

Abstract
The cyclic GMP–AMP synthase (cGAS)–stimulator of interferon genes (STING) signaling exert essential regulatory function in microbial-and onco-immunology through the induction of cytokines, primarily type I interferons. Recently, the aberrant and deranged signaling of the cGAS–STING axis is closely implicated in multiple sterile inflammatory diseases, including heart failure, myocardial infarction, cardiac hypertrophy, nonalcoholic fatty liver diseases, aortic aneurysm and dissection, obesity, etc. This is because of the massive loads of damage-associated molecular patterns (mitochondrial DNA, DNA in extracellular vesicles) liberated from recurrent injury to metabolic cellular organelles and tissues, which are sensed by the pathway. Also, the cGAS–STING pathway crosstalk with essential intracellular homeostasis processes like apoptosis, autophagy, and regulate cellular metabolism. Targeting derailed STING signaling has become necessary for chronic inflammatory diseases. Meanwhile, excessive type I interferons signaling impact on cardiovascular and metabolic health remain entirely elusive. In this review, we summarize the intimate connection between the cGAS–STING pathway and cardiovascular and metabolic disorders. We also discuss some potential small molecule inhibitors for the pathway. This review provides insight to stimulate interest in and support future research into understanding this signaling axis in cardiovascular and metabolic tissues and diseases.
KEY WORDS: STING, cGAS, Cardiovascular diseases, Metabolic diseases, Damage-associated molecular patterns, Inflammation, ER stress, Mitochondria
Abbreviations: AA, amino acids; AAD, aortic aneurysm and dissection; AKT, protein kinase B; AMPK, AMP-activated protein kinase; Ang II, angiotensin II; ATP, adenosine triphosphate; CBD, C-binding domain; CDG, c-di-GMP; CDNs, cyclic dinucleotides; cGAMP, 2′,3′-cyclic GMP–AMP; cGAS, cyclic GMP–AMP synthase; CTD, C-terminal domain; CTT, C-terminal tail; CVDs, cardiovascular diseases; Cys, cysteine; DAMPs, danger-associated molecular patterns; DsbA-L, disulfide-bond A oxidoreductase-like protein; dsDNA, double-stranded DNA; ER, endoplasmic reticulum; GTP, guanosine triphosphate; HAQ, R71H-G230A-R293Q; HFD, high-fat diet; hSTING, human stimulator of interferon genes; ICAM-1, intracellular adhesion molecule 1; IFN, interferon; IFNAR, interferon receptors; IFN-I, type 1 interferon; IFNIC, interferon-inducible cells; IKK, IκB kinase; IL, interleukin; IRF3, interferon regulatory factor 3; ISGs, IRF-3-dependent interferon-stimulated genes; LBD, ligand-binding pocket; LPS, lipopolysaccharides; MI, myocardial infarction; MLKL, mixed lineage kinase domain-like protein; MST1, mammalian Ste20-like kinases 1; mtDNA, mitochondrial DNA; mTOR, mammalian target of rapamycin; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; NF-κB, nuclear factor-kappa B; NLRP3, NOD-, LRR- and pyrin domain-containing protein 3; NO2-FA, nitro-fatty acids; NTase, nucleotidyltransferase; PDE3B/4, phosphodiesterase-3B/4; PKA, protein kinase A; Poly: I.C, polyinosinic-polycytidylic acid; PPI, protein–protein interface; ROS, reactive oxygen species; SAVI, STING-associated vasculopathy with onset in infancy; Ser, serine; SNPs, single nucleotide polymorphisms; STIM1, stromal interaction molecule 1; STING, stimulator of interferon genes; TAK1, transforming growth factor β-activated kinase 1; TBK1, TANK-binding kinase 1; TFAM, mitochondrial transcription factor A; TLR, Toll-like receptors; TM, transmembrane; TNFα, tumor necrosis factor-alpha; TRAF6, tumor necrosis factor receptor-associated factor 6; TREX1, three prime repair exonuclease 1; YAP1, Yes-associated protein 1
Graphical abstract
The review summarizes the current impact of hyperactivation of the cGAS–STING signaling, with emphasis on the link with cardiovascular and metabolic diseases and the emerged pathway's inhibitors for therapeutic prospects.
1. Introduction
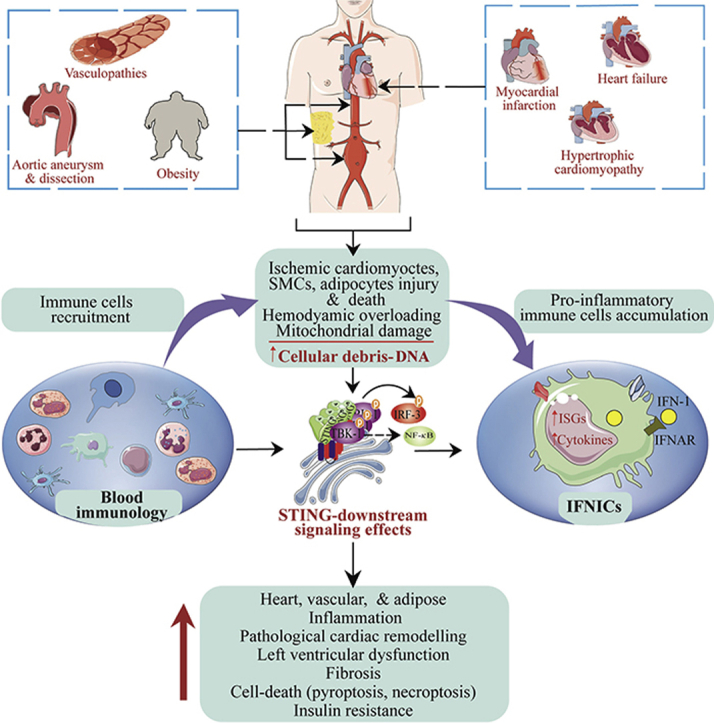
The prevalence and incidence of cardiovascular diseases (CVDs) and metabolic disorders, such as obesity, fatty liver diseases, and diabetes type two continue to accelerate exponentially in industrialized countries with devastating medical and economic consequences. Yearly, about 17.9 million mortalities from CVDs are recorded globally1. Furthermore, the increase in CVDs and metabolic disorders prevalence in developing countries has triggered a global shift towards alternative and complementary strategies, such as the call for reduction/mitigation in host lifestyle activities that impact metabolic dysfunction2. The global rise of these diseases demands thorough cellular understanding of molecular mechanisms that govern pathophysiological conditions. Centuries of research into the pathobiology of CVDs and metabolic diseases have gracefully led to the discoveries of diverse altered physiological processes, transports, and metabolic systems essential to host health. Recurrent sterile inflammation (chronic production of pro-inflammatory cytokines) triggered by metabolic noxiae is a classic example of pathophysiological conditions discovered to occur in patients with heart, vascular, and metabolic diseases3.
The derail and excessive activation of the immune system is a great instigator of chronic inflammation and human disease severity. In view of this, recently, diverse biomedical research findings have suggested varieties of immune cells (mast cells, macrophages, neutrophils, regulatory T cells, etc.) and macromolecules as targets to ameliorate chronic inflammatory diseases, including metabolic disorders and CVDs4, 5, 6. The stimulator of interferon gene (STING) is an innate immune macromolecule that was primarily discovered as a mediator of type I interferon (IFN-I) immune signaling to infections. It is an adaptor resident protein of the endoplasmic reticulum (ER), encoded by the TMEM173 gene and expressed ubiquitously in human tissues, predominantly the heart, spleen, peripheral leukocytes, lungs, and kidney7, 8, 9. Since its discovery, characterization studies have closely linked its baseline activation and hyperactivation to human diseases10, 11, 12, 13. STING is classified as an inflammatory protein that drives chronic systemic inflammation because STING's strongest links between immunity and human diseases sprung from chronic inflammation studies, which has later been confirmed in both de novo and mutational studies14, 15, 16. Moreover, several associative studies have linked the presence of circulating DNA (the precursor macromolecule used in the production of secondary messenger responsible for STING activation) to chronic vascular and metabolic inflammatory diseases17, 18, 19. This emphasizes the importance of STING as a potential chronic inflammatory protein that might be driving recurrent inflammatory disorders, including CVDs and metabolic diseases15,20.
The activation of STING signaling was initially thought to be characterized by the release and the immediate sense of pathogen-associated molecular patterns from microorganisms only. However, as studies on STING expanded, it was identified that nucleic acid pattern recognition receptor (cyclic GMP–AMP synthase, cGAS)21 was the upstream protein partly essential in STING activation. cGAS binds to double-stranded DNA (dsDNA) obtained from exogenous sources (microbial DNA, retroviruses DNA, DNA from dead cells, and extracellular vesicles enclosing DNA from the diverse origin, including but not limited to the tumor) and endogenous sources [chromatin fragments, defective DNA packaged into micronuclei and mitochondrial DNA (mtDNA)]22, 23, 24, 25, 26. After binding to dsDNA, cGAS utilizes adenosine triphosphate (ATP) and guanosine triphosphate (GTP) to catalyze the synthesis of a second messenger cyclic dinucleotide (CDN)–2′,3′-cyclic guanosine monophosphate-adenosine monophosphate (cGAMP). The synthesized cGAMP binds to STING active pocket site to trigger the activation of its downstream signaling22,27. Bacterial-derived CDNs, DNA damage, ER stress, and inherited gain-of-function mutations in the STING-encoding gene can directly induce STING activation independently of cGAMP28, 29, 30, 31. Subsequently, stimulated STING traffics and activates TANK-binding kinase 1 (TBK1) to induce the phosphorylation and nuclear trafficking of IFN regulatory factor 3 (IRF3) and nuclear factor-kappa B (NF-κB)32, 33, 34. IRF3 and NF-κB function to trigger the transcription of IFN-I and other pro-inflammatory cytokines, respectively. Fig. 1 illustrates the above projected molecular pathway and the template of focus for delineating the pathway's role in CVDs and metabolic diseases.
Figure 1.

Mechanism of activation of the cGAS–STING cytosolic DNA sensing pathway. Double-stranded DNA, RNA, and RNA–DNA hybrids from exogenous and endogenous sources are sensed by cGAS. After dsDNA/dsRNA binding on cGAS, cGAS utilizes ATP and GTP to synthesize cGAMP. cGAMP binds to STING at the ER. Upon cGAMP binding, STING traffics from the ER to the Golgi apparatus and finally interact with TBK1 and IRF3 and NF-κB, culminating in IFN-I and pro-inflammatory cytokines production. In addition, aside cGAMP, ER stress can initiate STING activation, leading to IFN-I and pro-inflammatory cytokines production.
Growing evidence demonstrates that the cGAS–STING–TBK1 signaling pathway is closely linked to multiple CVDs and metabolic diseases. Thus, this signaling pathway hyperactivation might be the underlying factor responsible for the recurrent inflammation and other pathologic features observed from CVDs and metabolic diseases. In fact, evidence shows that cGAS is not only localized to the plasma membrane but equally found in the nucleus and cytosol. Studies have also challenged the simplistic notion of the efficient recognition of non-self DNA over self-DNA by demonstrating that the location of cGAS at cytoplasmic and nuclear compartmentalization can efficiently recognize self-DNA if mechanisms keeping its inactive are disrupted, or the quantity of endogenous self-DNA exceeds the tolerated physiological threshold35, 36, 37. This shows that damage-associated molecular patterns (DAMPs), including mtDNA, can activate the cGAS–STING–TBK1 pathway when present abnormally in the cytosol. Indeed, in patients with heart and metabolic diseases, high circulating DAMPs are usually observed and often correlate with chronic inflammation and disease severity38. Consistently, studies have also shown that in myocardial infarction (MI) disease model, the massive ischemic cell death and the influx of cell-death debris by macrophages result in the activation of the cGAS–STING–IRF3 pathway and the subsequent expression of IFN-I. The pathway activation was discovered to contribute to MI-associated inflammation, thus, inflaming MI fatal responses39. Similarly, in heart failure, the cGAS–STING signaling is progressively activated, furthering pathological cardiac remodeling and left ventricular dysfunction40. Still and all, STING activation has been implicated in the furtherance of aortic aneurysm and dissection41, cardiac hypertrophy42, obesity-induced insulin resistance and inflammation43,44, cellular senescence45 and senescence-associated secretory phenotypes46, nonalcoholic steatohepatitis47,48, and vascular dysfunction49 through the direct and indirect recognition of abnormal self-DNA.
Thus, in the present review, we describe the latest basic molecular structure of cGAS and STING proteins and the molecular steps leading to the sensing of self-DNA, synthesis of cGAMP, STING activation, and cytokines production for basic cardiovascular researchers. Also, available evidence on the cGAS–STING signaling in the pathobiology of CVDs and metabolic diseases were highlighted, importantly focusing on the critical areas of the signaling pathway that crosslink with the cardiovascular and metabolic system. We also summarize recent research progress on inhibitor development based on these innate immune proteins. We aim that the review will spark interest and shape the intimate connection between this pathway and CVDs and metabolic diseases.
2. Basic structural features of cGAS and STING
2.1. Structure and molecular regulation of cGAS
2.1.1. Structure and localization
cGAS is a ∼522 amino acid (AA) protein belonging to the nucleotidyltransferase (NTase) family, which was identified as a sensor of dsDNA/DNA hydrides to mediate STING signaling50,51. Usually, it is an inactive protein consisting of a less-conserved N-terminal domain (1–160 AA) and highly conserved (globular) C-terminal NTase and Mab 21 domains (161–522 AA). The globular domain shows structural and sequence homology to the dsRNA sensing enzyme's catalytic domain, oligoadenylate synthase 122. The non-enzymatic N-terminal domain is believed to play a role in the stabilization or autoinhibition of cGAS protein, and in the nucleus, it determines functional prerogative association with centromeric DNA and innate immune activation52. The NTase domain also harbors the tethering surface of cGAS that sustains its reposing state and averts autoreactivity53. The Mab21 domain of cGAS comprises two lobes (C-lobe and N-lobe) separated by a deep cleft. The C-lobe has a canonical NTase fold with two-leaved, highly twisted core β-sheets, and some of the β-sheets harbor catalytic site residues54. On the other hand, the N-lobe is a bundle of four α-helices linked to the C-lobe by a long "spine” α-helix. It also contains two linker α-helices in proximity to the spine α-helix. Usually, these α-helices and the nucleotide-binding loop form the slightly concave molecular surface termed "platform”, which is opposite the active site. The deep groove between the C-lobe and N-lobe denotes the active site of cGAS55. The intriguing protrusion (367–382 AA) slightly shifted to the platform's top-end, represent the "Zn-thumb” loop—a region stabilized by Zn2+ ion. This protrusion has highly conserved histidine and cysteine residues (H367, C373, C374, and C381), responsible for coordinating the Zn2+ ion54. The Zn-thumb dimerization loop augments the formation of liquid-phase condensates crucial for cGAS catalytic activity, sensitivity, and efficiency. Besides Zn2+, other divalent metals such as manganese are believed to act similarly to promote cGAS optimal activation56, 57, 58. cGAS is localized in the nucleus, cytoplasm, and plasma membrane. First, the notion of its predominance in plasma membrane through the interaction between the N-terminal phosphoinositide-binding domain and phosphatidylinositol 4,5-bisphosphate protein reaffirmed the idea of cGAS recognizing only non-self DNA under physiological conditions. Nevertheless, present findings, suggesting cGAS as predominantly nuclear protein, have argued that quality control check mechanisms such as cGAS nuclear tethering (controlled by intact chromatin) operate to allow nuclear cGAS to differentiate self-DNA from foreign DNA within the same localization53. However, the detailed molecular understanding of the nuclear factors responsible for regulating the catalytic activity of cGAS is limited. It shows that identifying these cell-intrinsic mechanisms might pave the path to control cGAS activity. In addition, it has also been clarified that cGAS translocation from the nucleus to the cytoplasm or cGAS within the cytosol can efficiently recognize self-DNA from mitochondrial stress25.
2.1.2. Molecular mechanism of activation and catalytic formation of cGAMP
cGAS has three DNA binding sites (A, B, and C) that interact with the sugar-phosphate backbone of dsDNA/RNA–DNA hydrides in a sequence-independent pattern. After DNA binding, the stabilization of active cGAS–DNA complexes proceeds with higher-order conformational changes (oligomerization, clustering, and condensates) to trigger the catalytic activity of cGAS fully56,57. These higher-order conformational changes are distinct roles mediated by the DNA binding sites55. For instance, DNA-binding site C was discovered to be likely involved in enhancing multivalence-induced liquid-phase condensation of cGAS58. However, not all DNA lengths after binding to the DNA-binding sites of cGAS can trigger cGAS conformational changes. This suggests a DNA length-dependent mechanism for cGAS activation. Long DNA has been found to exhibit higher valence for cGAS, thereby promoting cGAS–DNA complexes’ stabilization better than short DNA, which readily binds to cGAS58,59. Accordingly, mitochondrial transcription factor A (TFAM), bacterial protein HU and high-mobility group box I protein are biological macromolecules found to stimulate cGAS sensing to long DNA species via introducing bends and turns in long DNA to nucleate cGAS dimers. This nucleation-cooperativity-based mechanism provides sensitive detection of pathogen DNA and mtDNA60. Once cGAS is active, it stems the generation of cGAMP through catalytically competent and accessible-binding pocket for substrates (ATP and GTP) in a two-step synthesis model: 1) production of linear heterodinucleoside phosphate [pppGp (2′–5′) A] intermediate; 2) Subsequent cyclization of the intermediate for the formation of cGAMP61,62 (Fig. 2).
Figure 2.

Basic features of cGAS structure and ligand binding dynamics. (A) Schematic diagram of human cGAS functional domains; (B) Left, the crystal structure of ligand-free apo human-cGAS (“side view,” PDB ID: 4MKP). Right, the crystal structure of ligand-free human-cGAS dimer (“upright view,” PDB ID: 4LEY); (C) Globular catalytic domain of human cGAS in complex with dsDNA, ATP, and GTP (PDB ID: 4KB6). PDB: Protein data bank; aa: amino acids.
2.1.3. Non-catalytic function, regulation, and posttranslational modifications of cGAS
Besides the catalytic function of cGAS for STING signaling, mounting evidence reveals additional cGAS-dependent functions. For instance, nuclear cGAS represses homologous recombination-mediated repair and promotes tumor growth63. More so, cGAS exhibited promising activity as a regulator of energy metabolism64 and a promoter of mitotic cell death65. Interestingly, cGAS-dependent sensing of cytoplasmic chromatin and micronuclei surveillance have been closely linked to inflammation and cellular senescence66,67. Its correlation to senescence projects cGAS as a prospective target for alleviating cellular senescence-associated human diseases, including CVDs, but a complementary role for antitumor immunity68. Moreover, multiple studies have also highlighted cellular posttranslational modifications mechanisms (phosphorylation, acetylation, ubiquitination, etc.) that could modulate cGAS activity and stability69, 70, 71. For instance, protein kinase B (AKT) phosphorylates Ser305 residue of the enzymatic domain of cGAS to negate cGAS catalytic activity72. Furthermore, similarly, cGAS acetylation at either Lys414 or Lys394/384 impedes cGAS activity73. In addition, transcription factor Sp1 and cAMP response element-binding protein, CREB, positively regulate and maintain cGAS transcription activity74.
