

Abstract
Hepatic ischemia/reperfusion injury (HIRI) is a serious complication that occurs following shock and/or liver surgery. Gut microbiota and their metabolites are key upstream modulators of development of liver injury. Herein, we investigated the potential contribution of gut microbes to HIRI. Ischemia/reperfusion surgery was performed to establish a murine model of HIRI. 16S rRNA gene sequencing and metabolomics were used for microbial analysis. Transcriptomics and proteomics analysis were employed to study the host cell responses. Our results establish HIRI was significantly increased when surgery occurred in the evening (ZT12, 20:00) when compared with the morning (ZT0, 08:00); however, antibiotic pretreatment reduced this diurnal variation. The abundance of a microbial metabolite 3,4-dihydroxyphenylpropionic acid was significantly higher in ZT0 when compared with ZT12 in the gut and this compound significantly protected mice against HIRI. Furthermore, 3,4-dihydroxyphenylpropionic acid suppressed the macrophage pro-inflammatory response in vivo and in vitro. This metabolite inhibits histone deacetylase activity by reducing its phosphorylation. Histone deacetylase inhibition suppressed macrophage pro-inflammatory activation and diminished the diurnal variation of HIRI. Our findings revealed a novel protective microbial metabolite against HIRI in mice. The potential underlying mechanism was at least in part, via 3,4-dihydroxyphenylpropionic acid-dependent immune regulation and histone deacetylase (HDAC) inhibition in macrophages.
KEY WORDS: Hepatic ischemia/reperfusion injury; Diurnal variation; 3,4-Dihydroxyphenylpropionic acid; Gut microbiota
Graphical abstract
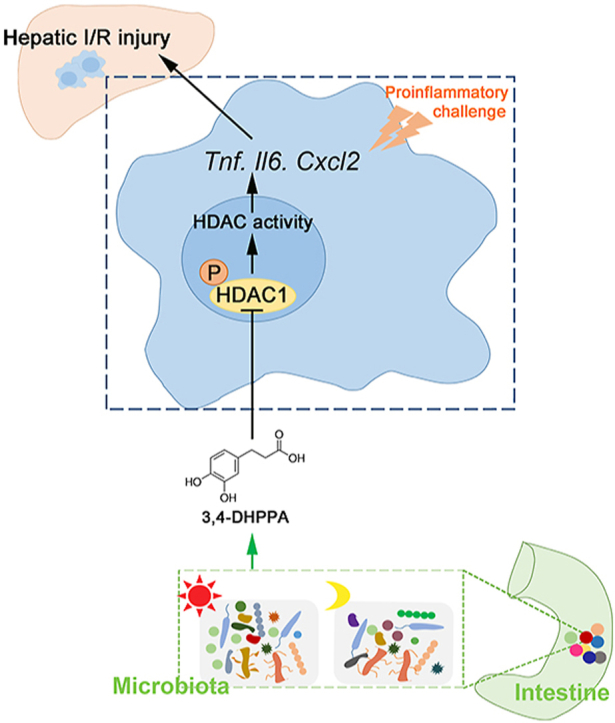
Hepatic ischemia/reperfusion injury (HIRI) exhibits diurnal variation, which is related to gut microbial 3,4-DHPPA production and accumulation at ZT0. 3,4-DHPPA inhibits macrophage inflammation and improves hepatic ischemia-reperfusion injury by inhibiting the phosphorylation and activity of histone deacetylase (HDAC).
1. Introduction
Hepatic ischemia/reperfusion injury (HIRI) is an important pathophysiologic cause of liver injury. It commonly occurs after liver surgery, such as liver transplantation and partial hepatectomy1. HIRI is a common cause for organ damage and potentially results in organ failure2,3. The pathogenesis of HIRI is complex; however, it generally comprises two stages. First, abnormal metabolic stress due to blood flow restriction leads to accumulation of an excess of reactive oxygen species (ROS) upon reperfusion, which directly damage hepatocytes, causing initial cytotoxicity. Second, injured liver cells trigger innate immune activation and amplify organ damage via pro-inflammatory responses4, 5, 6, 7. Macrophages are the dominant immune cells that drive inflammation during HIRI8, 9, 10. This injury is recognized as a major complication in liver surgery; however, limited interventions have been developed to date.
