

Abstract
Background
Accumulating evidences indicate that AXL overexpression or activation is associated with cancer progression and acquired resistance to targeted anti-cancer drugs such as epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKIs). Despite recent development of several drugs that target multiple receptor tyrosine kinases (RTKs), drugs that selectively target AXL signaling are extremely rare. Short nucleic acid aptamers are non-immunogenic molecules with high binding affinity and specificity to their target molecules that could potentially be used as a novel cancer treatment.
Methods
Modified-DNA aptamers were selected on the basis of its ability to bind recombinant human AXL. AXL aptamers were selected for their inhibition of AXL and then selected aptamers were tested for their use to overcome acquired resistant to EGFR-TKI on a lung cancer cell with acquired resistance to erlotinib.
Results
These new AXL aptamers inhibited cell viability to an extent of 30–40% in HCC827/ER cells with acquired resistance to erlotinib. The possible mechanism of overcoming the acquired resistance may be by inhibiting the activation of Akt and Erk. Although, aptamers effectively decreased cell viability of erlotinib‐resistant cell line, the combination of aptamers and erlotinib did not synergistically decrease the survival of the resistant cell line.
Conclusions
We developed newly modified DNA aptamers that selectively bind to AXL receptors, and assessed their efficacy in a human lung cancer cell with acquired resistance to EGFR-TKI.
Keywords: DNA aptamer, AXL, lung cancer, epidermal growth factor receptor-tyrosine kinase inhibitor (EGFR-TKI), acquired resistance
Introduction
AXL belongs to the TAM (Tyro-Axl-Mer) family of receptor tyrosine kinases (RTKs) which involves many biological functions including coagulation, immune response, and cancer progression (1-3). AXL consists of two immunoglobulin-like repeats with dual fibronectin type 3-like repeats in the extracellular domain and its intracellular kinase domain (4). AXL receptor activation occurs via ligand-dependent or ligand-independent receptor dimerization. Growth arrest-specific protein 6 (Gas6) is known to be the only ligand that binds to the extracellular domain of AXL, and receptor homo- or hetero-dimerization with other RTKs leads to the phosphorylation of AXL with subsequent activation of intracellular downstream signaling molecules (4-6). AXL receptor activation triggers the signaling of many downstream pathways such as PI3K/Akt, MAPK, and PKC, thus regulating cellular processes involved in proliferation, survival, and migration (7-10).
Overexpression of AXL has been reported in several human cancers including colon, esophageal, thyroid, breast, lung, and liver (7,11). Several previous studies showed that AXL overexpression or activation is associated with increased motility and invasiveness of cancer cells and further confers acquired resistance to conventional chemotherapy drugs and targeted drugs such as epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKIs), demonstrating its potential as a novel therapeutic target for cancer treatment (12-15).
Although few drugs that show efficacy against multiple RTKs including MET, VEGFR2, and AXL have been developed and are being tested in clinical trials in several types of cancers, drugs that selectively target AXL signaling are extremely rare (16-20). Nucleic acid aptamers are novel therapeutic molecules, composed of short structured single-stranded RNA or DNA with high selective binding affinity to their target molecules (21-24). Aptamers have several major advantages over other therapeutic molecules in terms of high specificity with less toxicity, low cost, convenient synthesis, no immunogenicity, long-term stability by chemical modification, and delivery capability of nanoparticles or chemotherapeutic agents (25-27).
In this context, we developed new and modified-DNA aptamers that selectively bind to AXL receptors, and evaluated their effects on human lung cancer cells that acquired resistance to EGFR-TKI via activation of AXL signaling. We present the following article in accordance with the MDAR checklist (available at http://dx.doi.org/10.21037/tcr-20-2447).
