

Abstract
Lysobacter are new biocontrol agents known for their prolific production of lytic enzymes and bioactive metabolites. L. enzymogenes is a predator of fungi and produces several structurally distinct antimicrobial compounds, such as the antifungal HSAF and analogs. The mechanism by which L. enzymogenes interacts with fungal preys is not well understood. Here, we found that the production of HSAF and analogs in L. enzymogenes OH11 was significantly induced in media supplemented with ground fungal mycelia or chitin. In OH11 genome, we identified a gene (LeLPMO10A) that was annotated to encode a chitin-binding protein. The stimulation of HSAF and analogs by chitin was diminished when LeLPMO10A was deleted. We expressed the gene in E. coli and demonstrated that purified LeLPMO10A oxidatively cleaved chitin into oligomeric products, including 1,5 δ-lactones and aldonic acids. The results revealed that LeLPMO10A encodes a lytic polysaccharide monooxygenase, which has not been reported in Lysobacter. Metabolite analysis, antifungal assay, and proteomic analysis showed that the antifungal compounds and the chitin-cleaving LeLPMO10A are co-localized in outer membrane vesicles (OMVs). The enzymatic products resulted from in vitro LeLPMO10A-cleaved chitin also significantly induced HSAF and analogs in OH11. Scanning electron microscopic analysis indicated that spherical vesicles were formed outside OH11 cells and fewer OH11 cells were observed to attach to fungal hyphae when LeLPMO10A was deleted. Together, the study revealed a previously uncharacterized synergistic strategy utilized by the predatory Lysobacter during interacting with fungal preys.
Keywords: Lysobacter enzymogenes, HSAF, lytic polysaccharide monooxygenases, outer membrane vesicles, predator-prey interaction
Graphical Abstract
INTRODUCTION
Lysobacter is a genus of Gram-negative gliding bacteria that have emerged as new biocontrol agents owing to the production of a large number of lytic enzymes and bioactive natural products.1 Several species of the genus are facultative predators of surrounding microorganisms.2 For example, L. enzymogenes exhibits potent activity against a broad spectrum of fungal and oomycete pathogens of plants, including Rhizoctonia solani, Bipolaris sorokiniana, Magnaporthe poae, Fusarium graminearum, Puccinia graminis, Uromyces appendiculatus, and Pythium ultimum.3-7 In addition, L. enzymogenes produces bioactive metabolites that show efficacy against the life-threatening human pathogens Aspergillus fumigatus and Candida albicans.8-10 The main metabolite conferring L. enzymogenes the potent antifungal and anti-oomycete activity is HSAF (Heat Stable Antifungal Factor), which is a polycyclic tetramate macrolactam (PoTeM) (Figure 1).11 Several HSAF analogs have been identified from L. enzymogenes, including alteramides, 3-deOH HSAF, and 3-deOH alteramides.12, 13 This group of natural products has a distinct chemical structure differing from any existing antifungal antibiotics or fungicides and probably possesses new modes of action.8-10 In the recent years, PoTeM has become one of the active areas in natural product research that has attracted researchers in natural product chemistry, chemical biology, and plant pathology.14-26
Figure 1.

Chemical structure of HSAF and its analogs produced by Lysobacter enzymogenes OH11.
Investigating the mechanism by which L. enzymogenes delivers the antifungal factors to microbial targets is important, because the studies could reveal new insights into the interactions between the predatory bacterium and its prey organisms. The understanding could also help devise new strategies for developing biocontrol agents with an improved efficacy. So far, few studies had been carried in this area. Meers and coworkers found that L. enzymogenes strain C3 was able to release outer membrane vesicles (OMVs) as delivery vehicles for fungal killing or growth inhibition.27 Their results indicated that the OMVs, with a diameter of approximately 130-150 nm, contained proteins, nucleic acids, phospholipids and lipopolysaccharides. Notably, nearly all of the extracellular heat-stable antifungal activity, most likely HSAF and analogs, was present in the OMVs. However, the molecular nature of the components in the OMVs was not very clear. Moreover, studies are needed to figure out potential functions of the macromolecules in the OMVs during the predatory interactions with fungi. In Lysobacter sp. XL1 and L. capsici VKM B-2533(T), OMVs were found to contain bacteriolytic enzymes, which could lyse surrounding bacteria and enable the producer to gain advantage in competition.28, 29
Gram-negative bacteria are known to produce OMVs under certain circumstances, often as a response to stress.30-32 As the vesicles bud from the outer membrane, cellular components encapsulated in OMVs, such as DNA, RNA, proteins, endotoxins and virulence factors, would also get delivered, which could mediate microbial interactions, pathogenesis, stress responses, and signaling.33 Interestingly, most reported OMV-mediated microbial interactions were found within bacterial communities and rarely inter-kingdom, such as between bacteria-fungi. Although Lysobacter species are not considered fungal pathogens, the OMVs secreted by L. enzymogenes strain C3 appeared to exert the antifungal activity through physical binding to the fungal cells and even probably entering into the target cells to transfer molecules.27 However, it is not clear how the OMVs would break into the fungal cells.
In this work, we aimed to identify if and what fungal components would have an impact on the production of the antifungal compounds in L. enzymogenes, to determine the main antifungal compounds in OMVs, and to identify factors that would facilitate the predatory bacterium to break into the recalcitrant polysaccharides, such as chitin, in fungal cell walls. Our results showed that HSAF and analogs in L. enzymogenes OH11 were exported through the vesicular delivery mechanism. We identified and characterized the first Lysobacter lytic polysaccharide monooxygenase (LPMO), LeLPMO10A, which oxidatively cleaves chitin of fungal cell walls. We showed that the LeLPMO10A-cleaved chitin products function as inducers for HSAF and analogs in L. enzymogenes OH11. Finally, bottom-up proteomic analysis by liquid chromatography and tandem mass spectrometry (LC-MS/MS) revealed the co-localization of the small antifungal compounds and the fungal cell wall-cleaving enzyme LeLPMO10A in OMVs of L. enzymogenes OH11. The OMV-mediated co-delivery of the antifungal compounds and the redox enzyme would provide a synergistic strategy for the predator. LeLPMO10A is able to break the recalcitrant polysaccharides in fungal cell walls for better access for other lytic enzymes produced by OH11 and meantime the LPMO-cleaved fungal cell wall products would serve as inducers for more HSAF and analogs production, which would in turn inhibit the fungal growth and provide more substrates for LeLPMO10A.
RESULTS AND DISCUSSION
Fungi stimulated production of HSAF and analogs in L. enzymogenes.
To improve the yield of HSAF and analogs in L. enzymogenes, we tested various growth conditions and potential stimulants. Considering that L. enzymogenes OH11 was originally isolated from plant rhizosphere and possesses antagonistic activity against a broad spectrum of fungi,34, 35 we chose several common fungal pathogens of plants, Fusarium graminearum, F. verticillioides, and Alternaria alternata, to test if the addition of the fungal extracts could stimulate the yield of the antifungal compounds. Strain OH11 was cultured in 1/10 TSB, a medium that is commonly used for production of HSAF and its analogs,11, 36 and the fungal mycelia were minced into powder and added as supplements to 1/10 TSB medium. The HPLC results showed that the yield of HSAF and its analogs in OH11 was significantly increased in a dose-dependent manner (Figure 2). HSAF increased 1.38, 2.70, 4.54, and 5.12 fold in the culture containing respectively 0.1%, 0.5%, 1%, and 1.5% of the F. graminearum supplements, when compared to that without the fungal supplements. For HSAF analogs, alteramides production increased 1.53, 2.58, 3.77 and 4.47 fold respectively, and 3-deOH analogs increased by 1.05, 1.42, 1.63 and 1.79 fold respectively (Figure 2A and 2B). Similar results were obtained when A. alternata and F. verticillioides were used as supplements. In cultures containing 1% of A. alternata and F. verticillioides, HSAF production increased 2.94 and 2.84 fold, alteramides production increased 2.31 and 2.36 fold, and 3-deOH analogs production increased 1.53 and 1.47 fold (Figure 2C and 2D). These results clearly showed that the fungal supplements contain factor(s) that can boost the Lysobacter species to produce the antifungal compounds.