Many hepatic physiologic activities and injury phenomena exhibit diurnal variation. For example, acetaminophen-induced hepatotoxicity is markedly increased at night when compared with the morning in mice11. In addition, hepatic bile acid metabolism displays rhythmic oscillation12,13. This variation is modulated by various factors, in which gut microbiota plays a key role14,15. Previously, we have reported that the intestinal microbial metabolite, 1-phenyl-1,2-propanedione, which depletes hepatic glutathione, participates in the regulation of diurnal variation of acetaminophen-induced acute liver injury16. In addition, gut microbial compositional and functional changes influences fatty liver progression17, 18, 19. However, whether HIRI exhibits diurnal variation and is related to gut microbiota remains unknown. In the present study, we showed that HIRI exhibited diurnal variation and identified a novel protective microbial metabolite. Hence, we found a direct association between gut microbiota and the diurnal variation of HIRI. Our findings enhance our understanding of the key modulatory effects of gut microbiota on liver pathophysiology.
2. Materials and methods
2.1. Experimental animals
Male C57BL/6J mice, aged 8–10 weeks (weight, 23–26 g), were housed in a temperature-controlled environment (22 ± 2 °C) under standard 12 h light/dark conditions (08:00, light on; 20:00, light off). All mice received food and water ad libitum. All animal procedures were approved by the Ethics Committee on the Care and Use of Laboratory Animals in Southern Medical University (Guangzhou, China).
Mice underwent 70% warm hepatic ischemia/reperfusion (I/R) surgery at ZT0 (08:00) or ZT12 (20:00). After mice were anesthetized, the blood supply to the left and median hepatic lobes was blocked with artery clamps. After 90 min of ischemia, the clamp was removed for 6, 12, 24, or 48 h of reperfusion. Following this, the mice were euthanized and the serum and tissues were collected for further analysis. For antibiotic (ABX) treatment, mice were administered neomycin sulfate (200 mg/kg), metronidazole (200 mg/kg), ampicillin (200 mg/kg), and vancomycin (100 mg/kg) by oral gavage once daily for 4 days. For 3,4-dihydroxyphenylpropionic acid (3,4-DHPPA) treatment, mice were administered 100 mg/kg 3,4-DHPPA (Macklin, Shanghai, China) by oral gavage once a day for 3 days. Control mice received the equivalent volume of ddH2O. Suberoylanilide hydroxamic acid (SAHA; 25 mg/kg; Aladdin, Shanghai, China) was intraperitoneally administered 30 min before HIRI. The 150 μL clodronate liposomes (LIPOSOMA, AMS, Netherland) were injected into mice 24 h before HIRI via the tail vein.
2.2. Fecal microbiota transplantation
Fecal microbiota transplantation (FMT) was performed as described previously20. Briefly, C57BL/6J mice (8- to 10-week-old male) were administered with ABX via oral gavage once a day for 4 days to eliminate the gut microbiota. Donor mice feces from the ZT0 and ZT12 groups were collected and resuspended in phosphate buffered saline (PBS) to a concentration of 0.125 g/mL. This suspension was administered to mice via oral gavage (0.15 mL) daily for 3 days. After 3 days, at ZT12, mice were underwent hepatic I/R surgery and euthanized for further analysis.
2.3. Hematoxylin and Eosin (HE) staining
The 4% paraformaldehyde was used to fix the mice liver specimens, and then specimens were embedded in paraffin blocks after dehydration. Next, blocks were sectioned (4 μm) and stained using a standard HE protocol. The staining was scanned on each slide and the degree of liver damage was graded using the Suzuki score system21. According to the Suzuki score system, the histological injury score of each sample was the sum of the individual scores given for 3 different parameters: congestion (none = 0, minimal = 1, mild = 2, moderate = 3, severe = 4), vacuolization (none = 0, minimal = 1, mild = 2, moderate = 3, severe = 4), and necrosis (none = 0, single cell necrosis = 1, <30% = 2, 30%–60% = 3, >60% = 4). Scores for each parameter ranged from 0 to 4, with a maximum possible score of 12.