Methods
Modified systematic evolution of ligands by exponential enrichment (SELEX)
The advanced SELEX technology was used as previously described (28). Briefly, aptamers were selected from a DNA library containing a 40-nucleotide randomized region in which 5-(N-benzylcarboxyamide)-2'deoxyuridine (Bz-dU) or 5-(N-naphthylcarboxyamide)-2'-deoxyuridine (Nap-dU) was substituted for dT (29,30). The oligonucleotides contained a central randomized region of 40 nucleotides flanked by two conserved flanking regions of nucleotides (5'-CGAGCGTCCTGCCTTTG-40N-CTGGGTGGCTGTCGGTG-3'). Twenty-five thermal cycles were conducted at 93 °C for 30 s, 52 °C for 20 s, and 72 °C for 60 s. A mixture of 1 nmole of aptamer library dissolved in a buffer solution (40 nM HEPES/pH 7.5, 120 mM NaCl, 5 mM KCl, 5 mM MgCl2, 0.002% Tween20) was heated at 95 °C for about 3 min and then slowly cooled to 30 °C at 0.1 °C/s for re-folding. The binding process was performed at 37 °C. Aptamer library was pre-incubated with His-tag magnetic bead (Invitrogen, Grand Island, NY, USA) to eliminate non-specific binder to magnetic bead. Aptamer library in supernatant were collected and incubated with purified 10 pmole of target protein for 30 min and then target protein was captured by contacting with His tag magnetic beads for 10 min. Aptamers bound to the target protein was eluted with 2 mM NaOH solution and amplified via PCR with normal NTPs, biotin-labeled reverse primer, and non-labeled forward primer, but not including Bz-dU nor Nap-dU. PCR products were collected and captured by streptavidin-coated beads, and sense-strands were eliminated with 2 mM NaOH solution. Modified aptamers were prepared by primer extension, which was performed with forward primer, and NTP mixture that dTTP was substituted with Bz-dU or Nap-dU. The resulting aptamers were used in the next SELEX rounds. The target protein used for aptamers selection were recombinant Human AXL extracellular domain (Met 1-Pro 449), fused with a polyhistidine tag at the C-terminus produced in Human Cell (Creative Biomart, Shirley, NY, USA).
Cloning and sequencing of selected aptamers
After 8 rounds of SELEX, eluted aptamers were amplified by PCR, and then cloned into TA cloning Kit (Solgent, Daejeon, Korea). Fifty clones were picked for each sample and the cloned part were sequenced by Solgent. Sequences were aligned using the ‘aptamer motif searcher’, an in-house program of Aptamer Sciences Inc. (Pohang, Korea).
Binding affinity assays
The aptamer-protein equilibrium dissociation constant (Kd) was determined by the nitrocellulose-filter binding method (31). For all binding assays, aptamers were dephosphorylated using alkaline phosphatase (New England Biolab, Berverly, MA, USA), 5-end labeled using T4 polynucleotide kinase (New England Biolabs) and [32P]-ATP (Amersham Pharmacia Biotech, Piscataway, NJ, USA) (32). Direct binding assays were carried out by incubating 32P-labeled aptamer at a concentration of less than 10 fM and protein at concentrations ranging from 1 mM to 10 fM in selection buffer at 37 °C. The fraction of bound aptamer was quantified with a PhosphorImager (Fuji FLA-5100 Image Analyzer, Tokyo, Japan).
Cell culture and reagents
The human non-small cell lung cancer (NSCLC) cell lines (HCC827/ER and H2009) were kindly provided by Lee JC (Asan Medical Center, Seoul, Korea). Cells were cultured in RPMI1640 medium containing 10% FBS, 2 mmol/L L-glutamine and 100 U/mL of penicillin and streptomycin, and maintained at 37 °C in a humidified chamber containing 5% CO2. Erlotinib and PHA-665752 were purchased from Selleck Chemicals. The cell line used was confirmed to be mycoplasma-free using standard methods.
Cell viability assay
To perform cell viability assay, cells were seeded at a concentration of 5×103 cells in 96-well sterile plastic plates overnight and then treated with relevant agents. After 72 hours, cell viability was determined by a Cell Counting Kit-8 (CCK-8, Dojindo, Kumamoto, Japan) according to the manufacturer’s instructions. The cell viability index was detected at the wavelength of 450 nm using a microplate reader.