To the end, cGAS is a vital protein, and its DNA responder role for STING signaling has recently attracted much attention. The current understanding of the biology of cGAS shows that cGAS could sense self-DNA that enters the cytosol from dying cells and dysfunctional organelle, particularly mitochondria. The excessive engagement of the DNA-sensing cGAS–STING signaling can be deleterious. Notably, in relevance to CVDs and metabolic disorders, chronic inflammation contributes to disease progression, and mitochondrial stress due to disoriented metabolic events promote the disorganization of mitochondrial genome75. This offers an avenue for targeting cGAS for therapeutic intervention in CVDs and metabolic diseases. However, aside from the DNA-sensing pathway, additional nodes of biological actions have emerged for cGAS on the cardiovascular and metabolic systems. For instance, recently, cGAS protected hepatocytes from ischemia–reperfusion injury via activating autophagy in STING-independent mechanism in vivo and in vitro76. This presents a platonic thought that several relevant biological roles of cGAS remain untapped regarding cardiovascular and metabolic health.
2.2. Structure and molecular regulation of STING
2.2.1. Structure and localization
STING is a 379 and 378 AA protein in human and murine cells, respectively. Human and murine STING shares 69% of AA residues identity (about 70% homology). The human STING (hSTING) comprises of C-terminal domain (CTD) and transmembrane domain (TM). The TM domain, also known as the N-terminal region, consists of four putative transmembrane domains, namely TM1–4. This N-terminal region is conserved among species with an essential role as ER membrane support77. On the other hand, the CTD comprises of the ligand-binding domain (LBD) or C-binding domain (CBD) and the C-terminal tail (CTT). ER-bound STING is a dimer whose CTD extends into the cytosol and forms a V-shaped ligand-binding pocket for CDNs binding. The CTT (constituting 40 AA) accommodates the most conserved TBK1-binding motifs that adjacently interacts with the pLxIS motif—a motif vital for IRF3 stimulation78,79. Therefore, STING's CTD is generally responsible for binding, protein interaction, and signal transduction80,81. STING is localized exclusively to the ER membrane, and it's inactive when its TM domain (mainly) and CTD (weakly) associates with Ca2+ sensor stromal interaction molecule 1 (STIM1). Also, Toll-interacting protein has been found to be stabilizer of STING, and it retains STING at the ER82. But after CDN binding, the STING–CDN complex traffics from ER via intermediate perinuclear compartments to the Golgi apparatus, and lastly to perinuclear microsomes. Following this translocation is the crucial posttranslational modification step (palmitoylation) necessary for STING downstream signaling activation.
2.2.2. Molecular mechanism of activation of STING downstream signaling
CDNs are the principal binding ligands of STING. Bacterial CDNs were first identified as STING activators before the mammalian endogenous synthesized cGAMP in response to DNA from myriad sources, such as the mitochondria, dead cells, etc30. Besides CDNs, STING is activated non-canonically by DNA damage83. Generally, after CDN binding to the LBD, STING's LBD undergoes a rotational switch to promote STING oligomerization, and the subsequent ER exist. STING-triggered oligomerization also promotes clustering and trans-autophosphorylation of TBK1 macromolecules within proximity. TBK1 is an elongated dimer made up of the kinase domain, ubiquitin-like domain, and scaffold and dimerization domain. Interestingly, it is postulated that TBK1 promotes STING polymerization by phosphorylating STING at Ser358, while palmitoylation at cysteine 88/91 facilitates STING clustering formation at the Golgi apparatus84. Subsequently, active TBK1 dimer binds two CTT (TBK1-binding motif) to phosphorylate LxIS motif residues of adjacent STING dimers85. Phosphorylated 363LxIS366 motif in STING CTT harbors IRF3-binding motif vital for recruiting IRF3 through its conserved positively charged phospho-binding domain. This allows TBK1 to phosphorylate IRF3. Subsequently, IRF3 dissociates and dimerizes to acquire transcriptional activity. IRF3 then traffics into the nucleus to trigger IRF3-dependent IFN-I response32,85,86 (Fig. 3).
Figure 3.

Basic features of STING structure and molecular mechanism of STING activation. (A) Schematic diagram of human STING functional domains; (B) Molecular mechanism model showing the binding and complex formation between cGAMP and human STING crystal structure dimer (PDB ID: 4LOH) (blue and red colors-STING monomers); (C) Full-length apo human STING crystal structure (PDB ID: 6NT5) (red-orange: TM1; golden yellow: TM2; lemon green: TM3; light green: TM4). PDB: protein data bank; CBD: C-binding domain; CTT: C-terminal tail; TM: transmembrane; aa: amino acids.
2.2.3. Isoforms and STING mutants
Studies on STING isoforms and inherited gain-of-function STING mutants have further confirmed the additional functional arm of STING as a driver of systemic inflammation. STING over-activation produces damaging inflammatory responses in patients15,16. Currently, two isoforms (MITA-related protein and STING-β) of hSTING have been identified with differences, and to some extent, similarities in functional domains. These isoforms exert a counteractive biological response to STING-innate signaling81,87. Also, about 20 non-synonymous single nucleotide polymorphisms (SNPs) have been pointed out from the TMEM173 coding sequence. R71H-G230A-R293Q (HAQ), R232H, G230A-R293Q, and R293Q are the most significant hSTING SNPs. The haplotypes HAQ, R232H, G230A-R293Q, and R293Q occur in 20.4%, 13.7%, 5.2%, and 1.5%, respectively, in the humans' population with differences in phylogenetic distribution88. HAQ is a loss-of-function STING haplotype with reduced basal and ligand-triggered STING signaling89. Interestingly, all the above hSTING SNPs show modest to dramatic induction of IFNβ or NF-κB promoters' activation in response to bacterial CDN88. Besides, various mutant STING variants have been identified and associated with STING gain-of-function. These variants include V155 L/M, N154S, V147L, R284 S/G, C206Y, R281Q, G207E, and G166E. These variants trigger uncontrolled STING activation leading to a myriad of pathological disorders termed type one interferonopathies (notably, STING-associated vasculopathy with onset in infancy (SAVI), familial inflammatory diseases, and other autoinflammatory diseases)15,90,91. However, how these gain-of-function STING mutants trigger STING over-activation in the absence of cGAMP remains controversial; researchers have proposed that mutations might disrupt the localization of STING to ER, or presumably due to defect in STING degradation92. Recently, it has been shown that gain-of-function STING mutants residing at CTT affect CTT release, leading to STING polymerization to switch STING into the hyperactive state93. Also, STING gain-of-function mutant N154S has been found to induce chronic ER stress and misfolded protein response via disrupting Ca2+ homeostasis. The authors identified a new STING CTD motif (unfolded protein response-motif) with R331 and R334 residues, which was responsible for modulating the misfolded protein response independently of IFN-I induction to mediate ER stress and cell death94. This observation agrees with other reports that suggest that STING's over-activation-induced deleterious responses, including chronic inflammation, and are independent of IFN-I induction94,95.
2.2.4. Regulation and posttranslational modifications of STING activation
Numerous regulatory mechanisms, including posttranslational modifications strategies, have been shown through various studies as a means to modulate STING-dependent signaling. STING-interacting fragment of STIM1 has been found to repress STING gain-of-function dominant mutants (V147L, N154S, and V155M) deleterious effects96 by restraining STING at ER. Also, inhibition of ADP ribosylation factor GTPases aborts STING trafficking while cytoplasmic coat protein complex II and inactive rhomboid protein two promote STING trafficking after ligand binding97. Mechanistically, inactive rhomboid protein two recruits translocon-associated protein and deubiquitination enzyme EIF3S5 to facilitate the trafficking and stability of STING, respectively98. Moreover, mutation of S366A to alanine blunts DNA-triggered IRF3 activation78,92. STING TM and CTD contain multiple cysteine residues (Cys64, 148, 309, 292, 88, and Cys91) crucial to IFN-I induction. Cys148 stabilizes STING dimer formation of disulfide bridge polymer93 while Cys88 and Cys91 are essential to STING palmitoylation at the Golgi apparatus. Mutation and alkylation of Cys88 and Cys91 render cells handicap to the induction of IFN-I response99. Glutathione peroxidase four inactivation enhances STING carbonylation at Cys88 residue leading to the suppression of STING trafficking to the Golgi complex100. Reactive oxygen species (ROS) oxidation of Cys148 inhibited STING polymerization and activation of downstream signaling, subsequently suppressing IFN-I response101. Also, STING phosphorylation at Ser366 by serine/threonine UNC-51-like kinase promotes STING degradation and negates IRF3 activation102. Likewise, protein phosphatase Mg2+/Mn2+-dependent-1A dephosphorylates STING and TBK1 at Ser358 and Ser172, respectively, to abolish STING aggregation and the subsequent interaction between STING oligomerization and TBK184. Though the molecular mechanism underlying STING degradation has not been clearly explained, the likely mechanism involves proteasomal and lysosomal processes, which negatively regulate STING and its downstream signaling82,103,104.
The role of STING as a driver of chronic systemic inflammation has directly implicated STING as an innate macromolecule involved in common chronic low-grade inflammatory diseases triggered by cytosolic DNA originating endogenously. In most CVDs and metabolic disorders involving recurrent low-grade inflammation and elevated levels of pro-inflammatory cytokines, targeting STING paves the path for ameliorating these pathological features. Also, growing evidence suggests STING as a signaling hub with a lot of intercellular crosslink functions. For instance, STING crosstalk with ER stress, inflammasome induction, lytic cell death signaling, etc. (see next section), providing additional molecular mechanisms by which aberrant STING regulation can elicit unwanted effects to drive CVDs and metabolic diseases progression.
3. STING serves as intracellular damage sensor and regulator of cellular metabolism and inflammation
3.1. Mitochondria DNA modulates the cGAS–STING signaling
Mitochondrion is a dynamic double-membrane organelle with special metabolic functions that include ATP production, intracellular Ca2+ regulation, ROS production and scavenging, metabolite production and metabolic flexibility, regulation of apoptotic cell death, and activation of the caspase family proteases, intracellular components communication, and mitophagy. In cells, mitochondria are the only cellular organelles with their genome (mtDNA). The genome of mitochondria is usually packaged into protein structures called mitochondrial nucleoids, typically regulated by TFAM. Due to its proximity to complexes and the insufficient robust safeguarding mechanisms, mtDNA is at risk of damage caused by ROS generated during respiration and pathological metabolic events. At physiologic level, damaged mitochondria are removed by mitophagy. But interestingly, in most metabolic disorders and CVDs, mitophagy is relatively impaired, the levels of regulators of mtDNA are decreased, and abnormal metabolic processes also impact mitochondria integrity and structure, leading to mitochondrial stress. Therefore, in most cases, for example, in obese, type two diabetic, and CVDs patients, plasma levels of mtDNA fragments and mtDNA damage are profoundly high and positively correlate with disease severity, chronic inflammation, oxidative stress, and insulin resistance105,106.
The biochemical and molecular mechanistic insights on the activation dynamics of cGAS and STING (see previous sections) had promoted understanding of signals involved in the activation of this pathway and how excessive stimulation of the pathway might influence human diseases. This cGAS–STING innate immune pathway is among several evolved pattern recognition receptors (retinoic acid-inducible gene 1, nucleotide-binding oligomerization domain-like receptor, TLR, etc.) vital in host defense against pathogenic invasion by recognizing pathogen-associated molecular patterns. However, the fascinating thing about the cGAS–STING signaling pathway is based on the ability of cGAS to sense cytosolic self-DNA under aberrant pathological conditions107. Notably, mtDNA released into cytosol can engage the cGAS–STING signaling and augments ISGs expression via IFN-I signaling. Interestingly, TFAM, the regulator of mtDNA replication and transcription machinery, has also been found to support cGAS in responding to long DNA by instituting U-turns and bends in DNA to nucleate cGAS dimers.
Moreover, heterozygosity of TFAM disorganizes and enhances mtDNA release into the cytosol, allowing mtDNA to activate the cGAS–STING pathway60. From this, it can be deduced that in CVDs and metabolic diseases, where TFAM levels are generally depleted due to mitochondrial stress in cardiovascular and metabolic cell-types, copious mtDNA release can excessively engage the cGAS–STING25 pathway to aggravate pathophysiological conditions. But the full understanding of how mtDNA leaks from the mitochondria into the cytosol is not fully understood and may differ under diverse pathophysiological conditions. However, available information indicates that BCL-2 associated X protein-induced permeability of the inner mitochondrial membrane and pore formation on the outer mitochondrial membrane, or it's interaction with, and the increased opening of the mitochondrial voltage-dependent anion channel, might be the mechanisms by which mtDNA escape from the mitochondria into the cytosol108,109. Nonetheless, emerging evidence points towards an important role for mitochondria in the regulation of the cGAS–STING pathway. Studies suggest that stimulation of mitochondrial proapoptotic caspases by cytochrome c release negates or suppresses the pathway110,111. This shows that mitochondria is crucial in cGAS–STING signaling and have bidirectional interplay role, but, likely, one will dominate to influence the signaling outcome under different physiological and pathological contexts.
3.2. ER stress and misfolded protein response regulates STING signaling
ER is a critical cellular organelle having essential cellular and metabolic functions, including protein synthesis, secretion and folding, lipid biosynthesis, and Ca2+ storage. ER stress occurs when there is a disruption to protein folding or Ca2+ homeostasis in the ER. This leads to the accumulation of unfolded proteins. The accretion of these misfolded proteins stimulates ER unfolded protein response through the activity of three primary ER-resident protein sensors (protein kinase R-like ER kinase, inositol-requiring enzyme 1α, and activating transcription factor 6). The sustained stimulation of ER stress sensors by intracellular pathologic stimulus can alter lipid enzymes, promote inflammation, apoptosis, etc. Thus, ER stress and unfolded protein response often correlates with diabetes, fatty liver disease, and CVDs progression. Interestingly, the attestation of STING as an ER-resident protein adaptor establishes a plausible relational influence of ER stress on the activation mechanics of STING either directly or indirectly. In fact, STIM1, the ER sensor responsible for regulating STING signaling, plays an essential role in Ca2+ homeostasis in the ER via association with plasma membrane sensor Orail96.
In aortic banding-induced dilated and hypertrophic heart and angiotensin II (Ang-II)-induced cardiomyocytes, ER stress protein sensors expression was sustained, and their activation led to STING–TBK1–NF-κB inflammatory pathway activation, contributing to heart inflammation and fibrosis. It was also observed that negation of STING restrains ER stress sensors and ER stress deleterious effects on heart42. Likewise, CDN-induced STING activation mediated stressed ER membranes, leading to ERphagy (degradation of ER via canonical autophagy pathway)112. In the study by Wu et al.94, it was shown that N145S STING mutant interferes with Ca2+ homeostasis, thereby triggering ER stress and cell death upon T cell receptor activation. Moreover, in other fatty liver and infectious disease models, ER stress induction was incredibly found to activate STING to potentiate inflammatory responses and fibrosis. From these studies, evidence suggests that STING might be sensitive to changes in ER Ca2+ homeostasis or becomes stimulated downstream of ER stress sensor, X-box binding protein-1113, 114, 115. However, the suppression of ER stress sensor (protein kinase R-like ER kinase) has been found to disorient nuclear factor erythroid 2-related factor 2-antioxidant signaling, subsequently impairing mitochondrial homeostasis leading to cytosolic mtDNA elevation and, consequently, STING activation116. The above evidence on ER stress–STING axis shows that ER stress–STING axis is an essential cellular regulatory axis. Although in one hand the activation of ER stress in some settings such as during infections could protect host, in the other hand maintenance of ER homeostasis is vital for cellular metabolic regulation. This shows that in CVDs and metabolic disorders, where ER homeostasis is important, ER stress–STING axis might offer many prospects on providing insight into ER stress-mediated inflammation and cellular metabolic dysregulation.
3.3. STING modulates cellular events, metabolism, and chronic inflammation
The widely known endpoint of STING signaling stimulation is the induction of IFN-I. Besides this mechanism, additional functional studies have reported crucial STING signaling outputs: autophagy, NF-κB pro-inflammatory activation, lytic cell death induction, inflammasome activation, apoptosis, etc. Interestingly, these signaling outputs are dependent or independent of its upstream cGAS and the secondary messenger cGAMP35. Below we discuss evidence that points towards an essential role for cGAS–STING in intracellular homeostasis events, metabolism and chronic inflammation.
3.3.1. STING and inflammation
Chronic activation of STING drives inflammation and contributes directly to common inflammatory diseases. The idea of STING as an inflammatory molecule emerged from studies on STING's gain-of-function mutants and three prime repair exonuclease (TREX1), a gene that encodes DNA exonuclease 1, loss-of-function mutant triggering autoinflammatory diseases. This definitive evidence from these studies undoubtedly projected over-activation of STING as a driver of systemic inflammation. For instance, in Trex1−/− animal models and autoinflammatory patients, it was reaffirmed that the knockout or mutation of Trex1 promotes the accumulation of cytoplasmic DNA. This switches the cGAS–STING pathway into a hyperactive state to continually release pro-inflammatory cytokines and increased expression of IRF3-dependent interferon-stimulated genes (ISG)117,118. Similarly, mice lacking Trex1 genes developed inflammatory myocarditis progressing to circulatory failure119. However, how excessive IFN-I signaling promotes CVDs and metabolic disease progression is unclear (see next section). But the molecular understanding of STING in inflammation aside IFN-I is gradually emerging. STING has been found to induce canonical and non-canonical activation of NF-κB inflammatory signaling. STING activation via upstream cGAS has been shown to stimulate NF-κB and mitogen-activated protein kinase pathway via downstream kinase TBK1, but to a weaker extent than other pattern recognition receptors like TLR signaling120,121. Evidence unfolding also shows that CTT is not involved in the activation of NF-κB, which rules out the involvement of TBK1122. DNA binding protein-IFN inducible 16 plus DNA damage response factors-kinase ataxia telangiectasia mutated and -poly (ADP-ribose) polymerase one could assemble DNA damage-induced STING complex, resulting in the non-canonical mode of STING activation independently of cGAS. This STING activation mechanism predominantly leads to the activation of NF-κB, rather than IRF3, and this was accomplished through tumor suppressor P53 and the E3 ubiquitin ligase tumor necrosis factor receptor-associated factor 6 (TRAF6)83. Furthermore, Balka et al.123 emphatically observed in vitro and in vivo that TBK1 is dispensable for STING downstream activation of NF-κB, and STING downstream kinases [TBK1 and IκB kinase ε (IKKε)] act redundantly to mediate STING-induced NF-κB activation. Intriguingly, the team found transforming growth factor β-activated kinase 1 (TAK1) activated downstream of STING-induced TBK1 and IKKε activity and upstream of IκB kinases (IKKα/IKKβ) activity. TAK1 and IKK complexes, but not TRAF6, are required for canonical NF-κB activation and the production of pro-inflammatory cytokines.
