

Abstract
Dietary intake is a major contributor to the global obesity epidemic and represents a complex behavioural phenotype that is partially affected by innate biological differences. Here, we present a multivariate genome-wide association analysis of overall variation in dietary intake to account for the correlation between dietary carbohydrate, fat and protein in 282,271 participants of European ancestry from the UK Biobank (n = 191,157) and Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium (n = 91,114), and identify 26 distinct genome-wide significant loci. Dietary intake signals map exclusively to specific brain regions and are enriched for genes expressed in specialized subtypes of GABAergic, dopaminergic and glutamatergic neurons. We identified two main clusters of genetic variants for overall variation in dietary intake that were differently associated with obesity and coronary artery disease. These results enhance the biological understanding of interindividual differences in dietary intake by highlighting neural mechanisms, supporting functional follow-up experiments and possibly providing new avenues for the prevention and treatment of prevalent complex metabolic diseases.
Dietary factors are of critical importance in the prevention and management of obesity, type 2 diabetes (T2D) and associated disorders, yet the underlying mechanisms influencing human food intake are poorly understood1–3. Preliminary evidence suggests that dietary intake is a complex behavioural phenotype shaped by the confluence of social and environmental factors acting on a background of inherited biological differences, as confirmed by moderate heritability4,5.
Advances in neuroscience have identified cortical and subcortical regions in the central nervous system (CNS) that play a pivotal role in eating behaviour and energy homoeostasis3,6,7. The best-understood regulatory signal is leptin, which is secreted by adipocytes in proportion to adipose stores and is sensed by the brain through specific neuronal receptors. Genetic deficiency of leptin or its receptor results in extreme hunger and obesity in both mice and humans8,9. The characterization of additional signals associated with dietary phenotypes, such as fibroblast growth factor 21 (FGF21), has enabled the development of a drug that reduces sugar intake and sweet taste preference10,11. Further genetic insights into eating behaviour in humans may inform future experimental approaches and generate new potential therapeutic avenues.
So far, genetic association studies for dietary intake traits have primarily focused on single phenotypes, including separate nutrients or foods5,12–14. However, dietary factors are largely correlated and increasing the consumption of one specific food or nutrient leads to eating less of other foods. Multivariate genome-wide association approaches have emerged to examine genetic associations for highly correlated traits15 and have successfully been implemented to unravel new genetic loci for highly correlated metabolic or behavioural phenotypes.16,17 In this study, we present a multivariate genome-wide association analysis of overall variation in dietary intake in 282,271 participants of European ancestry and identify 26 distinct genomic loci. Our findings provide insights into specific brain regions, cell types and CNS processes that influence dietary intake in humans. A key biological finding is the enrichment of dietary intake-associated signals for genes expressed in specific subtypes of GABAergic, dopaminergic and glutamatergic neurons distributed across several brain regions that have connections to the hypothalamus. In addition, we provide evidence for two main groups of dietary intake genetic variants with distinct associations with obesity and coronary artery disease (CAD) among individuals of European ancestry.
Results
Multivariate genetic analysis identifies 26 loci for diet.
We performed single-trait genetic association analyses for the proportion of total energy intake from carbohydrate, fat and protein in 191,157 participants from the UK Biobank (UKBB)18 (http://www.ukbiobank.ac.uk/; Supplementary Table 1 and Extended Data Fig. 1). Next, we meta-analysed single-trait genetic associations from the UKBB with summary statistics on the proportion of total energy intake from carbohydrate, fat and protein from the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium (n = 91,114)5. Single-trait meta-analysis estimates were then combined in a multivariate analysis using a cross-phenotype association analysis (CPASSOC; Methods)19. A flow chart of the study design is shown in Extended Data Fig. 2.
To distinguish between inflation and polygenic signal on dietary intake, we calculated lambda (λGC) and the linkage disequilibrium (LD) score intercept20. We found moderate inflation in single-trait and multivariate analyses (λGC ranging from 1.06 to 1.07) with an LD intercept of approximately 1 (s.e.m. = 0.01), indicating that most inflation could be explained by the polygenic signal (Extended Data Fig. 3 and Supplementary Table 2). Genome-wide single-nucleotide polymorphism (SNP)-based heritability was estimated at 4.1% (s.e.m. = 0.01), 3.5% (s.e.m. = 0.01) and 4.4% (s.e.m. = 0.02) for carbohydrate, fat and protein, respectively (Supplementary Table 2). Estimated SNP-based heritability was in line with other behavioural phenotypes, such as tobacco or alcohol use21, where variants that explain even a small proportion of phenotypic variance provided substantial insights into the underlying mechanisms affecting these behavioural phenotypes. We also examined whether reverse causation (that is, individuals at higher disease risk may have changed their diet) might have biased our results by testing associations of polygenic risk scores (PRS) for body mass index (BMI), T2D and CAD with dietary intake (Methods). We found no evidence of reverse causation effects, except for individuals with a higher number of BMI risk alleles that reported higher protein and lower carbohydrate intake (Supplementary Table 3).
In the multivariate analysis for overall dietary variation we identified 31 lead genome-wide significant variants in 26 distinct genomic loci (Fig. 1 and Supplementary Tables 4 and 5). All previously reported dietary intake signals from single-trait macronutrient intake genome-wide association studies (GWAS) were confirmed in the present analysis, except for six genetic variants recently reported by Meddens et al.14 that achieved suggestive evidence of association in our study (Supplementary Table 6). In addition, we provided evidence of association with dietary intake for 14 additional loci not described before, including, for example, a variant in the METLL15 locus (P = 5.6 × 10−12) associated with a combination of reduced protein and increased carbohydrate intake. The strongest signal for dietary intake was found for a cluster of four distinct genome-wide-significant variants at the FGF21 locus, which was associated with increased fat and protein and reduced carbohydrate intake. In a sensitivity analysis restricted to UKBB participants that was further adjusted for environmental factors associated with dietary intake, including the Townsend deprivation index, overall health rating, smoking, alcohol intake and sleep duration, the FGF21 results remained largely consistent with the primary results (Methods, Supplementary Tables 7 and 8 and Extended Data Fig. 4). Of the 15 loci that reached genome-wide significance in the UKBB alone, six loci (GCKR, TSPAN5, SLC39A8, CCDC171, PLEKHM1 and NSF) were no longer genome-wide significant after adjustment for additional environmental factors. Single-trait meta-analyses of carbohydrate, fat and protein intake identified seven, nine and seven genome-wide significant loci, respectively (Supplementary Tables 9–11).
Fig. 1 |. SNP-based association with overall variation in dietary intake in the multivariate genome-wide analysis of 282,271 individuals from CHARGe and the UKBB.
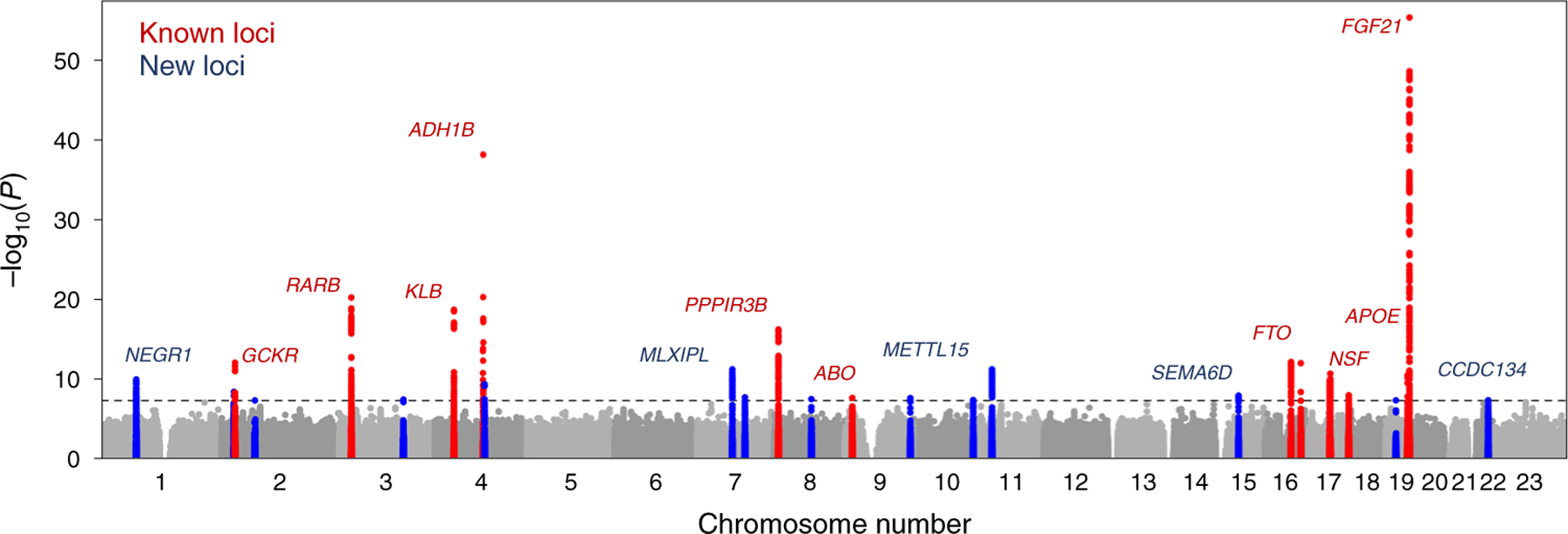
Manhattan plot showing the −log10 P values (y axis) for all genotyped and imputed SNPs that passed quality control in each GWAS, plotted by chromosome (x axis). The horizontal line denotes genome-wide significance (P = 5 × 10−8). The red labelling denotes previously identified variants and the blue labelling new variants.
Functional annotation of all genetic variants in associated loci (n = 7,636 variants) identified 17 non-synonymous variants in 4 distinct genomic loci (Supplementary Table 12). For example, we identified multiple non-synonymous variants in the KANSL1 gene, which has been previously linked with mood disorders22 and increased alcohol consumption23 (Supplementary Table 13). The non-synonymous variant in the ZNF789 gene (rs6962772), associated with reduced protein and carbohydrate intake in our study, has been associated with smoking initiation and alcohol consumption.21 The identification of missense variants for dietary intake associated with other behavioural or mental health phenotypes is consistent with recent evidence establishing causal links of dietary- and lifestyle-related domains with depression.24,25
Diet signals are enriched for genes expressed in the brain.
