

Summary
Centrioles comprise the heart of centrosomes, microtubule-organizing centers. To study the function of centrioles in lung and gut development, we genetically disrupted centrioles throughout the mouse endoderm. Surprisingly, removing centrioles from endoderm did not disrupt intestinal growth or development, but blocked lung branching. In the lung, acentriolar SOX2-expressing airway epithelial cells apoptosed. Loss of centrioles activated p53, and removing p53 restored survival of SOX2-expressing cells, lung branching and mouse viability. To investigate how endodermal p53 activation specifically killed acentriolar SOX2-expressing cells, we assessed ERK, a pro-survival cue. ERK was active throughout the intestine and in the distal lung buds, correlating with tolerance to centriole loss. Pharmacologically inhibiting ERK activated apoptosis in acentriolar cells, revealing that ERK activity protects acentriolar cells from apoptosis. Therefore, centrioles are largely dispensable for endodermal growth and the spatial distribution of ERK activity in the endoderm shapes the developmental consequences of centriolar defects and p53 activation.
Keywords: centriole, endoderm, lung branching, intestine development, ERK, p53, apoptosis
Graphical Abstract
eTOC Blurb
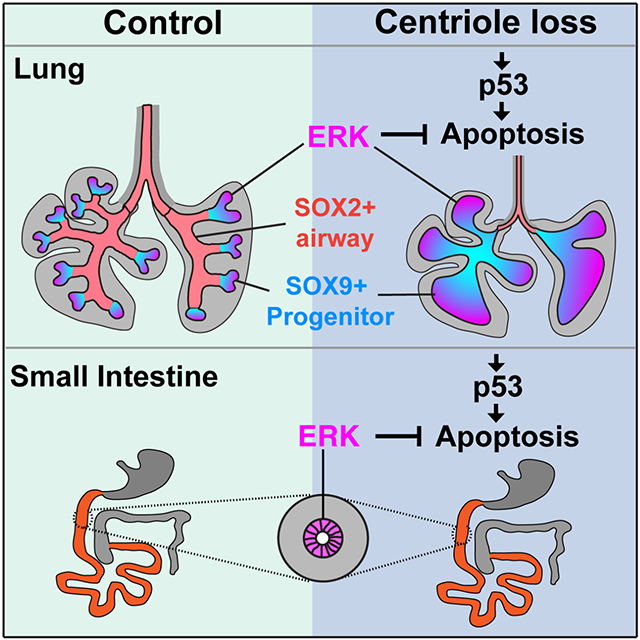
Chang et al. show that endoderm centrioles are dispensable for intestinal development but essential for lung branching. Throughout both intestine and lung loss of centrioles activates p53, but the lack of centrioles induces apoptosis only in domains of low ERK signaling.
Introduction
Each centriole comprises nine triplet microtubules arranged in a cylinder. Two centrioles and the pericentriolar materials make up the centrosome, the principal microtubule-organizing center in mammalian cells. Centrioles are at the heart of the mitotic poles during cell division and, in their roles as basal bodies, act as the cilium's foundation.
Much like chromosomes, centrioles are duplicated once and only once per cell cycle. Cenpj encodes SAS-4, also called CPAP, a core component of centrioles essential for centriole duplication (Gonczy, 2012). Human mutations in CENPJ cause microcephaly, a defect in brain growth, or Seckel syndrome, a general growth deficit (Al-Dosari et al., 2010; Bond et al., 2005). Consistent with the critical functions of centrioles in development, mouse embryos without CENPJ die at mid-gastrulation (Bazzi and Anderson, 2014). Pharmacologically inhibiting another protein required for centriole duplication, PLK4, causes non-cancer cells to undergo G1 arrest (Wong et al., 2015), suggesting that centrioles are broadly essential for mammalian cell division.
Loss of centrioles activates p53 (TP53), a tumor suppressor that interprets various cell stresses (Bazzi and Anderson, 2014; Kastenhuber and Lowe, 2017; Wong et al., 2015). One direct transcriptional target of p53 is p21 (CDKN1A), a cyclin-dependent kinase (CDK) inhibitor that induces cell-cycle arrest (el-Deiry et al., 1993; Harper et al., 1993). In cancer, p53 and p21 can be antagonized by activation of extracellular signal-regulated kinase (ERK) (Yang et al., 2017).
A diverse range of developmental syndromes activate p53, implicating increased p53 activity in the etiology of birth defects (Bowen and Attardi, 2019; Van Nostrand et al., 2014). In support of this possibility, activating mutations in p53 or attenuating a negative regulator of p53 cause developmental phenotypes in humans (Lessel et al., 2017; Toki et al., 2018). Moreover, deletion of p53 can partly or fully rescue defects in mouse genetic models of developmental syndromes (Bowen et al., 2019). For example, deletion of p53 reduces the increased cell death observed in Cenpj null embryos (Bazzi and Anderson, 2014).
The neural epithelium undergoes dramatic expansion during mid-gastrulation and is particularly dependent on CENPJ function (Bazzi and Anderson, 2014), suggesting that actively dividing cells are especially susceptible to centriole loss. Like the neural epithelium, the endodermal epithelium undergoes dramatic expansion during embryonic development (Zorn and Wells, 2009). We hypothesized that centrioles would, like for the neural epithelium, be critical for endodermal epithelial development and, consequently, the development of the alimentary tract and lungs.
To test this hypothesis, we removed Cenpj throughout the endoderm. Surprisingly, loss of centrioles had minimal effects on the growth and development of the small intestine. In contrast, in the lung, loss of centrioles severely attenuated lung branching. Within the developing acentriolar lungs, SOX9-expressing progenitors survived and expanded but failed to give rise to SOX2-expressing airway cells. We found that in the developing intestine ERK activity is broadly active but in the developing lung, ERK activity is restricted to the distal tip of lung buds where the SOX9-expressing reside. Pharmacological inhibition revealed that ERK is critical for the survival of SOX9-expressing lung progenitors and intestinal cells lacking centrioles. These results indicate that centrioles are differentially required for distinct endodermal lineages, and that differential ERK activity underlies the differences in phenotypic consequences caused by loss of centrioles.
Results
Epithelial centrioles are essential for lung branching but dispensable for intestinal development
To test whether centrioles are essential for the proliferation of quickly growing cells such as the endodermal epithelium, we used ShhCre to conditionally delete Cenpj, the ortholog of C. elegans sas-4. Centrioles were identified by staining for the centriolar protein FOP (FGFR1OP) and epithelia were identified by staining for E-cadherin (CDH1). Centrioles were present in mesenchymal and epithelial cells in control (ShhCre Cenpj+/lox) lungs and intestines but absent specifically from the endodermal epithelia of mutant (ShhCre Cenpj−/lox) lung and intestine (Figures 1A and 1B), indicating that CENPJ is essential for centriole biogenesis in the endoderm.
Figure 1. Endodermal centrioles are essential for lung branching, but not intestinal development.

(A, B) Immunostaining of E11.5 control (ShhCre Cenpj+/lox) and Cenpj loss of function (ShhCre Cenpj−/lox) lung (A) and small intestine (B) sections for centrioles (FOP, magenta), epithelium (CDH1, cyan) and nuclei (Hoechst, gray). Scale bar, 10 μm.
(C, D) Transmission electron microscopy of E16.5 control (ShhCre Cenpj+/lox) and Cenpj loss of function (ShhCre Cenpj−/lox) lung (A) and small intestine (B) epithelium. Other images from these serially sectioned lung cells are included in supplemental Figure S1 and Figure S2. No centrioles were detected in ShhCre Cenpj−/lox lung and intestinal epithelial cells. Scale bar, 2 μm.
(E) Gross appearance of the lung and intestine from the control (ShhCre Cenpj+/lox Rosa26tdTomato) and Cenpj loss of function (ShhCre Cenpj−/lox Rosa26tdTomato) embryos at E16.5.
(F) Statistics of the length of control (CTL, ShhCre Cenpj+/lox) and Cenpj loss of function (cKO, ShhCre Cenpj−/lox) small intestine. # denotes statical insignificance, p=0.1292, n=5 for CTL and n=4 for cKO.
(G) Wholemount immunostaining of control (ShhCre Cenpj+/lox) and Cenpj loss of function (ShhCre Cenpj−/lox) lungs for CDH1 at E11.5, E12.5, E13.5, E14.5 and E18.5. Dashed lines outline the major lobes including right cranial (RCr), right middle (RMd), right accessory (RAc), right caudal (RCd), and left (L) lobes. Scale bars, 1 mm.
(H) Measurement of EdU incorporation into replicating DNA of E12.5 control (ShhCre Cenpj+/lox) and Cenpj loss of function (ShhCre Cenpj−/lox) lung epithelium. # denotes statistical insignificance, p=0.1648. n=9, 11, 16 and 8 respectively.
To confirm that we had successfully removed centrioles from the endoderm, we used serial-section transmission electron microscopy (TEM). Control lung and intestinal epithelial cells contained centrioles, recognized by their characteristic 9-fold symmetry (Figures 1C and 1D). In contrast, ShhCre Cenpj−/lox lung and intestinal epithelial cells possessed no centrioles (Figures 1C and 1D and Figures S1 and S2). ShhCre induced recombination of the tdTomato reporter throughout the endoderm (Figure 1E), suggesting that the endoderm of ShhCre Cenpj−/lox embryos is acentriolar.
At E16.5, the gross appearance and length of ShhCre Cenpj−/lox small intestine were indistinguishable from those of controls (Figures 1E and 1F). In marked contrast, ShhCre Cenpj−/lox lung development was dramatically perturbed. Thus, endodermal centrioles are essential for lung development, but dispensable for intestine development.
During embryogenesis, the lung epithelium branches and ramifies into its surrounding mesenchyme (Metzger et al., 2008). Wholemount E11.5 lungs stained for CDH1 revealed that both control and mutant mouse embryos possessed four major lobes on the right side [right cranial (RCr), right middle (RMd), right accessory (RAc), right caudal (RCd)] and three main secondary branches on the left lobe (L1-L3). At E12.5, the major lobes of mutant lungs had failed to branch (Figure 1G).
Lungs grow throughout embryogenesis. Measuring EdU incorporation revealed that cell cycle engagement in the lung epithelium does not depend on centrioles and ShhCre Cenpj−/lox lungs grow throughout all lung developmental stages till birth (Figure 1H). Mutant pups died at birth, attributable to disrupted lung development. Therefore, endodermal centrioles are not essential for proliferation but are critical for lung branching.
Development of lung SOX2-expressing cells depends on CENPJ.
The embryonic lung epithelium is comprised of two main populations, the SOX2-expressing proximal airway epithelium (trachea, bronchi and bronchioles) and SOX9-expressing progenitors at the distal bud tips. During the pseudoglandular stage (E12-E16.5 in mouse), the lung distal tip cells proliferate rapidly to give rise to both the repetitively branching outgrowths and the proximal SOX2-expressing bronchiolar epithelium that form the conducting airways of the lung (Alanis et al., 2014; Rawlins et al., 2009).
To analyze cell differentiation during lung branching morphogenesis, we stained E12.5 to E15.5 wholemount lungs for SOX9 and SOX2. During control lung development, SOX9-expressing cells remained restricted to ramifying distal tips, and there was a concomitant increase in SOX2-expressing bronchioles (Figure 2A, upper panel). ShhCre Cenpj−/lox lungs developed major lobes, comparable to control lungs. However, in mutant lungs, SOX9-expressing cells expanded and, consistent with the absence of branching, were not constrained to distal tips (Figure 2A).
Figure 2. Centrioles are required for development of SOX2-expressing lung epithelium.

(A) Wholemount immunostaining of E12.5, E13.5, E14.5 and E15.5 control (ShhCre Cenpj+/lox) and Cenpj loss of function (ShhCre Cenpj−/lox) lungs for SOX2 (magenta), SOX9 (cyan). Schematic (right) illustrates the expression of SOX2 in the trachea, bronchi and bronchioles, and of SOX9 in the tracheal rings and distal tip progenitors of lung. Scale bars,1 mm.
(B) Quantification of the percentage of epithelium expressing SOX2. Maximum projection images of wholemount staining from (A) were analyzed. n=3.
