

Abstract
Cancer and the immune system share an intimate relationship. Chronic inflammation increases the risk of cancer occurrence and can also drive inflammatory mediators into the tumor microenvironment enhancing tumor growth and survival.
The p38 MAPK pathway is activated both acutely and chronically by stress, inflammatory chemokines, chronic inflammatory conditions, and cancer. These properties have led to extensive efforts to find effective drugs targeting p38, which have been unsuccessful. The immediate downstream serine/threonine kinase and substrate of p38 MAPK, mitogen-activated-protein-kinase-activated-protein-kinase-2 (MK2) protects cells against stressors by regulating the DNA damage response, transcription, protein and messenger RNA stability, and motility. The phosphorylation of downstream substrates by MK2 increases inflammatory cytokine production, drives an immune response, and contributes to wound healing.
By binding directly to p38 MAPK, MK2 is responsible for the export of p38 MAPK from the nucleus which gives MK2 properties that make it unique among the large number of p38 MAPK substrates. Many of the substrates of both p38 MAPK and MK2 are separated between the cytosol and nucleus and interfering with MK2 and altering this intracellular translocation has implications for the actions of both p38 MAPK and MK2.
The inhibition of MK2 has shown promise in combination with both chemotherapy and radiotherapy as a method for controlling cancer growth and metastasis in a variety of cancers. Whereas the current data are encouraging the field requires the development of selective and well tolerated drugs to target MK2 and a better understanding of its effects for effective clinical use.
Keywords: cancer, cell survival, inflammation, MAPKAPK2, metastasis, migration, MK2, p38 MAPK
1 |. INTRODUCTION
There exists an intimate relationship between inflammation and tumorigenesis. The presence of inflammatory or autoimmune conditions increases the likelihood of developing cancer.1,2 This chronic inflammation attracts fibroblasts, myeloid, and lymphoid cells to the tumor microenvironment (TME) which can then maintain the inflammatory phenotype and propagate tumorigenesis. Additionally, inflammatory signals secreted from the tumor itself, the inflammatory TME, and/or cancer therapies can also exacerbate inflammation and drive tumor progression.1,2 Given the well-documented contribution of inflammation to tumor growth and tumor progression, ongoing research targeting the mechanisms of tumor inflammation is invaluable and may lead to therapies that can be applied to myriad cancer types.
The p38 MAPK-MAPKAPK2 (mitogen-activated-protein-kinase-activated-protein-kinase-2 [MK2]) signaling axis is activated by cellular or environmental stressors and stimulates the expression of downstream effector proteins that activate inflammatory cytokines, chemokines, and transcription factors.3,4 MK2 also contributes to inflammatory processes via posttranscriptional regulation of various cytokines.5 This pathway has been implicated in multiple inflammatory conditions from fibrosis6–8 to arthritis4,9,10 and given the relationship among stress, inflammation, and cancer, it is likely that MK2 plays a significant role in cancer development and/or cancer progression.
Preclinical studies utilizing p38 MAPK inhibitors in multiple cancer types have shown success, but unfortunately no p38 MAPK inhibitors have been approved for use due to severe toxicity and side effects.3 As a downstream target of p38 MAPK activation, MK2 has been investigated for its role in tumorigenesis, cancer progression, and metastasis. Overall, results have been positive demonstrating that inhibition of MK2 can decrease tumor inflammation and epithelial-to-mesenchymal transition (EMT) in vitro, and tumor growth and progression in vivo in multiple cancer types.11–16 The purpose of this review is to highlight the downstream effectors of MK2 and their roles in both normal physiological and pathological processes, and to discuss the relevance of investigations of MK2 signaling in cancer.
2 |. p38 MAPK-MK2 MOLECULAR BIOLOGY
2.1 |. p38 MAPK and the pathway leading to MK2
The p38 MAPK pathway is considered a stress-response MAPK pathway due to its phosphorylation in response to environmental or cellular stressors such as ultraviolet (UV) radiation, inflammatory cytokines, heat shock, starvation, and osmotic shock.3,17 p38 MAPK is a MAPK that comes in several isoforms, p38α (MAPK14), P38β (MAPK11), p38γ (MAPK12), and p38δ (MAPK13). The different isoforms are characterized by their expression levels; p38α and P38β are ubiquitously expressed in all cells and tissues with higher expression of p38β in the lungs. p38γ is expressed in skeletal muscle and p38δ is expressed in lung and kidney.18–21 The functions of p38α are the most robustly studied among the isoforms. p38β is generally expressed at lower levels than p38α and appears to be redundant to the functions of the cell if p38α is present22 although it does seem to have a role in in vivo bone formation that is independent of p38α.23 p38γ and p38δ are sometimes known as alternative p38 isoforms and their roles involve posttranscriptional modification of cytokine release, mucus secretion, modulating insulin release, and amyloid plaque formation in neuronal tissue.24 Of the different isoforms, only p38α knockout results in a lethal embryonic phenotype,25,26 and p38β is unable to compensate for the deletion fully even when expressed by the same promoter. This demonstrates that even with expression normalized there are differences between the isoforms controlling downstream functions.27 It is assumed that the p38α (p38 MAPK) isoform is most relevant to this review as it is the isoform that controls the response to lipopolysaccharide (LPS) induced tumor necrosis factor α (TNFα) release and its interaction with MK2 is the most studied.28–30 Whereas we cannot discount any MK2 interactions with the other isoforms, to date no evidence to support their interaction exists.
The p38 MAPK pathway is one of the three highly conserved mammalian MAPK pathways that include the extracellular response kinase (ERK-1) and c-jun N-terminal kinases (JNK) signal transduction pathways. The ERK pathway is generally activated by extracellular mitogens whereas the JNK and p38 MAPK pathways were traditionally known as stress-activated protein kinases (SAPK) that respond to environmental stressors such as osmotic shock, UV radiation, and ischemic injury.31 The three signaling pathways are characterized by the canonical triple-tiered cascade of kinases beginning with the MAPK kinase kinase (MAP3K), followed by MAPK kinase (MAP2K), and finally MAPK. The MAP3Ks are relatively non-specific kinases that are classed as MAPK/ERK kinase (MEK) kinases, mixed lineage kinases (MLKs), and thousand and one kinases (TAOs).32 These factors are regulated by small GTPases or phosphorylation events such as the activation of transforming growth factor-β-activated kinase 1 (TAK1) which forms a complex with transforming growth factor binding proteins (TAB1, TAK1-binding protein 2 [TAB2]) to phosphorylate downstream MAP2Ks.33 The MAP3K responsible for upstream activation of the p38 MAPK signaling pathway are diverse and include MEKK1–4, ASK1, DLK1, TAK1, TAO 1/2, and ZAK1.3,34
Downstream from the MAP3K, the MAP2Ks that phosphorylate p38 MAPK specifically are MKK3 and MKK6. These two proteins share an 87% homology35,36 and activate p38 MAPK by phosphorylating Thr180 and Tyr182 in its activation loop. Although these MAP2Ks can activate other MAPKs (i.e., JNK37), they are vital for the canonical receptor activation of the p38 MAPK pathway. However, they are not the only MAP2K molecules capable of activating the p38 MAPK pathway. UV light has also been shown to cause the phosphorylation of p38 MAPK through the activity of MKK4 in mouse fibroblasts38 and there are other mechanisms of p38 MAPK signaling that appear to be separate from the MKK3/MKK6 signaling. G-protein coupled signaling has been implicated in the activation of p38 MAPK through the actions of E3 ubiquitin ligase neural precursor cell expressed developmentally downregulated 4–2 (NEDD4–2). Receptor binding causes the recruitment of NEDD4–2 which then ubiquitinates the receptor, orchestrating the binding of ubiquitin-binding adaptor protein TAB2. In turn, TAB2 then recruits TAB1, which binds and induces autophosphorylation of p38 MAPK.39,40 The generation of a TAB/p38 MAPK signaling complex by the actions of AMP-activated protein kinase (AMPK) in response to ischemia and hypoxia is another proposed noncanonical activation of p38 MAPK.41 The direct interaction of TAB1 with p38α induces a conformational change moving the active loop into the catalytic domain and enhancing ATP-binding, thus enabling cis-autophosphorylation of the active loop at Thr180 and Tyr182.40 Another method of MKK3/6 independent activation of p38 is through the actions of Src-family zeta-chain-associated protein kinase 70 (Zap70) in T cells. This activation is important for T-cell receptor-mediated activation of T cells. Zap70 causes phosphorylation of p38α and p38β at Tyr323. This results in dimerization of the p38 MAPK molecules and autophosphorylation at Thr180.42
p38 MAPK acts on many downstream targets that are involved in the stress response including embryonic development, immune response, cell cycle, cell differentiation, metabolism, senescence, and survival. The number of downstream targets currently exceeds more than 100 and that number will likely increase as a complete phospho-proteome on p38 MAPK targets has not been completed.3,43–45 The wide-reaching impact of p38 MAPK has made it an attractive target for treatment of immune disorders and cancer for the past two decades. However, the search for drugs that act on p38 MAPK has been hampered by a wide range of side effects including hepatic toxicity, cardiac toxicity, and central nervous system disorders, or a lack of efficacy.46–48 The lack of effective drugs may be due to the pleiotropic effects of p38 MAPK downstream signaling or off-target effects of the drugs. As a result, except for the weak and nonselective p38 MAPK inhibitor pir-fenidone, no approved drug that targets p38 MAPK inhibitors have passed Phase III clinical trials.48–50 Efforts continue as Ralimetinib (LY2228820) is in multistage Phase I and II testing and shows promise when combined with chemotherapy in female cancers and glioblastoma.3 A significant amount of effort was placed into finding drugs targeting p38, negatively impacting efforts to examine other kinases for suitable inhibitors. Although, in recent years the understanding of related signaling cascades has improved and efforts to look for targets that have fewer toxicities but comparable efficacy have advanced. MK2, a molecule that is directly downstream to p38 MAPK, is one such target that controls a significant portion of the inflammatory signaling post p38 MAPK phosphorylation. The inhibition of MK2 is not predicted to be as harmful as p38 MAPK inhibition because MK2 KO mice were viable and fertile, grew to normal size, and did not exhibit obvious behavioral defects whereas p38 MAPK knockout mice are embryonically lethal.26,28,51,52 Assuming the issues of p38 MAPK inhibition arise from its pleiotropic nature then MK2 may be a more attractive target for inhibition of inflammation and other cellular responses driven by the activation of the p38 MAPK-MK2 pathway.
2.2 |. MK2 structure and interaction with p38
MK2 is serine/threonine kinase that was identified in 1992 by Stokoe et al.53 who were looking for kinases that could phosphorylate glycogen synthase. MK2 was found to be expressed in several tissues including kidney, skeletal muscle, liver, testis, lung, and spleen.53 A recent large-scale proteome showed very little protein expression in muscle tissue but high expression in gastrointestinal tissue and immune organs like lymph nodes and bone marrow.54
Gene data show that there are two main transcripts to MK2; Isoform 1 (NM_004759.5, MK2s) is a 370 amino acid protein isolated by Zu et al.,55 whereas Isoform 2 (NM_032960.4, MK2) is a 400 amino acid in length similar to the protein isolated initially by Stokoe and colleagues.53 The latter variant uses an alternate splice junction at the 5ʹ end of the last exon compared to variant 1 and as a result, has a longer C-terminus (Figure 1). The full-length transcript (transcript 2, MK2) has a proline-rich N terminal domain (10–40) followed by XI catalytic subdomains (64–325) and a regulatory C-terminus domain (338–400). The c-terminal regulatory domain has an auto-inhibitory domain (339–353, a nuclear export sequence [NES; 356–365]), and a bipartite nuclear localization sequence [NLS; 371–374, 385–389]) that governs the activity and cellular location of the enzyme.56 For the enzyme to display kinase activity, two of three major phosphorylation sites (Thr222, Ser272, Thr334) must be phosphorylated with maximal activity if all three are phosphorylated.57 Two of these sites reside in the catalytic domain (Thr222 and Ser272), one of which is located between the catalytic domain and the auto-inhibitory domain (Thr334).
FIGURE 1.
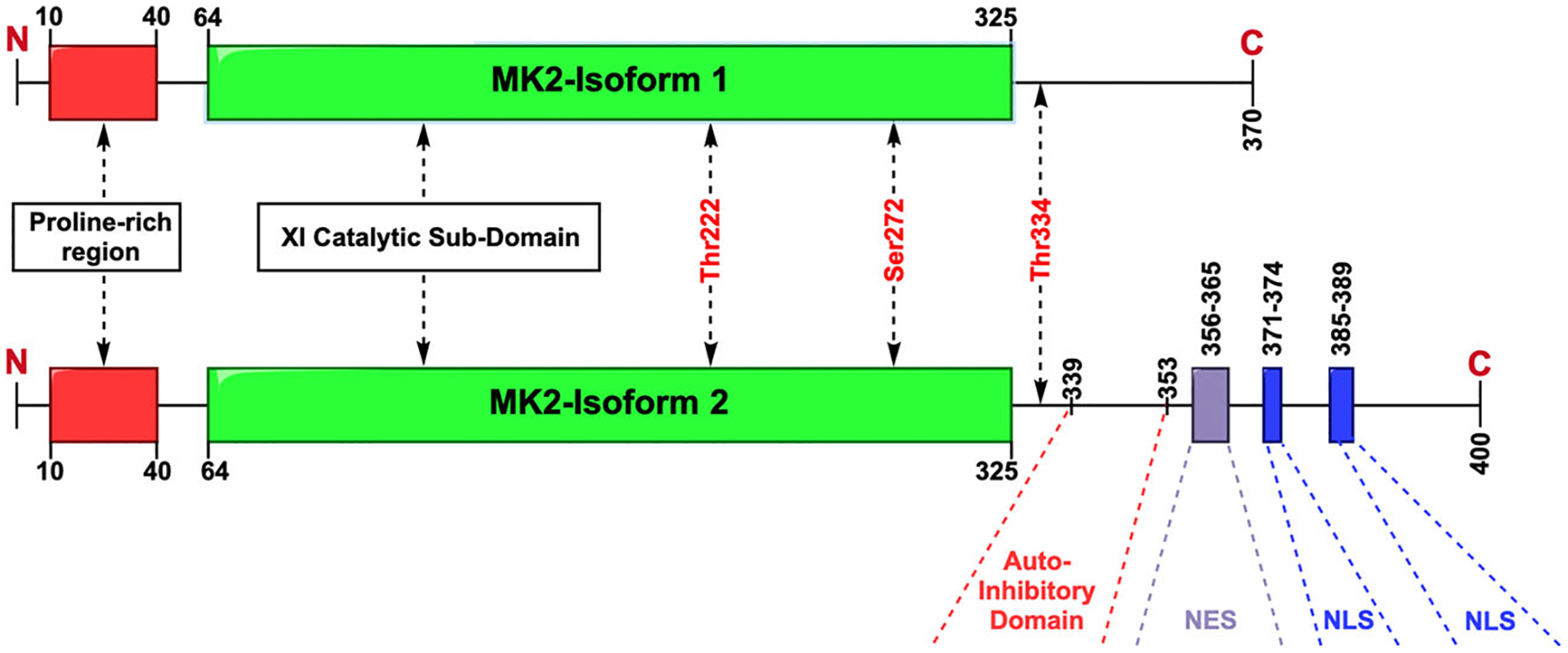
A schematic diagram of the two known transcripts for MK2. Isoform 1 is the shorter version shown above which contains an N-terminus proline rich region, the catalytic domain and a truncated C-terminus. The longer isoform 2 also contains the N-terminus proline rich region and the catalytic domain but also includes an auto-inhibitory domain, nuclear export sequence (NES), and two nuclear location sequences (NLS). The sites phosphorylated by p38, which is required for activation, are labeled in red. MK2, mitogen-activated-protein-kinase-activated-protein-kinase-2
The proline-rich N terminal domain was originally thought of as a potential binding site for SH3 binding domains58 and has been shown to be important to the motility of cells.59 Stokoe et al.,53 found that the catalytic subdomains require a minimum amino acid sequence of HYD–X–R–X–X–Ser–X–X, where HYD is a hydrophobic residue, for efficient phosphorylation of target kinases. A more precise optimal sequence based on several known substrates of MK2 was generated later ([Leu/Phe/Ile]–[X]–[Arg]–[Gln/Ser/Thr]–[Leu]–[Ser/Thr]–[Hydrophobic]).60
The catalytic subdomain contains two of the three phosphorylation sites required for kinase activity. Thr222 is of particular relevance because it sits in the activation loop (207–233) between catalytic subdomains VII and VIII and is thought to cause a conformational change in the catalytic domain to activate the kinase.61,62 The homology of the kinase domain makes it most similar to various calcium-activated kinases and myosin kinases.53,58 The C-terminal domain regulates the kinase activity of the cell, its cellular location, and its binding to its activating enzyme p38 MAPK. MK2 contains an inhibitory peptide that obscures both the catalytic domain and the ATP-binding site preventing activity.56 The phosphorylation of Thr334 weakens the binding of the autoinhibitory domain to the catalytic site, increasing kinase activity, and is also critical to the cellular localization of MK2 as it exposes the NES sequence that is otherwise hidden during its resting state.58,62,63
MK2 is a direct substrate of the p38 MAPK protein and has a unique relationship with the enzyme because of the physical partnership the two molecules display. While we have a strong understanding of the pairing of the two molecules, we do not fully understand its implications. There appears to be a mutual stabilization of the p38 MAPK and MK2 molecules as genetic deletion of either results in reduced expression of the other59,64 although how this stabilization occurs is not clear. In resting cells, p38 is located mainly in the cytoplasm and MK2 is located primarily in the nucleus63,65 upon phosphorylation of p38 it is translocated to the nucleus via importins.66 Unphosphorylated MK2 has a high affinity for p38 MAPK (Kd 2.5 nM) implying that upon p38 entering the nucleus these two molecules form a tight bond.67,68 The bond is formed by five connection points on the MK2 molecule; Gly73 and ILe74 form a stable backbone, Tyr288-Tyr240 binds to the catalytic site and may move on activation, Tyr264–284 is where regulatory phosphorylation domains from both molecules interact, Asp345-Val365 binding limits access to the MK2 substrate binding site and Asp366-Ala390 binds to p38 via extensive H-bonds, salt bridges, and a favorable docking groove on p38 MAPK (glutamate-aspartate, ED, and common docking, CD, regions common to MAP kinases).29,30 This latter interaction provides a significant contribution to the binding between these two molecules as demonstrated by inhibition of MK2 phosphorylation by a peptide mimic of the MK2 C-terminal sequence at a concentration of 60 nM.68 The binding of the two molecules come together in face to face fashion, where the ATP binding sites of both kinases are at the heterodimer interface. The C-terminus of MK2 wraps around p38 MAPK, inserts in the p38 MAPK docking groove and becomes sandwiched between the two molecules kinase domains.29,30 By binding in the docking groove, MK2 also binds to the same place as many other substrates of p38 MAPK. The implication is that other substrates may be able to disrupt p38 MAPK-MK2 interaction generating a fluid on/off pattern to the binding of the two molecules. It is worth mentioning that MK2 fits in the “reverse” configuration compared to other p38 MAPK docking substrates and thus may not be removed as easily as implied by the communal docking regions.
Upon MK2 phosphorylation by p38 MAPK, the affinity between the two proteins decreases slightly as MK2 changes conformation. This conformational change also exposes the NES, which overrides the NLS, and exports the complex to the cytoplasm.57,62,69 This conformational change occurs after the phosphorylation of Thr334 and opens the NES sequence for binding to exportin-1 that transports both MK2 and p38 to the cytoplasm.62 The transport of the complex out of the nucleus exposes the two kinases to different substrates, the nuclear substrates are more likely to be transcription factors whereas cytoplasmic targets include the small heat shock proteins (sHSP) (HSP25/27) and tristetraprolin (TTP). It is therefore an interesting facet of the kinase that Thr334 is phosphorylated at half the rate of Thr222 and Ser27268 possibly giving the complex an opportunity to activate nuclear substrates before moving to the cytoplasm for alternative functions.
The smaller Isoform 1, at 370 amino acids, does not include the NLS, the NES, and a significant portion of amino acids in the c-terminus that allows MK2 to bind tightly to p38. The kinase activity is 2 orders of magnitude weaker than the longer form and its binding to p38 is also much weaker. The significance of the shorter form is not well understood and macrophages expressing the shorter form exclusively show a reduced release of TNFα compared to macrophages expressing the full-length transcript.59 It is possible that the MK2s isoform is a nuclear resident isoform that is only concerned with the activity of nuclear-based transcription factors and that the main function of the longer MK2 isoform is to confer MK2/P38 MAPK signaling to the cytosol. As the loss of full-length MK2 is all that is required to reduce TNFα release we can conclude that the migration of MK2 to the cytosol is critical for its effects on TNFα release.
2.3 |. MK2 inhibitors and their mechanism of action
Although it was mentioned in a previous section that significant efforts to target p38 has left drug design studies on MK2 lacking, there have been several attempts at targeting MK2 or the p38-MK2 interaction through rational drug design approaches.
The majority of MK2 inhibitors designed have focused on molecular interactions of the ATP binding site between p38 and MK2.47,70 The ATP-binding site is formed by a flexible glycine rich group that connects the small N-terminus with a large α-helical domain and the catalytic group and this sits in the heterodimer interface of the p38-MK2 heterodimer.30 The difficulty in designing drugs for this site is increased because of the similarity of the ATP binding site to other similar kinases such as MK3, MK5, PKA, and CDK2 which affects the selectivity of the drugs for its preferred target. Furthermore, although many of these competitive inhibitors have high affinity for the ATP binding site, the affinity of ATP for the site (Km 2 μM) and its high cellular concentration in the cell (~2–5 mM) results in a discrepancy between the binding affinity and its cellular effectiveness (the ratio of a drugs binding affinity to its cellular activity is known as the biochemical efficiency or BE).71,72 Many of the current ATP competitive inhibitors show functional responses 10–100-fold greater than their affinity to the kinase (resulting in BE values of 0.1–0.01). This aspect of ATP competitive inhibitors and the issues with their solubility and cellular permeability have eroded enthusiasm of these drugs.47 Nevertheless, there are several available research grade drugs that can be used to probe the functions of MK2 while acknowledging the deficiencies in their selectivity and BE. PF3644022 is an ATP-competitive benzothiophene inhibitor of MK2 developed by Pfizer with kinase IC50 of 5.2 nM for the enzyme and IC50 of 160 nM for inhibition of TNFα from LPS stimulated whole blood.73 This is a widely used drug in preclinical settings but suffers from the low BE issues mentioned previously and has poor solubility in aqueous solutions (~5 μM). Other ATP-competitive MK2 inhibitors commonly available include PHA 767491 (IC50 171 nM) which also inhibits cyclin-dependent kinase (CDK1–5) at similar concentrations.74 CAS 1186648-22-5 (aka MK2 inhibitor III) is also available as an ATP-competitive inhibitor of MK2 with an IC50 of 8.5 nM for MK2 and an EC50 of TNFα release of 4.4 μM. However, it also inhibits MK3 and MK5 with IC50s of 81 and 210 nM, respectively, illustrating the difficulties of targeting a molecular location with similarities to other kinases.74 So far, no ATP-competitive inhibitors have made it to clinical trials.