2.4. Targeted metabolomic analysis
Mice feces at ZT0 and ZT12 were collected and homogenized with water. Acetonitrile/methanol (80:20, v/v) was used for metabolites extraction, and the samples were centrifuged for 20 min at 18,000 ×g. Next, the supernatant was examined via ultra-pressure liquid chromatography coupled to tandem mass spectrometry (UPLC–MS/MS) system (ACQUITY UPLC-Xevo TQ-S, Waters Corp., Milford, MA, USA) to quantify the microbial metabolites. ACQUITY UPLC BEH C18 1.7 μm VanGuard pre-columns (2.1 mm × 5 mm) and ACQUITY UPLC BEH C18 1.7 μm analytical columns (2.1 mm × 100 mm) were used for analysis. The column temperature was 40 °C. The flow rate was 0.4 mL/min; mobile phase A = water with 0.1% formic acid; and mobile phase B = acetonitrile/isopropyl alcohol (90:10, v/v). The analysis was conducted as follows: 0–1 min (5% B), 1–12 min (5%–80% B), 12–15 min (80%–95% B), 15–16 min (95%–100% B), 16–18 min (100% B), 18–20 min (100%–5% B). The raw data files were analyzed by QuanMET (v2.0; Metabo-Profile, Shanghai, China) to identify and quantify the metabolite.
2.5. Bacterial composition analysis
Mice feces at ZT0 and ZT12 were homogenized and resuspended in PBS containing 0.5% Tween-20 followed by three cycles of 80 °C/60 °C to damage the bacterial membrane. DNA was extracted as described previously22. DNA (5 ng/μL) was used for quantitative real-time polymerase chain reaction (qRT-PCR). For microbial diversity analysis, the 16S rRNA gene V4 region was amplified and sequenced by using the Illumina sequencing platform. The raw sequences were quality-controlled using QIIME (1.9.1). QIIME is an open-source project, developed primarily in the Knight and Caporaso labs. Next, they were demultiplexed and clustered into species-level (97% similarity) operational taxonomic units. Principal component, alpha diversity, and beta diversity analyses were performed by using QIIME.
2.6. 3,4-DHPPA analysis
The 3,4-DHPPA levels in mice samples were detected by liquid chromatography-tandem mass spectrometry (LC–MS/MS) system. For liver samples, 9-fold w/v of water was added to the liver samples followed by ultrasonic extraction for 10 min. Next, the homogenate was centrifuged at 13,000 ×g for 10 min at 4 °C after adding methanol. The supernatant was dried under nitrogen and re-dissolved into the mobile phase for analysis. Chromatographic separation of samples was conducted on a Prelude SPLC™ system (Thermo Fisher Scientific, Waltham, MA, USA). The Thermo TSQ Vantage triple quadrupole mass spectrometer was used for detection. The chromatographic column was Amide (Waters Corp., Milford, MA, USA), 2.1 mm × 100 mm, 1.7 μm; with a column temperature of 40 °C. The flow rate was 0.3 mL/min; mobile phase A = 50 mmol/L ammonium formate with 0.2% formic acid and mobile phase B = acetonitrile. The gradient elution was performed according to the following procedure: 0–0.5 min, 5% A and 95% B; 0.5–2.5 min, 50% A and 50% B; 2.5–4.0 min, 50% A and 50% B; and 4.0–5.5 min, 5% A and 95% B. Data acquisition and analysis were performed using TraceFinder™ (version 3.3 sp1; Thermo Fisher Scientific).
