

Abstract
KRAS‒PDEδ interaction is revealed as a promising target for suppressing the function of mutant KRAS. The bottleneck in clinical development of PDEδ inhibitors is the poor antitumor activity of known chemotypes. Here, we identified novel spiro-cyclic PDEδ inhibitors with potent antitumor activity both in vitro and in vivo. In particular, compound 36l (KD = 127 ± 16 nmol/L) effectively bound to PDEδ and interfered with KRAS–PDEδ interaction. It influenced the distribution of KRAS in Mia PaCa-2 cells, downregulated the phosphorylation of t-ERK and t-AKT and promoted apoptosis of the cells. The novel inhibitor 36l exhibited significant in vivo antitumor potency in pancreatic cancer patient-derived xenograft (PDX) models. It represents a promising lead compound for investigating the druggability of KRAS‒PDEδ interaction.
KEY WORDS: KRAS‒PDEδ interaction, PDX, Spiro-cyclic inhibitors, Lead optimization, SBDD, Anti-pancreatic cancer activity
Graphical abstract
Compound 36l is a novel spiro-cyclic PDEδ inhibitor (KD = 127 ± 16 nmol/L). It inhibited the growth of Mia PaCa-2 cells (IC50 = 6.67 ± 1.7 μmol/L) and exhibited significant in vivo antitumor potency in pancreatic cancer patient-derived xenograft (PDX) models.

1. Introduction
Pancreatic cancer is one of the most fatal solid tumor with a high recurrence rate and poor prognosis1,2. Mutations in KRAS gene are considered to be a key factor of pancreatic cancer progression3,4. However, the development of KRAS-targeting antitumor agents is highly challenging and a number of drug candidates failed in clinical trials5,6. Recently, the development of KRASG12C inhibitors has gained great attention7, 8, 9, which shows promising results in phase I clinical trial10, 11, 12. However, KRASG12C is a comparatively minority of the KRAS mutation spectrum (approximately 11%)13, inhibitors of other KRAS mutation subtypes, such as KRASG12D and KRASG12V, remain to be further investigated14,15.
Chaperone protein PDEδ plays an essential role in the function of KRAS protein in the cells16, 17, 18. PDEδ enhances the diffusion of KRAS in the cytoplasm by binding the farnesyl group, and promotes its distribution over intracellular membrane18. Down-modulation or inhibition of PDEδ protein suppresses KRAS signaling, and inhibits the growth and proliferation of cancer cells17.
KRAS‒PDEδ interaction has been proposed as a potential target for the development of novel antitumor agents. Currently, a series of KRAS‒PDEδ inhibitors have been reported (Fig. 1)19. Waldmann's group17 discovered the first KRAS‒PDEδ inhibitor deltarasin (1) by fragment based drug design (FBDD) at 2013. However, the selectivity of deltarasin was poor, which showed apparent cytotoxicity20. Subsequently, more selective inhibitors deltazinone (2) and deltasonamide (3) were designed20, 21, 22. Although these inhibitors generally showed high PDEδ binding affinity, further development was hampered by poor cellular potency and metabolic stability. More recently, triazole inhibitor (4)23, tetrahydrodibenzofuran inhibitor NHTD (5)24 and coumarin inhibitor deltaflexin (6)25 were reported. However, the antitumor efficacy of known PDEδ inhibitors remains to be significantly improved. Previously, our group reported the discovery and optimization of quinazolinone KRAS‒PDEδ inhibitors 7 and 8 based on structural biology guided FBDD26,27. Furthermore, we designed fluorescent probes and protein degraders of PDEδ28,29, which offered new chemical tools to investigate the biological function of KRAS‒PDEδ interaction. However, the known KRAS‒PDEδ inhibitors are generally limited by weak in vivo efficacy. For example, deltarasin was unspecific to PDEδ protein, leading to a “switch-like” response to cell death. Deltazinone was unable to exert in vivo antitumor activity due to poor metabolic stability20,21. In order to identify novel KRAS‒PDEδ inhibitors with enhanced antitumor potency, herein new spiro-cyclic PDEδ inhibitors were discovered from screening of an in-house library.
Figure 1.

Chemical structures of reported KRAS‒PDEδ inhibitors.
After structure-based hit optimization, compound 36l was identified to effectively inhibit the KRAS‒PDEδ interaction and disrupt the distribution of KRAS protein. Interestingly, inhibitor 36l showed potent anti-pancreatic cancer activity both in vitro and in vivo, which represented the first KRAS‒PDEδ inhibitor with in vivo efficacy in the patient-derived xenograft (PDX) models. Also, compound 36l is a promising lead compound for the treatment of pancreatic cancer.
2. Results and discussion
2.1. Discovery of a spiro-cyclic PDEδ inhibitor
Our in-house compound library containing diverse spiro-scaffolds30, 31, 32, 33 and approved drugs was screened using the fluorescence polarization (FP) assay26. First, the inhibitory ratio of all compounds was tested at 5 μmol/L (Supporting Information Table S1). Two compounds (atorvastatin and spiperone) were identified to have an inhibitory rate over 50%. Then, the KD value of the hit compounds was further determined. Atorvastatin has been reported to be a PDEδ inhibitor and is generally used as the probe in the FP assay17. Spiperone (9), which shared a spiro scaffold, exhibited moderate activity in a low micro-molar range to PDEδ (KD = 1471 ± 246 nmol/L, Fig. 2A and B). Subsequently, it was chosen for further structural optimization.
Figure 2.

Discovery and optimization of spiro-cyclic PDEδ inhibitors (A) Chemical structure of compound 9 identified from an in-house library. (B) The dose–response curve for compounds 9 (red) and 2 (blue) by the FP assay; (C) The proposed binding pose of compound 9 (green) to PDEδ protein (PDB: 5X7426). Hydrogen bonding interaction with Arg61, Gln78 and Tyr149 was represented by red dashed lines. (D) Optimization of compound 9 with SBDD. The figures were generated by Pymol (http://www.Pymol.org/).
2.2. Molecular docking and drug design
In order to determine the binding mode of compound 9 towards PDEδ protein, Glide docking with extra precision (XP) was performed. According to the predicted docking pose (Fig. 2C), the amide group within the spiro scaffold of compound 9 formed a hydrogen bond to Tyr149, while the p-fluorobenzoyl moiety binds to the Arg61 pocket through hydrogen bonding interactions between the ketone group and Arg61 and Gln78, respectively. The docking pose exhibited an obvious hydrophobic cavity in the Arg61 pocket, which suggested that the introduction of hydrophobic groups could improve the binding affinity. Guided by the binding conformation, structure-based drug design (SBDD) was applied to design a series of new inhibitors (Fig. 2D). First, various amides were introduced to replace the p-fluorobenzoyl moiety. Then N-monosubstitution or N-di-substitution was introduced by aromatic or aliphatic ring. The spiro moiety was reserved to maintain the binding conformation.
2.3. Chemistry
The synthetic route for the synthesis of target compounds 23a‒23e is shown in Scheme 1. The substituted primary amines reacted with 4-chlorobutyryl chloride to give intermediates 22, subsequently 22 reacted with intermediate 18 to afford target compounds 23a‒23e.
Scheme 1.

Reagent and conditions: (a) AcOH, rt, 2.5 h; (b) 98% H2SO4, rt, 18 h; (c) DMF-DMA, MeOH, 55 °C, 16 h; (d) NaBH4, MeOH, rt, 2 h; (e) H2, 20% Pd(OH)2, MeOH, rt, overnight; (f) 4-Chlorobutyryl chloride, TEA, DCM, rt, 2 h; (g) 18, TEA, KI, acetonitrile, 90 °C, 24 h.
The synthetic route for compounds 36a‒36o is shown in Scheme 2. Reductive amination was conducted to prepare intermediates 33 with substituted aldehydes 31 and aniline 32. Then, compounds 33 were substituted by γ-butyrolactone to obtain primary alcohols 34, which were oxidized to aldehydes 35. Finally, reductive amination was conducted between intermediates 35 and 18 to afforded target compounds 36a‒36o.
Scheme 2.

Reagents and conditions: (h) MgSO4, NaBH4, MeOH, rt, overnight; (i) γ-butyro-lactone, trimethylaluminum, toluene, 80 °C, 12 h; (j) DMP, DCM, rt, 2 h; (j) 18, MgSO4, NaBH4, MeOH, rt, overnight.
2.4. Biological evaluations and structure–activity relationships
Initially, compounds 23a‒23e with N-monosubstitutions were assayed. As shown in Table 1, these compounds generally showed modest inhibitory activities (KD range: 1538–2186 nmol/L), which were comparable to hit compound 9 (KD = 1471 ± 246 nmol/L). However, it was worth noting that introducing a large size group led to decreased PDEδ binding affinities. For example, naphthalen-1-yl derivative 23c showed the weakest activity (KD = 2186 ± 385 nmol/L).
Table 1.
Structures, binding affinity and anti-pancreatic cancer activities of PDEδ inhibitors.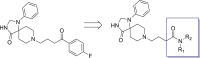
| Compd. | R1 | R2 | PDEδa (KD, nmol/L) | Mia PaCa-2b (IC50, μmol/L) |
|---|---|---|---|---|
| 23a | H | Ph | 1670 ± 260 | 46 ± 14 |
| 23b | H | 2,3-Dihydro-1H-inden-5-yl | 1538 ± 166 | 3.6 ± 2.1 |
| 23c | H | Naphthalen-1-ylmethyl | 2186 ± 385 | 4.8 ± 1.2 |
| 23d | H | Cyclohexyl | 1699 ± 178 | 64 ± 8.1 |
| 23e | H | Cyclopentyl | 1543 ± 244 | >100 |
| 36a | Ph | 2-F-Benzyl | 1311 ± 219 | 16 ± 0.10 |
| 36b | Ph | 4-Cl-Benzyl | 328 ± 59 | 4.3 ± 2.1 |
| 36c | Ph | 2-Br-Benzyl | 485 ± 35 | 12 ± 0.60 |
| 36d | Ph | 3-Br-Benzyl | 690 ± 198 | 4.9 ± 1.4 |
| 36e | Ph | 1H-Imidazole-4-ylmethyl | 1657 ± 436 | >100 |
| 36f | Ph | Thiophen-2-ylmethyl | 1475 ± 197 | 15 ± 5.4 |
| 36g | Ph | Thiophen-3-ylmethyl | 1240 ± 106 | 17 ± 8.3 |
| 36h | Ph | Pyridin-2-ylmethyl | 1373 ± 317 | 46 ± 21 |
| 36i | Ph | Pyridin-4-ylmethyl | 1482 ± 18 | 55 ± 2.8 |
| 36j | Ph | Quinolin-6-ylmethyl | 1343 ± 36 | 12 ± 4.0 |
| 36k | Ph | Quinolin-3-ylmethyl | 1328 ± 32 | 13 ± 2.8 |
| 36l | Ph | Cyclohexylmethyl | 127 ± 16 | 6.7 ± 1.7 |
| 36m | Ph | Cyclopentylmethyl | 159 ± 29 | 18 ± 1.5 |
| 36n | Ph | Cyclobutylmethyl | 393 ± 27 | 19 ± 4.0 |
| 36o | Ph | Cyclopropylmethyl | 1125 ± 140 | 26 ± 1.6 |
| 9 | / | / | 1471 ± 246 | >100 |
| 2 | / | / | 34 ± 4 | >100 |
Tested by fluorescent anisotropy assay.