3.3.2. STING and inflammasomes and lytic cell death
NLRP3 (NOD-, LRR- and pyrin domain-containing protein 3) inflammasome is another crucial arm of the innate immune system whose activation mediates pro-inflammatory cytokines secretion and caspase one activation. Evidence shows STING stimulates NLRP3 inflammasome via the efflux of K+ and membrane perturbation. Mechanistically, active STING traffics to lysosomes and induce a lytic form of lysosomal cell death by promoting K+ efflux. This subsequently leads to a decline in cytosolic K+, thereby triggering the classical mode of NLRP3 inflammasome activation, which mediates the release of IL-1β and IL-18 to promote sterile inflammation and pyroptosis124. Moreover, one of the cellular events mediated by STING is the induction of lytic forms of cell death (pyroptosis and necroptosis). Lytic forms of cell death usually provoke inflammation through the uncontrolled release of DAMPs, including extracellular vesicles containing DNA125. At the physiologic level, some ISGs are known to be regulators of lytic cell death, but the sustained expression of these ISGs can also promote lytic cell death necroptosis. Mixed lineage kinase domain-like protein (MLKL) is an ISG whose total levels are crucial checkpoints for necroptosis. When MLKL oligomerizes, it promotes the plasma membrane's rupture, leading to the liberation of mitochondria, ATP, and other DAMPs. Interestingly, it has been found that the inappropriate expression of ISGs via STING signaling triggers MLKL above its critical threshold, leading to MLKL oligomerization following MLKL phosphorylation-induced necroptosis126. Consistently, in SAVI mouse model, lytic forms of cell death contributed to the progression of the inflammatory disease, but surprisingly, it was independent of either IFNα/β receptor signaling or MLKL-dependent necroptotic cell death pathway127. This finding suggests that additional pathways might be involved in STNG-mediated necroptosis.
3.3.3. STING and mTOR
Mammalian target of rapamycin (mTOR) comprises of two complexes, mTOR complex 1 and mTOR complex 2. Both complexes have distinct functions essential to host metabolism, but generally, mTOR network is an integrative rheostat that couples intracellular nutritional status to inflammatory responses. Notably, mTOR complex 1 is a nutrient sensor whose signaling promotes cellular anabolic metabolism to regulate growth and survival, and suppress lipolysis and autophagy. In skeletal muscles, fat, and liver tissues of Trex1−/− background, chronic STING signaling downregulated mTOR complex 1, leading to deregulation of cellular metabolic phenotypes such as adiposity and energy balance128. This biological outcome was equated to STING-induced TBK1 activation independently of IRF3 since Irf3−/− rescued only inflammation but not altered metabolic phenotypes. Consistently, other reports have found that TBK1 activation inhibits mTOR complex 1 activation and signaling129,130. Furthermore, this has been confirmed in vitro in response to pathogen-associated molecules, where TBK1-induced activation phosphorylated a vital component of the mTOR complex 1, Raptor, at Ser877 to limit mTOR complex 1 activity131. However, in another study by Meade et al.132, it was observed that mTOR regulatory circuit prevents STING signaling. The authors identified a poxviruses-encoded protein that can sequester Raptor and Rictor (components of mTOR complex 1 and complex 2, respectively), leading to the disruption of STING activity and the facilitation of cGAS degradation. These findings establish a link between cellular metabolism regulator, mTOR, and STING–TBK1 signaling; however, STING–TBK1-induced activation or suppression of mTOR activity is cell type and context-dependent131. Therefore, future studies in metabolic tissues would be relevant for understanding STING–TBK1–mTOR signaling in CVDs and metabolic disorders.
3.3.4. STING and autophagy and apoptosis
Autophagy/mitophagy are intracellular quality control processes essential for the maintenance of metabolic homeostasis and tissue rejuvenation. Autophagy stimulation destroys damaged organelles, misfolded proteins, intracellular pathogens, etc., while mitophagy (a selective form of autophagy) “clean out” damaged mitochondria. In cardiometabolic disorders, dysregulation of autophagy/mitophagy contributes to the initiation and exacerbation of these disorders. Autophagy has been identified as a conserved function in evolution for hSTING since CTT deletion from hSTING, which cancel-out downstream TBK1/IRF3 involvement, still lead to autophagosome formation97. Interestingly however, this STING-induced autophagy was dependent on Trp–Asp (W–D) repeat domain phosphoinositide-interacting protein two and autophagy-related gene 5, but not the traditional autophagy initiators (UNC-51 like autophagy activating kinase 1, beclin 1). Furthermore, STING-mediated autophagy was found to be vital for the removal of cytosolic DNA after DNA damage. This finding demonstrates autophagy as a distinct role of STING in evolution alongside IFN-I induction. Nonetheless, aside STING-mediated autophagy, STING signaling upstream and downstream molecules and proteins (cGAMP, cGAS, and TBK1) can directly or indirectly activate autophagy/mitophagy. Interestingly, their autophagy/mitophagy mediation has been found to restrain STING signaling and its excessive responses102,133, 134, 135, 136. This indicates negative-feedback control for STING signaling, but how this cross-regulatory function is maintained in an on-going pathological context remains to be determined, particularly in cardiometabolic diseases, where the arm of mitophagy/autophagy is disrupted. In addition to autophagy/mitophagy, STING signaling induces apoptosis (programmed cell death process vital to organismal homeostasis)137, 138, 139. However, studies also suggest that apoptosis induction silence dying cells from triggering immune responses. By virtue of this, researchers suggest that STING signaling is suppressed during apoptosis via proapoptotic caspases stimulation111,140. Yet, the specific understanding of how caspases negatively regulate the cGAS–STING pathway is yet to be fully comprehended, but researchers and available findings suggest that cleavage and inactivation of component(s) of the pathway, caspases-induced degradation of cytosolic DNA (mtDNA) and suppression of gene expressions might be possible ways caspases pull-down the activation of the cGAS–STING pathway110,111,141.
4. STING–IFN-I signaling in cardiometabolic diseases
4.1. Effects of IFN-I and IRF3 reprograming on cardiometabolic diseases
IFN-I are polypeptides usually secreted by infected cells to boost cell-autonomous defense mechanisms in autocrine and paracrine patterns. IFN-I (IFNα, IFNβ, etc.) signals via Janus kinase/signal transducer and activator of transcription 1/2 signaling pathway after stimulating the heterodimeric interferon receptor (IFNAR). This typically leads to the induction of ISGs in both infected and neighboring cells. With respect to this, IFN-I functions to promote antigen presentation, immune cell functions, and chemokine and cytokines production. They can also activate additional pathways to promote effectors of T cell and adaptive immune responses142. Generally, optimal IFN-I induction by the cGAS–STING signaling cascade promotes antiviral and anticancer immunity143.
However, evidence from King et al.39 revealed that aberrant IFN-I production via cGAS–STING–IRF3 pathway and excessive IFN-I signaling have deleterious effect on cardiac healing following permanent MI in mice. The team observed that massive cardiomyocyte death due to ischemic myocardial injury activates the cGAS–STING signaling pathway through neighboring cells regardless of their state, but this only occurred in non-myocytes cell-types. Further single-cell analysis led to the identification of F4/80+ MHC II+CCR2+Ly6C– macrophages population termed interferon-inducible cells (IFNICs). This macrophage phenotype showed enrichment expression of ISGs and was responsible for the heightened cardiac inflammation and dysfunction upon dsDNA sensing following MI. The induction of MI in Irf3−/− and Ifnar–/– mice improved ventricular remodeling, contractile function, mortality, and survival by 98% and 100%, respectively, while in Cgas–/– and Stinggt/gt MI mice, less striking and no effect on cardiac healing, respectively, were observed (Fig. 4). The authors concluded that excessive IFN-I signaling is responsible for post-MI-associated inflammation and negatively affect cardiac healing following MI.
Figure 4.

STING signaling activation mediates heart diseases. In ischemic myocardial infarction, massive ischemic cardiomyocyte death leads to the aberrant liberation of DNA. This DNA is sensed by cGAS in macrophages, leading to the STING–TBK1–IRF3 signaling activation. Activated IRF3 translocates into the nucleus to induce the transcription expression of type one IFNs, causing recurrent expression of ISGs and subsequent recruitment and differentiation of leukocytes. On the other hand, in cardiac hypertrophy, ER stress occurs in cardiomyocytes and subsequently activates STING, leading to TBK1–IRF3 and -NF-κB downstream signaling pathways activation. Active IRF3 and NF-κB then enter the nucleus, where IRF3 function to switch on the expression of type one IFNs, while NF-κB promotes pro-inflammatory cytokines expression. Expressed pro-inflammatory cytokines act to enhance TGF-β1 levels leading to fibrosis.
In similar fate to this observation, Ter Horst et al.144 supported the finding that in rats the exogenous administration of IFN-I worsens cardiac healing following MI by exaggerating left ventricular dilatation and infarct size and promoting pro-inflammatory and alternative macrophage phenotypes in peripheral and myocardium, respectively. In cancer patients, IFN-I therapy was associated with subjects developing severe congestive cardiomyopathy with transient hypotension episodes and tachycardia145,146. In the above cases, the causal–effect relationship was unclear, but therapy discontinuation improved cardiovascular health despite the caveat. Also, for Trex1−/− mice model (a model known to promote aberrant IFN-I signaling via the cGAS–STING pathway), inflammatory myocarditis progressed, leading to dilated cardiomyopathy and circulatory failure119. In patients with insufficient coronary collateral artery growth, IFN-I signaling was enhanced, and suppression of IFN-I signaling promoted arteriogenesis and vascular smooth muscle proliferation in mice147,148. In contrast, Ter Horst et al.144 observed that in MI patients, monocyte-specific upregulation of IFN-I signaling is associated with beneficial cardiac healing post-MI. However, the authors posit that the observed deleterious response from the systemic administration of IFN-I in mice in their study cannot be overlooked and compared with the beneficial effects of monocyte-specific IFN-I signaling in MI patients. Because other contributing responses from non-myeloid cells are likely to be involved in the devastating effects of systemic IFN-I signaling.
Furthermore, IFN regulatory factors (IRF1-9) are family transcription factors responsible for regulating the inducible expressions of IFN. Specifically, IRF3 and IRF7 are involved in the generation of IFN-I through the upstream of DNA/RNA sensing receptors such as cGAS. Several studies have provided information on the individual roles of IRFs under diverse conditions such as infection, genotoxic stress, metabolic stress, etc. For instance, in relevance to cGAS–STING–IFN-I response signaling, IRF9 is known for regulating ISGs expression through Janus kinase/signal transducer and activator of transcription 1/2 pathway149. Moreover, multiple studies have elucidated the role of IRF family in cardiometabolic diseases150, 151, 152, 153. Findings strongly reaffirm the idea that the IRF family reprogram towards signaling pathways that are different from the canonical essential immune regulatory functions in response to hemodynamic overload, ischemia, mechanical injury, metabolic and shear stress150, 151, 152, 153, 154, and this influences the initiation and progression of CVDs and metabolic dysfunction154. In view of this, in 2014, Zhang et al.155 urgently suggested IFN regulatory factors, including IRF3 (stimulated by STING signaling), as therapeutic intervention target for the management of CVDs. Although these studies demonstrating the rewiring of IRF family, including IRF3, in cardiometabolic diseases were investigated independently of the STING signaling, it provides imminent information that excessive STING pathway activation can lead to IRF3 partially departing from its canonical immune signaling, thence impacting cardiovascular and metabolic regulatory functions. Besides, studies discussed in the next section have demonstrated IRF3 reprograming to STING-mediated IRF3 activation.
4.2. The role of the cGAS–STING pathway in cardiovascular and metabolic diseases
4.2.1. STING signaling in the pathophysiology of cardiac diseases
Hemodynamic overload and ischemia are crucial pathophysiological causal factors for most heart diseases, including heart failure and MI, in the elderly. Majority of studies have demonstrated that ischemia and chronic pressure overload usually damage heart tissues (cardiomyocytes, fibroblasts, and endothelial cells) leading to the massive expulsion of DAMPs156. In fact, hemodynamic overload can cause mitochondrial injury157. The extensive release of DAMPs or alarmins, which include self-DNA, mostly signals through pattern recognition receptors to contribute to adverse functional outcomes, disease severity and high mortality rates in individuals. In patients and mice with dilated hypertrophic hearts, STING expression was upregulated and correlated with profound macrophage infiltration, fibrosis, and inflammatory responses. A follow-up experiment using Ang II-treated cardiomyocytes and aortic banding-operated mice disclosed the activation of the STING–TBK1–IRF3 and -NF-κB signaling pathways, which were closely associated with ER stress activation and misfolded protein response. An intriguing finding was that the inhibition of ER stress or knocking out STING remarkably alleviated cardiac hypertrophy by reducing cardiomyocyte surface area, hypertrophic markers (ANP, BNP, and β-MHC), and pro-inflammatory and fibrosis factors (IL-6, IL-1β, MCP-1, TNFα, and IFNα/β, α-SMA, collagen I/III)42 (Fig. 4). The above mechanism of protection strongly links ER stress-STING axis as an essential regulatory axis for cardiac hypertrophy. In addition, in pressure overload-induced cardiac hypertrophy and heart failure, released self-DNA was found to engage the cGAS–STING signaling, contributing to the disease progression. Furthermore, the pathway's inactivation attenuated cardiac hypertrophy, fibrosis, apoptosis, immune inflammatory cells infiltration and cytokines production, and improved survival40.
Moreover, in cardiomyocytes stimulated by lipopolysaccharides (LPS), activated STING–IRF3 signaling was found to drive LPS-induced cardiac dysfunction, inflammation, apoptosis, and pyroptosis via increasing the expression of NLRP3158. In a similar vein, AMP-activated protein kinase (AMPK) and AKT double deletion provoked high-fat diet (HFD)-induced STING signaling activation, culminating in pathological cardiac remodeling and contractile anomalies. Interestingly, inhibition of cGAS or STING rescued HFD-induced cardiomyocyte contractile dysfunction. This suggests that AKT–AMPK signaling's insufficiency accentuates cardiac remodeling and contractile defects via chronic activation of STING signaling159. Furthermore, in myocardial ischemic injury, DNA sensing mediated by cGAS was involved in governing macrophage function during repair of a damaged heart. The pathway suppression safeguarded hearts by improving early survival, promoting angiogenesis, repressing pathological remodeling, and preserving cardiac function. Notably, it was observed, together with emerging finding that inhibiting cGAS accelerated repair by priming macrophages toward reparative subsets and eliminated vital inflammatory mediators (such as inducible nitric oxide synthase)11,39.
4.2.2. STING signaling in the pathophysiology of vascular diseases
Sporadic aortic aneurysm and dissection (AAD) is a lethal vascular condition characterized by continuous aortic smooth muscle cell loss and extracellular matrix degradation. Earlier established risk factors include atherosclerosis, aortic coarctation, high-intensity weight lifting, etc. Aside the progressive damage to the aortic structure, it's also unclear what pathobiological mechanisms are involved in driving aortic degeneration. Induction of sporadic AAD in Stinggt/gt mice showed reduced aortic enlargement and elastic fiber fragmentation together with a lower incidence of AAD in comparison to sporadic AAD in wild-type mice. This demonstrates that Stinggt/gt protects aortic architecture and preserves smooth muscle layer, confirming a positive involvement role for STING in aortic degeneration and sporadic AAD formation. Further, GO enrichment analysis, single-cell transcriptomic analysis of aorta, and in vitro experiments confirmed that STING is involved in multiple aortic smooth muscle cell stress responses (ROS, DNA-damage, inflammation, and cell death). And ROS-mediated DNA damage leads to the progressive release of DNA fragments, which subsequently activates the cGAS–STING–TBK1–IRF3 signaling pathway to promote aortic smooth muscle cell death. Interestingly, the team found that tissue monocytes are mainly responsible for sensing and engulfing dsDNA from damaged aortic smooth muscle cells to activate STING–IRF3 signaling, leading to increased matrix metallopeptidase 9 (a matrixin enzyme involved in extracellular matrix degradation) expression. Indeed, the use of pharmacological inhibitor targeting the blockage of TBK1 phosphorylation at STING's Ser366 rescued sporadic AAD by preserving aortic architecture, improving survival rate, reducing incidence and severity for AAD, etc.41.
DNA damage and extracellular vesicles containing DNA at the vascular site could contribute to the promotion of vascular disorders, particularly atherosclerosis, through calcification, plaque and thrombus formation progression. Indeed, DNA damage is a hallmark feature of many vascular diseases and other genetic vascular degenerative syndromes160. More so, earlier studies have long suggested and described microbial DNA's appearance in human atherosclerotic plaque with unclarified pathophysiological link to the vascular disease development161,162. However, the amount of bacterial DNA at the plaque site correlates with monocyte recruitment and accumulation, supporting the contention that DNA-mediated monocyte recruitment drives both vascular and systemic inflammation163. However, the pathobiological mechanism by which DNA damage and bacterial DNA contribute to atherosclerosis remains elusive. For instance, we do not fully know whether bacterial DNA functions as paracrine messengers to influence vascular dysfunction through stimulating vascular and immune cells, thereby intensifying inflammatory and oxidative responses during atherogenesis. But, if so, it is most likely that the cGAS–STING pathway will be activated at the injury site to expand immune-inflammatory responses. Besides, studies have already reported that invading bacterial pathogens, beneficial microbes harboring creatures, and microbial CDNs can activate STING dependently or independently of cGAS to initiate IRF3/NF-κB signaling17,19,164,165. But overall, to confirm the involvement of DNA-sensing cGAS–STING pathway at the vascular injury site and its role in human atherosclerotic CVDs requires future research.
Vascular endothelial dysfunction correlates with the exacerbation and progression of vascular diseases. Studies from our laboratory and others show that metabolic dysfunction-induced impairment of vascular endothelial cell function is a driver of microvascular and macrovascular complications in patients with type two diabetes166. Vascular endothelial cells strongly express STING, and when stimulated, initiate STING downstream signaling. STING-induced endothelial dysfunction has been observed in SAVI patients. And this led to vasculopathy, which is characterized by chronic inflammation and vaso-occlusive processes, as evidenced by elevated expression of markers of endothelial inflammation (inducible nitric oxide synthase), coagulation factor (tissue factor), endothelial-cell activation and-adhesion molecules [E-selectin and intercellular adhesion molecule (ICAM1)], IFN response genes, and increased endothelial cell death15. Equivalently, thrombotic coagulopathy in COVID-19 patients have been attributed to STING signaling over-activation-induced endothelial dysfunction in these patients167. Besides, prior studies also indicate that elevated IFN-I expression-induces vascular damage and suppresses vascular tissue repair by disrupting endothelial progenitor cells’ proliferation capacity. Thus, contributing to atherosclerosis development in systemic lupus erythematosus patients168.