Western-blot assay
Cells were lysed in buffer containing 137 mmol/L NaCl, 15 mmol/L EDTA, 0.1 mmol/L sodium orthovanadate, 15 mmol/L MgCl2, 0.1% Triton X-100, 25 mmol/L MOPS, 100 mmol/L phenylmethylsulfonyl fluoride, and 20 mmol/L leupeptin, adjusted to pH 7.2. The antibodies (Abs) used were anti-p-AXL(Tyr702) Ab (5724; Cell Signaling Technology, Beverly, MA, USA), anti-AXL Ab (sc-166269; Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-p-Akt (Ser473) Ab (9271; Cell Signaling Technology), anti-Akt Ab (sc-1618; Santa Cruz Biotechnology), anti-p-Erk (Thr202/Tyr204) Ab (9106; Cell Signaling Technology), anti-Erk Ab (sc-292838; Santa Cruz Biotechnology), and anti-actin Ab (sc-47778; Santa Cruz Biotechnology). Proteins were detected by a LAS-4000 (Fuji, Tokyo, Japan) imaging system using enhanced chemiluminescence reagents (PerkinElmer, Waltham, MA, USA).
Statistical analysis
All data were obtained by at least 3 independent experiments. Statistical analyses were carried out using SPSS Statistics version 24 (IBM Corp, Chicago, IL, USA). Differences between groups were tested using t-test. Data were processed as mean ± standard deviation (± SD), and P value <0.05 was regarded as statistically significant.
Results
Discovery of aptamers that selectively bind to AXL receptors and their effect on cell viability
Using differential advanced-SELEX, we identified modified-DNA aptamers that selectively bind recombinant human AXL. We ultimately selected and sequenced 5 different kinds of AXL aptamers (5, 9, 49, 55, and 81) with high affinity (Tables 1,2), and evaluated their effects on the cell viability of HCC827/ER cells with acquired resistance to erlotinib by the activation of AXL signaling (33). AXL was expressed and phosphorylated in HCC827/ER cells, but not in parental HCC827cells (33). Although no significant difference was noted between 5 different kinds of aptamers (200 nM), all selected aptamers 5, 9, 49, 55, and 81 showed 72.3%, 69.5%, 64.8%, 65.6%, and 75.5% of inhibitory effects on cell viability, respectively (Figure 1A). We investigated changes in previously known intracellular signaling pathway known to be associated with AXL (14,33), and found that phosphorylation of AXL, Akt and Erk were significantly inhibited by AXL aptamers 9, 49, 55, and 81 (Figure 1B). Despite the 72.3% inhibitory effect aptamer 5 had shown, it was excluded as it did not alter AXL, Akt, or Erk activations, implying a different pathway for the inhibition of cell viability. From the four candidates, we chose two aptamers (49 and 81) as aptamer 49 reduced phosphorylation level of AXL while AXL expression was maintained at a similar level to the control and aptamer 81 was selected as it reduced the expression of AXL the most. These two aptamers also demonstrated the strongest inhibition of Erk activation for further examination of its effects on the cell viability of HCC827/ER cell. For reference, selected aptamers, 49 and 81, did not affect phosphorylation levels of AXL and Erk in H2009 cell which also express phosphorylated form of AXL but do not represent acquired resistance (34,35) (Figure 1B), validating the specific activity of these aptamers on HCC827/ER cells.
Table 1. Sequence of aptamers.
| Aptamer | Sequence |
|---|---|
| 5 | 5’-CTGTAGCTTTAATCCGTACGTCGCTTTAGAAGCTAAGGCT-3’ |
| 9 | 5’-ACCTTTCTGCGTTTTAAATGGTTTGGGGACTAGAGCTATG-3’ |
| 49 | 5’-AACCCGCTGACTCGGACACGACATATTAGTTTAAAGGCAC-3’ |
| 55 | 5’-TTTTACAAAGGCGGGACTAGCTCGGAATACAGGACGCTCA-3’ |
| 81 | 5’-GGCTGCGAAGGGTGTATTTGCCCCTTAATAGGCCCTTCT-3’ |
Table 2. Binding affinity of aptamers to the AXL receptor.
| Aptamer | 5 | 9 | 49 | 55 | 81 |
|---|---|---|---|---|---|
| Aptamer-AXL equilibrium dissociation constant (Kd) | 40.6 nM | 6.8 nM | 85.5 nM | 7.5 nM | 4.2 nM |
Figure 1.