Tested by the CCK8 method.
When an additional phenyl group was added to the amine, most di-substituted compounds (36) showed comparable or superior activity to the lead compound. In particular, cyclohexyl (compound 36l, KD = 127 ± 16 nmol/L) and cyclopentyl (compound 36m, KD = 159 ± 29 nmol/L) derivatives showed excellent PDEδ binding affinity, which was about 7–9 fold more active than lead compound 9 (KD = 1471 ± 246 nmol/L). In contrast, less steric cyclobutyl (compound 36n) or cyclopropyl (compound 36o) derivative showed decreased activity. Replacement of the cycloalkyl-methyl substitions by benzyl (36a‒36d, KD range: 328–1311 nmol/L) or heterocyclic methyl group (36e‒36k, KD range: 1240–1657 nmol/L) generally led to reduced PDEδ binding affinity. The docking result (Fig. 3A) revealed that the amide substitutions of 36l could engage the hydrophobic pocket of PDEδ protein in which the cyclohexyl moiety formed hydrophobic interactions with residues Trp32, Val145 and Leu147. Moreover, the key hydrogen interactions with Arg61, Gln78 and Tyr149 were retained. The hydrophobic interaction between phenyl moiety and Leu38, Thr131 and Phe133 could be also observed, which was absent in the docking pose of compound 9 (Fig. 3B).
Figure 3.

Molecular docking of 36l with PDEδ protein. (A) Proposed-bonding mode of 36l with PDEδ protein. The red dash line represented hydrogen interaction with residues Arg61, Gln78 and Tyr149. (B) The overlay image binding mode of compounds 36l (yellow) and 9 (green) with PDEδ protein.
Furthermore, in vitro antitumor activity of the target compounds was assayed against human pancreatic cancer cell line Mia PaCa-2 (Table 1). PDEδ inhibitors 9 and 2 failed to exert cellular potency (IC50 > 100 μmol/L). Interestingly, most target compounds showed moderate to good antitumor activity. Among them, compound 36l (KD = 127 ± 16 nmol/L, IC50 = 6.3 ± 1.7 μmol/L) possessed balanced inhibitory activity both at the molecular and cellular level. Furthermore, patient-driven primary cells 098 and 099 were applied to evaluate the potential clinical application of compound 36l (Fig. 4). The 098 and 099 were both KRASG12D mutant pancreatic cancer cells. The result showed that 36l (IC50 range: 6.4–7.6 μmol/L) exhibited potent activity against primary pancreatic cancer cell line, while compound 2 (IC50 > 100 μmol/L) was totally inactive. Moreover, water solubility of inhibitor 36l (0.63 mmol/L) was about 20 fold higher than compound 2 (0.030 mmol/L). Considering the activity and physicochemical properties, compound 36l was chosen for further biological evaluations.
Figure 4.

In vitro inhibitory activity of compound 36l against two clinical pancreatic cancer cell lines. The primary cell lines 098 and 099 (KRASG12D mutation) were treated with 36l at various concentrations for 72 h. The percentages of viability were measured by CCK8 method. The studies with primary cell lines were approved by the Changhai Hospital Ethics Committee. Data were represented as means ± SEM, n = 3.
2.5. The cellular thermal shift assay (CETSA)
The CETSA assay is a useful method for the discovery of the intracellular target of inhibitors. The method evaluates the change in the stability when protein binds with the small molecules, which is fast and convenient without purifying the protein34,35. Mia Paca-2 cells were treated by compound 36l at 20 and 50 μmol/L, respectively, using compound 2 as a positive drug (Fig. 5A). Increased Tagg (aggregation temperature) values were observed in the group treated by compounds 2 and 36l. In addition, compound 36l introduced a higher Tagg value change than that of compound 2 at 50 μmol/L. The experiment verified PDEδ protein was the target of compound 36l in Mia Paca-2 cells.
Figure 5.

Mechanism study of compound 36l. (A) Cellular thermal shift assays of PDEδ. 2 (50 μmol/L) and 36l (20 and 50 μmol/L) were incubated with Mia PaCa-2 cells for 2 h. The results were shown with melt curves. (B) Co-IP assay of compound 36l. Immunoprecipitation antibody: KRAS and control rabbit IgG; Western blotting antibody: PDEδ. Statistical difference was indicated by gray intensity analysis. Data were represented as means ± SEM, n = 3. ∗∗∗P < 0.001.
2.6. Co-immunoprecipitation (Co-IP) experiment
Co-IP is a powerful technique for the identification of protein–protein interaction36,37. Co-IP experiment was further conducted to evaluate the inhibition of KRAS‒PDEδ by compound 36l (Fig. 5B). The cells were treated with compound 36l at 20 μmol/L for 24 h. Western blotting experiment was used to identify KRAS and PDEδ protein, respectively. The level of PDEδ/KRAS in the 36l treating group was lower than the control group. The results confirmed that the compound 36l effectively disrupted the interaction of KRAS‒PDEδ.
2.7. Compound 36l inhibited the phosphorylation of AKT in Mia PaCa-2 cells
The mutant KRAS protein activates downstream signal pathways, such as RAS-RAF-MAPK and PI3K-AKT-mTOR pathway, and then stimulates the growth, proliferation, and differentiation of cancer cells18. Thus, the effects of compound 36l on the phosphorylation of total extracellular signal-regulated kinase (t-ERK) and protein kinase B (t-AKT) were evaluated. Based on our previous work29, the epidermal growth factor (EGF) was applied to induce the expression and phosphorylation of t-ERK and t-AKT. Phosphorylation levels were determined by treating Mia PaCa-2 cells with compounds 36l for 4 h followed by stimulating for another 10 min with EGF. Compound 2 was used as positive control. As shown in Fig. 6, compounds 36l interfered with phosphorylation of t-ERK and t-AKT in a dose-dependent manner in Mia PaCa-2 cells, and significantly reduced their phosphorylation at 100 μmol/L, which was more potent than that of positive compound 2.
Figure 6.

Western blotting analysis of KRAS downstream signal pathways. (A) Phosphorylation levels of t-ERK and t-AKT by stimulated KRAS-dependent Mia PaCa-2 cells with EGF (125 ng/mL, 10 min). From top to bottom: phosphorylated ERK on Thr202 and Tyr204 (p-ERK), total level of ERK (t-ERK), phosphorylated AKT on S473 (p-AKT), total level of AKT (t-AKT), and loading control (GAPDH). (B and C) Gray intensity analysis of the blots in quantification of p-ERK/t-ERK ± SEM (top) and p-AKT/t-AKT ± SEM (bottom) was standardized to the EGF-stimulated control (1% DMSO). Data were represented as means ± SEM, n = 3, ∗∗∗P < 0.001.
2.8. Compound 36l increased KRAS-dependent apoptosis and influenced the distribution of KRAS protein in Mia PaCa-2 cells
The results of apoptosis assay demonstrated that positive control 2 induced cell apoptosis with an apoptotic rate of 28.55% at 50 μmol/L and 38.32% at 100 μmol/L respectively (Fig. 7). Similarly, compound 36l effectively promoted apoptosis of Mia PaCa-2 cells, and induced apoptosis in 19.42%, 31.22% and 54.28% at 5, 10 and 20 μmol/L, respectively. The results suggested that both compounds 2 and 36l induced apoptosis in a dose-dependent manner. Spiro-cyclic inhibitor 36l was more potent even at a lower concentration. Immunofluorescence staining was further applied to evaluate the KRAS distribution in Mia PaCa-2 cells (Fig. 8). Compound 36l was observed to accumulate PDEδ around the cell nucleus and disrupt the distribution of KRAS to endomembrane at 20 μmol/L, while compound 2 showed similar effects at the higher concentration (100 μmol/L).
Figure 7.

Apoptosis assay staining with Annexin V-FITC/PI in the Mia PaCa-2 cells. (A) Representative scatter plots of samples treated with 2 (50 and 100 μmol/L) and compound 36l (5, 10 and 20 μmol/L) for 48 h. (B) and (C) Bar charts show quantitative data of apoptosis assay in Mia PaCa-2 cells.
Figure 8.

Immunostaining of Mia PaCa-2 cells with anti-PDEδ (green) and anti-KRAS (red) by treated with tested compounds (2, 100 μmol/L; 36l, 20 μmol/L) for 4 h. 1% DMSO was used as the vehicle control. Leica confocal microscope was applied for the image collection. Scale bar: 10 μm.
2.9. Compound 36l inhibited proliferation of the pancreatic tumor in patient-derived xenograft (PDX) model
In order to evaluate the in vivo anti-pancreatic tumor activity, patient-derived primary cell lines were applied to detect the anti-proliferative effects of compound 36l (Fig. 9). Four KRAS mutant clinical primary pancreatic cancer cell lines [0001 (KRASG12A), 0034 (KRASG12R), 0037 (KRASG12D) and 0043 (KRASG12D)] were treated with 36l at different time. The results revealed that compound 36l dose-dependently inhibited the growth of primary cell lines. Moreover, 0001, 0037 and 0034 cell lines were more sensitive to inhibitor 36l than the 0043 cell line. Pharmacokinetic study and cellular permeability of 36l were further conducted in ICR mice. The results indicated that the spiro-cyclic inhibitor 36l exhibited acceptable plasma exposures (Cmax = 15.50 μg/mL, AUC0‒t = 59.23 h·μg/mL) and moderate cellular permeability (Supporting Information Fig. S1 and Table S3), which enabled further in vivo studies.
Figure 9.

The primary cell lines [0001 (KRASG12A), 0034 (KRASG12R), 0037 (KRASG12D) and 0043 (KRASG12D)] from Ruijin Hospital were treated with 36l at various concentrations for 24, 48 and 72 h, respectively. The percentages of viability were measured by CCK8 method. All the studies with primary cell lines were approved by the Ruijin Hospital Ethics Committee.
Based on the cellular potency of inhibitor 36l, PDX models were further applied to evaluate the in vivo antitumor potency. Guided by the antiproliferative assay to primary cells, the excised tumor tissue of patients produced 0034 primary cells was implanted into nude female mice. Consideration of the metabolic instability of compound 2, gemcitabine, a cytotoxic antitumor agent, was chosen as the positive drug. As depicted in Fig. 9, Compound 36l significantly inhibited the tumor growth with the tumor growth inhibition (TGI) rate of 57.3% (Fig. 10A and B). Moreover, the significant difference of tumor weight was also observed between compound 36l and the control group (Fig. 10C). Despite that the in vivo antitumor activity of compound 36l was lower than cytotoxic antitumor agent gemcitabine, it was the first KRAS‒PDEδ inhibitor with potent antitumor efficacy in PDX models. To further validate the in vivo antitumor potency, compound 36l (25, 50 and 75 mg/kg, QD) and positive control 2 (50 mg/kg, QD) were evaluated in Mia PaCa-2 pancreatic cancer mouse xenograft models. In consistent with the results from PDX models, compound 36l dose dependently inhibited the tumor growth without obvious adverse effects (Supporting Information Fig. S2). These results highlighted the therapeutic potential of this new class of PDEδ inhibitors in treating pancreatic cancer.