Moreover, shreds of evidence show that elevated free fatty acids are one essential link between obesity, type two diabetes, and other metabolic disorders. In diabetic patients and obese individuals, the physiologic elevation of free fatty acids and persistent rise in blood sugar signals to impair vascular mitochondria function, consequently contributing to endothelial dysfunction-induced vascular complications. Yuan and colleagues realized that damage to the mitochondria by free fatty acids mediates mtDNA release, which engages the cGAS–STING–IRF3 pathway to repress angiogenesis. Mechanistically, phosphorylated IRF3 binds directly to mammalian Ste20-like kinases 1 (MST1) and ICAM-1 promoter regions, inducing MST1 and ICAM1 expression, respectively. Increased ICAM-1 expression and monocyte–endothelial cell adhesion led to endothelial inflammation43 while MST1 increased expression deregulated the Hippo pathway by mainly inactivating yes-associated protein 1 (YAP1). The cGAS–STING–IRF3-induced suppression of Hippo pathway via MST1 led to inhibition of cell survival, proliferation, and angiogenesis49. Another team also demonstrated that aside free fatty acids, active N-terminal Gasdermin D protein (a pore-forming domain responsible for developing pores via oligomerization) impairs mitochondrial homeostasis by disrupting mtDNA compartmentalization. This leads to mtDNA engaging the cGAS–STING pathway, and the subsequent activation of the pathway inhibits the dephosphorylation and nuclear translocation of the transcriptional co-activators, YAP1 and WW domain-containing transcription regulator 1 (TAZ), by promoting large tumor suppressor kinase-mediated YAP1 phosphorylation and ubiquitin–proteasome mediated degradation, leading to the suppression of cyclin-D-mediated endothelial cell proliferation and vascular repair169 (Fig. 5). In all, these studies have demonstrated how STING signaling might contribute to vascular diseases-mediated by metabolic stress and injury. Nonetheless, evidence also indicates that optimal manipulation of the immune-vascular crosstalk with STING agonists, normalizes tumor vascular microenvironment, again supporting the positive immune role of STING on vascular systems170. Nonetheless, these partial views pave the way for detailed clinical-based studies on stimulation dynamics on STING signaling in human vascular diseases.
Figure 5.

The DNA-sensing cGAS–STING pathway activation-induces endothelial and vascular dysfunction. Metabolic stress-mediated upregulation of free fatty acids and the cytosolic accumulation of LPS triggers mitochondria damage. These actions destabilize mtDNA compartmentalization and promote mtDNA release into endothelial cytosol. In turn, the mtDNA is sensed by cGAS, which subsequently activates the STING–TBK-1 pathway, leading to the phosphorylation of IRF3 and LATS1/2. Active IRF3 translocate to the nucleus to promote transcription expression of ICAM1, MST1, and cytokines, including IFN-I. Expressed MST1 together with activated STING–TBK-1 function to suppress the Hippo pathway by stimulating phosphorylation of LATS1/2. This represses cyclin D-mediated endothelial cell proliferation and vascular repair. In addition, mutations in the STING-encoding gene can directly induce endothelial dysfunction through the progressive stimulation of endothelial activation and adhesion molecules, pro-inflammatory cytokines and factors, endothelial cell-death, etc., to drive pathological vascular conditions in SAVI patients.
4.2.3. STING signaling in the pathophysiology of obesity
Obesity is a metabolic disorder that fosters the occurrence and complication of diverse diseases. Patients with established atherosclerotic cardiovascular events and heart failure often benefit from interventions targeting weight reduction171. Adipose tissue consists of specialized brown and white adipocytes. This tissue is a critical regulator of whole-body energy metabolism172. In obesity, adipocytes exponentially increase in size to expand adipose tissue density. The abnormal expansion of adipose tissue (mainly adipocyte hypertrophy) is the genesis of fibrosis and inflammation observed in metabolic diseases and metabolic dysfunction-induced cardiovascular disorders173.
In HFD-fed obese murine and obese humans, Kumari et al.174 identified IRF3 as a critical early transcription factor whose level is highly expressed in the adipocytes, and was responsible for coordinating inflammation, macrophage infiltration, fat browning, and insulin sensitivity. Consistently, downstream activation of IRF3 via stimulation of TLR4 and TLR3 by LPS and polyinosinic-polycytidylic acid, respectively, led to insulin resistance in adipocytes, and the subsequent ablation of IRF3 reduced obesity-induced inflammation and improves insulin sensitivity. This was partly attributed to white fat's browning, reduced macrophage infiltration into fat pads, and the decreased expression of inflammatory genes.
Recently, Bai's research team44,46 has extensively studied how obesity contributes to the activation of the cGAS–cGAMP–STING pathway to mediate obesity-induced resistance, sterile inflammation, and energy dysregulation. The team found that obesity suppresses adipose tissue-specific over-expression of disulfide-bond A oxidoreductase-like protein (DsbA-L). DsbA-L is a mitochondrial matrix derived protein initially classified as a member of the glutathione transferase family. Later on, DsbA-L has been identified to be localized to the ER with chaperone function in adiponectin multimerization, and also partakes in the folding of essential mitochondrial proteins crucial to mtDNA replication175. Interestingly, the team observed that obesity suppression of DsbA-L destabilizes and promotes mtDNA's robust freeing, and as usual, mtDNA activated the cGAS–STING pathway and promoted the corresponding increased expression of TBK1, NF-κBP65, and IRF3. The elevated levels of TBK1, NF-κBP65, and IRF3 acted to suppress insulin signaling and promote inflammation. Also, phosphorylated TBK1 negatively regulated thermogenesis and energy expenditure via activation of phosphodiesterase-PDE3B/PDE4 to downregulate protein kinase A (PKA) signaling in adipocytes and HFD-fed mice44,64. Meanwhile, the over-expression of DsbA-L protected against mtDNA-induced stimulation of the cGAS–STING signaling pathway and subsequently repressed inflammatory factors through adiponectin-and ER localization-independent mechanism. Likewise, over-expression of DsbA-L restored the downregulated PKA signaling to increase energy expenditure44,64 (Fig. 6).
Figure 6.

Obesity-induced activation of DNA-sensing cGAS–STING signaling pathway mediates inflammation and metabolic abnormalities. The metabolic disorder (obesity) destabilizes mtDNA localization by inhibiting DsbA-L. Decreased DsbA-L promotes mtDNA liberation, which is sensed by cGAS and, in turn, activates the cGAS–STING–TBK-1 pathway. Besides active STING–TBK-1, LPS, and poly: I.C could activate TLR4 and TLR3 receptors to stimulate the phosphorylation of IRF3. IRF3 and NF-κB activation induce the transcription expression of pro-inflammatory cytokines (TNFα, IFNα, MCP-1, IL-18) while the phosphorylation of PDE3B/4 by active TBK-1 inhibits cellular cAMP levels. The increased expressions of pro-inflammatory cytokines contribute to inflammation-mediated metabolic dysfunction leading to insulin resistance. The reduced cAMP levels suppress PKA signaling contributing to the abrogation of thermogenesis (heat production) in adipose tissues.
In HFD-induced obesity, STING downstream kinase TBK1 activation dysregulated energy homeostasis by directly inhibiting the central energy homeostasis regulator AMPK at Thr142 of the AMPKα subunit176. While inhibition of TBK1, IRF3, and IKKε improved glucose tolerance, insulin sensitivity, energy expenditure, and decreased body weight in type two diabetes patients and adipocytes176,177. However, surprisingly, TBK1 shows to have bidirectional activity on inflammatory pathways in adipocytes44,176. This was explained on one hand that adipocyte-specific TBK1 knockout augments NF-κB activation, thereby exaggerating adipose tissue inflammation and insulin resistance. On the other hand, TBK1 activation suppresses inflammation by reducing the activity of atypical NF-κB via TBK1-mediated phosphorylation-dependent degradation of IKK kinase NF-κB-inducing kinase176. Notwithstanding, TBK1 activation could also prevent the cGAS–STING signaling via promoting STING ubiquitination and degradation135. These findings show that STING, importantly, its downstream kinase TBK1, has diverse and mayhap, conflicting roles depending on the metabolic tissue type and its activation (acute or chronic) in metabolic modulation.
Moreover, the STING 293Q allele (HAQ haplotype SNP) impaired STING function to protect against obesity-associated metabolic diseases and CVDs in aged subjects178,179. Likewise, loss of STING or its downstream transcription factor IRF3 prevented HFD-induced adipose inflammation, fibrosis, insulin resistance, and glucose intolerance in mice43,174. Moreover, Sting–/– or Irf3−/− improves free fatty acid-induced inflammation and apoptosis of pancreatic β cells, and even reverses impaired insulin synthesis, and upregulates the phosphoinositide 3–kinase–AKT signaling pathway, leading to insulin secretion in islets of db/db mice180. In all, STING activation induces a constellation of obesity-associated maladies, including insulin resistance, fibrosis, oxidative stress, and energy deregulation, fueling the development of obesity-dependent metabolic diseases and CVDs.
4.2.4. STING signaling in the pathophysiology of nonalcoholic fatty liver and steatohepatitis
The liver plays an essential role in the metabolism of diverse exogenous and endogenous biological substances, including fat, glucose, drugs and dietary products, etc. On that account, injuries to the liver crucially alter metabolic pathways driving the varied risk of developing liver metabolic diseases. Nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH) are the most common metabolic disorders that affect the liver. They are usually defined by progressive coexisting pathologic hallmarks such as hepatic inflammation, dyslipidemia, oxidative stress, ballooning hepatocytes, steatosis, and fibrosis181. Studies have revealed and associated STING signaling as an essential intracellular pathway worsening metabolic dysregulation in patients and mice with NASH and NAFLD.
In hepatic tissues from NAFLD patients, STING signaling was over-activated and was closely associated with severe macrophage-mediated hepatic steatosis, inflammation, and fibrosis. Notably, the team found that macrophages are the primary cellular source of STING in the liver, and STING over-stimulation regulates macrophages-mediated inflammatory responses and polarization, and hepatic steatosis and fibrosis. Interestingly, the systemic deletion of STING or myeloid-cell specific deletion of STING blunted liver fibrosis and inflammation, even in the presence of STING agonist. Also, transfer of bone marrow-derived macrophages from wild-type mice to Stinggt/gt mice caused severe steatosis, inflammation and fibrosis. This confirms that the enhanced metabolic phenotypes and inflammation are due to STING absence in liver-resident macrophages rather than hepatocytes47. Consistently, Qiao et al.182 also concluded that the recurrent activation of the STING–IRF3 signaling fosters NAFLD development by promoting inflammation, apoptosis, and disrupting glucose and lipid metabolism. But, surprisingly, the team observed STING–IRF3 signaling in free fatty acid-induced L-O2 normal human fetal hepatocytes. Also, remdesivir (a broad-spectrum antiviral nucleotide prodrug with anti-inflammatory activity) use demonstrated NAFLD protective activity via repressing STING signaling183.
On the other hand, NASH is characterized by abnormal lipid retention with evidence of inflammation, cellular damage, and varying fibrosis degrees, usually leading to hepatocellular carcinoma and cirrhosis. STING abrogation remarkably mitigated hepatic steatosis, insulin resistance, scarring, and inflammation. However, some studies on NASH have also shown that hepatocytes don't express STING protein. But instead, mtDNA freed from injured hepatocytes into the microenvironment are sensed by kupffer cells, accounting for DNA-sensing and the subsequent deleterious effects. Intriguingly, the team observed that both STING and TLR9 function synergistically to expand mtDNA-induced sterile inflammation in the liver48. The formation and accumulation of insoluble large protein inclusions and ubiquitinated proteins in hepatocytes is a pathologic feature of damaged liver and a marker for distinguishing NAFLD from NASH. Interestingly, the cGAS–STING-induced activation of TBK1 and the subsequent TBK1-mediated phosphorylation of P62/sequestosome one in hepatocytes has been identified as the mechanism responsible for hyper nutrition- and obesity-induced development of severe NASH through the facilitation formation of P62 insoluble inclusions184,185. Also, STING–IRF3 innate immune signaling modulates hepatocytes death in the liver to promote liver fibrosis115.
Collectively, the majority of the studies discussed here have established the pathophysiological implications and characteristics of STING signaling activation in NASH/NAFLD development. These findings suggest that targeting the pathway would help alleviate inflammation, fibrosis, steatosis, insulin resistance, and other impaired liver metabolic processes. Nonetheless, evidence shows that STING suppression is superior to rescuing inflammation and altered metabolic events, whereas Irf3–/–c rescue inflammation from Trex1−/− background128. Notwithstanding, most studies discussed here were mostly murine model-based studies; thus, human-based studies are urgently needed to cement the understanding of STING involvement in metabolic fatty liver diseases. Importantly, the subject of hepatocytes expressing STING remains controversial since some studies reported STING in hepatocytes115,182 while others said otherwise47,76. In part, some of the observation made (lack of STING in human hepatocytes) can explain why hepatitis virus escapes immune detection and often replicate in hepatocytes, usually resulting in liver diseases (cirrhosis and hepatocellular carcinoma) associated with chronic inflammation and altered metabolic dysfunctions (hyperglycemia and insulin resistance)186,187. Future studies employing single cell-transcriptomic analyses and bioinformatics tools would be of practical importance to simultaneously understand STING and STING signaling implications in the capacity of association to liver fatty diseases; thus, in the future, more robust studies would be apt on the subject field.
5. The developments on pharmacological small molecule inhibitors of cGAS and STING
The cGAS–STING pathway has emerged as an important therapeutic target for the induction of cytokines, including IFN-I. Several small-molecule agonists have been developed and are currently pursued in clinical trials (NCT03172936, NCT04144140)188. Despite the optimal beneficial effects of the cGAS–STING pathway in microbial control and tumor diseases, outstanding efforts are underway to develop novel compounds that will antagonize cGAS and STING to control the striking unwanted inflammation and acute tissue damage observed from their chronic stimulations. Currently, JAK inhibitor baricitinib and steroids are used clinically to manage patients presenting with chronic STING activation189. However, reports indicate that chronic STING activation-induced pathological features persist to an extent190. This suggests that novel pharmacological drugs targeting cGAS and STING directly are warranted.
Here, studies discussed have demonstrated the role of excessive cGAS–STING signaling in CVDs and metabolic diseases. The one incredible observation is that suppressing the excessive engagement of the cGAS–STING pathway, or IFN-I signaling, could effectively prevent chronic cardiometabolic inflammation and other pathological features contributing to the progression and development of CVDs and metabolic diseases. Thus, any potential pharmacological antagonist or anti-biologics against the pathway could have prospects in cardiovascular and metabolic conditions. Therefore, in the section, we highlight advances made so far on small molecule inhibitors that target: 1) the catalytic activity of cGAS; 2) competes for DNA-binding on cGAS; 3) directly inhibit or displaces cGAMP from the STING activation pocket; 4) STING palmitoylation (Table 1191, 192, 193, 194, 195, 196, 197, 198, 199, 200, 201).
Table 1.
Overview of cGAS and STING inhibitors.
| Compd. | Molecular mechanism | Ref. |
|---|---|---|
| cGAS inhibitors | ||
1. Antimalarial drugs (AMDS)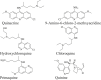
|
Target(s): Murine and human cGAS Pharmacological action: Disrupt cGAS/DNA binding complex interface via interacting with DNA binding sites A and B. |
191 |
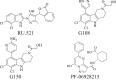
2.
|
Target(s): Murine cGAS for RU. 521, and human and murine cGAS for G108 and G150. Note, PF-06928215 displayed no cellular activity despite cGAS biochemical potency. Pharmacological action: Inhibit ATP and GTP catalytic active sites on cGAS, interfering with cGAMP production. |
192, 193, 194 |
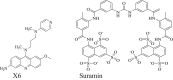
3.
|
Target(s): Human cGAS Pharmacological action: Competitive inhibition of cGAS's DNA binding sites. |
195,196 |
4.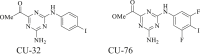 |
Target(s): Human and murine cGAS Pharmacological action: Inhibit the protein-protein interface of cGAS. |
197 |
| STING inhibitors | ||
1. Medicinal-derived compounds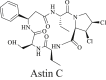
|
Target(s): Human STING Pharmacological action: Binds to the C-terminal activation pocket of STING and competitively displace CDNs to block the recruitment of IRF3 onto the STING signalosome. |
198 |
2. Nitrofuran derivatives
|
Target(s): Murine STING for C-176 and C-178; Human STING and murine STING for C-170, C-171, and H-151 Pharmacological action: Suppress palmitoylation-induce clustering of STING by covalently modifying Cys91 residue. |
199 |
3. Nitro-fatty acids (NO2-FAs) derivatives
|
Target(s): Murine and human STING Pharmacological action: Suppress palmitoylation-induce clustering of STING by covalently modifying Cys88 and Cys91 residues. |
200 |
4. Tetradroisoquinolone acetic acid
|
Target(s): human STING Pharmacological action: Displaces cGAMP from its binding site on STING |
201 |
5.1. Inhibitors of cGAS
cGAS serves as an alarm and an enzyme responsible for catalyzing the generation of second messenger-cGAMP for STING signaling. This makes it a favorable target to ameliorate cGAS-and STING-dependent inflammatory diseases. An in silico-based screening of chemical and drug libraries performed by An's group191, which was focused on murine cGAS–dsDNA crystal structure, led to the identification of antimalarial drugs (including quinacrine, hydroxychloroquine, etc.) that had the potential to suppress cGAS activity. The team found that the antimalarial drugs specifically bind to two drug sites on the 2:2 cGAS/dsDNA dimer, and in each site, the antimalarial drugs fit in the minor furrow of dsDNA between the cGAS/DNA interface (the interface linking dsDNA binding sites A/B on two adjacent monomers). This leads to the antimalarial drugs interacting with DNA binding sites (A and B) on two adjoining cGAS monomers, causing changes in the stability of the cGAS/dsDNA complex and subsequent suppression of activation of cGAS by dsDNA and its enzymatic activity. Follow-up studies show that IFNβ expression, cGAMP production, and some dsDNA-stimulated cytokines (IL-6 and TNFα) were all inhibited in the presence of the antimalarial drugs that were studied. Nonetheless, it is worth noting that different antimalarial drugs exhibited different inhibitory potencies against the above parameters. For instance, 9-amino-6-chloro-2-methoxyacridine was the most potent inhibitor of cGAMP production, followed by primaquine, chloroquine, and hydroxychloroquine, which all exerted intermediate inhibitory activities. Quinine exhibited the least inhibitory activity against cGAMP production.