(C) Immunostaining of CDH1 in frozen sections of E14.5 control (ShhCre Cenpj+/lox) and Cenpj loss of function (ShhCre Cenpj−/lox) lungs. Images are of section through the trachea and esophagus. White dashed lines outline the trachea. Scale bar, 100 μm.
(D) Immunostaining of E13.5 control (ShhCre Ift88+/lox) and Ift88 loss of function (ShhCre Ift88−/lox) lung sections for cilia (ARL13B, yellow), centrosomes (γTubulin, γTUB, magenta), epithelium (CDH1, cyan) and nuclei (Hoechst, gray). Scale bar, 10 μm.
(E) Wholemount immunostaining of E13.5 and E16.5 control (ShhCre Ift88+/lox) and Ift88 loss of function (ShhCre Ift88−/lox) lungs for CDH1. Dashed lines outline the major lobes. Scale bar, 1 mm.
(F) Wholemount immunostaining of E12.5 control (Sox2lox/lox) and Sox2 loss of function (ShhCre Sox2lox/lox) lungs for SOX2 (magenta), SOX9 (cyan) and epithelium (CDH1, yellow). Scale bar, 1 mm.
In addition to SOX9, distal epithelial cells express NKX2-1 (also known as TTF1) and SFTPC (Beers et al., 1992; Lazzaro et al., 1991). We examined their expression and found that ShhCre Cenpj−/lox lung epithelia expressed both (Figures S3A-C). These findings confirm that centrioles are not essential for the distal epithelial lung fate.
Early (E12.5) in lung development, mutant lungs contained fewer SOX2-expressing cells than control lungs, and mutant lungs were devoid of SOX2-expressing cells by E14.5 (Figure 2A). Measurement of the percentage of SOX2-expressing cells in the epithelium showed a gradual reduction from E12.5 and total loss of SOX2-expressing cells by E14.5 (Figure 2B). The absence of CDH1 at the trachea/bronchi confirmed the loss of SOX2-expressing epithelium by E14.5 (Figure 2C). Thus, CENPJ is essential for distal SOX9-expressing progenitors to give rise to SOX2-expressing bronchiolar epithelial cells.
Endodermal cilia are dispensable for lung branching morphogenesis
One critical function of centrioles is to support formation of the primary cilium, a cellular antenna important for lung development (Abdelhamed et al., 2015; Dyson et al., 2017). Primary cilia, identified by staining for the ciliary component ARL13B, were associated with centrosomes, identified by staining for γTubulin (γTUB) (Figure 2D). Both lung mesenchymal and epithelial cells possessed cilia during development (Figure 2D).
To investigate whether the role of centrioles in lung branching is mediated through ciliogenesis, we investigated whether lung branching depends on cilia. We removed IFT88, a component of intraflagellar transport machinery essential for ciliogenesis and ciliary maintenance (Davenport et al., 2007), throughout the endoderm by generating ShhCre Ift88−/lox embryos. In the ShhCre Ift88−/lox lungs, As expected, ShhCre Ift88−/lox lungs possessed cilia on their mesenchymal cells but not on their epithelial cells, indicating that removing IFT88 from the endoderm abrogates ciliogenesis in the developing lung epithelium (Figure 2D).
Wholemount staining of E13.5 and E16.5 control and ShhCre Ift88−/lox lungs revealed that branching was indistinguishable (Figure 2E). Impressively, ShhCre Ift88−/lox mice were viable through adulthood, indicating that endodermal cilia are not essential for lung function. Interestingly, limb digits 4 and 5 of ShhCre Ift88−/lox mice were atrophied (Figures S4B and S4D). As digits 4 and 5 are generated from the domain of Shh expression, the zone of polarizing activity (ZPA) (Ahn and Joyner, 2004), cilia transduce Hedgehog signals within the ZPA. As endodermal cilia are dispensable for lung branching, we conclude that centrioles function in lung branching independent of their role in ciliogenesis.
SOX2 is dispensable for lung branching morphogenesis
Deletion of SOX2 in part of the endoderm disrupts lung development (Que et al., 2007). Therefore, we hypothesized that the abrogation of SOX2 expression in ShhCre Cenpj−/lox lungs blocked branching in acentriolar lungs. To test this hypothesis, we removed SOX2 throughout the endoderm by generating ShhCre Sox2lox/lox embryos.
We stained wholemount lungs from ShhCre Sox2lox/lox and littermate control embryos for SOX2, SOX9 and CDH1. As expected, SOX2 was expressed in the developing trachea, bronchi and bronchioles of control lungs and was absent from ShhCre Sox2lox/lox lungs (Figure 2F). CDH1 and SOX9 staining revealed that lung branching was unaffected in ShhCre Sox2lox/lox lungs (Figure 2F). Thus, compromised expression of SOX2 does not underlie the lung branching defect caused by loss of CENPJ.
In addition to SOX2, bronchiolar epithelial cells express the transcription factor p73 (Marshall et al., 2016; Nemajerova et al., 2016). As expected, p73 and SOX2 were co-expressed in the bronchiolar epithelium of control E16.5 lungs (Figure S3E and S3F). In contrast, ShhCre Cenpj−/lox lungs were devoid of p73-expressing cells (Figure S3E and S3F). These results confirm that CENPJ is required for bronchiolar epithelial formation, and raise the possibility that SOX2 and p73-expressing cells critically depend on centrioles.
Loss of CENPJ in the endoderm activates p53 and apoptosis
As neither the absence of cilia nor loss of SOX2 function could account for how centriole loss disrupts lung branching, we analyzed the transcriptomes of control and ShhCre Cenpj−/lox lungs using RNAseq. To identify gene expression changes caused by loss of centrioles and not cilia, we compared RNAseq transcriptomes of control (ShhCre Cenpj+/lox), cilia-disrupted (ShhCre Ift88−/lox) and centriole-disrupted (ShhCre Cenpj−/lox) lungs at E11.5, a timepoint prior to the emergence of a branching defect in acentriolar lungs (Table S1).
Genes differentially expressed in acentriolar lungs as compared to control and cilia-disrupted lungs (in which branching is unaffected) are presented in a heatmap (Figure 3A). The fold changes and adjusted p values (FDR) of genes differentially expressed in acentriolar lungs are presented as a volcano plot (Figure 3B). Sox2 was the most downregulated protein-coding gene in ShhCre Cenpj−/lox lungs, providing an orthogonal confirmation of the decrease of SOX2-expressing cells. Target genes of the WNT pathway (e.g., Axin2, Bmp4) and FGF pathway (e.g., Etv4 and Etv5) were not significantly changed, suggesting that these developmental pathways were unaffected in Cenpj mutant lungs (Figure 3B).
Figure 3. Centriole loss in endoderm activates p53-dependent apoptosis.

(A) Heatmap of E11.5 control (ShhCre Cenpj+/lox), Ift88 loss of function (ShhCre Ift88−/lox) and Cenpj loss of function (ShhCre Cenpj−/lox) lung RNAseq. Significant differentially expressed genes from Cenpj loss of function (ShhCre Cenpj−/lox) lung against control (ShhCre Cenpj+/lox), Ift88 loss of function (ShhCre Ift88−/lox) lungs were listed in heatmap. Cdkn1a (p21) and several other p53-induced genes are specifically upregulated upon loss of CENPJ.
(B) Volcano plots comparing log2 fold change of normalized RNAseq reads for E11.5 Cenpj loss of function (ShhCre Cenpj−/lox) as compared to control (ShhCre Cenpj+/lox) lungs. Dashed line denotes the p=0.05 cutoff. Selected genes upregulated ≥2 folds were indicated in red, and selected genes down-regulated ≥2 folds were indicated in blue (FDR <0.05).
(C) RT-qPCR measurement of Cdkn1a (p21), Ccng1, Eda2r, and Sox2 expression in E13.5 control (ShhCre Cenpj+/lox) and Cenpj loss of function (ShhCre Cenpj−/lox) lungs. Sox2 was downregulated and Cdkn1a (p21), Ccng1 and Eda2r upregulated upon loss of CENPJ. n=4, 4, 4, 4, 3, 3 respectively for the control groups; n=3, 4, 4, 4, 3, 3 respectively for Cenpj loss of function groups.
(D-E) Staining (D) and quantification (E) of p53 in E12.5 control (CTL, ShhCre Cenpj+/lox) and Cenpj loss of function (cKO, ShhCre Cenpj−/lox) lung cryo-sections. Scale bar, 20 μm. 4 independent replicates were analyzed for statistics.
(F-G) Staining (F) and quantification (G) of p21 in E12.5 control (ShhCre Cenpj+/lox) and Cenpj loss of function (ShhCre Cenpj−/lox) lung cryo-sections. Scale bar, 20 μm. 4 and 3 independent replicates for SOX9+ and SOX2+ respectively were analyzed for statistics.
(H-I) Staining (H) and quantification (I) of p53 in E12.5 control (ShhCre Cenpj+/lox) and Cenpj loss of function (ShhCre Cenpj−/lox) small intestine cryo-sections. Scale bar, 50 μm. n=10 and 6 for CTL and cKO respectively.
(J-K) Staining (J) and quantification (K) of cleaved Caspase3 (CC3, green) in E12.5 control (ShhCre Cenpj+/lox) and Cenpj loss of function (ShhCre Cenpj−/lox) lungs. Epithelia (CDH1, red) were outlined by dashed line. Arrows indicate the distal region of Cenpj loss of function lung, and arrowheads indicate the proximal part. Scale bar, 1 mm. n=3.
(L) Wholemount immunostaining of E15.5 control (ShhCre Cenpj+/lox), Cenpj loss of function (ShhCre Cenpj−/lox), Cenpj and p53 combined loss of function (ShhCre Cenpj−/lox p53−/−) lungs for SOX2 (magenta) and SOX9 (cyan). Scale bar, 1 mm.
(M) Schematic showing proposed mechanism by which centrioles participate in lung branching. In the absence of centrioles, SOX2-expressing, but not SOX9-expressing, cells activate p53 to trigger apoptosis.
The genes most significantly upregulated in ShhCre Cenpj−/lox lungs included p53 target genes such as p21 (Cdkn1a), Eda2r, Tap, Ccng1, Zfp365, Ddit4l, Dcxr, and Klhdc7a (Allen et al., 2014; Bowen et al., 2019; Brosh et al., 2010; Forsberg et al., 2013; Turrell et al., 2017; Zhu et al., 1999) (Figure 3B). We confirmed the differential expression of p21, Eda2r, Ccng1 and Sox2 by RT-qPCR (Figure 3C), further suggesting that p53 is active in ShhCre Cenpj−/lox lungs. The expression of the gene encoding p53, Trp53, was unaffected in acentriolar lungs, consistent with p53 activity being principally controlled post-transcriptionally.
To determine whether p53 was activated in the endoderm, we stained control and ShhCre Cenpj−/lox lungs and small intestine for p53. p53 was nearly undetectable in control lungs and small intestine, and dramatically increased in nuclei of ShhCre Cenpj−/lox lung epithelial cells (Figures 3D and 3E). Similarly, the established p53 target, p21, was also upregulated in nuclei of ShhCre Cenpj−/lox lungs (Figures 3F and 3G).
In parallel with the analysis of lung transcriptomes, we conducted RNAseq analyses of control (ShhCre Cenpj+/lox), cilia-disrupted (ShhCre Iff88−/lox) and centriole-disrupted (ShhCre Cenpj−/lox) E11.5 small intestines. Compared to the lung, loss of CENPJ perturbed gene expression more modestly in the intestine (Table S2). Despite the less significant effect of loss of centrioles on intestinal gene expression, we examined whether p53 was activated in the acentriolar intestinal epithelium as it was in the lung epithelium. Nuclear p53 was present at negligible levels in control E13.5 intestines, and upregulated in the intestinal epithelium of ShhCre Cenpj−/lox embryos (Figures 3H and 3I). Thus, despite the dramatically different effects on lung and intestinal development, loss of centrioles activates p53 in both tissues.
p53 activation can induce apoptosis (Aubrey et al., 2018). To begin to assess whether p53-triggered apoptosis could account for the loss of SOX2-expressing cells and branching in ShhCre Cenpj−/lox lungs, we assessed the presence of cleaved Caspase3 (CC3). Cleaved Caspase3 was almost undetectable in control lungs and dramatically upregulated specifically in the epithelium of ShhCre Cenpj−/lox lungs (Figures 3J and 3K). Quantitating the spatial distribution of apoptotic cells revealed dramatically elevated levels in the proximal ShhCre Cenpj−/lox lungs and more mild increases in the distal lungs (Figure 3J and 3K). The high levels of apoptosis therefore correlate with the presence of SOX2-expressing cells in the developing trachea and bronchi (Figure 2A and 2B). Therefore, we hypothesized that loss of centrioles induced p53-mediated apoptosis in the proximal regions of the developing lung to disrupt branching.