Figure 10.

Therapeutic efficacy of compound 36l in the pancreatic tumor PDX model. When the tumor reached about 100 mm3 (set to day 0), the mice were divided into three group randomly. Then, the groups were respectively treated with the compound 36l (50 mg/kg, QD) intraperitoneally, gemcitabine (30 mg/kg, once every 3 days) and saline intravenously until the endpoint. (A) Effect of 36l on tumor volume growth in PDX models. (B) Effect of 36l on body weight in PDX models. (C) Weight of the PDX tumor treated with vehicle, gemcitabine and 36l. (D) Quantification of Ki-67 positive cells38. (E) Representative images of H&E staining and IHC staining for Ki-67 of tumor tissues treated by vehicle, gemcitabine and 36l. Scale bar: 200 μm. Data were represented as the mean ± standard deviation, n = 5. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001.
Furthermore, hematoxylin-eosin (H&E) staining and immunohisto-chemistry (IHC) for Ki-67 was performed for the investigation of the action mechanism of inhibitor 36l against the pancreatic tumor in vivo. As shown in Fig. 10D and E, hyalinization and reduction of cellularity was observed in the tumor treated with compound 36l (marked by dark arrow respectively) in the images of H&E staining and magnification image, while decreased proliferation was exhibited as shown by Ki-67 immunostaining. The same variation was visible in the gemcitabine group. The results supported the in vivo efficacy of 36l.
3. Conclusions
In summary, new spiro-cyclic KRAS‒PDEδ inhibitors were discovered by screening an in-house compound library and structure-based hit optimization. As compared with the hit compound, inhibitor 36l showed improved PDEδ binding affinity and antitumor activity. In KRAS-dependent Mia PaCa-2 cells, compound 36l promoted the apoptosis, induced down-regulation of t-AKT phosphorylation and disrupted the diffusion of KRAS in the cytoplasm. The spiro-cyclic KRAS‒PDEδ inhibitors revealed remarkable advantages over known ones due to the potent antitumor activity both in vitro and in vivo. In particular, compound 36l showed good therapeutic efficacy in the pancreatic cancer PDX models. Taken together, this study provides a promising lead compound for investigating the druggability of KRAS‒PDEδ protein–protein interaction. Further structural optimization of the spiro-cyclic inhibitors is in progress.
4. Experimental
4.1. Chemistry
All reagents were commercially available and used without further purification. 1H NMR and 13C NMR spectra were recorded on Bruker AVANCE300 or AVANCE600 spectrometer (Bruker Company, Germany). CDCl3 or DMSO-d6 were solvents with TMS as an internal standard. Chemical shifts (δ values) and coupling constants (J values) are given in ppm and Hz, respectively. HRMS spectra were detected on an Esquire 3000 LC‒MS mass spectrometer. Precoated plates GF-254 (Qingdao Haiyang Chemical, China) and silica gel 60G (Qindao Haiyang Chemical) were adopted for monitoring reaction and column chromatography respectively. The purities of final compounds were analyzed by HPLC (Agilent 1260), and were greater than 95%.
4.1.1. 1-Phenyl-1,3,8-triazaspiro[4.5]decan-4-one (18)
The synthesis of intermediate 18 was according to the literature39. White solid. 1H NMR (600 MHz, DMSO-d6) δ 8.91 (s, 1H), 7.21 (dd, 2H, J = 7.5, 8.7 Hz), 6.98 (d, 2H, J = 8.2 Hz), 6.76 (t, 1H, J = 7.5 Hz), 4.60 (s, 2H), 3.43–3.48 (m, 2H), 3.18–3.21 (m, 2H), 3.69–2.75 (m, 2H), 1.73 (d, 2H, J = 14.2 Hz).
4.1.2. 4-Chloro-N-phenylbutanamide (22a)
A mixture of aniline 21a (470 mg, 1 eq, 5 mmol), 4-chlorobutyryl chloride (850 mg, 1.2 eq, 6 mmol), TEA (1.01 g, 2eq, 10 mmol) in DCM (20 mL) was reacted at room temperature for 2 h. The mixture was washed with saturated Na2CO3 solution. The organic layer was combined and washed with brine, then it was evaporated to afford the crude product. Intermediate 22a was purified by flash column chromatography (hexane/ethyl acetate = 75:25) as white solid (yield 51.4%). 1H NMR (300 MHz, DMSO-d6) δ 9.98 (s, 1H), 7.59 (d, 2H, J = 7.7 Hz), 7.28 (t, 2H, J = 7.7 Hz), 7.02 (t, 1H, J = 7.7 Hz), 3.70 (t, 2H, J = 6.8 Hz), 2.48 (t, 2H, J = 6.8 Hz), 1.98–2.07 (m, 2H).
4.1.3. 4-(4-Oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-8-yl)-N-phenylbutanamide (23a)
Intermediate 22a (400 mg, 2 eq, 2 mmol), 18 (230 mg, 1 eq, 1 mmol) and KI (17 mg, 0.1 eq, 0.1 mmol) were added into acetonitrile (10 mL) and reacted under reflux. Additional 22a (1 eq, 1 mmol) was added to the reaction 2 h later. The mixture was reacted for another 24 h under reflux, and then concentrated under reduced pressure and purified by flash column chromatography (dichloromethane/methanol = 100:7) to give target molecules 23a (yield 36.4%). 1H NMR (600 MHz, DMSO-d6) δ 10.02 (s, 1H), 8.69 (s, 1H), 7.52 (d, 2H, J = 8.8 Hz), 7.27 (t, 2H, J = 8.2 Hz), 7.20 (t, 2H, J = 8.2 Hz), 7.11 (t, 1H, J = 7.2 Hz), 6.87 (d, 2H, J = 8.2 Hz), 6.78 (t, 1H, J = 7.6 Hz), 4.57 (s, 2H), 3.18 (br, 4H), 2.78 (t, 2H, J = 7.6 Hz), 2.61–2.66 (m, 2H), 2.37 (t, 2H, J = 6.9 Hz), 1.85–1.90 (m, 2H), 1.73 (d, 2H, J = 14.4 Hz). 13C NMR (150 MHz, DMSO-d6) δ 176.46, 171.46, 143.75, 139.81, 129.47, 129.10, 123.37, 119.50, 118.16, 114.79, 59.14, 58.51, 57.49, 49.59, 34.69, 28.48, 22.53. HRMS: m/z calcd. for C23H28N4O2 392.2212, found 393.2270 [M+H]+. HPLC purity: 95.3%, tR = 8.942 min.
4.1.4. N-(2,3-Dihydro-1H-inden-5-yl)-4-(4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-8-yl)butanamide (23b)
Pale solid, 135 mg, yield 31.3%. Synthesized by the protocol of compound 23a with 22b (475 mg, 2 mmol), 18 (230 mg, 1 mmol) and KI (17 mg, 0.1 mmol) and purified by flash column chromatography (dichloromethane/methanol = 100:7). 1H NMR (600 MHz, DMSO-d6) δ 9.82 (s, 1H), 8.74 (s, 1H), 7.52 (s, 1H), 7.28 (d, 1H, J = 7.9 Hz), 7.20 (t, 2H, J = 7.9 Hz), 7.11 (d, 1H, J = 7.9 Hz), 6.89 (d, 2H, J = 7.9 Hz), 6.75 (t, 1H, J = 7.9 Hz), 4.58 (s, 2H), 3.02 (br, 4H), 2.71–2.81 (m, 4H), 2.63–2.68 (m, 4H), 2.36 (t, 2H, J = 7.9 Hz), 1.96–2.01 (m, 2H), 1.84–1.86 (m, 2H), 1.67 (d, 2H, J = 13.3 Hz). 13C NMR (150 MHz, DMSO-d6) δ 176.20, 170.94, 144.47, 143.65, 138.59, 137.97, 129.50, 124.52, 118.27, 117.73, 115.81, 114.82, 59.20, 58.20, 57.15, 49.43, 34.39, 32.98, 32.23, 27.99, 25.64, 21.99. HRMS: m/z calcd. for C26H32N4O2 432.2525, found 433.2606 [M+H]+. HPLC purity: 99.1%, tR = 12.394 min.
4.1.5. N-(Naphthalen-1-ylmethyl)-4-(4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-8-yl)butanamide (23c)
Pale solid, 129 mg, yield 28.2%. Synthesized by the protocol of compound 23a with 22c (522 mg, 2 mmol), 18 (230 mg, 1 mmol) and KI (17 mg, 0.1 mmol) and purified by flash column chromatography (dichloromethane/methanol = 100:7). 1H NMR (600 MHz, DMSO-d6) δ 8.79 (s, 1H), 8.43 (t, 1H, J = 5.3 Hz), 8.08 (d, 1H, J = 8.8 Hz), 7.95 (d, 1H, J = 7.0 Hz),7.85–7.86 (m, 1H), 7.53–7.59 (m, 2H), 7.45–7.49 (m, 2H), 7.22 (t, 2H, J = 7.9 Hz), 6.90 (d, 2H, J = 8.8 Hz), 6.76 (d, 1H, J = 7.9 Hz), 4.76 (d, 2H, J = 5.3 Hz), 4.60 (s, 2H), 3.03 (br, 4H), 2.64–2.68 (m, 4H), 2.25 (t, 2H, J = 6.1 Hz), 1.81–1.86 (m, 2H), 1.68 (d, 2H, J = 14.1 Hz). 13C NMR (150 MHz, DMSO-d6) δ 175.70, 171.54, 143.16, 134.71, 133.32, 130.91, 129.02, 128.50, 127.58, 126.21, 125.79, 125.61, 125.38, 123.52, 117.86, 114.34, 58.74, 57.63, 56.49, 48.79, 40.23, 32.83, 27.39, 21.58. HRMS: m/z calcd. for C28H32N4O2 456.2525, found 457.2604 [M+H]+. HPLC purity: 99.3%, tR = 11.154 min.
4.1.6. N-Cyclohexyl-4-(4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-8-yl)butan-amide (23d)
White solid, 142 mg, yield 35.7%. Synthesized by the protocol of compound 23a with 22d (410 mg, 2 mmol), 18 (230 mg, 1 mmol) and KI (17 mg, 0.1 mmol) and purified by flash column chromatography (dichloromethane/methanol = 100:7). 1H NMR (600 MHz, DMSO-d6) δ 8.73 (s, 1H), 7.70 (d, 1H, J = 7.6 Hz), 7.25 (d, 2H, J = 8.2 Hz), 6.90 (d, 2H, J = 8.2 Hz), 6.78 (t, 1H, J = 7.2 Hz), 4.6 (s, 2H), 3.52–3.55 (m, 1H), 2.91–2.93 (m, 4H), 2.62–2.64 (m, 2H), 2.11 (t, 2H, J = 8.7 Hz), 1.55–1.75 (m, 9H), 1.23–1.29 (m, 2H), 1.09–1.17 (m, 3H). 13C NMR (150 MHz, CDCl3) δ 176.39, 171.24, 143.77, 129.55, 118.31, 114.87, 59.24, 58.38, 57.32, 49.48, 47.90, 33.70, 33.05, 28.26, 25.79, 25.14, 22.61. HRMS: m/z calcd. for C23H34N4O2 398.2782, found 399.2763 [M+H]+. HPLC purity: 99.2%, tR = 9.850 min.
