

Cancer predisposition syndromes are caused by inherited mutations in tumor suppressor genes. Remarkably, despite the presence of the faulty tumor suppressor gene in virtually every cell in the body, individuals with a cancer predisposition syndrome develop tumors in specific organs but not others. The underlying mechanisms of the tissue specificity varies depending on the mutated tumor suppressor gene. A recent study by He et al. (1) sheds new light on the tissue specificity of the BAP1 cancer syndrome, which is caused by inheritance of a germline mutation in the tumor suppressor gene BAP1. This familial cancer predisposition syndrome exhibits an increased incidence of specific types of cancers, including mesothelioma, melanocytic tumors, uveal melanoma, renal cell carcinoma and others (2-9). He et al. demonstrate that conditional Bap1 inactivation in mice resulted in cell death in many cell types, but not in mesothelium and melanocytes, the same cell types from which tumors arise in BAP1 mutation carriers in humans. Through experiments using CRISPR-based genetic screens, RNA sequencing (RNA-seq), and chromatin immunoprecipitation (ChIP), the authors provide new insight into the mechanism by which BAP1 loss causes apoptosis in certain tissues but not others.
BAP1 is a classic tumor suppressor gene that follows Knudson’s two hit hypothesis, in which carriers of a heterozygous mutation undergo loss of heterozygosity (LOH) to lose the functional allele in tumors. Paradoxically, despite the fact that BAP1 loss promotes tumorigenesis, BAP1 inactivation causes cell death in many cell types. He et al. showed that Bap1 knockout in mouse embryonic stem (ES) cells, fibroblasts, and keratinocytes caused cell death through the intrinsic apoptosis pathway (1), which is mediated by Bax/Bak oligomerization. Induced Bap1 inactivation in adult mice also caused Bax/Bak-dependent apoptosis in the liver and pancreas, leading to liver damage and pancreatic atrophy. Interestingly, Bap1 inactivation did not cause apoptosis in mesothelium and melanocytes. Because surviving BAP1 inactivation is prerequisite for development of BAP1-deficient tumors, the lack of apoptosis after Bap1 inactivation would provide an explanation for why tumors develop in mesothelium and melanocytes but not other cell types.
The BAP1 gene encodes a deubiquitinase, an enzyme that reverses post-translational modification of proteins with ubiquitin. BAP1 inactivation can occur as a consequence of either missense mutations in the ubiquitin C-terminal hydrolase (UCH) domain in the N-terminal region, or frameshift or nonsense mutations that cause truncation of the BAP1 protein. Because the C-terminal region encodes nuclear localization signals and a regulatory domain necessary for activation of the UCH domain, the truncated proteins often mislocalize in the cytoplasm or lack deubiquitinase activity. Among the substrates that have been reported to be deubiquitinated by BAP1, deubiquitination of histone H2A is best characterized. Histone H2A is monoubiquitinated at Lys119 by the ubiquitin ligase polycomb repressive complex 1 (PRC1) and deubiquitinated by BAP1 (10). Because H2AK119ub is generally a repressive mark for transcription, BAP1 inactivation is expected to cause PRC1-dependent gene repression.
To gain more insight into the apoptosis that follows Bap1 inactivation, He et al. performed a genome-wide CRISPR-Cas9 screen and identified Rnf2 as a gene that promotes the death of Bap1 knockout MEFs (1). Rnf2 is a ubiquitin E3 ligase component of PRC1, responsible for the deposition of the H2AK119ub mark. This finding suggests that apoptosis after Bap1 inactivation requires H2AK119 ubiquitination by Rnf2, an observation consistent with the antagonistic relationship between a ubiquitin ligase and a deubiquitinase for the H2AK119ub mark. Subsequent RNA-seq experiments revealed that Bap1 knockout in ES cells causes Rnf2-dependent down-regulation of the pro-survival genes Bcl2 and Mcl1. Ectopic expression of Bcl2 and Mcl1 prevented apoptotic cell death of Bap1 knockout ES cells, suggesting that the down-regulation of these pro-survival factors is the key event triggering cell death after Bap1 inactivation. This is consistent with the fact that the apoptosis after Bap1 inactivation is Bax/Bak-dependent, given that Bcl2 and Mcl1 promote cell survival by restraining the activity of cell death mediators Bax and Bak, respectively. Subsequently, He et al. employed ChIP-seq to demonstrate that the Rnf2 and Bap1 proteins localize to the Bcl2 promoter in ES cells, and Bap1 knockout leads to an Rnf2-dependent increase in the H2AK119ub mark at the Bcl2 promoter. Therefore, in many cell types, it seems that the basal expression levels of Bcl2 and Mcl1 are maintained through an equilibrium of Rnf2-dependent H2AK119 ubiquitination and Bap1-dependent deubiquitination at their promoters (Figure 1A). When this equilibrium is disturbed by Bap1 inactivation, it results in down-regulation of the pro-survival factors and apoptotic cell death.