We next tested multivariate dietary intake signals for enrichment of gene expression in 54 tissue types obtained from the Genotype-Tissue Expression (GTEx) project v.8.0 (https://www.gtexportal.org/home/)26. Dietary intake signals were significantly enriched for genes predominantly expressed in several brain tissues including the cerebellum (PBonferroni-adjusted = 4.2 × 10−5), cortex (PBonferroni-adjusted = 8.3 × 10−5), frontal cortex (PBonferroni-adjusted = 7.1 × 10−3), anterior cingulate cortex (PBonferroni-adjusted = 3.4 × 10−2) and nucleus accumbens (PBonferroni-adjusted = 3.9 × 10−2; Fig. 2a and Supplementary Table 14). To replicate the brain expression findings, we used expression quantitative trait loci (eQTL) data from the UK Brain Expression Consortium,27 a dataset that provides eQTL results from 10 brain regions from 134 individuals of European ancestry free of neurodegenerative disorders (Methods). We found widespread eQTLs among all brain regions; the cerebellum was the region with the highest enrichment with a total of 3,054 significant eQTLs (P = 1.9 × 10−9; Supplementary Table 15). Two dietary intake variants in the MAPT region (17q21)—rs62065393 and rs1999535—were associated with the expression of KANSL1, CRHR1 or LRRC37A2/A3 in these brain regions. We further queried the BrainSeq eQTL database28, which includes frontal cortex eQTL information from 484 donors and found 7,123 significant eQTLs (P = 6.5 × 10−9) associated with the expression of 13 genes, including confiding in others KLB, PTCD1, METTL15 or LRRC37A2/A3 (Supplementary Table 15). Previous studies linked these genes with neurological disorders and CNS signal transduction pathways29,30.
Fig. 2 |. Implicated tissue and cell expression profiles for dietary intake in the multivariate genome-wide analysis of 282,271 individuals from CHARGe and the UKBB.
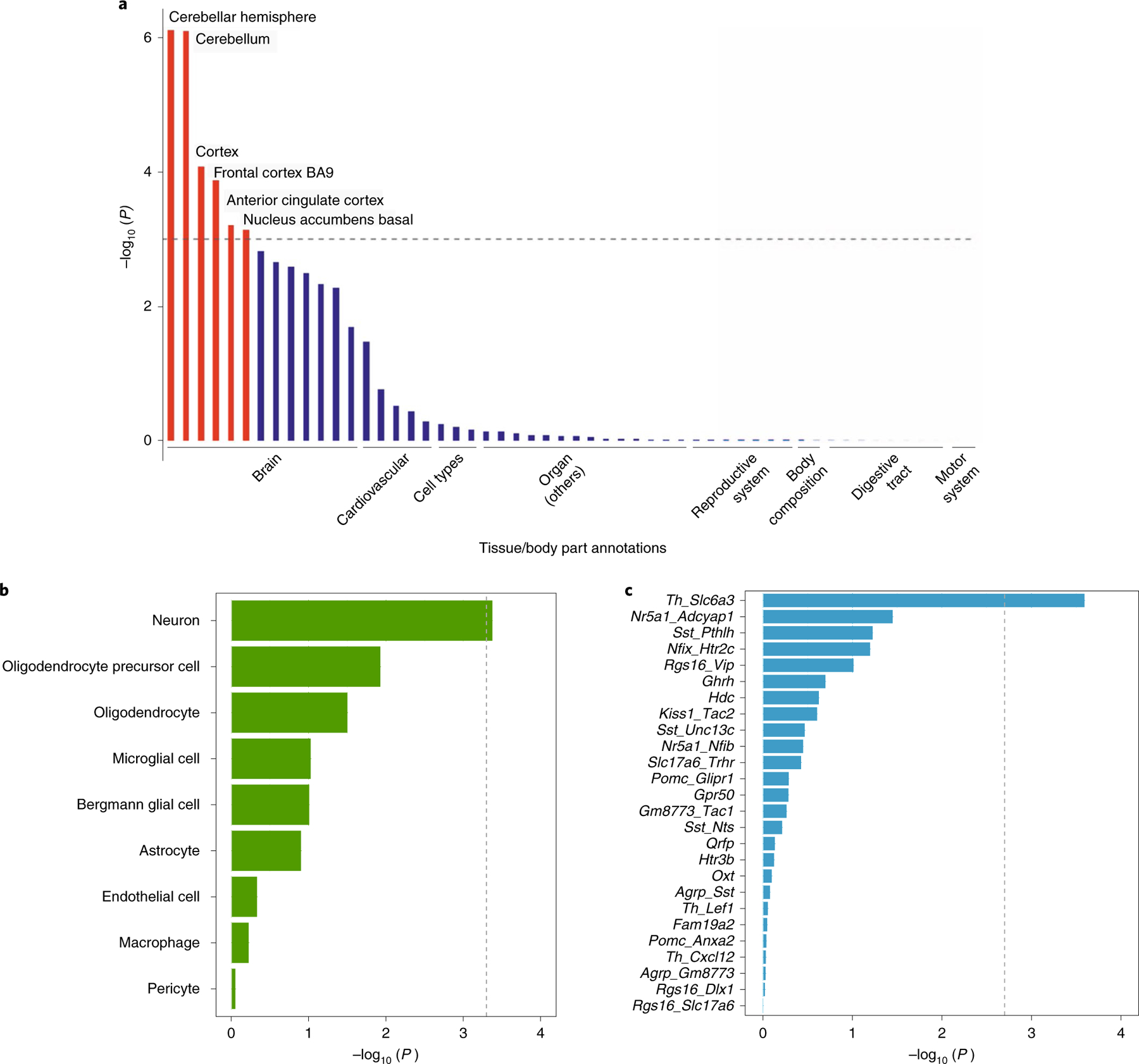
a, MAGMA tissue expression enrichment analysis using gene expression per tissue based on GTEx RNA-seq data for 54 specific tissue types for dietary intake signals. Expression values (RPKM) were log2-transformed with winsorization at 50 and averaged per tissue (Methods). The dashed line indicates the threshold for significance (P < 0.05/54). b, Single-cell expression analysis of genes related to dietary intake in 20 tissue/organ datasets from 44,949 annotated cells corresponding to 119 whole-body cell types from the Tabula Muris Consortium (Methods). The x axis shows the − log10-transformed two-tailed P value of association of multivariate analysis with cell-specific gene expression levels in a linear model. The dashed grey line indicates the Bonferroni-corrected significance threshold (P < 0.05/119 cell types). c, Single-cell expression analysis of genes related to dietary intake using Drop-seq data from 20,921 individual cells in and around the adult mouse Arc-ME (Methods). The x axis shows the −log10-transformed two-tailed P value of association of multivariate analysis with cell-specific gene expression levels in a linear model. The dashed grey line denotes the Bonferroni-corrected significance threshold (P < 0.05/28 Arc-ME cell types) derived by MAGMA.
Single-cell analyses prioritize cell types for dietary intake.
To evaluate whether the genomic loci implicated in dietary intake map onto specific brain cell types, we integrated a battery of publicly available single-cell RNA sequencing (scRNA-seq) datasets using MAGMA v.1.0.8 (http://ctg.cncr.nl/software/magma) (Methods). Whole-body single-cell mouse expression datasets from the Tabula Muris Consortium (https://tabula-muris.ds.czbiohub.org)31 showed that dietary intake signals were enriched for genes exclusively expressed in neurons (PBonferroni-adjusted = 4.9 × 10−2; Fig. 2b and Supplementary Table 16), findings that were mainly driven by fat intake preference (PBonferroni-adjusted = 4.8 × 10−2; Supplementary Table 17). We next investigated which neuronal populations were enriched for dietary intake signals using human scRNA-seq datasets. We showed that genes associated with dietary intake were enriched for genes predominantly expressed in GABAergic (PBonferroni-adjusted = 9.2 × 10−3), dopaminergic (PBonferroni-adjusted = 2.7 × 10−2) and glutamatergic neurons (PBonferroni-adjusted = 2.1 × 10−3; Supplementary Table 18) in different brain areas. Using data from the Mouse Nervous System dataset (http://mousebrain.org)32, we showed that two subpopulations of highly correlated GABAergic neurons located in the parvocellular reticular nuclei of the medulla, HBINH3 (PBonferroni-adjusted = 1.8 × 10−3) and HBINH9 (PBonferroni-adjusted = 1.8 × 10−2), which express the dopamine-transported gene and are distinguished from each other by the expression of SLC6A5 (encoding a glycine transporter), were enriched for association. We also identified two specific glutamatergic neuronal subtypes enriched for association with diet signals, DEGLU3 (PBonferroni-adjusted = 3.6 × 10−2) in the paraventricular nucleus of the thalamus and HBGLU3 (PBonferroni-adjusted = 3.6 × 10−2) in the nucleus of the solitary tract (Supplementary Table 18).
Given the relevance of the hypothalamus for eating behaviour, we also performed cell-type prioritization using 50 transcriptionally distinct cell populations in and around the nucleus of the hypothalamic arcuate–median eminence complex (Arc-ME). We showed an enrichment of dietary-linked gene expression in Th/Slc6a3 neurons, a highly specialized type of neuron that expresses the dopamine-transporter gene SLC6A3 and the prolactin receptor gene PRLR (PBonferroni-adjusted = 6.6 × 10−3; Fig. 2c and Supplementary Table 19). An additional analysis was performed with CELLEX v.1.0.0 (https://github.com/perslab/CELLEX) and CELLECT v.1.3.0 (https://github.com/perslab/CELLECT/tree/90ba133) (Methods) to leverage the availability of estimated effect sizes for genetic variants associated with specific nutrients and integrate them with single-cell expression from Tabula Muris, the Mouse Nervous System, the hypothalamic Arc-ME and five additional hypothalamic datasets. This analysis recapitulated the importance of neurons in the Tabula Muris dataset, driven again by fat intake preference (PBonferroni-adjusted = 4.8 × 10−3; Supplementary Table 20), and further highlighted a subtype of GABAergic somatostatin Sst+/Opt+-expressing neuron in the lateral hypothalamic area that was enriched for carbohydrate preference (PBonferroni-adjusted = 8.9 × 10−3; Supplementary Table 21). Overall, these results provide evidence of interrelated cortical and subcortical regions and cell types in the CNS involved in food intake and nutrient preference and might support functional follow-up experiments.
Clusters of diet signals affect metabolic traits differently.
To group dietary intake signals into clusters of genetic variants with potential similarities, we used a Bayesian non-negative matrix factorization (bNMF) clustering approach33. The input for the bNMF was based on effect sizes from the single-trait GWAS derived from this study and 19 additional previously published dietary intake GWAS phenotypes (Supplementary Table 22 and Extended Data Fig. 5). We aligned variants by their alleles associated with the increased proportion of fat intake (the single macronutrient with the highest number of significant signals) and their respective associations with other dietary traits.
The defining features of each identified cluster were determined by the strongest trait and variant associations after 1,000 iterations. Clustering of variant-trait associations led to the identification of two clusters present in 82% of iterations. The two clusters denoted different associations with specific dietary features including a cluster characterized by increased proportion of fat, protein and polyunsaturated fat intake (‘increased fat and protein’) and a cluster characterized by increased fat and reduced carbohydrate intake (‘reduced carbohydrate diet’). The loci and traits defining each cluster are described in Extended Data Fig. 6 and Supplementary Tables 23 and 24.