Noncompetitive inhibitors for MK2 were available as early as 2004 with CMPD-1 functioning as an inhibitor of p38 preventing the phosphorylation of MK2 without inhibiting the phosphorylation of other p38 substrates such as ATF-2 and MBP. The binding of CMPD-1 to p38 appeared to alter the active site region of p38 causing the suboptimal positioning of substrates and selectively inhibiting p38 phosphorylation of MK2.75 With a IC50 of 330 nM the drug appears not as effective as some of the ATP-competitive inhibitors and has off target effects that cause cytotoxicity by inhibiting tubulin formation.76 The MK2 inhibitor, MK-25 (also known as MK2 inhibitor IV), is a noncompetitive inhibitor of MK2 with an IC50 of 110 nM and and EC50 of 4 μM for TNFα release from LPS treated THP-1 cells.72
The peptide inhibitor of MK2, MMI-0100, has been studied in a number of different systems.77–80 This peptide drug targets the substrate-binding site of MK2, is carried into cells via cell-permeant domains and is rapidly taken up by macropinocytosis and targeted to endosomal compartments.8 The sequence of the peptide was designed from the consensus sequence of phosphorylation of HSP-27 and inhibits the phosphorylation of the protein as a result. It showed efficacy in reducing pulmonary and cardiac fibrosis and inflammation in animal models at micromolar concentrations and had entered clinical trials in 2014 but has not appeared to progress since.
A recently developed MK2 inhibitor, ATI-450 (also known as CDD-450) selectively blocks p38α activation of MK2 while sparing the inhibition of other effectors of p38α. The inhibitor was designed to interact with the binding surfaces interface near the p38α ATP site and a natural cleft in MK2. The presence of ATI-450 prevents phosphorylation of MK2 by p38 due to this in a manner 700-fold greater than its ability to inhibit phosphorylation of ATF-2 and PRAK.10 The drug has shown efficacy in preclinical trials, inhibiting interleukins (IL)-1β and TNFα at 1–10 μM concentrations. The drug was demonstrated to be safe and well tolerated following completion of a Phase 1 clinical trial.81 Current Phase 2a trials for rheumatoid arthritis (NCT04247815) and COVID-19 (NCT04481685) have both recently completed patient accrual and safety and efficacy results from both of these trials are forthcoming.
3 |. DOWNSTREAM TARGETS AND CELLULAR FUNCTION OF MK2
With the activation of p38 MAPK, MK2 becomes activated via phosphorylation and in turn phosphorylates downstream substrates that mediate migration, cell growth, differentiation, inflammation, and apoptosis in downstream pathways (Figure 2). Whereas some of the downstream targets have well-known functions, there are other targets that we can only speculate about their cellular effects based on the understanding we have of the physiological response to the phosphorylation and activation of the relevant protein species. Typically, examining the consequences of signaling cascades results in protein blots and kinetic experiments that capture early events in what are often processes that occur over days, weeks, or months in vivo. Hence, we are left trying to extrapolate from intracellular signaling into whole-body physiology that may leave many of the details up to interpretation.
FIGURE 2.
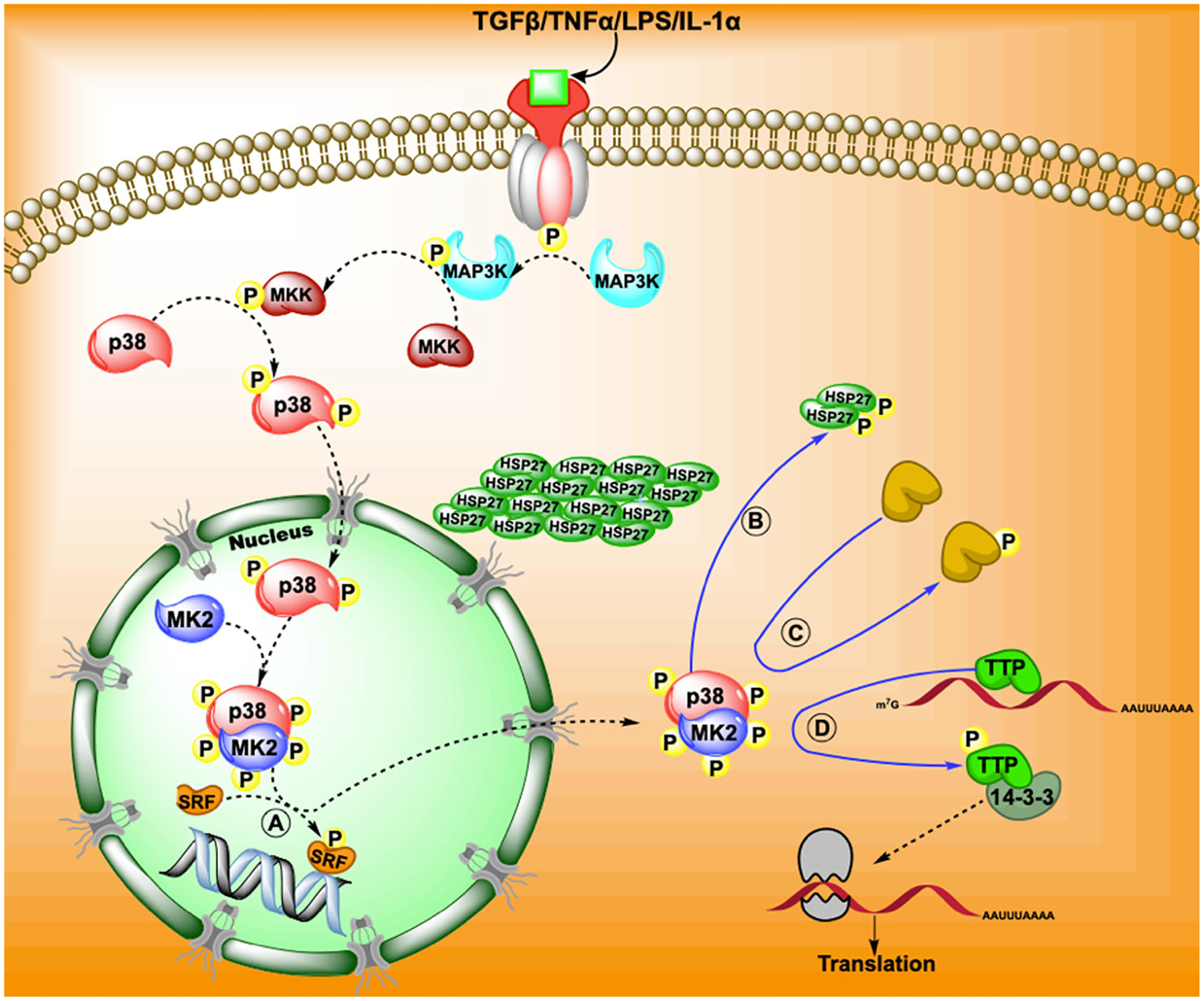
A brief overview of the canonical pathway for p38 MAPK-MK2 pathway and the downstream effects following MK2 phosphorylation. The pathway may involve the binding of a cytokine or growth factor to a cell membrane receptor which results in a sequence of kinase chains through MAP3K to MKK to p38 MAPK. The phosphorylation of p38 MAPK stimulates its migration into the cytosol where it binds and phosphorylates MK2. Phosphorylated MK2 is then activated and may further phosphorylate (A) transcription factors and co-factors that initiate transcription. Phosphorylation of MK2 results in exposing its nuclear export sequence which facilitates the movement of p38 MAPK-MK2 complex into the cytosol. Here, MK2 further phosphorylates targets such as (B) heat shock proteins, (C) enzymes (e.g., 5-lipoxygenase) and (D) binding proteins and chaperones (TTP or 14-3-3) to elicit responses. The figure shows that the migration of MK2 and thus p38 are required for their full function
We list the known substrates for MK2 based on experiments that have shown direct MK2 phosphorylation (Table 1), and attempt to delineate what this might mean for the fate of cells responding to MK2 activation below. When considering the different types of substrates phosphorylated by MK2 a pattern of activity is seen. Under stress conditions, MK2 action slows the cell cycle to enable repair, stabilizing necessary protein structures and eliminating potential hazardous proteins, increasing transcription of immediate early genes for rapid cellular reaction, stabilizing messenger RNA (mRNA) for rapid translation, regulating cellular motility and initiating immune signals for leukocyte influx and tissue repair. Many of these mechanisms have been explored in detail and are covered below.
TABLE 1.
The known phosphorylation substrates of MK2
| Molecule name | Function of MK2 phosphorylation | Phosphorylation site | Reference |
|---|---|---|---|
| HSP25/HSP27 | Protein Stabilization, chaperoning and motility | HSP25-Ser15 Ser86 | Stokoe et al. (1992)53 |
| HSP27-Ser15 Ser78 Ser82 | Rouse et al. (1994)82 | ||
| LIMK1 | Cell motility | Ser323 | Kobayashi et al. (2006)83 |
| LSP1 | cell motility | Ser243 | Wu et al. (2007)84 |
| NOGO-B | Motility | Ser107 | Rousseau et al. (2005)85 |
| KRT18, KRT20, Vimentin | Cell structure and motility | Keratin 18 (Ser52) and 20 (Ser20) | Menon et al. (2010)86 |
| RCSD1/CAPZIP | Motility | Ser179 and Ser244 | Eyers et al. (2005)87 |
| SRF | Transcription factor | Ser103 | Heidenreich et al. (1999)88 |
| SRC-3 | Transcription cofactor | Ser857 | Shrestha et al. (2020)89 |
| MRTF-A | Transcription cofactor | Ser312 and Ser333 | Ronkina et al. (2016)90 |
| CREB | Transcription factor | Ser133 | Tan et al. (1996),91 Iordanov et al. (1997),92 Faust et al. (2012)93 |
| HSF1 | Transcription factor | Ser121 | Wang et al. (2006)94 |
| ALOX5 | Immune response | Ser271 | Werz et al. (2000),95 Flamand et al. (2009)96 |
| PDE4A | Cell signaling, immune response | Ser147 | MacKenzie et al. (2011),97 Houslay et al. (2017),98 (2019)99 |
| RPS6KA3 | Cell signaling | Ser386 | Zaru et al. (2007)100 |
| TAB3 | Cell signaling | Ser506 | Mendoza et al. (2008)101 |
| RIPK-1 | Signaling and cell death | Ser321 | Jaco et al. (2017)102 |
| ZFP36/TTP | RNA binding protein | Ser52 and Ser178 | Mahtani et al. (2001),103 Ronkina et al. (2019)104 |
| HNRNP A0 | RNA binding protein | Ser84 | Rousseau et al. (2002)105 |
| NELFE | RNA binding protein | Ser51, Ser115, and Ser251 | Borisova et al. (2018)106 |
| RBM7 | RNA binding protein | Ser136 | Tiedje et al. (2015)107 |
| PABPC1 | RNA binding protein | unknown | Bollig et al. (2003)108 |
| Dazl | RNA binding protein | Ser65 | Williams et al. (2016)109 |
| 14-3-3ζ | Accessory binding protein | Ser58 | Powell et al. (2003)110 |
| PARN | mRNA stability and cell cycle control | Ser557 | Reinhardt et al. (2010)87, Duan et al., 2020)111 |
| CDC25B, CDC25C | Cell cycle control | Ser169, Ser249, Ser323, Ser353 and Ser375 | Lemaire et al. (2006)112 |
| HDM2/MDM2 | Cell cycle and DNA repair | Ser157 and Ser166 | Weber et al. (2005)51 |
| ATDC/TRIM29 | Cell signaling | Ser550 | Wang et al. (2014)113 |
| UBE2J1 ubiquitin conjugating enzyme | Posttranslational modification | Ser184 | Menon et al. (2013)114 |
| CEP131 | Protein stability | Ser47 and Ser78 | Tollenaere et al. (2015)115 |
| Beclin-1 | Autophagy | Ser90 | Wei et al. (2015)116 |
| RSK | macropinocytosis | Ser386 | Zaru et al. (2007)100 |
Note: Each protein listed is confirmed to have sites phosphorylated by MK2.
Abbreviations: LSP1, lymphocyte specific protein 1; SRC-3, steroid receptor coactivator 3; SRF, serum response factor.
3.1 |. Cell cycle and the DNA damage response
The DNA damage response results in a series of steps that are designed to halt the cell cycle to facilitate repair of DNA before cell division occurs. Damage to the DNA results in the activation of the proteins ATM and ATR and subsequent phosphorylation of a range of proteins including p38, MK2, p53, MDM2, ChK1, and ChK2.117,118
The tumor suppressor, p53, is typically regarded as one of the main effector molecules of the DNA damage response by transcribing the protein p21 that inhibits CDK2-cyclin and CDK1-cyclin activities leading to an arrest of the cell cycle in either the G1/S or G2/M phases, respectively.119 In the absence of an effective p53 response, the cell short circuits and begins to rely on MK2 for cell cycle arrest117 because several its substrates are involved in the regulation of the cell cycle during the DNA damage response.
The cell cycle phosphatases M-phase inducer phosphatase 2 and 3 (CDC25B, CDC25C) play an important role in determining the transitions between the different phases of the cell cycle. Various CDK/cyclin complexes are activated in rhythmic patterns as the cell cycle progresses. The CDC25 proteins dephosphorylate CDK proteins and activate them, progressing the cell cycle forward. CDC25B and CDC25C are both involved in regulating the G2/M transition by activating CDK1-cyclinB causing the cycle to advance.120 MK2 phosphorylates CDC25B and CDC25C in response to UV radiation112 increasing the binding to 14-3-3 proteins, altering the cellular location of CDC25, marking the CDC25 molecules for degradation, and arresting the cell cycle in the G2/M phase60,117,121 (see Figures 3 and 4). Further, MK2 phosphorylates 14-3-3ζ as determined by computer modeling and mutagenesis.110 These proteins are ubiquitously expressed in nature and serve as scaffold proteins that form complexes with themselves and other molecules to regulate processes in the cell such as the cell cycle and RNA stability.122 The phosphorylation of 14-3-3 alters their ability to dimerize and bind other substrate proteins where they act as chaperones and stabilization proteins.123 There are 7 known 14-3-3 isoforms and, although we know 14-3-3ζ is the molecule targeted by MK2 isoforms, 14-3-3β, 14-3-3ε, and 14-3-3γ contain a serine in a MK2 consensus phosphorylation site at a location similar to that of 14-3-3ζ. Thus, MK2 may regulate the function of several 14-3-3 isoforms and a wide range of cellular processes including cell cycle, apoptosis and autophagy.110,124 In this case, the phosphorylation of 14-3-3 proteins by MK2 enhances the arrest of the cell cycle by targeting CDC25C/B. It is through this mechanism that MK2 can act as a checkpoint molecule allowing the cell to halt division before adequate DNA repair has taken place.
FIGURE 3.
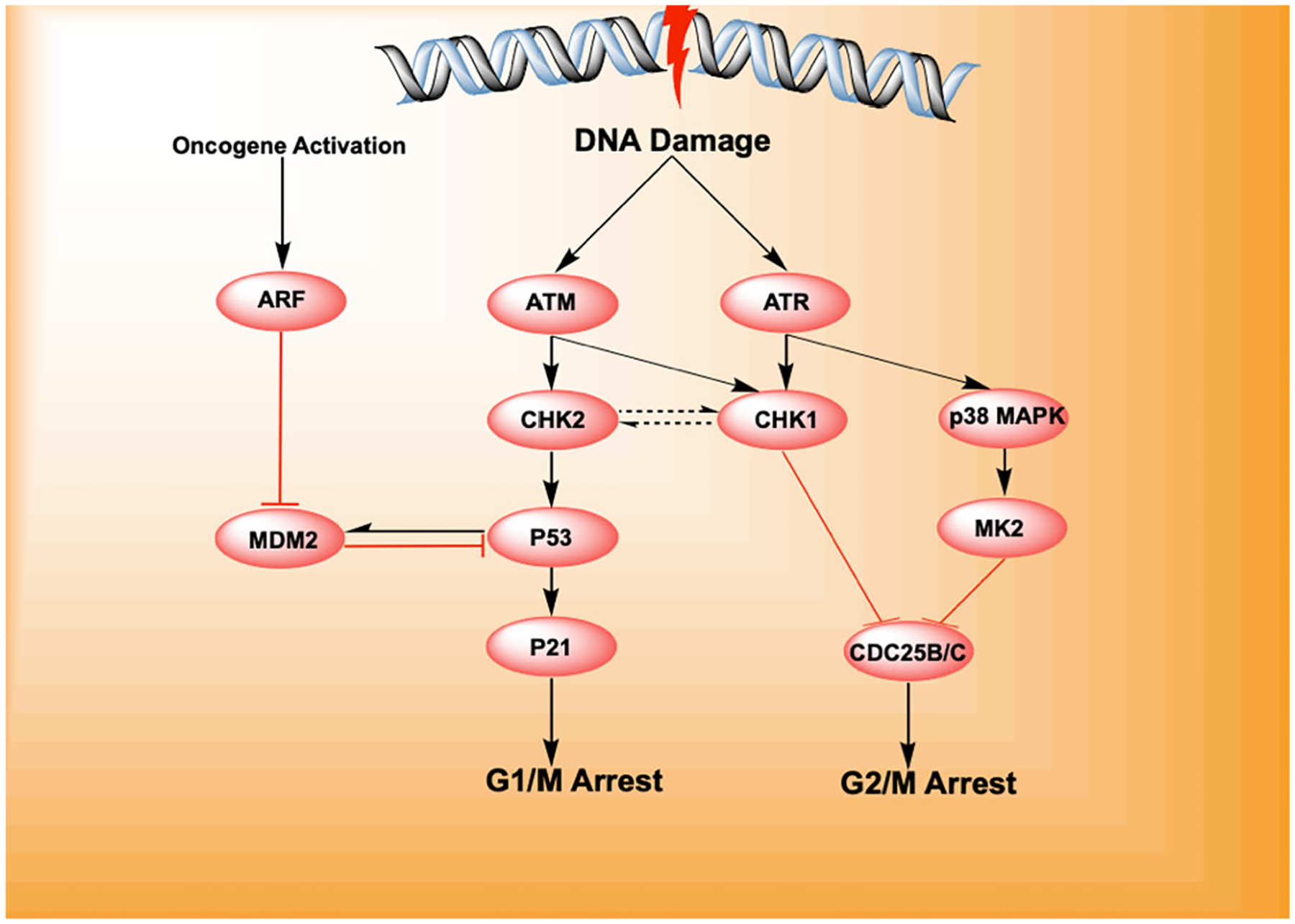
DNA damage initiates the DNA damage response through the activation of the kinases ATM and ATR that activate the checkpoint proteins CHK1 and CHK2. These proteins initiate cell cycle arrest by activating p53 (which dissociates from its inhibitory factor (HDM2/MDM2) followed by p21 that causes arrest in the G1/M phase of the cell cycle. In the absence of p53, it was noticed that signaling through p38 MAPK activated MK2, which lead to the phosphorylation of the CDC25 phosphatases arresting the cell cycle in the G2/M checkpoint. Cancers with inactive p53 were susceptible to death by mitotic catastrophe when exposed to DNA damage in the presence of CHK1 and MK2 inhibition.119
FIGURE 4.