Effects of aptamers in lung cancer cell lines. (A) Cell viability after the treatment of aptamers that selectively bind to AXL receptors in HCC827/ER cells. Cell viability was measured after 72-hr treatment of each aptamer in the concentration of 200 nM. Cell viability was measured using the CCK-8 assay. Results are representative of at least 3 independent experiments, and the error bars signify standard deviations (± SD). (B) Immunoblot analysis of AXL, phospho-AXL, Akt, phospho-Akt, Erk, and phospho-Erk. HCC827/ER and H2009 cells were treated with each aptamers (200 nM) for 72 hrs. Actin serves as a loading control.
Aptamer treatment has no effect on the sensitivity of HCC827/ER cells to EGFR-TKIs
HCC827/ER cells treated with increasing doses of AXL aptamers 49 and 81 showed maximal inhibitory effect on cell viability (approximately 40%) with 400 nM of aptamers with no significant differences between the two aptamers (Figure 2A). The decrease in cell viability was much greater compared to previous studies where inhibition effects of AXL aptamer or AXL inhibitory drugs were less than 20–50% (14,23,27,33). A previous study showed that the combined treatment of erlotinib and NPS-1034 effectively inhibited AXL tyrosine phosphorylation and overcame erlotinib resistance via a complete inhibition of EGFR downstream signaling (33). Therefore, we also evaluated the combined effects of erlotinib and AXL aptamers. HCC827/ER cells were treated with increasing doses of AXL aptamers, 49 or 81, and a fixed dose of erlotinib (1 µM), which resulted in a maximal inhibitory effect with 400 nM of aptamer treatment (Figure 2B). With a fixed dose of AXL aptamers, inhibition of cell viability was approximately 30%, independent of the dose of erlotinib (Figure 2C). In conclusion, no significant additive or synergistic effects were noted compared to the effects of AXL aptamer alone (Figure 2). In the same sense, a previous study of Cerchia et al. also showed that treatment with chemotherapeutic agents observed no measurable synergy when combined with AXL aptamers (27).
Figure 2.

Efficacy of aptamers and their combined treatment of erlotinib in HCC827/ER cells. (A) Cell viability after treatment with the aptamers targeting AXL (49 and 81). Cells were treated with aptamers at indicated doses for 72 hrs. (B) Efficacy of the combination of erlotinib with an aptamer. Cells were treated with multiple incremental doses of each aptamer in combination with the fixed dose (1 µM) of erlotinib for 72 hrs. (C) Efficacy of the combination of erlotinib with an aptamer. Cells were treated with multiple incremental doses of erlotinib in combination with the fixed dose (200 nM) of each aptamer for 72 hrs. Cell viability was measured using the CCK-8 assay. Results are representative of at least 3 independent experiments, and the error bars signify standard deviations (± SD).
Aptamer treatment does not enhance sensitivity to EGFR-TKIs and/or MET inhibitor in HCC827/ER cells
HCC827/GR cells with acquired resistance to gefitinib exhibits MET overexpression as a mechanism of resistance to EGFR-TKI, and thus treatment with MET inhibitor such as PHA-665752 in combination with gefitinib could effectively overcome gefitinib resistance (33). However, PHA-665752 in combination with erlotinib did not overcame erlotinib resistance in HCC827/ER cells (33). To further explore combined activity of aptamers with other drug, we evaluated the effects of PHA-665752 in combination with AXL aptamers and/or erlotinib in HCC827/ER cells with acquired resistance to erlotinib. While combination of aptamer and drug had no negative effect on the cells, combination of aptamers with drugs had no significant additive or synergistic effects on decreasing the cell viability (Figure 3).
Figure 3.