We used the set of the strongest-weighted variants from each cluster to generate cluster-specific PRS. We first showed that PRS were significantly associated with the traits defining each cluster (Supplementary Table 25). We next assessed PRS associations with BMI, T2D and CAD using individual-level data from the UKBB (Methods). The PRS for the ‘increased fat and protein’ cluster was associated with lower BMI with adjusted estimated effect sizes of −0.04 kg m−2 (s.e.m. = 0.007, P = 2.3 × 10−8) per 1 s.d. increase in the score. The PRS for ‘reduced carbohydrate diet’ cluster was associated with lower CAD, with an adjusted estimated odds ratio (OR) of 0.97 (95% confidence interval (CI) 0.95–0.99, P = 8.0 × 10−3) per 1 s.d. increase in the score (Fig. 3a). We detected no significant associations with T2D. We further investigated the association between PRS for diet clusters and obesity, T2D and CAD using the summary statistics from large-scale genetic consortia from the GIANT Consortium for BMI (n = 339,224),34 the DIAGRAM Consortium for T2D (n = 26,676 T2D cases, n = 132,532 controls)35 and the CARDIOGRAM Consortium for CAD (n = 60,801 CAD cases, n = 123,504 controls)36. We found an association between the PRS for ‘increased fat and protein’ and BMI (β = −0.002, s.e.m. = 0.001, P = 0.036; Fig. 3b) but no evidence of significant associations for the ‘reduced carbohydrate diet’ cluster and CAD (OR = 1.00, 95% CI 0.99–1.01, P = 0.14; Fig. 3b). As an additional replication analysis, we analysed data from the Partners HealthCare Biobank37, a multi-ethnic electronical health record-linked dataset, and showed no evidence of significant associations (Fig. 3c). These results might suggest a beneficial association of genetic variants for increased fat and protein with low BMI, which is mainly observed among relatively healthy individuals of European ancestry.
Fig. 3 |. Association of cluster-specific PRS and cardiometabolic phenotypes.
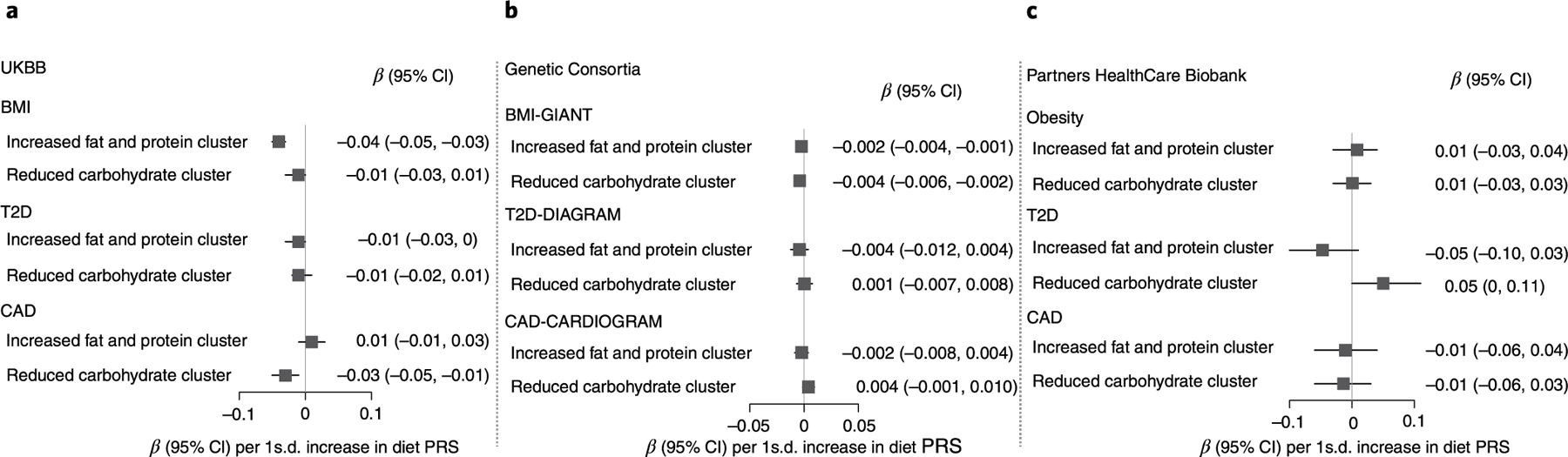
Cluster-specific PRS were defined by the top set of strongest-weighted variants for each cluster using a cut-off weighting of 0.94 (Methods). In brief, loci associated to a cluster included: cluster 1: NEGR1, RARB, ADCYAP1, METTL15 and TMEM108; cluster 2: PLEKHM1, NSF, KLB, TSPAN5, FGF21, ADH1A, GCKR, PPP1R3B, RP11–696N14.1, FUT1, FUT2 and DNMT3A. Individual participant scores were created by summing the number of alleles at each genetic variant weighted by the respective effect sizes on cluster pertinence. a, Individual-level data from the UKBB was used to test the association of cluster-specific polygenic risk scores with BMI in 450,981 participants, T2D (n = 18,292 cases, n = 429,183 controls) and CAD (n = 10,384 cases, n = 442,059 controls) after adjusting for age, sex, principal components, genotyping array and BMI (only in the analyses for CAD and T2D) (Methods). b, Publicly available summary statistics from the GIANT Consortium for BMI (n = 339,224), the DIAGRAM Consortium for T2D (n = 26,676 T2D cases, n = 132,532 controls) and the CARDIOGRAM Consortium for CAD (n = 60,801 CAD cases, n = 123,504 controls) were used to test the association of diet PRS with BMI, T2D and CAD (Methods). For the BMI analyses, nine genetic variants were included in the PRS for increased fat and protein PRS due to the lack of available good quality proxies. c, Individual-level data from the Partners HealthCare Biobank were used to investigate the extent to which previous observed associations were replicated in an independent dataset. Sample size in the Partners HealthCare Biobank was obesity (n = 5,039 cases and 14,557 controls), T2D (n = 1,497 cases and 18,099 controls) and CAD (n = 2,673 cases and 16,293 controls) (Methods). Estimated effect sizes are provided by 1 s.d. increase in PRS. Associations for UKBB analyses were considered significant at a Bonferroni-corrected threshold of significance (0.05/6, 2 clusters and 3 outcomes).
Discussion
In the present study, we expanded the genetic landscape of overall variation in dietary intake by identifying 26 distinct genomic loci. Our findings provide insights into the biological mechanisms that influence dietary intake, highlighting the relevance of brain-expressed genes, brain cell types and neural processes. Furthermore, we showed that genetic variants associated with overall variation in dietary intake converge into two main groups of genetic variants that are differently associated with obesity among relatively healthy individuals of European ancestry.
Our results align with findings from tissue expression and single-cell enrichment analyses of BMI-associated loci that pinpoint the CNS as a critical regulator of energy homoeostasis and body weight34. Our findings provide evidence that metabolically diverse and specialized GABAergic, dopaminergic and glutamatergic neurons distributed across different brain regions are enriched for dietary intake signals. For example, we identified a subtype of glutamatergic neuronal population, HBGLU3, in the nucleus of the solitary tract, a heterogeneous nucleus that integrates direct signals from the gut and taste receptors and indirect input from leptin signalling in the hypothalamus38–41, that might be important for eating behaviour and appetite modulation. The identification of HBGLU3, together with findings from other hindbrain regions, suggests that neuronal subpopulations in these regions are as relevant as hypothalamic neurons in modulating nutrient preference and eating behaviour, as demonstrated recently for brain cell types enriched for obesity genes42. Collectively, this knowledge might serve to guide future functional neuroimaging studies to provide insight into physiological, functional and connectional properties of food intake-related signals.
The multivariate approach implemented in this study allowed us to increase the power for genomic locus discovery, as has been the case for other highly correlated traits16,17. The bNMF clustering algorithm enabled us to dissect dietary intake genetic heterogeneity and identify two main domains of genetic variants characterized by different nutritional profiles that may have distinct association with obesity and CAD. Our findings showing that genetic variability associated with increased proportion of fat and protein associates with lower BMI among healthy participants of European ancestry is aligned with previous evidence suggesting that low-carbohydrate diets might outperform low-fat diets for obesity outcomes43,44. However, caution is needed when interpreting our data, since results were not replicated when studying individuals from a hospital-based biobank with marked demographic and clinical differences, suggesting that residual confounding might still exist. In addition, considering the complex network of biological and non-biological determinants of dietary intake and body weight trajectories, caution must be exercised in drawing strong conclusions from our findings, which should be interpreted in the context of healthy individuals of European ancestry.
While the present study was limited to self-reported dietary intake, more precise measurements, including evaluating age and social, cultural and economic factors as mediators or effect modifiers of a dynamic phenotype such as dietary intake, may help to refine our findings. We further conducted a sensitivity analysis in the UKBB to investigate the effect of environmental factors on reported associations and showed that our genetic estimates were largely consistent with our primary results. In our study we were not able to account for potential confounders such as stress or emotional eating, which have been shown to have an impact on eating behaviour and food preference45. Replication of dietary intake genetic variants in an independent dataset is missing but, to the best of our knowledge, no other studies of similar magnitude are available in individuals of European ancestry. While it is possible that single-cell expression data from mice might not be optimal to perform inference in humans, recent scRNA-seq studies assessing the convergence of mouse and human CNS gene expression have found that cell types and gene expression levels were generally conserved across species46. In addition, cis-eQTL association results from brain tissues for multivariate genome-wide significant SNPs do not imply signal overlap. Thus, future colocalization analyses are needed to test the concordance of the eQTL with the association signal. It is possible that some inflation exists in our estimated effect sizes for the association between cluster-specific PRS and BMI, T2D and CAD if a correlation exists between diet and these outcomes. However, this is likely to be minimal because macronutrient preference was not associated with T2D and CAD. In addition, expanding these analyses to identify genetic variants associated with food preferences in non-European ancestries is warranted. The present findings enhance our understanding of the biological variability of a complex and disease-relevant behaviour and provide a starting point for functional research that might aid in the discovery of new and more efficient therapeutic avenues to curb the obesity epidemic.
Methods
Study design and population.
The current study was conducted using publicly available summary statistics from the CHARGE Consortium5 and individual-level data from the UKBB18 under UKBB application no. 27892 (J.C.F. was the principal investigator). The UKBB received ethical approval from the National Research Ethics Service Committee North West Haydock (reference no. 11/NW/0382); all procedures were performed in accordance with the World Medical Association guidelines. We have complied with all relevant ethical regulations for work with the UKBB; all participants provided written informed consent.