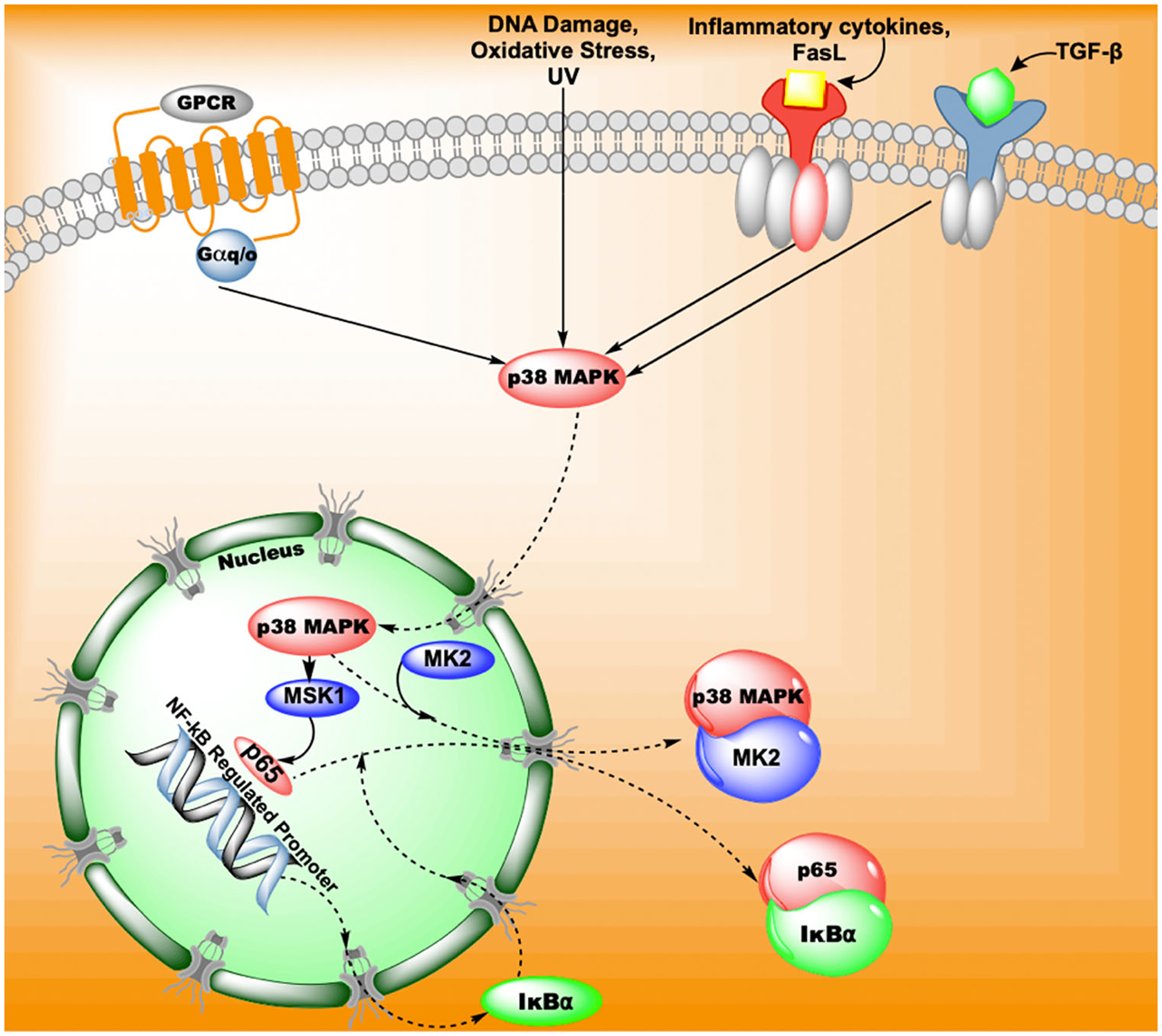
MK2 regulates nuclear factor kappa B (NF-κB) transcription by altering the location of p38 MAPK. When p38 MAPK is activated, it translocates to the nucleus and regulates NF-κB activity through phosphorylating MSK1. MSK-1 activation initiates IF-κB transcription and the translocation of p65 out of the nucleus preventing NF-κB gene transcription. However, MK2 activity removes p38 MAPK from the nucleus, separating it from MSK-1 and preventing the inhibitory actions MSK-1 on NF-κB activity.125
The phosphorylation of poly(A)-specific ribonuclease (PARN) by MK2 is another mechanism by which MK2 regulates the cell cycle. The protein is an 3′-exoribonuclease that deadenylates mRNA, reducing their stability and facilitating their degradation. The protein favors binding to poly(A) tails of mRNA and exonucleolytic degradation of the tail is often the first step in the decay of eukaryotic mRNAs. The protein is found in nucleoli, cajal bodies and the endoplasmic reticulum (ER) where it regulates the fate of ER associated mRNA. A significant number of the ER associated mRNA were found to code for proteins involved in the cell cycle and DNA damage response.111 MK2 phosphorylates PARN resulting in the stabilization of mRNA, such as GADD45α which also inhibits the Chk1-cyclin checkpoint responsible for G2/M progression in the face of cellular stress.121 Without this phosphorylation, cells were able to initiate but not maintain cell cycle arrest due to DNA damage by doxorubicin.121 This MK2-dependent mechanism of action required MK2 to translocate to the cytoplasm, as the stabilization of GADD45α also required the phosphorylation of the RNA binding protein hnRNP A0 (Figure 4).
A loss or inhibition of MK2 in p53 deficient cells that were damaged by cytotoxic DNA stress resulted in death due to mitotic catastrophe,121 whereas cytotoxic damage to wild type p53 cells resulted in no increased cell death. This revealed an avenue for the generation of synthetic lethality (SL) mutants in cancer cells with p53 mutations.117,121,126–130 KRAS and BRAF cancers commonly express p53 mutations or CDKN2A mutations that destabilize cell cycle checkpoints through p53 and instead become reliant on MK2 and CHK1 for cell cycle stability. Inhibition of CHK1 and MK2 led to apoptosis in KRAS and BRAF mutant cancers, showing the importance of MK2 in cell cycle regulation under these conditions.129 Later studies described a concept of augmented SL in tumors with defective p53 by targeting MK2 and the DNA repair protein XPA with small interfering RNA (siRNA) peptides. The tumors from these mice exhibited an enhanced cisplatin response and in turn the mice lived longer, particularly when these tumors were treated with both MK2 and XPA compared to MK2 inhibition alone.130 It remains to be discovered if there are other lethal combinations between MK2 and other molecules that may target mutant p53 cells while causing little effects to p53 positive cells.
P53 plays a large role in the DNA damage response by activating repair proteins, arresting the cell cycle in G1/M and causing apoptosis if the damage is too severe.131 Under resting conditions, the activity of p53 is kept low by its interaction with the protein HDM2 which signals the p53 for degradation through its E3 ubiquitin ligase activity. Phosphorylation of p53 causes a dissociation with HDM2 enabling the accumulation of cellular p53 through enhanced protein stability. MK2 was found to phosphorylate HDM2 at locations that increased HDM2 affinity for p53 thereby enhancing its degradation in what could be described as a blunting of p53 function. In contrast, loss of MK2 in murine embryonic fibroblasts (MEFs) led to reduced MDM2 (HDM2 in humans) and increased p53 levels following UV irradiation. This apparently paradoxical role MK2 is possibly a form of negative feedback of cell cycle arrest and DNA damage sensing.51
An additional mechanism of cell cycle control by MK2 is through TRIM29/ATDC which is a member of the tripartite motif (TRIM) protein family that consists of 70 members. The TRIM family of proteins has been implicated in a variety of physiologic processes, such as development, oncogenesis, apoptosis, and antiviral defense. TRIM is highly expressed in pancreatic cancer cells, promoting pancreatic tumor growth via stimulation of the β-catenin pathway. MK2 was shown to phosphorylate TRIM29 which correlated with an increased resistance of pancreatic tumor cells to radiation.113 The mechanism of this action is unclear but TRIM has been shown to act as a regulator of the assembly of DNA-damage proteins and so phosphorylation may alter the conformation of the assembly to promote repair.132
3.2 |. Transcription
Several transcription factors are known to be phosphorylated by MK2 activating genes that code for immediate early genes, heat shock proteins and inflammatory mediators.89,91,93,94
Serum response factor (SRF) is a protein that belongs to the MADS superfamily of transcription factors that binds to the serum response element of promoters of immediate early genes.133 Immediate early genes are a collection of genes such as c-fos, c-jun, and c-myc that need to be activated for later responses to occur. Many of these genes are transcription factors themselves and they activate genes involved in changes to the cell cycle, cell growth, cell differentiation, apoptosis, and the immune response.134,135 Heidenreich et al., (1999)88 showed the MK2 phosphorylated SRF in both in vitro and in vivo conditions. Whereas the role of phosphorylation is currently unknown, the study demonstrated an increase in the affinity and rate of SRF to the binding to serum response element suggesting activation of gene transcription.
Myocardin-related transcription factor A (MRTFA) is a known actin-regulated transcriptional coactivator of SRF. Actin polymerization activates MRTF-A by releasing it from G-actin and thus allowing it to bind to and activate SRF. MRTF-A is phosphorylated by MK2 at in a stress but not mitogen dependent manner. However, the phosphorylation led to no increase in dimerization, no change in subcellular localization and translocation or interaction with actin or SRF. Therefore, the physiological role of this phosphorylation is unknown.90
Heat shock transcription factor-1 (HSF-1) is a highly evolutionary conserved transcription factor responsible for initiating the transcription of heat shock proteins during times of cellular stress. HSF-1 upregulates the transcription of several heat shock proteins such as HSP27, HSP40, HSP70, and HSP90. As with HSP27, these proteins refold misfolded proteins and confer resistance against cellular stress. Under normal conditions, HSF-1 is inactive and found in monomeric form in cellular complexes with HSP proteins. Activation of HSF-1 involves binding of misfolded proteins with the complex and relieving the binding and inhibition of HSF-1. It then undergoes homo-trimerization and activates transcription by binding to heat shock elements (HSE) on the DNA.136 HSP-1 also plays a role in inhibiting the transcription of proinflammatory genes such as IL-1β, IL-6, and TNFα.137,138 Wang et al.,136 showed that MK2 can phosphorylate HSF-1 reducing the ability to bind to HSE and increasing its affinity to HSP90 thus inhibiting HSP1 function. Therefore, MK2 influences transcription by inhibiting repressors of inflammatory signaling.
Steroid receptor coactivator 3 (SRC-3) is a transcriptional cofactor from the p160 family that binds to steroid hormone receptors in a ligand-dependent manner serving as a coactivator of transcription.139 It is a cofactor to steroid nuclear receptors and as a number of transcription factors including E2F transcription factor 1 (E2F1), polyomavirus enhancer activator 3 (PEA3), activator protein-1 (AP-1), and nuclear factor-κB (NF-κB). Its activity has been implicated in cell proliferation, development, survival, metabolism and is linked to the severity of both hormone-dependent and independent cancers. The function of this gene is heavily regulated by phosphorylation which alters the transcription of hormone receptors and NF-κB. One of its primary phosphorylation sites was shown to be phosphorylated by MK2 implicating a role for the p38/MK2 pathway in modulating the transcriptional activation of NF-κB and releasing IL-6 in A549 cells.89
The transcriptional changes stimulated by MK2 are not well characterized but likely enhance the role of MK2 in directing cells toward a path of survival and resilience in the face of cell stress. Although MK2 activates several transcription factors directly, there is also evidence that its signaling affects other transcription elements, particularly NF-κB, in endothelial cells.125 Part of the effects of MK2 on NF-κB signaling is governed by the trafficking of p38 from the nucleus to the cytosol. When NF-κB is activated, its activity is checked by IκBα activity that mediates nuclear export of p65, a component of the NF-κB family.140 The export of p65 results in reduced expression of NF-κB genes and a blunted inflammatory response. Phosphorylation of p65, increased expression of IκBα and extracellular transport of p65 is initiated by p38 MAPK phosphorylation of nuclear MSK1. The activation of MK2 results in the trafficking of p38 out of the nucleus causing a reduced phosphorylation of MSK1 and a more sustained activation of NF-κB genes.125 Thus, MK2 can alter transcription of genes by altering the location of p38 MAPK and altering cellular function as a result (Figure 5).
FIGURE 5.
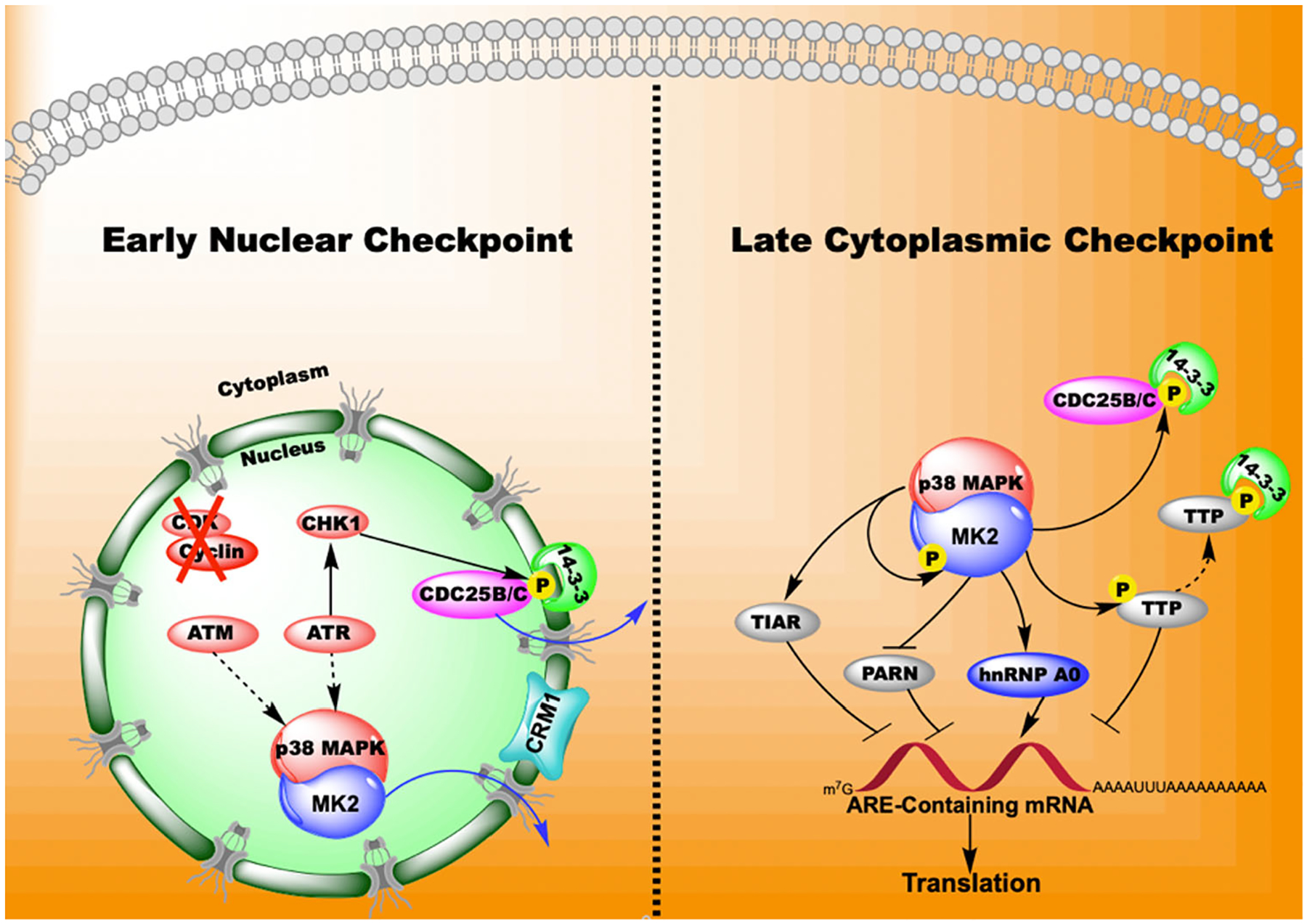
p38 MAPK-MK2 pathway mediates the late cytoplasmic G2/M checkpoint through its actions on RBPs and 14-3-3 accessory proteins in the cytosol. ATM and ATR cause the activation of p38 MAPK that forms a complex with MK2 and translocates out of the nucleus. At this stage, CHK1 phosphorylates 14-3-3 and CDC25 proteins initiating arrest at the G2/M checkpoint. In the cytoplasm, MK2 phosphorylates PARN, hnRNP A0, TTP and 14-3-3 proteins causing the stabilization of mRNA and the subsequent transcription of genes such as GADD45α that maintain the checkpoint status. The phosphorylation of 14-3-3 also maintains the sequestration of CDC25B/C contributing to the arrest at G2/M phase.121
MK2 also supports the activity of NF-κB through the activation of SRC-3 by phosphorylation.89 The phosphorylation site was dependent onTNFα activation of MK2 as it was blocked by the inhibitor PF3644022 that blocks MK2 kinase activity. Phosphorylation caused the translocation of SRC-3 into the nucleus, creation of the NF-κB-SRC-3 complex and transcription of inflammatory genes such as IL-6. The indirect action of MK2 on NF-κB demonstrates the role of MK2 in stimulating the expression inflammatory gene transcription in addition to the direct activation of genes due to the phosphorylation of direct targets such as SRF, HSF1, and CREB.73
The transcription factor cyclic adenosine monophosphate (cAMP) response element-binding protein (CREB) is a nuclear transcription factor that binds to CRE elements on the genome to promote transcription of genes such as somatostatin, c-fos and genes regulating the circadian clock. It was shown to be phosphorylated by MK2 after stimulation of cells by fibroblast growth factor in vitro suggesting a role for MK2 in CREB activation91,92 that was contradicted later with the knowledge that CREB activation was far more likely to be dependent on mitogen and stress activated kinase (MSK1) due to the much lower Km MSK1 had for CREB.141 However, a subsequent study concluded that when cells are exposed to mitogens, p38 MAPK is activated, but the response is transient and not able to activate CREB, but with cell stress (i.e., anisomycin) the activation of p38 MAPK is sustained resulting in MK2 and subsequent CREB phosphorylation.93
3.3 |. Role in RNA-binding-protein function
MK2 has a central role in the regulation of mRNA binding proteins by stabilizing transcripts for inflammatory cytokines, growth factors and cell cycle checkpoint stabilizers.5 Through its action of stabilizing transcripts, the translation response to MK2 activation is accelerated compared to stimulating transcription of genes. Therefore, it appears the p38/MK2 axis is evolved for rapid response to stress by stabilizing inflammatory mRNA and initiating an appropriate response from the cell.
Several RBPs are known to be direct phosphorylation targets of MK2.5,103–109 These mRNA binding proteins are structured to bind to adenylate and uridylate-rich elements (ARE) found on the 3′-untranslated regions of mRNAs. The binding of mRNA to RBPs results in either the degradation or stabilization of these mRNAs depending on the RBP species and ARE binding competition. This facet of control over the fate of mRNA by binding to different RBPs results in posttranscriptional gene regulation to control the translation and expression of effector genes. A census of RBPs shows that there are over 1500 of these molecules in the human genome with different abilities to regulate the binding of RNA to effector proteins that govern the fate of the transcript.142 Many of these RNA binding proteins compete for the same targets and confer opposite fates to the bound mRNAs. Therefore, MK2’s ability to phosphorylate certain RBP will allow for transcriptional-translational modulation.143
TTP is one RBP that is directly phosphorylated by MK2.103,144 Phosphorylation of TTP increases its binding to 14-3-3 proteins that prevent the recruitment of deadenylases by TTP thereby prolonging the half-life of cytokine mRNA such as TNFα (Figure 4).145,146 The mRNA that would be bound to TTP then binds to other RBPs like Human antigen R (HuR or ELAVL1) or hnRNP A0 and these transcripts are stabilized and go forward to translation.147,148 The end result of mRNA stabilization by the sequestration of TTP results in increased production of many inflammatory mRNAs such asTNFα, IL-2, GC-MSF, COX2, and Nitric oxide synthase.149–152 The effect of downregulation of TTP was shown in studies examining TTP−/− mice that demonstrated an overexpression of TNFα by macrophages causing severe arthritis and cachexia.149,153,154 Inhibition of MK2 in LPS treated macrophages caused a reduction in TNFα production but no change in TNFα transcript concentration, giving further evidence for the critical role of MK2 in posttranscriptional control of inflammatory cytokines.28,155 Thus, MK2 effects on RBPs is a potent way of regulating inflammation in response to stress. The activation of MK2 also results in feedback mechanisms as TTP expression is enhanced by MK2 signaling demonstrating tight control of this cellular mechanism.147,156
The subcellular location of HuR changes with stress and cytoplasmic accumulation of HuR occurs in cells that are subjected to oxidative stress, or cells that transiently overexpressed constitutively active MK2.148,157,158 Although HuR and TTP are highly interconnected, there does not appear to be a sequence within HuR that MK2 phosphorylates.159 It has been reported that p38 phosphorylates HuR, but the shuttling of HuR from the nucleus to the cytoplasm, required for mRNA stability of IL-6, TNFα etc., is MK2 dependent. However, the specific interactions that allow this mechanism to function are not clear.160–162 Although it is not known how MK2 is involved in the translocation of HuR, the cycling of molecules out of the nucleus appears to be a mechanism that MK2 regulates in several cell systems. Its role in cytoplasmic translocation of p38 MAPK can alter NF-κB signaling and the MK2 functions that occur in the cytoplasm such as the phosphorylation of hnRNP A0 for the stabilization of GADD45α in the DNA damage response121 suggest that much of MK2 signaling relies on its cycling out of the nucleus (Figures 4 and 5). This is exemplified by the reduction in TNFα release in murine macrophages that only express the shorter form of MK2 without the NES sequence preventing cytosolic MK2 accumulation.59
Bollig et al.,108 demonstrated that MK2 can phosphorylate the RBP polyadenylate-binding protein 1 which is also involved in the regulation of RNA stability by TTP as part of a complex that must dissociate in order for HuR to bind and stabilize mRNAs. The RBP HNRNP A0 binds to mRNA sequences for TNFα, COX2, GADD25α and macrophage inhibitory protein-2 when phosphorylated by MK2.105,121 This resulted in the stabilization of these mRNA moieties and subsequent translation of their target proteins.
The nuclear exosome targeting complex is required for degradation of nuclear noncoding RNAs endowed with 3′–5′ exo- and endoribonuclease lytic activities. In the nucleoplasm, this complex is composed of hMTR4, ZCCHC8, and RBM7. RBM7 is phosphorylated by MK2 leading to a reduction in affinity of this complex and a stabilization of promoter upstream transcripts (PROMPTS) that normally undergo rapid nucleolysis.107
MK2 has been shown to phosphorylate the negative elongation factor complex (NELF) subunit E (NELFE), the RBP part of the complex, at serine positions 51, 115, and 251. This phosphorylation enhances the binding of NELF to 14-3-3 proteins. The binding of 14-3-3 proteins to NELFE causes its dissociation from the complex allowing gene transcription by RNA polymerase II. The function of these genes includes telomere maintenance, RNA metabolism, cell cycle, and DNA repair.106 In addition to this direct measure of MK2 on NELFE, the study demonstrated the phosphorylation of 122 proteins by the p38 MAPK-MK2 axis suggesting a large list of unknown targets for MK2 phosphorylation.
3.4 |. Cytoskeletal activity, motility, and migration
Genetic depletion of MK2 has profound effects on motility as seen in MK2 KO mouse embryonic fibroblasts and smooth muscle cells that displayed reduced migration,59 whereas MK2 KO neutrophils showed a loss of directionality but higher migration rate.51,163 This implies a complicated regulatory role for MK2 in motility and migration. Further, the wide array of targets affecting actin polymerization and other structural proteins suggests a role in regulating intracellular trafficking in addition to motility.