Figure 2.
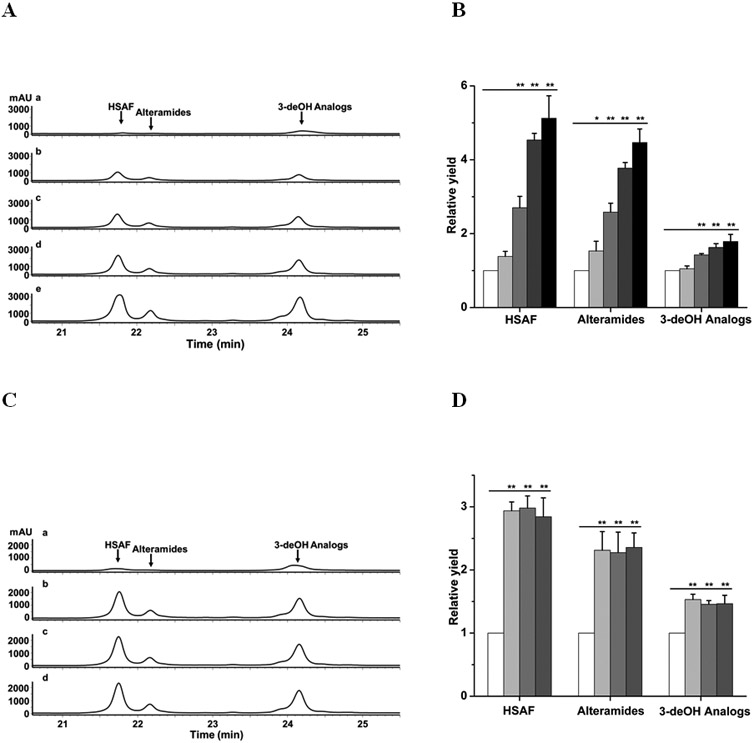
Induction of the antifungal HSAF and analogs in L. enzymogenes OH11 by fungi. (A) HPLC analysis of HSAF and analogs from OH11 cultured for 48 h in 1/10 TSB supplemented with an increased amount (0%, 0.1%, 0.5%, 1% and 1.5%, w/v) of mycelia preparation of Fusarium graminearum. Traces a-e indicate HPLC of cultures supplemented with 0%, 0.1%, 0.5%, 1% and 1.5% (w/v), respectively, of the mycelia preparation. (B) Quantitative analysis of HSAF and analogs in these cultures. White column, light-gray column, medium-gray column, dark-gray column, and black column indicate cultures supplemented with 0%, 0.1%, 0.5%, 1% and 1.5% (w/v), respectively, of the mycelia preparation. (C) HPLC analysis of HSAF and analogs from OH11 cultured for 48 h in 1/10 TSB supplemented with 1% mycelia preparation of different fungi. Traces a-d indicate HPLC of cultures with no supplement, with Alternaria alternata, with F. graminearum, and with F. verticillioides, respectively. (D) Quantitative analysis of HSAF and analogs in these cultures. White column, light-gray column, medium-gray column, and dark-gray column indicate cultures with no supplement, with A. alternata, with F. graminearum, and with F. verticillioides, respectively. Data are presented as averages of three independent experiments, each conducted in triplicates, with * p < 0.05, ** p < 0.01.
Chitin stimulated production of HSAF and analogs in L. enzymogenes.
We figured that fungal cell walls would be a main component of the mycelia powder that was added into the L. enzymogenes OH11 cultures. Fungal cell walls are comprised of glycoproteins and polysaccharides, mainly β-1,3-glucan and chitin.37 To test if β-1,3-glucan and chitin are involved in the stimulation of HSAF and analogs, we used β-1,3-glucan and chitin as a supplement in 1/10 TSB cultures of OH11. The results showed that β-1,3-glucan had no obvious effect on HSAF and its analogs (data not shown). However, supplementing 1% (w/v) chitin to the OH11 cultures led to a significant enhancement of HSAF and analogs (Figure 3A). The increased level of HSAF and analogs was similar to that from the cultures supplemented with 1% (w/v) fungal mycelia powder. The results suggest that chitin in the fungal cell walls could serve as a stimulating factor for production of the antifungal compounds in L. enzymogenes OH11.
Figure 3.
Induction of the antifungal HSAF and analogs in L. enzymogenes OH11 and in mutant ΔLeLPMO10A by chitin. (A) OH11 (a and b) and ΔLeLPMO10A (c) were cultured for 48 h in 1/10 TSB supplemented with 1% of mycelia preparation of F. graminearum (a) or 1% chitin (b and c). (B) Quantitative analysis of HSAF and analogs in OH11 (white column) and ΔLeLPMO10A (light-grey column) with the 1% chitin supplement. Data are presented as averages of three independent experiments, each conducted in triplicates, with ** p < 0.01.
Deletion of LeLPMO10A diminished the chitin-induced production of HSAF and analogs in L. enzymogenes.
To understand the potential mechanism by which chitin induces L. enzymogenes OH11 to produce the antifungal compounds, we searched the genome sequence of strain OH11 genome for genes that might be involved in the utilization of chitin. Aside from chitinase genes which had been reported previously,38, 39 we found a gene (named LeLPMO10A in this study) that was predicted to code for a “chitin-binding protein”. The sequence analysis indicated that the protein LeLPMO10A consists of two domains, with the N-terminal domain homologous to lytic polysaccharide monooxygenases (LPMO) of the AA10 family and C-terminal domain homologous to cellulose binding domains (CBDs, also known as CBMs, carbohydrate-binding modules) (Figure S1A).40-42 LPMO is a relatively new family of monooxygenases that play an important role in the enzymatic depolymerization of recalcitrant polysaccharides such as chitin and cellulose.43-45 Different from the hydrolytic enzymes such as β-1,3-glucanases and chitinases, LPMO is a mono-copper enzyme that oxidatively cleaves the glycosidic bonds of polysaccharides. Several families of LPMOs have been identified from various microorganisms, but so far none has been identified from Lysobacter. LeLPMO10A in L. enzymogenes OH11 contains the three highly conserved amino acid residues (His-His-Phe) for coordinating the copper ion and shares other conserved residues with LPMOs known to use chitin as substrate (Figure S1B). To find out whether LeLPMO10A plays a role in the chitin-induced production of HSAF and analogs, we performed in-frame deletion of the gene in strain OH11 and confirmed the identity of the mutant strain ΔLeLPMO10A (Figure S2). The production of HSAF and analogs in the mutant was compared to OH11, which showed that deletion of this gene significantly decreased the production of HSAF, alteramides and 3-deOH analogs by 53%, 41% and 47%, respectively (Figure 3B). The results suggest that LeLPMO10A may play an important role in the chitin-induced production of HSAF and analogs.