4.1.7. N-Cyclopentyl-4-(4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-8-yl)butanamide (23e)
Pale solid, 135 mg, yield 35.2%. Synthesized by the protocol of compound 23a with 22e (380 mg, 2 mmol), 18 (230 mg, 1 mmol) and KI (17 mg, 0.1 mmol) and purified by flash column chromatography (dichloromethane/methanol = 100:7). 1H NMR (600 MHz, DMSO-d6) δ 8.71 (s, 1H), 7.78 (d, 1H, J = 7.6 Hz), 7.25 (t, 2H, J = 7.6 Hz), 6.90 (d, 2H, J = 7.6 Hz), 6.78 (t, 1H, J = 7.6 Hz), 4.60 (s, 2H), 3.98–4.01 (m, 1H), 2.86–2.90 (m, 4H), 2.58–2.63 (m, 2H), 2.47–2.50 (m, 2H), 2.10 (t, 2H, J = 7.6 Hz), 1.77–1.81 (m, 2H), 1.70–1.75 (m, 2H), 1.60–1.65 (m, 4H), 1.49–1.51 (m, 2H), 1.33–1.39 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ176.39, 171.70, 143.74, 129.49, 118.26, 114.85, 59.171, 58.402, 57.402, 50.624, 49.491, 33.625, 32.773, 28.338, 23.905, 22.686. HRMS: m/z calcd. for C22H32N4O2 384.2525, found 385.2602 [M+H]+. HPLC purity: 98.9%, tR = 9.887 min.
4.1.8. N-(2-Fluorobenzyl)aniline (33a)
The synthesis of secondary amines 33a was applied the reductive amination procedure according to the literature procedure I22. In brief, a solution of 2-fluorobenzaldehyde 31a (465 mg, 1 eq, 5 mmol) and aniline 32 (0.47 g, 1 eq, 5 mmol) in methanol (25 mL) was treated with MgSO4 (1.2 g, 10 mmol) at room temperature overnight. Sodium borohydride (95 mg, 0.5 eq, 2.5 mmol) was added into the reaction under ice bath and reacted for another 2 h. The mixture was filtered. The methanol kept and removed in vacuo to give the secondary amines. The product 33a (825 mg) was used directly in the next step without purification. 1H NMR (300 MHz, DMSO-d6) δ 7.35 (t, 1H, J = 7.5 Hz), 7.22–7.29 (m, 1H), 7.08–7.17 (m, 2H), 7.01 (t, 2H, J = 7.8 Hz), 6.46–6.55 (m, 2H), 6.16 (t, 2H, J = 5.9 Hz), 4.26 (d, 2H, J = 6.2 Hz).
4.1.9. N-(2-Fluorobenzyl)-4-hydroxy-N-phenylbutanamide (34a)
The secondary amines 33a (600 mg, 1 eq, 3 mmol) was dissolved into dry toluene (10 mL) and protected by nitrogen. The reaction was cooled down to 0 °C, and trimethylaluminum (2 mol/L in toluene, 2 eq, 3 mL) was added to the solution dropwise. The mixture was reacted for 2 h and γ-butyrolactone (510 mg, 2 eq, 6 mmol) was added. The solution was reacted at 65 °C for 24 h. The reaction mixture was moved to ice bath. Then, methanol (5 mL) was added dropwise followed by saturated sodium tartrate solution. Ethyl acetate (30 mL) was used to wash the solution and combined. The crude product was afford by removing the ethyl acetate and purified by flash column chromatography (hexane/ethyl = 40:60–0:100) to give intermediate 34a (105 mg, 12.2% yield). 1H NMR (300 MHz, DMSO-d6) δ 7.05–7.39 (m, 9H), 4.90 (s, 2H), 4.36 (t, 1H, J = 5.1 Hz), 3.28 (t, 2H, J = 6.3 Hz), 2.08 (t, 2H, J = 7.2 Hz), 1.57–1.66 (m, 2H).
4.1.10. N-(2-Fluorobenzyl)-4-oxo-N-phenylbutanamide (35a)
A solution of 34a (145 mg, 1 eq, 0.5 mmol) in dry DCM (15 mL) were stirred at 0 °C added Dess‒Martin reagent (425 mg, 2 eq, 1 mmol). The resulting suspension was reacted for 1 h at room temperature. After completion of the reaction, the solution was added saturated sodium thiosulfate solution followed by saturated Na2CO3 solution. The water phase was washed with DCM twice. The organic layer was combined, washed with brine and evaporated under reduced pressure to afford crude product. The crude product was purified by column chromatography (hexane/ethyl = 75:25) to give target molecules 35a (86 mg, 60.3% yield). 1H NMR (300 MHz, DCCl3) δ 9.80 (s, 1H), 7.31–7.37 (m, 4H), 7.17–7.22 (m, 1H), 7.05–7.10 (m, 3H), 6.92 (t, 1H, J = 8.8 Hz), 4.96 (s, 2H), 2.78 (t, 2H, J = 6.4 Hz), 2.36 (t, 2H, J = 6.4 Hz).
4.1.11. N-(2-Fluorobenzyl)-4-(4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-8-yl)-N-phenylbutanamide (36a)
Target compounds 36a were prepared by the reductive amination by using 35a (142 mg, 1 eq, 0.5 mmol), 18 (115 mg, 1 eq, 0.5 mmol) and MgSO4 (300 mg, 2.5 mmol) in dry methanol (10 mL) as the procedure of preparation of 33a. The product was purified by flash C18 column chromatography [H2O (0.1% TFA)/methanol = 40:60–80:20] to give target molecules 36a (pale solid, 76 mg, yield 30.4%). 1H NMR (600 MHz, DMSO-d6) δ 8.60 (s, 1H), 7.32–7.35 (m, 3H), 7.24–7.27 (m, 2H), 7.18–7.21 (m, 4H), 7.11–7.13 (m, 1H), 7.06–7.09 (m, 1H), 6.75–6.78 (m, 3H), 4.91 (s, 2H), 4.55 (s, 2H), 2.58 (br, 4H), 2.35–2.40 (m, 2H), 2.21 (t, 2H, J = 6.9 Hz), 2.11 (t, 2H, J = 6.7 Hz), 1.65–1.67 (m, 2H), 1.59 (d, 2H, J = 13.5 Hz). 13C NMR (150 MHz, DMSO-d6) δ 163.53, 161.33, 159.69, 157.18, 153.61, 147.75, 135.13, 135.01, 134.64, 132.52, 131.97, 131.92, 131.29, 130.27, 129.67, 129.44, 129.10, 128.57, 128.48, 125.77, 125.65, 118.80, 117.96, 117.10, 116.96, 115.86, 115.82, 115.53, 114.00, 69.18, 66.93, 45.82. MS (ESI): m/z calcd. for C30H33FN4O2 500.2588, found 501.2658 [M+H]+. HPLC purity: 98.7%, tR = 13.036 min.
4.1.12. N-(4-Chlorobenzyl)-4-(4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-8-yl)-N-phenylbutanamide (36b)
Pale solid, 86 mg, yield 33.2%. Synthesized by the protocol of compound 36a with 35b (150 mg, 0.5 mmol) and 18 (115 mg, 0.5 mmol) and purified by flash C18 column chromatography [H2O (0.1% TFA)/methanol = 40:60–80:20]. 1H NMR (600 MHz, DMSO-d6) δ 8.65 (s, 1H), 7.34–7.39 (m, 4H), 7.30 (t, 1H, J = 7.1 Hz), 7.18–7.24 (m, 6H), 6.78–6.82 (m, 3H), 4.86 (s, 2H), 4.58 (s, 2H), 2.71 (d, 4H, J = 7.8 Hz), 2.41–2.46 (m, 2H), 2.23 (t, 2H, J = 6.5 Hz), 2.14 (t, 2H, J = 8.4 Hz), 1.69–1.74 (m, 2H), 1.56 (d, 2H, J = 13.6 Hz). 13C NMR (150 MHz, DMSO-d6) δ 176.00, 171.88, 143.28, 142.14, 136.79, 131.62, 129.82, 129.48, 128.98, 128.27, 128.16, 117.83, 114.48, 58.64, 58.08, 56.64, 51.30, 48.97, 31.23, 28.08, 22.12. HRMS (ESI): m/z calcd. for C30H33ClN4O2 516.2292, found 517.2381 [M+H]+. HPLC purity: 96.9%, tR = 14.829 min.
4.1.13. N-(2-Bromobenzyl)-4-(4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-8-yl)-N-phenylbutanamide (36c)
Yellow solid, 99 mg, yield 35.3%. Synthesized by the protocol of compound 36a with 35c (172 mg, 0.5 mmol) and 18 (115 mg, 0.5 mmol) and purified by flash C18 column chromatography [H2O (0.1% TFA)/methanol = 50:50–80:20]. 1H NMR (600 MHz, DMSO-d6) δ 8.62 (s, 1H), 7.55 (d, 1H, J = 8.1 Hz), 7.27–7.39 (m, 7H), 7.16–7.22 (m, 3H), 6.76–6.79 (m, 3H), 4.93 (s, 2H), 4.56 (s, 2H), 2.56–2.59 (m, 4H), 2.36–2.41 (m, 2H), 2.24 (t, 2H, J = 7.1 Hz), 2.18 (t, 2H, J = 7.7 Hz), 1.66–1.71 (m, 2H), 1.56 (d, 2H, J = 14.1 Hz). 13C NMR (150 MHz, DMSO-d6) δ 176.15, 172.05, 143.34, 142.17, 136.13, 132.47, 129.71, 129.41, 129.09, 128.96, 128.04, 127.73, 122.58, 117.71, 114.40, 58.61, 58.29, 56.82, 52.20, 49.11, 48.58, 31.22, 28.42, 22.47. HRMS (ESI): m/z calcd. for C30H33BrN4O2 560.1787, found 561.1846 [M+H]+. HPLC purity:99.3%, tR = 14.700 min.