Figure 1.
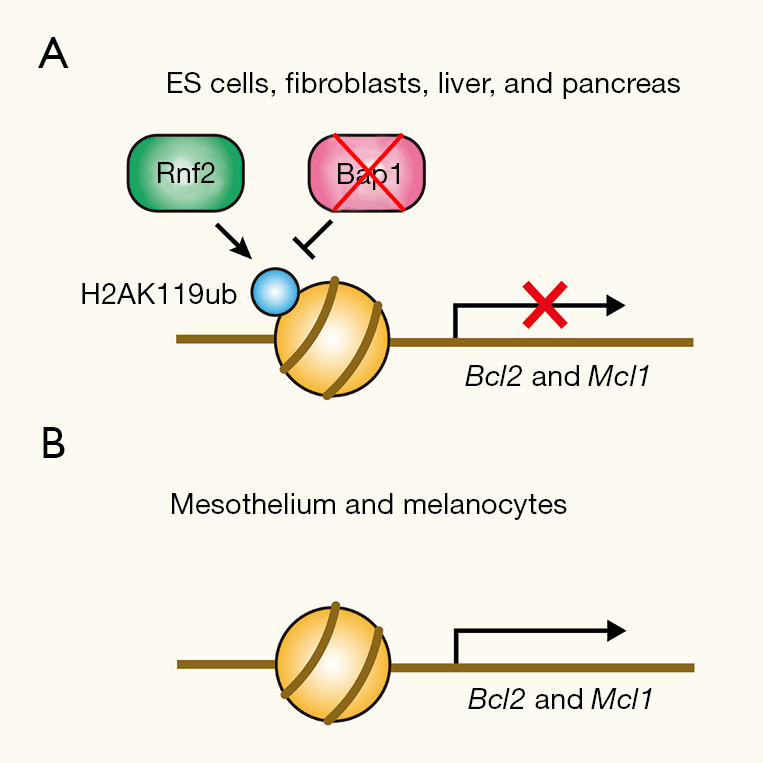
Tissue-specific effects of Bap1 inactivation on pro-survival gene expression. (A) In many tissues, Bcl2 and Mcl1 are regulated by histone H2A monoubiquitination (H2AK119ub), which is maintained by the equilibrium between ubiquitination by Rnf2 and deubiquitination by Bap1. Bap1 inactivation results in elevated H2AK119ub levels at the promoter and causes repression of the pro-survival genes; (B) in mesothelium and melanocytes, Rnf2 and Bap1 are not engaged in the regulation of Bcl2 and Mcl1 expression; therefore, Bap1 inactivation has minimum effect on the pro-survival gene expression.