Purified LeLPMO10A bound to chitin and catalyzed oxidative degradation of chitin in vitro.
To further understand the function of LeLPMO10A in Lysobacter-fungi interactions, we expressed the gene in E. coli. The protein was produced in the periplasmic space to help the protein’s posttranslational processing, as described for heterologous expression of several LPMO proteins.41 To make sure LeLPMO10A could be translocated to periplasmic space of E. coli cells, we replaced its original signal peptide sequence with the PelB signal sequence (Figure S3). The PelB N-terminal signal peptide is a part of the pET-26b vector, which will be cleaved off during translocation of the protein to the periplasmic space. This cleavage results in the conserved histidine becoming the first amino acid residue in the mature protein, which is essential for the copper binding in the active site. The purified LeLPMO10A gave a band at ~34 kDa (calculated 34.02 kDa) on SDS-PAGE (Figure S3). With the purified protein, we tested its binding to chitin and cellulose. The result showed that the LeLPMO10A band on SDS-PAGE after interacting with chitin was markedly more intense than the LeLPMO10A band interacting with cellulose (Figure S4), indicating that LeLPMO10A has a binding preference for chitin than cellulose. As a control, BSA did not exhibit a clear binding band to chitin or cellulose.
To confirm LeLPMO10A is indeed a lytic polysaccharide monooxygenase, we carried in vitro enzyme activity assays using chitin as substrate. MALDI-MS analysis of the reaction products showed that LeLPMO10A oxidatively cleaved the C-1 glycosidic bond in the insoluble chitin polymers, as evidenced by the soluble chitin oligosaccharides with different degrees of polymerization (DP), including DP3ox, DP4ox, DP5ox and DP6ox (Figure 4A). Both the lactone form and the hydrolyzed form (aldonic acid) were detected for the DPox products, and the aldonic acid form was the majority of the soluble products, as exemplified in the more detailed MS analysis of DP6ox (Figure 4B-C).
Figure 4.

Products of the reaction catalyzed by LeLPMO10A with chitin as substrate. (A) MALDI-MS analysis of the reaction products, shown as degree of polymerization of oxidized oligosaccharides (DPox). (B) An enlarged view of the ions in DP6ox complex. (C) Chemical structures and predicted masses of the ions in the DP6ox complex, including the sodium adduct of the 1,5 δ-lactone, the sodium adduct, potassium adduct, and sodium/potassium adduct of the aldonic acid.
OMVs mediated the delivery of HSAF and analogs as well as LeLPMO10A during the Lysobacter-fungi interactions.
To understand the relation between the small molecule antifungal compounds and the chitin-lytic LeLPMO10A during the Lysobacter-fungi interactions, we prepared metabolite extracts from the cultures of strains OH11, ΔHSAF, and ΔLeLPMO10A. The HPLC results and antifungal assays, using F. graminearum as the testing organism, showed that HSAF and analogs were clearly present in both the extracellular fraction and intracellular fraction of OH11 and ΔLeLPMO10A, but not of ΔHSAF (Figure S5). Although a lower level of HSAF and analogs was produced by ΔLeLPMO10A than by OH11, the amount of the antifungal compounds produced in ΔLeLPMO10A was sufficient to generate a clear inhibition zone in the disk-based assay.
Since LeLPMO10A is expected to be exported into the culture media, we prepared protein extracts from the extracellular fraction of the cultures of strains OH11, ΔHSAF, and ΔLeLPMO10A. The protein bands around ~34 kDa on SDS-PAGE were excised and subjected to LC-MSE analysis. The E. coli-produced, purified LeLPMO10A was used as positive control (Figure S6). The results clearly showed that LeLPMO10A was present in strains OH11 and ΔHSAF, but absent in strain ΔLeLPMO10A. The protein sequencing results using LC-MSE showed that the purified LPMO had a sequence coverage of 89.7%, and the extracellular proteins from OH11, ΔHSAF, and ΔLeLPMO10A gave a sequence coverage of 60.9%, 35.0%, and 0%, respectively.
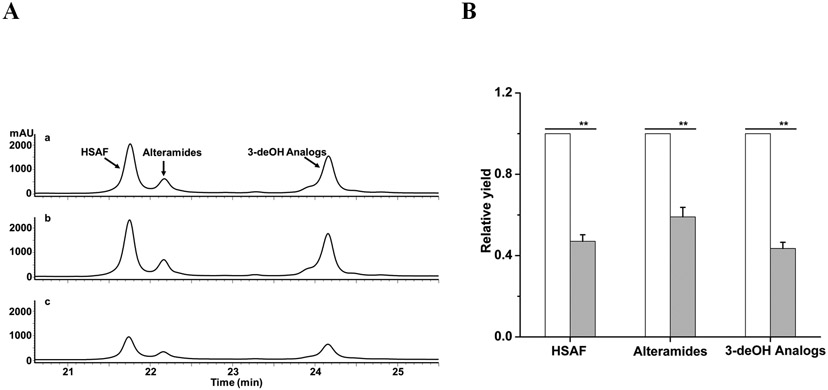
A recent study indicated that essentially all of the heat-stable antifungal activity in the culture supernatant of L. enzymogenes strain C3, a close relative of OH11,14 was found in OMVs. Thus, we set out to isolate OMVs from strains OH11, ΔHSAF, and ΔLeLPMO10A, and examined whether LeLPMO10A was coexist with the antifungal compounds in OMVs. HPLC analysis showed that HSAF and analogs were clearly present in the OMVs from OH11 and ΔLeLPMO10A, but not in the OMVs from ΔHSAF (Figure 5A). Correspondingly, the OMVs from OH11 and ΔLeLPMO10A, but not that from ΔHSAF, exhibited clear antifungal activity against F. graminearum (Figure 5B). The protein sequencing results using LC-MS/MSE showed that the purified LPMO had a sequence coverage of 50.3%, and the OMVs from OH11, ΔHSAF, and ΔLeLPMO10A gave a sequence coverage of 48.3%, 42.0%, and 0%, respectively (Figure 5C-D). Together, the results clearly showed that the small molecule antifungal compounds are co-localized with the chitin-cleaving redox enzyme in OMVs.
Figure 5.


Analysis of OMVs prepared from various strains of L. enzymogenes grown in M813 medium. (A) HPLC of the metabolites in OMVs from the wild type OH11 (a), mutant ΔHSAF (b), and mutant ΔLeLPMO10A (c). Note that in M813 medium, which is suitable for OMV preparation, the main antifungal compounds were HSAF and alteramides, and the 3-deOH compounds were not produced as much as in 10% TSB medium. (B) Antifungal activity assay of the metabolites in OMVs using F. graminearum as testing organism. Each filter paper was added with 5 μl of extract from the OMVs. Filter 1 added with methanol as a negative control; 2 added with total culture extract of OH11; 3, 4 and 5 added with OMVs extracts from OH11, ΔHSAF, and ΔLeLPMO10A, respectively. (C) SDS-PAGE analysis of the OMVs. Lane 1 is purified LeLPMO10A from E. coli BL21(DE3)/LeLPMO10A, as positive control. Lanes 2, 3 and 4 are OMVs from OH11, ΔHSAF, and ΔLeLPMO10A, respectively. M is protein marker. (D) LC-MSE sequencing of the proteins in the excised gel fragments at ~34.02 kDa. Purified LPMO had a sequence coverage of 50.3%, OH11 OMVs gave 48.3%, ΔHSAF OMVs gave 42.0%, and ΔLeLPMO10A OMV had zero sequence coverage. Underlines indicate the exact peptides that were observed and matched to the amino acid sequence of LeLPMO10A.