We conducted a multivariate genome-wide analysis of overall variation in dietary intake by accounting for the correlation between dietary fat, protein and carbohydrate intake among 282,271 participants of European ancestry from the UKBB and CHARGE Consortium (http://www.chargeconsortium.com). The UKBB is a large population-based cohort established primarily to allow detailed investigations of the genetic and lifestyle determinants of a wide range of phenotypes18. Between 2006 and 2010, the UKBB recruited more than 500,000 individuals aged 37–73 years (99.5% aged 40–69 years) from across the UK living within 25 miles of one of 22 assessment centres. We used data released by the UKBB in March 2018; filtering (described below) resulted in a final sample size of 191,157 unrelated individuals of White European ancestry who had genetic and dietary intake data available.
Dietary intake summary statistics from the CHARGE dietary composition GWAS were extracted. The CHARGE dietary composition GWAS included up to 91,114 participants of European ancestry from 24 cohorts5.
Dietary intake assessment.
In the UKBB, dietary data were collected from 211,036 participants using the Oxford WebQ, a Web-based 24-h diet recall that asks participants to self-report on the frequency of intake of about 200 commonly consumed foods and drinks for the preceding 24 h47,48. The reliability and internal consistency of the Oxford WebQ has been investigated before showing that it performs well when compared against dietary intake biomarkers measured from 24-h urine samples49. The recall was administered at baseline visits and in person at assessment centres (towards the end of recruitment for the last 70,000 participants) and later administered electronically by email on 4 separate occasions over a 16-month period (February 2011–April 2012) to approximately 320,000 participants with known email addresses. Email invitations were sent on variable days of the week to capture variability in daily dietary intake. Daily intake of nutrients was calculated by multiplying the frequency of consumption of each food or drink by a standard portion size and the nutrient composition of that particular item derived from the McCance and Widdowson’s Composition of Foods database50. The following analysis was limited to carbohydrate (f.100005), fat (f.100004) and protein (f.100003) intake, all originally obtained in grams per day and transformed to the proportion of total energy intake (f.100002) from carbohydrate, fat and protein. We focused on relative rather than absolute intake because the total amount of carbohydrate, fat or protein is highly determined by BMI or physical activity51. Participants with unlikely estimated total energy intake in more than 50% of recalls were excluded (<500 or >3,500 kcal day−1 for women and <800 or >4,200 kcal day−1 for men). For participants who completed more than one recall, the averages of each macronutrient were calculated. The use of averages for participants with more than one recall has been shown to improve the accuracy of dietary intake assessment49. The final sample size for this analysis was 191,157 UKBB participants.
In the CHARGE Consortium, dietary intake was evaluated using validated food frequency questionnaires, diet history and diet records5. Based on the responses to each dietary assessment tool and study-specific nutrient database, habitual nutrient consumption was estimated. Over- and under-reporters were excluded by standard cut-offs determined by each study cohort as part of quality control. The final sample size for this analysis was 91,114 CHARGE participants.
Genotyping, imputation and quality control.
We used genotype data released by the UKBB in March 2018. A total of 502,536 UKBB participants were genotyped on two customized genetic arrays (the UK BiLEVE Axiom Array and the UKBB Axiom Array) covering 812,428 unique genetic markers with 95% overlap in variant content52. Genotyping, imputation and initial quality control on the genetic dataset has been described previously52. In brief, genotypes were phased using SHAPEIT3 and the 1000 Genomes phase 3 panel and imputed using the IMPUTE4 program with the Haplotype Reference Consortium reference panel or with a merged UK10K and 1000 Genomes phase 3 reference panel when a variant was not present in Haplotype Reference Consortium resulting in approximately 96 million genetic variants. After exclusion of participants who did not pass quality control filtering and those who withdrew consent, imputed and quality-controlled genotype data were available for 485,580 individuals and 92,693,895 genetic variants. Population structure was captured by principal component analysis on the samples and by self-reported ancestry identifying 451,660 participants of European ancestry included in the present analysis, of which 191,157 had available dietary intake data and thus remained in the final analysis. A flow chart summarizing sample selection and more detailed description of the criteria on each step is provided in Extended Data Fig. 1.
Genotyping, imputation and quality control methods for the CHARGE Consortium dietary intake GWAS has been detailed elsewhere5. In brief, each participating cohort performed quality control for genotyped variants based on minor allele frequency, call rate and departure from the Hardy–Weinberg equilibrium. Phased haplotypes from 1000 Genomes were used to impute approximately 38 million autosomal variants using a Hidden Markov model algorithm implemented in MACH 1.0/minimac (http://csg.sph.umich.edu//abecasis/MaCH/)53 or SHAPEIT v2.17/IMPUTE v2.3.2 (https://mathgen.stats.ox.ac.uk/genetics_software/shapeit/shapeit.html)54. Variants with low imputation quality (<0.4) were removed. The number of autosomal genetic variants analysed in this study was approximately 11.8 million.
Single-trait genome-wide association meta-analysis and multivariate analysis.
Single-trait genetic association analyses were performed separately for carbohydrate, fat and protein as percentages of total energy in 191,157 UKBB participants using a generalized mixed model implemented in SAIGE. (Scalable and Accurate Implementation of GEneralized mixed model) v.0.35.8.8 (https://github.com/weizhouUMICH/SAIGE/).55 Models were adjusted for age, sex, BMI, 20 principal components of ancestry, genotyping array and accounted for sample relatedness. We used a minor allele count threshold of 20 to select the genetic variants. Meta-analysis of UKBB and CHARGE single-trait associations was performed using METAL (https://genome.sph.umich.edu/wiki/METAL) by weighting effect size estimates using the inverse of the corresponding standard errors squared (version released on 25 March 2011)56. Heterogeneity in genetic effects across studies was evaluated using the I2 heterogeneity index57.
Single-trait meta-analyses estimates for carbohydrate, fat and protein were combined in a multivariate analysis using CPASSOC19. In brief, the method is similar to the sample size weighted fixed effect meta-analysis using summary-level data from single variant-trait associations from GWAS assuming homogeneity while accounting for trait correlations (provided in the SHomstatistic). CPASSOC also provides an extension approach (SHet) allowing for heterogeneous and opposite association effects as exhaustively testing all possible subsets of traits and provides the statistic on the subset with the strongest associations. The significance of SHet is estimated assuming a gamma distribution under the null hypothesis. In this study, we assumed heterogeneity effects, which are relevant for dietary composition. We provided as input for CPASSOC the macronutrient intake correlation matrix estimated in the UKBB (carbohydrate and fat, r2 = −0.36; carbohydrate and protein, r2 = −0.14; fat and protein, r2 = 0.007).
In a sensitivity analysis restricted to UKBB participants, we examined associations between environmental factors and the percentage of energy intake from carbohydrates, fat and protein. Environmental factors considered for this additional analysis included the Townsend deprivation index as a measure of socio-economic status, overall health rating, smoking, alcohol intake and sleep duration. Definitions about these environmental factors are provided in Supplementary Table 7. Next, we performed single-trait genetic association analyses for the proportion of total energy intake from carbohydrate, fat and protein after adjusting our primary model for these environmental factors in addition to the covariates originally included in our model. Then, single-trait genetic association estimates were combined into a multivariate analysis.
Bioinformatic analyses.
Trait SNP-based heritability was calculated as the proportion of trait variance due to additive genetic factors using the BOLT-REML algorithm from the BOLT-LMM software v.2.3. Genomic control lambda (λGC) values were calculated using GenABEL v1.8 in R using postquality control GWAS results. LD Score regression was used to estimate the cross-cohort genetic correlations calculated using the 1000 Genomes phase 3 panel for the European population20. The number of associated loci was determined by the clumping of genome-wide significant variants (P < 5 × 10−8) that were in low LD at r2 < 0.1 (based on LD information from UKBB genotype data) within 500-kilobase windows. The method ‘functional mapping and annotation of genetic associations’ (FUMA v.1.3.6; http://fuma.ctglab.nl/)58 was applied to explore the functional consequences of all genetic variants that were in LD (r2 ≥ 0.6) with one of the distinct significant variant and that had P < 5 × 10−8 and minor allele frequency > 0.0001, which included ANNOVAR v.20191024 categories59, combined annotation dependent depletion scores60, RegulomeDB v.1.1 scores61 and chromatin states62,63.
To assess whether the identified dietary intake loci were influenced by the possibility that individuals at high metabolic risk had made changes to their diet (reverse causation), we compared the mean proportion of total energy intake from carbohydrate, fat and protein among quartiles of PRS for BMI, T2D and CAD. The weights to generate these PRS were extracted from published meta-analyses of GWAS that did not use UKBB participants, including data from the GIANT Consortium for BMI (n = 339,224)34, the DIAGRAM Consortium for T2D (n = 26,676 T2D cases, n = 132,532 controls)35 and the CARDIOGRAM Consortium for CAD (n = 60,801 CAD cases, n = 123,504 controls)36. Individual participant scores were computed by summing the number of alleles at each genetic variant weighted by the respective effect sizes on BMI, T2D and CAD. If SNPs were not directly genotyped or imputed, we used available proxies based on r2 > 0.8. We compared the mean proportion of total energy intake from carbohydrate, fat and protein among the quartiles of each PRS using analysis of variance.
Tissue enrichment analyses in 54 tissue types obtained from the GTEx project v.8.0 (ref. 26) were conducted using MAGMA v.1.0.864. For these analyses, P values from the multivariate analysis were used as input. Gene expression values were log2-transformed reads per kilobase per million (RPKM) per tissue type after winsorization at 50 based on GTEx RNA-seq data. MAGMA was performed using the result of gene analysis (gene-based P) and tested for one side (greater) with conditioning on average expression across all tissue types. For brain eQTL analyses, we used data from the UK Brain Expression Consortium and the BrainSeq datasets27,28. The UK Brain Expression Consortium dataset comprises data from 10 brain regions from 134 individuals of European ancestry free of neurodegenerative disorders. The BrainSeq dataset includes frontal cortex eQTL information from 484 donors. cis-eQTL association results from brain tissues were obtained for multivariate genome-wide significant SNPs; expression enrichment and P values were retrieved from FUMA. Only eQTLs reaching the corrected threshold of significance of P < 1 × 10−6 were considered.
To connect genomic loci implicated in dietary intake with relevant brain cell types defined by gene expression profiles, we used independent brain scRNA-seq datasets. We first used information from the Tabula Muris Consortium, which includes single-cell transcriptomic data from 100,605 cells isolated from 20 organs from 3-month-old C57BL/6NJ mice31. Because dietary signals were exclusively enriched for association in different brain tissues, we next used brain scRNA-seq data from the Mouse Nervous System dataset32. In brief, the Mouse Nervous System dataset comprises 265 cell-type clusters from 19 nervous system regions represented by 160,796 high-quality single-cell transcriptomes. Given the relevance of the hypothalamic Arc-ME for feeding behaviour, we also included Drop-seq data from 50 transcriptionally distinct Arc-ME cell populations6. To connect evidence from mouse single-cell expression profiles with human single-cell expression, we complemented our analyses using human brain scRNA-seq datasets including single-cell expression profiles from the prefrontal cortex65, neocortex66, midbrain46 and hippocampus67. These gene sets were tested for association with gene-based test statistics using P values from the multivariate analysis in MAGMA (GWAS association P cut-off <10−5).