Removing p53 restores branching to acentriolar lungs
To test whether p53 activation underlies the lung branching defect, we generated ShhCre Cenpj−/lox p53−/− lungs. Mice lacking p53 are viable and did not display discernible differences in lung morphology, indicating that p53 is not required for lung development (data not included). We stained wholemount lungs from E15.5 control, ShhCre Cenpj−/lox and ShhCre Cenpj−/lox p53−/− embryos for SOX2 and SOX9. As observed previously, loss of CENPJ disrupted lung branching and led to loss of SOX2-expressing epithelial cells. In stark contrast, ShhCre Cenpj−/lox p53−/− lungs displayed lung branching indistinguishable from those of control mice (Figure 3L). In addition, removal of p53 restored SOX2-expressing airway cells to ShhCre Cenpj−/lox lungs. Impressively, ShhCre Cenpj−/lox p53−/lox mice were viable through adulthood (Figure S4A and S4C). As removing p53 entirely rescued lung development in ShhCre Cenpj−/lox p53−/− mice, we conclude that, in the absence of p53, centrioles are dispensable throughout the endoderm for lung development and animal viability.
Interestingly, ShhCre Cenpj−/lox p53−/lox mice exhibited atrophic digits 4 and 5, similar to those of ShhCre Ift88−/lox mice (Figure S4A-S4D). The finding that both centrioles and cilia are essential in limb bud cells to generate digits, regardless of whether p53 is active, are consistent with an essential role for centrioles in generating the primary cilia required to interpret the Hedgehog cues essential for digit development (Huangfu and Anderson, 2005).
The finding that inhibiting p53 restores lung branching to acentriolar lungs reveals that p53 activation disrupts branching. Lungs branch as the distal fields of SOX9-expressing progenitors are bifurcated and as proximal field progenitors differentiate into SOX2-expressing airway epithelium (Alanis et al., 2014; Rawlins et al., 2009). Centriole loss activates p53 to induce apoptosis preferentially depleting the SOX2-expressing cells. We propose that the loss of these SOX2-expressing cells in the distal airway prevents bifurcation of the progenitor field and thereby disrupts branching (schematized in Figure 3M). These findings raise the interesting question of why centrioles are essential for SOX2-expressing lung cells but dispensable for SOX9-expressing lung cells and intestinal epithelial cells, despite the shared activation of p53.
In the lung, ERK activity is restricted distally, whereas in the intestine, it is widespread
The loss of SOX2-expressing epithelial cells with the concomitant expansion of SOX9-expressing epithelial cells in acentriolar lungs suggested that some factors conferred tolerance to loss of centrioles in the SOX9-expressing progenitors at the distal developing lung buds. The signaling factor FGF10 is present specifically at the distal developing lung buds where it maintains the progenitor pool and drives bud growth (Arman et al., 1999; Bellusci et al., 1997; Min et al., 1998; Sekine et al., 1999; Volckaert et al., 2013). FGF signaling phosphorylates and activates ERK at the distal lung buds (Liu et al., 2004). Like FGF10, ERK function in the lung epithelium is critical for lung development (Boucherat et al., 2014). Therefore, we investigated the possibility that ERK protects acentriolar cells from p53-mediated apoptosis.
As expected, phosphorylated ERK (pERK) was present in the distal tips of developing control lung buds overlapping with the SOX9-expressing progenitors (Figures 4A and 4C). pERK was not detected in the SOX2-expressing airway epithelium of control lungs (Figures 4B and 4C). In contrast, pERK was distributed throughout the epithelium of ShhCre Cenpj−/lox lungs (Figures 4A and 4C). Immunoblot for pERK confirmed that E13.5 ShhCre Cenpj−/lox lungs possess increased ERK activity (Figure 4H). Thus, ERK is active in surviving acentriolar cells and, in ShhCre Cenpj−/lox lungs, corresponding with the expansion of SOX9-expressing progenitors and the loss of SOX2-expressing epithelial cells.
Figure 4. ERK activity correlates with survival of acentriolar cells.

(A-C) Immunostaining (A, B) and quantification (C) of phospho-ERK (pERK) in E12.5 control (ShhCre Cenpj+/lox), Cenpj loss of function (ShhCre Cenpj−/lox) and Cenpj and p53 combined loss of function (ShhCre Cenpj−/lox p53−/−) lung sections. pERK was co-stained with SOX9 (A) or SOX2 (B), respectively. Scale bar, 50 μm. Data from 4 independent experiments for control and Cenpj loss of function were analyzed in the super plot; n=6 for ShhCre Cenpj−/lox p53−/− from one experiment.
(D) RT-qPCR measurement of Fgf9, Fgf10 and Fgfr2 expression in E13.5 control (ShhCre Cenpj+/lox) and Cenpj loss of function (ShhCre Cenpj−/lox) lungs. Fgf10 was up-regulated. Data from 3 independent experiments were analyzed in super plot.
(E) In situ hybridization of Fgf10 in E12.5 control (ShhCre Cenpj+/lox) and Cenpj loss of function (ShhCre Cenpj−/lox) lungs. Scale bar, 1 mm.
(F-G) Immunostaining of phospho-ERK (pERK) in E13.5 (F) and E17.5 (G) control (ShhCre Cenpj+/lox) and Cenpj loss of function (ShhCre Cenpj−/lox) small intestine sections. Inlets depicted amplified view of epithelium. Scale bar denotes 50 μm and 100 μm, respectively.
(H) Immunoblots of phospho-ERK (pERK) and total ERK (ERK) in control (ShhCre Cenpj+/lox) and Cenpj loss of function (ShhCre Cenpj−/lox) lung and small intestine at E13.5. We quantified the ratio of pERK:ERK densitometry. n=3.
(I) Schematic showing the distribution of ERK activity (magenta) in developing lung and intestine.
In ShhCre Cenpj−/lox p53−/− lungs, pERK distribution pattern is restored to that of control lungs (Figure 4A and 4C). Thus, removing p53 both restores SOX2-expressing cells to acentriolar lungs and restores pERK-negative cells, consistent with a role for ERK activity in protecting against p53-mediated apoptosis.
Interestingly, Fgf10 expression was also expanded in ShhCre Cenpj−/lox lungs and restricted to the distal lung buds of control lungs (Figures 4D and 4E). Thus, the preferential survival of Fgf10-expressing distal lung cells may account for the expansion of pERK in ShhCre Cenpj−/lox lungs.
Unlike the lung, both control and ShhCre Cenpj−/lox intestinal epithelia displayed comparable broad pERK distribution at both E13.5 and E17.5 (Figures 4F and 4G). Therefore, unlike the developing lung where ERK activity is spatially restricted, ERK activity is widespread in the epithelium during intestinal development. Also unlike the case for the lung, immunoblot for pERK revealed no difference in ERK activity in E13.5 ShhCre Cenpj−/lox intestines (Figure 4H). The distribution of ERK activity in both developing lung and small intestine are schematized in Figure 4I, highlighting that ERK activity is restricted to the distal buds in the lung and widespread in the developing intestine and raising the possibility that increased pERK underlies the selective survival of acentriolar cells.
ERK suppresses p53-dependent apoptosis in acentriolar lung progenitors
To test whether different ERK activity levels explain the different responses to loss of centrioles, we used SCH772984, a well-characterized and highly selective ERK antagonist (Chaikuad et al., 2014; Morris et al., 2013). Consistent with its ability to inhibit ERK, SCH772984 dramatically inhibited the growth and branching of epithelium of cultured lungs (Figures 5A-5C). In contrast, other kinase inhibitors, such as p38 MAPK inhibitor BIRB 796 (Pargellis et al., 2002) or JNK inhibitor JNK-IN-8 (Zhang et al., 2012) did not block the growth of cultured lungs (Figure 5A-5C). Thus, pharmacological inhibitors of ERK, like genetic inactivation (Boucherat et al., 2015b), disrupt lung branching. Although branching is already compromised in ShhCre Cenpj−/lox lungs, the addition of SCH772984 inhibited their growth, similar to control lungs (Figures 5A-5C), further suggesting that acentriolar cells depend on ERK activity.
Figure 5. ERK activity protects acentriolar cells from p53-dependent apoptosis.

(A) Control (ShhCre Cenpj+/lox) and Cenpj loss of function (ShhCre Cenpj−/lox) lungs were isolated at E11.5 and cultured on a semi-permeable membrane. Control lungs were treated with DMSO, SCH772984, BIRB 796 or JNK-IN-8 for 48 hours. Cenpj loss of function lungs were treated with DMSO or SCH772984 for 48 hours. Images were taken every 24 hours. Scale bar, 1 mm.
(B-C) We measured the number of branches (B) and epithelial extension (C) of the lungs from (A). The fold change of epithelial extension was normalized to that immediately before the addition of the indicated compounds (i.e., at 0 hours). DMSO treatment was compared to BIRB 796, JNK-IN-8 or SCH772984 treatment within the same genotype using unpaired t-test. n=3.
(D) Visualization of GFP and tdTomato fluorescence in control (ShhCre Cenpj+/lox R26tdTomato Sox9GFP) and Cenpj loss of function (ShhCre Cenpj−/lox R26tdTomato Sox9GFP) lung organoid culture cryo-sections. Scale bar, 50 μm. Note: parameters of GFP fluorescence were adjusted in each group for better visualization.
(E-F) Staining (E) and quantification (F) of cleaved Casp3 (CC3) in control (ShhCre Cenpj+/lox R26tdTomato Sox9GFP) and Cenpj loss of function (ShhCre Cenpj−/lox R26tdTomato Sox9GFP) lung organoid culture cryo-sections. Inlets show the magnified view of cleaved Casp3 staining. Scale bar, 50 μm. DMSO treatment was compared to SCH772984 treatment within the same genotype, and different genotype with the same treatment was compared using unpaired t-test. n=6, 12, 8, 5 respectively.
(G-H) Staining (G) and quantification (H) of Cleaved Casp3 (CC3) stained in control (ShhCre Cenpj+/lox R26tdTomato) and Cenpj loss of function (ShhCre Cenpj−/lox R26tdTomato) intestinal organoid culture cryo-sections. Inlets show cleaved Casp3 staining. Scale bar, 20 μm. DMSO treatment was compared to SCH772984 treatment within the same genotype, and different genotype with the same treatment was compared using unpaired t-test. n=5, 4, 4, 5 respectively.
(I) Schematic illustrating the differential responses of lung epithelial cells to loss of centrioles. Loss of centrioles induced p53 activation in the endodermal epithelium. Acentriolar lung cells with low ERK activity (SOX2-expressing cells) apoptose, whereas acentriolar lung SOX9-expressing progenitors with higher ERK activity survive and grow.
See also Figure S5.
To more quantitatively assess whether ERK activity sustains acentriolar lung and intestinal epithelial cells, we generated lung and intestinal epithelial organoids. More specifically, we generated ShhCre Cenpj+/lox Rosa26tdTomato Sox9GFP (control) and ShhCre Cenpj−/lox Rosa26tdTomato Sox9GFP (mutant) embryos that selectively express tdTomato throughout the endoderm. Progenitors from isolated lungs of both E12.5 control and mutants (Nichane et al., 2017) grew into sphere-shaped organoids (Figures 5D and S5A). SCH772984 reduced the growth of both control and acentriolar organoids (Figures 5D, S5A, S5B, S5D, S5E, S5F, S5G, S5H and S5I). In addition to reducing organoid size, SC772984 blocked the ability of acentriolar progenitors to grow into sphere-like structures (Figure 5D). We also observed the reduction of Sox9-IRES-GFP expression in acentriolar organoids and SCH772984-treated control organoids, indicating the role of ERK activity in maintaining the expression of progenitor markers (Figure 5D, S5A and S5C). Inhibiting ERK with SCH772984 did not affect apoptosis in control lung organoids (Figures 5E and 5F). However, in acentriolar lung organoids, SCH772984 dramatically activated apoptosis throughout the epithelium (Figures 5E and 5F). Thus, ERK activity is essential for lung progenitors to survive without centrioles.