HSP27/25 was one of the earliest found substrates of MK2 and its function in cell motility is widely researched. In unstressed cells, it provides cytoskeletal stability by capping actin filaments and this activity is altered by phosphorylation during times of stress. Although the precise mechanisms of HSP27 on motility is unknown, the capping of actin filaments by HSP27 likely alters the polymerization of actin filaments and HSP27 phosphorylation changes what kinds of actin filaments are capped. When HSP27 was knocked down genetically, the result was reduced motility in a monolayer wound healing assay. HSP27 has also been immunoprecipitated with actin after heat shock demonstrating its colocalization with actin fibers.164
Heat shock proteins are divided into five families identified by size; HSP100, HSP90, HSP70, HSP60, and sHSP and the members phosphorylated by MK2 are the sHSPs (HSP27 and HSP25).58,82,165 These are highly evolutionarily conserved proteins that are part of the alpha-crystallin family, which share a conserved 80–100 amino acid sequence.166,167 These proteins have conserved β-sheet regions in their secondary structure that allows these sHSPs to form large oligomers in cells.168,169 In addition to interacting with actin, the oligomers stabilize peptides and act as molecular chaperones that modulate protein degradation in an ATP-independent manner (induced by thermal, radiation, dehydration, redox, or other forms of stress).170
HSP27 exists in both the cytosol and nucleus and is found as monomers, dimers, and large oligomers. Nonphosphorylated HSPs have been shown to exist as monomers in vitro. However, the nonphosphorylated HSPs combine to form large oligomeric structures (~24mer) that bind to proteins, preventing excessive protein aggregation and refolding of denatured proteins.171–173 When the cell is exposed to heat or other stressors the cell responds by phosphorylating the sHSPs causing a reduction in the oligomer size.174 The phosphorylation alters the ability of the sHSPs to function as molecular chaperones and facilitate protein folding. Under these conditions, the sHSPs dissociate to form dimers that comprise the building blocks of the oligomers. In addition to the reduction in size, the location of the HSPs change when phosphorylated. Non-phosphorylated HSP27 is found predominantly in the cytosol, but migrates to the nucleus upon stress and phosphorylation.175 There is evidence that these oligomers can form multimers with other sHSPs, altering their ability to chaperone.176 In resting cells, the expression of sHSP is maintained in oligomers of different sizes at a certain ratio (38% <150 kDa, 14% 150–400 KDa, 52% 400 KDa). These ratios have been shown to fluctuate with different cellular states such as starvation, serum feeding, apoptosis, oxidative stress, and heat shock. With increased cellular stress, HSP27 forms more of the smaller complexes to increase the number of substrates it binds to and stabilizes. Alternatively, the smaller complexes may bind to different types of substrates in the nucleus. Another mechanism that HSP27 uses to facilitate cell survival is through its antioxidant properties. HSP27 can stimulate glucose-6-phosphate dehydrogenase by the small and highly phosphorylated HSP27 oligomers. It also maintains reduced glutathione and displays an iron-chelating activity helping to maintain a reduced oxidative state in the cell.177
As well as serving as regulators of motility, protein stability and aggregation the sHSP also have functions in normal development, cell signaling, and apoptosis.178,179 HSPs interact with actin, in both their large oligomeric and smaller phosphorylated forms, altering the motility of the cell after cellular stimulation by factors’ such as VEGF.180 HSP27 has antiapoptotic functions through binding of its non-phosphorylated or phosphorylated oligomeric form to the apoptotic factors’ cytochrome-C and DAXX. Although named as small molecules that respond to heat stresses, their functions have been implicated in motility, differentiation, apoptosis, heat tolerance, chaperoning, stress, and the immune response, and are a major effector molecule of MK2 signaling.181
LIMK1 is a serine/threonine kinase that regulates actin polymerization via phosphorylation and inactivation of the actin binding factor cofilin. This protein is involved in the VEGF stimulated actin remodeling by depolymerizing and severing actin filaments. The downstream effects lead to migration of endothelial cells and angiogenesis.182 Kobayashi et al.,83 showed that MK2 phosphorylated LIMK1 causing activation of the kinase, which resulted in stress fiber formation, cell migration and tube formation. This is an additional mechanism of MK2 to regulate cell motility and possibly angiogenesis.
In conjunction with HSP27, LIMK1 is phosphorylated by MK2 and activates various GTPases that function at the leading edge assembly of actin and cell migration.83,183 LLIMK1 was shown to act in parallel with HSP27 to cause actin rearrangements, by affecting the actin depolymerizing protein cofilin to increase cell motility after stimulation by bone morphogenic proteins.184 MK2 knockdown in endothelial cells reduced VEGF induced migration, actin formation and tubule formation.83
The lymphocyte specific protein 1 (LSP1) gene encodes an intracellular F-actin binding protein and is expressed in lymphocytes, neutrophils, macrophages, and endothelium.185 The function of the molecule is believed to regulate migration and chemotaxis by polarizing F-actin.186 MK2 phosphorylates LSP1, resulting in the colocalization of LSP1 with polarized F-actin at the leading edge of polarized neutrophils, contributing to the directionality and polarization of migrating cells.84
Capping protein (CP) binds to the barbed head of fast-growing actin regulating its assembly by preventing addition or removal of actin subunits as the dynamic actin remodels cellular structure.187 CapZ-interacting protein (CAPZIP) is a binding protein that is localized at the membrane of cells where it can bind to CP and regulate its function. CAPZIP itself has several phosphorylation sites that regulate its function and were found to be phosphorylated by MK2 after osmotic shock. While the specific role of CAPZIP is still unknown, phosphorylation of CAPZIP caused dissociation from the CP complex. Therefore, the stress-induced phosphorylation of CAPZIP may regulate the ability of F-actin-CP to remodel actin filament assembly.87
Reticulon 4 (Isoform B) or Neurite outgrowth inhibitor protein B (NOGO-B), is a protein from the reticulon family of proteins that were originally found to inhibit axonal regeneration in the CNS. Since they have been found to function in endothelial cells, vascular smooth muscle cells and cells of the immune system. It has been shown to be a modulator of vascular remodeling, promoting the migration and lipid synthesis of endothelial cells but inhibits the migration of vascular smooth muscle cells. NOGO-B promotes macrophage homing and functions as a cytokine in angiogenesis, arteriogenesis and tissue repair.188 It also mediates ICAM1 induced transendothelial migration of leukocytes such as monocytes and neutrophils in acute inflammation.189 The protein of NOGO-B was found to be phosphorylated by MK2, although how it modulates cellular function by this mechanism is not understood.85
Ribosomal Protein S6 Kinase (RSK) acts as a downstream kinase of the MAPK ERK (MAPK1/ERK2 and MAPK3/ERK1) and mediates mitogenic and stress-induced activation of several transcription factors through phosphorylation. The enzyme also regulates translation, and mediates cellular proliferation, survival, and differentiation by modulating mTOR signaling.190 However, in LPS-stimulated dendritic cells RSK proteins were phosphorylated by TLR dependent mechanisms. Absence of MK2 prevented this phosphorylation and reduced dendritic cell macropinocytosis.100 Therefore, in dendritic cells, MK2 can regulate TLR responses through crosstalk with kinases usually phosphorylated by pathways other than p38 MAPK.
Intermediate filaments (IF) are found preferentially expressed in epithelial cells that provide structural support for epithelial cells and respond to external stresses and transduce signals into the cells. The filaments’ basic structure are coiled-coils of two different proteins formed from different types of keratins and other proteins that combine to produce six different types of filaments (Types I–VI).191,192 The filaments assemble into lateral tetramers with N and C termini that are non-helical, have vimentin caps, and can bind DNA. There are a wide range of IF molecules (~70) but they all show the same polymer characteristics to serve as building blocks for the filaments. MK2 was found to phosphorylate the IF proteins Keratin 18 and 20. Vimentin was also phosphorylated, but the phosphorylation site is unknown.86,193 The IF proteins function as stress induced proteins where they are upregulated and phosphorylated in times of cellular stress. While the implications of phosphorylation are still unknown, it is suggested that MK2 contributes to the stress and cell cycle dependent reorganization of the keratin cytoskeleton, possibly altering the stiffness and mobility of the cells.
There are indications that the potential of MK2 to regulate motility is affected by its protein structure. Kotlyarov et al.,59 demonstrated that MK2 KO reduced the formation of filopodia in macrophages, and reduced migration of fibroblasts and smooth muscle cells. The restoration of this required not only a functional kinase, but also the proline rich N terminus. It is not well understood what role the N-terminus plays in facilitating migration, but it is interesting to note that the splice variant, with a longer N-terminus that includes an additional phosphorylation, site was not able to phosphorylate HSP27 or support migration.194 Another nonkinase activity altering the ability of MK2 to affect cell migration concerns SUMOylation, a unique posttranslational modification akin to ubiquitination that conjugates small ubiquitin-like proteins called SUMO (Small Ubiquitin-like MOdifier) to proteins, which was shown to affect the motility regulated by MK2. A SUMOlation site on MK2 was found (Lys339) to reduce the phosphorylation of HSP27 and its effects on actin polymerization.195
These findings along with the ability of MK2 to phosphorylate many molecules that are involved in cytoskeletal and actin stability demonstrate a significant role for MK2 in regulating motility. As the activation of p38 MAPK-MK2 pathway can occur via proteases like matrix metalloproteinase 2 (MMP2), that have a well-defined role in migration. It suggests that MK2 may be a key player in regulating migration and possibly metastasis.196
3.5 |. Other cell responses
Outside of the more established role of MK2 in cellular functions (DNA damage response, transcription activation, inflammatory mediator production and migration), there are several emerging processes that the signaling of MK2 may control.
TNFα signaling can result in inflammation, proliferation, differentiation, survival, and cell death and activates the p38 MAPK-MK2 pathway.197 The binding of TNFα to its receptor (TNFR1) forms a complex composed of TRADD, TRAF, RIPK1, and cIAP1/2, which is involved in the activation of NF-κB (Complex I) and activation of an immune response. Once the immune response is initiated many of the proteins in Complex I will reorganize to stimulate cell death. The second complex (Complex II) forms from RIPK1, RIPK3, TRADD, FADD, TRAF2, Caspase 8, and cIAP1/2, and stimulates apoptosis via caspase 8 or stimulating RIPK3, which leads to necroptosis.198–200 The formation of Complex II depends on cytosolic RIPK1 and partially from the dissociation of RIPK1 from complex I and binding to FADD and caspase 8. MK2 phosphorylates RIPK1, reducing its affinity to bind to FADD and inhibiting TNFα induced cell death.102,201 Hence, MK2 controls inflammation by increasing cytokine expression and inhibiting RIPK1-dependent cytotoxicity.
Experiments in Drosophila have shown an antiapoptotic effect of p38 MAPK-MK2 by downregulating JNK mediated apoptosis in gut epithelium.202 However, a proapoptosis effect of MK2 was seen in human lung microvascular endothelial cells stimulated with LPS, causing nuclear translocation of cleaved caspase 3 and apoptosis, which were both prevented by MK2 silencing.203 The discrepancy in the pro/antiapoptotic activity of MK2 possibly relies on the strength of the stimuli and the cells and tissue of origin.
The ability of MK2 to affect cell motility may also contribute to a growing body of evidence that the kinase has some control over angiogenesis as implied by the LMK1 data referred to in the motility section above. IL-1β is a known angiogenic cytokine that stimulates inflammation and angiogenesis in vascular pathophysiological processes such as atherosclerosis and tumor neo-angiogenesis.204 IL-1 stimulation assembles a proangiogenic signaling module consisting of caveolin-1, TRAF6, p38 MAPK, and MK2 in endothelial cells.205 MK2 KO mice were used to examine the effect of MK2 on angiogenesis in mouse retina and the study revealed MK2 had no effect on physiological retinal angiogenesis but lowered arterial area and altered smooth muscle cell genetic profiles.206 The authors concluded a cell specific mechanism of MK2 regulation of angiogenesis.
Two recent studies by the Yaffe group implicated MK2 in the role of angiogenesis stimulation by tumor resident macrophages. Using MK2 KO mice, Suarez-Lopez et al.,207 showed that MK2 supported tumor associated macrophage polarization and transformation into proangiogenic M2-like macrophages. This ability was reversed by chemical inhibition of MK2. A follow-up study concluded that these effects occurred through MK2 regulation of the angiogenic factor CXCL-12/SDF-1 secreted by tumor associated-macrophages, in addition to MK2-dependent regulation of Serpin-E1/PAI-1 by several cell types within the TME.208
5-lipoxygenase (5LO) catalyzes the 1st step in the synthesis of leukotrienes, which are mediators of inflammation. They are released from activated leukocytes and exhibit several biological effects such as contraction of bronchial smooth muscles, stimulation of vascular permeability, and attraction and activation of leukocytes. Although necessary for a complete immune response, the pathophysiological role of leukotrienes has been realized by therapies that inhibit leukotriene function seen in patients suffering from asthma, allergies and chronic obstructive pulmonary disease.209 The activity of 5LO depends on the concentration of it in the nucleus, the greater the concentration of 5LO in the nucleus, the greater the concentration of leukotriene released. The activity and the cellular location of the enzyme is modulated by phosphorylation. MK2 has been shown to phosphorylate the protein which alters the cellular location of 5LO by inhibiting its export from the nucleus.95,96 This suggests a role of MK2 in potentiating leukotriene release by keeping the enzyme in its most active location and consistent with the role of MK2 potentiating the inflammatory response.
The mechanisms of MK2 on cell function can be broadly considered to be an adaptation to cell stress that activates immune cell function for clean up and repair and supports cell survival through DNA checkpoint regulation and migration. Under conditions of stress, the regulation of autophagy would appear to be in line with the cellular programming of the MK2 pathway, as it is a method of fuel generation and protein recycling suitable for a damaged cell attempting to repair and survive. Autophagy is a cellular process where cellular proteins are broken down and recycled to provide energy and new substrates for growth. It is both a method of cellular recycling and clean up where damaged and dysfunctional proteins and organelles are removed, as well as a means for cells to survive and maintain energy when external nutrient sources are scarce.210 Beclin-1 is an essential protein for autophagy and its complexing with Bcl-2/Bcl-xL stabilizes homodimerization of beclin-1 and prevents its interaction with PI3KC3 complexes inhibiting autophagy. Activation of beclin-1, and thus autophagy, involves its phosphorylation which disrupts its binding to Bcl-2. Starvation caused phosphorylation of Beclin-1 by MK2, disruption of its binding to Bcl-2, and initiation of autophagy.116 Thus, MK2 may regulate the initiation of autophagy under certain conditions.
Phosphodiesterase 4A (PDE4) is part of the cyclic nucleotide phosphodiesterase (PDE) family and the PDE4 subfamily is encoded by 4 genes making 20 different isoforms. Long forms of PDE4 are regulated by phosphorylation that fine tunes their activity. PDE hydrolyzes the second messenger, cAMP, which is a regulator and mediator of many cellular functions including inflammatory signaling.211 Therefore, the regulation of the cellular concentration of cAMP plays a key role in many important physiological processes. MK2 phosphorylates PDE4 with the resulting effect of attenuating the activity of PKA activation of PDE4. The attenuation of PKA activity on PDE4 causes cAMP levels to be maintained and fine tunes the ability of PKA to activate PDE4. Further, the phosphorylation of PDE4 by MK2 enhances its binding to p75NTR (p75 neurotrophin receptor) reducing the breakdown of fibrin.97–99
Ubiquitination is a method for cells to mark proteins for degradation, alter their cellular location, affect their activity, or for protein-protein interactions. UBE2j1 (ubiquitin-conjugating enzyme) is a noncanonical ubiquitin-conjugating E2 family of proteins. It is an ER-bound protein that has been shown to affect the proteosomal degradation of TCRα (T cell receptor), mutant CFTR (cystic fibrosis transmembrane conductance factor), TRAF2 (TNF associated factor 2), and MHC I. MK2 was shown to phosphorylate UBE2J1, and genetic silencing of the UBE2J1 reduced the release of TNFα from immortalized macrophages.114 This implies the involvement of UBE2J1 in the activation of TNFα and MHC1 translation in LPS stimulated macrophages.
Centrosomal satellites (CS) are small granular clusters of proteins that cluster around centrosomes and traffic along microtubules. While their role is poorly defined, it is suggested that they act as mobile packages that facilitate cell processes. UV stress causes abrupt placement of CS constituents in the cytoplasm, one of which is the protein CEP131. Once CEP131 is released into the cytoplasm, MK2 phosphorylates the protein catalyzing the binding of CEP131 to 14-3-3 proteins leading to a rapid clearance of the protein and other moieties in the CS.115 The implications of this effect are not well understood, but the actions of MK2 on protein released from CSs suggests a form of cytosolic clean up.
The TAB proteins are protein binding subunits that are involved in the signaling activation of Toll-like receptors, IL-1 and TNFα receptors. Upon receptor binding, molecules, such as TRAF6 promote the formation of the TAB-TAK complex resulting in autophosphorylation and activation of TAK1 that stimulates the activation of NF-κB and AP1 transcription factors.212 TheTAB binding proteins are extensively phosphorylated in this process and MK2 phosphorylates TAB3 (transforming growth factor [TGF]-beta activated kinase 3 [MAP3K7]) possibly contributing to the activation and binding of TAB3 to the TAB/TAK process.101 However, the mechanistic effect of this phosphorylation event is currently unknown.
4 |. PHYSIOLOGICAL ROLES OF MK2
4.1 |. Immune modulation
One of the main functions of the p38 MAPK-MK2 pathway is to regulate the immune response through the activation of transcription, and stabilization of mRNA, which increases the release of inflammatory mediators. Activation of the p38 MAPK-MK2 pathway can also be maintained by inflammatory cytokine signaling. For example, MK2 activation has been shown to increase TNFα levels,197 and conversely TNFα can activate p38 MAPK and subsequently MK2.89,102 This suggests that cytokine release can serve as a positive feedback signal for continued p38 MAPK-MK2 activation and could be one of several mechanisms for the maintenance of an inflammatory phenotype in a variety of biological or medical conditions.
The inflammatory function of MK2 was shown in experiments of MK2−/− mice where the splenic cells from these mice showed reduced release of the cytokines TNFα, IL-1β, IL-6, IL-10, and inter-feron gámma (IFNγ) compared to WT and these mice were resistant to LPS induced endotoxic shock, showing a 90% reduction in serum TNFα levels.52,59 LPS-induced IFN-β and IL-10 gene expression is downregulated in MK2-deficient macrophages.213 Later studies showed that in the absence of MK2, MK3 negatively regulates IFN-γ and delayed the nuclear translocation of NF-κB by delaying the ubiquitination and subsequent degradation of Iκ-Bβ.214
MK2 contributes to airway inflammation by signaling a Th2 response, as MK2 knockout mice are unable to mount localized Th2-type inflammation and development of experimental asthma. These effects were partially due to reduced endothelial permeability caused by a reduction in NF-κB mediated cytokines and mediators due to aberrant MK2 signaling. These animals displayed significant reduction in airway inflammation, mucus production and extracellular matrix (ECM) deposition. The reduced inflammation was associated with significantly decreased levels of many cytokines, (such as IL-5, IL-9, IL-13, and IL-25) in lung mRNA.125
The increased activation of MK2 is responsible for the elevated and post transcriptionally regulated TNFα protein expression in psoriatic skin (as described by the effect of MK2 on human kerati-nocytes).215 When MK2 knockout mice were used to look at oxalone-induced lesional psoriasis it was found that skin inflammation was reduced and IL-1β, TNFα, and IFNγ expression were decreased in MK2 knockout mice compared with wild-type mice.216 However, later studies concluded that only certain chemically induced skin inflammation involves MK2.217
Examining MK2 KO mice in a model of experimental colitis indicated that the MK2 deletion in mice reduces colitis by dextran sodium sulfate (DSS).218 Another study of colitis showed a reduction of IL-6, TNFα and reactive oxygen species (ROS) in DSS lesions from MK2 KO mice compared to control. The neutrophils from these mice had reduced capacity to produce ROS in response to stimulation, suggesting a role of MK2 in NADPH oxidase activity.219 A role for MK2 in neutrophil ROS production was further supported by experiments examining liver reperfusion injury. The number of infiltrating neutrophils were reduced in MK2 KO mice compared to control, and NADPH oxidase phosphorylation was reduced in C5a stimulated mice from MK2 KO neutrophils.220
Microglia from MK2 KO mice showed reduced release of TNFα and macrophage inflammatory protein 1 after stimulation with LPS or amyloid peptides compared with wild-type microglia. Cortical neurons cocultured with these same factors and microglia were protected from microglial-mediated neuronal cell toxicity.221
The effects of MK2 on the inflammatory response is seen across an array of conditions including skin, colon, airway, and neuroinflammation. Due its pivotal role in stress induced cytokine release, it will likely be found to play a role in other inflammatory conditions as research continues.
4.2 |. Wound healing and fibrosis
Wound healing is an integral part of self-repair following injury for complex organisms and can be characterized into four major phases: (1) hemostasis, (2) inflammation and innate and adaptive immune response, (3) epithelial and mesenchymal proliferation, (4) tissue remodeling. Immediately following hemostasis, growth factors and cytokines are released from damaged epithelial tissue and platelets, which lead to recruitment of diverse innate myeloid and adaptive immune response.222,23,223 This immune component (i.e., macrophages, neutrophils, leukocytes) is necessary to limit pathogenic infection while further recruiting cells to initiate localized tissue fibrosis.224–226 As the localized infection resolves, additional proangiogenic factors and growth cytokines are released, leading to epithelial and mesenchymal cell proliferation as inflammation begins to resolve in the wound.227,228 Finally, the stromal component, consisting of endothelial cells, fibroblast and myofibroblasts, secrete new ECM proteins providing a new scaffold for a new epithelial layer to be generated.226,228
While wound healing is critical for normal tissue repair, this process can go awry following aberrant stimulation (i.e., infection, chronic inflammation). This can lead to myofibroblast over-stimulation, excessive ECM deposition and increased collagen production (as opposed to turnover) thereby resulting in the formation of fibrotic scarring.229 Given that MK2 pathway regulates many of the same inflammatory factors that are triggered during chronic inflammation, its role in dysregulated tissue fibrosis and remodeling are currently being explored. However, conflicting results between different MEFs, murine animal models and human cell lines regarding the role of MK2 and myofibroblast activation have been reported. One group had demonstrated that MK2 knockout MEFs upon TGFβ1 stimulation had reduced alpha-smooth muscle actin expression compared to wild-type MEFs.230 This same group went on to demonstrate that MK2 disruption exacerbated fibrosis rather than ameliorate it. When mice lacking MK2 were exposed to bleomycin, increased collagen deposition, an exacerbated fibroblast response and reduced myofibroblast presence, was observed compared to wild-type mice. Their findings suggest that MK2 is necessary for myofibroblast development and for regulating tissue fibrosis.231 The impact of genetic loss of MK2 in these murine models did not affect macrophage or neutrophil intravasation into wounds but did lead to substantially reduced inflammatory cytokine production and impaired wound healing. The impaired wound healing could be rescued by introducing MK2 wild-type macrophages highlighting the importance of intact MK2 in macrophages and its role in cutaneous wound healing.232 Using a novel peptide-mimetic oligomer of HSP27 capable of selectively inhibiting MK2, Vittal and colleagues demonstrated that their pharmacologic agent could inhibit TGFβ1-mediated myofibroblast development similar to what Sousa and Liu had shown. However, using a similar lung-directed bleomycin fibrosis model, this group using their novel MK2 inhibitor could block the excessive collagen and fibronectin deposition in the lungs as well as reduce SMAD3, serpine1, IL-1β expression, and plasma IL-6 levels. This is a direct contrast to Liu’s findings which demonstrated genetic loss of MK2 led to an exacerbation of the fibrotic response. Similarly, Wang et al.,10 demonstrated using a novel reversible MK2 inhibitor (CDD-450) that it could selectively inhibit MK2, leading to a reduction in inflammatory cytokine activation from rheumatoid arthritis synovial fibroblasts. Furthermore, treatment with this drug could substantially reduce rat joint destruction in an inflammatory arthritis model. The observed discrepancy between the genetic and the pharmacologic models may suggest potential off-target interactions (pharmacologic), leading to reduced myofibroblast development and ECM deposition versus embryonic-physiologic compensation leading to an altered wound healing-fibrotic response in vivo (genetic). Additional work in this area is needed to improve our understanding of the temporal and cell compartmental contribution of MK2 inhibition on wound healing and fibrosis.