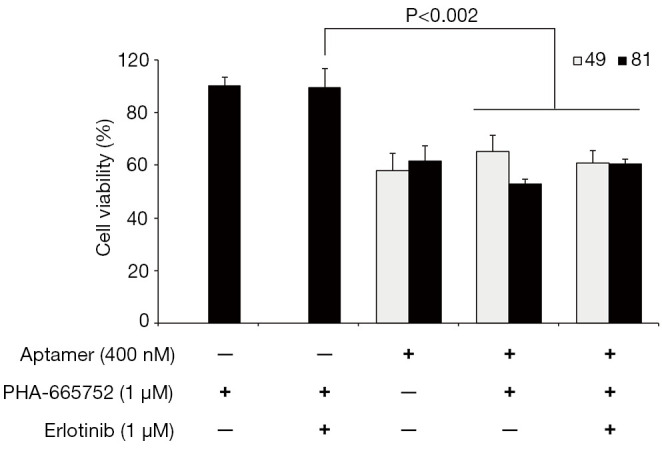
Efficacy of aptamers and their combined treatment with erlotinib and/or PHA-665752 in HCC827/ER cells. Cells were treated with an aptamer (400 nM), erlotinib (1 µM), or PHA-665752 (1 µM), separately, or in a combination of aptamer (400 nM) with erlotinib (1 µM) and/or PHA-665752 (1 µM). Cell viability was measured using the CCK-8 assay. Results are representative of at least 3 independent experiments, and the error bars signify standard deviations (± SD).
Discussion
In this present study, we investigated the efficacy of newly developed modified-DNA aptamers that selectively bind to AXL receptors in human lung cancer cells with acquired resistance to EGFR-TKI via activation of AXL signaling. Increased expression and activation of AXL accompanied by epithelial to mesenchymal transition (EMT) are known to be associated with acquired resistance to chemotherapeutic agents including both antimitotic drugs and targeted drugs (36-38). Previous studies revealed that the sole overexpression of AXL was observed in cells from acute myeloid leukemia patients with progression on chemotherapy (36) and that acquired cisplatin refractoriness with subsequent resistance to EGFR-TKI in NSCLC cell lines was closely related with AXL tyrosine kinase-mediated EMT (37). Moreover, Liu et al. reported that AXL overexpression confers lapatinib resistance in HER2-positive breast cancer with restoration of the sensitivity to lapatinib by siRNA transfection against AXL (38).
We demonstrated that the two newly developed aptamers exhibiting high selective affinity to AXL receptors inhibited 30–40% of cell growth and viability in human lung cancer cells with acquired resistance to erlotinib, possibly through mechanisms of inhibiting AXL and also its downstream components, Akt and Erk (Figure 1). This is in line with previous study by Cerchia et al. (27), which showed that the selective binding of the RNA-based aptamer, GL21.T to AXL significantly reduced RTK activity and the subsequent activation of two main downstream effector molecules Akt and Erk. However, treatment with GL21.T alone exhibited poor efficacy on inhibiting cell viability and proliferation, only reducing ~20% of viable cells in four different cell lines. In contrast, interfering with AXL expression by a specific shRNA against AXL displayed a much stronger effect in reducing cell viability (to the extent of 50%) than the aptamer treatment alone, possibly suggesting a limited potential of AXL aptamers in suppressing AXL receptors or signaling pathway downstream of AXL. Furthermore, other previous studies demonstrated that the single inhibition of AXL via XL880, a small molecule inhibitor of AXL and MET or siRNA transfection was not successful in the sufficient suppression of cell viability in erlotinib-resistant cells (14,33). In this sense, a further study is required in identifying ways to increase the effectiveness of aptamers.