Single-cell enrichment analyses for separate nutrients were tested using two computational toolkits, CELLEX and CELLECT, which integrate multiple expression specificity metrics and LD Score regression annotations to prioritize cell types enriched for associations42. CELLEX and CELLECT were run to leverage the availability of estimated effect sizes for genetic variants associated with specific nutrients. Similarly to MAGMA, cell-type prioritization analysis with Tabula Muris, Mouse Nervous System and five hypothalamic datasets were processed with CELLEX and CELLECT.
Clustering of dietary intake signals and PRS for outcome associations.
We applied a bNMF algorithm33 with the aim of grouping the identified dietary intake genetic loci into subgroups of variants based on potential clinical and physiological similarities (Supplementary Information). The input for the bNMF algorithm was the set of the dietary intake-associated variants identified in this study and their respective association with single-trait GWAS from this study and publicly available summary statistics for 19 additional dietary intake traits from the UKBB. Given that the C/G and A/T alleles are ambiguous and can lead to errors in aligning alleles across the GWAS, we avoided inclusion of ambiguous alleles, choosing proxies instead. We generated standardized effect sizes for variant-trait associations by dividing the estimated regression coefficient by the standard error. The defining features of each cluster were determined by the most highly associated traits, a natural output of the bNMF approach. The bNMF algorithm was performed in R for 1,000 iterations as previously suggested; the maximum posterior solution at the most probable number of clusters was selected for downstream analysis.
The variants and traits defining each cluster were based on a cut-off of weighting of 0.94, which was determined by the optimal threshold to define the beginning of the long tail of the distribution of clusters’ weights across all clusters (the top 5% were considered to be significant). Cluster-specific PRS were created by summing the number of alleles at each genetic variant weighted by the respective effect sizes on cluster pertinence. Scaling of the individual scores was performed to allow interpretation of the effects as a per 1 s.d. increase in the PRS for each trait (division by twice the sum of the effect sizes and multiplication by twice the square of the SNP count representing the maximum number of risk alleles). We tested the association of each cluster-specific PRS with BMI, T2D and CAD after adjusting for age, sex, 20 principal components, genotyping array and BMI (only in analyses for CAD and T2D). Outcome definition for BMI was based on data field f.21001 and BMI analyses included a total of 450,981 participants. We used a conservative definition of T2D that was determined in a manner similar to previous described algorithms (n = 18,292 cases, n = 429,183 controls)68. CAD definition included self-reported history of myocardial infarction with information on coronary procedures (n = 10,384 cases, n = 442,059 controls).
Summary statistics from large-scale genetic consortia that did not include participants from the UKBB were used to assess diet PRS associations with BMI, T2D and CAD. For each dietary intake variant included in one of the two clusters, we used summary-level results from the GIANT Consortium for BMI (n = 339,224)34, the DIAGRAM Consortium for T2D (n = 26,676 T2D cases, n = 132,532 controls)35 and the CARDIOGRAM Consortium for CAD (n = 60,801 CAD cases, n = 123,504 controls)36 If some of the variants included in the diet PRS were not available in the summary statistics, we used available proxies based on r2 > 0.8. To obtain estimated effect sizes of diet PRS with BMI, T2D and CAD, we used the grs.summary function from the Genetics ToolboX v2.1.6 R package (https://cran.r-project.org/src/contrib/Archive/gtx/).
Finally, individual-level data from the Partners HealthCare Biobank (currently Mass General Brigham Biobank)37, a multi-ethnic hospital-linked electronical medical record dataset, was used to investigate associations of diet PRS with cardiometabolic phenotypes. Approval for analysis of the Partners HealthCare Biobank data was obtained by the Mass General Brigham institutional review board, protocol no. 2018P002276. Disease prevalences for obesity (n = 5,039 cases and 14,557 controls), T2D (n = 1,497 cases and 18,099 controls) and CAD (n = 2,673 cases and 16,293 controls) were determined from electronic medical records. Logistic regression models were adjusted for age, sex, five principal components and obesity (only in the analyses for CAD and T2D). PRS associations with cardiometabolic phenotypes were considered significant at a Bonferroni-corrected threshold of significance of P < 8.3 × 10−3 (0.05/6; 2 clusters and 3 traits).
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. UK Biobank sample selection.
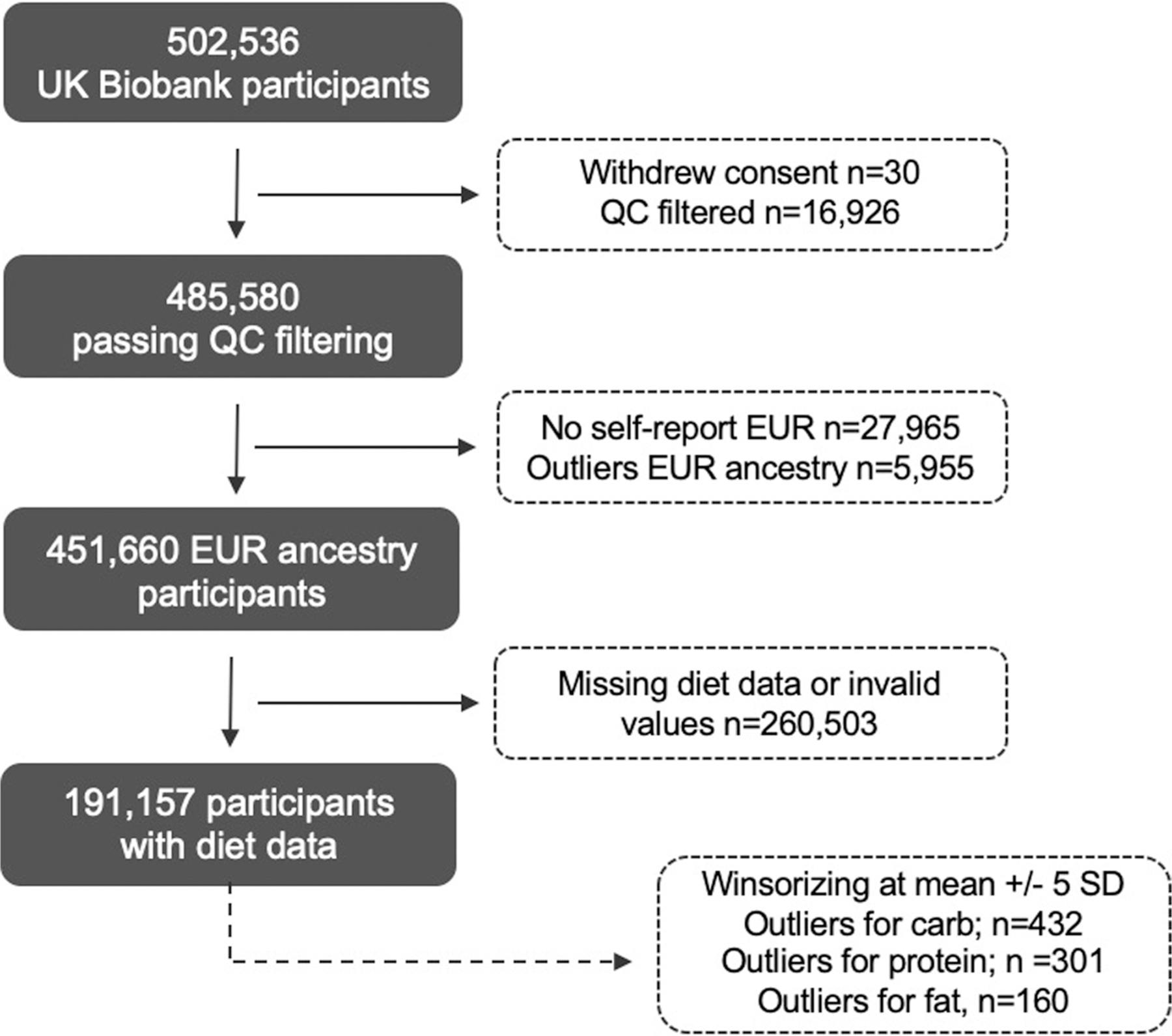
A total of 502,536 participants were available in UK Biobank at the beginning of this study. Thirty participants withdrew consent during the implementation of the analysis plan. We excluded 892 participants with invalid diet data based on previously defined quality control filtering criteria for diet data. A total of 16,034 participants did not pass quality control criteria based on UK Biobank quality control definitions (high heterozygosity & high missing rate, sex aneuploidy, submitted sex different from inferred sex). We excluded 27,965 participants based on a non-European self-reported ancestry, and 5,955 European ancestry outliers based on + /−6SD from the mean in the subset of 192,025 participants based on the first 4 PCs. Among 451,660 participants remaining for the discovery of genetic variants for dietary intake, 260,503 had missing diet data (n = 258,393) or invalid values (n = 2,110). Due to the skewed distribution of macronutrient intake, we winsorized at mean + /− 5 SD for each phenotype.
Extended Data Fig. 2 |. Schematic of the study design in the multi-trait genome-wide association meta-analysis for dietary intake in 282,271 individuals.