Investigations into the role of MK2 in remodeling and re-endothelialization after arterial injury in genetically modified mice in vivo showed MK2-deficiency nearly completely prevented hypertrophy and lumen loss after injury which was accompanied by reduced proliferation and migration of MK2-deficient smooth muscle cells. In addition, MK2-deficiency severely reduced monocyte adhesion to the arterial wall because of reduced expression of the chemokine ligands CCL2 and CCL5. MK2 KO also significantly reduced the infiltration of monocytes, neutrophils, and lymphocytes in the arterial wall. The authors concluded that deficiency of MK2 prevents adverse remodeling and promotes endothelial healing of the arterial wall after injury.233 While the exact role for MK2 in wound healing is evasive its cellular functions suggest a regulatory influence on tissue repair and together with its effects on cell survival and the immune response, it is well suited to help aberrant cells survive hypoxia, acidosis and other stressors created in the microenvironment.
5 |. MK2 IN CANCER
MK2 and its downstream cytokine, and transcription factor targets (i.e., EMT) have been associated with disease progression, metastasis, and poor survival outcomes in a variety of cancers including gastrointestinal, bladder, breast, lung and head and neck. Though the precise mechanisms by which MK2 mediates cancer progression, metastasis, and response to therapies is still not clear, pharmacologic or biologic inhibition of MK2 pathway signaling has shown promise in decreasing proliferation and slowing tumor progression in multiple cancer models.11,13,14,234–237
5.1 |. Gastrointestinal cancers
MK2 and its downstream targets have been implicated in gastric and colorectal cancers in human and mouse models.13,14,234,235,238 Human gastric tumor tissues had higher levels of MK2-mediated inflammatory cytokines compared to normal tissues, and high levels of MK2 in gastric cancer tumors were associated with a significantly increased rate of metastasis. Furthermore, levels of cytokines IL-1β, IL-6, and TNFα are higher in primary tumors that have metastasized to the lymphatic system compared to non-metastatic primary tumors.234
Inflammation in the colon contributes to higher rates of colorectal cancer among patients with inflammatory bowel diseases compared to healthy individuals.239 Some data suggest that inflammatory cytokine signaling by way of MK2 may contribute to colorectal cancer development and progression.13,14 In an azoxy-methane (AOM)/DSS mouse model of colon cancer, MK2 knockout mice had decreased levels of tumor derived inflammatory cytokines IL-1α, IL-1β, IL-6, and TNFα. Compared to control mice, MK2 knockout mice,13 or mice treated with an MK2 inhibitor14 had decreased tumor incidence and tumor volumes compared to control mice. These authors also found decreased cytokine response and decreased macrophage production in response to AOM/DSS treatment in MK2 knockout mice compared to wild-type mice.14 These findings suggest that the reduction in MK2-mediated inflammatory signaling decreases tumor incidence and proliferation and points toward cytokine signaling as a potential mechanism by which MK2 mediates tumor growth in colorectal cancer models.
The Apc mutant mouse spontaneously develops several intestinal tumors and is often used as a model of colorectal cancer.240 When crossed with MK2 knockout mice, Apc mutant mice had decreased tumor incidence and decreased tumor volumes compared to Apc/MK2 wild-type mice.235 Furthermore, inhibition of MK2 in Apc mutant mice reduced tumor formation, abrogated tumor proliferation, and increased expression of the apoptotic marker cleaved caspase-3.235 Similar findings in the AOM/DSS mouse model of colon cancer demonstrated that MK2 inhibition decreased tumor volumes and cytokine levels in vivo, and suppressed colon cancer cell invasion in vitro.14
An important stromal cell type which contributes to colon cancer tumorigenesis is the macrophage. Colon cancer cells when treated with an MK2 inhibitor had significantly decreased expression of the chemokines MCP-1, Mip-1α, and Mip-2α,238 all of which are involved in macrophage recruitment.241,242 Similarly, decreased chemokine levels were detected in colon cancer cell-derived tumors treated with MK2 inhibitor compared to vehicle, and macrophage recruitment to the tumors was significantly decreased in MK2 inhibitor treated mice. Tumor chemokine levels were rescued in MK2 inhibitor-treated tumors upon the addition of macrophages. Furthermore, treatment with an MK2 inhibitor decreased tumor growth in vivo whereas treatment with MCP-1, Mip-1α, and Mip-2α increased tumor volumes. Interestingly, in tumors treated with MK2 inhibitor the addition of these chemokines rescued tumor growth.238 These findings suggest that MK2 may regulate macrophage chemokine expression and macrophage recruitment in colon cancer. While this work demonstrates that functional MK2 is necessary for stromal-mediated tumorigenesis, it remains unknown whether overexpression of tumor MK2 can compensate, thereby allowing these tumors to continue growing
5.2 |. Head and neck cancers
MK2 has recently been shown to be a potential mediator of disease in head and neck cancers.12,243,244 In a small clinical study, high levels of phosphorylated MK2 (p-MK2) were associated with lower overall survival.243 A larger tissue microarray analysis showed that in p16-negative disease, high levels of p-MK2 were associated with worse cancer-specific survival compared to patients with low p-MK2 levels.12 Compared to normal tissues and noncancerous cell lines, levels of p-MK2 protein expression and expression of RNA binding proteins such as HuR are increased in head and neck squamous cell carcinoma (HNSCC) tumor specimens and in HNSCC cell lines.244
As mentioned previously, chronic-tonic activation of the p38-MK2 signaling axis through a continued positive inflammatory cytokine feedback can allow for a persistently inflamed, protumorigenic microenvironment. This in turn may drive tumor progression and/or resistance to treatment in head and neck cancers. However, the precise mechanisms by which MK2 potentially mediates disease progression and ultimately patient outcomes in HNSCC, have yet to be elucidated. There is evidence that cytokines contribute to tumor cell migration and invasion in HNSCC; and mRNA levels of several inflammatory cytokines (IL-6, IL-1α, IL-1β, TNFα) and angiogenic factors (HIF-1α, VEGF) are increased in cancer cells and tumor tissues.244–246 The inflammatory cytokines IL-1α, IL-1β, IL-6, and TNFα are regulated in part by MK2.10 High expression levels of IL-6 are associated with worse overall survival in oropharyngeal squamous cell carcinoma (OSCC) patients. A larger dataset of HNSCC patients confirmed the finding that high IL-6 expression was associated with worse overall survival.247 High pretreatment blood IL-6 levels served as a predictor of worse survival outcomes and greater incidence of cancer recurrence in HNSCC patients.248 In Cal27 HNSCC cells, overexpression of IL-6 exacerbates tumor growth and metastasis compared to nontransfected Cal27 cells,249 whereas inhibition of IL-6 with the chemotherapeutic drug Bazedoxifene (BZA) sensitized HNSCC cells to cisplatin treatment and radiation therapy (RT) in vitro and in vivo.250 The regulation of IL-6 by MK2, and the involvement of IL-6 in head and neck cancers suggests that cytokine activation and expression may be a mechanism by which MK2 is involved in cancer progression and poor clinical outcomes.
MK2 also drives expression of IL-1α, as seen by MK2 knockout mice having significantly decreased IL-1α compared to wild-type mice in a mouse model of colorectal cancer.13 Therefore, IL-1α is another cytokine that may drive cancer progression by way of the p38/MK2 pathway. High tumoral levels of IL-1α mRNA is associated with significantly reduced distant metastasis-free survival compared to tumors expressing low levels of IL-1α in HNSCC. Furthermore, tumors from patients with distant metastasis secreted significantly more IL-1α protein compared to patients without distant metastasis.245 When cocultured with IL-1α-expressing OSCC cell lines, cancer associated fibroblasts (CAFs) express significantly greater levels of IL-1α receptor (IL-1αR), and CAFs treated with IL-1α proliferate significantly more than untreated CAFs and this proliferation was blocked upon treatment with an IL-1α neutralizing antibody.251 Additionally, cocultured OSCC cell lines and CAFs secreted high levels of other cytokines including CXCL1 and IL-8, with the effect shown to be dependent upon IL-1α.251 These findings suggest that IL-1α activity in tumor and stromal compartments in HNSCC may contribute to enhanced cytokine activity and disease progression in HNSCC.
TNFα is another cytokine whose synthesis is regulated by MK2 and is relevant in head and neck cancer progression and outcomes. High levels of plasma TNFα were correlated with increased incidence of cachexia,252 and is associated with worse overall survival in head and neck cancer patients.253 Treatment with TNFα resulted in migration and invasion in HNSCC cells in vitro.254 TNFα synthesis is significantly decreased in MK2 knockout mice,28,155,255 likely due to loss of inhibitory regulation of TTP. Conversely, in the presence of MK2 activity, MK2 phosphorylates TTP preventing its binding of the ARE of TNFα mRNA, allowing for TNFα protein translation.103,144
Inflammatory signaling by downstream targets of MK2 such as TNFα, IL-6, and IL-1β are involved in EMT.256–259 Overexpression of EMT markers is associated with disease progression260 and decreased survival in head and neck cancers. Zheng254 and colleagues found that expression of both SNAI1 (also known as Snail) and SNAI2 (also known as Slug) was associated with worse survival in tongue cancers. Knockdown of either SNAI1, Slug, or both, decreased migration and invasion in Cal27 HNSCC cells. The Notch signaling pathway, known to be involved in EMT200 and in migration and invasion of HNSCC cancer cells, was found to be upregulated at later disease stages, and was overexpressed in metastatic tissue from HSNCC patients.261 Zhou256 and colleagues found increases in the mesenchymal cell marker vimentin and decreased epithelial marker E-cadherin following treatment with TNFα in HNSCC cell lines. Furthermore, they found that TNFα stabilized the EMT transcription factor SNAI1 through activation of NF-κB. Similarly, TNFα treatment was found to stabilize Slug, also by way of NF-κB activation, and blocking NF-κB destabilized Slug.124 Treatment of Cal27 cells with IL-6 induced increased expression of the EMT markers vimentin (VIM) and Snail; overexpression of IL-6 in Cal27 HNSCC cells resulted in enhanced migration in vitro, and increased locoregional (lymph node) and distant (lung) metastasis in vivo.262 These data suggest a potential mechanism by which activation of MK2 contributes not only to tumor inflammation, but also to migration, invasion, and metastasis through cytokine induced EMT signaling in HNSCC.
Treatment resistance and loco-regional disease failure contribute to the poor long-term overall survival for patients with p16-negative, smoking associated head and neck cancer and remains a significant unaddressed problem.263 RT is a mainstay in the treatment for early stage head and neck cancers and in cases of locoregional disease combined chemoradiotherapy is often indicated to improve patient outcomes.264 However, some data suggest that RT can activate the TME to promote tumor inflammation,265 EMT12,266 and rapid tumor repopulation,266–268 which can drive locoregional and/or distant metastasis. Therefore, a better understanding of the mechanisms underlying radiation-induced treatment failure in head and neck cancer is needed.
In vitro and in vivo models of head and neck cancer demonstrate that radiation induced EMT may be a potential mechanism of tumor repopulation in head and neck cancers.12,266 Irradiation of Tu167 and HN11 head and neck cancer cells significantly increased the EMT markers vimentin, ZEB2 and Snail, and chronic irradiation resulted in increased ZEB2, VIM, and TWIST1 gene expression.266 This group found that following irradiation of either the tumor or its stromal compartment could induce rapid HNC tumor cell repopulation in vivo. Blockade with a hedgehog pathway inhibitor could abrogate this radiation-induced tumor repopulation effect.
RT activates the p38/MK2 pathway and mediates radiation-induced inflammation and EMT gene expression in head and neck cancers. RT has been shown to increase MK2 phosphorylation in established head and neck cancer cell lines in vitro and similarly in vivo in multiple head and neck PDX lines. Radiation therapy led to significant MK2-mediated gene expression of inflammatory cytokines (IL-1α, IL-1β, and IL-6), and the EMT markers (SNAl1, SNAI2, VIM). Both pharmacologic (i.e., PF3644022) and genetic (i.e. siRNA) inhibition led to an abrogation of the radiation-induced increases in cytokine and EMT gene levels.12 Finally, treatment with combined RT and MK2 inhibitors decreased tumor volumes and increased survival in p16-positive and p16-negative PDX HNSCC models compared to monotherapy. Interestingly, levels of tumor-derived IL-1α, IL-1β, and IL-6 were also decreased with combined treatment compared to treatment with either RT or MK2 inhibitor alone12 and may suggest these same inflammatory cytokines may stimulate tumor (re)growth in vivo similar to rectal cancer.14 These findings suggest that MK2 is a mediator of radiation-induced inflammatory and EMT signaling in head and neck cancers.
5.3 |. Breast, bladder, and other cancers
The p38/MK2 pathway has also been implicated in preclinical models of breast cancer and breast cancer metastasis, with some work showing that this involvement is particularly crucial in the TME. Alspach and colleagues demonstrated that inhibition of p38/MAPK factors in CAFs decreased the growth of HaCaT cells in coculture with CAFs.269 Furthermore, they showed that inhibition of p38 in patient derived CAFs coinjected with breast cancer tumor cells inhibited tumor growth in vivo.269 These findings suggest that specific interactions between the TME and the p38 pathway, and possibly downstream molecules such as MK2, contribute to tumor growth and metastasis in breast cancer models.
Despite the success of p38 inhibitors in preclinical models of breast cancer, the side effect profile of p38 MAPK inhibitors prevent their application for widespread clinical use. Therefore, downstream effectors of p38 MAPK, including MK2, have been examined in breast cancer models. In agreement with previous studies of p38 MAPK inhibition and decreased tumor progression, Murali and colleagues (2018)11 inhibited p38 and found significantly decreased bone and visceral metastasis in mouse models of breast cancer. Interestingly, however, they found that neither pharmacological inhibition nor short hairpin RNA (shRNA) knockdown of p38 MAPK resulted in decreased tumor growth. To determine if MK2 inhibition, rather than p38 inhibition, could similarly reduce metastasis and to examine the effect of MK2 on tumor proliferation, mice were treated with the MK2 inhibitor ATI-450. Inhibition of MK2 did not abrogate tumor growth in vitro or in vivo; similarly, MK2 knockdown in tumor cells did not decrease tumor cells growth in vitro or in vivo.11 However, MK2 inhibition or MK2 knockdown did significantly decrease bone and visceral metastasis to a similar extent compared to pharmaceutical or genetic manipulation of p38 MAPK.11 These authors conclude that because inhibition or knockdown of MK2 or p38 did not lead to decreased tumor growth but did decrease metastasis, the mechanism by which MK2 or p38 inhibition decreases metastasis resides in the activity of this pathway in the TME/stroma specifically.11 That MK2 inhibition successfully decreased metastatic burden in this model is advantageous from a translational standpoint because it points toward the ability to avoid the toxicity associated with p38 MAPK inhibition while still achieving positive outcomes.
The p38 MAPK-MK2 pathway may also mediate bladder cancer proliferation and invasion. Kumar and colleagues16 demonstrated that inhibition of p38 decreased bladder cancer cell proliferation and DNA synthesis. Furthermore, inhibition of p38 MAPK caused G2/M arrest and G1 arrest in the bladder cancer cell lines HTB9 and HTB5, respectively. To further elucidate the mechanism by which inhibition of p38 MAPK interfered with bladder cancer cell proliferation, these authors examined the roles of MK2 and MMPs known to be involved in tumor progression270 and which are downstream of p38 MAPK in bladder cancer cell invasion and proliferation.16 Inhibition of p38 MAPK inhibited MMP2 and MMP9 activity in HTB9 bladder cancer cells, and blockade of MMP2 decreased cancer cell invasion. Furthermore, MMP transcript stability was decreased in cells treated with a p38 MAPK inhibitor. In investigating whether MK2 played a role in bladder cancer invasion, Kumar and colleagues16 showed that cells expressing an MK2 kinase-inactive mutant had significantly decreased MMP mRNA levels, and significantly decreased invasion compared to wild-type MK2 cells. These findings suggest that the inhibitory effects of p38 blockade on bladder cancer cell invasion are mediated through MMPs via MK2.
MK2 may also be relevant in hematologic cancers. High MK2 expression is associated with worse event-free and overall survival in multiple myeloma.237 MK2 knockdown or overexpression significantly decreased and increased proliferation in multiple myeloma cell lines, respectively. When combined with common systemic treatments for multiple myeloma, MK2 inhibitor further abrogated cell death in vitro. Furthermore, mice treated with an MK2 inhibitor had increased survival compared to control mice.237
Common cancers harboring KRAS-mutations include lung, colorectal, and pancreatic.271,272 These cancers have poor prognosis, and MK2 may play a role in tumorigenesis and cancer outcome. Using a computer software-based analysis, Dietlein and colleagues examined a panel of 96 cancer lines and identified those cell lines that would be most susceptible to combined checkpoint inhibition and MK2 inhibition. The software predicted that cell lines harboring KRAS mutations had the greatest sensitivity of dual checkpoint and MK2 inhibition.129 To experimentally confirm these analyses, KRAS mutant cell lines were treated with a Chk1 inhibitor, an MK2 inhibitor, or both; survival was significantly decreased in dual-treated cells compared to treatment with Chk1 or MK2 inhibitor alone. Furthermore, shRNA experiments also demonstrated that cell survival was significantly decreased when both Chk1 and MK2 were knocked down, but not when either was knocked down individually. These findings were confirmed in vivo in xenograft models of KRAS-mutant tumors with combined Chk1 and MK2 inhibition decreasing tumor volumes compared to monotherapy.129
MK2 may also mediate treatment resistance in pancreatic cancer. Kopper273 and colleagues found that MK2 is a mediator of gemcitabine sensitivity in pancreatic cancer cell lines via the DNA damage response. Inhibition of MK2 decreased activation of the DNA damage response marker H2AX (histone variant 2AX) and resulted in increased cell proliferation in gemcitabine-sensitive pancreatic cancer cell lines. Similarly, MK2 has been shown to mediate radiation sensitivity in pancreatic cancer by way of DNA damage response mechanisms. Ataxia-telangiectasia group D-associated gene (ATDC, also known asTRIM29) is overexpressed in pancreatic cancer274 and confers radiation and chemotherapy (gemcitabine) resistance in pancreatic cells lines, but knockdown of ATDC in xenograft models resulted in enhanced radiation sensitivity.113 MK2 phosphorylates ATDC by way of upstream activation of the DNA damage response effector ATM (and its subsequent activation of p38 MAPK, and MK2 phosphorylation-site mutants) or knockdown of MK2 abrogated radiation resistance in ATDC overexpressing cell lines.113
Finally, there is evidence that MK2 may be involved in preventing the immune system from mounting a defense through supporting the expression of PDL1 and an immunosuppressive phenotype. In colorectal and lung tumors, KRAS associated ROS accumulation led to enhanced TTP phosphorylation at MK2 inhibitory sites. This suggested persistent MK2 activation inhibited TTP, which increased the stability of PDL1 mRNA and reduced antitumor activity.275 It is therefore possible that in addition to modulating survival through checkpoint arrest, enhancing cytokine release and promoting migration, MK2 may also provide tumor cells protection from attack by the immune system.
5.4 |. Potential therapeutic uses of MK2
Acute and chronic Inflammation both contribute to the pathobiology of many oncologic and non-oncologic human diseases.1 While p38 MAPK is considered a central player in regulating inflammation, cell differentiation, proliferation, apoptosis, senescence and RNA biology, attempts to regulate p38 MAPK clinically have largely been unsuccessful.3 However, an immediate downstream substrate of p38 MAPK is MK2. With its powerful effects on the inflammatory process, cellular migration, and cell survival, this protein appears to be an attractive target for intervention. However, to date, there is a substantial dearth of MK2 specific clinical inhibitors being explored in the oncologic domain.
The data is promising that targeting MK2 (pharmacologic or genetic) causes inhibitory effects on the release of cytokines that may aid the maintenance and growth of cancer tissues to stimulate a persistent inflammatory microenvironment. Since many of the cytokines released through MK2 actions support its own stimulation, it appears that MK2 may function in a positive feedback loop exacer-bating inflammation. The consequence of aberrant MK2 pathway activation may promote metastasis through increased motility, cell survival and angiogenesis. Therefore, one might expect that reducing the persistent activity of MK2 would counteract the positive feedback and reduce tumor inflammation and growth. Several preclinical studies have demonstrated efficacy with research grade MK2 inhibitors thus providing proof of principle of MK2 as an attractive target for limiting tumor inflammation.
