Toker L, Toldo S, Tooze SA, Topisirovic I, Topisirovic I, Torgersen ML, Torgersen ML, Torgersen ML, Torosantucci L, Torriglia A, Torrisi MR, Tournier C, Towns R, Trajkovic V, Travassos LH, Triola G, Tripathi DN, Trisciuoglio D, Troncoso R, Troncoso R, Trougakos IP, Truttmann AC, Tsai KJ, Tschan MP, Tseng YH, Tsukuba T, Tsung A, Tsvetkov AS, Tu S, Tuan HY, Tucci M, Tumbarello DA, Turk B, Turk V, Turner RFB, Tveita AA, Tyagi SC, Ubukata M, Uchiyama Y, Udelnow A, Ueno T, Umekawa M, Umemiya-Shirafuji R, Underwood BR, Ungermann C, Ureshino RP, Ushioda R, Uversky VN, Uzcátegui NL, Vaccari T, Vaccaro MI, Váchová L, Vakifahmetoglu-Norberg H, Valdor R, Valente EM, Vallette F, Valverde AM, Van den Berghe G, Van Den Bosch L, van den Brink GR, van der Goot FG, van der Klei IJ, van der Laan LJW, van Doorn WG, van Egmond M, van Golen KL, van Golen KL, van Golen KL, Van Kaer L, Campagne M van L, Vandenabeele P, Vandenberghe W, Vandenberghe W, Vanhorebeek I, Varela-Nieto I, Vasconcelos MH, Vasconcelos MH, Vasconcelos MH, Vasko R, Vavvas DG, Vega-Naredo I, Velasco G, Velentzas AD, Velentzas PD, Vellai T, Vellenga E, Vendelbo MH, Venkatachalam K, Ventura N, Ventura N, Ventura S, Veras PST, Verdier M, Vertessy BG, Viale A, Vidal M, Vieira HLA, Vierstra RD, Vigneswaran N, Vij N, Vila M, Vila M, Vila M, Villar M, Villar VH, Villarroya J, Vindis C, Viola G, Viscomi MT, Vitale G, Vogl DT, Voitsekhovskaja O V, von Haefen C, von Schwarzenberg K, Voth DE, Vouret-Craviari V, Vuori K, Vyas JM, Waeber C, Walker CL, Walker MJ, Walter J, Wan L, Wan L, Wan X, Wang B, Wang C, Wang CY, Wang C, Wang C, Wang C, Wang D, Wang F, Wang F, Wang G, Wang HJ, Wang H, Wang HG, Wang H, Wang HD, Wang J, Wang J, Wang M, Wang MQ, Wang PY, Wang P, Wang RC, Wang S, Wang TF, Wang X, Wang XJ, Wang XW, Wang X, Wang X, Wang Y, Wang Y, Wang Y, Wang YJ, Wang Y, Wang Y, Wang YT, Wang Y, Wang ZN, Wappner P, Ward C, Ward DMV, Warnes G, Watada H, Watanabe Y, Watase K, Weaver TE, Weekes CD, Wei J, Weide T, Weihl CC, Weindl G, Weis SN, Wen L, Wen X, Wen X, Wen Y, Wen Y, Westermann B, Weyand CM, White AR, White E, Whitton JL, Whitworth AJ, Wiels J, Wild F, Wildenberg ME, Wileman T, Wilkinson DS, Wilkinson S, Willbold D, Willbold D, Williams C, Williams C, Williams K, Williamson PR, Winklhofer KF, Witkin SS, Wohlgemuth SE, Wollert T, Wolvetang EJ, Wong E, Wong GW, Wong RW, Wong VKW, Woodcock EA, Wright KL, Wu C, Wu D, Wu GS, Wu J, Wu J, Wu M, Wu M, Wu S, Wu WKK, Wu Y, Wu Z, Xavier CPR, Xavier CPR, Xavier RJ, Xia GX, Xia T, Xia W, Xia W, Xia Y, Xiao H, Xiao J, Xiao S, Xiao W, Xie CM, Xie Z, Xie Z, Xilouri M, Xiong Y, Xu C, Xu C, Xu F, Xu H, Xu H, Xu J, Xu J, Xu J, Xu J, Xu L, Xu X, Xu Y, Xu Y, Xu ZX, Xu Z, Xu Z, Xue Y, Yamada T, Yamamoto A, Yamanaka K, Yamashina S, Yamashiro S, Yan B, Yan B, Yan X, Yan Z, Yanagi Y, Yang DS, Yang JM, Yang L, Yang M, Yang PM, Yang P, Yang Q, Yang W, Yang WY, Yang X, Yang Y, Yang Y, Yang Z, Yang Z, Yao MC, Yao PJ, Yao X, Yao Z, Yao Z, Yao Z, Yasui LS, Ye M, Yedvobnick B, Yeganeh B, Yeh ES, Yeyati PL, Yi F, Yi L, Yin XM, Yip CK, Yoo YM, Yoo YH, Yoon SY, Yoshida KI, Yoshimori T, Young KH, Yu H, Yu JJ, Yu JT, Yu J, Yu L, Yu WH, Yu XF, Yu Z, Yuan J, Yuan ZM, Yue BYJT, Yue J, Yue Z, Zacks DN, Zacksenhaus E, Zaffaroni N, Zaglia T, Zakeri Z, Zecchini