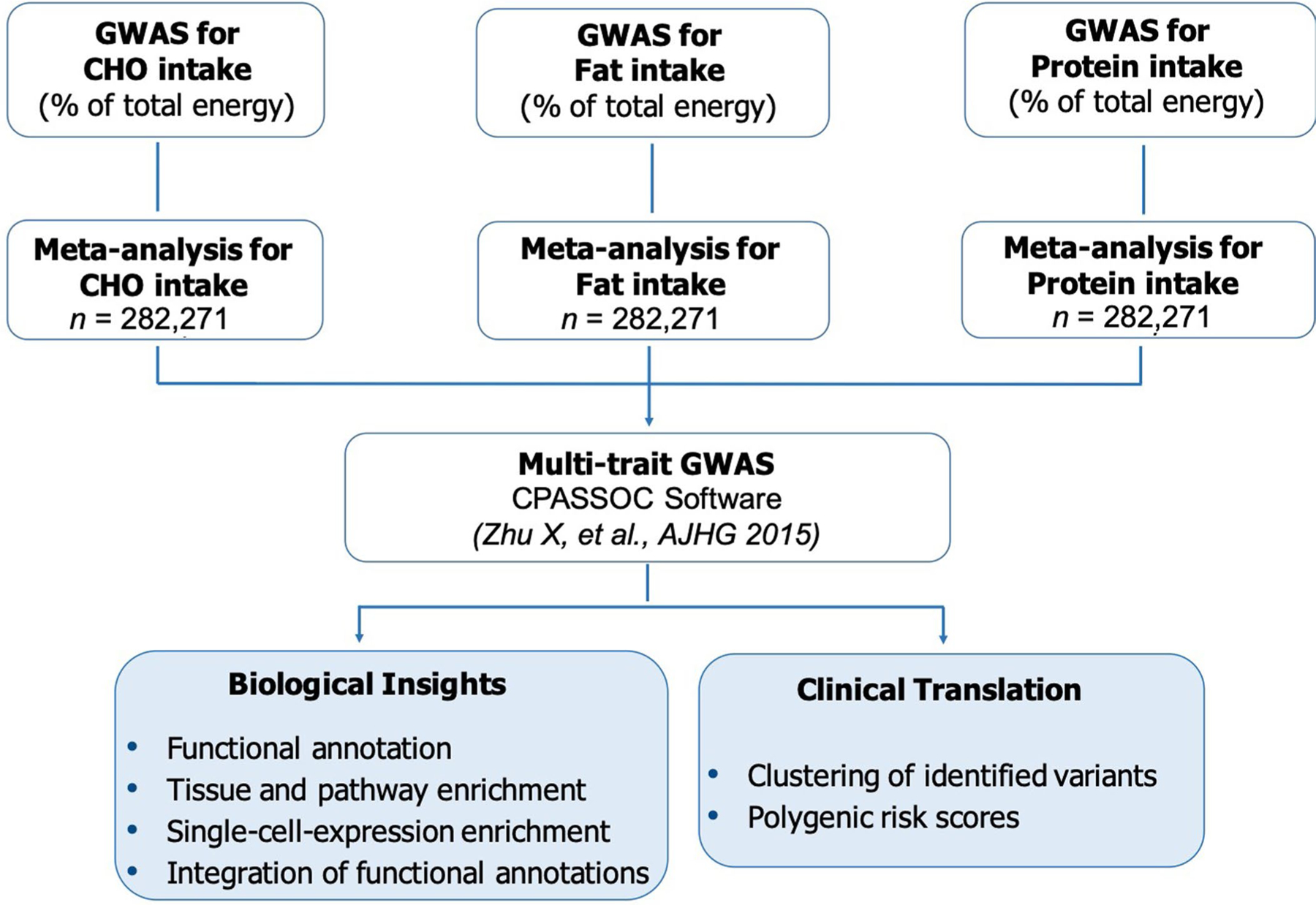
The genome-wide association meta-analysis of dietary intake comprised data from 191,157 participants from the UK Biobank and 91,114 participants from the CHARGE Consortium. Single-trait macronutrient GWAS from the UK Biobank and CHARGE Consortium were meta-analyzed using METAL and then combined into a multi-trait GWAS using the multi-trait CPASSOC method. Downstream in silico analyses were conducted to identify biological features of identified loci including functional annotation, tissue and pathway enrichment, and single-cell RNA expression analyses. The Bayesian nonnegative matrix factorization clustering algorithm was used to classify dietary intake genetic loci into subgroups based on potential functional and clinical similarities. Cluster-based polygenic risk scores were built to investigate patterns of metabolic risk.
Extended Data Fig. 3 |. Quantile-quantile plot of the SNP-based associations with single-trait and multi-trait genome-wide association meta-analyses of 282,271 individuals.
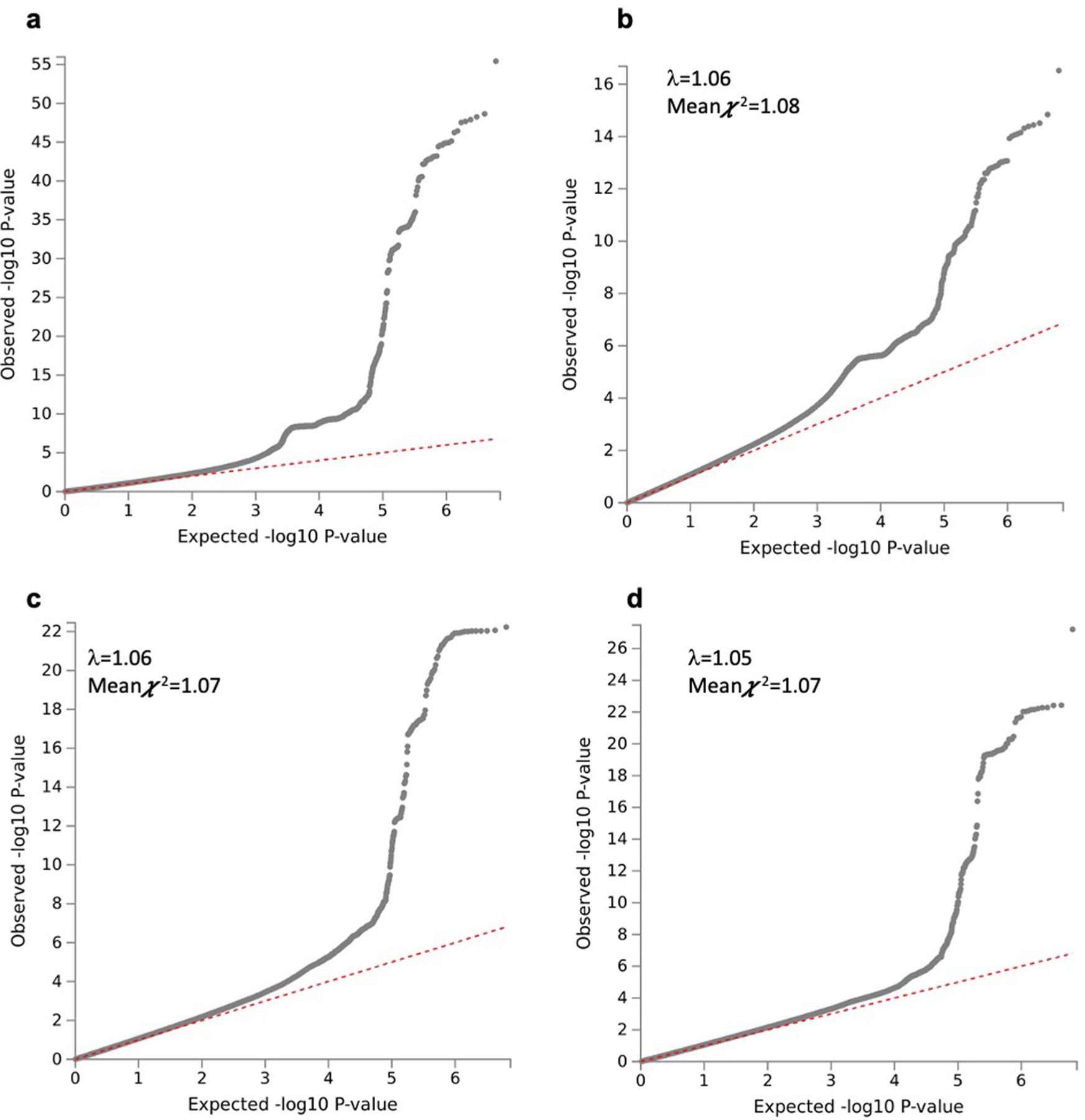
Quantile–quantile plot of the SNP-based associations with multivariate (a), carbohydrate (b), fat (c), and protein intake (d). SNP P values were computed in METAL by weighting effect size estimates using the inverse of the corresponding standard errors.
Extended Data Fig. 4 |. Associations between environmental factors and macronutrient intake.
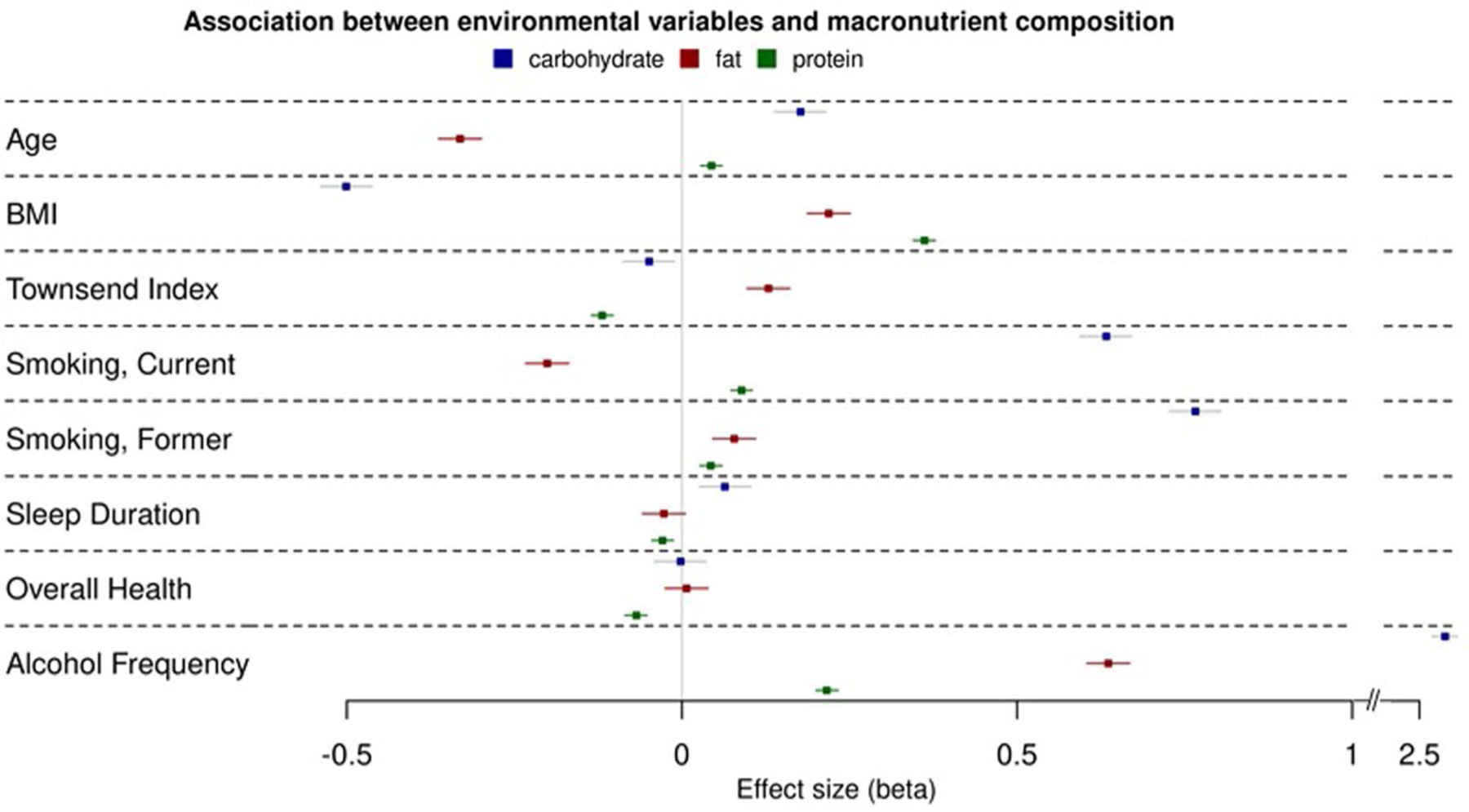
Shown are effect estimates (betas) and 95% confidence intervals for the association between environmental factors and macronutrient intake among UK Biobank participants. Environmental factors were all added to the initial model used for the primary main UK Biobank analyses. A null model was first run using SAIGE to evaluate the association of each environmental factor with each macronutrient intake. Only significant factors were retained in the model. Genetic association analyses were then conducted, similarly to the primary main UK Biobank analyses, using SAIGE.