2.7. Cell culture
Bone marrow-derived macrophages (BMDMs) were isolated from the femurs of mice and cultured in DMEM supplemented with 1% penicillin-streptomycin, 10% fetal bovine serum (FBS) and 20 ng/mL macrophage colony-stimulating factor (M-CSF, R&D Systems, Minneapolis, MN, USA). BMDMs were incubated at 37 °C with 5% CO2 and matured after 7 days. RAW264.7 cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin. Cultures were incubated at 37 °C with 5% CO2. The lipopolysaccharides (LPS; 100 ng/mL; Merck, NJ, USA) was incubated with BMDMs or RAW264.7 for 6 h to simulate an inflammatory response. The 20 μmol/L 3,4-DHPPA, 20 μmol/L 4,5,6,7-tetrabromobenzotriazole (TBB, Aladdin), 5 μmol/L SAHA, or 5 μmol/L trichostatin A (TSA, Aladdin) was used to treat cells in subsequent experiments.
2.8. Transcriptome analysis
Hepatic RNA was extracted from HIRI mice using the TRIzol extraction method, according to manufacturer's instructions. RNA concentration and purity was measured using the NanoDrop Spectrophotometer (Thermo Fisher Scientific) and Labchip GX Touch HT Nucleic Acid Analyzer (PerkinElmer, USA). Following this, the mRNA was enriched with oligo (dT) beads. According to the manufacturer's protocol, RNA sequencing libraries were generated using the KAPA Stranded RNA-Seq Kit for Illumina with multiplexing primers. Next, sequencing was performed on the Illumina Nova sequencer (Bioacme Biological Technologies Corporation, Wuhan, China). The gene ontology (GO) enrichment analysis of differentially expressed genes (DEGs) was implemented using the GOseq R package and GO terms. The genes with a P value < 0.05 and a fold change (FC) > 1.5 when compared between groups were considered as DEGs.
2.9. Proteomic LC–MS/MS
The 100 μg of total protein from individual samples was prepared. The protein samples were digested into peptides as previously described23. Briefly, dithiothreitol was added to samples to a final concentration of 10 mmol/L for reducing the proteins at 56 °C for 30 min. Then a final concentration of 20 mmol/L iodoacetamide was added and incubated for 30 min at room temperature without light expose. To decrease the urea concentration below 1 mol/L, samples were diluted in 100 mmol/L NH4HCO3. Lastly, MS grade trypsin (trypsin:protein = 1:50) was added to samples for overnight digestion at 37 °C (Promega, Madison, WI, USA). The digested peptides were loaded on the C18 column and bounded peptides were eluted with step gradient of 80 μL of 6%, 9%, 12%, 15%, 18%, 21%, 25%, 30%, and 35% acetonitrile (pH = 10) and combined into 3 pools [(1) 6% eluent combined with 15%, 25% eluent; (2) 9% plus 18%, 30%; (3) 12% plus 21%, 35%] and vacuum-dried for LC–MS/MS. Based on an Orbitrap Fusion (Thermo Fisher Scientific) mass spectrometer interfaced with an Easy-nLC 1000 nanoflow liquid chromatography system (Thermo Fisher Scientific), peptide mixtures were analyzed. The data-dependent acquisition (DDA) with full scans (m/z 300–1400) was performed.
Raw data were searched against the mouse reviewed uniprot protein database (https://www.uniprot.org/uniprot/?query=reviewed:yes%20taxonomy:10090) with Proteome Discoverer (Thermo Fisher Scientific, version 2.2). Carbamidomethylation of cysteine was set as fixed modifications while variable modifications were N-terminal protein acetylation and methionine oxidation. The data were also searched against a decoy database so that protein identifications were accepted at a false discovery rate of 1%. The identified proteins with a FC > 1.2 and a P value < 0.05 when compared between groups were considered as differentially expressed proteins (DEPs).