V, Zeng J, Zeng M, Zeng Q, Zervos AS, Zhang DD, Zhang F, Zhang G, Zhang GC, Zhang H, Zhang H, Zhang H, Zhang H, Zhang H, Zhang J, Zhang J, Zhang J, Zhang J, Zhang JP, Zhang L, Zhang L, Zhang L, Zhang L, Zhang L, Zhang MY, Zhang X, Zhang XD, Zhang Y, Zhang Y, Zhang Y, Zhang Y, Zhang Y, Zhang Y, Zhao M, Zhao WL, Zhao WL, Zhao WL, Zhao X, Zhao YG, Zhao Y, Zhao Y, Zhao YX, Zhao Z, Zhao ZJ, Zheng D, Zheng XL, Zheng X, Zhivotovsky B, Zhivotovsky B, Zhong Q, Zhong Q, Zhou GZ, Zhou G, Zhou H, Zhou SF, Zhou XJ, Zhou XJ, Zhou XJ, Zhu H, Zhu H, Zhu WG, Zhu W, Zhu XF, Zhu Y, Zhuang SM, Zhuang X, Ziparo E, Zois CE, Zoladek T, Zong WX, Zorzano A, Zorzano A, Zorzano A & Zughaier SM (2016) Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Denton D, Nicolson S & Kumar S (2012) Cell death by autophagy: Facts and apparent artefacts. Cell Death Differ. 19, 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Doherty J & Baehrecke EH (2018) Life, death and autophagy. Nat. Cell Biol 20, 1110–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kroemer G & Levine B (2008) Autophagic cell death: the story of a misnomer. Nat. Rev. Mol. Cell Biol 9, 1004–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lee C-Y, Cooksey BAK & Baehrecke EH (2002) Steroid Regulation of Midgut Cell Death during Drosophila Development. Dev. Biol 250, 101–111. [DOI] [PubMed] [Google Scholar]
- 102.Wang H, Lu Q, Cheng S, Wang X & Zhang H (2013) Autophagy activity contributes to programmed cell death in Caenorhabditis elegans. Autophagy 9, 1975–1982. [DOI] [PubMed] [Google Scholar]
- 103.Mesquita A, Cardenal-Muñoz E, Dominguez E, Muñoz-Braceras S, Nuñez-Corcuera B, Phillips BA, Tábara LC, Xiong Q, Coria R, Eichinger L, Golstein P, King JS, Soldati T, Vincent O & Escalante R (2017) Autophagy in Dictyostelium: Mechanisms, regulation and disease in a simple biomedical model. Autophagy 13, 24–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Liu Y, Shoji-Kawata S, Sumpter RM, Wei Y, Ginet V, Zhang L, Posner B, Tran KA, Green DR, Xavier RJ, Shaw SY, Clarke PGH, Puyal J & Levine B (2013) Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl. Acad. Sci 110, 20364–20371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Liu Y & Levine B (2015) Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ. 22, 367–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kriel J & Loos B (2019) The good, the bad and the autophagosome: exploring unanswered questions of autophagy-dependent cell death. Cell Death Differ. 26, 640–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB & Tsujimoto Y (2004) Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell Biol 6, 1221–1228. [DOI] [PubMed] [Google Scholar]
- 108.Arakawa S, Tsujioka M, Yoshida T, Tajima-Sakurai H, Nishida Y, Matsuoka Y, Yoshino I, Tsujimoto Y & Shimizu S (2017) Role of Atg5-dependent cell death in the embryonic development of Bax/Bak double-knockout mice. Cell Death Differ. 24, 1598–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shen S, Kepp O, Michaud M, Martins I, Minoux H, Métivier D, Maiuri MC, Kroemer RT & Kroemer G (2011) Association and dissociation of autophagy, apoptosis and necrosis by systematic chemical study. Oncogene 30, 4544–4556. [DOI] [PubMed] [Google Scholar]