Reducing inflammation on its own is unlikely to inhibit tumor growth completely but the effect of MK2 on checkpoint arrest offers other avenues to combat tumor growth. The idea of SL of certain tumor models with adjuvant MK2 suggests a pathway into the clinic using combination therapies against p53 mutant tumors, which account for over 50% of all tumors.276 It was shown in p53 mutant cancers that utilizing MK2 in combination with CHK1 inhibition caused death by mitotic catastrophe. Potentially using an MK2 inhibitor to further short circuit a tumors already compromised repair mechanism in combination with traditional chemotherapy and/or radiotherapy would be a novel way to destroy rapidly proliferating cells to improve therapeutic outcome.126 Studies will hopefully discover more checkpoint molecules that could be used in conjunction with MK2 as a therapy. For example, in addition to CHK1, the DNA repair protein XPA has shown efficacy in enhancing the SL of MK2 in p53 negative tumors.130 As we increase our knowledge of the cell cycle and DNA repair further, combined therapies either with traditional cytotoxic chemotherapies or novel targeted agents may enhance this killing depending on the molecular mechanisms of the tumor.
MK2 inhibition has also shown efficacy with radiotherapy in preclinical work demonstrating that the combination reduces tumor growth more than monotherapy indicating synergy.12 It is possible that radiotherapy along with the combinatorial treatment of MK2 inhibition with CHK1 or XPA antagonists might improve responses even further. Certainly, radiotherapy is a very efficient method of introducing DNA damage and blocking cell cycle arrest after radiotherapy may improve current treatment modalities. Given the propensity of radiotherapy to generate fibrosis and the emerging roles of MK2 in wound healing, it is not clear whether it would exacerbate or reduce unwanted side effects. Conflicting reports leaves the role of MK2 in wound healing and collagen deposition uncertain.10,231,277 A better understanding of MK2 function in fibrosis and wound healing is necessary to direct its use in treatments and whether it is better suited for aiding chemotherapy or radiotherapy.
The action of MK2 on migration and EMT gene changes implicate its ability to control metastasis by increasing its invasive potential through motility and differentiation. Because metastasis is the main cause of mortality in cancer finding treatments to block this hallmark of cancer biology is paramount. Preclinical models have demonstrated the potential benefit of blocking MK2 and reducing cancer metastases.11,234 However, whether this is applicable to one group of cancers versus all cancers has not yet been realized. It is reasonable to assume that MK2 is likely to contribute globally to tumor migration, invasion and metastases and therapeutically targeting MK2 may be an important future clinical direction that will need to be explored.
6 |. CONCLUSION
The effects of MK2 are diverse and are centered around responding to stress, facilitating survival, migration, repair, and regrowth. Many aspects of its function arise from its ability to modulate mRNA stability for cytokines and other factors that control cell survival. The observation that the stability mRNA for PDL1 begs the question of how many more mRNA moieties are regulated in this fashion and what that means for the mechanism of MK2 action.275 The regulation of PDL1 expression by MK2 hints at a role for MK2 in immunosuppression and tumor survival leading to potential clinical applications in immunological tumors that have evaded killing mechanisms of the immune system. As we learn more about the functions of MK2 in inflammation, wound healing, and tumor growth we will be better served to understand how to deploy pharmacological intervention of MK2 in the patient setting. Much of this will also rely on the development of specific inhibitors of MK2 that have high efficacy and are well tolerated.
ACKNOWLEDGEMENT
This work was supported by the Kansas Institute for Precision Medicine NIGMS COBRE grant (P20GM130423) (G.N. Gan, A.K. Godwin).
Funding information
Kansas Institute for Precision Medicine NIGMS, Grant/Award Number: COBRE grant (P20GM130423)
DATA AVAILABILITY STATEMENT
No datasets were generated or analyzed during the current study.
REFERENCES
- 1.Greten FR, Grivennikov SI. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity. 2019;51(1):27–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mantovani A, Marchesi F, Portal C, Allavena P, Sica A. Linking inflammation reactions to cancer: novel targets for therapeutic strategies. Adv Exp Med Biol. 2008;610:112–127. [DOI] [PubMed] [Google Scholar]
- 3.Martinez-Limon A, Joaquin M, Caballero M, Posas F, de Nadal E. The p38 pathway: from biology to cancer therapy. Int J Mol Sci. 2020;21(6):1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh RK, Najmi AK, Dastidar SG. Biological functions and role of mitogen-activated protein kinase activated protein kinase 2 (MK2) in inflammatory diseases. Pharmacol Rep. 2017;69(4):746–756. [DOI] [PubMed] [Google Scholar]
- 5.Soni S, Anand P, Padwad YS. MAPKAPK2: the master regulator of RNA-binding proteins modulates transcript stability and tumor progression. J Exp Clin Cancer Res. 2019;38(1):121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liang J, Liu N, Liu X, et al. Mitogen-activated protein kinase-activated protein kinase 2 inhibition attenuates fibroblast invasion and severe lung fibrosis. Am J Respir Cell Mol Biol. 2019;60(1): 41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vittal R, Fisher A, Gu H, et al. Peptide-mediated inhibition of mitogen-activated protein kinase-activated protein kinase-2 ameliorates bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2013;49(1):47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu L, Yates CC, Lockyer P, et al. MMI-0100 inhibits cardiac fibrosis in myocardial infarction by direct actions on cardiomyocytes and fibroblasts via MK2 inhibition. J Mol Cell Cardiol. 2014;77:86–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hegen M, Gaestel M, Nickerson-Nutter CL, Lin LL, Telliez JB. MAPKAP kinase 2-deficient mice are resistant to collagen-induced arthritis. J Immunol. 2006;177(3):1913–1917. [DOI] [PubMed] [Google Scholar]
- 10.Wang C, Hockerman S, Jacobsen EJ, et al. Selective inhibition of the p38alpha MAPK-MK2 axis inhibits inflammatory cues including inflammasome priming signals. J Exp Med. 2018;215(5):1315–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murali B, Ren Q, Luo X, et al. Inhibition of the stromal p38MAPK/MK2 pathway limits breast cancer metastases and chemotherapy-induced bone Loss. Cancer Res. 2018;78(19):5618–5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berggren KL, Restrepo Cruz S, Hixon MD, et al. MAPKAPK2 (MK2) inhibition mediates radiation-induced inflammatory cytokine production and tumor growth in head and neck squamous cell carcinoma. Oncogene. 2019;38(48):7329–7341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ray AL, Castillo EF, Morris KT, et al. Blockade of MK2 is protective in inflammation-associated colorectal cancer development. Int J Cancer. 2016;138(3):770–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ray AL, Berggren KL, Restrepo Cruz S, Gan GN, Beswick EJ. Inhibition of MK2 suppresses IL-1beta, IL-6, and TNF-alpha-dependent colorectal cancer growth. Int J Cancer. 2018;142(8): 1702–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar B, Sinclair J, Khandrika L, Koul S, Wilson S, Koul HK. Differential effects of MAPKs signaling on the growth of invasive bladder cancer cells. Int J Oncol. 2009;34(6):1557–1564. [DOI] [PubMed] [Google Scholar]
- 16.Kumar B, Koul S, Petersen J, et al. p38 mitogen-activated protein kinase-driven MAPKAPK2 regulates invasion of bladder cancer by modulation of MMP-2 and MMP-9 activity. Cancer Res. 2010;70(2): 832–841. [DOI] [PubMed] [Google Scholar]
- 17.Han J, Wu J, Silke J. An overview of mammalian p38 mitogen-activated protein kinases, central regulators of cell stress and receptor signaling. F1000Res. 2020:9. 10.12688/f1000research.22092.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang Y, Chen C, Li Z, et al. Characterization of the structure and function of a new mitogen-activated protein kinase (p38beta). J Biol Chem. 1996;271(30):17920–17926. [DOI] [PubMed] [Google Scholar]
- 19.Li Z, Jiang Y, Ulevitch RJ, Han J. The primary structure of p38 gamma: a new member of p38 group of MAP kinases. Biochem Biophys Res Commun. 1996;228(2):334–340. [DOI] [PubMed] [Google Scholar]
- 20.Jiang Y, Gram H, Zhao M, et al. Characterization of the structure and function of the fourth member of p38 group mitogen-activated protein kinases, p38delta. J Biol Chem. 1997;272(48): 30122–30128. [DOI] [PubMed] [Google Scholar]
- 21.Zhang YY, Mei ZQ, Wu JW, Wang ZX. Enzymatic activity and substrate specificity of mitogen-activated protein kinase p38alpha in different phosphorylation states. J Biol Chem. 2008;283(39): 26591–26601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beardmore VA, Hinton HJ, Eftychi C, et al. Generation and characterization of p38beta (MAPK11) gene-targeted mice. Mol Cell Biol. 2005;25(23):10454–10464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Greenblatt MB, Shim JH, Zou W, et al. The p38 MAPK pathway is essential for skeletogenesis and bone homeostasis in mice. J Clin Invest. 2010;120(7):2457–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cuenda A, Sanz-Ezquerro JJ. p38gamma and p38delta: from spectators to key physiological players. Trends Biochem Sci. 2017; 42(6):431–442. [DOI] [PubMed] [Google Scholar]
- 25.Adams RH, Porras A, Alonso G, et al. Essential role of p38alpha MAP kinase in placental but not embryonic cardiovascular development. Mol Cell. 2000;6(1):109–116. [PubMed] [Google Scholar]
- 26.Mudgett JS, Ding J, Guh-Siesel L, et al. Essential role for p38alpha mitogen-activated protein kinase in placental angiogenesis. Proc Natl Acad Sci USA. 2000;97(19):10454–10459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.del Barco Barrantes I, Coya JM, Maina F, Arthur JS, Nebreda AR. Genetic analysis of specific and redundant roles for p38alpha and p38beta MAPKs during mouse development. Proc Natl Acad Sci USA. 2011;108(31):12764–12769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kotlyarov A, Neininger A, Schubert C, et al. MAPKAP kinase 2 is essential for LPS-induced TNF-alpha biosynthesis. Nat Cell Biol. 1999;1(2):94–97. [DOI] [PubMed] [Google Scholar]
- 29.White A, Pargellis CA, Studts JM, Werneburg BG, Farmer BT 2nd. Molecular basis of MAPK-activated protein kinase 2:p38 assembly. Proc Natl Acad Sci USA. 2007;104(15):6353–6358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haar ET, Prabakhar P, Liu X, Lepre C. Crystal structure of the p38 alpha-MAPKAP kinase 2 heterodimer. J Biol Chem. 2007;282(13): 9733–9739. [DOI] [PubMed] [Google Scholar]
- 31.Arthur JS, Ley SC. Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol. 2013;13(9):679–692. [DOI] [PubMed] [Google Scholar]
- 32.Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81(2):807–869. [DOI] [PubMed] [Google Scholar]
- 33.Xu YR, Lei CQ. TAK1-TABs complex: a central signalosome in inflammatory responses. Front Immunol. 2020;11:608976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morrison DK. MAP kinase pathways. Cold Spring Harb Perspect Biol. 2012;4(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raingeaud J, Whitmarsh AJ, Barrett T, Derijard B, Davis RJ. MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol Cell Biol. 1996;16(3):1247–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moriguchi T, Kuroyanagi N, Yamaguchi K, et al. A novel kinase cascade mediated by mitogen-activated protein kinase kinase 6 and MKK3. J Biol Chem. 1996;271(23):13675–13679. [DOI] [PubMed] [Google Scholar]
- 37.Lin A, Minden A, Martinetto H, et al. Identification of a dual specificity kinase that activates the Jun kinases and p38-Mpk2. Science. 1995;268(5208):286–290. [DOI] [PubMed] [Google Scholar]
- 38.Brancho D, Tanaka N, Jaeschke A, et al. Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 2003;17(16):1969–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ge B, Gram H, Di Padova F, et al. MAPKK-independent activation of p38alpha mediated by TAB1-dependent autophosphorylation of p38alpha. Science. 2002;295(5558):1291–1294. [DOI] [PubMed] [Google Scholar]
- 40.DeNicola GF, Martin ED, Chaikuad A, et al. Mechanism and consequence of the autoactivation of p38alpha mitogen-activated protein kinase promoted by TAB1. Nat Struct Mol Biol. 2013;20(10): 1182–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li J, Miller EJ, Ninomiya-Tsuji J, Russell RR 3rd, Young LH. AMP-activated protein kinase activates p38 mitogen-activated protein kinase by increasing recruitment of p38 MAPK to TAB1 in the ischemic heart. Circ Res. 2005;97(9):872–879. [DOI] [PubMed] [Google Scholar]
- 42.Salvador JM, Mittelstadt PR, Guszczynski T, et al. Alternative p38 activation pathway mediated by T cell receptor-proximal tyrosine kinases. Nat Immunol. 2005;6(4):390–395. [DOI] [PubMed] [Google Scholar]
- 43.Trempolec N, Dave-Coll N, Nebreda AR. SnapShot: p38 MAPK substrates. Cell. 2013;152(4):924–924 e921. [DOI] [PubMed] [Google Scholar]
- 44.Iida N, Fujita M, Miyazawa K, Kobayashi M, Hattori S. Proteomic identification of p38 MAP kinase substrates using in vitro phosphorylation. Electrophoresis. 2014;35(4):554–562. [DOI] [PubMed] [Google Scholar]
- 45.Maik-Rachline G, Lifshits L, Seger R. Nuclear P38: roles in physiological and pathological processes and regulation of nuclear translocation. Int J Mol Sci. 2020;21(17):6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Norman P. Investigational p38 inhibitors for the treatment of chronic obstructive pulmonary disease. Expert Opin Investig Drugs. 2015;24(3):383–392. [DOI] [PubMed] [Google Scholar]
- 47.Fiore M, Forli S, Manetti F. Targeting mitogen-activated protein kinase-activated protein kinase 2 (MAPKAPK2, MK2): Medicinal chemistry efforts to lead small molecule inhibitors to clinical trials. J Med Chem. 2016;59(8):3609–3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ferguson FM, Gray NS. Kinase inhibitors: the road ahead. Nat Rev Drug Discov. 2018;17(5):353–377. [DOI] [PubMed] [Google Scholar]
- 49.Lee N, Kim D. Cancer metabolism: fueling more than just growth. Mol Cells. 2016;39(12):847–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haller V, Nahidino P, Forster M, Laufer SA. An updated patent review of p38 MAP kinase inhibitors (2014–2019). Expert Opin Ther Pat. 2020;30(6):453–466. [DOI] [PubMed] [Google Scholar]
- 51.Weber HO, Ludwig RL, Morrison D, Kotlyarov A, Gaestel M, Vousden KH. HDM2 phosphorylation by MAPKAP kinase 2. Oncogene. 2005;24(12):1965–1972. [DOI] [PubMed] [Google Scholar]
- 52.Hitti E, Iakovleva T, Brook M, et al. Mitogen-activated protein kinase-activated protein kinase 2 regulates tumor necrosis factor mRNA stability and translation mainly by altering tristetraprolin expression, stability, and binding to adenine/uridine-rich element. Mol Cell Biol. 2006;26(6):2399–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stokoe D, Campbell DG, Nakielny S, et al. MAPKAP kinase-2; a novel protein kinase activated by mitogen-activated protein kinase. EMBO J. 1992;11(11):3985–3994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Uhlén M, Fagerberg L, Hallström BM, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347(6220): 1260419. [DOI] [PubMed] [Google Scholar]
- 55.Zu YL, Wu F, Gilchrist A, Ai Y, Labadia ME, Huang CK. The primary structure of a human MAP kinase activated protein kinase 2. Biochem Biophys Res Commun. 1994;200(2):1118–1124. [DOI] [PubMed] [Google Scholar]
- 56.Zu YL, Ai Y, Huang CK. Characterization of an autoinhibitory domain in human mitogen-activated protein kinase-activated protein kinase 2. J Biol Chem. 1995;270(1):202–206. [DOI] [PubMed] [Google Scholar]
- 57.Ben-Levy R, Leighton IA, Doza YN, et al. Identification of novel phosphorylation sites required for activation of MAPKAP kinase-2. EMBO J. 1995;14(23):5920–5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stokoe D, Engel K, Campbell DG, Cohen P, Gaestel M. Identification of MAPKAP kinase 2 as a major enzyme responsible for the phosphorylation of the small mammalian heat shock proteins. FEBS Lett. 1992;313(3):307–313. [DOI] [PubMed] [Google Scholar]
- 59.Kotlyarov A, Yannoni Y, Fritz S, et al. Distinct cellular functions of MK2. Mol Cell Biol. 2002;22(13):4827–4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Manke IA, Nguyen A, Lim D, Stewart MQ, Elia AE, Yaffe MB. MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Mol Cell. 2005;17(1):37–48. [DOI] [PubMed] [Google Scholar]
- 61.Engel K, Schultz H, Martin F, et al. Constitutive activation of mitogen-activated protein kinase-activated protein kinase 2 by mutation of phosphorylation sites and an A-helix motif. J Biol Chem. 1995;270(45):27213–27221. [DOI] [PubMed] [Google Scholar]
- 62.Engel K, Kotlyarov A, Gaestel M. Leptomycin B-sensitive nuclear export of MAPKAP kinase 2 is regulated by phosphorylation. EMBO J. 1998;17(12):3363–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ben-Levy R, Hooper S, Wilson R, Paterson HF, Marshall CJ. Nuclear export of the stress-activated protein kinase p38 mediated by its substrate MAPKAP kinase-2. Curr Biol. 1998;8(19):1049–1057. [DOI] [PubMed] [Google Scholar]
- 64.Sudo T, Kawai K, Matsuzaki H, Osada H. p38 mitogen-activated protein kinase plays a key role in regulating MAPKAPK2 expression. Biochem Biophys Res Commun. 2005;337(2):415–421. [DOI] [PubMed] [Google Scholar]
- 65.Gong X, Ming X, Deng P, Jiang Y. Mechanisms regulating the nuclear translocation of p38 MAP kinase. J Cell Biochem. 2010;110(6): 1420–1429. [DOI] [PubMed] [Google Scholar]
- 66.Zehorai E, Seger R. Beta-like importins mediate the nuclear translocation of MAPKs. Cell Physiol Biochem. 2019;52(4):802–821. [DOI] [PubMed] [Google Scholar]
- 67.Smith JA, Poteet-Smith CE, Lannigan DA, Freed TA, Zoltoski AJ, Sturgill TW. Creation of a stress-activated p90 ribosomal S6 kinase. The carboxyl-terminal tail of the MAPK-activated protein kinases dictates the signal transduction pathway in which they function. J Biol Chem. 2000;275(41):31588–31593. [DOI] [PubMed] [Google Scholar]
- 68.Lukas SM, Kroe RR, Wildeson J, et al. Catalysis and function of the p38 alpha.MK2a signaling complex. Biochemistry. 2004;43(31): 9950–9960. [DOI] [PubMed] [Google Scholar]
- 69.Meng W, Swenson LL, Fitzgibbon MJ, et al. Structure of mitogen-activated protein kinase-activated protein (MAPKAP) kinase 2 suggests a bifunctional switch that couples kinase activation with nuclear export. J Biol Chem. 2002;277(40): 37401–37405. [DOI] [PubMed] [Google Scholar]
- 70.Schlapbach A, Huppertz C. Low-molecular-weight MK2 inhibitors: a tough nut to crack! Future Med Chem. 2009;1(7):1243–1257. [DOI] [PubMed] [Google Scholar]
- 71.Olsson H, Sjo P, Ersoy O, Kristoffersson A, Larsson J, Norden B. 4-Anilino-6-phenyl-quinoline inhibitors of mitogen activated protein kinase-activated protein kinase 2 (MK2). Bioorg Med Chem Lett. 2010;20(16):4738–4740. [DOI] [PubMed] [Google Scholar]
- 72.Huang X, Shipps GW Jr Jr., Cheng CC, et al. Discovery and Hit-to-lead optimization of non-ATP competitive MK2 (MAPKAPK2) inhibitors. ACS Med Chem Lett. 2011;2(8):632–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mourey RJ, Burnette BL, Brustkern SJ, et al. A benzothiophene inhibitor of mitogen-activated protein kinase-activated protein kinase 2 inhibits tumor necrosis factor alpha production and has oral anti-inflammatory efficacy in acute and chronic models of inflammation. J Pharmacol Exp Ther. 2010;333(3):797–807. [DOI] [PubMed] [Google Scholar]
- 74.Anderson DR, Meyers MJ, Vernier WF, et al. Pyrrolopyridine inhibitors of mitogen-activated protein kinase-activated protein kinase 2 (MK-2). J Med Chem. 2007;50(11):2647–2654. [DOI] [PubMed] [Google Scholar]
- 75.Davidson W, Frego L, Peet GW, et al. Discovery and characterization of a substrate selective p38alpha inhibitor. Biochemistry. 2004;43(37):11658–11671. [DOI] [PubMed] [Google Scholar]
- 76.Gurgis F, Åkerfeldt MC, Heng B, et al. Cytotoxic activity of the MK2 inhibitor CMPD1 in glioblastoma cells is independent of MK2. Cell Death Discov. 2015;1:15028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Muto A, Panitch A, Kim N, et al. Inhibition of mitogen activated protein kinase activated protein kinase II with MMI-0100 reduces intimal hyperplasia ex vivo and in vivo. Vascul Pharmacol. 2012; 56(1–2):47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Brown DI, Cooley BC, Quintana MT, Lander C, Willis MS. Nebulized Delivery of the MAPKAP kinase 2 peptide inhibitor MMI-0100 protects against ischemia-induced systolic dysfunction. Int J Pept Res Ther. 2016;22(3):317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jiang J, Wang Z, Liang X, et al. Intranasal MMI-0100 attenuates abeta1-42- and LPS-induced neuroinflammation and memory impairments via the MK2 signaling pathway. Front Immunol. 2019;10: 2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang Z, Liang XY, Chang X, et al. MMI-0100 ameliorates dextran sulfate sodium-induced colitis in mice through targeting MK2 pathway. Molecules. 2019;24(15):2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gordon D, Hellriegel ET, Hope HR, Burt D, Monahan JB. Safety, tolerability, pharmacokinetics, and pharmacodynamics of the MK2 inhibitor ATI-450 in healthy subjects: a placebo-controlled, randomized Phase 1 study. Clin Pharmacol. 2021;13:123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rouse J, Cohen P, Trigon S, et al. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell. 1994;78(6): 1027–1037. [DOI] [PubMed] [Google Scholar]