4.1.14. N-(3-Bromobenzyl)-4-(4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-8-yl)-N-phenylbutanamide (36d)
Yellow solid, 101 mg, yield 36.3%. Synthesized by the protocol of compound 36a with 35d (172 mg, 0.5 mmol) and 18 (115 mg, 0.5 mmol) and purified by flash C18 column chromatography [H2O (0.1% TFA)/methanol = 50:50–80:20]. 1H NMR (600 MHz, DMSO-d6) δ 8.59 (s, 1H), 7.42 (d, 1H, J = 8.0 H), 7.35–7.37 (m, 3H), 7.28 (t, 1H, J = 7.4 H), 7.25 (t, 1H, J = 7.9 Hz), 7.18–7.21 (m, 5H), 6.75–6.78 (m, 3H), 4.84 (s, 2H), 4.54 (s, 2H), 2.55–2.60 (m, 4H), 2.34–2.39 (m, 2H), 2.22 (t, 2H, J = 7.8 Hz), 2.13 (t, 2H, J = 6.8 Hz), 1.64–1.69 (m, 2H), 1.50 (d, 2H, J = 13.2 Hz). 13C NMR (150 MHz, DMSO-d6) δ 176.15, 172.05, 143.34, 142.17, 141.12, 130.96, 130.40, 129.95, 129.43, 128.55, 128.21, 127.37, 122.02, 118.26, 114.99, 59.08, 58.75, 57.26, 51.87, 49.58, 31.68, 28.88, 22.96. HRMS (ESI): m/z calcd. for C30H33BrN4O2 560.1787, found 561.1865 [M+H]+. HPLC purity: 97.9%, tR = 14.361 min.
4.1.15. N-((1H-Imidazole-4-yl)methyl)-4-(4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-8-yl)-N-phenylbutanamide (36e)
Yellow solid, 61 mg, yield 25.8%. Synthesized by the protocol of compound 36a with 35e (129 mg, 0.5 mmol) and 18 (115 mg, 0.5 mmol) and purified by flash C18 column chromatography [H2O (0.1% TFA)/methanol = 50:50–80:20]. 1H NMR (600 MHz, DMSO-d6) δ 8.97 (s, 1H), 7.20–7.40 (m, 7H), 6.94–6.99 (m, 4H), 6.78 (t, 1H, J = 7.5 Hz), 4.89 (s, 2H), 4.62 (s, 2H), 3.31–3.43 (m, 4H), 2.79–2.95 (m, 4H), 2.16 (br, 2H), 1.90 (br, 2H), 1.79 (d, 2H, J = 14.2 Hz). 13C NMR (150 MHz, DMSO-d6) δ 175.58, 171.52, 144.01, 143.34, 142.44, 129.95, 129.53, 128.64, 128.38, 122.23, 118.43, 114.71, 59.29, 57.42, 56.01, 48.79, 46.59, 31.46, 26.72, 20.18. HRMS (ESI): m/z calcd. for C27H32N6O2 472.2587, found 473.2679 [M+H]+. HPLC purity: 98.0%, tR = 7.85 min.
4.1.16. 4-(4-Oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-8-yl)-N-phenyl-N-(thio-phen-2-ylmethyl)butanamide (36f)
Brown solid, 71 mg, yield 29.1%. Synthesized by the protocol of compound 36a with 35f (136 mg, 0.5 mmol) and 18 (115 mg, 0.5 mmol) and purified by flash C18 column chromatography [H2O (0.1% TFA)/methanol = 50:50–80:20]. 1H NMR (600 MHz, DMSO-d6) δ 8.62 (s, 1H), 7.37–7.41 (m, 3H), 7.31 (t, 1H, J = 7.0 Hz), 7.21 (t, 1H, J = 7.8 Hz), 7.15 (d, 1H, J = 7.4 Hz), 6.88–6.90 (m, 1H), 6.76–6.80 (m, 4H), 4.97 (s, 2H), 4.55 (s, 2H), 2.57 (br, 4H), 2.34–2.39 (m, 2H), 2.20 (t, 2H, J = 6.7 Hz), 2.07 (t, 2H, J = 7.4 Hz), 1.63–1.66 (m, 2H), 1.90 (br, 2H), 1.49 (d, 2H, J = 13.3 Hz). 13C NMR (150 MHz, DMSO-d6) δ 178.01, 173.47, 145.17, 143.78, 141.96, 131.28, 130.97, 130.81, 129.99, 129.68, 128.68, 128.24, 127.82, 119.58, 116.29, 60.46, 60.13, 58.67, 50.94, 48.81, 33.10, 30.25, 24.36. HRMS (ESI): m/z calcd. for C28H32N4O2S 488.2251, found 489.2323 [M+H]+. HPLC purity: 98.9%, tR = 12.334 min.
4.1.17. 4-(4-Oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-8-yl)-N-phenyl-N-(thio-phen-3-ylmethyl)butanamide (36g)
Brown solid, 77 mg, yield 31.5%. Synthesized by the protocol of compound 36a with 35g (136 mg, 0.5 mmol) and 18 (115 mg, 0.5 mmol) and purified by flash C18 column chromatography [H2O (0.1% TFA)/methanol = 50:50–80:20]. 1H NMR (600 MHz, DMSO-d6) δ 8.71 (s, 1H), 7.46–7.47 (m, 1H), 7.38 (t, 2H, J = 7.4 Hz), 7.31 (t, 1H, J = 6.5 Hz), 7.18–7.24 (m, 5H), 6.98 (d, 1H, J = 5.1 Hz), 6.84 (d, 2H, J = 7.8 Hz), 6.79 (d, 1H, J = 7.4 Hz), 4.84 (s, 2H), 4.58 (s, 2H), 2.83–2.86 (m, 4H), 2.49–2.54 (m, 2H), 2.42 (t, 2H, J = 6.9 Hz), 2.12 (t, 2H, J = 6.0 Hz), 1.72–1.76 (m, 2H), 1.60 (d, 2H, J = 13.3 Hz). 13C NMR (150 MHz, DMSO-d6) δ 176.31, 171.82, 143.69, 142.71, 138.94, 129.88, 129.46, 128.63, 128.18, 126.78, 123.33, 118.32, 114.88, 59.16, 58.33, 56.89, 49.29, 47.95, 31.70, 28.21, 22.15. HRMS (ESI): m/z calcd. for C28H32N4O2S 488.2251, found 489.2322 [M+H]+. HPLC purity: 99.7%, tR = 12.682 min.
4.1.18. 4-(4-Oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-8-yl)-N-phenyl-N-(pyridin-2-ylmethyl)butanamide (36h)
Brown solid, 63 mg, yield 26.1%. Synthesized by the protocol of compound 36a with 35h (134 mg, 0.5 mmol) and 18 (115 mg, 0.5 mmol) and purified by flash C18 column chromatography [H2O (0.1% TFA)/methanol = 50:50–80:20]. 1H NMR (600 MHz, DMSO-d6) δ 8.65 (s, 1H), 7.38 (d, 1H, J = 2.9 Hz), 7.71–7.74 (m, 1H), 7.32–7.36 (m, 5H), 7.27 (t, 1H, J = 6.9 Hz), 7.19–7.23 (m, 3H), 6.82 (d, 2H, J = 7.5 Hz), 6.79 (t, 1H, J = 6.9 Hz), 4.93 (s, 2H), 4.56 (s, 2H), 2.71–2.79 (m, 4H), 2.34–2.46 (m, 4H), 2.18 (br, 2H), 1.71 (br, 2H), 1.56 (d, 2H, J = 12.9 Hz). 13C NMR (150 MHz, DMSO-d6) δ 176.41, 172.30, 157.70, 149.36, 143.73, 143.31, 137.08, 129.88, 129.45, 128.53, 128.09, 122.64, 122.26, 118.27, 114.89, 59.13, 58.50, 57.11, 54.77, 49.47, 31.64, 28.49, 22.54. HRMS (ESI): m/z calcd. for C29H32N5O2 483.2634, found 484.2707 [M+H]+. HPLC purity: 97.7%, tR = 9.197 min.
4.1.19. 4-(4-Oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-8-yl)-N-phenyl-N-(pyridin-4-ylmethyl)butanamide (36i)
Brown solid, 68 mg, yield 28.1%. Synthesized by the protocol of compound 36a with 35i (134 mg, 0.5 mmol) and 18 (115 mg, 0.5 mmol) and purified by flash C18 column chromatography [H2O (0.1% TFA)/methanol = 50:50–80:20]. 1H NMR (600 MHz, DMSO-d6) δ 8.72 (s, 1H), 8.50 (br, 1H), 7.17–7.38 (m, 10H), 6.82 (d, 2H, J = 8.7 Hz), 6.75 (t, 1H, J = 7.5 Hz), 4.85 (s, 2H), 4.55 (s, 2H), 2.86–2.90 (m, 4H), 2.50–2.54 (m, 2H), 2.17–2.19 (m, 2H), 1.74–1.76 (m, 2H), 1.60 (d, 2H, J = 13.7 Hz). 13C NMR (150 MHz, DMSO-d6) δ 176.23, 172.45, 150.01, 143.65, 142.69, 130.06, 129.46, 128.48, 128.31, 118.36, 114.91, 59.16, 58.26, 56.78, 49.26, 31.51, 28.09, 21.95. HRMS (ESI): m/z calcd. for C29H33N5O2 483.2634, found 484.2708 [M+H]+. HPLC purity: 99.4%, tR = 8.833 min.
4.1.20. 4-(4-Oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-8-yl)-N-phenyl-N-(quinolin-6-ylmethyl)butanamide (36j)
Brown solid, 88 mg, yield 33.0%. Synthesized by the protocol of compound 36a with 35j (159 mg, 0.5 mmol) and 18 (115 mg, 0.5 mmol) and purified by flash C18 column chromatography [H2O (0.1% TFA)/methanol = 50:50–80:20]. 1H NMR (600 MHz, DMSO-d6) δ 8.84 (dd, 1H, J = 1.6, 4.0 Hz), 8.62 (s, 1H), 8.27 (d, 1H, J = 8.3 Hz), 7.70 (s, 1H), 7.96 (d, 1H, J = 8.8 Hz), 7.63 (dd, 1H, J = 1.9, 8.6 Hz), 7.46–7.48 (m, 1H), 7.30–7.33 (m, 2H), 7.17–7.25 (m, 5H), 6.73–6.77 (m, 3H), 5.05 (s, 2H), 4.54 (s, 2H), 2.55–2.59 (m, 4H), 2.34–2.39 (m, 2H), 2.22 (t, 2H, J = 6.4 Hz), 2.16 (t, 2H, J = 6.4 Hz), 1.60–1.72 (m, 2H), 1.49 (d, 2H, J = 13.6 Hz). 13C NMR (150 MHz, DMSO-d6) δ 176.64, 172.60, 150.75, 147.50, 143.80, 142.73, 136.57, 136.24, 130.15, 129.91, 129.55, 129.41, 128.67, 128.16, 128.07, 127.00, 122.05, 118.22, 114.95, 59.09, 58.78, 57.40, 52.31, 49.60, 31.85, 28.91, 23.07. HRMS (ESI): m/z calcd. for C33H35N5O2 533.2791, found 534.2875 [M+H]+. HPLC purity: 98.9%, tR = 10.733 min.