- 110.Chang TK, Shravage BV., Hayes SD, Powers CM, Simin RT, Wade Harper J & Baehrecke EH (2013) Uba1 functions in Atg7- and Atg3-independent autophagy. Nat. Cell Biol 15, 1067–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fulda S & Kögel D (2015) Cell death by autophagy: Emerging molecular mechanisms and implications for cancer therapy. Oncogene 34, 5105–5113. [DOI] [PubMed] [Google Scholar]
- 112.Towers CG, Wodetzki D & Thorburn A (2019) Autophagy and cancer: Modulation of cell death pathways and cancer cell adaptations. J. Cell Biol 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shi J, Gao W & Shao F (2017) Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci 42, 245–254. [DOI] [PubMed] [Google Scholar]
- 114.Yuan J, Najafov A & Py BF (2016) Roles of Caspases in Necrotic Cell Death. Cell 167, 1693–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ruan J, Xia S, Liu X, Lieberman J & Wu H (2018) Cryo-EM structure of the gasdermin A3 membrane pore. Nature 557, 62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Monteleone M, Stanley AC, Chen KW, Brown DL, Bezbradica JS, von Pein JB, Holley CL, Boucher D, Shakespear MR, Kapetanovic R, Rolfes V, Sweet MJ, Stow JL & Schroder K (2018) Interleukin-1β Maturation Triggers Its Relocation to the Plasma Membrane for Gasdermin-D-Dependent and -Independent Secretion. Cell Rep. 24, 1425–1433. [DOI] [PubMed] [Google Scholar]
- 117.Wang Y, Gao W, Shi X, Ding J, Liu W, He H, Wang K & Shao F (2017) Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547, 99–103. [DOI] [PubMed] [Google Scholar]
- 118.Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G & Alnemri ES (2017) Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun 8, 14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yokomizo K, Harada Y, Kijima K, Shinmura K, Sakata M, Sakuraba K, Kitamura Y, Shirahata A, Goto T, Mizukami H, Saito M, Kigawa G, Nemoto H & Hibi K (2012) Methylation of the DFNA5 gene is frequently detected in colorectal cancer. Anticancer Res. 32, 1319–1322. [PubMed] [Google Scholar]
- 120.Kim MS, Lebron C, Nagpal JK, Chae YK, Chang X, Huang Y, Chuang T, Yamashita K, Trink B, Ratovitski EA, Califano JA & Sidransky D (2008) Methylation of the DFNA5 increases risk of lymph node metastasis in human breast cancer. Biochem. Biophys. Res. Commun 370, 38–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Akino K, Toyota M, Suzuki H, Imai T, Maruyama R, Kusano M, Nishikawa N, Watanabe Y, Sasaki Y, Abe T, Yamamoto E, Tarasawa I, Sonoda T, Mori M, Imai K, Shinomura Y & Tokino T (2007) Identification of DFNA5 as a target of epigenetic inactivation in gastric cancer. Cancer Sci. 98, 88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, Rongvaux A, Bunnell SC, Shao F, Green DR & Poltorak A (2018) Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl. Acad. Sci 115, E10888–E10897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, Brooks A, Xia S, Wu H, Kelliher MA, Berger SB, Gough PJ, Bertin J, Proulx MM, Goguen JD, Kayagaki N, Fitzgerald KA & Lien E (2018) Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 362, 1064–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Biasutto L, Azzolini M, Szabò I & Zoratti M (2016) The mitochondrial permeability transition pore in AD 2016: An update. Biochim. Biophys. Acta - Mol. Cell Res 1863, 2515–2530. [DOI] [PubMed] [Google Scholar]