- 83.Kobayashi M, Nishita M, Mishima T, Ohashi K, Mizuno K. MAPKAPK-2-mediated LIM-kinase activation is critical for VEGF-induced actin remodeling and cell migration. EMBO J. 2006;25(4): 713–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wu Y, Zhan L, Ai Y, et al. MAPKAPK2-mediated LSP1 phosphorylation and FMLP-induced neutrophil polarization. Biochem Biophys Res Commun. 2007;358(1):170–175. [DOI] [PubMed] [Google Scholar]
- 85.Rousseau S, Peggie M, Campbell DG, Nebreda AR, Cohen P. Nogo-B is a new physiological substrate for MAPKAP-K2. Biochem J. 2005;391(Pt 2):433–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Menon MB, Schwermann J, Singh AK, et al. p38 MAP kinase and MAPKAP kinases MK2/3 cooperatively phosphorylate epithelial keratins. J Biol Chem. 2010;285(43):33242–33251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Eyers CE, McNeill H, Knebel A, et al. The phosphorylation of CapZ-interacting protein (CapZIP) by stress-activated protein kinases triggers its dissociation from CapZ. Biochem J. 2005;389(Pt 1): 127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Heidenreich O, Neininger A, Schratt G, et al. MAPKAP kinase 2 phosphorylates serum response factor in vitro and in vivo. J Biol Chem. 1999;274(20):14434–14443. [DOI] [PubMed] [Google Scholar]
- 89.Shrestha A, Bruckmueller H, Kildalsen H, et al. Phosphorylation of steroid receptor coactivator-3 (SRC-3) at serine 857 is regulated by the p38(MAPK)-MK2 axis and affects NF-kappaB-mediated transcription. Sci Rep. 2020;10(1):11388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ronkina N, Lafera J, Kotlyarov A, Gaestel M. Stress-dependent phosphorylation of myocardin-related transcription factor A (MRTF-A) by the p38(MAPK)/MK2 axis. Sci Rep. 2016;6:31219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tan Y, Rouse J, Zhang A, Cariati S, Cohen P, Comb MJ. FGF and stress regulate CREB and ATF-1 via a pathway involving p38 MAP kinase and MAPKAP kinase-2. EMBO J. 1996;15(17): 4629–4642. [PMC free article] [PubMed] [Google Scholar]
- 92.Iordanov M, Bender K, Ade T, et al. CREB is activated by UVC through a p38/HOG-1-dependent protein kinase. EMBO J. 1997; 16(5):1009–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Faust D, Schmitt C, Oesch F, et al. Differential p38-dependent signalling in response to cellular stress and mitogenic stimulation in fibroblasts. Cell Commun Signal. 2012;10:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang X, Khaleque MA, Zhao MJ, Zhong R, Gaestel M, Calderwood SK. Phosphorylation of HSF1 by MAPK-activated protein kinase 2 on serine 121, inhibits transcriptional activity and promotes HSP90 binding. J Biol Chem. 2006;281(2):782–791. [DOI] [PubMed] [Google Scholar]
- 95.Werz O, Klemm J, Samuelsson B, Radmark O. 5-lipoxygenase is phosphorylated by p38 kinase-dependent MAPKAP kinases. Proc Natl Acad Sci USA. 2000;97(10):5261–5266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Flamand N, Luo M, Peters-Golden M, Brock TG. Phosphorylation of serine 271 on 5-lipoxygenase and its role in nuclear export. J Biol Chem. 2009;284(1):306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.MacKenzie KF, Wallace DA, Hill EV, et al. Phosphorylation of cAMP-specific PDE4A5 (phosphodiesterase-4A5) by MK2 (MAP-KAPK2) attenuates its activation through protein kinase A phosphorylation. Biochem J. 2011;435(3):755–769. [DOI] [PubMed] [Google Scholar]
- 98.Houslay KF, Christian F, MacLeod R, Adams DR, Houslay MD, Baillie GS. Identification of a multifunctional docking site on the catalytic unit of phosphodiesterase-4 (PDE4) that is utilised by multiple interaction partners. Biochem J. 2017; 474(4):597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Houslay KF, Fertig BA, Christian F, et al. Phosphorylation of PDE4A5 by MAPKAPK2 attenuates fibrin degradation via p75 signalling. J Biochem. 2019;166(1):97–106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 100.Zaru R, Ronkina N, Gaestel M, Arthur JS, Watts C. The MAPK-activated kinase Rsk controls an acute Toll-like receptor signaling response in dendritic cells and is activated through two distinct pathways. Nat Immunol. 2007;8(11):1227–1235. [DOI] [PubMed] [Google Scholar]
- 101.Mendoza H, Campbell DG, Burness K, et al. Roles for TAB1 in regulating the IL-1-dependent phosphorylation of the TAB3 regulatory subunit and activity of the TAK1 complex. Biochem J. 2008; 409(3):711–722. [DOI] [PubMed] [Google Scholar]
- 102.Jaco I, Annibaldi A, Lalaoui N, et al. MK2 Phosphorylates RIPK1 to Prevent TNF-Induced Cell Death. Mol Cell. 2017;66(5): 698–710 e695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mahtani KR, Brook M, Dean JL, Sully G, Saklatvala J, Clark AR. Mitogen-activated protein kinase p38 controls the expression and posttranslational modification of tristetraprolin, a regulator of tumor necrosis factor alpha mRNA stability. Mol Cell Biol. 2001; 21(19):6461–6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ronkina N, Shushakova N, Tiedje C, et al. The role of TTP phosphorylation in the regulation of inflammatory cytokine production by MK2/3. J Immunol. 2019;203(8):2291–2300. [DOI] [PubMed] [Google Scholar]
- 105.Rousseau S, Morrice N, Peggie M, Campbell DG, Gaestel M, Cohen P. Inhibition of SAPK2a/p38 prevents hnRNP A0 phosphorylation by MAPKAP-K2 and its interaction with cytokine mRNAs. EMBO J. 2002;21(23):6505–6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Borisova ME, Voigt A, Tollenaere MAX, et al. p38-MK2 signaling axis regulates RNA metabolism after UV-light-induced DNA damage. Nat Commun. 2018;9(1):1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tiedje C, Lubas M, Tehrani M, et al. p38MAPK/MK2-mediated phosphorylation of RBM7 regulates the human nuclear exosome targeting complex. RNA. 2015;21(2):262–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bollig F, Winzen R, Gaestel M, Kostka S, Resch K, Holtmann H. Affinity purification of ARE-binding proteins identifies polyA-binding protein 1 as a potential substrate in MK2-induced mRNA stabilization. Biochem Biophys Res Commun. 2003;301(3):665–670. [DOI] [PubMed] [Google Scholar]
- 109.Williams PA, Krug MS, McMillan EA, et al. Phosphorylation of the RNA-binding protein Dazl by MAPKAP kinase 2 regulates spermatogenesis. Mol Biol Cell. 2016;27(15):2341–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Powell DW, Rane MJ, Joughin BA, et al. Proteomic identification of 14-3-3zeta as a mitogen-activated protein kinase-activated protein kinase 2 substrate: role in dimer formation and ligand binding. Mol Cell Biol. 2003;23(15):5376–5387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Duan TL, Jiao H, He GJ, Yan YB. Translation efficiency and degradation of ER-associated mRNAs modulated by ER-anchored poly(A)-specific ribonuclease (PARN). Cells. 2020;9(1):162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lemaire M, Froment C, Boutros R, et al. CDC25B phosphorylation by p38 and MK-2. Cell Cycle. 2006;5(15):1649–1653. [DOI] [PubMed] [Google Scholar]
- 113.Wang L, Yang H, Palmbos PL, et al. ATDC/TRIM29 phosphorylation by ATM/MAPKAP kinase 2 mediates radioresistance in pancreatic cancer cells. Cancer Res. 2014;74(6):1778–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Menon MB, Tiedje C, Lafera J, et al. Endoplasmic reticulum-associated ubiquitin-conjugating enzyme Ube2j1 is a novel substrate of MK2 (MAPKAP kinase-2) involved in MK2-mediated TNFalpha production. Biochem J. 2013;456(2):163–172. [DOI] [PubMed] [Google Scholar]
- 115.Tollenaere MAX, Villumsen BH, Blasius M, et al. p38- and MK2-dependent signalling promotes stress-induced centriolar satellite remodelling via 14-3-3-dependent sequestration of CEP131/AZI1. Nat Commun. 2015;6:10075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wei Y, An Z, Zou Z, et al. The stress-responsive kinases MAP-KAP-K2/MAPKAPK3 activate starvation-induced autophagy through Beclin 1 phosphorylation. eLife. 2015;4:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell. 2007;11(2):175–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Donzelli M, Draetta GF. Regulating mammalian checkpoints through Cdc25 inactivation. EMBO Rep. 2003;4(7):671–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Reinhardt HC, Yaffe MB. Phospho-Ser/Thr-binding domains: navigating the cell cycle and DNA damage response. Nat Rev Mol Cell Biol. 2013;14(9):563–580. [DOI] [PubMed] [Google Scholar]
- 120.Sur S, Agrawal DK. Phosphatases and kinases regulating CDC25 activity in the cell cycle: clinical implications of CDC25 overexpression and potential treatment strategies. Mol Cell Biochem. 2016;416(1–2):33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Reinhardt HC, Hasskamp P, Schmedding I, et al. DNA damage activates a spatially distinct late cytoplasmic cell-cycle checkpoint network controlled by MK2-mediated RNA stabilization. Mol Cell. 2010;40(1):34–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sluchanko NN, Gusev NB. Oligomeric structure of 14-3-3 protein: what do we know about monomers? FEBS Lett. 2012;586(24): 4249–4256. [DOI] [PubMed] [Google Scholar]
- 123.Sun L, Stoecklin G, Van Way S, et al. Tristetraprolin (TTP)-14-3-3 complex formation protects TTP from dephosphorylation by protein phosphatase 2a and stabilizes tumor necrosis factor-alpha mRNA. J Biol Chem. 2007;282(6):3766–3777. [DOI] [PubMed] [Google Scholar]
- 124.Liu J, Cao S, Ding G, et al. The role of 14-3-3 proteins in cell signalling pathways and virus infection. J Cell Mol Med. 2021;25(9): 4173–4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gorska MM, Liang Q, Stafford SJ, et al. MK2 controls the level of negative feedback in the NF-kappaB pathway and is essential for vascular permeability and airway inflammation. J Exp Med. 2007; 204(7):1637–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Reinhardt HC, Jiang H, Hemann MT, Yaffe MB. Exploiting synthetic lethal interactions for targeted cancer therapy. Cell Cycle. 2009; 8(19):3112–3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Reinhardt HC, Yaffe MB. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr Opin Cell Biol. 2009;21(2):245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Morandell S, Reinhardt HC, Cannell IG, et al. A reversible gene-targeting strategy identifies synthetic lethal interactions between MK2 and p53 in the DNA damage response in vivo. Cell Rep. 2013; 5(4):868–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dietlein F, Kalb B, Jokic M, et al. A synergistic interaction between Chk1- and MK2 inhibitors in KRAS-mutant cancer. Cell. 2015; 162(1):146–159. [DOI] [PubMed] [Google Scholar]
- 130.Kong YW, Dreaden EC, Morandell S, et al. Enhancing chemotherapy response through augmented synthetic lethality by co-targeting nucleotide excision repair and cell-cycle checkpoints. Nat Commun. 2020;11(1):4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hafner A, Bulyk ML, Jambhekar A, Lahav G. The multiple mechanisms that regulate p53 activity and cell fate. Nat Rev Mol Cell Biol. 2019;20(4):199–210. [DOI] [PubMed] [Google Scholar]
- 132.Masuda Y, Takahashi H, Sato S, et al. TRIM29 regulates the assembly of DNA repair proteins into damaged chromatin. Nat Commun. 2015;6:7299. [DOI] [PubMed] [Google Scholar]
- 133.Norman C, Runswick M, Pollock R, Treisman R. Isolation and properties of cDNA clones encoding SRF, a transcription factor that binds to the c-fos serum response element. Cell. 1988;55(6): 989–1003. [DOI] [PubMed] [Google Scholar]
- 134.Lanahan A, Worley P. Immediate-early genes and synaptic function. Neurobiol Learn Mem. 1998;70(1–2):37–43. [DOI] [PubMed] [Google Scholar]
- 135.Bahrami S, Drablos F. Gene regulation in the immediate-early response process. Adv Biol Regul. 2016;62:37–49. [DOI] [PubMed] [Google Scholar]
- 136.Wang S, You M, Wang C, Zhang Y, Fan C, Yan S. Heat shock pretreatment induced cadmium resistance in the nematode Caenorhabditis elegans is depend on transcription factors DAF-16 and HSF-1. Environ Pollut. 2020;261:114081. [DOI] [PubMed] [Google Scholar]
- 137.Cahill CM, Waterman WR, Xie Y, Auron PE, Calderwood SK. Transcriptional repression of the prointerleukin 1beta gene by heat shock factor 1. J Biol Chem. 1996;271(40):24874–24879. [PubMed] [Google Scholar]
- 138.Xie Y, Zhong R, Chen C, Calderwood SK. Heat shock factor 1 contains two functional domains that mediate transcriptional repression of the c-fos and c-fms genes. J Biol Chem. 2003;278(7): 4687–4698. [DOI] [PubMed] [Google Scholar]
- 139.Gojis O, Rudraraju B, Alifrangis C, Krell J, Libalova P, Palmieri C. The role of steroid receptor coactivator-3 (SRC-3) in human malignant disease. Eur J Surg Oncol. 2010;36(3):224–229. [DOI] [PubMed] [Google Scholar]
- 140.Napetschnig J, Wu H. Molecular basis of NF-kappaB signaling. Annu Rev Biophys. 2013;42:443–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Deak M, Clifton AD, Lucocq LM, Alessi DR. Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J. 1998;17(15):4426–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gerstberger S, Hafner M, Tuschl T. A census of human RNA-binding proteins. Nat Rev Genet. 2014;15(12):829–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.García-Mauriño SM, Rivero-Rodríguez F, Velázquez-Cruz A, et al. RNA binding protein regulation and cross-talk in the control of AU-rich mRNA fate. Front Mol Biosci. 2017;4:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Chrestensen CA, Schroeder MJ, Shabanowitz J, et al. MAPKAP kinase 2 phosphorylates tristetraprolin on in vivo sites including Ser178, a site required for 14-3-3 binding. J Biol Chem. 2004; 279(11):10176–10184. [DOI] [PubMed] [Google Scholar]
- 145.Clement SL, Scheckel C, Stoecklin G, Lykke-Andersen J. Phosphorylation of tristetraprolin by MK2 impairs AU-rich element mRNA decay by preventing deadenylase recruitment. Mol Cell Biol. 2011;31(2):256–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Stoecklin G, Stubbs T, Kedersha N, et al. MK2-induced tristetraprolin: 14-3-3 complexes prevent stress granule association and ARE-mRNA decay. EMBO J. 2004;23(6):1313–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tiedje C, Ronkina N, Tehrani M, et al. The p38/MK2-driven exchange between tristetraprolin and HuR regulates AU-rich element-dependent translation. PLoS Genet. 2012;8(9): e1002977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Tran H, Maurer F, Nagamine Y. Stabilization of urokinase and urokinase receptor mRNAs by HuR is linked to its cytoplasmic accumulation induced by activated mitogen-activated protein kinase-activated protein kinase 2. Mol Cell Biol. 2003;23(20):7177–7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Carballo E, Lai WS, Blackshear PJ. Evidence that tristetraprolin is a physiological regulator of granulocyte-macrophage colony-stimulating factor messenger RNA deadenylation and stability. Blood. 2000;95(6):1891–1899. [PubMed] [Google Scholar]
- 150.Raghavan A, Robison RL, McNabb J, Miller CR, Williams DA, Bohjanen PR. HuA and tristetraprolin are induced following T cell activation and display distinct but overlapping RNA binding specificities. J Biol Chem. 2001;276(51):47958–47965. [DOI] [PubMed] [Google Scholar]
- 151.Phillips K, Kedersha N, Shen L, Blackshear PJ, Anderson P. Arthritis suppressor genes TIA-1 and TTP dampen the expression of tumor necrosis factor alpha, cyclooxygenase 2, and inflammatory arthritis. Proc Natl Acad Sci USA. 2004;101(7):2011–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Fechir M, Linker K, Pautz A, et al. Tristetraprolin regulates the expression of the human inducible nitric-oxide synthase gene. Mol Pharmacol. 2005;67(6):2148–2161. [DOI] [PubMed] [Google Scholar]
- 153.Carballo E, Gilkeson GS, Blackshear PJ. Bone marrow transplantation reproduces the tristetraprolin-deficiency syndrome in recombination activating gene-2 (−/−) mice. Evidence that monocyte/macrophage progenitors may be responsible for TNFalpha over-production. J Clin Invest. 1997;100(5):986–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Taylor GA, Carballo E, Lee DM, et al. A pathogenetic role for TNF alpha in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity. 1996;4(5): 445–454. [DOI] [PubMed] [Google Scholar]
- 155.Neininger A, Kontoyiannis D, Kotlyarov A, et al. MK2 targets AU-rich elements and regulates biosynthesis of tumor necrosis factor and interleukin-6 independently at different post-transcriptional levels. J Biol Chem. 2002;277(5):3065–3068. [DOI] [PubMed] [Google Scholar]
- 156.Fowler T, Sen R, Roy AL. Regulation of primary response genes. Mol Cell. 2011;44(3):348–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Doller A, Akool e, Huwiler A, et al. Posttranslational modification of the AU-rich element binding protein HuR by protein kinase Cdelta elicits angiotensin II-induced stabilization and nuclear export of cyclooxygenase 2 mRNA. Mol Cell Biol. 2008;28(8):2608–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Velazquez-Cruz A, Banos-Jaime B, Diaz-Quintana A, De la Rosa MA, Diaz-Moreno I. Post-translational control of RNA-binding proteins and disease-related dysregulation. Front Mol Biosci. 2021;8:658852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Doller A, Pfeilschifter J, Eberhardt W. Signalling pathways regulating nucleo-cytoplasmic shuttling of the mRNA-binding protein HuR. Cell Signal. 2008;20(12):2165–2173. [DOI] [PubMed] [Google Scholar]
- 160.Lafarga V, Cuadrado A, Lopez de Silanes I, Bengoechea R, Fernandez-Capetillo O, Nebreda AR. p38 Mitogen-activated protein kinase- and HuR-dependent stabilization of p21(Cip1) mRNA mediates the G(1)/S checkpoint. Mol Cell Biol. 2009;29(16): 4341–4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lin FY, Chen YH, Lin YW, et al. The role of human antigen R, an RNA-binding protein, in mediating the stabilization of toll-like receptor 4 mRNA induced by endotoxin: a novel mechanism involved in vascular inflammation. Arterioscler Thromb Vasc Biol. 2006; 26(12):2622–2629. [DOI] [PubMed] [Google Scholar]
- 162.Gurgis FM, Yeung YT, Tang MX, et al. The p38-MK2-HuR pathway potentiates EGFRvIII-IL-1beta-driven IL-6 secretion in glioblastoma cells. Oncogene. 2015;34(22):2934–2942. [DOI] [PubMed] [Google Scholar]
- 163.Hannigan MO, Zhan L, Ai Y, Kotlyarov A, Gaestel M, Huang CK. Abnormal migration phenotype of mitogen-activated protein kinase-activated protein kinase 2−/− neutrophils in Zigmond chambers containing formyl-methionyl-leucyl-phenylalanine gradients. J Immunol. 2001;167(7):3953–3961. [DOI] [PubMed] [Google Scholar]
- 164.Doshi BM, Hightower LE, Lee J. HSPB1. actin filament dynamics, and aging cells. Ann N Y Acad Sci. 2010;1197:76–84. [DOI] [PubMed] [Google Scholar]
- 165.Freshney NW, Rawlinson L, Guesdon F, et al. Interleukin-1 activates a novel protein kinase cascade that results in the phosphorylation of Hsp27. Cell. 1994;78(6):1039–1049. [DOI] [PubMed] [Google Scholar]
- 166.Merck KB, Groenen PJ, Voorter CE, et al. Structural and functional similarities of bovine alpha-crystallin and mouse small heat-shock protein. A family of chaperones. J Biol Chem. 1993;268(2): 1046–1052. [PubMed] [Google Scholar]
- 167.Kim KK, Kim R, Kim SH. Crystal structure of a small heat-shock protein. Nature. 1998;394(6693):595–599. [DOI] [PubMed] [Google Scholar]
- 168.van Montfort RL, Basha E, Friedrich KL, Slingsby C, Vierling E. Crystal structure and assembly of a eukaryotic small heat shock protein. Nat Struct Biol. 2001;8(12):1025–1030. [DOI] [PubMed] [Google Scholar]
- 169.Van Montfort R, Slingsby C, Vierling E. Structure and function of the small heat shock protein/alpha-crystallin family of molecular chaperones. Adv Protein Chem. 2001;59:105–156. [DOI] [PubMed] [Google Scholar]
- 170.Nakamoto H, Vigh L. The small heat shock proteins and their clients. Cell Mol Life Sci. 2007;64(3):294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Jakob U, Gaestel M, Engel K, Buchner J. Small heat shock proteins are molecular chaperones. J Biol Chem. 1993;268(3):1517–1520. [PubMed] [Google Scholar]
- 172.Horwitz J. Alpha-crystallin can function as a molecular chaperone. Proc Natl Acad Sci USA. 1992;89(21):10449–10453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lambert H, Charette SJ, Bernier AF, Guimond A, Landry J. HSP27 multimerization mediated by phosphorylation-sensitive inter-molecular interactions at the amino terminus. J Biol Chem. 1999; 274(14):9378–9385. [DOI] [PubMed] [Google Scholar]
- 174.Rogalla T, Ehrnsperger M, Preville X, et al. Regulation of Hsp27 oligomerization, chaperone function, and protective activity against oxidative stress/tumor necrosis factor alpha by phosphorylation. J Biol Chem. 1999;274(27):18947–18956. [DOI] [PubMed] [Google Scholar]
- 175.Arrigo AP, Suhan JP, Welch WJ. Dynamic changes in the structure and intracellular locale of the mammalian low-molecular-weight heat shock protein. Mol Cell Biol. 1988;8(12):5059–5071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Mymrikov EV, Riedl M, Peters C, Weinkauf S, Haslbeck M, Buchner J. Regulation of small heat-shock proteins by hetero-oligomer formation. J Biol Chem. 2020;295(1):158–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Yan LJ, Christians ES, Liu L, Xiao X, Sohal RS, Benjamin IJ. Mouse heat shock transcription factor 1 deficiency alters cardiac redox homeostasis and increases mitochondrial oxidative damage. EMBO J. 2002;21(19):5164–5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Sun Y, MacRae TH. Small heat shock proteins: molecular structure and chaperone function. Cell Mol Life Sci. 2005;62(21):2460–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kamradt MC, Lu M, Werner ME, et al. The small heat shock protein alpha B-crystallin is a novel inhibitor of TRAIL-induced apoptosis that suppresses the activation of caspase-3. J Biol Chem. 2005; 280(12):11059–11066. [DOI] [PubMed] [Google Scholar]