4.1.21. 4-(4-Oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-8-yl)-N-phenyl-N-(quinolin-3-ylmethyl)butanamide (36k)
Brown solid, 70 mg, yield 26.2%. Synthesized by the protocol of compound 36a with 35k (159 mg, 0.5 mmol) and 18 (115 mg, 0.5 mmol) and purified by flash C18 column chromatography [H2O (0.1% TFA)/methanol = 50:50–80:20]. 1H NMR (600 MHz, DMSO-d6) δ 8.74 (d, 1H, J = 1.7 Hz), 8.67 (s, 1H), 8.10 (s, 1H), 7.98 (d, 1H, J = 8.7 Hz), 7.93 (d, 2H, J = 7.7 Hz), 7.71–7.74 (m, 1H), 7.57–7.58 (m, 1H), 7.33 (t, 2H, J = 7.7 Hz), 7.18–7.31 (m, 5H), 6.75–6.82 (m, 3H), 5.08 (s, 2H), 4.57 (s, 2H), 2.75–2.80 (m, 4H), 2.42–2.46 (m, 4H), 2.17 (t, 2H, J = 6.3 Hz), 1.73–1.78 (m, 2H), 1.59 (d, 2H, J = 13.5 Hz). 13C NMR (150 MHz, DMSO-d6) δ 176.36, 172.44, 151.47, 147.26, 143.70, 142.43, 135.02, 131.19, 130.05, 129.76, 129.45, 129.12, 128.76, 128.40, 128.31, 127.83, 127.25, 118.36, 114.99, 59.14, 58.41, 57.00, 50.37, 49.40, 31.73, 28.35, 22.30. HRMS (ESI): m/z calcd. for C33H35N5O2 533.2791, found 534.2876 [M+H]+. HPLC purity: 96.8%, tR = 11.406 min.
4.1.22. N-(Cyclohexylmethyl)-4-(4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-8-yl)-N-phenylbutanamide (36l)
Pale solid, 85 mg, yield 34.8%. Synthesized by the protocol of compound 36a with 35l (136 mg, 0.5 mmol) and 18 (115 mg, 0.5 mmol) and purified by flash C18 column chromatography [H2O (0.1% TFA)/methanol = 50:50–80:20]. 1H NMR (600 MHz, DMSO-d6) δ 8.59 (s, 1H), 7.43 (t, 2H, J = 7.5 Hz), 7.29–7.33 (m, 3H), 7.21–7.24 (m, 2H), 6.77–6.79 (m, 3H), 4.93 (s, 2H), 3.51 (d, 2H, J = 7.5 Hz), 2.53–2.55 (m, 4H), 2.33–2.39 (m, 2H), 2.17 (t, 2H, J = 6.2 Hz), 2.02 (t, 2H, J = 7.5 Hz), 1.56–1.64 (m, 7H), 1.50 (d, 2H, J = 13.0 Hz), 1.33–1.35 (m, 1H), 1.06–1.11 (m, 3H), 0.87–0.92 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 176.23, 171.75, 143.39, 142.96, 129.58, 129.02, 128.24, 127.55, 117.80, 114.49, 58.66, 58.36, 56.98, 54.03, 49.14, 35.73, 31.50, 30.27, 28.45, 26.06, 25.37, 22.62. HRMS (ESI): m/z calcd. for C30H40N4O2 488.3151, found 489.3231 [M+H]+. HPLC purity: 99.0%, tR = 16.157 min.
4.1.23. N-(Cyclopentylmethyl)-4-(4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-8-yl)-N-phenylbutanamide (36m)
Pale solid, 86 mg, yield 36.3%. Synthesized by the protocol of compound 36a with 35m (130 mg, 0.5 mmol) and 18 (115 mg, 0.5 mmol) and purified by flash C18 column chromatography [H2O (0.1% TFA)/methanol = 50:50–80:20]. 1H NMR (600 MHz, DMSO-d6) δ 8.70 (s, 1H), 7.49 (t, 2H, J = 8.1 Hz), 7.30–7.35 (m, 3H), 7.22 (t, 2H, J = 7.9 Hz), 6.77–6.82 (m, 3H), 4.57 (s, 2H), 3.61 (d, 2H, J = 7.9 Hz), 2.76 (br, 4H), 2.48–2.50 (m, 2H), 2.35 (t, 2H, J = 6.0 Hz), 2.02 (t, 2H, J = 6.9 Hz), 1.87–1.92 (m, 1H), 1.66–1.68 (m, 2H), 1.54–1.60 (m, 6H), 1.43–1.45 (m, 2H), 1.15–1.19 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 175.90, 171.34, 143.25, 142.47, 129.53, 128.99, 128.32, 127.59, 117.80, 114.38, 58.67, 57.94, 56.59, 52.48, 48.87, 37.75, 31.42, 29.72, 27.86, 24.73, 21.89. HRMS (ESI): m/z calcd. for C29H38N4O2 474.2995, found 475.3070 [M+H]+. HPLC purity: 99.2%, tR = 15.359 min.
4.1.24. N-(Cyclobutylmethyl)-4-(4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-8-yl)-N-phenylbutanamide (36n)
White solid, 75 mg, yield 32.6%. Synthesized by the protocol of compound 36a with 35n (122 mg, 0.5 mmol) and 18 (115 mg, 0.5 mmol) and purified by flash C18 column chromatography [H2O (0.1% TFA)/methanol = 50:50–80:20]. 1H NMR (600 MHz, DMSO-d6) δ 8.67 (s, 1H), 7.43 (t, 2H, J = 7.6 Hz), 7.34 (t, 2H, J = 7.3 Hz), 7.21–7.26 (m, 4H), 6.77–6.81 (m, 3H), 4.56 (s, 2H), 3.70 (d, 1H, J = 7.6 Hz), 2.71–2.72 (m, 4H), 2.42–2.47 (m, 2H), 2.29–2.37 (m, 3H), 2.00 (t, 2H, J = 7.1 Hz), 1.85–1.91 (m, 2H), 1.73–1.79 (m, 2H), 1.63–1.65 (m, 2H), 1.54–1.61 (m, 4H). 13C NMR (150 MHz, DMSO-d6) δ 175.95, 171.31, 143.26, 142.43, 129.47, 128.98, 128.33, 127.63, 117.79, 114.40, 58.65, 57.99, 56.68, 53.04, 48.94, 33.61, 31.35, 27.99, 25.67, 22.05, 17.80. HRMS (ESI): m/z calcd. for C28H36N4O2 460.2838, found 461.2914 [M+H]+. HPLC purity: 99.2%, tR = 14.265 min.
4.1.25. N-(Cyclopropylmethyl)-4-(4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-8-yl)-N-phenylbutanamide (36o)
White solid, 74 mg, yield 33.2%. Synthesized by the protocol of compound 36a with 35o (116 mg, 0.5 mmol) and 18 (115 mg, 0.5 mmol) and purified by flash C18 column chromatography [H2O (0.1% TFA)/methanol = 50:50–80:20]. 1H NMR (600 MHz, DMSO-d6) δ 8.65 (s, 1H), 7.45 (t, 2H, J = 6.9 Hz), 7.31–7.37 (m, 3H), 7.21–7.24 (m, 2H), 6.77–6.83 (m, 3H), 4.57 (s, 2H), 3.51 (d, 1H, J = 6.9 Hz), 2.69 (br, 4H), 2.45–2.48 (m, 2H), 2.29 (t, 2H, J = 8.1 Hz), 2.00 (t, 2H, J = 6.9 Hz), 1.64–1.69 (m, 2H), 1.55 (d, 2H, J = 13.0 Hz), 0.85–0.91 (m, 1H), 0.36–0.39 (m, 2H), 0.04–0.05 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 176.48, 171.71, 143.75, 143.17, 129.91, 129.45, 128.99, 128.12, 118.25, 114.89, 59.13, 58.57, 57.26, 52.99, 49.47, 31.90, 28.57, 22.65, 10.34, 3.90. HRMS (ESI): m/z calcd. for C27H34N4O2 446.2682, found 447.2757 [M+H]+. HPLC purity: 97.2%, tR = 12.547 min.
4.2. Fluorescence anisotropy assay
The assay was carried out using our established protocol previously26. In Brief, 80 nmol/L PDEδ protein and 25 nmol/L Atorvastatin-FITC probe in PBS buffer were mix in the plate (Corning 3650). The compounds with various concentrations were added to each well. PBS buffer was supplied until the volume reached 200 μL. The plate was incubated at 30 °C for 10 h. The fluorescence anisotropy and fluorescence intensity were detected by Bioteck Synergy H2 with 485 nm excitation and 528 nm emission. The results were calculated by Mathematic 9.0 (Wolfram Research).
4.3. Molecular docking
The crystal complex 5X7426 was chosen for the docking study. Firstly, the reliability of the docking method was established with the following procedure: 1) the protocol of protein preparation was according to previous works. The water molecules were removed, hydrogen atoms were complemented and energy minimized with OPLS2005 to the RMSD reaching 0.30 Å; 2) the program LigPrep was used for ligand preparation with OPLS2005 force-field. Other settings follow the default parameters. 20 low energy ring conformations were generated for each ligand; 3) the protein grid was generated by Receptor Grid Generation using H-bond constraints with Agr61, Gln78, Glu88 and Try149. Extra Precision (XP) and H-bond constraints were applied in the docking study. The figures were generated using PyMol academic version (http://pymol.sourceforge.net/).
4.4. Determination of the solubility of compounds in water
The solubility assay was conducted according to the literature40. In brief, 5 mg compound was dissolved in 1 mL PBS (pH = 7.4) for 24 h in 37 °C. Then the suspension was filtered to afford the saturated solution. Standards in acetonitrile (0.2 mL) with concentration at 1, 0.5, 0.25, 0.125, and 0.0625 mg/mL and saturated solution were added into a quartz 96-well plate. Acetonitrile and PBS solution was added as blank respectively. UV absorbance value at 254 nm was determined with Biotek Synergy H2. The solubility was calculated using the absorbance value and concentration of standards according to Lambert–Beer law.
4.5. Cell culture
Mia PaCa-2 cells were cultured in DMEM (Hyclone) containing 10% fetal bovine serum (Gibco), 2.5% horse serum (Biosharp), 100 U/mL penicillin and 100 μg/mL streptomycin (Gibco). The primary cell lines were cultured in 1640 (Hyclone) containing 10% fetal bovine serum (Gibco), 100 U/mL penicillin and 100 μg/mL streptomycin (Gibco). Cells were maintained in a humidified incubator with 95% air/5% CO2 at 37 °C.
4.6. In vitro antiproliferative assay
Mia PaCa-2 cell line and primary cell lines (0001, 0034, 0037 and 0043) were chosen to determine the inhibitory activity towards pancreatic cancer. The density was 5000 per well in 96-well plates. After cultured for 24 h, each compound was added to triplicate wells at different concentrations and 0.5% DMSO for control. After 72 h of incubation, the medium was removed and 10% CCK8 in DMEM was added to each well following incubating further for the suitable time at 37 °C. The absorbance (OD) at 450 nm was quantitated with Biotek Synergy H2. Wells with no cells and drugs were used as blanks. The concentration inhibited cell growth by 50% (IC50) was calculated by GraphPad Prism 5.0.
4.7. Cell apoptosis assay
Mia PaCa-2 cells were seeded at a density of 3 × 105 cells per well in 6-well plates. After 24 h, the cells were treated with compounds of different concentrations for the proper time. Cells were collected and treated with Annexin V-FITC and PI according to the manufacture's protocol. The stained cells were analyzed by a flow cytometer (BD Accuri C6).