LeLPMO10A-cleaved chitin products and N-acetylglucosamine induced production of HSAF and analogs in L. enzymogenes.
The fact that OH11 uses OMVs to co-deliver the chitin-cleaving redox enzyme and the antifungal compounds encouraged us to search for potential connections between LeLPMO10A and the observed stimulation of HSAF and analogs by fungal mycelia and chitin. First, we prepared the oligomeric products resulted from the in vitro chitin-lytic reaction catalyzed by the purified LeLPMO10A (Figure 4). The oligomeric products were then added into Lysobacter cultures as supplements. In strain ΔLeLPMO10A, the production of HSAF increased approximately 8 fold upon supplementing the LeLPMO10A-catalyzed reaction products (Figure 6). The production of other analogs increased approximately 2-5 fold. Interestingly, non-lysed chitin could also stimulate strain ΔLeLPMO10A to produce the antifungal compounds, but at a lower level (~4 fold for HSAF and ~1-3 fold for analogs). Since LeLPMO10A gene had been deleted in strain ΔLeLPMO10A, the results imply that other chitin-degrading enzymes, such as chitinases, might also have contributed to the observed stimulation of HSAF and analogs. As control experiments, the production of HSAF and analogs increased significantly in the wild type strain OH11, but not in strain ΔHSAF, upon adding non-lysed chitin or the LeLPMO10A-cleave chitin products (Figure 6). In OH11, the level of the antifungal compounds stimulated by non-lysed chitin was notably higher than that stimulated by the LeLPMO10A-cleaved chitin products. The result is consistent with that from ΔLeLPMO10A, which suggests that other enzymes in the wild type, such as chitinases, also have contributed to the observed stimulation of the antifungal compounds. To provide evidence for this notion, we tested the effect of N-acetylglucosamine (GlcNAc), the ultimate product (monomer) of chitin hydrolysis by chitinases, on HSAF and its analogs. When GlcNAc was utilized as a supplement in cultures, the production of HSAF, alteramides, and the 3-deOH analogs in OH11 increased 3.1, 2.5 and 1.6 fold, respectively, when compared to the culture without GlcNAc. In ΔLeLPMO10A, the production of HSAF, alteramides, and the 3-deOH analogs slightly increased (1.5, 1.6 and 1.1 fold, respectively), when compared the culture without GlcNAc (Figure S7). The difference observed in OH11 and in ΔLeLPMO10A is consistent with the observations that both GlcNAc and the oxidized products have inductive effects on HSAF and analogs. OH11 can release GlcNAc (by chitinases) as well as the oxidized products (by LeLPMO10A), while ΔLeLPMO10A can only release GlcNAc.
Figure 6.

Induction of HSAF and analogs in L. enzymogenes OH11 by the LeLPMO10A-cleaved chitin oligomeric products. White, light-gray and dark-gray columns: wild type OH11 in 1/10 TSB, in 1/10 TSB supplemented with 1% chitin, and in 1/10 TSB supplemented with 1% chitin cleaved by LeLPMO10A, respectively. Light-yellow, yellow and dark-yellow columns: mutant ΔHSAF in 1/10 TSB, in 1/10 TSB supplemented with 1% chitin, and in 1/10 TSB supplemented with 1% chitin cleaved by LeLPMO10A, respectively. Light-purple, purple and dark-purple columns: ΔLeLPMO10A in 1/10 TSB, in 1/10 TSB supplemented with 1% chitin, and in 1/10 TSB supplemented with 1% chitin cleaved by LeLPMO10A, respectively. Data is presented as averages of three independent experiments, each conducted in triplicates, with ** p < 0.01.
SEM images indicated extracellular vesicles and interactions with fungal hyphae.
Extracellular spherical structures with a diameter of approximately 50-150 nm were observed in SEM images of L. enzymogenes OH11 (Figure S8A). The vesicles tended to cluster together to form network-like structures outside of the rod-shaped Lysobacter cells. When the wild type strain was incubated with the hyphae of F. graminearum, SEM images indicated that a large number of OH11 cells attached to the fungal hyphae. The longer incubation time led to more OH11 cells on the surface of fungal hyphae. When the mutant strain ΔLeLPMO10A incubated with the hyphae of F. graminearum, the number of Lysobacter cells attached to the fungal hyphae was clearly lower than that of the wild type (Figure S8B). Physical interactions between L. enzymogenes and oomycetes were observed previously. Strain SB-K88 of L. enzymogenes could perpendicularly attach to and densely colonize on the hyphae surface of Aphanomyces cochlioides.46 Strain OH11 could attach, penetrate and lyse the hyphae of P. aphanidermatum.47 Our results here provided evidence for production of OMVs in strain OH11 and further supported the in vivo and in vitro studies describe above, which showed that LeLPMO10A plays an important role during the Lysobacter-fungi interactions.
Conclusions.
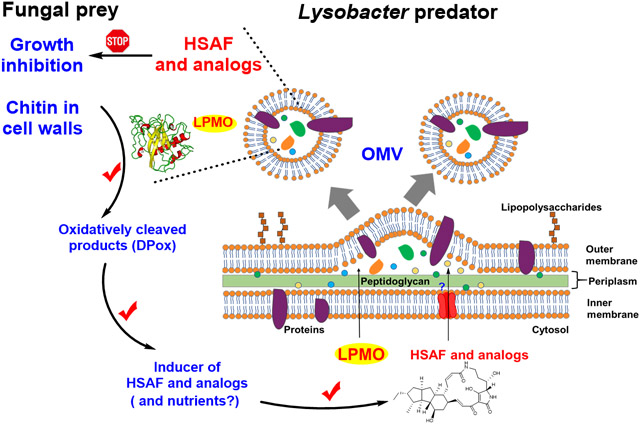
L. enzymogenes has attracted much attention from researchers for its potent antifungal and anti-oomycete activity and potential applications in agricultural, environmental, and pharmaceutical industries. The activity and application are attributed to the production of the structurally intriguing antifungal compounds HSAF and analogs, as well as the production of a large number of lytic enzymes. Many studies have shown that OMVs play an important role in stress response and can deliver virulence factors to host cells.33 One main research interest in OMVs has been on pathogenesis. L. enzymogenes OH11 is not a plant pathogen; it is an environmental bacterium originally isolated from the rhizosphere soil of pepper plants.34 The benefits of OMVs for non-pathogenic bacteria like Lysobacter are not well appreciated. Moreover, the mechanisms by which OMVs mediate microbial interactions still remain unclear. The study presented here revealed a synergistic strategy utilized by the Lysobacter species during interactions with the fungi-preys (Figure 7). Our results show that this predatory bacterium has evolved a smart mechanism, in which a “wall opener” (the chitin-cleaving LeLPMO10A) and “warheads” (the small molecule antifungal compounds) are packaged together into a lethal cargo (OMV) to coordinately kill the fungal preys. The cleaved products of the fungal cell walls serve as inducers for the antifungal compounds in L. enzymogenes OH11 that will execute the fungal growth inhibition and killing. The more fungal cell walls are broken down, the more antifungal compounds are induced, which in turn leads to the more fungal attack. The inhibited fungal cells then become available for degradation by the wall-breaking enzymes in OMVs (including LPMO, chitinases, and 1,3-glucansases). Meantime, as more fungi are “captured” as preys, OH11 would acquire more nutrients to support a larger population for growth and secrete more HSAF and analogs for further attack on the fungal preys.