We returned to consider the mystery of why CENPJ is critical for lung development but dispensable for intestinal development and hypothesized that widespread ERK activity protects acentriolar intestinal cells. To test whether ERK protects acentriolar intestinal cells from apoptosis, we generated intestinal epithelial organoids from E13.5 ShhCre Cenpj+/lox Rosa26tdTomato and ShhCre Cenpj−/lox Rosa26tdTomato embryos by culturing the epithelial progenitors in Matrigel (Andersson-Rolf et al., 2014). As with lung organoids, inhibiting ERK with SCH772984 did not affect apoptosis in control intestinal organoids (Figures 5G and 5H). And again, as with lung organoids, SCH772984 induced apoptosis specifically in acentriolar intestinal organoids (Figures 5G and 5H). The growth of control and acentriolar intestinal organoids were similar to lung organoids with/without SCH772984 treatment (Figures S5C and S5D). These results demonstrate that ERK activity protects both intestinal and lung epithelium from apoptosis in the absence of centrioles.
Loss of centrioles activates p53 throughout the lung (Figure 3D and 3F) whereas ERK is activated specifically in the distal lung, suggesting that ERK activity does not affect the levels of p53. ERK activation is similar in control and ShhCre Cenpj−/lox p53−/− lungs (Figure 4A), indicating that p53 does not directly affect ERK activity. Therefore, we propose that ERK and p53 act in parallel, with loss of centrioles inducing p53 activation and cell death specifically when ERK activity is low (Figure 5I). Thus, in the endoderm, the developmental consequences of centriole loss and p53 activation are shaped by ERK. In the context of ERK activation, as in SOX9-expressing lung epithelium and intestinal epithelium, cells tolerate loss of centrioles and p53 activation. However, without ERK activity, as in SOX2-expressing lung epithelium, loss of centrioles activates p53 to induce apoptosis.
Discussion
Centrioles are essential for the proliferation of many animal cells in vitro and in vivo (Bazzi and Anderson, 2014; Doxsey et al., 2005; Faisst et al., 2002; Hudson et al., 2001; Izraeli et al., 1999; Rieder et al., 2001; Stevens et al., 2009; Tang et al., 2011; Wong et al., 2015). For example, centrioles are critical for early divisions in both C. elegans and Drosophila development (Rodrigues-Martins et al., 2008; Schwarz et al., 2018; Stevens et al., 2007). However, not all cell division requires centrioles. For example, the earliest divisions of mouse development occur without centrioles (Bangs et al., 2015; Dumont and Desai, 2012), later Drosophila divisions do not depend on centrioles (Basto et al., 2006), planaria cells divide without centrioles (Azimzadeh et al., 2012) and plants do not have centrioles (Brown and Lemmon, 2011).
Previous work shows the absence of centrioles causes cell death in rapidly proliferating mammalian lineages (Bazzi and Anderson, 2014). Therefore, we examined the function of the roles of centrioles in the endoderm, focusing on the lung and small intestine epithelium, rapidly proliferating lineages (Miller et al., 1999). To our surprise, genetically removing centrioles from the endoderm had profound effects on one lineage (lung) but not another (intestine). Loss of centrioles in the lung epithelium blocked all branching after the formation of the major divisions, whereas the equivalent perturbation did not disrupt intestinal development.
Investigating why centrioles are differentially required for lung and intestine development, we discovered that, while loss of centrioles activated p53 in both, the phenotypic consequences of p53 activation were distinct. Indeed, even within the lung, activated p53 disrupted formation of SOX2-expressing airway cells, but not of SOX9-expressing progenitors.
SOX2-expressing cells differentiate from SOX9-expressing progenitors and are present more proximal to the progenitors in the developing trachea, bronchi and bronchioles. While we demonstrated that SOX2 itself is dispensable for lung branching, we propose that the SOX2-expressing cells themselves are critical to bifurcate the progenitor populations and create individualized buds. Consistent with this proposal, other mutation that decrease the number of SOX2-expressing cells also cause branching defects, although less profoundly than that caused by loss of CENPJ (Boucherat et al., 2015a; Li et al., 2018; Lin et al., 2017; Mahoney et al., 2014). Therefore, we propose that, in acentriolar lungs, the loss of SOX2-expressing cells prevents division of the progenitor pool and the main lung divisions develop as large SOX9-expressing cysts. Perhaps the decreased ERK activity in SOX2-expressing cells causes branching by altering growth at branchpoints.
Removing p53 dramatically rescued the development of acentriolar lungs, even restoring viability. In contrast, limb development was not restored by removing p53 and the limb patterning defect recapitulated by removing cilia, indicating that cilia are essential for limb development independent of p53 activity. Moreover, removing cilia in the endoderm did not affect lung morphogenesis. Together, these results confirm that centrioles are essential for cilium-mediated signaling, but that cilia and ciliary signaling are not critical for lung patterning. In brain development, removing p53 only partially rescued phenotypes caused by loss of centrioles (Lin et al., 2020). In contrast, the critical function of centrioles in lung morphogenesis is to restrain p53 activity.
Activation of p53 has different developmental consequences in different tissues (Bowen et al., 2019). The factors that dictate this differential impact are not understood but could include differences in timing of activation, differences in degree of activation, or differences in transcriptional context. Removing MDM2 caused a transient upregulation of p53 during early lung development in part of the epithelium and modestly increased apoptosis, disrupted separation of the esophagus and trachea, and delayed but did not block branching and differentiation of the airway epithelium (Bowen et al., 2019; Sui et al., 2019). In contrast, we found that p53 activation caused by loss of centrioles is more extensive and blocked branching and the differentiation of lung SOX9+ progenitors into SOX2+ bronchiolar cells. We surmise that loss of centrioles activates p53 to a greater extent than loss of MDM2.
As we observed that SOX9-expressing cells are relatively resistant to p53-mediated apoptosis in acentriolar lungs, we also considered whether their position at the distal bud tips may be relevant. FGF10 is expressed specifically at the distal lung buds where it activates ERK (Arman et al., 1999; Bellusci et al., 1997; Liu et al., 2004; Min et al., 1998; Sekine et al., 1999; Volckaert et al., 2013). We found that ERK was specifically active in a subset of the SOX9-expressing progenitors, raising the possibility that ERK activation shapes the response to p53 activation. Consistent with that possibility, pharmacologically inhibiting ERK induced apoptosis specifically in acentriolar SOX9-expressing cells. Unlike the lung, ERK activity in the intestine is widely distributed, but in this tissue too, inhibiting ERK specifically induced apoptosis in acentriolar cells. Therefore, we propose that the spatial distribution of ERK activity shapes the outcome of loss of centrioles. We predict that ERK activity will also shape the phenotypic outcomes of other cell stresses that activate p53, and may explain the differential impacts of centriolar abnormalities and p53 activation outside of the endoderm. For example, centrioles and ERK activity are both important for brain growth (Basson et al., 2008; Bazzi and Anderson, 2014; Meyers et al., 1998; Pucilowska et al., 2012; Raballo et al., 2000; Shi et al., 2014). Perhaps one reason microcephaly, associated with centriolar defects, affects cerebral growth more than the midbrain is that FGF8 protects midbrain cells from apoptosis.
Mutation of p53 is an extremely common hallmark of diverse cancers, particularly lung cancers, many of which express SOX2 (Gibbons et al., 2014; Karachaliou et al., 2013). Centriolar abnormalities are also common in cancer cells (Marteil et al., 2018). Our finding that, during development, SOX2-expressing lung cells apoptose without CENPJ but are rescued by loss of p53, is consistent with the possibility that, in tumor suppression, p53 removes cells with centriolar abnormalities. Of course, activated ERK is another hallmark of diverse cancers, including some lung cancers (Feldser et al., 2010; Junttila et al., 2010; Vicent et al., 2004). One prediction of this work on non-cancerous tissues is that cancers that retain wild type p53, like hepatocellular carcinoma, will either have unaltered centrioles or will possess oncogenic mutations that activate ERK. Our work further suggests that ERK activity inhibition could be particularly efficacious in cancers with retained p53 activity.
Our work reveals that centrioles are differentially required by different lineages during endoderm development, and spatial distribution of ERK activity shapes the phenotypic outcomes of centriole-loss induced p53 activation.
Limitations of study
The role of ERK activity in shaping the acentriolar output is revealed by the correlation of ERK activity distribution and survival pattern, and tested by chemically inhibiting ERK in lung and intestinal organoid cultures that have high ERK activity. Confirming that high ERK activity can protect SOX2+ expressing airway cells from apoptosis in vivo will require activating ERK activity in the proximal acentriolar lung.
STAR★Methods
RESOURCE AVAILABILITY
Lead contact
Requests for further information or reagents can be directed to and will be fulfilled by the Lead Contact, Jeremy Reiter (Jeremy.Reiter@ucsf.edu ), Department of Biochemistry and Biophysics, Cardiovascular Research Institute, University of California, San Francisco.
Materials availability
The Cenpj+/− and Cenpjlox/lox mice generated in this study will be made available upon request. We may require a completed materials transfer agreement and reasonable compensation by the requestor for its processing and shipping.
Data and code availability
RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Original western blot images have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
This manuscript did not generate new code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
ShhCre [Shhtm1(E.G.F.P./Cre)Cjt], Ift88lox (Ift88tm1Bky), Sox2lox (Sox2tm1.1Lan), R26tdTomato [Rosa26tm14(CAG-CAGtdTomato)Hze/J], Sox9GFP (Sox9IRES-EGFP), p53 KO (B6.129S2-Trp53tm1Tyj/J), and p53LoxP (B6.129P2-Trp53tm1Brn/J) alleles have been described previously (Chan et al., 2011; Davenport et al., 2007; Harfe et al., 2004; Jacks et al., 1994; Madisen et al., 2010; Marino et al., 2000; Shaham et al., 2009). The knockout first Cenpjtm1a(EUCOMM.)Wtsi ES cells were purchased from the European Mutant Mice Repository. The conditional allele (Cenpjlox) was generated by crossing Cenpjtm1a(EUCOMM.)Wtsi mice to A.C.T.B.:FLPe mice (JAX Stock No: 005703). The null allele (Cenpj−) was generated by crossing the Cenpjlox/+ mice to β-actin-Cre mice (JAX Stock No: 019099). C57BL/6J mice (JAX Stock No: 000664) were purchased from Jackson Laboratory. 6-24 week old male ShhCre Cenpj+/−, or ShhCre Cenpj+/− Sox9GFP/GFP, or ShhCre Cenpj+/− p53+/− mice were mated with 6-24 week old female Cenpjlox/lox, or Cenpjlox/lox Rosa26tdTomato/tdTomato, or Cenpjlox/lox p53+/−, or Cenpj1ox/lox p53lox/lox mice. 6-24 week old male ShhCre Ift88+/− mice were mated with 6-24 week old female Ift88lox/lox mice. 6-24 week old male ShhCre Sox2+/lox mice were mated with 6-24 week old female Sox2lox/lox mice. Embryos were collected in a gender-unbiased manner.
All mice were maintained under specific pathogen-free conditions at the UCSF animal care facility. Mice were cared for, and all experiments were approved by the Administrative Panel on Laboratory Care, and the Institutional Animal Care and Use Committee (IACUC) of UCSF Mice were maintained on a normal chow diet.
METHOD DETAILS
Lung and intestinal culture
Lung progenitors from E12.5 control (ShhCre Cenpj+/lox R26tdTomato Sox9GFP or ShhCre Cenpj+/lox), Cenpj loss of function (ShhCre Cenpj−/lox R26tdTomato Sox9GFP or ShhCre Cenpj−/lox), and Cenpj and p53 combined loss of function (ShhCre Cenpj−/lox P53−/−) embryos were isolated and cultured as described (Nichane et al., 2017). Briefly, lungs were digested with collagenase I and II, mixed with 10% FBS/DMEM and filtered and washed twice in DMEM/F12. GFP+ cells were collected or all the cells were collected and counted. About 1000 cells were seeded into 40 μl Matrigel in 24-well plate. Twenty minutes after Matrigel solidified, 500 μl culture medium was added. Twenty-four hours later, the organoid culture was incubated with DMSO or 2.5 μM SCH772984 for 24 hours and then imaged directly, or subjected to frozen section, immunostaining and imaged using a Zeiss LSM800 confocal laser scanning microscope.