- 125.Bernardi P, Rasola A, Forte M & Lippe G (2015) The Mitochondrial Permeability Transition Pore: Channel Formation by F-ATP Synthase, Integration in Signal Transduction, and Role in Pathophysiology. Physiol. Rev 95, 1111–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Izzo V, Bravo-San Pedro JM, Sica V, Kroemer G & Galluzzi L (2016) Mitochondrial Permeability Transition: New Findings and Persisting Uncertainties. Trends Cell Biol. 26, 655–667. [DOI] [PubMed] [Google Scholar]
- 127.Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park H-A, Licznerski P, Li H, Nabili P, Hockensmith K, Graham M, Porter GA & Jonas EA (2014) An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc. Natl. Acad. Sci 111, 10580–10585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabó I, Lippe G & Bernardi P (2013) Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. U. S. A 110, 5887–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chinopoulos C (2018) Mitochondrial permeability transition pore: Back to the drawing board. Neurochem. Int 117, 49–54. [DOI] [PubMed] [Google Scholar]
- 130.Urbani A, Giorgio V, Carrer A, Franchin C, Arrigoni G, Jiko C, Abe K, Maeda S, Shinzawa-Itoh K, Bogers JFM, McMillan DGG, Gerle C, Szabò I & Bernardi P (2019) Purified F-ATP synthase forms a Ca2+-dependent high-conductance channel matching the mitochondrial permeability transition pore. Nat. Commun 10, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mnatsakanyan N, Llaguno MC, Yang Y, Yan Y, Weber J, Sigworth FJ & Jonas EA (2019) A mitochondrial megachannel resides in monomeric F1FO ATP synthase. Nat. Commun 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Davies KM, Anselmi C, Wittig I, Faraldo-Gómez JD & Kühlbrandt W (2012) Structure of the yeast F 1F o-ATP synthase dimer and its role in shaping the mitochondrial cristae. Proc. Natl. Acad. Sci. U. S. A 109, 13602–13607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Marzo I, Brenner C, Zamzami N, Jürgensmeier JM, Susin SA, Vieira HL, Prévost MC, Xie Z, Matsuyama S, Reed JC & Kroemer G (1998) Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science 281, 2027–31. [DOI] [PubMed] [Google Scholar]
- 134.Tsujimoto Y, Shimizu S, Narita M & Tsujimoto Y (1999) Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 399, 483–487. [DOI] [PubMed] [Google Scholar]
- 135.Whelan RS, Konstantinidis K, Wei A-C, Chen Y, Reyna DE, Jha S, Yang Y, Calvert JW, Lindsten T, Thompson CB, Crow MT, Gavathiotis E, Dorn GW, O’Rourke B & Kitsis RN (2012) Bax regulates primary necrosis through mitochondrial dynamics. Proc. Natl. Acad. Sci. U. S. A [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Karch J, Kwong JQ, Burr AR, Sargent MA, Elrod JW, Peixoto PM, Martinez-Caballero S, Osinska H, Cheng EH-Y, Robbins J, Kinnally KW & Molkentin JD (2013) Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. Elife 2, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chen Y, Aon MA, Hsu Y-T, Soane L, Teng X, McCaffery JM, Cheng W-C, Qi B, Li H, Alavian KN, Dayhoff-Brannigan M, Zou S, Pineda FJ, O’Rourke B, Ko YH, Pedersen PL, Kaczmarek LK, Jonas EA & Hardwick JM (2011) Bcl-x L regulates mitochondrial energetics