Figure 7.

Schematic illustration of the OMV-mediated co-delivery of the antifungal HSAF metabolites (warheads) and the copper-containing lytic polysaccharide monooxygenase (cell wall opener) of the predatory L. enzymogenes OH11 during interactions with fungal preys.
Furthermore, this OMV-mediated co-delivery mechanism provides an explanation to the previous observation in strain C3, in which the fungal inhibitory activity was dependent on physical contact between OMVs and the fungal cells.27 As metal-containing monooxygenases, LPMOs are able to cleave recalcitrant polysaccharides such as crystalline chitin in fungal cell walls.41-45 Hydrolases, such as β-1,3-glucanases and chitinases, are not efficient in utilizing the recalcitrant substrates with crystalline packing. On the other hand, LPMOs can bind to the surface of the crystalline polysaccharides and use its catalytic copper ion to activate molecular oxygen to disrupt the recalcitrant packing of the cell walls. Once broken by the wall-opener enzyme, the cell wall polymers would become more accessible by the hydrolases. A physical contact of the OMVs with the fungal cells would facilitate the process. The physical contact and fungal cell wall breaking could also lead to some of OMV components entering into the fungal cells as observed previously.27
EXPERIMENTAL METHODS
Microbial strains and growth conditions.
The bacterial strains and plasmids utilized in this study are included in Table S1. Escherichia coli strain XL-1 Blue was used for plasmids propagation, strain BL21(DE3) for production of LeLPMO10A, and strain S17 for intergeneric conjugation. The wild type L. enzymogenes strain OH11 (CGMCC No. 1978) was used for production of HSAF and its analogs.36 Plasmid pJQ200SK was used to construct vector to obtain the gene deletion mutant ΔLeLPMO10A from L. enzymogenes OH11.11 Luria-Bertani (LB) broth medium was used for growth of OH11. M813 modified medium (3 g K2HPO4, 1.38 g NaH2PO4·H2O, 1 g NH4Cl, 0.15 g KCl, 2.5 mg FeSO4, 0.15 g MgSO4, 10 mg CaCl2, and 4 g glucose, per liter) and 1/10 Tryptic Soy Broth (3g TSB, per liter) were used for production of HSAF and its analogs. Martin medium (50 g peptone, 10 g KH2PO4, 5 g MgSO4·7H2O, 100 g glucose, per liter) was used to prepare fungal cultures, which grew at 30 °C with shaking at 200 rpm for 7 days. Fungal mycelia were collected by vacuum filtration via filter paper and washed thrice with deionized water. The mycelia were freeze-dried in a Labconco FreeZone 1 lyophilizer and minced into powder, which was added as culture supplements to 1/10 TSB that was used for production of HSAF and its analogs. When chitin was used as a culture supplement, chitin power from shrimp shells (Sigma-Aldrich, USA) was added to 1/10 TSB to make a 1% (w/v) supplement for production of HSAF and its analogs.
Construction of in-frame gene deletion mutant ΔLeLPMO10A.
The flanking regions of the target gene LeLPMO10A were amplified by PCR using primers LeLPMO10A-UF/UR and LeLPMO10A-DF/DR listed in Table S2, respectively. The upstream fragment was 547 bp, and the downstream fragment was 582 bp. After treating the upstream fragment with XbaI/BamHI and the downstream fragment with BamHI/XhoI, the two DNA fragments were ligated into the XhoI/XbaI sites of the plasmid pJQ200SK, to generate the recombination plasmid pJQ200SK::LeLPMO10A. The plasmid was transformed into L. enzymogenes OH11 through intergeneric conjugation with E. coli S17 that contains the construct. After antibiotic and sucrose screening, the putative transformants were confirmed by diagnostic PCR using primers LeLPMO10A-VF and LeLPMO10A-VR listed in Table S2.
Expression of LeLPMO10A in E. coli BL21(DE3).
LeLPMO10A gene was cloned into pET26b using a restriction enzyme-free cloning method.48 Plasmid pET26b contains a signal peptide that is responsible for guiding the produced protein to the periplasmic space of the host cells. The first step was to use PCR to amplify the gene from the OH11 genomic DNA using primers LeLPMO10A-amp1 and LeLPMO10A-amp2 (Table S2). The second step was to use the PCR product as the primer and plasmid pET26b as the template to amplify the entire expression vector, pET26b-LeLPMO10A. Restriction enzyme DpnI was used to digest the methylated parental plasmid, and the DpnI-treated vector pET26b-LeLPMO10A was transformed into E. coli XL-1 Blue. The expression construct was confirmed by DNA sequencing (Eurofins MWG Operon LLC) and then transformed into E. coli BL21(DE3). Strain BL21(DE3)/pET26b-LeLPMO10A was cultured in 1 mL LB medium containing 50 μg/ml kanamycin at 37 °C, with shaking at 200 rpm for 12 h. The culture was transferred to 50 mL fresh LB medium and, when the cell density reached OD600 0.6, added with IPTG (Isopropyl β-D-1-thiogalactopyranoside, 0.5 mM). After growing at 16 °C overnight, the cells were collected and treated by cold osmotic shock to extract the periplasmic proteins from the cells.49 The periplasmic extract was adjusted to 1 M (NH4)2SO4 and 50 mM Tris-HCl (pH 8.0), and LeLPMO10A was purified using a Ni-NTA (nickel-nitrilotriacetic acid) column (GE Healthcare). The purity and size of LeLPMO10A was analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
Chitin binding assay and enzymatic activity assay of LeLPMO10A.
The binding assay of LeLPMO10A was conducted in 1.5 mL micro-centrifuge tubes containing 1% (w/v) of chitin (from shrimp shells, Sigma-Aldrich, USA) or fibrous cellulose (Sigma-Aldrich, USA). LeLPMO10A (100 μg/mL) or BSA (Sigma-Aldrich, USA, 100 μg/mL) in 20 mM Bis-Tris buffer (pH 6.0) was added to the tubes. The mixtures were incubated in a 30°C shaker for 24 h. To prevent nonspecific binding, the substrate was pelleted by centrifugation and washed thrice with 1 mL 20 mM Bis-Tris buffer. The pellets were resuspended in SDS-PAGE loading buffer and boiled for 10 min. An aliquot (10 μL) of each of the samples was analyzed by SDS-PAGE.
To measure the enzymatic activity of LeLPMO10A, the reaction was performed in 2 mL microcentrifuge tubes containing 0.1% (w/v) of substrate in 20 mM Bis-Tris buffer (pH 6.0), 1 mM ascorbic acid as reducing agent, and 1 μM LeLPMO10A. The reaction mixture was incubated at 30°C for 24 h. The reaction was stopped, and the supernatant was collected by centrifugation (14,000 × g) for 10 min. To prepare the samples for MALDI-MS analysis, an aliquot of 0.75 μL sample was mixed with an equal amount of 10 mg/mL 2,5-dihydroxybenzoic acid (DHB) in 40% acetonitrile/0.1% trifluoroacetic acid (TFA) on an Opti-TOF 384 target plate. The spotted samples were dried with air flow and analyzed on a MALDI-MS instrument (15 T Bruker Solarix XR).