Extended Data Fig. 5 |. Schematic overview of the Bayesian nonnegative matrix factorization clustering algorithm.
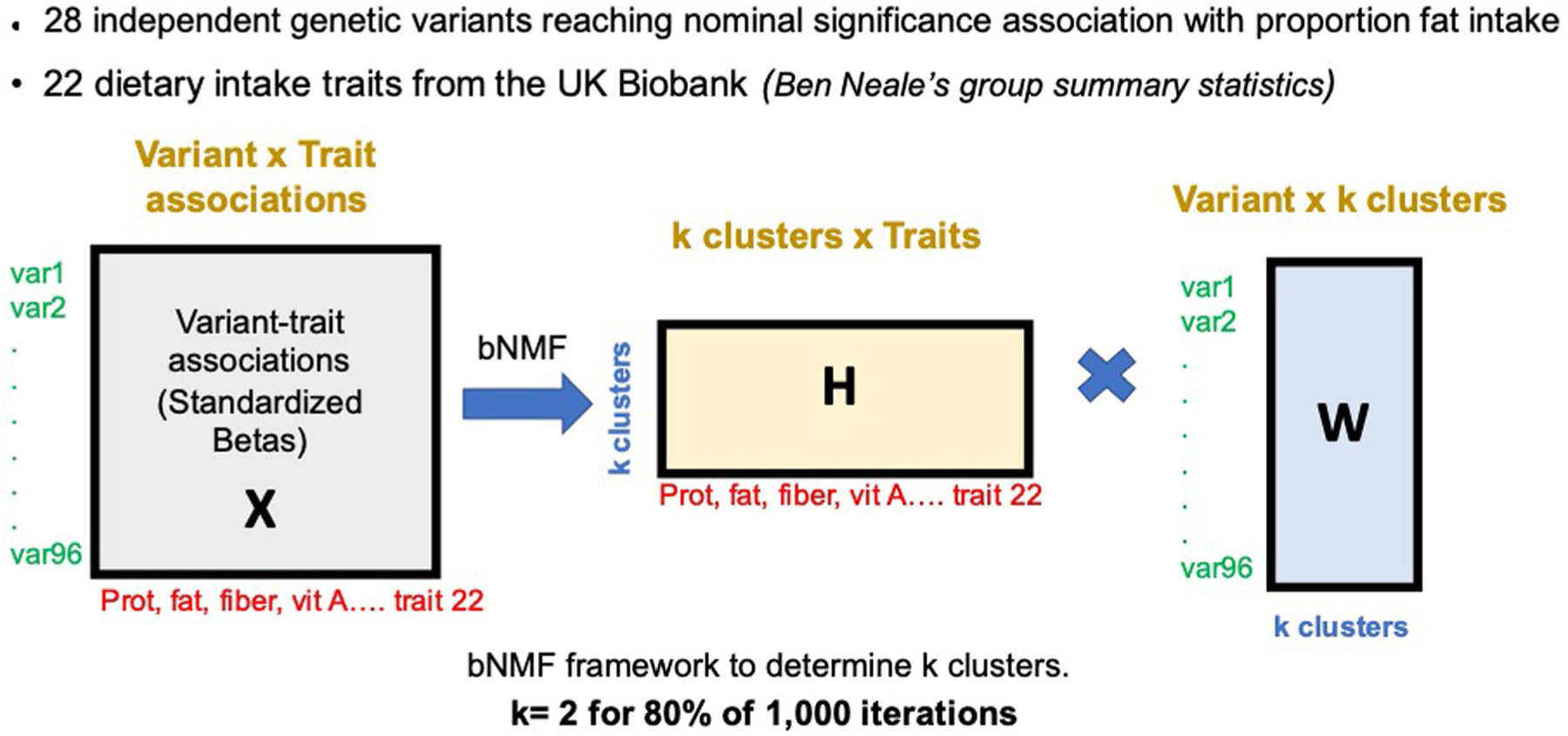
The input for the Bayesian nonnegative matrix factorization clustering algorithm (bNMF) was the set of 31 genetic variants reaching nominal significance association with proportion fat intake. Next summary association statistics for 22 dietary intake traits from the UK Biobank were aggregated for each dietary intake variant. Our analyses involved variants aligned by their alleles associated with increased fat intake. We generated standardized effect sizes for variant trait associations from GWAS by dividing the estimated regression coefficient beta by the standard error, using the UK Biobank summary statistic results (variant-trait association matrix (31 by 22)). The defining features of each cluster were determined by the most highly associated traits, which is a natural output of the bNMF approach. bNMF algorithm was performed in R for 1,000 iterations with different initial conditions, and the maximum posterior solution at the most probable number of clusters was selected for downstream analysis.
Extended Data Fig. 6 |. Trait and loci association to clusters.
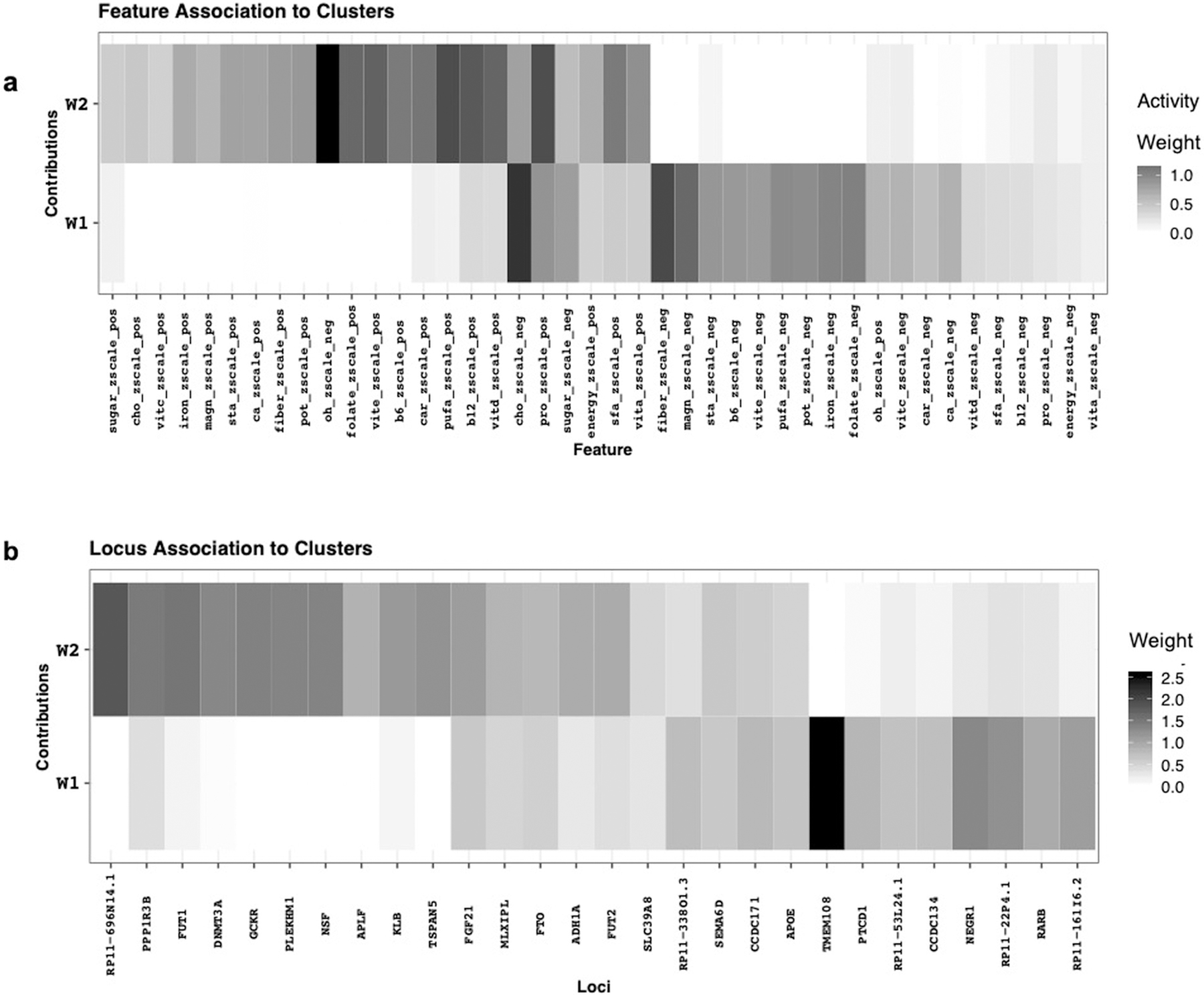
Clustering of variant-trait associations was performed for 31 genetic variants reaching nominal significance association with proportion fat intake and 22 nutritional traits derived from GWAS using the Bayesian nonnegative matrix factorization clustering algorithm, with identification of two clusters present on 80% of iterations. Loci and traits defining each cluster were based on a cut-off of weighting of 0.94 (Methods). a) trait association to cluster, b) loci association to cluster: 1 NEGR1, RARB, RP11.161I6.2, RP11.22P4.1, TMEM108. Cluster 2: ADH1A, DNMT3A, FGF21, FUT1, FUT2, GCKR, KLB, PLEKHM1, PPP1R3B, RP11.696N14.1, TSPAN5.