2.10. SPRi experiment
The PlexArray® HT surface plasmon resonance imaging platform (SPRi) was used for testing the binding affinity of 3,4-DHPPA with HDAC proteins, and SAHA was used as a positive control. The sensors were fixed on 0.5 mg/mL HDAC1, HDAC3, and HDAC5 (Abnova, Jhongli, Taiwan, China) in acetate acid buffer, pH 5.5. Next, these were activated with different concentrations of 3,4-DHPPA and SAHA (0.25, 0.5, and 1 mmol/L) in PBS. 3,4-DHPPA and SAHA were in the mobile phases at a rate of 30 μmol/L/min for 120 s. The data was obtained and analyzed with PlexArray® HT specialized software (Plexera Bioscience, Seattle, WA, USA).
2.11. Phosphoproteomics LC–MS/MS
Two milligram of nucleoprotein from individual samples was digested into peptides following procedures of total protein treatment. Digested peptide fragments were desalted with C18 column according to the manufacturer's protocol (#S181001; Agela Technologies, Torrance, CA, USA). Then, phosphopeptides of above desalted peptides were enriched with High-Select™ TiO2 Phosphopeptide Enrichment Kit (A32993; Thermo Fisher Scientific). Phosphopeptide and peptide mixtures were analyzed on a C18 column (50 μm × 15 cm, 3 μm) at 50 °C using an EASY-nLC1200 connected to an Orbitrap fusion mass spectrometer (Thermo Fisher Scientific). RAW files from all MS analyses were searched by MaxQuant v.1.6.10.43 (https://www.maxquant.org/) against the mouse reviewed uniprot protein database (https://www.uniprot.org/uniprot/?query=reviewed:yes%20taxonomy:10090) allowing for two missed trypsin-cleavage sites and variable modifications for phosphorylation (Ser, Thr, and Tyr residues), oxidation (Met), and acetylation (N-terminal). Carbamidomethylation was set as a fixed modification on cysteine residues. A false-discovery cutoff of 0.01 was set to filter for identified candidate peptides and phosphopeptides based on the searching of reverse-sequence decoy mode.
2.12. Data availability of microbial diversity and omics analysis
Raw sequencing data associated with microbial diversity analysis is accessible at https://www.ncbi.nlm.nih.gov/bioproject and the accession numbers: PRJNA659591; Raw sequencing data associated with transcriptome analysis is accessible at https://www.ncbi.nlm.nih.gov/bioproject and the accession numbers: PRJNA659337; The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the iProX database (https://iprox.org/) and is available via ProteomeXchange with identifier PXD021195.
2.13. Statistical analysis
All results are expressed as mean ± standard error of the mean (SEM). Statistical differences between two groups were analyzed using the two-tailed Student's t-test by GraphPad Prism 5 software (La Jolla, USA). Sample size (n) was listed in each figure legend. Differences were considered statistically significant when P < 0.05.
3. Results
3.1. Intestinal microbes participate in the regulation of the diurnal variation of murine HIRI
We established a murine HIRI model at ZT0 (08:00) and ZT12 (20:00). Data presented in Fig. 1A–E and Supporting Information Fig. S1A showed that liver damage, including serum transaminase levels [alanine aminotransferase (ALT), aspartate aminotransferase (AST), and hepatic myeloperoxidase (MPO) activity], histopathologic changes (Suzuki score), and representative inflammatory factors in HIRI24,25, was markedly exacerbated at ZT12 when compared with that at ZT0 after ischemia reperfusion challenge, while no difference was observed after sham operation. These results demonstrated that HIRI in mice displayed diurnal variation. In addition, oral ABX (metronidazole +neomycin + vancomycin + ampicillin) pretreatment obviously reduced intestinal total bacterial load (Supporting Information Fig. S1B) and meanwhile enhanced liver injury after HIRI at ZT0, but did not influence HIRI at ZT12, indicating that gut microbiota depletion diminished the liver injury oscillation during HIRI development (Fig. 1A–E). More importantly, the FMT experiment revealed that feces from ZT0 independently mitigated HIRI when compared with ZT12 feces at ZT12 (Fig. 1F–I). Thus, gut microbiota was involved in the modulation of the diurnal variation of HIRI in mice. Specifically, gut microbiota at ZT0 protected against HIRI when compared with gut microbiota at ZT12.