4.8. Western blotting
Mia PaCa-2 cells were seeded at a density of 6 × 105 cells per well in 6-well plates. Then cells were starved and treated with EGF followed by compounds for 2 h as the procedure reported. Then, the protein was extracted, denatured and run on SDS-PAGE gels. Each sample was loaded with 30 μg total protein. The gels were electroblotted onto polyvinylidene fluoride membrane (0.22 μm) and blocked for 2 h at room temperature. The primary antibodies used for western blotting were: p44/42 MAPK (ERK1/2) (Cell Signaling Technology # 9107, 1:1000), anti-Phospho AKT (Cell Signaling Technology # 4060, 1:800), anti-AKT1 (Cell Signaling Technology, 1:1000), GAPDH (Abcam, 1:1000), Phospho p44/42 MAPK (ERK1/2) (Thr202/Tyr204) (Cell Signaling Technology # 4370, 1:1000). And secondary FITC-labeled antibodies including goat anti rabbit IgG (Abcam, 1:5000) and goat anti mouse IgG (Abcam, 1:8000) were applied. The blots were scanned on the LI-COR Odyssey imaging system. The gray values of the bands were applied for quantification of the protein level while the control group as the standard.
4.9. CETSA41,42
Prepared the cell in 4 separate 10 cm flasks. Complete medium containing the different concentrations of compounds were added, and the control group containing 1% DMSO. Then cultured the cells for another 2 h. Harvested and re-suspended the cells with 1 mL PBS buffer with protease inhibitors, then divided into 10 tubes. Heated the tubes successively at different temperatures for 3 min, followed by incubation at room temperature for 3 min. Snap-freeze the tube in liquid nitrogen for 3 circles. The cell lysates were reserved on ice after the third snap-freeze. Centrifuge the tubes at 4 °C, 12,000 rpm, 15 min. The supernatant was kept and the loading buffer was added. Then heated the mixture at 70 °C for 10 min. Western blotting experiments and gray-scale statistics were conducted to quantify the protein.
4.10. Co-IP43,44
Six dishes of Mia PaCa-2 cells were prepared. Renewed the medium with 20 μmol/L compound 36l and 1% DMSO to every 3 dishes respectively and incubated for another 2 h. Harvested and combined the cells, then added 500 μL of IP lysate (Beyotime, containing 1% protease inhibitor) and lysed the cells on ice for 30 min. The prepared Protein A/G beads (Santa Cruz) were added into the cell lysates. Shacked the tubes at 4 °C for 1 h followed by a centrifuge at 4 °C, 12,000 rpm, 15 min. Quantify the supernatant by the BCA method and adjust the total protein concentration to 5 μg/mL. Respectively added KRAS primary antibody (Abcam #ab275876) and IgG (Abcam #ab125938) antibody to 100 μL the above lysates followed by rotating at 4 °C for 1 h. Add the Protein A/G beads 20 μL respectively into the supernatant above, and rotated on the shaker overnight. Centrifuged and collected the precipitate followed by washed it with 500 μL PBS buffer 3 times. Centrifuged and collected the precipitate followed by denaturation in 2 × loading buffer 60 μL at 95 °C for 10 min. Western blotting experiments and gray-scale statistics were conducted to quantify the KRAS and PDEδ protein.
4.11. Immunostaining
After 24 h, the cells were incubated with either 1% DMSO or compounds for 2 h, and then treated as following steps: 1) fixated with 4% paraformaldehyde for 10 min, 2) permeabilized with PBS/0.1% Triton-X for 10 min, 3) blocked with donkey serum blocking buffer [10% donkey serum (Solarbio) in PBS] at a period of 60 min, washed the cells with PBS three times between each steps. The anti-Pan-RAS mouse monoclonal antibody (Calbiochem # OP40-100UG; 1:40) and anti-PDEδ antibody (Genetax # GTX109240, 1:500) were mixed and incubated in blocking buffer overnight at 4 °C. Afterwards, the cells were washed with PBS three times. The secondary antibody, Dylight 649 nm goat anti-Mouse antibody (Abbkine; 1:1000) and Alexa 488 nm goat anti-rabbit antibody (abcam #ab150077, 1:1000) was incubated for another 2 h. The cells were washed three times with PBST, and the images were collected by confocal microscopy (Leica TCS SP5 equipped with Argon-Heli-umNenon laser).
4.12. Pharmacokinetic study of compound 36lin vivo
The animal use and care protocols and the experimental procedures were approved by the Committee on Ethics of Biomedicine, Second Military Medical University (Shanghai, China). The compound 36l were chosen for pharmacokinetic study with male ICR mice. The compounds were dissolved in saline containing 1% DMSO and 0.05% CMC-Na and administered intraperitoneally at a dose of 100 mg/kg. Blood samples about 0.05 mL were collected at 1, 2, 4, and 8 h in micro-tubes containing 0.1% heparin. The plasma were afforded by centrifuging the blood samples for 10 min at 4000 rpm, which were further precipitated protein with methanol, centrifuged for 5 min at 12,000 rpm. The supernatant was kept and detected by HPLC. HPLC condition: water (0.1 TFA)/methanol = 40:60–0:100 (15 min), flow rate = 0.6 mL/min. Peak areas of each sample were detected and converted to concentration according to the standard curve. The bioavailability was calculated by GraphPad Prism 5.0.
4.13. Study of in vivo efficacy in PDX mouse model
The PDX model was constructed as the literature45. All the studies with mice were approved by the Ruijin Hospital Ethics Committee. In brief, the nude female mice were implanted with passaged PDXs tumors using a trocar. When the tumor reached 100 mm3, the mice were divided into three group randomly. The mice were respectively treated with the compound gemcitabine (30 mg/kg, once every 3 days), saline intravenously and 36l (50 mg/kg, QD) intraperitoneally until the endpoint. The body weight were evaluated. The tumor size was calculated using formula (L × W2) × (π/6), where L is the long axis and W is the width axis. Tumor size and body weight were recorded every 3 days over the course of study. Mice were sacrificed on Day 14 and all tumors were harvested and analyzed.
4.14. H&E staining and immunochemical staining
All tumor samples were fixed in 10% formaldehyde overnight, then dehydrated in graded concentrations of ethanol. For hematoxylin and eosin (H&E) staining, slides were stained with Mayer's hematoxylin for 2 min, blued in 0.1% sodium bicarbonate for 1 min, washed in water and counterstained with Eosin Y solution for 1 min. For immunohistochemistry, sections were fixed onto poly-l-lysine coated slides. The slides were incubated with rabbit anti-Ki67 antibody (1:500, GB111141, Servicebio, China) in a humid incubator at 4 °C overnight. The secondary antibody system (PV9000, Golden Bridge International, Beijing, China) was followed according to the manufacturer's instructions. A positive staining result was recorded when the nucleus of cells was stained yellow or brown. Per slide were randomly chosen to measure the proportion of immunopositive cells. Quantification of Ki-67 positive drug was calculated by ImageJ 1.51.
4.15. Statistical analysis
The Student's t-test and one-way analysis of variance were used for comparison among all different groups represented with the mean values ± standard errors. A probability of 0.05 or less (∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001) was considered statistically significant.
Acknowledgments
This work was supported by the National Key R&D Program of China (Grant No. 2020YFA0509100), the National Natural Science Foundation of China (Grants 21738002, 82030105, 81725020 and 81903436).
Author contributions
Long Chen synthesized the target compounds and completed most biological and cell-based assays. Jing Zhang and Xinjing Wang constructed the (PDX) models and completed the in vivo antitumor assays. Yu Li and Lu Zhou participated in research design and conducted experiments. Xiongxiong Lu, Guoqiang Dong and Chunquan Sheng proposed the project, performed data analysis and contributed to the writing–review&editing of the manuscript. All authors have given approval to the final version of the manuscript.
Conflicts of interest
The authors have no conflicts of interest to declare.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting information to this article can be found online at https://doi.org/10.1016/j.apsb.2021.07.009.
Contributor Information
Xiongxiong Lu, Email: simone515night@126.com.
Guoqiang Dong, Email: gdong@smmu.edu.cn.
Chunquan Sheng, Email: shengcq@hotmail.com, shengcq@smmu.edu.cn.