Preparation of extracellular, intracellular proteins, and OMVs from L. enzymogenes.
The bacterial strains were cultured in 1 mL LB medium at 30 °C overnight. An aliquot (1%) of the cultures was transferred to 50 mL M813 modified medium and grew at 30 °C with shaking at 200 rpm for 48 h. The culture supernatant and bacterial cells were separated by centrifugation (14,000 × g) for 30 min. Bacterial cells were added with 50 mL of the M813 modified medium and sonicated to release the proteins within. The intracellular fraction was collected by centrifugation (14,000 × g) of 30 min and gradually added with (NH4)2SO4 (22.5 g, 75% saturation), to precipitate intracellular proteins at 4 °C overnight. Similarly, the cell-free culture broth was gradually added with (NH4)2SO4 (22.5 g), to precipitate extracellular proteins at 4 °C overnight. Both fractions were centrifuged (14,000 × g) for 30 min to obtain the proteins pellets, which were then redissolved in PBS for further analysis.
To prepare OMVs, the bacterial strains were cultured in 1 mL LB medium at 30 °C overnight. An aliquot (1%) of the cultures was transferred to 50 mL M813 modified medium and grew at 30 °C with shaking at 200 rpm for 48 h. Bacterial cells were removed by centrifugation (14,000 × g) for 30 min. The remaining cell-free supernatants were undergone ultracentrifugation at 140,000 × g for 2 h in a 45ti fixed-angle titanium rotor using a Beckman Coulter Ultracentrifuge. The OMV pellets were collected from the bottom of the polycarbonate ultracentrifuge tubes.
In-gel trypsin digestion and tandem mass spectrometry of proteins.
Protein gels were stained with Coomassie Brilliant Blue (CBB) G-250 prior to in-gel trypsin digestion. The digestion was performed according to a previously reported method.50 Gel slats containing proteins of approximately 34 kDa, from E. coli BL21(DE3)/LeLPMO10A or various strains of L. enzymogenes, were excised from SDS-PAGE gels and cut into approximately 1 mm squares. The gel pieces were de-stained with a freshly prepared acetonitrile with 100 mM NH4HCO3 (1:1 v/v) and allowed to shake for 10 min. The solution was removed, and the de-staining process was repeated until CBB was removed. Acetonitrile was added to dehydrate the gel pieces until completely opaquely white. The gel pieces were added with 20 mM DTT in 100 mM NH4HCO3 and incubated at 56 °C for 45 min to reduce the proteins. The gel pieces were then alkylated using 20 μL of 55 mM iodoacetamide in 100 mM NH4HCO3 in the dark at room temperature for 40 min. All solution was removed, and acetonitrile was added to dehydrate the gel pieces until any residual CBB was removed. The gel pieces were dried in a Speed-vac and bathed in 40 μL of 15 ng/μL trypsin in 50 mM NH4HCO3 to rehydrate for 30 minutes. NH4HCO3 (50 mM) was added to cover in the digestion mixture which was incubated at 37 °C overnight. After digestion, 15 μL of 5% formic acid in 50% acetonitrile (v/v) was used to quench the digestion. The supernatant was collected in a new microcentrifuge tube. The gel pieces were then rinsed with 50 μL of 1% FA in 60% methanol (v/v) at 40 °C for 15 minutes while vortexing occasionally, and the liquid was collected and combined with the previously extracted supernatant. Then the gel pieces were rinsed again with 50 μL of 1% formic acid in 50% acetonitrile (v/v) at 40°C for 15 minutes while vortexing occasionally, and the supernatant was combined with previous extracts. The gel pieces were dried by adding enough acetonitrile and allowed to sit for 15 minutes while vortexing occasionally, and the supernatant was extracted and added to the combined extracts. U-C18 ZipTips with tip size P10 (Millipore, USA) were used following the manufacturer’s instructions to purify and desalt peptides. The samples were dried in a Speed-vac and stored at −20 °C for further analysis.
For protein identification, the peptide samples were subject to LC-MS/MS on a nanoACQUITY UPLC coupled to Xevo G2-XS QToF MS system (Waters, Manchester, UK) using instrumental and experimental parameters reported previously.51 Dried samples were reconstituted with 12 μL of 3% acetonitrile plus 0.1% formic acid prior to MS analysis. Samples were injected onto a nano-LC reverse phase column HSS T3 (75 μm x 150 mm, 1.8 μm particle size, Waters) and separated over a 24 min gradient from 3-40% of B. Mobile phase A was nanowater, with 0.1% formic acid, and mobile phase B was acetonitrile, with 0.1% formic acid. The flow rate was set to 0.35 μL/min and column temperature kept at 35 °C. Peptides were ionized by nanoelectrospray with capillary voltage of 2.1 kV, sample cone of 35.0 V, source temperature of 80 °C, and analyzed by QToF mass spectrometer in sensitivity and positive mode. Data collection was in data independent acquisition mode (MSE), and collision energy between 19-45 V was applied after MS spectrum is collected to provide both MS and MS/MS spectrum of intact peptide ions and peptide fragment ions. Lockspray with [Glu1] fibrinopeptide solution (1 fM/μL) with a reference mass of m/z 785.8427 was utilized to allow for continuous calibration during MSE data acquisitions.
For MS data processing, raw LC-MSE files were processed by ProteinLynx Global Server (PLGS) version 3.0.3. PLGS workflow parameters were set to have an m/z tolerance of 1 ppm, fixed modification of carbamidomethyl of cysteines and variable modification of oxidation of methionine. The database was created from unverified UniProt for L. enzymogenes OH11 (created on December 19, 2019). For the PLGS processing parameters, the lock mass was set to 785.843 amu with a lock mass tolerance of 0.25 amu. The primary digest enzyme was set as trypsin. The minimum amino acids allowed for a peptide fragment was set to 3, with the minimum peptides per protein assignment set at 7. The maximum false positive rate was set at 1. The maximum protein mass was set at 250,000. For peptide product fragments, the ratio of minimum product ion intensity to precursor ion intensity was set to 0.03. For the variable modification (oxidation), the maximum allowed simultaneous modifications was 3. The maximum number of missed cleavages was 1. All peptides determined by processing parameters were manually verified to check that signal to noise of selected ions was over 3 and that the selected monoisotopic mass was not overlapping with other fragment ions.
SEM examination of Lysobacter cells and Lysobacter-fungal hyphae interactions.
Lysobacter cells were fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.2) and post-fixed using 1% osmium tetroxide at room temperature for 1 h respectively. The cells were then processed through a series of ethanol dehydration and critical point dry. After sputter coating with a thin layer of chromium, the samples were imaged using a Hitachi field-emission scanning electron microscope (Hitachi S4700). In experiments to examine interactions with fungal hyphae, Lysobacter cells were collected, washed with water, and re-suspended in water to obtain a solution with a cell density of OD600 1.0. Fusarium graminearum was inoculated to Martin medium and allowed to grow at 30°C in a 200 rpm shaker for 7 days. Fungal mycelia were collected by vacuum filtration through a polyamide filter. The polyamide filter was carefully immersed into the Lysobacter solution for 2 h or 6 h. The samples were fixed and washed with sterile water, followed by the drying process as described above. SEM images were obtained by using Hitachi S4700 Field-Emission SEM.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful for the SEM work by Y. Zhou at the Microscopy Core Research Facility, Center for Biotechnology at University of Nebraska–Lincoln. We thank K. Wulser at Nebraska Center for Mass Spectrometry for technical assistance. We thank V. Eijsink and A.C. Bunæs, Norwegian University of Life and Sciences, for generously providing the construct pRSETB-CBP21 as the positive control for the LeLPMO10A expression and in vitro enzyme activity assays.