RU.521 affected cGAS activity by occupying the catalytic site of cGAS and reducing its affinity for ATP and GTP without directly interfering with dsDNA binding. Nonetheless, RU.521 had a vigorous mouse activity but a weak recombinant human cGAS inhibitory activity192. This is probably because only 60% of the amino acids of human and murine cGAS are identical. Thus, treatments focused on human cGAS are indispensable. Correspondingly, a study found that G108 and G150 small molecules could co-crystallize with human cGAS and occupy the ATP- and GTP-binding active site to suppress cGAS activity, similar to RU.521. Moreover, their inhibitory potencies and specificity activities were further confirmed in follow-up experiments in primary human macrophages193.
Furthermore, high-throughput biochemical screening of cGAS inhibitors led to the identification of PF-06928215. Later on, it was noted that PF-06928215 could also bind to cGAS active site with a high-affinity value of 0.2 μmol/L and suppress cGAS activity with inhibitory potency of IC50 value of 4.9 μmol/L. However, PF-06928215 failed to exhibit activity in cellular cGAS assay194. Suramin, oligodeoxynucleotides A151, and X6 prevented cGAS activation in a competitive pattern through dsDNA displacement from cGAS195,196. Recently, reports indicated and demonstrated that CU-32 and CU-76 small molecules could inhibit the protein−protein interface (PPI) of human cGAS in human monocyte THP-1 cells. The PPI interface is required for IRF3 activation and downstream IFN-I induction197. Lastly, the role of posttranslational modification, such as acetylation, ubiquitination, and sumoylation of the cGAS protein, are equally important potential regulatory targets to repress cGAS activation. Thus, therapeutic agents targeting posttranslational modifications of cGAS may be feasible in regulating cGAS activity. For instance, aspirin could robustly enforce the acetylation of cGAS, hence suppressing cGAS activity202. Indeed, aspirin has been suggested for use in COVID-19 (STING disorder) patients to prevent thrombotic coagulopathy observed in these patients167. In all, small molecules and posttranslational modifications aimed at cGAS would be of great value in the modulation of cGAS activity to control over-activation of STING signaling cascade events.
5.2. Inhibitors of STING
STING is the critical signaling molecule for the DNA-sensing cGAS–STING pathway. STING-dependent activation remains crucial in diverse intracellular signaling pathways; therefore, efforts to develop STING inhibitors have been undertaken based on its crystal structure dynamics and regulatory factors, as discussed previously. For example, astin C is a natural cyclopeptide derived from Aster tataricus (a traditional Chinese medicinal plant). It was identified by Li et al.198 as a potent bioactive compound that suppresses the cGAS–STING signaling to mitigate autoinflammatory response in Trex1−/− murine model and macrophage cells. Although the specific mechanism of astin C remains to be resolved, the team found that astin C binds specifically to the C-terminal activation pocket of STING to prevent the recruitment and activation of IRF3 onto the STING signalosome through the displacement of CDN after CDN-induced STING conformational rearrangements. Interestingly, astin C could dose-dependently reduce DNA-induced type IFN-I expression and comparably bind to STING H232 like cGAMP.
STING palmitoylation is one of the critical molecular processes identified to induce STING activation by promoting STING clustering at the Golgi apparatus and subsequent recruitment of STING downstream signaling factors99. 2-Bromopalmitate, a non-selective antagonist of palmitoylation, is the most commonly used compound in experimental studies for in vitro palmitoylation studies. However, Haag et al.199 identified that nitrofuran derivatives (C-176, C-178, C-170, C-171, and H-151) could block palmitoylation-induced clustering of STING by covalently modifying Cys91 residue of STING. Notably, C-176 and C-178 show murine STING activity but not hSTING. However, their derivatives (C-170 and-C-171 and H-151) actively suppressed hSTING palmitoylation, with H-151 being the most robust and profound. Indeed, the abrogation of palmitoylation-induced assembly of STING clusters by the nitrofuran derivatives markedly blunted IFN-I responses, TBK1 phosphorylation, and pro-inflammatory parameters both in Trex1−/− murine model and human cells. Nitro-fatty acids (NO2-FA) are fatty acids formed endogenously, and previous reports classify them as anti-inflammatory lipids mediators203. Similarly, NO2-FAs have been identified to act in the same fate as nitrofuran derivatives to suppress STING signaling. NO2-FAs block STING palmitoylation via S-nitro-alkylation reaction. Specifically, they nitro-alkylate the thiol groups of Cys88 and Cys99 located at the N-terminal region of STING. NO2-FA exerted the above activity in both human and murine cells. Also, NO2-FAs-induced suppression of STING palmitoylation was observed in SAVI patient-derived fibroblasts. Moreover, NO2-FA treatment in Trex1−/− model decreased inflammatory markers in heart tissues as well199,200. Interestingly, a point worth mentioning is that CXA-10 (a safe and well-tolerated NO2-FAs) is currently being investigated in phase II clinical trials for pulmonary arterial hypertension treatment (NCT04125745, NCT04053543, and NCT03449524).
Tetradroisoquinolone acetic acid (compound 18) stabilizes the open, inactive conformation of STING and binds to the cGAMP binding site in a 2:1 ratio to displace cGAMP from its binding site on STING. Compound 18 binds to Thr263 and Thr267 residues, displays slow dissociation kinetics, good oral bioavailability, and was potent with suppressing cGAMP-dependent signaling in vitro201. Besides, butenolide heterodimer 13 has also been identified to exert a potent inhibitory activity on STING pathway, but the exact target remains unclear204. Nonetheless, posttranslational modifications of STNG and downstream kinases are equally plausible and feasible approaches to repress STING activity. For instance, amlexanox is an FDA-approved drug with anti-inflammatory properties and used clinically to manage asthma and recurrent aphthous ulcer205. This drug blocks full activation of STING by inhibiting TBK1-induced phosphorylation of STING at Ser366 because of its high affinity and specificity for TBK1205,206. Interestingly, treatment with amlexanox in sporadic AAD mice and human sporadic ascending thoracic AAD tissues prevented aortic degeneration and AAD development by improving elastic fiber architecture, preserving aortic structure, mitigating rupture, reducing extracellular matrix degradation, suppressing STING and IRF3 phosphorylation, etc.41. Indeed, amlexanox also improved obesity-related metabolic dysfunction in mice206. This shows that other TBK1 inhibitors, such as aminopyrimidines207 and GSK8612208, could suppress STING to improve STING-associated diseases. For the detailed synthesis pathways and the clinical status of cGAS–STING–TBK1 small molecule modulators, check this extensive review209.
6. Discussion: Summary, caveats, outstanding questions, and future directions
Hitherto, the discussed works have provided the first glimpse into the role of the cGAS–STING pathway activation in the development and progression of CVDs and metabolic diseases. The studies have shown that in patients with CVDs and metabolic disorders, such as cardiac hypertrophy, sporadic AAD, NAFLD, etc., STING expression levels are high, and its excessive signaling might contribute to human CVDs and metabolic diseases progression. Under the discussed CVDs and metabolic disorders, cGAS–STING activation principally promotes the following defining characteristic endpoints: chronic sterile inflammation, steatosis, fibrosis, insulin resistance, cardiac remodeling, aortic degeneration, left ventricular dysfunction, coagulation, impaired endothelial function, senescence, and lytic cell death. Since the pathway discovery, STING is recognized as a vital sensor of dsDNA and induces downstream inflammation response. In spite of this, majority of the studies have revealed and affirmed the notion of released DAMPs, including mtDNA, under pathophysiological conditions, such as obesity, fatty liver, pressure overload, ischemia, etc., could switch the cGAS–STING–IRF3 signaling pathway into a continual active state, leading to the above stated pathologic endpoints and subsequently contributing to CVDs and metabolic diseases development and progression41,138,210.
Again, the multifaceted functions of STING have placed this pathway at the epicenter between immunity and metabolism, inflammation, cellular stress responses, and homeostasis processes. Among the discussed mounted evidence, disruption of STING signaling impacts cellular metabolism, ER stress, misfolded protein response, and intracellular homeostasis processes141. The ER is a functionally disparate podium for nutrient sensing and nutrient metabolism, which is critical component in maintaining and restoring metabolic health. Because of this, ER stress and unfolded protein responses usually dysregulate lipid and carbohydrate metabolism, contributing to CVDs and metabolic diseases. Notably, the crosstalk between STING signaling and ER stress has been identified. Studies show that ER stress could activate STING and its downstream signalings, and reciprocal. This provides a critical axis for the appraisal of STING in cellular stress responses. In addition to the ER stress-STING signaling axis, STING signaling is at the crossroad with apoptosis18,110,111,137, autophagy/mitophagy97,134, and central metabolism regulator mTOR complex 1128. These findings add to the growing body of literature showing that STING signaling has evolved to be an important regulator of cellular metabolism that connects inflammation to nutrient metabolism. This provides a powerful scientific breakthrough lens through which the role of STING signaling in cardiometabolic diseases should be investigated.
Therefore, finding out whether the chronic activation of downstream STING signaling, as seen in CVDs and metabolic diseases, would dysregulate anabolic and catabolic metabolism balance to suppress cellular survival in patients with CVDs and metabolic diseases, would offer an additional avenue for understanding the pathological implication of STING signaling in cardiovascular and metabolic health. But before clinical trials commence, several steps need to be taken. First, it would be of interest to evaluate self-DNA sensing via the cGAS–STING pathway in other forms of cardiometabolic sterile tissue injuries and the pathophysiological relevance to the development or progression of the diseases. Second, it would be useful to study the step-by-step mechanisms and STING role in upregulating ER stress, lytic cell death and misfolded protein response pathways in metabolic tissues, samples and cultured systems to assist in the generalizability of the findings.
Generally, autophagy and mitophagy are defective in most CVDs and metabolic diseases211. However, the detailed understanding of how STING signaling regulates autophagy and mitophagy in cells of metabolic and cardiovascular origin (cardiomyocytes, adipocytes, endothelial cells, arterial smooth muscle cells, hepatocytes, etc.) remain unknown entirely. Answering the following questions (What role does STING-induced autophagy or mitophagy play in metabolic disorders and CVDs? To what extent, and in what ways, does acute or chronic stimulation of STING downstream signaling influence the induction of autophagy or mitophagy to hijack STING signaling in metabolic tissues?) may provide useful data sources for reasoning into how the pathway affects these intracellular homeostasis processes to impact CVDs and metabolic diseases.
Notwithstanding, the immune system is vital in cardiovascular and metabolic health. Generally, immunostimulatory pathways’ normal functioning supports cardiovascular and metabolic health, but the dysregulation or uncontrolled engagement of immunostimulatory responses can be detrimental. IFN-I, the primary molecules secreted by the activation of the cGAS–STING pathway, role on CVDs and metabolic dysfunction are primarily understudied and unclear. However, studies show that systemic IFN-I signaling and IFN-I transcriptional factors reprograming are involved in CVDs and metabolic disease progression. In this context, one can say that the chronic stimulation of the cGAS–STING pathway could predict the role and effect of IFN-I signaling on CVDs and metabolic diseases. However, this might not be straightforward; therefore, answers to the following outstanding questions (what relative proportions of IFN-I responses could be deleterious to cardiovascular and metabolic health? What pathways does IFN-I mediate to affect cardiovascular and metabolic health? How do IFN-I crosstalk with other intracellular signaling pathways relevant to cardiovascular and metabolic health? What additional cardiometabolic cell types are involved in IFN-I signaling?) would provide a better understanding of the role of the cardiometabolic–immune axis in cardiometabolic health.
Away from the immunity–metabolic axis, STING proves to be a significant chronic inflammatory molecule. The excessive engagement of STING sparks devastating inflammatory response popularly termed cytokine storm. This response is a life-threatening condition with ravaging heart problems, hypotension, and even organ failure consequences212. Moreover, it is worth mentioning that the still ongoing COVID-19 global pandemic is regarded as a STING disorder because of the observed aberrant activation of STING, with pathological features similar to SAVI. Intriguingly, despite the over-stimulation of STING and its correlation with elevated cytokines signatures (TNFα, IL-6, etc.), disease severity (mainly heart diseases), vaso-occlusive process, and endothelial dysfunction in COVID-19 aged patients, IFN-I responses are impaired, following excessive stimulation of the immune system in end stages of the disease213,214. Studies have also shown that loss-of-function in the enzyme (TREX1) drives the cGAS–STING into a hyperactive state to promote chronic inflammation. Linking STING to autoimmune inflammatory diseases, including systemic lupus erythematosus. Furthermore, deficiency within metabolic quality control mechanisms and mitochondrial stress promote chronic sterile low-grade inflammation via cGAS–STING signaling. In all cases, STING deletion attenuates the chronic inflammatory response134.
However, some concerns, such as how does cardiometabolic-derived dsDNA escape the phagosome or lysosome, and also some cardiometabolic-origin cells expressing the integral protein STING, remain contended. Specifically, STING and its signaling activation in hepatocytes remain a bone of contention. As discussed, studies presented conflicting results, which could not be explained by species variation because different findings report STING in murine and human hepatocytes. The utilization of modern tools such as single-cell RNA sequencing with highly multiplexed genomic and proteomic data could help resolve the discrepancies. It will also be of importance to examine how under cardiometabolic pathophysiological conditions, liberated dsDNA escape degradation. Furthermore, it will be crucial to investigate other dsDNA sensors’ involvement in the heart and metabolic tissues to determine whether specific self-DNA elements drive the cGAS–STING pathway better than different dsDNA sensing pathways. Reliable tools for measuring STING activation-dependent biomarkers such as IFN-I and cGAMP in patients are needed to provide further evidence of its pertinence to chronic inflammatory CVDs and metabolic diseases.
In recent times the apprehending of microbes and their biological components DNA in CVDs and metabolic diseases have rapidly surged again, particularly in the current era of microbiome science215. Microbial DNA is overwhelmingly linked to crucial metabolic phenotypes, particularly platelet reactivity, vascular/metabolic inflammation, glucose and lipid metabolism, adiposity, insulin sensitivity, etc. In reported cases of human atherosclerotic CVDs, high microbial DNA levels are observed at the plaque site without a well-defined pathological link to intracellular signaling mechanism. Meanwhile, microbial DNA from both pathogenic and beneficial microbes harboring within cavities activate STING signaling17, 18, 19. Also, STING activation disrupts the gut intestinal barrier, increases bacteria translocation, and promotes intestinal inflammation216. Is STING signaling involved in the pathological metabolic phenotypes mediated by microbial DNA at the vasculature? One conceivable scenario is that STING disruption of gut intestinal barrier will promote not only translocation of commensal bacteria but also the trafficking of bacteria inflammatory metabolites such as LPS, which signals through TLR to trigger systemic inflammation. In the future, comparing the relative contributions and the synergistic effects of other dsDNA-sensing pathways with the STING signaling could provide thorough insights into the role of dsDNA-sensing signaling cascades in vascular biology and systemic inflammation.
On therapeutics, generally, the interventional role of cGAS and STING on CVDs and metabolic diseases by pharmacological inhibition has not been carried out extensively except by genetic modulation strategies. However, it is becoming clear that targeting the cGAS–STING pathway would be of importance in the fight against common sterile inflammatory diseases such as CVDs and metabolic diseases. Therefore, cGAS and STING pharmacological antagonists might be useful therapeutic tools against CVDs and metabolic diseases. Hopefully, the “STING” in the tail of cardiovascular and metabolic diseases will be solved in the end.
7. Concluding remarks
The cGAS–STING innate immune signaling is rapidly gaining attention as a significant contributor to CVDs and metabolic diseases that usually involve chronic low-grade sterile inflammation. Moreover, the pathway provides the link between immunity and metabolism. Nonetheless, while current findings seem promising, several key issues remain to be determined, including the tissue-specific crosstalk functions of this pathway with metabolic processes, epigenetic effects of this pathway in CVDs and metabolic diseases, STING- and cGAS-dependent functions on the cardiovascular and metabolic system, etc. And, evidence of the activation of STING-induced chronic inflammation in patients with CVDs and metabolic diseases remains insufficient, but findings discussed open an exciting new aisle for further translational studies in CVDs and metabolic diseases.
Acknowledgments
This study was supported by National Key R&D Program of China (2018YFC1704500), the National Program for NSFC (81973624, 81873192, 81830112, China), the Natural Science Foundation of Tianjin City (19JCYBJC28200, China), Important Drug Develop of MOST (2019ZX09201005-002-007, China), and Science and Technology Program of Tianjin (No. 20ZYJDJC00070, China).
Author contributions
All authors have read and approved the final manuscript. Qilong Wang, Mei Du, Patrick Kwabena Oduro, and Xianxian Zheng conceived and designed this review. Patrick Kwabena Oduro and Xianxian Zheng searched, analyzed, and summarized the literature results. Patrick Kwabena Oduro designed all figures. Jinna Wei, Yanze Yang, Yuefei Wang, Han Zhang, Erwei Liu, and Xiumei Gao reviewed and edited this review. Qilong Wang, Mei Du, and Patrick Kwabena Oduro revised this review.
Conflicts of interest
The authors have no conflicts of interest to declare.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Contributor Information
Mei Du, Email: dumei@tmu.edu.cn.
Qilong Wang, Email: wangqilong_00@tjutcm.edu.cn.