by stabilizing the inner membrane potential. J. Cell Biol 195, 263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Alavian KN, Li H, Collis L, Bonanni L, Zeng L, Sacchetti S, Lazrove E, Nabili P, Flaherty B, Graham M, Chen Y, Messerli SM, Mariggio MA, Rahner C, McNay E, Shore GC, Smith PJS, Hardwick JM & Jonas EA (2011) Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat. Cell Biol 13, 1224–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Vander Heiden MG, Li XX, Gottleib E, Hill RB, Thompson CB & Colombini M (2001) Bcl-x l Promotes the Open Configuration of the Voltage-dependent Anion Channel and Metabolite Passage through the Outer Mitochondrial Membrane. J. Biol. Chem 276, 19414–19419. [DOI] [PubMed] [Google Scholar]
- 140.Vander Heiden MG, Chandel NS, Schumacker PT & Thompson CB (1999) Bcl-xL Prevents Cell Death following Growth Factor Withdrawal by Facilitating Mitochondrial ATP/ADP Exchange. Mol. Cell 3, 159–167. [DOI] [PubMed] [Google Scholar]
- 141.Chen Q, Xu H, Xu A, Ross T, Bowler E, Hu Y & Lesnefsky EJ (2015) Inhibition of Bcl-2 Sensitizes Mitochondrial Permeability Transition Pore (MPTP) Opening in Ischemia-Damaged Mitochondria. PLoS One 10, e0118834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yu T, Chen C, Sun Y, Sun H, Li T, Meng J & Shi X (2015) ABT-737 sensitizes curcumin-induced anti-melanoma cell activity through facilitating mPTP death pathway. Biochem. Biophys. Res. Commun 464, 286–291. [DOI] [PubMed] [Google Scholar]
- 143.Zamzami N, El Hamel C, Maisse C, Brenner C, Muñoz-Pinedo C, Belzacq AS, Costantini P, Vieira H, Loeffler M, Molle G & Kroemer G (2000) Bid acts on the permeability transition pore complex to induce apoptosis. Oncogene 19, 6342–50. [DOI] [PubMed] [Google Scholar]
- 144.Oakes SA, Scorrano L, Opferman JT, Bassik MC, Nishino M, Pozzan T & Korsmeyer SJ (2005) Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc. Natl. Acad. Sci. U. S. A 102, 105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Palmer AE, Jin C, Reed JC & Tsien RY (2004) Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc. Natl. Acad. Sci. U. S. A 101, 17404–17409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Schulman JJ, Wright FA, Kaufmann T & Wojcikiewicz RJH (2013) The Bcl-2 protein family member Bok binds to the coupling domain of inositol 1,4,5-trisphosphate receptors and protects them from proteolytic cleavage. J. Biol. Chem 288, 25340–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Carpio MA, Michaud M, Zhou W, Fisher JK, Walensky LD & Katz SG (2015) BCL-2 family member BOK promotes apoptosis in response to endoplasmic reticulum stress. Proc. Natl. Acad. Sci 112, 7201–7206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Dixon SJ & Stockwell BR (2019) The Hallmarks of Ferroptosis. Annu. Rev. Cancer Biol 3, 35–54. [Google Scholar]
- 149.Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R & Tang D (2016) Ferroptosis: process and function. Cell Death Differ. 23, 369–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB & Jiang X (2018) Role of Mitochondria in Ferroptosis. Mol. Cell 73, 354–363.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Magtanong L, Ko P-J, To M, Cao JY, Forcina GC, Tarangelo A, Ward CC, Cho K, Patti GJ, Nomura DK, Olzmann JA & Dixon SJ (2019) Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem. Biol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Roh JL, Kim EH, Jang HJ, Park JY & Shin D (2016) Induction of ferroptotic cell death for overcoming cisplatin resistance of head and neck cancer. Cancer Lett. 381, 96–103. [DOI] [PubMed] [Google Scholar]