In stark contrast to ES cells, however, primary melanocytes and mesothelial cells maintained normal levels of Bcl2 and Mcl1 proteins after Bap1 inactivation and did not undergo apoptosis (1). He et al. found that melanocytes and mesothelial cells had minimal Rnf2 binding to the Bcl2 promoter regardless of the Bap1 gene status. Therefore, in melanocytes and mesothelial cells, it appears that Rnf2 does not engage in the regulation of Bcl2 and Mcl1 expression, and as a result, neither does Bap1 (Figure 1B). The authors propose that this difference in the engagement of Rnf2 and Bap1 in the regulation of the pro-survival gene expression determines the cellular fate after Bap1 inactivation. When a cell spontaneously loses functional BAP1 through LOH in BAP1 mutation carriers, that precancerous cell vanishes due to apoptotic cell death in the majority of tissues. In mesothelial and melanocytic tissues, however, such a precancerous cell with inactive BAP1 would persist because apoptosis is not triggered in those cell types. Having survived BAP1 loss, those precancerous cells in melanocytic tissue and mesothelium are poised to respond to additional tumor-promoting effects of BAP1 inactivation.
In fact, He et al. also provided insight into the tumor-promoting effect of BAP1 inactivation in melanocytes (1). The authors showed that melanocyte-inducing transcription factor (Mitf), a melanoma oncogene, is upregulated after Bap1 knockout or inactivation in melanocytes. The molecular mechanism for this upregulation is not provided in this study, but it will be worth investigating whether Mitf is directly regulated by Bap1 and whether the upregulation involves histone H2AK119ub or ubiquitination of other Bap1 substrates such as the transcriptional regulator HCF-1 (11,12) or O-linked N-acetylglucosaminyl transferase (OGT) (13), both of which could influence transcription. The function of BAP1 is also not limited to transcriptional regulation. BAP1 regulates Ca2+ release from endoplasmic reticulum and mitochondria by deubiquitinating and stabilizing inositol 1,4,5-trisphosphate receptor type 3 (IP3R3), promoting apoptosis after genotoxic stress (14). BAP1 is also involved in DNA repair (15,16) although the relevant substrate remains unclear. Given the diversity of the cellular processes that BAP1 regulates and the possibility that BAP1 influences expression of many target genes, it is possible that the tumor-promoting effect of BAP1 inactivation is also tissue-specific. Therefore, it will be important to investigate the mechanism underlying the tumor-promoting effect of BAP1 inactivation in each cell type.
In summary, this study demonstrated that Bap1 loss promotes tumorigenesis only in the tissues in which Bap1 is not engaged in pro-survival gene expression, providing an explanation for the tissue specificity of the BAP1 cancer predisposition syndrome. Somatic mutations in BAP1 have been reported in the sporadic form of mesothelioma, uveal melanoma, and renal cell carcinoma (2,4,17-19), suggesting that the reported mechanism for the tissue specificity applies to the sporadic form as well. However, somatic BAP1 mutations have also been reported in other types of spontaneous tumors including cholangiocarcinoma (20,21), hepatocellular carcinoma (22), thymic epithelial tumors (23), uterine corpus endometrial carcinoma (24), and others. Therefore, a key question will be how spontaneous tumors in other tissues bypass cell death when BAP1 is inactivated. Given that resisting apoptosis is a hallmark of cancer (25), it will be important to understand the preceding oncogenic events that promote survival of BAP1-deficient cancer cells.
Acknowledgments
I thank Scott H. Kaufmann for critical reading of the manuscript.
Funding: None.
Ethical Statement: The author is accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Footnotes
Provenance and Peer Review: This article was commissioned and reviewed by the Section Editor Chen Qian, PhD (Center for Inflammation & Epigenetics, Houston Methodist Hospital Research Institute, Houston, TX, USA).
Conflicts of Interest: The author has completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/tcr.2019.06.41). The author has no conflicts of interest to declare.