Funding
This work was supported in part by University of Nebraska Collaboration Initiative Seed Grant and through the use of core facilities sponsored by the Nebraska Center for Integrated Biomolecular Communication (NIH P20GM113126) and the Nebraska Research Initiative (High-Field FTICR-MS).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/
Microbial strains and plasmids, primer and protein sequences, DNA manipulation and gene deletion, heterologous expression of LeLPMO10A in E. coli, transformation of Lysobacter, extraction and analysis of HSAF and analogs, and antifungal assays, which are included in Tables S1-S2 and Figures S1-S8.
The authors declare no competing financial interest.
REFERENCES
- (1).Xie Y, Wright S, Shen Y, and Du L (2012) Bioactive natural products from Lysobacter. Nat. Prod. Rep 19, 1277–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Seccareccia I, Kost C, and Nett M (2015) Quantitative analysis of Lysobacter predation. Appl. Environ. Microb 81, 7098–7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Zhang Z, and Yuen GY (1999) Biological control of Bipolaris sorakiniana on tall fescue by Stenotrophomonas maltophilia strain C3. Phytopathol. 89, 817–822. [DOI] [PubMed] [Google Scholar]
- (4).Giesler LJ, and Yuen GY (1998) Evaluation of Stenotrophomonas maltophilia strain C3 for biocontrol of brown patch disease. Crop Prot. 17, 509–513. [Google Scholar]
- (5).Jochum CC, Osborne LE, and Yuen GY (2006) Fusarium head blight biological control with Lysobacter enzymogenes. Biol. Control 39, 336–344. [Google Scholar]
- (6).Kobayashi DY, Reedy RM, Palumbo JD, Zhou JM, and Yuen GY (2005) A clp gene homologue belonging to the Crp gene family globally regulates lytic enzyme production, antimicrobial activity, and biological control activity expressed by Lysobacter enzymogenes strain C3. Appl. Environ. Microb 71, 261–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Yuen GY, Steadman JR, Lindgren DT, Schaff D, and Jochum C (2001) Bean rust biological control using bacterial agents. Crop Prot. 20, 395–402. [Google Scholar]
- (8).Li S, Du L, Yuen G, and Harris SD (2006) Distinct ceramide synthases regulate polarized growth in the filamentous fungus Aspergillus nidulans. Mol. Biol. Cell 17, 1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ding Y, Li Z, Li Y, Lu C, Wang H, Shen Y, and Du L (2016) HSAF-induced antifungal effects in Candida albicans through ROS-mediated apoptosis. RSC Adv. 6, 30895–30904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ding Y, Li Y, Li Z, Zhang J, Lu C, Wang H, Shen Y, and Du L (2016) Alteramide B is a microtubule antagonist of inhibiting Candida albicans. Biochim. Biophys. Acta 1860, 2097–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Yu F, Zaleta-Rivera K, Zhu X, Huffman J, Millet JC, Harris SD, Yuen G, Li XC, and Du L (2007) Structure and biosynthesis of heat-stable antifungal factor (HSAF), a broad-spectrum antimycotic with a novel mode of action. Antimicrob. Agents Chemother 51, 64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Xu L, Wu P, Wright SJ, Du L, and Wei X (2015) Bioactive polycyclic tetramate macrolactams from Lysobacter enzymogenes and their absolute configurations by theoretical ECD calculations. J. Nat. Prod 78, 1841–1847. [DOI] [PubMed] [Google Scholar]
- (13).Li Y, Huffman J, Li Y, Du L, and Shen Y (2012) 3-Hydroxylation of the polycyclic tetramate macrolactam in the biosynthesis of antifungal HSAF from Lysobacter enzymogenes C3. MedChemComm 9, 982–986. [Google Scholar]
- (14).Lou L, Qian G, Xie Y, Hang J, Chen H, Zaleta-Rivera K, Li Y, Shen Y, Dussault PH, Liu F, and Du L (2011) Biosynthesis of HSAF, a tetramic acid-containing macrolactam from Lysobacter enzymogenes. J. Am. Chem. Soc 133, 643–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Lou L, Chen H, Cerny RL, Li Y, Shen Y, and Du L (2012) Unusual activities of the thioesterase domain for the biosynthesis of the polycyclic tetramate macrolactam HSAF in Lysobacter enzymogenes C3. Biochem. 51, 4–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Blodgett JA, Oh DC, Cao S, Currie CR, Kolter R, and Clardy J (2010) Common biosynthetic origins for polycyclic tetramate macrolactams from phylogenetically diverse bacteria. Proc. Natl. Acad. Sci. U S A 107, 11692–11697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Luo Y, Huang H, Liang J, Wang M, Lu L, Shao Z, Cobb RE, and Zhao H (2013) Activation and characterization of a cryptic polycyclic tetramate macrolactam biosynthetic gene cluster. Nat. Commun 4, 2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zhang G, Zhang W, Zhang Q, Shi T, Ma L, Zhu Y, Li S, Zhang H, Zhao YL, Shi R, and Zhang C (2014) Mechanistic insights into polycycle formation by reductive cyclization in ikarugamycin biosynthesis. Angew. Chem. Int. Ed. Engl 53, 4840–4844. [DOI] [PubMed] [Google Scholar]
- (19).Saha S, Zhang W, Zhang G, Zhu Y, Chen Y, Liu W, Yuan C, Zhang Q, Zhang H, Zhang L, Zhang W, and Zhang C (2017) Activation and characterization of a cryptic gene cluster reveals a cyclization cascade for polycyclic tetramate macrolactams. Chem. Sci 8, 1607–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Antosch J, Schaefers F, and Gulder TA (2014) Heterologous reconstitution of ikarugamycin biosynthesis in E. coli. Angew. Chem. Int. Ed. Engl 53, 3011–3014. [DOI] [PubMed] [Google Scholar]
- (21).Greunke C, Antosch J, and Gulder TA (2015) Promiscuous hydroxylases for the functionalization of polycyclic tetramate macrolactams--conversion of ikarugamycin to butremycin. Chem. Commun 51, 5334–5336. [DOI] [PubMed] [Google Scholar]
- (22).Greunke C, Glöckle A, Antosch J, and Gulder TAM (2017) Biocatalytic total synthesis of ikarugamycin. Angew. Chem. Int. Ed. Engl 56, 4351–4355. [DOI] [PubMed] [Google Scholar]