Figure 1.

Gut bacteria were involved in the regulation of the diurnal variation of murine HIRI. (A–E) Mice were administered ABX by oral gavage once daily for 4 days and underwent hepatic I/R surgery at ZT0 or ZT12. Serum transaminase levels and MPO activity of HIRI mice at ZT0 and ZT12 (n = 7, A), representative photographs of mice livers (B), HE staining and Suzuki scores (n = 7, C), the mRNA levels of key cytokines and chemokine in whole liver and liver macrophage (n = 5–7, D), and the mRNA levels of hepatic Cd68 (n = 7, E) were shown. (F–I) Mice were transplanted with fecal microbiota and then underwent hepatic I/R surgery at ZT12. Serum transaminase levels and hepatic MPO activity (n = 7, F), the mRNA levels of key cytokines and chemokine in the liver (n = 7, G), the mRNA levels of hepatic Cd68 (n = 7, H), and HE staining and Suzuki scores (n = 7, I) were shown. Scale bar: 50 μm. Data were expressed as mean ± SEM. ∗P < 0.05; n.s. indicates nonsignificant.
3.2. The metabolite of gut microbiota, 3,4-DHPPA, protects mice against HIRI
Bacterial metabolites are major modulators of gut bacteria on host pathophysiology. In order to explore the protective mechanism of gut microbiota, we sought to detail the compositional differences between ZT0 and ZT12 and identify the potential beneficial product generated from gut microbiota at ZT0. Thus, we performed 16S rRNA gene sequencing and metabolomics analysis for feces from ZT0 and ZT12. Fig. 2A and Supporting Information Fig. S1C–E showed that the overall microbial composition between the two time points was different, which was in agreement with our previous findings16. Fig. 2B shows the main different metabolites detected in the feces between ZT0 and ZT12. We focused on 3,4-DHPPA (Fig. 2C) because its abundance was significantly higher in ZT0 when compared with ZT12, we found 3,4-DHPPA was also enriched in the livers of mice which underwent hepatic I/R surgery at ZT0 (Fig. 2D). In line with our hypothesis, 3,4-DHPPA treatment significantly diminished the diurnal variation of HIRI by rescuing liver damage and the recruitment of macrophages at ZT12 (Fig. 2E–I). But this metabolite did not affect liver injury in sham group (Supporting Information Fig. S1F). These data demonstrated that this compound may be important in regulating the diurnal variation of HIRI.
Figure 2.

The metabolite 3,4-DHPPA protects mice against HIRI. (A–D) The differences between ZT0 and ZT12 feces were tested, and 3,4-DHPPA was identified. Scatter plots of pearson PCoA for the microbial composition of mice feces at ZT0 and ZT12 (n = 10) and the differences between ZT0 and ZT12 feces in beta diversity were tested by permutational multivariate analysis of variance (Adonis) (A); The heat map of gut microbial differential metabolites in mice at ZT0 and ZT12 was shown. Red represents high relative abundance, while blue represents low relative abundance (n = 6, B); The structure of 3,4-DHPPA (C); The 3,4-DHPPA contents of livers from mice that underwent hepatic I/R surgery at ZT0 and ZT12 (n = 7–8, D) were shown. (E–I) Mice were treated with 100 mg/kg 3,4-DHPPA for three days and then were underwent hepatic I/R surgery. Serum transaminase levels and hepatic MPO activity (n = 5–6, E), HE staining and Suzuki scores (n = 5, F), the mRNA levels of key cytokines and chemokine in whole liver (n = 5–6, G), the mRNA levels of hepatic Cd68 (n = 5–6, H), and the protein levels and quantification results of hepatic CD68 (n = 5, I) were shown. Scale bar: 50 μm. Data were expressed as mean ± SEM. ∗P < 0.05.