References
- 1.Meier T., Gräfe K., Senn F., Sur P., Stangl G.I., Dawczynski C., et al. Cardiovascular mortality attributable to dietary risk factors in 51 countries in the WHO European Region from 1990 to 2016: a systematic analysis of the Global Burden of Disease Study. Eur J Epidemiol. 2019;34:37–55. doi: 10.1007/s10654-018-0473-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kivimäki M., Steptoe A. Effects of stress on the development and progression of cardiovascular disease. Nat Rev Cardiol. 2018;15:215–229. doi: 10.1038/nrcardio.2017.189. [DOI] [PubMed] [Google Scholar]
- 3.Donath M.Y., Meier D.T., Böni-Schnetzler M. Inflammation in the pathophysiology and therapy of cardiometabolic disease. Endocr Rev. 2019;40:1080–1091. doi: 10.1210/er.2019-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chinetti-Gbaguidi G., Colin S., Staels B. Macrophage subsets in atherosclerosis. Nat Rev Cardiol. 2015;12:10–17. doi: 10.1038/nrcardio.2014.173. [DOI] [PubMed] [Google Scholar]
- 5.Shi G.P., Bot I., Kovanen P.T. Mast cells in human and experimental cardiometabolic diseases. Nat Rev Cardiol. 2015;12:643–658. doi: 10.1038/nrcardio.2015.117. [DOI] [PubMed] [Google Scholar]
- 6.Sreejit G., Abdel-Latif A., Athmanathan B., Annabathula R., Dhyani A., Noothi S.K., et al. Neutrophil-derived S100A8/A9 amplify granulopoiesis after myocardial infarction. Circulation. 2020;141:1080–1094. doi: 10.1161/CIRCULATIONAHA.119.043833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhong B., Yang Y., Li S., Wang Y.Y., Li Y., Diao F., et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008;29:538–550. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 8.Ishikawa H., Barber G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun W., Li Y., Chen L., Chen H., You F., Zhou X., et al. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc Natl Acad Sci U S A. 2009;106:8653–8658. doi: 10.1073/pnas.0900850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lemos H., Mohamed E., Huang L., Chandler P.R., Ou R., Pacholczyk R., et al. Stimulator of interferon genes agonists attenuate type I diabetes progression in NOD mice. Immunology. 2019;158:353–361. doi: 10.1111/imm.13122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao D.J., Schiattarella G.G., Villalobos E., Jiang N., May H.I., Li T., et al. Cytosolic DNA sensing promotes macrophage transformation and governs myocardial ischemic injury. Circulation. 2018;137:2613–2634. doi: 10.1161/CIRCULATIONAHA.117.031046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quan Y., Xin Y., Tian G., Zhou J., Liu X. Mitochondrial ROS-modulated mtDNA: a potential target for cardiac aging. Oxid Med Cell Longev. 2020;2020:9423593. doi: 10.1155/2020/9423593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He C., Qin M., Sun X. Highly pathogenic coronaviruses: thrusting vaccine development in the spotlight. Acta Pharm Sin B. 2020;10:1175–1191. doi: 10.1016/j.apsb.2020.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hansen A.L., Mukai K., Schopfer F.J., Taguchi T., Holm C.K. STING palmitoylation as a therapeutic target. Cell Mol Immunol. 2019;16:236–241. doi: 10.1038/s41423-019-0205-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Y., Jesus A.A., Marrero B., Yang D., Ramsey S.E., Sanchez G.A.M., et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. 2014;371:507–518. doi: 10.1056/NEJMoa1312625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jeremiah N., Neven B., Gentili M., Callebaut I., Maschalidi S., Stolzenberg M.C., et al. Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J Clin Invest. 2014;124:5516–5520. doi: 10.1172/JCI79100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andrade W.A., Agarwal S., Mo S., Shaffer S.A., Dillard J.P., Schmidt T., et al. Type I interferon induction by Neisseria gonorrhoeae: dual requirement of cyclic GMP–AMP synthase and toll-like receptor 4. Cell Rep. 2016;15:2438–2448. doi: 10.1016/j.celrep.2016.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petrasek J., Iracheta-Vellve A., Csak T., Satishchandran A., Kodys K., Kurt-Jones E.A., et al. STING–IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc Natl Acad Sci U S A. 2013;110:16544–16549. doi: 10.1073/pnas.1308331110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gutierrez-Merino J., Isla B., Combes T., Martinez-Estrada F., Maluquer De Motes C. Beneficial bacteria activate type-I interferon production via the intracellular cytosolic sensors STING and MAVS. Gut Microb. 2020;11:771–788. doi: 10.1080/19490976.2019.1707015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Munoz J., Rodière M., Jeremiah N., Rieux-Laucat F., Oojageer A., Rice G.I., et al. Stimulator of interferon genes-associated vasculopathy with onset in infancy: a mimic of childhood granulomatosis with polyangiitis. JAMA Dermatol. 2015;151:872–877. doi: 10.1001/jamadermatol.2015.0251. [DOI] [PubMed] [Google Scholar]
- 21.Liang Q., Seo G.J., Choi Y.J., Ge J., Rodgers M.A., Shi M., et al. Autophagy side of MB21D1/cGAS DNA sensor. Autophagy. 2014;10:1146–1147. doi: 10.4161/auto.28769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun L., Wu J., Du F., Chen X., Chen Z.J. Cyclic GMP–AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma Z., Jacobs S.R., West J.A., Stopford C., Zhang Z., Davis Z., et al. Modulation of the cGAS–STING DNA sensing pathway by gammaherpesviruses. Proc Natl Acad Sci U S A. 2015;112:E4306–E4315. doi: 10.1073/pnas.1503831112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barber G.N. STING-dependent cytosolic DNA sensing pathways. Trends Immunol. 2014;35:88–93. doi: 10.1016/j.it.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 25.West A.P., Khoury-Hanold W., Staron M., Tal M.C., Pineda C.M., Lang S.M., et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520:553–557. doi: 10.1038/nature14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Q., Guo N., Zhou Y., Chen J., Wei Q., Han M. The role of tumor-associated macrophages (TAMs) in tumor progression and relevant advance in targeted therapy. Acta Pharm Sin B. 2020;10:2156–2170. doi: 10.1016/j.apsb.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu J., Sun L., Chen X., Du F., Shi H., Chen C., et al. Cyclic GMP–AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339:826–830. doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moretti J., Roy S., Bozec D., Martinez J., Chapman J.R., Ueberheide B., et al. STING senses microbial viability to orchestrate stress-mediated autophagy of the endoplasmic reticulum. Cell. 2017;171 doi: 10.1016/j.cell.2017.09.034. 809-23.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yin Q., Tian Y., Kabaleeswaran V., Jiang X., Tu D., Eck M.J., et al. Cyclic di-GMP sensing via the innate immune signaling protein STING. Mol Cell. 2012;46:735–745. doi: 10.1016/j.molcel.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burdette D.L., Monroe K.M., Sotelo-Troha K., Iwig J.S., Eckert B., Hyodo M., et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature. 2011;478:515–518. doi: 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Woodward J.J., Iavarone A.T., Portnoy D.A. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science. 2010;328:1703–1705. doi: 10.1126/science.1189801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu S., Cai X., Wu J., Cong Q., Chen X., Li T., et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science. 2015;347:aaa2630. doi: 10.1126/science.aaa2630. [DOI] [PubMed] [Google Scholar]
- 33.Abe T., Barber G.N. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-kappaB activation through TBK1. J Virol. 2014;88:5328–5341. doi: 10.1128/JVI.00037-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fitzgerald K.A., McWhirter S.M., Faia K.L., Rowe D.C., Latz E., Golenbock D.T., et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 35.Hopfner K.P., Hornung V. Molecular mechanisms and cellular functions of cGAS–STING signalling. Nat Rev Mol Cell Biol. 2020;21:501–521. doi: 10.1038/s41580-020-0244-x. [DOI] [PubMed] [Google Scholar]
- 36.Roers A., Hiller B., Hornung V. Recognition of endogenous nucleic acids by the innate immune system. Immunity. 2016;44:739–754. doi: 10.1016/j.immuni.2016.04.002. [DOI] [PubMed] [Google Scholar]
- 37.Uggenti C., Lepelley A., Depp M., Badrock A.P., Rodero M.P., El-Daher M.T., et al. cGAS-mediated induction of type I interferon due to inborn errors of histone pre-mRNA processing. Nat Genet. 2020;52:1364–1372. doi: 10.1038/s41588-020-00737-3. [DOI] [PubMed] [Google Scholar]
- 38.Nakahira K., Hisata S., Choi A.M. The roles of mitochondrial damage-associated molecular patterns in diseases. Antioxidants Redox Signal. 2015;23:1329–1350. doi: 10.1089/ars.2015.6407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.King K.R., Aguirre A.D., Ye Y.X., Sun Y., Roh J.D., Ng J.r., RP., et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat Med. 2017;23:1481–1487. doi: 10.1038/nm.4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu D., Cui Y.X., Wu M.Y., Li L., Su L.N., Lian Z., et al. Cytosolic DNA sensor cGAS plays an essential pathogenetic role in pressure overload-induced heart failure. Am J Physiol Heart Circ Physiol. 2020;318:H1525–H1537. doi: 10.1152/ajpheart.00097.2020. [DOI] [PubMed] [Google Scholar]
- 41.Luo W., Wang Y., Zhang L., Ren P., Zhang C., Li Y., et al. Critical role of cytosolic DNA and its sensing adaptor STING in aortic degeneration, dissection, and rupture. Circulation. 2020;141:42–66. doi: 10.1161/CIRCULATIONAHA.119.041460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Y., Chen W., Wang Y. STING is an essential regulator of heart inflammation and fibrosis in mice with pathological cardiac hypertrophy via endoplasmic reticulum (ER) stress. Biomed Pharmacother. 2020;125:110022. doi: 10.1016/j.biopha.2020.110022. [DOI] [PubMed] [Google Scholar]
- 43.Mao Y., Luo W., Zhang L., Wu W., Yuan L., Xu H., et al. STING–IRF3 triggers endothelial inflammation in response to free fatty acid-induced mitochondrial damage in diet-induced obesity. Arterioscler Thromb Vasc Biol. 2017;37:920–929. doi: 10.1161/ATVBAHA.117.309017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bai J., Cervantes C., Liu J., He S., Zhou H., Zhang B., et al. DsbA-L prevents obesity-induced inflammation and insulin resistance by suppressing the mtDNA release-activated cGAS–cGAMP–STING pathway. Proc Natl Acad Sci U S A. 2017;114:12196–12201. doi: 10.1073/pnas.1708744114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lan Y.Y., Heather J.M., Eisenhaure T., Garris C.S., Lieb D., Raychowdhury R., et al. Extranuclear DNA accumulates in aged cells and contributes to senescence and inflammation. Aging Cell. 2019;18 doi: 10.1111/acel.12901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Niu H., Qian L., Sun B., Liu W., Wang F., Wang Q., et al. Inactivation of TFEB and NF-κB by marchantin M alleviates the chemotherapy-driven pro-tumorigenic senescent secretion. Acta Pharm Sin B. 2019;9:923–936. doi: 10.1016/j.apsb.2019.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luo X., Li H., Ma L., Zhou J., Guo X., Woo S.L., et al. Expression of STING ss increased in liver tissues from patients with NAFLD and promotes macrophage-mediated hepatic inflammation and fibrosis in mice. Gastroenterology. 2018;155:1971–19784.e4. doi: 10.1053/j.gastro.2018.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu Y., Liu Y., An W., Song J., Zhang Y., Zhao X. STING-mediated inflammation in kupffer cells contributes to progression of nonalcoholic steatohepatitis. J Clin Invest. 2019;129:546–555. doi: 10.1172/JCI121842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yuan L., Mao Y., Luo W., Wu W., Xu H., Wang X.L., et al. Palmitic acid dysregulates the Hippo–YAP pathway and inhibits angiogenesis by inducing mitochondrial damage and activating the cytosolic DNA sensor cGAS–STING–IRF3 signaling mechanism. J Biol Chem. 2017;292:15002–15015. doi: 10.1074/jbc.M117.804005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gentili M., Lahaye X., Nadalin F., Nader G.P.F., Puig Lombardi E., Herve S., et al. The N-terminal domain of cGAS determines preferential association with centromeric DNA and innate immune activation in the nucleus. Cell Rep. 2019;26 doi: 10.1016/j.celrep.2019.01.105. 2377-93.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barnett K.C., Coronas-Serna J.M., Zhou W., Ernandes M.J., Cao A., Kranzusch P.J., et al. Phosphoinositide interactions position cGAS at the plasma membrane to ensure efficient distinction between self- and viral DNA. Cell. 2019;176 doi: 10.1016/j.cell.2019.01.049. 1432-46.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gentili M., Lahaye X., Nadalin F., Nader G.P.F., Lombardi E.P., Herve S., et al. The N-terminal domain of cGAS determines preferential association with centromeric DNA and innate immune activation in the nucleus. Cell Rep. 2019;26:3798. doi: 10.1016/j.celrep.2019.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Volkman H.E., Cambier S., Gray E.E., Stetson D.B. Tight nuclear tethering of cGAS is essential for preventing autoreactivity. Elife. 2019;8 doi: 10.7554/eLife.47491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Civril F., Deimling T., de Oliveira Mann C.C., Ablasser A., Moldt M., Witte G., et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature. 2013;498:332–337. doi: 10.1038/nature12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li X., Shu C., Yi G., Chaton C.T., Shelton C.L., Diao J., et al. Cyclic GMP–AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity. 2013;39:1019–1031. doi: 10.1016/j.immuni.2013.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hooy R.M., Massaccesi G., Rousseau K.E., Chattergoon M.A., Sohn J. Allosteric coupling between Mn2+ and dsDNA controls the catalytic efficiency and fidelity of cGAS. Nucleic Acids Res. 2020;48:4435–4447. doi: 10.1093/nar/gkaa084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Du M., Chen Z.J. DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science. 2018;361:704–709. doi: 10.1126/science.aat1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xie W., Lama L., Adura C., Tomita D., Glickman J.F., Tuschl T., et al. Human cGAS catalytic domain has an additional DNA-binding interface that enhances enzymatic activity and liquid-phase condensation. Proc Natl Acad Sci U S A. 2019;116:11946–11955. doi: 10.1073/pnas.1905013116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Luecke S., Holleufer A., Christensen M.H., Jønsson K.L., Boni G.A., Sørensen L.K., et al. cGAS is activated by DNA in a length-dependent manner. EMBO Rep. 2017;18:1707–1715. doi: 10.15252/embr.201744017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Andreeva L., Hiller B., Kostrewa D., Lässig C., de Oliveira Mann C.C., Jan Drexler D., et al. cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein–DNA ladders. Nature. 2017;549:394–398. doi: 10.1038/nature23890. [DOI] [PubMed] [Google Scholar]
- 61.Gao P., Ascano M., Wu Y., Barchet W., Gaffney B.L., Zillinger T., et al. Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP–AMP synthase. Cell. 2013;153:1094–1107. doi: 10.1016/j.cell.2013.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ablasser A., Goldeck M., Cavlar T., Deimling T., Witte G., Röhl I., et al. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature. 2013;498:380–384. doi: 10.1038/nature12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu H., Zhang H., Wu X., Ma D., Wu J., Wang L., et al. Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature. 2018;563:131–136. doi: 10.1038/s41586-018-0629-6. [DOI] [PubMed] [Google Scholar]
- 64.Bai J., Cervantes C., He S., He J., Plasko G.R., Wen J., et al. Mitochondrial stress-activated cGAS–STING pathway inhibits thermogenic program and contributes to overnutrition-induced obesity in mice. Commun Biol. 2020;3:257. doi: 10.1038/s42003-020-0986-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zierhut C., Yamaguchi N., Paredes M., Luo J.D., Carroll T., Funabiki H. The cytoplasmic DNA sensor cGAS promotes mitotic cell death. Cell. 2019;178 doi: 10.1016/j.cell.2019.05.035. 302-15.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mackenzie K.J., Carroll P., Martin C.A., Murina O., Fluteau A., Simpson D.J., et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 2017;548:461–465. doi: 10.1038/nature23449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dou Z., Ghosh K., Vizioli M.G., Zhu J., Sen P., Wangensteen K.J., et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 2017;550:402–406. doi: 10.1038/nature24050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang H., Wang H., Ren J., Chen Q., Chen Z.J. cGAS is essential for cellular senescence. Proc Natl Acad Sci U S A. 2017;114 doi: 10.1073/pnas.1705499114. E4612-e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ning S., Wang L. The multifunctional protein P62 and its mechanistic roles in cancers. Curr Canc Drug Targets. 2019;19:468–478. doi: 10.2174/1568009618666181016164920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang Q., Huang L., Hong Z., Lv Z., Mao Z., Tang Y., et al. The E3 ubiquitin ligase RNF185 facilitates the cGAS-mediated innate immune response. PLoS Pathog. 2017;13 doi: 10.1371/journal.ppat.1006264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang Q., Tang Z., An R., Ye L., Zhong B. USP29 maintains the stability of cGAS and promotes cellular antiviral responses and autoimmunity. Cell Res. 2020;30:914–927. doi: 10.1038/s41422-020-0341-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Seo G.J., Yang A., Tan B., Kim S., Liang Q., Choi Y., et al. Akt kinase-mediated checkpoint of cGAS DNA sensing pathway. Cell Rep. 2015;13:440–449. doi: 10.1016/j.celrep.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dai J., Huang Y.J., He X., Zhao M., Wang X., Liu Z.S., et al. Acetylation blocks cGAS activity and inhibits self-DNA-induced autoimmunity. Cell. 2019;176:1447–1460. doi: 10.1016/j.cell.2019.01.016. e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen H.Y., Pang X.Y., Xu Y.Y., Zhou G.P., Xu H.G. Transcriptional regulation of human cyclic GMP–AMP synthase gene. Cell Signal. 2019;62:109355. doi: 10.1016/j.cellsig.2019.109355. [DOI] [PubMed] [Google Scholar]
- 75.Pal S., Rao G.N., Pal A. High glucose-induced ROS accumulation is a critical regulator of ERK1/2-Akt-tuberin-mTOR signalling in RGC-5 cells. Life Sci. 2020;256:117914. doi: 10.1016/j.lfs.2020.117914. [DOI] [PubMed] [Google Scholar]