Regarding resistance to RTK inhibitors, AXL is known to interact and dimerize with other cell surface receptors such as HER2, HER3, and EGFR in several tumor types, thus making it possible for cancer cells to survive through activation of bypass signaling pathway even in the presence of targeted agents blocking those receptors (6,14,38-40). Concerning this, AXL overexpression and activation with EMT-related phenotypical change were demonstrated in EGFR-mutant lung cancer xenografts with acquired resistance to EGFR-TKI (14). Similarly, AXL overexpression and its dimerization with EGFR played a pivotal role in maintaining EGFR signaling pathway despite the presence of EGFR-blocking antibody cetuximab (40), and AXL dimerization with HER3 bypassed inhibition of HER2 signaling in breast cancer cells (38). These results provide reasonable evidence for the treatment of AXL inhibitors in combination with targeted agents at the time of acquired resistance by activation of AXL signaling. In support of this, Rho et al. demonstrated that a combination of EGFR-TKI with NPS-1034 that targets both MET and AXL synergistically inhibited cell proliferation and induced cell death in human lung cancer cells with acquired resistance to EGFR-TKI via AXL activation (33). Also, Zhang and colleagues showed that genetic or pharmacological inhibition of AXL with concurrent treatment with erlotinib significantly inhibited tumor growth in in vivo EGFR-mutant lung cancer models with acquired resistance to erlotinib (14). In contrast, our study indicated that aptamers targeting AXL partly inhibited cell viability independent of the effects of combined EGFR-TKI. However, previous studies have shown that the treatment with AXL aptamers alone significantly inhibited in vivo tumor growth in a mouse xenograft model (23,27). Therefore, further studies should demonstrate whether our newly developed aptamers with selective affinity to AXL also suppress in vivo tumor growth or exhibit significant antitumor efficacy when combined with EGFR-TKIs in a in vivo study.
Conclusions
To sum up, our newly developed modified-DNA aptamers selectively targeting AXL showed partial inhibitory effects in human lung cancer cells with acquired resistance to EGFR-TKI via activation of AXL signaling.
Acknowledgments
Funding: This study was supported by a grant from the Asan Institute for Life Sciences, Asan Medical Center, Seoul, Korea [2016-654].
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Footnotes
Reporting Checklist: The authors have completed the MDAR checklist. Available at http://dx.doi.org/10.21037/tcr-20-2447
Data Sharing Statement: Available at http://dx.doi.org/10.21037/tcr-20-2447
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/tcr-20-2447). The authors have no conflicts of interest to declare.
References
- 1.Janssen JW, Schulz AS, Steenvoorden AC, et al. A novel putative tyrosine kinase receptor with oncogenic potential. Oncogene 1991;6:2113-20. [PubMed] [Google Scholar]
- 2.Lemke G. Biology of the TAM receptors. Cold Spring Harb Perspect Biol 2013;5:a009076. 10.1101/cshperspect.a009076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsou WI, Nguyen KQ, Calarese DA, et al. Receptor tyrosine kinases, TYRO3, AXL, and MER, demonstrate distinct patterns and complex regulation of ligand-induced activation. J Biol Chem 2014;289:25750-63. 10.1074/jbc.M114.569020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Linger RM, Keating AK, Earp HS, et al. TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv Cancer Res 2008;100:35-83. 10.1016/S0065-230X(08)00002-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Varnum BC, Young C, Elliott G, et al. Axl receptor tyrosine kinase stimulated by the vitamin K-dependent protein encoded by growth-arrest-specific gene 6. Nature 1995;373:623-6. 10.1038/373623a0 [DOI] [PubMed] [Google Scholar]