Since the substrate for 3,4-DHPPA formation was mainly from diet, we then fasted mice to check whether food intake could influence the fecal 3,4-DHPPA level. As presented in Supporting Information Fig. S2A, fasting could significantly reduce fecal 3,4-DHPPA level at both ZT0 and ZT12, indicating the fecal level of 3,4-DHPPA is dependent on food intake. However, fasted mice at ZT0 showed decreased liver injury during HIRI compared with ad libitum mice (Supporting Information Fig. S2B and C), which suggested starving may influence many other key pathways that contribute to HIRI beside 3,4-DHPPA.
3.3. Macrophages mediate the diurnal variation of HIRI in mice
To further explore the underlying mechanism of the diurnal variation of HIRI, we then investigated the host response at different chronological time points. Transcriptome analysis was performed on liver tissue harvested from ZT0 and ZT12 mice after HIRI. Fig. 3A–C shows the different gene expression profile between the ZT0 and ZT12 groups. Notably, we found differences in many inflammatory-associated pathways, such as chemokine signaling pathway, cytokine–cytokine receptor interaction, and nuclear factor kappa-B (NF-κB) signaling pathways. These data indicated the pro-inflammatory responses may contribute to differential liver damage after I/R challenge between ZT0 and ZT12 in mice.
Figure 3.

Macrophages mediate the diurnal variation of HIRI in mice. (A–C) The differences between HIRI mice livers at ZT0 and ZT12 were analyzed by transcriptome. The heat map was performed to display the transcriptional level which was significant differentially expressed of inflammation-related genes in HIRI mice livers at ZT0 and ZT12. Red represents high relative expression and blue represents low relative expression (n = 4, A); The volcano plots of gene expression in HIRI mice livers (n = 4, B); KEGG signaling pathway analysis reveal inflammation related functional terms enriched in HIRI mice at ZT0 and ZT12 (C). (D–F) Mice underwent hepatic I/R surgery at ZT0 after injected with 150 μL clodronate liposomes via the tail vein. Serum transaminase levels and hepatic MPO activity in HIRI mice with macrophages depletion at ZT0 (n = 7, D), the mRNA levels of key cytokines and chemokine in whole liver (n = 7, E), and HE staining and Suzuki scores (n = 7, F) were shown. Scale bar: 50 μm. Data were expressed as mean ± SEM. ∗P < 0.05.
Macrophages are recognized as key player in the pro-inflammatory response during HIRI. Therefore, we explored the involvement of macrophages in the regulation of HIRI by gut microbiota. Macrophage accumulation and pro-inflammatory factors expression in the liver were significantly enhanced at ZT12 when compared with ZT0 (Fig. 1D and E). Next, we used clodronate liposomes to remove mice macrophages. As expected, clodronate liposomes significantly cleared the hepatic macrophages (Supporting Information Fig. S3A), and in antibiotics pretreated mice we found significantly attenuated HIRI in macrophage-cleared mice than non-cleared controls (Fig. 3D–F), which indicated that the macrophage may be one of key cells that mediate the protective effect of gut microbiota on HIRI.
3.4. 3,4-DHPPA suppresses the pro-inflammatory activation of macrophages in vivo and in vitro
To directly link the bacterial product and host response, we next sought to determine if 3,4-DHPPA influenced host cell physiologic activity. Since we proposed macrophages were involved in the diurnal variation of HIRI, we then focused on the linkage between macrophage and 3,4-DHPPA. We confirmed the anti-inflammatory effect of 3,4-DHPPA on BMDMs and RAW264.7 cells (Fig. 4A and Supporting Information Fig. S3B). Moreover, Fig. 4B shows that 3,4-DHPPA administration significantly suppressed expression of pro-inflammatory factors in isolated hepatic macrophages from mice that underwent hepatic I/R surgery at ZT12 and also decreased the level of representative pro-inflammatory factors in serum of HIRI mice (Supporting Information Fig. S3C). In order to investigate whether the effect of 3,4-DHPPA on reducing hepatic I/R injury is dependent on macrophages in vivo. Liposomes combined and 3,4-DHPPA were employed together in HIRI mice and we found 3,4-DHPPA did not further improve the hepatic I/R injury after the elimination of macrophage in mice (Fig. 4C–E), suggesting that macrophages may be one of the key targets of 3,4-DHPPA. Thus, our data clearly demonstrated that 3,4-DHPPA suppresses the pro-inflammatory response of macrophages in vivo and in vitro.