- 180.Doshi BM, Hightower LE, Lee J. The role of Hsp27 and actin in the regulation of movement in human cancer cells responding to heat shock. Cell Stress Chaperones. 2009;14(5):445–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Singh MK, Sharma B, Tiwari PK. The small heat shock protein Hsp27: present understanding and future prospects. J Therm Biol. 2017;69:149–154. [DOI] [PubMed] [Google Scholar]
- 182.Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev. 2004;25(4):581–611. [DOI] [PubMed] [Google Scholar]
- 183.Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Dev Biol. 2004;265(1):23–32. [DOI] [PubMed] [Google Scholar]
- 184.Gamell C, Susperregui AG, Bernard O, Rosa JL, Ventura F. The p38/MK2/Hsp25 pathway is required for BMP-2-induced cell migration. PLoS One. 2011;6(1):e16477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Jongstra-Bilen J, Jongstra J. Leukocyte-specific protein 1 (LSP1): a regulator of leukocyte emigration in inflammation. Immunol Res. 2006;35(1–2):65–74. [DOI] [PubMed] [Google Scholar]
- 186.Huang CK, Zhan L, Ai Y, Jongstra J. LSP1 is the major substrate for mitogen-activated protein kinase-activated protein kinase 2 in human neutrophils. J Biol Chem. 1997;272(1):17–19. [DOI] [PubMed] [Google Scholar]
- 187.Edwards M, Zwolak A, Schafer DA, Sept D, Dominguez R, Cooper JA. Capping protein regulators fine-tune actin assembly dynamics. Nat Rev Mol Cell Biol. 2014;15(10):677–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kondo Y, Jadlowiec CC, Muto A, et al. The Nogo-B-PirB axis controls macrophage-mediated vascular remodeling. PLoS One. 2013;8(11):e81019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Di Lorenzo A, Manes TD, Davalos A, Wright PL, Sessa WC. Endothelial reticulon-4B (Nogo-B) regulates ICAM-1-mediated leukocyte transmigration and acute inflammation. Blood. 2011;117(7): 2284–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Hauge C, Frodin M. RSK and MSK in MAP kinase signalling. J Cell Sci. 2006;119(Pt 15):3021–3023. [DOI] [PubMed] [Google Scholar]
- 191.Schweizer J, Bowden PE, Coulombe PA, et al. New consensus nomenclature for mammalian keratins. J Cell Biol. 2006;174(2): 169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Datta A, Deng S, Gopal V, et al. Cytoskeletal dynamics in epithelial-mesenchymal transition: insights into therapeutic targets for cancer metastasis. Cancers (Basel). 2021;13(8):1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Cheng TJ, Lai YK. Identification of mitogen-activated protein kinase-activated protein kinase-2 as a vimentin kinase activated by okadaic acid in 9L rat brain tumor cells. J Cell Biochem. 1998;71(2): 169–181. [PubMed] [Google Scholar]
- 194.Trulley P, Snieckute G, Bekker-Jensen D, et al. Alternative Translation initiation generates a functionally distinct isoform of the stress-activated protein kinase MK2. Cell Rep. 2019;27(10): 2859–2870 e2856. [DOI] [PubMed] [Google Scholar]
- 195.Chang E, Heo KS, Woo CH, et al. MK2 SUMOylation regulates actin filament remodeling and subsequent migration in endothelial cells by inhibiting MK2 kinase and HSP27 phosphorylation. Blood. 2011;117(8):2527–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Das K, Prasad R, Ansari SA, Roy A, Mukherjee A, Sen P. Matrix metalloproteinase-2: a key regulator in coagulation proteases mediated human breast cancer progression through autocrine signaling. Biomed Pharmacother. 2018;105:395–406. [DOI] [PubMed] [Google Scholar]
- 197.Menon MB, Gaestel M. MK2-TNF-signaling comes full circle. Trends Biochem Sci. 2018;43(3):170–179. [DOI] [PubMed] [Google Scholar]
- 198.Van Quickelberghe E, De Sutter D, van Loo G, Eyckerman S, Gevaert K. A protein-protein interaction map of the TNF-induced NF-kappaB signal transduction pathway. Sci Data. 2018;5:180289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517(7534):311–320. [DOI] [PubMed] [Google Scholar]
- 200.Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008;133(4):693–703. [DOI] [PubMed] [Google Scholar]
- 201.Menon MB, Gropengießer J, Fischer J, et al. p38(MAPK)/MK2-dependent phosphorylation controls cytotoxic RIPK1 signalling in inflammation and infection. Nat Cell Biol. 2017;19(10):1248–1259. [DOI] [PubMed] [Google Scholar]
- 202.Seisenbacher G, Hafen E, Stocker H. MK2-dependent p38b signalling protects drosophila hindgut enterocytes against JNK-induced apoptosis under chronic stress. PLoS Genet. 2011;7(8): e1002168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Damarla M, Parniani AR, Johnston L, et al. Mitogen-activated protein kinase-activated protein kinase 2 mediates apoptosis during lung vascular permeability by regulating movement of cleaved caspase 3. Am J Respir Cell Mol Biol. 2014;50(5):932–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Voronov E, Carmi Y, Apte RN. The role IL-1 in tumor-mediated angiogenesis. Front Physiol. 2014;5:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Jagielska J, Kapopara PR, Salguero G, et al. Interleukin-1 assembles a proangiogenic signaling module consisting of caveolin-1, tumor necrosis factor receptor-associated factor 6, p38-mitogen-activated protein kinase (MAPK), and MAPK-activated protein kinase 2 in endothelial cells. Arterioscler Thromb Vasc Biol. 2012; 32(5):1280–1288. [DOI] [PubMed] [Google Scholar]
- 206.Napp LC, Jabs O, Höckelmann A, et al. Normal endothelial but impaired arterial development in MAP-Kinase activated protein kinase 2 (MK2) deficient mice. Vasc Cell. 2016;8:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Suarez-Lopez L, Sriram G, Kong YW, et al. MK2 contributes to tumor progression by promoting M2 macrophage polarization and tumor angiogenesis. Proc Natl Acad Sci USA. 2018;115(18): E4236–E4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Suarez-Lopez L, Kong YW, Sriram G, et al. MAPKAP kinase-2 drives expression of angiogenic factors by tumor-associated macrophages in a model of inflammation-induced colon cancer. Front Immunol. 2021;11:607891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Flamand N, Mancuso P, Serezani CH, Brock TG. Leukotrienes: mediators that have been typecast as villains. Cell Mol Life Sci. 2007;64(19–20):2657–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Vega-Rubin-de-Celis S, Kinch L, Pena-Llopis S. Regulation of beclin 1-mediated autophagy by oncogenic tyrosine kinases. Int J Mol Sci. 2020;21(23):1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Tasken K, Aandahl EM. Localized effects of cAMP mediated by distinct routes of protein kinase A. Physiol Rev. 2004;84(1): 137–167. [DOI] [PubMed] [Google Scholar]
- 212.Hirata Y, Takahashi M, Morishita T, Noguchi T, Matsuzawa A. Post-translational modifications of the TAK1-TAB complex. Int J Mol Sci. 2017;18(1):205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Ehlting C, Ronkina N, Böhmer O, et al. Distinct functions of the mitogen-activated protein kinase-activated protein (MAPKAP) kinases MK2 and MK3: MK2 mediates lipopolysaccharide-induced signal transducers and activators of transcription 3 (STAT3) activation by preventing negative regulatory effects of MK3. J Biol Chem. 2011;286(27):24113–24124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Ehlting C, Rex J, Albrecht U, et al. Cooperative and distinct functions of MK2 and MK3 in the regulation of the macrophage transcriptional response to lipopolysaccharide. Sci Rep. 2019;9(1): 11021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Johansen C, Funding AT, Otkjaer K, et al. Protein expression of TNF-alpha in psoriatic skin is regulated at a posttranscriptional level by MAPK-activated protein kinase 2. J Immunol. 2006;176(3): 1431–1438. [DOI] [PubMed] [Google Scholar]
- 216.Funding AT, Johansen C, Gaestel M, et al. Reduced oxazolone-induced skin inflammation in MAPKAP kinase 2 knockout mice. J Invest Dermatol. 2009;129(4):891–898. [DOI] [PubMed] [Google Scholar]
- 217.Schottelius AJ, Zügel U, Döcke WD, et al. The role of mitogen-activated protein kinase-activated protein kinase 2 in the p38/TNF-alpha pathway of systemic and cutaneous inflammation. J Invest Dermatol. 2010;130(2):481–491. [DOI] [PubMed] [Google Scholar]
- 218.Li YY, Yuece B, MH C, et al. Inhibition of p38/Mk2 signaling pathway improves the anti-inflammatory effect of WIN55 on mouse experimental colitis. Lab Invest. 2013;93(3):322–333. [DOI] [PubMed] [Google Scholar]
- 219.Zhang T, Jiang J, Liu J, et al. MK2 is required for neutrophil-derived ROS production and inflammatory bowel disease. Front Med (Lausanne). 2020;7:207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Sun L, Wu Q, Nie Y, et al. A Role for MK2 in enhancing neutrophil-derived ROS production and aggravating liver ischemia/reperfusion injury. Front Immunol. 2018;9:2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Culbert AA, Skaper SD, Howlett DR, et al. MAPK-activated protein kinase 2 deficiency in microglia inhibits pro-inflammatory mediator release and resultant neurotoxicity. Relevance to neuroinflammation in a transgenic mouse model of Alzheimer disease. J Biol Chem. 2006;281(33):23658–23667. [DOI] [PubMed] [Google Scholar]
- 222.Clark RA, DellaPelle P, Manseau E, Lanigan JM, Dvorak HF, Colvin RB. Blood vessel fibronectin increases in conjunction with endothelial cell proliferation and capillary ingrowth during wound healing. J Invest Dermatol. 1982;79(5):269–276. [DOI] [PubMed] [Google Scholar]
- 223.Clark RAF. Wound repair: Overview and general considerations. In: Clark RAF, ed. The Molecular, Cellular Biology of Wound Repair. Plenum Press; 1996:3–35. [Google Scholar]
- 224.Gosain A, DiPietro LA. Aging and wound healing. World J Surg. 2004;28(3):321–326. [DOI] [PubMed] [Google Scholar]
- 225.Broughton G 2nd, Janis JE, Attinger CE. The basic science of wound healing. Plast Reconstr Surg. 2006;117(7 Suppl):12S–34S. [DOI] [PubMed] [Google Scholar]
- 226.Campos AC, Groth AK, Branco AB. Assessment and nutritional aspects of wound healing. Curr Opin Clin Nutr Metab Care. 2008; 11(3):281–288. [DOI] [PubMed] [Google Scholar]
- 227.Meszaros AJ, Reichner JS, Albina JE. Macrophage-induced neutrophil apoptosis. J Immunol. 2000;165(1):435–441. [DOI] [PubMed] [Google Scholar]
- 228.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Pardo A, Selman M. Matrix metalloproteases in aberrant fibrotic tissue remodeling. Proc Am Thorac Soc. 2006;3(4):383–388. [DOI] [PubMed] [Google Scholar]
- 230.Sousa AM, Liu T, Guevara O, et al. Smooth muscle alpha-actin expression and myofibroblast differentiation by TGFbeta are dependent upon MK2. J Cell Biochem. 2007;100(6):1581–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Liu T, Warburton RR, Guevara OE, et al. Lack of MK2 inhibits myofibroblast formation and exacerbates pulmonary fibrosis. Am J Respir Cell Mol Biol. 2007;37(5):507–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Thuraisingam T, Xu YZ, Eadie K, et al. MAPKAPK-2 signaling is critical for cutaneous wound healing. J Invest Dermatol. 2010; 130(1):278–286. [DOI] [PubMed] [Google Scholar]
- 233.Kapopara PR, von Felden J, Soehnlein O, et al. Deficiency of MAPK-activated protein kinase 2 (MK2) prevents adverse remodelling and promotes endothelial healing after arterial injury. Thromb Haemost. 2014;112(6):1264–1276. [DOI] [PubMed] [Google Scholar]
- 234.Qeadan F, Bansal P, Hanson JA, Beswick EJ. The MK2 pathway is linked to G-CSF, cytokine production and metastasis in gastric cancer: a novel intercorrelation analysis approach. J Transl Med. 2020;18(1):137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Henriques A, Koliaraki V, Kollias G. Mesenchymal MAPKAPK2/HSP27 drives intestinal carcinogenesis. Proc Natl Acad Sci USA. 2018;115(24):E5546–E5555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Pomerance M, Quillard J, Chantoux F, Young J, Blondeau JP. High-level expression, activation, and subcellular localization of p38-MAP kinase in thyroid neoplasms. J Pathol. 2006;209(3):298–306. [DOI] [PubMed] [Google Scholar]
- 237.Guo M, Sun D, Fan Z, et al. Targeting MK2 Is a novel approach to interfere in multiple myeloma. Front Oncol. 2019;9:722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Phinney BB, Ray AL, Peretti AS, et al. MK2 regulates macrophage chemokine activity and recruitment to promote colon tumor growth. Front Immunol. 2018;9:1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Terzic J, Grivennikov S, Karin E, Karin M. Inflammation and colon cancer. Gastroenterology. 2010;138(6):2101–2114. [DOI] [PubMed] [Google Scholar]
- 240.Taketo MM, Edelmann W. Mouse models of colon cancer. Gastroenterology. 2009;136(3):780–798. [DOI] [PubMed] [Google Scholar]
- 241.Hori M, Nobe H, Horiguchi K, Ozaki H. MCP-1 targeting inhibits muscularis macrophage recruitment and intestinal smooth muscle dysfunction in colonic inflammation. Am J Physiol Cell Physiol. 2008; 294(2):C391–C401. [DOI] [PubMed] [Google Scholar]
- 242.Argyle D, Kitamura T. Targeting macrophage-recruiting chemokines as a novel therapeutic strategy to prevent the progression of solid tumors. Front Immunol. 2018;9:2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Seiwert TY, Wang X, Heitmann J, et al. DNA repair biomarkers XPF and phospho-MAPKAP kinase 2 correlate with clinical outcome in advanced head and neck cancer. PLoS One. 2014;9(7):e102112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Soni S, Saroch MK, Chander B, Tirpude NV, Padwad YS. MAP-KAP-K2 plays a crucial role in the progression of head and neck squamous cell carcinoma by regulating transcript stability. J Exp Clin Cancer Res. 2019;38(1):175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.León X, Bothe C, García J, et al. Expression of IL-1alpha correlates with distant metastasis in patients with head and neck squamous cell carcinoma. Oncotarget. 2015;6(35):37398–37409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Swartz JE, Wegner I, Noorlag R, et al. HIF-1a expression and differential effects on survival in patients with oral cavity, larynx, and oropharynx squamous cell carcinomas. Head Neck. 2021;43(3): 745–756. [DOI] [PubMed] [Google Scholar]
- 247.You GR, Cheng AJ, Lee LY, et al. Prognostic signature associated with radioresistance in head and neck cancer via transcriptomic and bioinformatic analyses. BMC Cancer. 2019;19(1):64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Duffy SA, Taylor JM, Terrell JE, et al. Interleukin-6 predicts recurrence and survival among head and neck cancer patients. Cancer. 2008;113(4):750–757. [DOI] [PubMed] [Google Scholar]
- 249.Yadev NP, Murdoch C, Saville SP, Thornhill MH. Evaluation of tissue engineered models of the oral mucosa to investigate oral candidiasis. Microb Pathog. 2011;50(6):278–285. [DOI] [PubMed] [Google Scholar]
- 250.Yadav A, Kumar B, Teknos TN, Kumar P. Bazedoxifene enhances the anti-tumor effects of cisplatin and radiation treatment by blocking IL-6 signaling in head and neck cancer. Oncotarget. 2017; 8(40):66912–66924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Bae JY, Kim EK, Yang DH, et al. Reciprocal interaction between carcinoma-associated fibroblasts and squamous carcinoma cells through interleukin-1alpha induces cancer progression. Neoplasia. 2014;16(11):928–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Powrozek T, Mlak R, Brzozowska A, Mazurek M, Golebiowski P, Malecka-Massalska T. Relationship between TNF-alpha −1031T/C gene polymorphism, plasma level of TNF-alpha, and risk of cachexia in head and neck cancer patients. J Cancer Res Clin Oncol. 2018;144(8):1423–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Powrozek T, Porgador A, Malecka-Massalska T. Detection, prediction, and prognosis: blood circulating microRNA as novel molecular markers of head and neck cancer patients. Expert Rev Mol Diagn. 2020;20(1):31–39. [DOI] [PubMed] [Google Scholar]
- 254.Zheng M, Jiang YP, Chen W, et al. Snail and Slug collaborate on EMT and tumor metastasis through miR-101-mediated EZH2 axis in oral tongue squamous cell carcinoma. Oncotarget. 2015;6(9): 6797–6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Ronkina N, Kotlyarov A, Dittrich-Breiholz O, et al. The mitogen-activated protein kinase (MAPK)-activated protein kinases MK2 and MK3 cooperate in stimulation of tumor necrosis factor biosynthesis and stabilization of p38 MAPK. Mol Cell Biol. 2007;27(1): 170–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Zhou JP, Gao ZL, Zhou ML, et al. Snail interacts with Id2 in the regulation of TNF-alpha-induced cancer cell invasion and migration in OSCC. Am J Cancer Res. 2015;5(5):1680–1691. [PMC free article] [PubMed] [Google Scholar]
- 257.Lee CH, Chang JS, Syu SH, et al. IL-1beta promotes malignant transformation and tumor aggressiveness in oral cancer. J Cell Physiol. 2015;230(4):875–884. [DOI] [PubMed] [Google Scholar]
- 258.Liu S, Shi L, Wang Y, et al. Stabilization of Slug by NF-kappaB is Essential for TNF-alpha-induced migration and epithelial-mesenchymal transition in head and neck squamous cell carcinoma cells. Cell Physiol Biochem. 2018;47(2):567–578. [DOI] [PubMed] [Google Scholar]
- 259.Kumar B, Yadav A, Brown NV, et al. Nuclear PRMT5, cyclin D1 and IL-6 are associated with poor outcome in oropharyngeal squamous cell carcinoma patients and is inversely associated with p16-status. Oncotarget. 2017;8(9):14847–14859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Joseph JP, Harishankar MK, Pillai AA, Devi A. Hypoxia induced EMT: a review on the mechanism of tumor progression and metastasis in OSCC. Oral Oncol. 2018;80:23–32. [DOI] [PubMed] [Google Scholar]
- 261.Mk H, Prince S, Mohan AM, Krishnan KV, Devi A. Association of Notch4 with metastasis in human oral squamous cell carcinoma. Life Sci. 2016;156:38–46. [DOI] [PubMed] [Google Scholar]
- 262.Yadav A, Kumar B, Datta J, Teknos TN, Kumar P. IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol Cancer Res. 2011;9(12):1658–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Pignon JP, le Maitre A, Maillard E, Bourhis J. Group M-, NC Meta-analysis of chemotherapy in head and neck cancer (MACH-NC): an update on 93 randomised trials and 17,346 patients. Radiother Oncol. 2009;92(1):4–14. [DOI] [PubMed] [Google Scholar]
- 264.Alterio D, Marvaso G, Ferrari A, Volpe S, Orecchia R, Jereczek-Fossa BA. Modern radiotherapy for head and neck cancer. Semin Oncol. 2019;46(3):233–245. [DOI] [PubMed] [Google Scholar]
- 265.Principe S, Dikova V, Bagan J. Salivary cytokines in patients with head and neck cancer (HNC) treated with radiotherapy. J Clin Exp Dent. 2019;11(11):e1072–e1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Gan GN, Eagles J, Keysar SB, et al. Hedgehog signaling drives radioresistance and stroma-driven tumor repopulation in head and neck squamous cancers. Cancer Res. 2014;74(23):7024–7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Marcu LG, Bezak E. Radiobiological modeling of interplay between accelerated repopulation and altered fractionation schedules in head and neck cancer. J Med Phys. 2009;34(4):206–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Wilson GD, Thibodeau BJ, Fortier LE, et al. Cancer stem cell signaling during repopulation in head and neck cancer. Stem Cells Int. 2016;2016:1894782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Alspach E, Flanagan KC, Luo X, et al. p38MAPK plays a crucial role in stromal-mediated tumorigenesis. Cancer Discov. 2014;4(6): 716–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Gialeli C, Theocharis AD, Karamanos NK. Roles of matrix metallo-proteinases in cancer progression and their pharmacological targeting. FEBS J. 2011;278(1):16–27. [DOI] [PubMed] [Google Scholar]
- 271.Timar J, Kashofer K. Molecular epidemiology and diagnostics of KRAS mutations in human cancer. Cancer Metastasis Rev. 2020; 39(4):1029–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Uprety D, Adjei AA. KRAS: from undruggable to a druggable Cancer Target. Cancer Treat Rev. 2020;89:102070. [DOI] [PubMed] [Google Scholar]
- 273.Kopper F, Binkowski AM, Bierwirth C, Dobbelstein M. The MAPK-activated protein kinase 2 mediates gemcitabine sensitivity in pancreatic cancer cells. Cell Cycle. 2014;13(6):884–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Logsdon CD, Simeone DM, Binkley C, et al. Molecular profiling of pancreatic adenocarcinoma and chronic pancreatitis identifies multiple genes differentially regulated in pancreatic cancer. Cancer Res. 2003;63(10):2649–2657. [PubMed] [Google Scholar]
- 275.Coelho MA, de Carné Trécesson S, Rana S, et al. Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD-L1 mRNA. Immunity. 2017;47(6):1083–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2(1):a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Souza EG, De Lorenzo A, Huguenin G, Oliveira GM, Tibirica E. Impairment of systemic microvascular endothelial and smooth muscle function in individuals with early-onset coronary artery disease: studies with laser speckle contrast imaging. Coron Artery Dis. 2014;25(1):23–28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No datasets were generated or analyzed during the current study.