Supplementary Material
Acknowledgements
This study was designed and carried out by the CHARGE Consortium Nutrition Working Group. Part of this work was conducted using the UKBB resource under application no. 27892. This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No 703787. J.M. is supported by the National Institutes of Health (NIH) grant no. P30 DK040561. H.S.D. and R.S. are supported by NIH grant nos. R01 DK105072 and DK107859. R.S. is supported by NIH grant no. R01 DK102696 and the MGH Research Scholar Fund. J.M.L. is supported by NIH grant nos. F32 DK102323 and T32 HL007901. C.S., J.C.F. and J.D. are supported by NIH grant no. U01 DK078616. J.C.F. is supported by NIH grant no. K24 DK110550. J.C. is supported by the American Diabetes Association Pathway to Stop Diabetes award no. 1–18-INI-14. T.H.P. and P.V.T. acknowledge the Novo Nordisk Foundation (no. NNF16OC0021496). T.H.P. acknowledges the Lundbeck Foundation (no. R19020143904). This research was supported in part by the Intramural Research Program of the National Institute on Aging. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Footnotes
Code availability
The code used to reproduce the analyses for this manuscript will be made available on publication at http://sites.bu.edu/fhspl/publications/ and https://github.com/perslab/Merino_2020. The CELLEX precomputed expression specificity files are available at https://github.com/perslab/CELLECT/wiki/Precomputed-CELLEX-datasets.
Competing interests
A.Y.C. is currently employed by Merck Research Laboratories. M.K.R. reports receiving research funding from Novo Nordisk, consultancy fees from Novo Nordisk and Roche Diabetes Care and modest owning of shares in GlaxoSmithKline. All other authors declare no competing interests.
Additional information
Extended data is available for this paper at https://doi.org/10.1038/s41562-021-01182-w.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41562-021-01182-w.
Peer review information Nature Human Behaviour thanks the anonymous reviewers for their contribution to the peer review of this work.
Data availability
The summary GWAS statistics will be publicly available at the UKBB website (http://biobank.ctsu.ox.ac.uk/), database of Genotypes and Phenotypes (accession no. phs000930) and Type 2 Diabetes Knowledge Portal (http://www.kp4cd.org/dataset_downloads/t2d). The single-cell expression datasets can be found at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc= under accession nos. GSE93374, GSE104276 and GSE763816,46,65. The ldsc command line tool can be found at https://github.com/bulik/ldsc20.
References
- 1.Atasoy D, Betley JN, Su HH & Sternson SM Deconstruction of a neural circuit for hunger. Nature 488, 172–177 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van der Klaauw AA & Farooqi IS The hunger genes: pathways to obesity. Cell 161, 119–132 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Andermann ML & Lowell BB Toward a wiring diagram understanding of appetite control. Neuron 95, 757–778 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Polderman TJC et al. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat. Genet 47, 702–709 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Merino J et al. Genome-wide meta-analysis of macronutrient intake of 91,114 European ancestry participants from the cohorts for heart and aging research in genomic epidemiology consortium. Mol. Psychiatry 24, 1920–1932 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campbell JN et al. A molecular census of arcuate hypothalamus and median eminence cell types. Nat. Neurosci 20, 484–496 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Livneh Y et al. Homeostatic circuits selectively gate food cue responses in insular cortex. Nature 546, 611–616 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Farooqi IS et al. Leptin regulates striatal regions and human eating behavior. Science 317, 1355 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lowell BB New neuroscience of homeostasis and drives for food, water, and salt. N. Engl. J. Med 380, 459–471 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Gaich G et al. The effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metab 18, 333–340 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Søberg S et al. FGF21 is a sugar-induced hormone associated with sweet intake and preference in humans. Cell Metab 25, 1045–1053.e6 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Chu AY et al. Novel locus including FGF21 is associated with dietary macronutrient intake. Hum. Mol. Genet 22, 1895–1902 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhong VW et al. A genome-wide association study of bitter and sweet beverage consumption. Hum. Mol. Genet 28, 2449–2457 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meddens SFW et al. Genomic analysis of diet composition finds novel loci and associations with health and lifestyle. Mol. Psychiatry 10.1038/s41380-020-0697-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vroom C-R, de Leeuw C, Posthuma D, Dolan CV & van der Sluis S The more the merrier? Multivariate approaches to genome-wide association analysis. Preprint at bioRxiv 10.1101/610287 (2019). [DOI] [Google Scholar]
- 16.Lane JM et al. Genome-wide association analyses of sleep disturbance traits identify new loci and highlight shared genetics with neuropsychiatric and metabolic traits. Nat. Genet 49, 274–281 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turley P et al. Multi-trait analysis of genome-wide association summary statistics using MTAG. Nat. Genet 50, 229–237 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sudlow C et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 12, e1001779 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu X et al. Meta-analysis of correlated traits via summary statistics from GWASs with an application in hypertension. Am. J. Hum. Genet 96, 21–36 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bulik-Sullivan BK et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet 47, 291–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu M et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat. Genet 51, 237–244 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagel M, Watanabe K, Stringer S, Posthuma D & van der Sluis S Item-level analyses reveal genetic heterogeneity in neuroticism. Nat. Commun 9, 905 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karlsson Linnér R et al. Genome-wide association analyses of risk tolerance and risky behaviors in over 1 million individuals identify hundreds of loci and shared genetic influences. Nat. Genet 51, 245–257 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi KW et al. Assessment of bidirectional relationships between physical activity and depression among adults: a 2-sample Mendelian randomization study. JAMA Psychiatry 76, 399–408 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi KW et al. An exposure-wide and Mendelian randomization approach to identifying modifiable factors for the prevention of depression. Am. J. Psychiatry 177, 944–954 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ardlie KG et al. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramasamy A et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat. Neurosci 17, 1418–1428 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jaffe AE et al. Developmental and genetic regulation of the human cortex transcriptome illuminate schizophrenia pathogenesis. Nat. Neurosci 21, 1117–1125 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schumann G et al. KLB is associated with alcohol drinking, and its gene product β-Klotho is necessary for FGF21 regulation of alcohol preference. Proc. Natl Acad. Sci. USA 113, 14372–14377 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Z et al. Association of corticotropin-releasing hormone receptor1 gene SNP and haplotype with major depression. Neurosci. Lett 404, 358–362 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Schaum N et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zeisel A et al. Molecular architecture of the mouse nervous system. Cell 174, 999–1014.e22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tan VYF & Févotte C Automatic relevance determination in nonnegative matrix factorization with the β-divergence. IEEE Trans. Pattern Anal. Mach. Intell 35, 1592–1605 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Locke AE et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 518, 197–206 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scott RA et al. An expanded genome-wide association study of type 2 diabetes in Europeans. Diabetes 66, 2888–2902 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nikpay M et al. A comprehensive 1000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet 47, 1121–1130 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karlson EW, Boutin NT, Hoffnagle AG & Allen NL Building the Partners HealthCare Biobank at Partners Personalized Medicine: informed consent, return of research results, recruitment lessons and operational considerations. J. Pers. Med 6, 2 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blouet C & Schwartz GJ Brainstem nutrient sensing in the nucleus of the solitary tract inhibits feeding. Cell Metab 16, 579–587 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hayes MR et al. Endogenous leptin signaling in the caudal nucleus tractus solitarius and area postrema is required for energy balance regulation. Cell Metab 23, 744 (2016). [DOI] [PubMed] [Google Scholar]
- 40.D’Agostino G et al. Nucleus of the solitary tract serotonin 5-HT2C receptors modulate food intake. Cell Metab 28, 619–630.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holt MK et al. Preproglucagon neurons in the nucleus of the solitary tract are the main source of brain GLP-1, mediate stress-induced hypophagia, and limit unusually large intakes of food. Diabetes 68, 21–33 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Timshel PN, Thompson JJ & Pers TH Genetic mapping of etiologic brain cell types for obesity. eLife 9, e55851 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shai I et al. Weight loss with a low-carbohydrate, Mediterranean, or low-fat diet. N. Engl. J. Med 359, 229–241 (2008). [DOI] [PubMed] [Google Scholar]
- 44.Ludwig DS, Willett WC, Volek JS & Neuhouser ML Dietary fat: from foe to friend? Science 362, 764–770 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Gibson EL Emotional influences on food choice: sensory, physiological and psychological pathways. Physiol. Behav 89, 53–61 (2006). [DOI] [PubMed] [Google Scholar]
- 46.La Manno G et al. Molecular diversity of midbrain development in mouse, human, and stem cells. Cell 167, 566–580.e19 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu B et al. Development and evaluation of the Oxford WebQ, a low-cost, web-based method for assessment of previous 24 h dietary intakes in large-scale prospective studies. Public Health Nutr 14, 1998–2005 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Bradbury KE, Young HJ, Guo W & Key TJ Dietary assessment in UK Biobank: an evaluation of the performance of the touchscreen dietary questionnaire. J. Nutr. Sci 7, e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Greenwood DC et al. Validation of the Oxford WebQ online 24-hour dietary questionnaire using biomarkers. Am. J. Epidemiol 188, 1 858–1867 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McCance RA & Widdowson EM McCance and Widdowson’s the Composition of Foods (Royal Society of Chemistry, 2002). [Google Scholar]
- 51.Mifflin MD et al. A new predictive equation for resting energy expenditure in healthy individuals. Am. J. Clin. Nutr 51, 241–247 (1990). [DOI] [PubMed] [Google Scholar]
- 52.Bycroft C et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li Y, Willer CJ, Ding J, Scheet P & Abecasis GR MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet. Epidemiol 34, 816–834 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Howie B, Fuchsberger C, Stephens M, Marchini J & Abecasis GR Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat. Genet 44, 955–959 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou W et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet 50, 1335–1341 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Willer CJ, Li Y & Abecasis GR METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Higgins JPT & Thompson SG Quantifying heterogeneity in a meta-analysis. Stat. Med 21, 1539–1558 (2002). [DOI] [PubMed] [Google Scholar]
- 58.Watanabe K, Taskesen E, van Bochoven A & Posthuma D Functional mapping and annotation of genetic associations with FUMA. Nat. Commun 8, 1826 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang H & Wang K Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc 10, 1556–1566 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kircher M et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet 46, 310–315 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boyle AP et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res 22, 1790–1797 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ernst J & Kellis M ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods 9, 215–216 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kundaje A et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.de Leeuw CA, Mooij JM, Heskes T & Posthuma D MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol 11, e1004219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhong S et al. A single-cell RNA-seq survey of the developmental landscape of the human prefrontal cortex. Nature 555, 524–528 (2018). [DOI] [PubMed] [Google Scholar]
- 66.Hodge RD et al. Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang D et al. Comprehensive functional genomic resource and integrative model for the human brain. Science 362, eaat8464 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eastwood SV et al. Algorithms for the capture and adjudication of prevalent and incident diabetes in UK Biobank. PLoS ONE 11, e0162388 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The summary GWAS statistics will be publicly available at the UKBB website (http://biobank.ctsu.ox.ac.uk/), database of Genotypes and Phenotypes (accession no. phs000930) and Type 2 Diabetes Knowledge Portal (http://www.kp4cd.org/dataset_downloads/t2d). The single-cell expression datasets can be found at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc= under accession nos. GSE93374, GSE104276 and GSE763816,46,65. The ldsc command line tool can be found at https://github.com/bulik/ldsc20.