Embryonic intestinal cells were isolated from E13.5 control (ShhCre Cenpj+/lox R26tdTomato) and Cenpj loss of function (ShhCre Cenpj−/lox R26tdTomato) embryos and cultured as described (Andersson-Rolf et al., 2014). Briefly, small intestine were cut longitudinally and incubated with 2 mM EDTA for 30 min at 4°C and then digested in 1 ml Trypsin buffer at 37°C for 10 min. Then the samples were changed to 10% FBS/DMEM and pipetted up and down, then filtered. The cells were washed twice, then about 2000 cells were seeded into Matrigel. Twenty-four hours after the seeding in Matrigel, the intestinal culture was incubated with DMSO or 2.5 μM SCH772984 for 24 hours before being imaged for tdTomato fluorescence. Intestinal organoids isolated from control (ShhCre Cenpj+/lox), Cenpj loss of function (ShhCre Cenpj−/lox), and Cenpj and p53 combined loss of function (ShhCre Cenpj−/lox p53−/−) embryos were cultured and treated with DMSO or 2.5 μM SCH772984 for 24 hours, then subjected to frozen section and immunostaining.
Wholemount lungs isolated from E11.5 control (ShhCre Cenpj+/lox) and Cenpj loss of function (ShhCre Cenpj−/lox) embryos were isolated and cultured as described (Carraro et al., 2010). Briefly, lungs were dissected and isolated at E11.5. Then lungs were placed on the top of semi-permeable membrane in 24-well plate loaded with 10% FBS/DMEM. The concentrations of the chemical compounds were used as follows: ERK1/2 inhibitor SCH772984 at 2.5 μM, p38 MAPK inhibitor Doramapimod (BIRB 796) at 10 μM, JNK1/2/3 inhibitor JNK-IN-8 at 10 μM. Images were taken every 24 hours with Zeiss Discovery V12 stereomicroscope.
Immunostaining
Embryos were immediately fixed in 4% paraformaldehyde at 4°C after dissection. Lung tissue was isolated and further fixed in 4% paraformaldehyde at 4°C for 2 hours. After fixation, the tissue was washed three times with ice-cold PBS. Then samples were dehydrated in 30% sucrose at 4°C overnight before being embedded in OCT and stored at −80°C. 12 μm frozen sections were washed three times with PBS and incubated with primary antibodies diluted in IF buffer (5% donkey serum and 0.3% Triton X-100 in PBS) at 4°C overnight. Then slides were washed five times with IF buffer and incubated with fluorescent secondary antibodies and Hoechst 33342 at room temperature for 2 hours, followed by five washes with IF buffer. Slides were mounted with FluorSave. Confocal images were obtained with Zeiss LSM800 confocal laser scanning microscope.
For EdU labeling, 600 μl 2.5 mg/ml EdU/D.P.B.S. solution was intraperitoneally injected into pregnant female mice at E11.5. After 15 minutes, the injected females were euthanized, and the embryonic lungs were dissected and fixed in 4% paraformaldehyde at 4°C for 1 hour. Then the fixed lungs were washed with ice-cold PBS three times and embedded in OCT. Frozen sections were first stained with primary antibodies, fluorescent secondary antibodies. Then EdU staining was performed according to the manufacturer's protocol (C10269, Thermo Fisher Scientific). Slides were washed three times with PBS, and Hoechst was stained for 5 minutes before being mounted with FluorSave mounting medium.
Wholemount lung staining
Wholemount lung staining was carried out as described previously (Metzger et al., 2008). Briefly, samples were fixed in 4% paraformaldehyde for 1 hour. Then, samples were washed in PBS for 3 times and gradually dehydrated to pure methanol. After incubation in 5% H2O2 in methanol for 5 hours at room temperature, samples were subjected to blocking and permeabilization in 5% dankey serum/1% Triton/PBS, then incubated with primary antibodies overnight at 4°On blocking buffer, washed with blocking buffer for 5 times. Then samples were incubated with fluorescent secondary antibodies overnight at 4°C Samples were washed and further fixed in 4% paraformaldehyde for 30 min at 4°C after the staining and then cleared according to the iDISCO protocol. Samples were stored in DBE and imaged with AZ100 wide field light-sheet microscope at Nikon Imaging Center at UCSF. Images were processed with Micro-Manager 2.0 gamma and Image J. Max projection of the z-stacks were presented.
Next-generation RNA sequencing and analysis
E11.5 lungs and intestines from control (ShhCre/+ Cenpj+/lox), Cenpj loss of function (ShhCre/+ Cenpj−/lox) and Ift88 loss of function (ShhCre/+ Ift88−/lox) embryos were isolated and homogenized in 1 ml Trizol, followed by mixing with 200 μl chloroform and centrifugation at 12,000 rpm at 4°C for 15 min. The water phase was transferred to new tubes and mixed with an equal volume of pure ethanol. Then the samples were processed with the QIAGEN RNeasy Micro kit for RNA extraction, followed by library construction at UCSF Functional Genomics Core Facility. Paired-end 150 bp (PE150) sequencing data were generated on an Illumina NovaSeq 6000. Differential gene expression among Control, Cenpj loss of function, and Ift88 loss of function lung and intestine were assessed. Differential gene expression was called significant if FDR (adjusted p-value) < 0.05.
The common list of differentially expressed genes between control (ShhCre/+ Cenpj+/lox) and Cenpj loss of function (ShhCre/+ Cenpj−/lox) lungs, and between Cenpj loss of function (ShhCre/+ Cenpj−/lox) and Ift88 loss of function (ShhCre/+ Ift88−/lox) lungs were chosen for heatmap analysis if FDR <0.05 and FC (fold change) >1.5 or FC < 0.67. Gene expression was normalized to the control group.
Volcano plot was generated from the log2(FC) and FDR of gene expression calculated from Cenpj loss of function (ShhCre/+ Cenpj−/lox) versus control (ShhCre/+ Cenpj+/lox) lungs. Upregulated genes were highlighted in red if the FDR<0.0.5 and Log2(FC)>=1 (FC>=2); downregulated genes were highlighted in blue if the FDR<0.0.5 and Log2(FC)<=−1 (FC<=1/2).
Quantitative real-time PCR
RNA was extracted using the Trizol and RNeasy Kit (QIAGEN), and cDNA was synthesized using BioRad iScript cDNA synthesis kit (BioRad, 1708890). Quantitative real-time PCR was performed using PowerUp™ SYBRTM Green Master Mix (Applied Biosystems, A25742) in TempPlate polypropylene 384-well PCR plates (USA Scientific, 1438-4700) on QuantStudio 5 qPCR machine. Expression levels were normalized to the geometric average expression of Dolk, Hprt and Pde12.
Immunoblotting
Lung and small intestine were isolated from E13.5 control (ShhCre/+ Cenpj+/lox) and Cenpj loss of function (ShhCre/+ Cenpj−/lox) embryos in ice-cold PBS. Then samples were lysed in RIPA buffer supplemented with sufficient protease inhibitor (Roche, complete™ Protease Inhibitor Cocktail, #11697498001) and phosphatase inhibitor (Roche, PhosSTOP, #04906845001). The supernatant was mixed with 4X loading buffer and heated at 100°C for 10 minutes before being loaded onto PAGE gel for electrophoresis. Then proteins were transferred from PAGE gel to PVDF membrane and incubated with primary antibodies ERK (1:1000) and pERK (1:1000) at 4°C overnight. After five washes with TBST, HRP conjugated secondary antibodies were incubated at room temperature for 1 hour and washed five times in TBST before being developed for ECL imaging with BioRad ChemiDoc Imager. SuperSignal™ West Pico PLUS Chemiluminescent Substrate for ERK and SuperSignal™ West Femto Maximum Sensitivity Substrate for pERK were used in the ECL reaction.
QUANTIFICATION AND STATISTICAL ANALYSIS
Gut isolated from E16.5 control (ShhCre Cenpj+/lox Rosa26tdTomato) and Cenpj loss of function (ShhCre Cenpj−/lox Rosa26tdTomato) embryos were dissected and placed onto a petri dish. Tissue was linearized to the petri dish by gently removing PBS. Images were taken along with a ruler under the ZEISS Discovery V12 stereomicroscope. Then the length of the small intestine, as well as ruler, were measured using the segmented line tool of ImageJ. Pixel length was then transformed into millimeters.
The branches of cultured wholemount lungs from control (ShhCre Cenpj+/lox) and Cenpj loss of function (ShhCre Cenpj−/lox) embryos were counted based on the images taken every 24 hours. The dimension between left and right edges of epithelium was measured with ImageJ. Each measurement was normalized to that of the lungs at 0 hours immediately before the incubation of indicated compounds.
Quantification of the immunostaining images was carried out in CellProfiler (version 3.1.9). First, primary objects of "Epithelium," “Nuclei,” “SOX2” or“SOX9” were identified from images stained with CDH1, Hoechst, SOX2 or SOX9 respectively, with “Method to distinguish clumped objects” and “Method to draw dividing lines between clumped objects” set as “Intensity”. Then RelateObjects was used to identify SOX2-expressing epithelium or SOX9-expressing epithelium by setting “Epithelium” as the Parent Objects and “SOX2” or “SOX9” as the Child Objects. The newly identified objects were the nuclei of cells positive in SOX2 (nuclear) and CDH1, or SOX9 (nuclear) and CDH1. Mean intensity of p53 and p21 was measured in the objects of SOX2-expressing epithelium or SOX9-expressing epithelium. The average mean intensity was calculated and used for statistics.
For cleaved Caspase3 and phospho-ERK quantification, RelateObjects was used to identify SOX2-expressing epithelium or SOX9-expressing epithelium by setting “Epithelium” as the “Child Objects”. The related objects were outlined by “Epithelium” (CDH1) and included the whole cells. Mean intensity of phospho-ERK, the number of cleaved Caspase3-positive cells were measured in these objects. The average mean intensity was calculated and used for statistics.
For wholemount staining of cleaved Caspase 3 quantification, SOX2+ epithelium (trachea & bronchi) were manually outlined according to the structure. The proximal and distal SOX9+ epithelium were divided by midline. The mean intensity of cleaved Caspase 3 was measured by Image J.
For EdU quantification, primary objects “EdU labeled” were identified from EdU staining channel. Objects of “Epithelial nuclei” were identified with MaskObjects by masking primary objects “Epithelium” from primary objects “Nuclei”. Then EdU labeled epithelial nuclei were identified with RelateObjects by setting “Epithelial Nuclei” as the Child Objects and “EdU labeled” as the Parent Objects. Then EdU positive nuclei were counted against the total nuclei in “Epithelium”.
For the size of organoid cultures, the primary objects were identified from tdTomato fluorescence with “Method to distinguish clumped objects” and “Method to draw dividing lines between clumped objects” set as “Shape” so that objects were whole sphere-shaped organoids. MajorAxisLength was measured for each sphere-shaped organoid.
Then, the counted number or the averaged mean intensity from each image were calculated and used as one dot in the plots. Super plots were generated from 3-4 independent experiments with the average of each experiment as one datum. Data are represented as the mean±standard deviation. One-way ANOVA analysis was performed to check whether the data were significant. Then p-value between every two groups was determined using unpaired two-tailed t-tests for single plots or paired two-tailed t-tests for super plots. Unless otherwise stated, statistical analyses were performed in GraphPad Prism. A p-value less than 0.05 was considered statistically significant and denoted as follows: *<0.05, **<0.01, ***<0.001.
Supplementary Material
Table S1: RNAseq of control (ShhCre Cenpj+/lox), Ift88 loss of function (ShhCre Ift88−/lox) (cilia-disrupted) and Cenpj loss of function (ShhCre Cenpj−/lox) (centriole-disrupted) lungs, Related to Figure 3.