- 153.Yu Y, Xie Y, Cao L, Yang L, Yang M, Lotze MT, Zeh HJ, Kang R & Tang D (2015) The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol. Cell. Oncol 2, e1054549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Huo H, Zhou Z, Qin J, Liu W, Wang B & Gu Y (2016) Erastin Disrupts Mitochondrial Permeability Transition Pore (mPTP) and Induces Apoptotic Death of Colorectal Cancer Cells. PLoS One 11, e0154605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Neitemeier S, Jelinek A, Laino V, Hoffmann L, Eisenbach I, Eying R, Ganjam GK, Dolga AM, Oppermann S & Culmsee C (2017) BID links ferroptosis to mitochondrial cell death pathways. Redox Biol. 12, 558–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Jiang L, Kon N, Li T, Wang S-J, Su T, Hibshoosh H, Baer R & Gu W (2015) Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Aubrey BJ, Kelly GL, Janic A, Herold MJ & Strasser A (2018) How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 25, 104–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ou Y, Wang SJ, Li D, Chu B & Gu W (2016) Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc. Natl. Acad. Sci. U. S. A 113, E6806–E6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Murphy ME (2016) Ironing out how p53 regulates ferroptosis. Proc. Natl. Acad. Sci. U. S. A 113, 12350–12352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hong SH, Lee D-H, Lee Y-S, Jo MJ, Jeong YA, Kwon WT, Choudry HA, Bartlett DL & Lee YJ (2017) Molecular crosstalk between ferroptosis and apoptosis: emerging role of ER stress-induced p53-independent PUMA expression. Oncotarget 8, 115164–115178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lee Y-S, Lee D-H, Choudry HA, Bartlett DL & Lee YJ (2018) Ferroptosis-Induced Endoplasmic Reticulum Stress: Cross-talk between Ferroptosis and Apoptosis. Mol. Cancer Res 16, 1073–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wang GX, Tu H-C, Dong Y, Skanderup AJ, Wang Y, Takeda S, Ganesan YT, Han S, Liu H, Hsieh JJ & Cheng EH (2017) ΔNp63 Inhibits Oxidative Stress-Induced Cell Death, Including Ferroptosis, and Cooperates with the BCL-2 Family to Promote Clonogenic Survival. Cell Rep. 21, 2926–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Chauhan D, Bartok E, Gaidt MM, Bock FJ, Herrmann J, Seeger JM, Broz P, Beckmann R, Kashkar H, Tait SWG, Müller R & Hornung V (2018) BAX/BAK-Induced Apoptosis Results in Caspase-8-Dependent IL-1β Maturation in Macrophages. Cell Rep. 25, 2354–2368.e5. [DOI] [PubMed] [Google Scholar]
- 164.Vince JE, De Nardo D, Gao W, Vince AJ, Hall C, McArthur K, Simpson D, Vijayaraj S, Lindqvist LM, Bouillet P, Rizzacasa MA, Man SM, Silke J, Masters SL, Lessene G, Huang DCS, Gray DHD, Kile BT, Shao F & Lawlor KE (2018) The Mitochondrial Apoptotic Effectors BAX/BAK Activate Caspase-3 and −7 to Trigger NLRP3 Inflammasome and Caspase-8 Driven IL-1β Activation. Cell Rep. 25, 2339–2353.e4. [DOI] [PubMed] [Google Scholar]