- 76.Lei Z., Deng M., Yi Z., Sun Q., Shapiro R.A., Xu H., et al. cGAS-mediated autophagy protects the liver from ischemia–reperfusion injury independently of STING. Am J Physiol Gastrointest Liver Physiol. 2018;314:G655–G667. doi: 10.1152/ajpgi.00326.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ouyang S., Song X., Wang Y., Ru H., Shaw N., Jiang Y., et al. Structural analysis of the STING adaptor protein reveals a hydrophobic dimer interface and mode of cyclic di-GMP binding. Immunity. 2012;36:1073–1086. doi: 10.1016/j.immuni.2012.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tanaka Y., Chen Z.J. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal. 2012;5:ra20. doi: 10.1126/scisignal.2002521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhao B., Shu C., Gao X., Sankaran B., Du F., Shelton C.L., et al. Structural basis for concerted recruitment and activation of IRF-3 by innate immune adaptor proteins. Proc Natl Acad Sci U S A. 2016;113:E3403–E3412. doi: 10.1073/pnas.1603269113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen Q., Sun L., Chen Z.J. Regulation and function of the cGAS–STING pathway of cytosolic DNA sensing. Nat Immunol. 2016;17:1142–1149. doi: 10.1038/ni.3558. [DOI] [PubMed] [Google Scholar]
- 81.Wang P.H., Fung S.Y., Gao W.W., Deng J.J., Cheng Y., Chaudhary V., et al. A novel transcript isoform of STING that sequesters cGAMP and dominantly inhibits innate nucleic acid sensing. Nucleic Acids Res. 2018;46:4054–4071. doi: 10.1093/nar/gky186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pokatayev V., Yang K., Tu X., Dobbs N., Wu J., Kalb R.G., et al. Homeostatic regulation of STING protein at the resting state by stabilizer TOLLIP. Nat Immunol. 2020;21:158–167. doi: 10.1038/s41590-019-0569-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dunphy G., Flannery S.M., Almine J.F., Connolly D.J., Paulus C., Jønsson K.L., et al. Non-canonical activation of the DNA sensing adaptor STING by ATM and IFI16 mediates NF-κB signaling after nuclear DNA damage. Mol Cell. 2018;71:745–760. doi: 10.1016/j.molcel.2018.07.034. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li Z., Liu G., Sun L., Teng Y., Guo X., Jia J., et al. PPM1A regulates antiviral signaling by antagonizing TBK1-mediated STING phosphorylation and aggregation. PLoS Pathog. 2015;11 doi: 10.1371/journal.ppat.1004783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang C., Shang G., Gui X., Zhang X., Bai X.C., Chen Z.J. Structural basis of STING binding with and phosphorylation by TBK1. Nature. 2019;567:394–398. doi: 10.1038/s41586-019-1000-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shu C., Yi G., Watts T., Kao C.C., Li P. Structure of STING bound to cyclic di-GMP reveals the mechanism of cyclic dinucleotide recognition by the immune system. Nat Struct Mol Biol. 2012;19:722–724. doi: 10.1038/nsmb.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen H., Pei R., Zhu W., Zeng R., Wang Y., Wang Y., et al. An alternative splicing isoform of MITA antagonizes MITA-mediated induction of type I IFNs. J Immunol. 2014;192:1162–1170. doi: 10.4049/jimmunol.1300798. [DOI] [PubMed] [Google Scholar]
- 88.Yi G., Brendel V.P., Shu C., Li P., Palanathan S., Cheng Kao C. Single nucleotide polymorphisms of human STING can affect innate immune response to cyclic dinucleotides. PLoS One. 2013;8 doi: 10.1371/journal.pone.0077846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jin L., Xu L.G., Yang I.V., Davidson E.J., Schwartz D.A., Wurfel M.M., et al. Identification and characterization of a loss-of-function human MPYS variant. Gene Immun. 2011;12:263–269. doi: 10.1038/gene.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Saldanha R.G., Balka K.R., Davidson S., Wainstein B.K., Wong M., Macintosh R., et al. A mutation outside the dimerization domain causing atypical STING-associated vasculopathy with onset in infancy. Front Immunol. 2018;9:1535. doi: 10.3389/fimmu.2018.01535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Keskitalo S., Haapaniemi E., Einarsdottir E., Rajamäki K., Heikkilä H., Ilander M., et al. Novel TMEM173 mutation and the role of disease modifying alleles. Front Immunol. 2019;10:2770. doi: 10.3389/fimmu.2019.02770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Melki I., Rose Y., Uggenti C., Van Eyck L., Frémond M.L., Kitabayashi N., et al. Disease-associated mutations identify a novel region in human STING necessary for the control of type I interferon signaling. J Allergy Clin Immunol. 2017;140:543–552.e5. doi: 10.1016/j.jaci.2016.10.031. [DOI] [PubMed] [Google Scholar]
- 93.Ergun S.L., Fernandez D., Weiss T.M., Li L. STING polymer structure reveals mechanisms for activation, hyperactivation, and inhibition. Cell. 2019;178:290–301.e10. doi: 10.1016/j.cell.2019.05.036. [DOI] [PubMed] [Google Scholar]
- 94.Wu J., Chen Y.J., Dobbs N., Sakai T., Liou J., Miner J.J., et al. STING-mediated disruption of calcium homeostasis chronically activates ER stress and primes T cell death. J Exp Med. 2019;216:867–883. doi: 10.1084/jem.20182192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Warner J.D., Irizarry-Caro R.A., Bennion B.G., Ai T.L., Smith A.M., Miner C.A., et al. STING-associated vasculopathy develops independently of IRF3 in mice. J Exp Med. 2017;214:3279–3292. doi: 10.1084/jem.20171351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Srikanth S., Woo J.S., Wu B., El-Sherbiny Y.M., Leung J., Chupradit K., et al. The Ca2+ sensor STIM1 regulates the type I interferon response by retaining the signaling adaptor STING at the endoplasmic reticulum. Nat Immunol. 2019;20:152–162. doi: 10.1038/s41590-018-0287-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gui X., Yang H., Li T., Tan X., Shi P., Li M., et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature. 2019;567:262–266. doi: 10.1038/s41586-019-1006-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Luo W.W., Li S., Li C., Lian H., Yang Q., Zhong B., et al. iRhom2 is essential for innate immunity to DNA viruses by mediating trafficking and stability of the adaptor STING. Nat Immunol. 2016;17:1057–1066. doi: 10.1038/ni.3510. [DOI] [PubMed] [Google Scholar]
- 99.Mukai K., Konno H., Akiba T., Uemura T., Waguri S., Kobayashi T., et al. Activation of STING requires palmitoylation at the Golgi. Nat Commun. 2016;7:11932. doi: 10.1038/ncomms11932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jia M., Qin D., Zhao C., Chai L., Yu Z., Wang W., et al. Redox homeostasis maintained by GPX4 facilitates STING activation. Nat Immunol. 2020;21:727–735. doi: 10.1038/s41590-020-0699-0. [DOI] [PubMed] [Google Scholar]
- 101.Tao L., Lemoff A., Wang G., Zarek C., Lowe A., Yan N., et al. Reactive oxygen species oxidize STING and suppress interferon production. Elife. 2020;9 doi: 10.7554/eLife.57837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Konno H., Konno K., Barber G.N. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell. 2013;155:688–698. doi: 10.1016/j.cell.2013.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gonugunta V.K., Sakai T., Pokatayev V., Yang K., Wu J., Dobbs N., et al. Trafficking-mediated STING degradation requires sorting to acidified endolysosomes and can be targeted to enhance anti-tumor response. Cell Rep. 2017;21:3234–3242. doi: 10.1016/j.celrep.2017.11.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Xia T., Yi X.M., Wu X., Shang J., Shu H.B. PTPN1/2-mediated dephosphorylation of MITA/STING promotes its 20S proteasomal degradation and attenuates innate antiviral response. Proc Natl Acad Sci U S A. 2019;116:20063–20069. doi: 10.1073/pnas.1906431116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yuzefovych L.V., Pastukh V.M., Ruchko M.V., Simmons J.D., Richards W.O., Rachek L.I. Plasma mitochondrial DNA is elevated in obese type 2 diabetes mellitus patients and correlates positively with insulin resistance. PLoS One. 2019;14 doi: 10.1371/journal.pone.0222278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nie S., Lu J., Wang L., Gao M. Pro-inflammatory role of cell-free mitochondrial DNA in cardiovascular diseases. IUBMB Life. 2020;72:1879–1890. doi: 10.1002/iub.2339. [DOI] [PubMed] [Google Scholar]
- 107.Zierhut C., Funabiki H. Regulation and consequences of cGAS activation by self-DNA. Trends Cell Biol. 2020;30:594–605. doi: 10.1016/j.tcb.2020.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kim J., Gupta R., Blanco L.P., Yang S., Shteinfer-Kuzmine A., Wang K., et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science. 2019;366:1531–1536. doi: 10.1126/science.aav4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.McArthur K., Whitehead L.W., Heddleston J.M., Li L., Padman B.S., Oorschot V., et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science. 2018;359 doi: 10.1126/science.aao6047. [DOI] [PubMed] [Google Scholar]
- 110.Rongvaux A., Jackson R., Harman C.C., Li T., West A.P., de Zoete M.R., et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell. 2014;159:1563–1577. doi: 10.1016/j.cell.2014.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.White M.J., McArthur K., Metcalf D., Lane R.M., Cambier J.C., Herold M.J., et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell. 2014;159:1549–1562. doi: 10.1016/j.cell.2014.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Moretti J., Roy S., Bozec D., Martinez J., Chapman J.R., Ueberheide B., et al. STING senses microbial viability to orchestrate stress-mediated autophagy of the endoplasmic reticulum. Cell. 2017;171 doi: 10.1016/j.cell.2017.09.034. 809-23.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hu F., Yu X., Wang H., Zuo D., Guo C., Yi H., et al. ER stress and its regulator X-box-binding protein-1 enhance polyIC-induced innate immune response in dendritic cells. Eur J Immunol. 2011;41:1086–1097. doi: 10.1002/eji.201040831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liu Y.P., Zeng L., Tian A., Bomkamp A., Rivera D., Gutman D., et al. Endoplasmic reticulum stress regulates the innate immunity critical transcription factor IRF3. J Immunol. 2012;189:4630–4639. doi: 10.4049/jimmunol.1102737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Iracheta-Vellve A., Petrasek J., Gyongyosi B., Satishchandran A., Lowe P., Kodys K., et al. Endoplasmic reticulum stress-induced hepatocellular death pathways mediate liver injury and fibrosis via stimulator of interferon genes. J Biol Chem. 2016;291:26794–26805. doi: 10.1074/jbc.M116.736991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mohamed E., Sierra R.A., Trillo-Tinoco J., Cao Y., Innamarato P., Payne K.K., et al. The unfolded protein response mediator PERK governs myeloid cell-driven immunosuppression in tumors through Inhibition of STING signaling. Immunity. 2020;52:668–682. doi: 10.1016/j.immuni.2020.03.004. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ablasser A., Hemmerling I., Schmid-Burgk J.L., Behrendt R., Roers A., Hornung V. TREX1 deficiency triggers cell-autonomous immunity in a cGAS-dependent manner. J Immunol. 2014;192:5993–5997. doi: 10.4049/jimmunol.1400737. [DOI] [PubMed] [Google Scholar]
- 118.Rice G.I., Forte G.M., Szynkiewicz M., Chase D.S., Aeby A., Abdel-Hamid M.S., et al. Assessment of interferon-related biomarkers in Aicardi-Goutières syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study. Lancet Neurol. 2013;12:1159–1169. doi: 10.1016/S1474-4422(13)70258-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Morita M., Stamp G., Robins P., Dulic A., Rosewell I., Hrivnak G., et al. Gene-targeted mice lacking the Trex1 (DNase III) 3′–>5′ DNA exonuclease develop inflammatory myocarditis. Mol Cell Biol. 2004;24:6719–6727. doi: 10.1128/MCB.24.15.6719-6727.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Abe T., Barber G.N. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J Virol. 2014;88:5328–5341. doi: 10.1128/JVI.00037-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cavlar T., Deimling T., Ablasser A., Hopfner K.P., Hornung V. Species-specific detection of the antiviral small-molecule compound CMA by STING. EMBO J. 2013;32:1440–1450. doi: 10.1038/emboj.2013.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.de Oliveira Mann C.C., Orzalli M.H., King D.S., Kagan J.C., Lee A.S.Y., Kranzusch P.J. Modular architecture of the STING C-terminal tail allows interferon and NF-κB signaling adaptation. Cell Rep. 2019;27:1165–1175. doi: 10.1016/j.celrep.2019.03.098. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Balka K.R., Louis C., Saunders T.L., Smith A.M., Calleja D.J., D'Silva D.B., et al. TBK1 and IKKepsilon act redundantly to mediate STING-induced NF-kappaB responses in myeloid cells. Cell Rep. 2020;31:107492. doi: 10.1016/j.celrep.2020.03.056. [DOI] [PubMed] [Google Scholar]
- 124.Gaidt M.M., Ebert T.S., Chauhan D., Ramshorn K., Pinci F., Zuber S., et al. The DNA inflammasome in human myeloid cells is initiated by a STING-cell death program upstream of NLRP3. Cell. 2017;171 doi: 10.1016/j.cell.2017.09.039. 1110-24.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Liu S., Guan W. STING signaling promotes apoptosis, necrosis, and cell death: an overview and update. Mediat Inflamm. 2018;2018:1202797. doi: 10.1155/2018/1202797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sarhan J., Liu B.C., Muendlein H.I., Weindel C.G., Smirnova I., Tang A.Y., et al. Constitutive interferon signaling maintains critical threshold of MLKL expression to license necroptosis. Cell Death Differ. 2019;26:332–347. doi: 10.1038/s41418-018-0122-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Motwani M., Pawaria S., Bernier J., Moses S., Henry K., Fang T., et al. Hierarchy of clinical manifestations in SAVI N153S and V154M mouse models. Proc Natl Acad Sci U S A. 2019;116:7941–7950. doi: 10.1073/pnas.1818281116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hasan M., Gonugunta V.K., Dobbs N., Ali A., Palchik G., Calvaruso M.A., et al. Chronic innate immune activation of TBK1 suppresses mTORC1 activity and dysregulates cellular metabolism. Proc Natl Acad Sci U S A. 2017;114:746–751. doi: 10.1073/pnas.1611113114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kim J.K., Jung Y., Wang J., Joseph J., Mishra A., Hill E.E., et al. TBK1 regulates prostate cancer dormancy through mTOR inhibition. Neoplasia. 2013;15:1064–1074. doi: 10.1593/neo.13402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yu J., Zhou X., Chang M., Nakaya M., Chang J.H., Xiao Y., et al. Regulation of T-cell activation and migration by the kinase TBK1 during neuroinflammation. Nat Commun. 2015;6:6074. doi: 10.1038/ncomms7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Antonia R.J., Castillo J., Herring L.E., Serafin D.S., Liu P., Graves L.M., et al. TBK1 limits mTORC1 by promoting phosphorylation of raptor Ser 877. Sci Rep. 2019;9:13470. doi: 10.1038/s41598-019-49707-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Meade N., Furey C., Li H., Verma R., Chai Q., Rollins M.G., et al. Poxviruses evade cytosolic sensing through disruption of an mTORC1-mTORC2 regulatory circuit. Cell. 2018;174:1143–1157. doi: 10.1016/j.cell.2018.06.053. e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Richter B., Sliter D.A., Herhaus L., Stolz A., Wang C., Beli P., et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A. 2016;113:4039–4044. doi: 10.1073/pnas.1523926113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sliter D.A., Martinez J., Hao L., Chen X., Sun N., Fischer T.D., et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature. 2018;561:258–262. doi: 10.1038/s41586-018-0448-9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 135.Prabakaran T., Bodda C., Krapp C., Zhang B.C., Christensen M.H., Sun C., et al. Attenuation of cGAS–STING signaling is mediated by a P62/SQSTM1-dependent autophagy pathway activated by TBK1. EMBO J. 2018;37 doi: 10.15252/embj.201797858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Liang Q., Seo G.J., Choi Y.J., Kwak M.J., Ge J., Rodgers M.A., et al. Crosstalk between the cGAS DNA sensor and Beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe. 2014;15:228–238. doi: 10.1016/j.chom.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gulen M.F., Koch U., Haag S.M., Schuler F., Apetoh L., Villunger A., et al. Signalling strength determines proapoptotic functions of STING. Nat Commun. 2017;8:427. doi: 10.1038/s41467-017-00573-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Tang C.H., Zundell J.A., Ranatunga S., Lin C., Nefedova Y., Del Valle J.R., et al. Agonist-mediated activation of STING induces apoptosis in malignant B cells. Canc Res. 2016;76:2137–2152. doi: 10.1158/0008-5472.CAN-15-1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Weiss J.M., Guérin M.V., Regnier F., Renault G., Galy-Fauroux I., Vimeux L., et al. The STING agonist DMXAA triggers a cooperation between T lymphocytes and myeloid cells that leads to tumor regression. OncoImmunology. 2017;6 doi: 10.1080/2162402X.2017.1346765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wang Y., Ning X., Gao P., Wu S., Sha M., Lv M., et al. Inflammasome activation triggers caspase-1-mediated cleavage of cGAS to regulate responses to DNA virus infection. Immunity. 2017;46:393–404. doi: 10.1016/j.immuni.2017.02.011. [DOI] [PubMed] [Google Scholar]
- 141.Bai J., Liu F. The cGAS–cGAMP–STING pathway: a molecular link between immunity and metabolism. Diabetes. 2019;68:1099–1108. doi: 10.2337/dbi18-0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ivashkiv L.B., Donlin L.T. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Aricò E., Castiello L., Capone I., Gabriele L., Belardelli F. Type I interferons and cancer: an evolving story demanding novel clinical applications. Cancers. 2019;11:1943. doi: 10.3390/cancers11121943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ter Horst E.N., Krijnen P.A.J., Hakimzadeh N., Robbers L., Hirsch A., Nijveldt R., et al. Elevated monocyte-specific type I interferon signalling correlates positively with cardiac healing in myocardial infarct patients but interferon alpha application deteriorates myocardial healing in rats. Basic Res Cardiol. 2018;114:1. doi: 10.1007/s00395-018-0709-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zimmerman S., Adkins D., Graham M., Petruska P., Bowers C., Vrahnos D., et al. Irreversible, severe congestive cardiomyopathy occurring in association with interferon alpha therapy. Canc Biother. 1994;9:291–299. doi: 10.1089/cbr.1994.9.291. [DOI] [PubMed] [Google Scholar]
- 146.Khakoo A.Y., Halushka M.K., Rame J.E., Rodriguez E.R., Kasper E.K., Judge D.P. Reversible cardiomyopathy caused by administration of interferon alpha. Nat Clin Pract Cardiovasc Med. 2005;2:53–57. doi: 10.1038/ncpcardio0069. [DOI] [PubMed] [Google Scholar]