Table S2: RNAseq of control (ShhCre Cenpj+/lox), Ift88 loss of function (ShhCre Ift88−/lox) (cilia-disrupted) and Cenpj loss of function (ShhCre Cenpj−/lox) (centriole-disrupted) small intestine, Related to Figure 3.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-FGFR1OP (FOP, Rabbit) | Proteintech | Cat#11343-1-AP, PRID: AB_2103362 |
| Anti-ARL13B (Rabbit) | Proteintech | Cat#17711-1-AP, PRID: AB_2060867 |
| Anti-CDH1 (Rat) | Invitrogen | Cat#13-1900, PRID: AB_2533005 |
| Anti-γTubulin (Goat) | Santa Cruz | Cat#sc-7396, PRID: AB_2211262 |
| Anti-p53 (Rabbit) | Proteintech | Cat#10442-1-AP, PRID: AB_2206609 |
| Anti-p21 (Rabbit) | Proteintech | Cat#28248-1-AP, PRID: AB_2881097 |
| Anti-Cleaved Caspase-3 (Rabbit) | Cell Signaling Technology | Cat#9661S, PRID: AB_2341188 9664S |
| Anti-SOX2 (Rabbit) | Cell Signaling Technology | Cat#23064S, PRID: AB_2714146 |
| Anti-SOX2 (Goat) | R&D | Cat#AF2018, PRID: AB_355110 |
| Anti-SOX9 (Goat) | R&D | Cat#AF3075, PRID: AB_2194160 |
| Anti-TTF-1 (Nkx2.1, Rabbit) | Cell Signaling Technology | Cat#12373S, PRID: AB_2797895 |
| Anti-Prosurfactant Protein C (SFTPC, Rabbit) | Sigma | Cat#AB3786, PRID: AB_91588 |
| Anti-p73 (Rabbit) | Abcam | Cat#ab40658, PRID: AB_776999 |
| Anti-Phospho-p44/42 MAPK (pErk1/2, Rabbit) | Cell Signaling Technology | Cat#9101S, PRID: AB_331646 |
| Anti-Phospho-p44/42 MAPK (pErk1/2, Rabbit) | Cell Signaling Technology | Cat#4370S, PRID: AB_2315112 |
| Anti-p44/42 MAPK (Erk1/2, Rabbit) | Cell Signaling Technology | Cat#4695S, PRID: AB_390779 |
| Anti-Ki67 (Rabbit) | Abcam | Cat#ab15580, RRID: AB_443209 |
| Donkey anti-rat Alexa Fluor 488 | Invitrogen | Cat#A-21208, PRID: AB_2535794 |
| Donkey anti-goat Alexa Fluor 647 | Invitrogen | Cat#A-21447, PRID: AB_2535864 |
| Donkey anti-rabbit Alexa Fluor 568 | Invitrogen | Cat#A-10042, PRID: AB_2534017 |
| Goat Anti-Rabbit IgG HRP. | Jackson ImmunoResearch | Cat#111-035-144, PRID: AB_2307391 |
| Anti-Cdh1 e660 | eBioscience | Cat#50-3249-82, PRID: AB_11040003 |
| Anti-Pecam1/CD31 e450 | eBioscience | Cat#480311, PRID: AB_10598807 |
| Anti-Ptprc/CD45 e450 | eBioscience | Cat#480451, PRID: AB_1518807 |
| Anti-Ter119 e450 | eBioscience | Cat#485921, PRID: AB_1518809 |
| Bacterial and Virus Strains | ||
| Stable Competent E. coli | NEB | Cat#C3040H |
| Chemicals, Peptides, and Recombinant Proteins | ||
| SCH772984 | MedChemExpress | Cat#HY-50846 |
| Doramapimod (BIRB 796) | MedChemExpress | Cat#HY-10320 |
| JNK-IN-8 | MedChemExpress | Cat#HY-13319 |
| FluorSave | E.M.D. Millipore Calbiochem | Cat#34-578-920ML |
| Hoechst 33342 | Tdermo Fisher | Cat#H21492 |
| EdU | Tdermo Fisher | Cat#E10187 |
| PhosSTOP | Roche | Cat#04906845001 |
| cOmplete™ EDTA-free Protease Inhibitor Cocktail | Roche | Cat#11873580001 |
| Phenol red-free Leibovitz L-15 | Tdermo Fisher | Cat#21083027 |
| Advanced DMEM/F12 | Tdermo Fisher | Cat#12634010 |
| Collagenase I | Tdermo Fisher | Cat#17100017 |
| Collagenase II | Tdermo Fisher | Cat#17101015 |
| Dnase I | Tdermo Fisher | Cat#04716728001 |
| Trypsin | Tdermo Fisher | Cat#27250018 |
| FBS | Gibco | Cat#10439-024 |
| BSA | Sigma | Cat#A9418 |
| HEPES | Tdermo Fisher | Cat#15630080 |
| EDTA | Tdermo Fisher | Cat#15575020 |
| RNasin Ribonuclease Inhibitor | Promega | Cat#N2115 |
| Growtd Factor Reduced, Phenol Red-Free Matrigel 10 mg/mL | Corning | Cat#356231 |
| Fgf10 | R&D | Cat#6224-FG-025 |
| Fgf9 | R&D | Cat#7399-F9-025 |
| EGF | R&D | Cat#2028-EG-200 |
| CHIR99021 | Tocris | Cat#4423 |
| Y27632 | Tocris | Cat#1254 |
| A8301 | Tocris | Cat#2939 |
| Heparin | Sigma | Cat#H3149 |
| Insulin | Roche | Cat#11376497001 |
| Transferrin | Roche | Cat#10652202001 |
| PenStrep | Tdermo Fisher | Cat#15140122 |
| Glutamine | Tdermo Fisher | Cat#25030081 |
| Anti-Anti | Tdermo Fisher | Cat#15240062 |
| Dispase II | Tdermo Fisher | Cat#17105041 |
| Collagenase IV | Tdermo Fisher | Cat#17104019 |
| Papain | Wortdington | Cat#LS003118 |
| Noggin | Peprotech | Cat#10772-456 |
| R-Spondin 1 | R&D | Cat#7150-RS-025/CF |
| TRI reagent | Sigma | Cat#T9424 |
| SuperSignal™ West Pico PLUS Chemiluminescent Substrate | Tdermo Fisher | Cat#PI34577 |
| SuperSignal™ West Femto Maximum Sensitivity Substrate | Tdermo Fisher | Cat#34095 |
| Critical Commercial Assays | ||
| PowerUp™ SYBRTM Green Master Mix | Applied Biosystems | Cat#A25742 |
| iScript cDNA syntdesis kit | BioRad | Cat#1708890 |
| RNeasy Mini Kit (250) | Qiagen | Cat#74106 |
| Zymo RNA Clean & Concentrator-5 columns | Zymo Research | Cat#1016 |
| Deposited Data | ||
| Raw and processed RNAseq data | Tdis paper | GEO: GSE188585 |
| Raw Western blot images | Tdis paper | DOI:10.17632/tdwstr46bb.1 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | Tde Jackson Laboratory | Jax stock 000664 |
| Mouse: ShhCre: B6.Cg-Shhtm1(E.G.F.P./cre)Cjt/J | Tde Jackson Laboratory | Jax stock 005622 |
| Mouse: Ift88lox: B6.129P2-Ift88tm1Bky/J | Tde Jackson Laboratory | Jax stock 022409 |
| Mouse: Sox2lox:Sox2tm1.1Lan/J | Tde Jackson Laboratory | Jax stock 013093 |
| Mouse: R26tdTomato: Rosa26tm14(CAG-tdTomato)Hze/J | Tde Jackson Laboratory | Jax stock 007914 |
| Mouse: Sox9GFP:Sox9IRES-EGFP | Tde Jackson Laboratory | Jax stock 030137 |
| Mouse ES cell: Cenpjtm1a(EUCOMM.)Wtsi | European Mouse Mutant Cell Repository | EPD0028_7_G05 |
| Mouse: p53 KO: B6.129S2-Trp53tm1Tyj/J | Tde Jackson Laboratory | Jax stock 002101 |
| Mouse: p53LoxP: B6.129P2-Trp53tm1Brn/J | Tde Jackson Laboratory | Jax stock 008462 |
| Mouse: ACTB:FLPe: B6;SJL-Tg(ACTFLPe)9205Dym/J | Tde Jackson Laboratory | Jax stock 005703 |
| Mouse: β-actin-cre: B6N.FVB-Tmem163Tg(ACTB-cre)2Mrt/CjDswJ | Tde Jackson Laboratory | Jax stock 019099 |
| Oligonucleotides | ||
| qPCR-Fgf9-F: GGAGTTGGATATACCTCGCCT | Tdis paper | N/A |
| qPCR-Fgf9-R: TGATCCATACAGCTCCCCCT | Tdis paper | N/A |
| qPCR-Fgf10-F: GGAGATGTCCGCTGGAGAAG | Tdis paper | N/A |
| qPCR-Fgf10-R: GCTGTTGATGGCTTTGACGG | Tdis paper | N/A |
| qPCR-Fgfr2-F: TTTCCTCCGAGATCATCGCC | Tdis paper | N/A |
| qPCR-Fgfr2-R: ACCATGGGCTACCTGCAATG | Tdis paper | N/A |
| qPCR-TAp63-F: ATTCAGTTGGAGCAAGGGGG | Tdis paper | N/A |
| qPCR-TAp63-R: CACGAGAAATGAGCTGGGGT | Tdis paper | N/A |
| qPCR-DNp63-F: CAGCCTTGACCAGTCTCACT | Tdis paper | N/A |
| qPCR-DNp63-R: TGTCTTCGCAATCTGGCAGT | Tdis paper | N/A |
| qPCR-Dolk-F: CAGTGTGGGACCGATACTCCT | (Kopinke et al., 2017) | N/A |
| qPCR-Dolk-R: CCAAGCAAAGGCATGACCA | (Kopinke et al., 2017) | N/A |
| qPCR-Hprt-F: CATAACCTGGTTCATCATCGC | (Kopinke et al., 2017) | N/A |
| qPCR-Hprt-R: TCCTCCTCAGACCGCTTTT | (Kopinke et al., 2017) | N/A |
| qPCR-Pde12-F: ACCTTTTGGGTGCCAGTAGA | (Kopinke et al., 2017) | N/A |
| qPCR-Pde12-R: CCAGAGGTCATCTGTCCTTCA | (Kopinke et al., 2017) | N/A |
| Software and Algoritdms | ||
| SDS. Software version 2.3 | Tdermo Fisher | https://www.tdermofisher.com/us/en/home/technical-resources/software-downloads/applied-biosystems-7500-real-time-pcr-system.html |
| QuantStudio™ Design & Analysis 1.3.1 | Tdermo Fisher | https://www.tdermofisher.com/us/en/home/global/forms/life-science/quantstudio-3-5-software.html |
| Prism 8 for Windows | GraphPad Software | www.graphpad.com |
| CellProfiler version 3.1.9 | (McQuin et al., 2018) | https://cellprofiler.org/releases/ |
| R Studio | Rstudio | https://rstudio.com/products/rstudio/download/ |
| R | Tde R Foundation | https://cran.r-project.org/mirrors.html |
| Galaxy | (Afgan et al., 2018) | https://usegalaxy.org/ |
| FIJI | (Schindelin et al., 2012) | https://imagej.net/Fiji |
| Zen version 3.1 blue edition | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
Highlights.
Endodermal centrioles are critical for lung branching but not for intestine development
Centriole loss activates p53 to induce apoptosis in lung SOX2-expressing cells
ERK activity throughout the small intestine protects it from p53-mediated apoptosis
A gradient of ERK activity in the lung shapes which cells die to block branching
Acknowledgement
We are grateful to Ace Lewis and Jeffrey Bush at UCSF for generating Sox2 conditional knockout embryos. We thank David Erle, Andrea Barczak, Matthew Aber and Joshua Rudolph at the UCSF Functional Genomics Core Facility, and Lauren Byrnes for help with RNAseq sequencing and analysis. We thank E Yu for husbandry and genotyping of the mice used in this study. We thank Ross J. Metzger, Francisco Hernan Espinoza and Mark A. Krasnow at Stanford University, Nan Tang at the National Institute of Biological Sciences, Beijing, and Gail R. Martin at UCSF for thoughtful discussions. We also thank the members of the Reiter lab for critical comments and suggestions on the study. This work was funded by NIH R01GM095941, R01AR054396, and R01HD089918 to JFR and GVA PROMETEO/2019/075 to JMGV.