Appendix ASupplementary data
The following is the Supplementary data to this article:
References
- 1.Kamerkar S., LeBleu V.S., Sugimoto H., Yang S., Ruivo C.F., Melo S.A., et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017;546:498–503. doi: 10.1038/nature22341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu Z., Xiao S., Hao H., Hou Q., Fu X. Kirsten rat sarcoma viral oncogene homologue (kras) mutations in the occurrence and treatment of pancreatic cancer. Curr Top Med Chem. 2019;19:2176–2186. doi: 10.2174/1568026619666190828160804. [DOI] [PubMed] [Google Scholar]
- 3.Gillson J., Ramaswamy Y., Singh G., Gorfe A.A., Pavlakis N., Samra J., et al. Small molecule KRAS inhibitors: the future for targeted pancreatic cancer therapy?. Cancers (Basel) 2020;12:1341–1349. doi: 10.3390/cancers12051341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bryant K.L., Mancias J.D., Kimmelman A.C., Der C.J. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci. 2014;39:91–100. doi: 10.1016/j.tibs.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nagasaka M., Li Y., Sukari A., Ou S.I., Al-Hallak M.N., Azmi A.S. KRAS G12C game of thrones, which direct KRAS inhibitor will claim the iron throne?. Cancer Treat Rev. 2020;84:101974. doi: 10.1016/j.ctrv.2020.101974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Misale S., Fatherree J.P., Cortez E., Li C., Bilton S., Timonina D., et al. KRAS G12C NSCLC models are sensitive to direct targeting of KRAS in combination with PI3K inhibition. Clin Cancer Res. 2019;25:796–807. doi: 10.1158/1078-0432.CCR-18-0368. [DOI] [PubMed] [Google Scholar]
- 7.Nollmann F.I., Ruess D.A. Targeting mutant KRAS in pancreatic cancer: futile or promising?. Biomedicines. 2020;8:281–294. doi: 10.3390/biomedicines8080281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seton-Rogers S. KRAS-G12C in the crosshairs. Nat Rev Cancer. 2020;20:3. doi: 10.1038/s41568-019-0228-3. [DOI] [PubMed] [Google Scholar]
- 9.Canon J., Rex K., Saiki A.Y., Mohr C., Cooke K., Bagal D., et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575:217–223. doi: 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- 10.Hata A.N., Shaw A.T. Resistance looms for KRAS(G12C) inhibitors. Nat Med. 2020;26:169–170. doi: 10.1038/s41591-020-0765-z. [DOI] [PubMed] [Google Scholar]
- 11.Veluswamy R., Mack P.C., Houldsworth J., Elkhouly E., Hirsch F.R. KRAS G12C-mutant non-small cell lung cancer: biology, developmental therapeutics, and molecular testing. J Mol Diagn. 2021;23:507–520. doi: 10.1016/j.jmoldx.2021.02.002. [DOI] [PubMed] [Google Scholar]
- 12.Kim D., Xue J.Y., Lito P. Targeting KRAS(G12C): from inhibitory mechanism to modulation of antitumor effects in patients. Cell. 2020;183:850–859. doi: 10.1016/j.cell.2020.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grapsa D., Syrigos K. Direct KRAS inhibition: progress, challenges, and a glimpse into the future. Expert Rev Anticancer Ther. 2020;20:437–440. doi: 10.1080/14737140.2020.1760093. [DOI] [PubMed] [Google Scholar]
- 14.Khan I., Rhett J.M., O'Bryan J.P. Therapeutic targeting of RAS: new hope for drugging the "undruggable". Biochim Biophys Acta Mol Cell Res. 2020;1867:118570–118585. doi: 10.1016/j.bbamcr.2019.118570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones G.D., Caso R., Tan K.S., Mastrogiacomo B., Sanchez-Vega F., Liu Y., et al. KRAS (G12C) mutation is associated with increased risk of recurrence in surgically resected lung adenocarcinoma. Clin Cancer Res. 2021;27:2604–2612. doi: 10.1158/1078-0432.CCR-20-4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suladze S., Ismail S., Winter R. Thermodynamic, dynamic and solvational properties of PDEdelta binding to farnesylated cystein: a model study for uncovering the molecular mechanism of PDEdelta interaction with prenylated proteins. J Phys Chem B. 2014;118:966–975. doi: 10.1021/jp411466r. [DOI] [PubMed] [Google Scholar]
- 17.Zimmermann G., Papke B., Ismail S., Vartak N., Chandra A., Hoffmann M., et al. Small molecule inhibition of the KRAS–PDEdelta interaction impairs oncogenic KRAS signalling. Nature. 2013;497:638–642. doi: 10.1038/nature12205. [DOI] [PubMed] [Google Scholar]
- 18.Chandra A., Grecco H.E., Pisupati V., Perera D., Cassidy L., Skoulidis F., et al. The GDI-like solubilizing factor PDEdelta sustains the spatial organization and signalling of Ras family proteins. Nat Cell Biol. 2011;14:148–158. doi: 10.1038/ncb2394. [DOI] [PubMed] [Google Scholar]
- 19.Martin-Gago P., Fansa E.K., Wittinghofer A., Waldmann H. Structure-based development of PDEdelta inhibitors. Biol Chem. 2017;398:535–545. doi: 10.1515/hsz-2016-0272. [DOI] [PubMed] [Google Scholar]
- 20.Papke B., Murarka S., Vogel H.A., Martin-Gago P., Kovacevic M., Truxius D.C., et al. Identification of pyrazolopyridazinones as PDEdelta inhibitors. Nat Commun. 2016;7:11360–11368. doi: 10.1038/ncomms11360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murarka S., Martin-Gago P., Schultz-Fademrecht C., Al Saabi A., Baumann M., Fansa E.K., et al. Development of pyridazinone chemotypes targeting the pdedelta prenyl binding site. Chemistry (Easton) 2017;23:6083–6093. doi: 10.1002/chem.201603222. [DOI] [PubMed] [Google Scholar]
- 22.Martin-Gago P., Fansa E.K., Klein C.H., Murarka S., Janning P., Schurmann M., et al. A PDE6delta-KRas inhibitor chemotype with up to seven H-bonds and picomolar affinity that prevents efficient inhibitor release by Arl2. Angew Chem Int Ed Engl. 2017;56:2423–2428. doi: 10.1002/anie.201610957. [DOI] [PubMed] [Google Scholar]
- 23.Chen D., Chen Y., Lian F., Chen L., Li Y., Cao D., et al. Fragment-based drug discovery of triazole inhibitors to block PDEdelta-RAS protein‒protein interaction. Eur J Med Chem. 2019;163:597–609. doi: 10.1016/j.ejmech.2018.12.018. [DOI] [PubMed] [Google Scholar]
- 24.Leung E.L., Luo L.X., Li Y., Liu Z.Q., Li L.L., Shi D.F., et al. Identification of a new inhibitor of KRAS‒PDEdelta interaction targeting KRAS mutant nonsmall cell lung cancer. Int J Cancer. 2019;145:1334–1345. doi: 10.1002/ijc.32222. [DOI] [PubMed] [Google Scholar]
- 25.Siddiqui F.A., Alam C., Rosenqvist P., Ora M., Sabt A., Manoharan G.B., et al. PDE6D inhibitors with a new design principle selectively block K-Ras activity. ACS Omega. 2020;5:832–842. doi: 10.1021/acsomega.9b03639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang Y., Zhuang C., Chen L., Lu J., Dong G., Miao Z., et al. Structural biology-inspired discovery of novel KRAS-PDEdelta inhibitors. J Med Chem. 2017;60:9400–9406. doi: 10.1021/acs.jmedchem.7b01243. [DOI] [PubMed] [Google Scholar]
- 27.Chen L., Zhuang C., Lu J., Jiang Y., Sheng C. Discovery of novel KRAS-PDEdelta inhibitors by fragment-based drug design. J Med Chem. 2018;61:2604–2610. doi: 10.1021/acs.jmedchem.8b00057. [DOI] [PubMed] [Google Scholar]
- 28.Dong G., Chen L., Zhang J., Liu T., Du L., Sheng C., et al. Discovery of turn-on fluorescent probes for detecting PDEdelta protein in living cells and tumor slices. Anal Chem. 2020;92:9516–9522. doi: 10.1021/acs.analchem.0c00335. [DOI] [PubMed] [Google Scholar]
- 29.Cheng J., Li Y., Wang X., Dong G., Sheng C. Discovery of novel PDEdelta degraders for the treatment of KRAS mutant colorectal cancer. J Med Chem. 2020;63:7892–7905. doi: 10.1021/acs.jmedchem.0c00929. [DOI] [PubMed] [Google Scholar]
- 30.Wang S., Zhang Y., Dong G., Wu S., Zhu S., Miao Z., et al. Asymmetric synthesis of chiral dihydrothiopyrans via an organocatalytic enantioselective formal thio [3 + 3] cycloaddition reaction with binucleophilic bisketone thioethers. Org Lett. 2013;15:5570–5573. doi: 10.1021/ol4027705. [DOI] [PubMed] [Google Scholar]
- 31.Wang S., Jiang Y., Wu S., Dong G., Miao Z., Zhang W., et al. Meeting organocatalysis with drug discovery: asymmetric synthesis of 3,3ʹ-spirooxindoles fused with tetrahydrothiopyrans as novel P53-MDM2 inhibitors. Org Lett. 2016;18:1028–1031. doi: 10.1021/acs.orglett.6b00155. [DOI] [PubMed] [Google Scholar]
- 32.Wu S., Li Y., Xu G., Chen S., Zhang Y., Liu N., et al. Novel spiropyrazolone antitumor scaffold with potent activity: design, synthesis and structure‒activity relationship. Eur J Med Chem. 2016;115:141–147. doi: 10.1016/j.ejmech.2016.03.039. [DOI] [PubMed] [Google Scholar]
- 33.Wang S., Chen S., Guo Z., He S., Zhang F., Liu X., et al. Synthesis of spiro-tetrahydrothiopyran-oxindoles by Michael-aldol cascade reactions: discovery of potential P53-MDM2 inhibitors with good antitumor activity. Org Biomol Chem. 2018;16:625–634. doi: 10.1039/c7ob02726e. [DOI] [PubMed] [Google Scholar]
- 34.Chambers M., Delport A., Hewer R. The use of the cellular thermal shift assay for the detection of intracellular beta-site amyloid precursor protein cleaving enzyme-1 ligand binding. Mol Biol Rep. 2021;48:2957–2962. doi: 10.1007/s11033-021-06229-9. [DOI] [PubMed] [Google Scholar]
- 35.Chernobrovkin A.L., Cazares-Korner C., Friman T., Caballero I.M., Amadio D., Martinez Molina D. A tale of two tails: efficient profiling of protein degraders by specific functional and target engagement readouts. SLAS Discov. 2021;26:534–546. doi: 10.1177/2472555220984372. [DOI] [PubMed] [Google Scholar]
- 36.Husain A., Begum N.A., Kobayashi M., Honjo T. Native co-immunoprecipitation assay to identify interacting partners of chromatin-associated proteins in mammalian cells. Bio Protoc. 2020;10:e3837. doi: 10.21769/BioProtoc.3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang J., He S. Co-immunoprecipitation assay for blue light-dependent protein interactions in plants. Methods Mol Biol. 2021;2297:141–146. doi: 10.1007/978-1-0716-1370-2_14. [DOI] [PubMed] [Google Scholar]
- 38.Del Rosario Taco Sanchez M., Soler-Monso T., Petit A., Azcarate J., Lasheras A., Artal C., et al. Digital quantification of KI-67 in breast cancer. Virchows Arch. 2019;474:169–176. doi: 10.1007/s00428-018-2481-3. [DOI] [PubMed] [Google Scholar]
- 39.Morciano G., Preti D., Pedriali G., Aquila G., Missiroli S., Fantinati A., et al. Discovery of novel 1,3,8-triazaspiro[4.5]decane derivatives that target the c subunit of f1/fo-adenosine triphosphate (ATP) synthase for the treatment of reperfusion damage in myocardial infarction. J Med Chem. 2018;61:7131–7143. doi: 10.1021/acs.jmedchem.8b00278. [DOI] [PubMed] [Google Scholar]
- 40.Roy D., Ducher F., Laumain A., Legendre J.Y. Determination of the aqueous solubility of drugs using a convenient 96-well plate-based assay. Drug Dev Ind Pharm. 2001;27:107–109. doi: 10.1081/ddc-100000135. [DOI] [PubMed] [Google Scholar]
- 41.Jafari R., Almqvist H., Axelsson H., Ignatushchenko M., Lundback T., Nordlund P., et al. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protoc. 2014;9:2100–2122. doi: 10.1038/nprot.2014.138. [DOI] [PubMed] [Google Scholar]
- 42.Seashore-Ludlow B., Axelsson H., Lundback T. Perspective on CETSA literature: toward more quantitative data interpretation. SLAS Discov. 2020;25:118–126. doi: 10.1177/2472555219884524. [DOI] [PubMed] [Google Scholar]
- 43.Tang Z., Takahashi Y. Analysis of protein‒protein interaction by co-ip in human cells. Methods Mol Biol. 2018;1794:289–296. doi: 10.1007/978-1-4939-7871-7_20. [DOI] [PubMed] [Google Scholar]
- 44.Lin J.S., Lai E.M. Protein‒protein interactions: co-immunoprecipitation. Methods Mol Biol. 2017;1615:211–219. doi: 10.1007/978-1-4939-7033-9_17. [DOI] [PubMed] [Google Scholar]
- 45.Wen C.L., Huang K., Jiang L.L., Lu X.X., Dai Y.T., Shi M.M., et al. An allosteric PGAM1 inhibitor effectively suppresses pancreatic ductal adenocarcinoma. Proc Natl Acad Sci U S A. 2019;116:23264–23273. doi: 10.1073/pnas.1914557116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.