- 147.Schirmer S.H., Fledderus J.O., Bot P.T., Moerland P.D., Hoefer I.E., Baan J., Jr., et al. Interferon-beta signaling is enhanced in patients with insufficient coronary collateral artery development and inhibits arteriogenesis in mice. Circ Res. 2008;102:1286–1294. doi: 10.1161/CIRCRESAHA.108.171827. [DOI] [PubMed] [Google Scholar]
- 148.Schirmer S.H., Bot P.T., Fledderus J.O., van der Laan A.M., Volger O.L., Laufs U., et al. Blocking interferon β stimulates vascular smooth muscle cell proliferation and arteriogenesis. J Biol Chem. 2010;285:34677–34685. doi: 10.1074/jbc.M110.164350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Jefferies C.A. Regulating IRFs in IFN driven disease. Front Immunol. 2019;10:325. doi: 10.3389/fimmu.2019.00325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lu J., Bian Z.Y., Zhang R., Zhang Y., Liu C., Yan L., et al. Interferon regulatory factor 3 is a negative regulator of pathological cardiac hypertrophy. Basic Res Cardiol. 2013;108:326. doi: 10.1007/s00395-012-0326-9. [DOI] [PubMed] [Google Scholar]
- 151.Jiang D.S., Luo Y.X., Zhang R., Zhang X.D., Chen H.Z., Zhang Y., et al. Interferon regulatory factor 9 protects against cardiac hypertrophy by targeting myocardin. Hypertension. 2014;63:119–127. doi: 10.1161/HYPERTENSIONAHA.113.02083. [DOI] [PubMed] [Google Scholar]
- 152.Eguchi J., Kong X., Tenta M., Wang X., Kang S., Rosen E.D. Interferon regulatory factor 4 regulates obesity-induced inflammation through regulation of adipose tissue macrophage polarization. Diabetes. 2013;62:3394–3403. doi: 10.2337/db12-1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wang X.A., Zhang R., Zhang S., Deng S., Jiang D., Zhong J., et al. Interferon regulatory factor 7 deficiency prevents diet-induced obesity and insulin resistance. Am J Physiol Endocrinol Metab. 2013;305:E485–E495. doi: 10.1152/ajpendo.00505.2012. [DOI] [PubMed] [Google Scholar]
- 154.Zhang Y., Li H. Reprogramming interferon regulatory factor signaling in cardiometabolic diseases. Physiology. 2017;32:210–223. doi: 10.1152/physiol.00038.2016. [DOI] [PubMed] [Google Scholar]
- 155.Zhang X.J., Jiang D.S., Li H. The interferon regulatory factors as novel potential targets in the treatment of cardiovascular diseases. Br J Pharmacol. 2015;172:5457–5476. doi: 10.1111/bph.12881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Condorelli G., Morisco C., Stassi G., Notte A., Farina F., Sgaramella G., et al. Increased cardiomyocyte apoptosis and changes in proapoptotic and antiapoptotic genes bax and bcl-2 during left ventricular adaptations to chronic pressure overload in the rat. Circulation. 1999;99:3071–3078. doi: 10.1161/01.cir.99.23.3071. [DOI] [PubMed] [Google Scholar]
- 157.Bugger H., Schwarzer M., Chen D., Schrepper A., Amorim P.A., Schoepe M., et al. Proteomic remodelling of mitochondrial oxidative pathways in pressure overload-induced heart failure. Cardiovasc Res. 2010;85:376–384. doi: 10.1093/cvr/cvp344. [DOI] [PubMed] [Google Scholar]
- 158.Li N., Zhou H., Wu H., Wu Q., Duan M., Deng W., et al. STING–IRF3 contributes to lipopolysaccharide-induced cardiac dysfunction, inflammation, apoptosis and pyroptosis by activating NLRP3. Redox Biol. 2019;24:101215. doi: 10.1016/j.redox.2019.101215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Gong Y., Li G., Tao J., Wu N.N., Kandadi M.R., Bi Y., et al. Double knockout of Akt 2 and AMPK accentuates high fat diet-induced cardiac anomalies through a cGAS–STING–mediated mechanism. Biochim Biophys Acta Mol Basis Dis. 2020;1866:165855. doi: 10.1016/j.bbadis.2020.165855. [DOI] [PubMed] [Google Scholar]
- 160.Uryga A., Gray K., Bennett M. DNA damage and repair in vascular disease. Annu Rev Physiol. 2016;78:45–66. doi: 10.1146/annurev-physiol-021115-105127. [DOI] [PubMed] [Google Scholar]
- 161.Hayashi C., Viereck J., Hua N., Phinikaridou A., Madrigal A.G., Gibson F.C., 3rd, et al. Porphyromonas gingivalis accelerates inflammatory atherosclerosis in the innominate artery of ApoE deficient mice. Atherosclerosis. 2011;215:52–59. doi: 10.1016/j.atherosclerosis.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Calandrini C.A., Ribeiro A.C., Gonnelli A.C., Ota-Tsuzuki C., Rangel L.P., Saba-Chujfi E., et al. Microbial composition of atherosclerotic plaques. Oral Dis. 2014;20:e128–e134. doi: 10.1111/odi.12205. [DOI] [PubMed] [Google Scholar]
- 163.Koren O., Spor A., Felin J., Fåk F., Stombaugh J., Tremaroli V., et al. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc Natl Acad Sci U S A. 2011;108 Suppl 1:4592–4598. doi: 10.1073/pnas.1011383107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hansen K., Prabakaran T., Laustsen A., Jørgensen S.E., Rahbæk S.H., Jensen S.B., et al. Listeria monocytogenes induces IFNβ expression through an IFI16-, cGAS- and STING-dependent pathway. EMBO J. 2014;33:1654–1666. doi: 10.15252/embj.201488029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hahn W.O., Butler N.S., Lindner S.E., Akilesh H.M., Sather D.N., Kappe S.H., et al. cGAS-mediated control of blood-stage malaria promotes Plasmodium-specific germinal center responses. JCI Insight. 2018;3 doi: 10.1172/jci.insight.94142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Oduro P.K., Fang J., Niu L., Li Y., Li L., Zhao X., et al. Pharmacological management of vascular endothelial dysfunction in diabetes: TCM and western medicine compared based on biomarkers and biochemical parameters. Pharmacol Res. 2020;158:104893. doi: 10.1016/j.phrs.2020.104893. [DOI] [PubMed] [Google Scholar]
- 167.Berthelot J.M., Drouet L., Lioté F. Kawasaki-like diseases and thrombotic coagulopathy in COVID-19: delayed over-activation of the STING pathway? Emerg Microb Infect. 2020;9:1514–1522. doi: 10.1080/22221751.2020.1785336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Denny M.F., Yalavarthi S., Zhao W., Thacker S.G., Anderson M., Sandy A.R., et al. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J Immunol. 2010;184:3284–3297. doi: 10.4049/jimmunol.0902199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Huang L.S., Hong Z., Wu W., Xiong S., Zhong M., Gao X., et al. mtDNA activates cGAS signaling and suppresses the YAP-mediated endothelial cell proliferation program to promote inflammatory injury. Immunity. 2020;52:475–486.e5. doi: 10.1016/j.immuni.2020.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zhao Y., Yu X., Li J. Manipulation of immune‒vascular crosstalk: new strategies towards cancer treatment. Acta Pharm Sin B. 2020;10:2018–2036. doi: 10.1016/j.apsb.2020.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ghosh-Swaby O.R., Goodman S.G., Leiter L.A., Cheng A., Connelly K.A., Fitchett D., et al. Glucose-lowering drugs or strategies, atherosclerotic cardiovascular events, and heart failure in people with or at risk of type 2 diabetes: an updated systematic review and meta-analysis of randomised cardiovascular outcome trials. Lancet Diabetes Endocrinol. 2020;8:418–435. doi: 10.1016/S2213-8587(20)30038-3. [DOI] [PubMed] [Google Scholar]
- 172.Lee J.H., Park A., Oh K.J., Lee S.C., Kim W.K., Bae K.H. The role of adipose tissue mitochondria: regulation of mitochondrial function for the treatment of metabolic diseases. Int J Mol Sci. 2019;20:4924. doi: 10.3390/ijms20194924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Merrick D., Sakers A., Irgebay Z., Okada C., Calvert C., Morley M.P., et al. Identification of a mesenchymal progenitor cell hierarchy in adipose tissue. Science. 2019;364 doi: 10.1126/science.aav2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kumari M., Wang X., Lantier L., Lyubetskaya A., Eguchi J., Kang S., et al. IRF3 promotes adipose inflammation and insulin resistance and represses browning. J Clin Invest. 2016;126:2839–2854. doi: 10.1172/JCI86080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zhou L., Liu M., Zhang J., Chen H., Dong L.Q., Liu F. DsbA-L alleviates endoplasmic reticulum stress-induced adiponectin downregulation. Diabetes. 2010;59:2809–2816. doi: 10.2337/db10-0412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Zhao P., Wong K.I., Sun X., Reilly S.M., Uhm M., Liao Z., et al. TBK1 at the crossroads of inflammation and energy homeostasis in adipose tissue. Cell. 2018;172:731–743. doi: 10.1016/j.cell.2018.01.007. e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Oral E.A., Reilly S.M., Gomez A.V., Meral R., Butz L., Ajluni N., et al. Inhibition of IKKɛ and TBK1 improves glucose control in a subset of patients with type 2 diabetes. Cell Metabol. 2017;26:157–170. doi: 10.1016/j.cmet.2017.06.006. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Hamann L., Ruiz-Moreno J.S., Szwed M., Mossakowska M., Lundvall L., Schumann R.R., et al. STING SNP R293Q ss associated with a decreased risk of aging-related diseases. Gerontology. 2019;65:145–154. doi: 10.1159/000492972. [DOI] [PubMed] [Google Scholar]
- 179.Hamann L., Szwed M., Mossakowska M., Chudek J., Puzianowska-Kuznicka M. First evidence for STING SNP R293Q being protective regarding obesity-associated cardiovascular disease in age-advanced subjects—a cohort study. Immun Ageing. 2020;17:7. doi: 10.1186/s12979-020-00176-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hu H.Q., Qiao J.T., Liu F.Q., Wang J.B., Sha S., He Q., et al. The STING–IRF3 pathway is involved in lipotoxic injury of pancreatic β cells in type 2 diabetes. Mol Cell Endocrinol. 2020;518:110890. doi: 10.1016/j.mce.2020.110890. [DOI] [PubMed] [Google Scholar]
- 181.Paul S., Davis A.M. Diagnosis and management of nonalcoholic fatty liver disease. J Am Med Assoc. 2018;320:2474–2475. doi: 10.1001/jama.2018.17365. [DOI] [PubMed] [Google Scholar]
- 182.Qiao J.T., Cui C., Qing L., Wang L.S., He T.Y., Yan F., et al. Activation of the STING–IRF3 pathway promotes hepatocyte inflammation, apoptosis and induces metabolic disorders in nonalcoholic fatty liver disease. Metabolism. 2018;81:13–24. doi: 10.1016/j.metabol.2017.09.010. [DOI] [PubMed] [Google Scholar]
- 183.Li Y.N., Su Y. Remdesivir attenuates high fat diet (HFD)-induced NAFLD by regulating hepatocyte dyslipidemia and inflammation via the suppression of STING. Biochem Biophys Res Commun. 2020;526:381–388. doi: 10.1016/j.bbrc.2020.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Cho C.S., Park H.W., Ho A., Semple I.A., Kim B., Jang I., et al. Lipotoxicity induces hepatic protein inclusions through TANK binding kinase 1-mediated P62/sequestosome 1 phosphorylation. Hepatology. 2018;68:1331–1346. doi: 10.1002/hep.29742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Brown G.T., Kleiner D.E. Histopathology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Metabolism. 2016;65:1080–1086. doi: 10.1016/j.metabol.2015.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Thomsen M.K., Nandakumar R., Stadler D., Malo A., Valls R.M., Wang F., et al. Lack of immunological DNA sensing in hepatocytes facilitates hepatitis B virus infection. Hepatology. 2016;64:746–759. doi: 10.1002/hep.28685. [DOI] [PubMed] [Google Scholar]
- 187.Desbois A.C., Cacoub P. Diabetes mellitus, insulin resistance and hepatitis C virus infection: a contemporary review. World J Gastroenterol. 2017;23:1697–1711. doi: 10.3748/wjg.v23.i9.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Zhu H.F., Li Y. Small-molecule targets in tumor immunotherapy. Nat Prod Bioprospect. 2018;8:297–301. doi: 10.1007/s13659-018-0177-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Lin B., Berard R., Al Rasheed A., Aladba B., Kranzusch P.J., Henderlight M., et al. A novel STING1 variant causes a recessive form of STING-associated vasculopathy with onset in infancy (SAVI) J Allergy Clin Immunol. 2020;146:1204–1208. doi: 10.1016/j.jaci.2020.06.032. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Sanchez G.A.M., Reinhardt A., Ramsey S., Wittkowski H., Hashkes P.J., Berkun Y., et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J Clin Invest. 2018;128:3041–3052. doi: 10.1172/JCI98814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.An J., Woodward J.J., Sasaki T., Minie M., Elkon K.B. Cutting edge: antimalarial drugs inhibit IFN-beta production through blockade of cyclic GMP–AMP synthase–DNA interaction. J Immunol. 2015;194:4089–4093. doi: 10.4049/jimmunol.1402793. [DOI] [PubMed] [Google Scholar]
- 192.Vincent J., Adura C., Gao P., Luz A., Lama L., Asano Y., et al. Small molecule inhibition of cGAS reduces interferon expression in primary macrophages from autoimmune mice. Nat Commun. 2017;8:750. doi: 10.1038/s41467-017-00833-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Lama L., Adura C., Xie W., Tomita D., Kamei T., Kuryavyi V., et al. Development of human cGAS-specific small-molecule inhibitors for repression of dsDNA-triggered interferon expression. Nat Commun. 2019;10:2261. doi: 10.1038/s41467-019-08620-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Hall J., Brault A., Vincent F., Weng S., Wang H., Dumlao D., et al. Discovery of PF-06928215 as a high affinity inhibitor of cGAS enabled by a novel fluorescence polarization assay. PLoS One. 2017;12 doi: 10.1371/journal.pone.0184843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Steinhagen F., Zillinger T., Peukert K., Fox M., Thudium M., Barchet W., et al. Suppressive oligodeoxynucleotides containing TTAGGG motifs inhibit cGAS activation in human monocytes. Eur J Immunol. 2018;48:605–611. doi: 10.1002/eji.201747338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Wang M., Sooreshjani M.A., Mikek C., Opoku-Temeng C., Sintim H.O. Suramin potently inhibits cGAMP synthase, cGAS, in THP1 cells to modulate IFN-β levels. Future Med Chem. 2018;11:1301–1317. doi: 10.4155/fmc-2017-0322. [DOI] [PubMed] [Google Scholar]
- 197.Padilla-Salinas R., Sun L., Anderson R., Yang X., Zhang S., Chen Z.J., et al. Discovery of small-molecule cyclic GMP–AMP synthase inhibitors. J Org Chem. 2020;85:1579–1600. doi: 10.1021/acs.joc.9b02666. [DOI] [PubMed] [Google Scholar]
- 198.Li S., Hong Z., Wang Z., Li F., Mei J., Huang L., et al. The cyclopeptide astin C specifically inhibits the innate immune CDN sensor STING. Cell Rep. 2018;25:3405–3421. doi: 10.1016/j.celrep.2018.11.097. e7. [DOI] [PubMed] [Google Scholar]
- 199.Haag S.M., Gulen M.F., Reymond L., Gibelin A., Abrami L., Decout A., et al. Targeting STING with covalent small-molecule inhibitors. Nature. 2018;559:269–273. doi: 10.1038/s41586-018-0287-8. [DOI] [PubMed] [Google Scholar]
- 200.Hansen A.L., Buchan G.J., Ruhl M., Mukai K., Salvatore S.R., Ogawa E., et al. Nitro-fatty acids are formed in response to virus infection and are potent inhibitors of STING palmitoylation and signaling. Proc Natl Acad Sci U S A. 2018;115:E7768–E7775. doi: 10.1073/pnas.1806239115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Siu T., Altman M.D., Baltus G.A., Childers M., Ellis J.M., Gunaydin H., et al. Discovery of a novel cGAMP competitive ligand of the inactive form of STING. ACS Med Chem Lett. 2019;10:92–97. doi: 10.1021/acsmedchemlett.8b00466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Dai J., Huang Y.J., He X., Zhao M., Wang X., Liu Z.S., et al. Acetylation blocks cGAS activity and inhibits self-DNA-induced autoimmunity. Cell. 2019;176:1447–1460.e14. doi: 10.1016/j.cell.2019.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Delmastro-Greenwood M., Hughan K.S., Vitturi D.A., Salvatore S.R., Grimes G., Potti G., et al. Nitrite and nitrate-dependent generation of anti-inflammatory fatty acid nitroalkenes. Free Radic Biol Med. 2015;89:333–341. doi: 10.1016/j.freeradbiomed.2015.07.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Huffman B.J., Chen S., Schwarz J.L., Plata R.E., Chin E.N., Lairson L.L., et al. Electronic complementarity permits hindered butenolide heterodimerization and discovery of novel cGAS/STING pathway antagonists. Nat Chem. 2020;12:310–317. doi: 10.1038/s41557-019-0413-8. [DOI] [PubMed] [Google Scholar]
- 205.Ozasa K., Temizoz B., Kusakabe T., Kobari S., Momota M., Coban C., et al. Cyclic GMP–AMP triggers asthma in an IL-33-dependent manner that is blocked by amlexanox, a TBK1 inhibitor. Front Immunol. 2019;10:2212. doi: 10.3389/fimmu.2019.02212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Reilly S.M., Chiang S.H., Decker S.J., Chang L., Uhm M., Larsen M.J., et al. An inhibitor of the protein kinases TBK1 and IKK-ɛ improves obesity-related metabolic dysfunctions in mice. Nat Med. 2013;19:313–321. doi: 10.1038/nm.3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Li J., Huang J., Jeong J.H., Park S.J., Wei R., Peng J., et al. Selective TBK1/IKKi dual inhibitors with anticancer potency. Int J Canc. 2014;134:1972–1980. doi: 10.1002/ijc.28507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Thomson D.W., Poeckel D., Zinn N., Rau C., Strohmer K., Wagner A.J., et al. Discovery of GSK8612, a highly selective and potent TBK1 inhibitor. ACS Med Chem Lett. 2019;10:780–785. doi: 10.1021/acsmedchemlett.9b00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Ding C., Song Z., Shen A., Chen T., Zhang A. Small molecules targeting the innate immune cGAS‒STING‒TBK1 signaling pathway. Acta Pharm Sin B. 2020;10:2272–2298. doi: 10.1016/j.apsb.2020.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Kim E.H., Wong S.W., Martinez J. Programmed necrosis and disease: we interrupt your regular programming to bring you necroinflammation. Cell Death Differ. 2019;26:25–40. doi: 10.1038/s41418-018-0179-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Bravo-San Pedro J.M., Kroemer G., Galluzzi L. Autophagy and mitophagy in cardiovascular Disease. Circ Res. 2017;120:1812–1824. doi: 10.1161/CIRCRESAHA.117.311082. [DOI] [PubMed] [Google Scholar]
- 212.Gangadhar T.C., Vonderheide R.H. Mitigating the toxic effects of anticancer immunotherapy. Nat Rev Clin Oncol. 2014;11:91–99. doi: 10.1038/nrclinonc.2013.245. [DOI] [PubMed] [Google Scholar]
- 213.Berthelot J.M., Lioté F. COVID-19 as a STING disorder with delayed over-secretion of interferon-beta. EBioMedicine. 2020;56:102801. doi: 10.1016/j.ebiom.2020.102801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Gruber C. Impaired interferon signature in severe COVID-19. Nat Rev Immunol. 2020;20:353. doi: 10.1038/s41577-020-0335-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Zhao X., Oduro P.K., Tong W., Wang Y., Gao X., Wang Q. Therapeutic potential of natural products against atherosclerosis: targeting on gut microbiota. Pharmacol Res. 2021;163:105362. doi: 10.1016/j.phrs.2020.105362. [DOI] [PubMed] [Google Scholar]
- 216.Hu Q., Ren H., Li G., Wang D., Zhou Q., Wu J., et al. STING-mediated intestinal barrier dysfunction contributes to lethal sepsis. EBioMedicine. 2019;41:497–508. doi: 10.1016/j.ebiom.2019.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]