Footnotes
Declaration of interests: The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdelhamed ZA, Natarajan S, Wheway G, Inglehearn CF, Toomes C, Johnson CA, and Jagger DJ (2015). The Meckel-Gruber syndrome protein TMEM67 controls basal body positioning and epithelial branching morphogenesis in mice via the non-canonical Wnt pathway. Dis Model Mech 8, 527–541. 10.1242/dmm.019083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afgan E, Baker D, Batut B, van den Beek M, Bouvier D, Cech M, Chilton J, Clements D, Coraor N, Gruning BA, et al. (2018). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res 46, W537–W544. 10.1093/nar/gky379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn S, and Joyner AL (2004). Dynamic changes in the response of cells to positive hedgehog signaling during mouse limb patterning. Cell 118, 505–516. 10.1016/j.cell.2004.07.023. [DOI] [PubMed] [Google Scholar]
- Al-Dosari MS, Shaheen R, Colak D, and Alkuraya FS (2010). Novel CENPJ mutation causes Seckel syndrome. J Med Genet 47, 411–414. 10.1136/jmg.2009.076646. [DOI] [PubMed] [Google Scholar]
- Alanis DM, Chang DR, Akiyama H, Krasnow MA, and Chen J (2014). Two nested developmental waves demarcate a compartment boundary in the mouse lung. Nat Commun 5, 3923. 10.1038/ncomms4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen MA, Andrysik Z, Dengler VL, Mellert HS, Guarnieri A, Freeman JA, Sullivan KD, Galbraith MD, Luo X, Kraus WL, et al. (2014). Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. Elife 3, e02200. 10.7554/eLife.02200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson-Rolf A, Fink J, Mustata RC, and Koo BK (2014). A video protocol of retroviral infection in primary intestinal organoid culture. J Vis Exp, e51765. 10.3791/51765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arman E, Haffner-Krausz R, Gorivodsky M, and Lonai P (1999). Fgfr2 is required for limb outgrowth and lung-branching morphogenesis. Proc Natl Acad Sci U S A 96, 11895–11899. 10.1073/pnas.96.21.11895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubrey BJ, Kelly GL, Janic A, Herold MJ, and Strasser A (2018). How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ 25, 104–113. 10.1038/cdd.2017.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azimzadeh J, Wong ML, Downhour DM, Sanchez Alvarado A, and Marshall WF (2012). Centrosome loss in the evolution of planarians. Science 335, 461–463. 10.1126/science.1214457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangs FK, Schrode N, Hadjantonakis AK, and Anderson KV (2015). Lineage specificity of primary cilia in the mouse embryo. Nat Cell Biol 17, 113–122. 10.1038/ncb3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basson MA, Echevarria D, Ahn CP, Sudarov A, Joyner AL, Mason IJ, Martinez S, and Martin GR (2008). Specific regions within the embryonic midbrain and cerebellum require different levels of FGF signaling during development. Development 135, 889–898. 10.1242/dev.011569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basto R, Lau J, Vinogradova T, Gardiol A, Woods CG, Khodjakov A, and Raff JW (2006). Flies without centrioles. Cell 125, 1375–1386. 10.1016/j.cell.2006.05.025. [DOI] [PubMed] [Google Scholar]
- Bazzi H, and Anderson KV (2014). Acentriolar mitosis activates a p53-dependent apoptosis pathway in the mouse embryo. Proc Natl Acad Sci U S A 111, E1491–1500. 10.1073/pnas.1400568111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers MF, Wali A, Eckenhoff MF, Feinstein SI, Fisher JH, and Fisher AB (1992). An antibody with specificity for surfactant protein C precursors: identification of pro-SP-C in rat lung. Am J Respir Cell Mol Biol 7, 368–378. 10.1165/ajrcmb/7.4.368. [DOI] [PubMed] [Google Scholar]
- Bellusci S, Grindley J, Emoto H, Itoh N, and Hogan BL (1997). Fibroblast growth factor 10 (FGF10) and branching morphogenesis in the embryonic mouse lung. Development 124, 4867–4878. [DOI] [PubMed] [Google Scholar]
- Bond J, Roberts E, Springell K, Lizarraga SB, Scott S, Higgins J, Hampshire DJ, Morrison EE, Leal GF, Silva EO, et al. (2005). A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat Genet 37, 353–355. 10.1038/ng1539. [DOI] [PubMed] [Google Scholar]
- Boucherat O, Landry-Truchon K, Berube-Simard FA, Houde N, Beuret L, Lezmi G, Foulkes WD, Delacourt C, Charron J, and Jeannotte L (2015a). Epithelial inactivation of Yy1 abrogates lung branching morphogenesis. Development 142, 2981–2995. 10.1242/dev.120469. [DOI] [PubMed] [Google Scholar]
- Boucherat O, Nadeau V, Berube-Simard FA, Charron J, and Jeannotte L (2014). Crucial requirement of ERK/MAPK signaling in respiratory tract development. Development 141, 3197–3211. 10.1242/dev.110254. [DOI] [PubMed] [Google Scholar]
- Boucherat O, Nadeau V, Berube-Simard FA, Charron J, and Jeannotte L (2015b). Crucial requirement of ERK/MAPK signaling in respiratory tract development. Development 142, 3801. 10.1242/dev.131821. [DOI] [PubMed] [Google Scholar]
- Bowen ME, and Attardi LD (2019). The role of p53 in developmental syndromes. J Mol Cell Biol 11, 200–211. 10.1093/jmcb/mjy087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen ME, McClendon J, Long HK, Sorayya A, Van Nostrand JL, Wysocka J, and Attardi LD (2019). The Spatiotemporal Pattern and Intensity of p53 Activation Dictates Phenotypic Diversity in p53-Driven Developmental Syndromes. Dev Cell 50, 212–228 e216. 10.1016/j.devcel.2019.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosh R, Sarig R, Natan EB, Molchadsky A, Madar S, Bornstein C, Buganim Y, Shapira T, Goldfinger N, Paus R, and Rotter V (2010). p53-dependent transcriptional regulation of EDA2R and its involvement in chemotherapy-induced hair loss. FEBS Lett 584, 2473–2477. 10.1016/j.febslet.2010.04.058. [DOI] [PubMed] [Google Scholar]
- Brown RC, and Lemmon BE (2011). Dividing without centrioles: innovative plant microtubule organizing centres organize mitotic spindles in bryophytes, the earliest extant lineages of land plants. AoB Plants 2011, plr028. 10.1093/aobpla/plr028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carraro G, del Moral PM, and Warburton D (2010). Mouse embryonic lung culture, a system to evaluate the molecular mechanisms of branching. J Vis Exp. 10.3791/2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaikuad A, Tacconi EM, Zimmer J, Liang Y, Gray NS, Tarsounas M, and Knapp S (2014). A unique inhibitor binding site in ERK1/2 is associated with slow binding kinetics. Nat Chem Biol 10, 853–860. 10.1038/nchembio.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan HY, V S, Xing X, Kraus P, Yap SP, Ng P, Lim SL, and Lufkin T (2011). Comparison of IRES and F2A-based locus-specific multicistronic expression in stable mouse lines. PLoS One 6, e28885. 10.1371/journal.pone.0028885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport JR, Watts AJ, Roper VC, Croyle MJ, van Groen T, Wyss JM, Nagy TR, Kesterson RA, and Yoder BK (2007). Disruption of intraflagellar transport in adult mice leads to obesity and slow-onset cystic kidney disease. Curr Biol 17, 1586–1594. 10.1016/j.cub.2007.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doxsey S, McCollum D, and Theurkauf W (2005). Centrosomes in cellular regulation. Annu Rev Cell Dev Biol 21, 411–434. 10.1146/annurev.cellbio.21.122303.120418. [DOI] [PubMed] [Google Scholar]
- Dumont J, and Desai A (2012). Acentrosomal spindle assembly and chromosome segregation during oocyte meiosis. Trends Cell Biol 22, 241–249. 10.1016/j.tcb.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson JM, Conduit SE, Feeney SJ, Hakim S, DiTommaso T, Fulcher AJ, Sriratana A, Ramm G, Horan KA, Gurung R, et al. (2017). INPP5E regulates phosphoinositide-dependent cilia transition zone function. J Cell Biol 216, 247–263. 10.1083/jcb.201511055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, and Vogelstein B (1993). WAF1, a potential mediator of p53 tumor suppression. Cell 75, 817–825. 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- Faisst AM, Alvarez-Bolado G, Treichel D, and Gruss P (2002). Rotatin is a novel gene required for axial rotation and left-right specification in mouse embryos. Mech Dev 113, 15–28. 10.1016/s0925-4773(02)00003-5. [DOI] [PubMed] [Google Scholar]
- Feldser DM, Kostova KK, Winslow MM, Taylor SE, Cashman C, Whittaker CA, Sanchez-Rivera FJ, Resnick R, Bronson R, Hemann MT, and Jacks T (2010). Stage-specific sensitivity to p53 restoration during lung cancer progression. Nature 468, 572–575. 10.1038/nature09535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg K, Wuttke A, Quadrato G, Chumakov PM, Wizenmann A, and Di Giovanni S (2013). The tumor suppressor p53 fine-tunes reactive oxygen species levels and neurogenesis via PI3 kinase signaling. J Neurosci 33, 14318–14330. 10.1523/JNEUROSCI.1056-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons DL, Byers LA, and Kurie JM (2014). Smoking, p53 mutation, and lung cancer. Mol Cancer Res 12, 3–13. 10.1158/1541-7786.MCR-13-0539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonczy P (2012). Towards a molecular architecture of centriole assembly. Nat Rev Mol Cell Biol 13, 425–435. 10.1038/nrm3373. [DOI] [PubMed] [Google Scholar]
- Harfe BD, Scherz PJ, Nissim S, Tian H, McMahon AP, and Tabin CJ (2004). Evidence for an expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell 118, 517–528. 10.1016/j.cell.2004.07.024. [DOI] [PubMed] [Google Scholar]
- Harper JW, Adami GR, Wei N, Keyomarsi K, and Elledge SJ (1993). The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75, 805–816. 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- Huangfu D, and Anderson KV (2005). Cilia and Hedgehog responsiveness in the mouse. Proc Natl Acad Sci U S A 102, 11325–11330. 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson JW, Kozarova A, Cheung P, Macmillan JC, Swallow CJ, Cross JC, and Dennis JW (2001). Late mitotic failure in mice lacking Sak, a polo-like kinase. Curr Biol 11, 441–446. 10.1016/s0960-9822(01)00117-8. [DOI] [PubMed] [Google Scholar]
- Izraeli S, Lowe LA, Bertness VL, Good DJ, Dorward DW, Kirsch IR, and Kuehn MR (1999). The SIL gene is required for mouse embryonic axial development and left-right specification. Nature 399, 691–694. 10.1038/21429. [DOI] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, and Weinberg RA (1994). Tumor spectrum analysis in p53-mutant mice. Curr Biol 4, 1–7. 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- Junttila MR, Karnezis AN, Garcia D, Madriles F, Kortlever RM, Rostker F, Brown Swigart L, Pham DM, Seo Y, Evan GI, and Martins CP (2010). Selective activation of p53-mediated tumour suppression in high-grade tumours. Nature 468, 567–571. 10.1038/nature09526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karachaliou N, Rosell R, and Viteri S (2013). The role of SOX2 in small cell lung cancer, lung adenocarcinoma and squamous cell carcinoma of the lung. Transl Lung Cancer Res 2, 172–179. 10.3978/j.issn.2218-6751.2013.01.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastenhuber ER, and Lowe SW (2017). Putting p53 in Context. Cell 170, 1062–1078. 10.1016/j.cell.2017.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopinke D, Roberson EC, and Reiter JF (2017). Ciliary Hedgehog Signaling Restricts Injury-Induced Adipogenesis. Cell 170, 340–351 e312. 10.1016/j.cell.2017.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzaro D, Price M, de Felice M, and Di Lauro R (1991). The transcription factor TTF-1 is expressed at the onset of thyroid and lung morphogenesis and in restricted regions of the foetal brain. Development 113, 1093–1104. [DOI] [PubMed] [Google Scholar]