- 6.Meyer AS, Miller MA, Gertler FB, et al. The receptor AXL diversifies EGFR signaling and limits the response to EGFR-targeted inhibitors in triple-negative breast cancer cells. Sci Signal 2013;6:ra66. 10.1126/scisignal.2004155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Graham DK, DeRyckere D, Davies KD, et al. The TAM family: phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat Rev Cancer 2014;14:769-85. 10.1038/nrc3847 [DOI] [PubMed] [Google Scholar]
- 8.Shankar SL, O'Guin K, Kim M, et al. Gas6/Axl signaling activates the phosphatidylinositol 3-kinase/Akt1 survival pathway to protect oligodendrocytes from tumor necrosis factor alpha-induced apoptosis. J Neurosci 2006;26:5638-48. 10.1523/JNEUROSCI.5063-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sayan AE, Stanford R, Vickery R, et al. Fra-1 controls motility of bladder cancer cells via transcriptional upregulation of the receptor tyrosine kinase AXL. Oncogene 2012;31:1493-503. 10.1038/onc.2011.336 [DOI] [PubMed] [Google Scholar]
- 10.Dufies M, Jacquel A, Belhacene N, et al. Mechanisms of AXL overexpression and function in Imatinib-resistant chronic myeloid leukemia cells. Oncotarget 2011;2:874-85. 10.18632/oncotarget.360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Verma A, Warner SL, Vankayalapati H, et al. Targeting Axl and Mer kinases in cancer. Mol Cancer Ther 2011;10:1763-73. 10.1158/1535-7163.MCT-11-0116 [DOI] [PubMed] [Google Scholar]
- 12.Zhang YX, Knyazev PG, Cheburkin YV, et al. AXL is a potential target for therapeutic intervention in breast cancer progression. Cancer Res 2008;68:1905-15. 10.1158/0008-5472.CAN-07-2661 [DOI] [PubMed] [Google Scholar]
- 13.Lay JD, Hong CC, Huang JS, et al. Sulfasalazine suppresses drug resistance and invasiveness of lung adenocarcinoma cells expressing AXL. Cancer Res 2007;67:3878-87. 10.1158/0008-5472.CAN-06-3191 [DOI] [PubMed] [Google Scholar]
- 14.Zhang Z, Lee JC, Lin L, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet 2012;44:852-60. 10.1038/ng.2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang F, Liu X, Bartholdy BA, et al. Blockade of AXL activation overcomes acquired resistance to EGFR tyrosine kinase inhibition in non-small cell lung cancer. Translational Cancer Research 2019;8:2425-38. 10.21037/tcr.2019.09.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Myers SH, Brunton VG, Unciti-Broceta A. AXL Inhibitors in Cancer: A Medicinal Chemistry Perspective. J Med Chem 2016;59:3593-608. 10.1021/acs.jmedchem.5b01273 [DOI] [PubMed] [Google Scholar]
- 17.Logan TF. Foretinib (XL880): c-MET inhibitor with activity in papillary renal cell cancer. Curr Oncol Rep 2013;15:83-90. 10.1007/s11912-013-0299-3 [DOI] [PubMed] [Google Scholar]
- 18.Yau TC, Lencioni R, Sukeepaisarnjaroen W, et al. A Phase I/II Multicenter Study of Single-Agent Foretinib as First-Line Therapy in Patients with Advanced Hepatocellular Carcinoma. Clin Cancer Res 2017;23:2405-13. 10.1158/1078-0432.CCR-16-1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neal JW, Dahlberg SE, Wakelee HA, et al. Erlotinib, cabozantinib, or erlotinib plus cabozantinib as second-line or third-line treatment of patients with EGFR wild-type advanced non-small-cell lung cancer (ECOG-ACRIN 1512): a randomised, controlled, open-label, multicentre, phase 2 trial. Lancet Oncol 2016;17:1661-71. 10.1016/S1470-2045(16)30561-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tolaney SM, Ziehr DR, Guo H, et al. Phase II and Biomarker Study of Cabozantinib in Metastatic Triple-Negative Breast Cancer Patients. Oncologist 2017;22:25-32. 10.1634/theoncologist.2016-0229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cerchia L, de Franciscis V. Nucleic acid aptamers against protein kinases. Curr Med Chem 2011;18:4152-8. 10.2174/092986711797189592 [DOI] [PubMed] [Google Scholar]