References
- 1.He M, Chaurushiya MS, Webster JD, et al. Intrinsic apoptosis shapes the tumor spectrum linked to inactivation of the deubiquitinase BAP1. Science 2019;364:283-5. [DOI] [PubMed] [Google Scholar]
- 2.Testa JR, Cheung M, Pei J, et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet 2011;43:1022-5. 10.1038/ng.912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wiesner T, Obenauf AC, Murali R, et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet 2011;43:1018-21. 10.1038/ng.910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harbour JW, Onken MD, Roberson ED, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 2010;330:1410-3. 10.1126/science.1194472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abdel-Rahman MH, Pilarski R, Cebulla CM, et al. Germline BAP1 mutation predisposes to uveal melanoma, lung adenocarcinoma, meningioma, and other cancers. J Med Genet 2011;48:856-9. 10.1136/jmedgenet-2011-100156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wadt K, Choi J, Chung JY, et al. A cryptic BAP1 splice mutation in a family with uveal and cutaneous melanoma, and paraganglioma. Pigment Cell Melanoma Res 2012;25:815-8. 10.1111/pcmr.12006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Njauw CN, Kim I, Piris A, et al. Germline BAP1 inactivation is preferentially associated with metastatic ocular melanoma and cutaneous-ocular melanoma families. PLoS One 2012;7:e35295. 10.1371/journal.pone.0035295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Farley MN, Schmidt LS, Mester JL, et al. A novel germline mutation in BAP1 predisposes to familial clear-cell renal cell carcinoma. Mol Cancer Res 2013;11:1061-71. 10.1158/1541-7786.MCR-13-0111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Popova T, Hebert L, Jacquemin V, et al. Germline BAP1 mutations predispose to renal cell carcinomas. Am J Hum Genet 2013;92:974-80. 10.1016/j.ajhg.2013.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scheuermann JC, de Ayala Alonso AG, Oktaba K, et al. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature 2010;465:243-7. 10.1038/nature08966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Machida YJ, Machida Y, Vashisht AA, et al. The deubiquitinating enzyme BAP1 regulates cell growth via interaction with HCF-1. J Biol Chem 2009;284:34179-88. 10.1074/jbc.M109.046755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Misaghi S, Ottosen S, Izrael-Tomasevic A, et al. Association of C-terminal ubiquitin hydrolase BRCA1-associated protein 1 with cell cycle regulator host cell factor 1. Mol Cell Biol 2009;29:2181-92. 10.1128/MCB.01517-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dey A, Seshasayee D, Noubade R, et al. Loss of the tumor suppressor BAP1 causes myeloid transformation. Science 2012;337:1541-6. 10.1126/science.1221711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bononi A, Giorgi C, Patergnani S, et al. BAP1 regulates IP3R3-mediated Ca(2+) flux to mitochondria suppressing cell transformation. Nature 2017;546:549-53. 10.1038/nature22798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu H, Pak H, Hammond-Martel I, et al. Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc Natl Acad Sci U S A 2014;111:285-90. 10.1073/pnas.1309085110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ismail IH, Davidson R, Gagne JP, et al. Germline mutations in BAP1 impair its function in DNA double-strand break repair. Cancer Res 2014;74:4282-94. 10.1158/0008-5472.CAN-13-3109 [DOI] [PubMed] [Google Scholar]
- 17.Bott M, Brevet M, Taylor BS, et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat Genet 2011;43:668-72. 10.1038/ng.855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peña-Llopis S S, Vega-Rubin-de-Celis S, Liao A, et al. BAP1 loss defines a new class of renal cell carcinoma. Nat Genet 2012;44:751-9. 10.1038/ng.2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo G, Gui Y, Gao S, et al. Frequent mutations of genes encoding ubiquitin-mediated proteolysis pathway components in clear cell renal cell carcinoma. Nat Genet 2011;44:17-9. 10.1038/ng.1014 [DOI] [PubMed] [Google Scholar]
- 20.Chan-On W, Nairismagi ML, Ong CK, et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat Genet 2013;45:1474-8. 10.1038/ng.2806 [DOI] [PubMed] [Google Scholar]
- 21.Jiao Y, Pawlik TM, Anders RA, et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat Genet 2013;45:1470-3. 10.1038/ng.2813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Woo HG, Choi JH, Yoon S, et al. Integrative analysis of genomic and epigenomic regulation of the transcriptome in liver cancer. Nat Commun 2017;8:839. 10.1038/s41467-017-00991-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petrini I, Meltzer PS, Kim IK, et al. A specific missense mutation in GTF2I occurs at high frequency in thymic epithelial tumors. Nat Genet 2014;46:844-9. 10.1038/ng.3016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature 2013;502:333-9. 10.1038/nature12634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646-74. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]