- (23).Li Y, Chen H, Ding Y, Xie Y, Wang H, Cerny RL, Shen Y, and Du L (2014) Iterative assembly of two separate polyketide chains by the same single-module bacterial polyketide synthase in the biosynthesis of HSAF. Angew. Chem. Int. Ed. Engl 53, 7524–7530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Li Y, Wang H, Liu Y, Jiao Y, Li S, Shen Y, and Du L (2018) Biosynthesis of the polycyclic system in the antifungal HSAF and analogues from Lysobacter enzymogenes. Angew. Chem. Int. Ed. Engl 57, 6221–6225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Li X, Wang H, Shen Y, Li Y, and Du L (2019) OX4 is an NADPH-dependent dehydrogenase catalyzing an extended Michael addition reaction to form the six-membered ring in the antifungal HSAF. Biochem. 58, 5245–5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Li X, Wang HX, Li YY, and Du LC (2019) Construction of a hybrid gene cluster to reveal coupled ring formation-hydroxylation in the biosynthesis of HSAF and analogues from Lysobacter enzymogenes. MedChemComm 10, 907–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Meers PR, Liu C, Chen R, Bartos W, Davis J, Dziedzic N, Orciuolo J, Kutyla S, Pozo MJ, Mithrananda D, Panzera D, and Wang S (2018) Vesicular delivery of the antifungal antibiotics of Lysobacter enzymogenes C3. Appl. Environ. Microbiol 84, e01353–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kudryakova IV, Shishkova NA, and Vasilyeva NV (2016) Outer membrane vesicles of Lysobacter sp XL1: biogenesis, functions, and applied prospects. Appl. Microbiol. Biotechnol 100, 4791–4801. [DOI] [PubMed] [Google Scholar]
- (29).Afoshin AS, Kudryakova IV, Borovikova AO, Suzina NE, Toropygin IY, Shishkova NA, and Vasilyeva NV (2020) Lytic potential of Lysobacter capsici VKM B-2533(T): bacteriolytic enzymes and outer membrane vesicles, Sci Rep 10, 9944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kulp A, and Kuehn MJ (2010) Biological functions and biogenesis of secreted bacterial outer membrane vesicles. Annu. Rev. Microbiol 64, 163–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ellis TN, and Kuehn MJ (2010) Virulence and immunomodulatory toles of bacterial outer membrane vesicles. Microbiol. Mol. Biol. Rev 74, 81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Kaparakis-Liaskos M, and Ferrero RL (2015) Immune modulation by bacterial outer membrane vesicles. Nat. Rev. Immunol 15, 375–387. [DOI] [PubMed] [Google Scholar]
- (33).Schwechheimer C, and Kuehn MJ (2015) Outer-membrane vesicles from Gram-negative bacteria: biogenesis and functions. Nat. Rev. Microbiol 13, 605–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Qian GL, Hu BS, Jiang YH, and Liu FQ (2009) Identification and characterization of Lysobacter enzymogenes as a biological control agent against some fungal pathogens. Agr. Sci. China 8, 68–75. [Google Scholar]
- (35).Li S, Jochum CC, Yu F, Zaleta-Rivera K, Du L, Harris SD, and Yuen GY (2008) An antibiotic complex from Lysobacter enzymogenes strain C3: antimicrobial activity and role in plant disease control. Phytopathol. 98, 695–701. [DOI] [PubMed] [Google Scholar]
- (36).Wang Y, Zhao Y, Zhang J, Zhao Y, Shen Y, Su Z, Xu G, Du L, Huffman JM, Venturi V, Qian G, and Liu F (2014) Transcriptomic analysis reveals new regulatory roles of Clp signaling in secondary metabolite biosynthesis and surface motility in Lysobacter enzymogenes OH11. Appl. Microbiol. Biotechnol 98, 9009–9020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Bowman SM, and Free SJ (2006) The structure and synthesis of the fungal cell wall. Bioessays 28, 799–808. [DOI] [PubMed] [Google Scholar]
- (38).Zhang Z, Yuen GY, Sarath G, and Penheiter AR (2001) Chitinases from the plant disease biocontrol agent, Stenotrophomonas maltophilia C3. Phytopathol. 91, 204–211. [DOI] [PubMed] [Google Scholar]
- (39).Kobayashi DY, Reedy RM, Bick J, and Oudemans PV (2002) Characterization of a chitinase gene from Stenotrophomonas maltophilia strain 34S1 and its involvement in biological control. Appl. Environ. Microbiol 68, 1047–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Hamre AG, Stromnes AGS, Gustavsen D, Vaaje-Kolstad G, Eijsink VGH, and Sorlie M (2019) Treatment of recalcitrant crystalline polysaccharides with lytic polysaccharide monooxygenase relieves the need for glycoside hydrolase processivity. Carbohyd. Res 473, 66–71. [DOI] [PubMed] [Google Scholar]
- (41).Vaaje-Kolstad G, Westereng B, Horn SJ, Liu ZL, Zhai H, Sorlie M, and Eijsink VGH (2010) An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science 330, 219–222. [DOI] [PubMed] [Google Scholar]
- (42).Forsberg Z, Sorlie M, Petrovic D, Courtade G, Aachmann FL, Vaaje-Kolstad G, Bissaro B, Rohr AK, and Eijsink VGH (2019) Polysaccharide degradation by lytic polysaccharide monooxygenases. Curr. Opin. Struc. Biol 59, 54–64. [DOI] [PubMed] [Google Scholar]
- (43).Hemsworth GR, Henrissat B, Davies GJ, and Walton PH (2014) Discovery and characterization of a new family of lytic polysaccharide monooxygenases. Nat. Chem. Biol 10, 122–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Frandsen KEH, Simmons TJ, Dupree P, Poulsen JCN, Hemsworth GR, Ciano L, Johnston EM, Tovborg M, Johansen KS, von Freiesleben P, Marmuse L, Fort S, Cottaz S, Driguez H, Henrissat B, Lenfant N, Tuna F, Baldansuren A, Davies GJ, Lo Leggio L, and Walton PH (2016) The molecular basis of polysaccharide cleavage by lytic polysaccharide monooxygenases. Nat. Chem. Biol 12, 298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Urresti S, Hemsworth GR, Screeton HC, Walton PH, and Davies GJ (2015) Structure and function of lytic polysaccharide monooxygenases. Acta Crystallogr. A 71, S225–S225. [Google Scholar]
- (46).Islam MT, Hashidoko Y, Deora A, Ito T, and Tahara S (2005) Suppression of damping-off disease in host plants by the rhizoplane bacterium Lysobacter sp strain SB-K88 is linked to plant colonization and antibiosis against soilborne peronosporomycetes. Appl. Environ. Microbiol 71, 3786–3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Zhao Y, Qian G, Chen Y, Du L, and Liu F (2017) Transcriptional and antagonistic responses of biocontrol strain Lysobacter enzymogenes OH11 to the plant pathogenic oomycete Pythium aphanidermatum. Front. Microbiol 8, 1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).van den Ent F, and Lowe J (2006) RF cloning: A restriction-free method for inserting target genes into plasmids. J. Biochem. Bioph. Meth 67, 67–74. [DOI] [PubMed] [Google Scholar]
- (49).Leal TF, and de Sa-Nogueira I (2004) Purification, characterization and functional analysis of an endo-arabinanase (AbnA) from Bacillus subtilis. FEMS Microbiol. Lett 241, 41–48. [DOI] [PubMed] [Google Scholar]
- (50).Shevchenko A, Tomas H, Havlis J, Olsen JV, and Mann M (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc 1, 2856–2860. [DOI] [PubMed] [Google Scholar]
- (51).Leite AL, Lobo JGVM, Pereira HABD, Fernandes MS, Martini T, Zucki F, Sumida DH, Rigalli A, and Buzalaf MAR (2014) Proteomic analysis of gastrocnemius muscle in rats with streptozotocin-induced diabetes and chronically exposed to fluoride. PLoS One 9, e106646. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.