- 22.Chen F, Zhang X, Zhou J, et al. Aptamer inhibits Mycobacterium tuberculosis (H37Rv) invasion of macrophage. Mol Biol Rep 2012;39:2157-62. 10.1007/s11033-011-0963-3 [DOI] [PubMed] [Google Scholar]
- 23.Kanlikilicer P, Ozpolat B, Aslan B, et al. Therapeutic Targeting of AXL Receptor Tyrosine Kinase Inhibits Tumor Growth and Intraperitoneal Metastasis in Ovarian Cancer Models. Mol Ther Nucleic Acids 2017;9:251-62. 10.1016/j.omtn.2017.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jain KK. Nanotechnology-based drug delivery for cancer. Technol Cancer Res Treat 2005;4:407-16. 10.1177/153303460500400408 [DOI] [PubMed] [Google Scholar]
- 25.Esposito CL, Catuogno S, de Franciscis V, et al. New insight into clinical development of nucleic acid aptamers. Discov Med 2011;11:487-96. [PubMed] [Google Scholar]
- 26.Cerchia L, de Franciscis V. Targeting cancer cells with nucleic acid aptamers. Trends Biotechnol 2010;28:517-25. 10.1016/j.tibtech.2010.07.005 [DOI] [PubMed] [Google Scholar]
- 27.Cerchia L, Esposito CL, Camorani S, et al. Targeting Axl with an high-affinity inhibitory aptamer. Mol Ther 2012;20:2291-303. 10.1038/mt.2012.163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990;249:505-10. 10.1126/science.2200121 [DOI] [PubMed] [Google Scholar]
- 29.Gold L, Ayers D, Bertino J, et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS One 2010;5:e15004. 10.1371/journal.pone.0015004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rohloff JC, Gelinas AD, Jarvis TC, et al. Nucleic Acid Ligands With Protein-like Side Chains: Modified Aptamers and Their Use as Diagnostic and Therapeutic Agents. Mol Ther Nucleic Acids 2014;3:e201. 10.1038/mtna.2014.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.White R, Rusconi C, Scardino E, et al. Generation of species cross-reactive aptamers using "toggle" SELEX. Mol Ther 2001;4:567-73. 10.1006/mthe.2001.0495 [DOI] [PubMed] [Google Scholar]
- 32.Fitzwater T, Polisky B. A SELEX primer. Methods Enzymol 1996;267:275-301. 10.1016/S0076-6879(96)67019-0 [DOI] [PubMed] [Google Scholar]
- 33.Rho JK, Choi YJ, Kim SY, et al. MET and AXL inhibitor NPS-1034 exerts efficacy against lung cancer cells resistant to EGFR kinase inhibitors because of MET or AXL activation. Cancer Res 2014;74:253-62. 10.1158/0008-5472.CAN-13-1103 [DOI] [PubMed] [Google Scholar]
- 34.Linger RM, Cohen RA, Cummings CT, et al. Mer or Axl receptor tyrosine kinase inhibition promotes apoptosis, blocks growth and enhances chemosensitivity of human non-small cell lung cancer. Oncogene 2013;32:3420-31. 10.1038/onc.2012.355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choi YJ, Kim JH, Rho JK, et al. AXL and MET receptor tyrosine kinases are essential for lung cancer metastasis. Oncol Rep 2017;37:2201-8. 10.3892/or.2017.5482 [DOI] [PubMed] [Google Scholar]
- 36.Hong CC, Lay JD, Huang JS, et al. Receptor tyrosine kinase AXL is induced by chemotherapy drugs and overexpression of AXL confers drug resistance in acute myeloid leukemia. Cancer Lett 2008;268:314-24. 10.1016/j.canlet.2008.04.017 [DOI] [PubMed] [Google Scholar]
- 37.Kurokawa M, Ise N, Omi K, et al. Cisplatin influences acquisition of resistance to molecular-targeted agents through epithelial-mesenchymal transition-like changes. Cancer Sci 2013;104:904-11. 10.1111/cas.12171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu L, Greger J, Shi H, et al. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: activation of AXL. Cancer Res 2009;69:6871-8. 10.1158/0008-5472.CAN-08-4490 [DOI] [PubMed] [Google Scholar]
- 39.Bose R, Molina H, Patterson AS, et al. Phosphoproteomic analysis of Her2/neu signaling and inhibition. Proc Natl Acad Sci U S A 2006;103:9773-8. 10.1073/pnas.0603948103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brand TM, Iida M, Stein AP, et al. AXL mediates resistance to cetuximab therapy. Cancer Res 2014;74:5152-64. 10.1158/0008-5472.CAN-14-0294 [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]