- Lessel D, Wu D, Trujillo C, Ramezani T, Lessel I, Alwasiyah MK, Saha B, Hisama FM, Rading K, Goebel I, et al. (2017). Dysfunction of the MDM2/p53 axis is linked to premature aging. J Clin Invest 127, 3598–3608. 10.1172/JCI92171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Jiao J, Li H, Wan H, Zheng C, Cai J, and Bao S (2018). Histone arginine methylation by Prmt5 is required for lung branching morphogenesis through repression of BMP signaling. J Cell Sci 131. 10.1242/jcs.217406. [DOI] [PubMed] [Google Scholar]
- Lin C, Yao E, Zhang K, Jiang X, Croll S, Thompson-Peer K, and Chuang PT (2017). YAP is essential for mechanical force production and epithelial cell proliferation during lung branching morphogenesis. Elife 6. 10.7554/eLife.21130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YN, Lee YS, Li SK, and Tang TK (2020). Loss of CPAP in developing mouse brain and its functional implication for human primary microcephaly. J Cell Sci 133. 10.1242/jcs.243592. [DOI] [PubMed] [Google Scholar]
- Liu Y, Stein E, Oliver T, Li Y, Brunken WJ, Koch M, Tessier-Lavigne M, and Hogan BL (2004). Novel role for Netrins in regulating epithelial behavior during lung branching morphogenesis. Curr Biol 14, 897–905. 10.1016/j.cub.2004.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13, 133–140. 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney JE, Mori M, Szymaniak AD, Varelas X, and Cardoso WV (2014). The hippo pathway effector Yap controls patterning and differentiation of airway epithelial progenitors. Dev Cell 30, 137–150. 10.1016/j.devcel.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino S, Vooijs M, van Der Gulden H, Jonkers J, and Berns A (2000). Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev 14, 994–1004. [PMC free article] [PubMed] [Google Scholar]
- Marshall CB, Mays DJ, Beeler JS, Rosenbluth JM, Boyd KL, Santos Guasch GL, Shaver TM, Tang LJ, Liu Q, Shyr Y, et al. (2016). p73 Is Required for Multiciliogenesis and Regulates the Foxj1-Associated Gene Network. Cell Rep 14, 2289–2300. 10.1016/j.celrep.2016.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marteil G, Guerrero A, Vieira AF, de Almeida BP, Machado P, Mendonca S, Mesquita M, Villarreal B, Fonseca I, Francia ME, et al. (2018). Over-elongation of centrioles in cancer promotes centriole amplification and chromosome missegregation. Nat Commun 9, 1258. 10.1038/s41467-018-03641-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuin C, Goodman A, Chernyshev V, Kamentsky L, Cimini BA, Karhohs KW, Doan M, Ding L, Rafelski SM, Thirstrup D, et al. (2018). CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol 16, e2005970. 10.1371/journal.pbio.2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger RJ, Klein OD, Martin GR, and Krasnow MA (2008). The branching programme of mouse lung development. Nature 453, 745–750. 10.1038/nature07005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers EN, Lewandoski M, and Martin GR (1998). An Fgf8 mutant allelic series generated by Cre- and Flp-mediated recombination. Nat Genet 18, 136–141. 10.1038/ng0298-136. [DOI] [PubMed] [Google Scholar]
- Miller SA, Adornato M, Briglin A, Cavanaugh M, Christian T, Jewett K, Michaelson C, Monoson T, Price F, Tignor J, and Tyrell D (1999). Domains of differential cell proliferation suggest hinged folding in avian gut endoderm. Dev Dyn 216, 398–410. . [DOI] [PubMed] [Google Scholar]
- Min H, Danilenko DM, Scully SA, Bolon B, Ring BD, Tarpley JE, DeRose M, and Simonet WS (1998). Fgf-10 is required for both limb and lung development and exhibits striking functional similarity to Drosophila branchless. Genes Dev 12, 3156–3161. 10.1101/gad.12.20.3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris EJ, Jha S, Restaino CR, Dayananth P, Zhu H, Cooper A, Carr D, Deng Y, Jin W, Black S, et al. (2013). Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov 3, 742–750. 10.1158/2159-8290.CD-13-0070. [DOI] [PubMed] [Google Scholar]
- Nemajerova A, Kramer D, Siller SS, Herr C, Shomroni O, Pena T, Gallinas Suazo C, Glaser K, Wildung M, Steffen H, et al. (2016). TAp73 is a central transcriptional regulator of airway multiciliogenesis. Genes Dev 30, 1300–1312. 10.1101/gad.279836.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichane M, Javed A, Sivakamasundari V, Ganesan M, Ang LT, Kraus P, Lufkin T, Loh KM, and Lim B (2017). Isolation and 3D expansion of multipotent Sox9(+) mouse lung progenitors. Nat Methods 14, 1205–1212. 10.1038/nmeth.4498. [DOI] [PubMed] [Google Scholar]
- Pargellis C, Tong L, Churchill L, Cirillo PF, Gilmore T, Graham AG, Grob PM, Hickey ER, Moss N, Pav S, and Regan J (2002). Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat Struct Biol 9, 268–272. 10.1038/nsb770. [DOI] [PubMed] [Google Scholar]
- Pucilowska J, Puzerey PA, Karlo JC, Galan RF, and Landreth GE (2012). Disrupted ERK signaling during cortical development leads to abnormal progenitor proliferation, neuronal and network excitability and behavior, modeling human neuro-cardio-facial-cutaneous and related syndromes. J Neurosci 32, 8663–8677. 10.1523/JNEUROSCI.1107-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Que J, Okubo T, Goldenring JR, Nam KT, Kurotani R, Morrisey EE, Taranova O, Pevny LH, and Hogan BL (2007). Multiple dose-dependent roles for Sox2 in the patterning and differentiation of anterior foregut endoderm. Development 134, 2521–2531. 10.1242/dev.003855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raballo R, Rhee J, Lyn-Cook R, Leckman JF, Schwartz ML, and Vaccarino FM (2000). Basic fibroblast growth factor (Fgf2) is necessary for cell proliferation and neurogenesis in the developing cerebral cortex. J Neurosci 20, 5012–5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlins EL, Clark CP, Xue Y, and Hogan BL (2009). The Id2+ distal tip lung epithelium contains individual multipotent embryonic progenitor cells. Development 136, 3741–3745. 10.1242/dev.037317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder CL, Faruki S, and Khodjakov A (2001). The centrosome in vertebrates: more than a microtubule-organizing center. Trends Cell Biol 11, 413–419. 10.1016/s0962-8924(01)02085-2. [DOI] [PubMed] [Google Scholar]
- Rodrigues-Martins A, Riparbelli M, Callaini G, Glover DM, and Bettencourt-Dias M (2008). From centriole biogenesis to cellular function: centrioles are essential for cell division at critical developmental stages. Cell Cycle 7, 11–16. 10.4161/cc.7.1.5226. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz A, Sankaralingam P, O'Connell KF, and Muller-Reichert T (2018). Revisiting Centrioles in Nematodes-Historic Findings and Current Topics. Cells 7. 10.3390/cells7080101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine K, Ohuchi H, Fujiwara M, Yamasaki M, Yoshizawa T, Sato T, Yagishita N, Matsui D, Koga Y, Itoh N, and Kato S (1999). Fgf10 is essential for limb and lung formation. Nat Genet 21, 138–141. 10.1038/5096. [DOI] [PubMed] [Google Scholar]
- Shaham O, Smith AN, Robinson ML, Taketo MM, Lang RA, and Ashery-Padan R (2009). Pax6 is essential for lens fiber cell differentiation. Development 136, 2567–2578. 10.1242/dev.032888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Lin Q, and Su B (2014). Human-specific hypomethylation of CENPJ, a key brain size regulator. Mol Biol Evol 31, 594–604. 10.1093/molbev/mst231. [DOI] [PubMed] [Google Scholar]
- Stevens NR, Dobbelaere J, Wainman A, Gergely F, and Raff JW (2009). Ana3 is a conserved protein required for the structural integrity of centrioles and basal bodies. J Cell Biol 187, 355–363. 10.1083/jcb.200905031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens NR, Raposo AA, Basto R, St Johnston D, and Raff JW (2007). From stem cell to embryo without centrioles. Curr Biol 17, 1498–1503. 10.1016/j.cub.2007.07.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui P, Li R, Zhang Y, Tan C, Garg A, Verheyden JM, and Sun X (2019). E3 ubiquitin ligase MDM2 acts through p53 to control respiratory progenitor cell number and lung size. Development 146. 10.1242/dev.179820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang CJ, Lin SY, Hsu WB, Lin YN, Wu CT, Lin YC, Chang CW, Wu KS, and Tang TK (2011). The human microcephaly protein STIL interacts with CPAP and is required for procentriole formation. EMBO J 30, 4790–4804. 10.1038/emboj.2011.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toki T, Yoshida K, Wang R, Nakamura S, Maekawa T, Goi K, Katoh MC, Mizuno S, Sugiyama F, Kanezaki R, et al. (2018). De Novo Mutations Activating Germline TP53 in an Inherited Bone-Marrow-Failure Syndrome. Am J Hum Genet 103, 440–447. 10.1016/j.ajhg.2018.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrell FK, Kerr EM, Gao M, Thorpe H, Doherty GJ, Cridge J, Shorthouse D, Speed A, Samarajiwa S, Hall BA, et al. (2017). Lung tumors with distinct p53 mutations respond similarly to p53 targeted therapy but exhibit genotype-specific statin sensitivity. Genes Dev 31, 1339–1353. 10.1101/gad.298463.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand JL, Brady CA, Jung H, Fuentes DR, Kozak MM, Johnson TM, Lin CY, Lin CJ, Swiderski DL, Vogel H, et al. (2014). Inappropriate p53 activation during development induces features of CHARGE syndrome. Nature 514, 228–232. 10.1038/nature13585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicent S, Lopez-Picazo JM, Toledo G, Lozano MD, Torre W, Garcia-Corchon C, Quero C, Soria JC, Martin-Algarra S, Manzano RG, and Montuenga LM (2004). ERK1/2 is activated in non-small-cell lung cancer and associated with advanced tumours. Br J Cancer 90, 1047–1052. 10.1038/sj.bjc.6601644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volckaert T, Campbell A, Dill E, Li C, Minoo P, and De Langhe S (2013). Localized Fgf10 expression is not required for lung branching morphogenesis but prevents differentiation of epithelial progenitors. Development 140, 3731–3742. 10.1242/dev.096560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YL, Anzola JV, Davis RL, Yoon M, Motamedi A, Kroll A, Seo CP, Hsia JE, Kim SK, Mitchell JW, et al. (2015). Reversible centriole depletion with an inhibitor of Polo-like kinase 4. Science 348, 1155–1160. 10.1126/science.aaa5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HW, Chung M, Kudo T, and Meyer T (2017). Competing memories of mitogen and p53 signalling control cell-cycle entry. Nature 549, 404–408. 10.1038/nature23880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Inesta-Vaquera F, Niepel M, Zhang J, Ficarro SB, Machleidt T, Xie T, Marto JA, Kim N, Sim T, et al. (2012). Discovery of potent and selective covalent inhibitors of JNK. Chem Biol 19, 140–154. 10.1016/j.chembiol.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu K, Wang J, Zhu J, Jiang J, Shou J, and Chen X (1999). p53 induces TAP1 and enhances the transport of MHC class I peptides. Oncogene 18, 7740–7747. 10.1038/sj.onc.1203235. [DOI] [PubMed] [Google Scholar]
- Zorn AM, and Wells JM (2009). Vertebrate endoderm development and organ formation. Annu Rev Cell Dev Biol 25, 221–251. 10.1146/annurev.cellbio.042308.113344. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: RNAseq of control (ShhCre Cenpj+/lox), Ift88 loss of function (ShhCre Ift88−/lox) (cilia-disrupted) and Cenpj loss of function (ShhCre Cenpj−/lox) (centriole-disrupted) lungs, Related to Figure 3.
Table S2: RNAseq of control (ShhCre Cenpj+/lox), Ift88 loss of function (ShhCre Ift88−/lox) (cilia-disrupted) and Cenpj loss of function (ShhCre Cenpj−/lox) (centriole-disrupted) small intestine, Related to Figure 3.
Data Availability Statement
RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Original western blot images have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
This manuscript did not generate new code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.