Figure 4.

The anti-inflammatory effect of 3,4-DHPPA was dependent on HDAC activity inhibition. (A) The mRNA levels of key cytokines and chemokine in LPS-activated BMDMs and RAW264.7 cells treated with 20 μmol/L 3,4-DHPPA (n = 4). (B) The mRNA levels of key cytokines and chemokine in hepatic macrophages of mice treated with 100 mg/kg 3,4-DHPPA and underwent hepatic I/R surgery at ZT12 (n = 5). (C–E) Mice were gavaged with 100 mg/kg 3,4-DHPPA for three days and then were underwent hepatic I/R surgery at ZT12 after injected with 150 μL clodronate liposomes via the tail vein. Serum transaminase levels and hepatic MPO activity in HIRI mice with macrophages depletion at ZT12 (n = 7–9, C), the mRNA levels of key cytokines and chemokine in whole liver (n = 6, D), and HE staining and Suzuki scores (n = 5–6, E) were shown. Scale bar: 50 μm. (F) Clustering analysis was performed to display the proteins which were significant differentially expressed both in LPS treated group and LPS+3,4-DHPPA treated group. Red represents high expression, while blue represents low expression. (G) Cytoscape software combined with ClueGO App was used to reveal signaling pathways enriched by the DEPs, and the networks identified a serious of pathways associated with histone deacetylation. (H, I) HDAC activity and the protein expression of BMDMs and RAW264.7 cells stimulated by LPS with 3,4-DHPPA. HDAC activity of BMDMs and RAW264.7 cells stimulated by 100 ng/mL LPS for 6 h with 20 μmol/L 3,4-DHPPA (n = 5, H), and the protein expression and quantification results of acetyl-H3K9 in LPS-activated BMDMs with 20 μmol/L 3,4-DHPPA (n = 3, I) were shown. Data were expressed as mean ± SEM. ∗P < 0.05; n.s. indicates nonsignificant.
Since hepatocyte damage was also the main contributor for liver dysfunction, we then evaluated the impact of 3,4-DHPPA on hepatocytes. We found that 3,4-DHPPA had limited protective effects on hepatocytes damage under hypoxia/reoxygenation challenge (Supporting Information Fig. S4), which suggested that this metabolite may not directly target hepatocytes during HIRI. This finding further strengthened the conclusion that macrophages may be the targeted cell type of 3,4-DHPPA during HIRI.
3.5. The anti-inflammatory effect of 3,4-DHPPA may be dependent on HDAC activity inhibition
To further explore the anti-inflammatory mechanisms of 3,4-DHPPA, proteomic analysis was performed on LPS-activated BMDMs treated with or without 3,4-DHPPA. Fig. 4F shows that 3,4-DHPPA administration altered the protein expression profile in macrophages following LPS challenge. In addition, we found that inflammation- and immune-related signaling pathways were significantly enriched through the function analysis of these DEPs between 3,4-DHPPA treated and non-treated groups (Supporting Information Fig. S5). Functional analysis of differential proteins also revealed significant enrichment of biological processes associated with histone acetylation (Fig. 4G). Specifically, the results of functional analysis of different proteins have shown that HDAC associated pathways may be shifted in the presence of 3,4-DHPPA. To further confirm this finding, we determined HDAC enzymatic activity in macrophages. Fig. 4H shows that 3,4-DHPPA pretreatment significantly suppressed total HDAC activity. Furthermore, the HDAC substrate, H3K9 histone, showed reduced acetylation following LPS challenge but was increased in the presence of 3,4-DHPPA treatment (Fig. 4I). Moreover, the relative expression of ac-H3K9 histone was also rescued by 3,4-DHPPPA when normalized to total level of histone H3, which indicated that 3,4-DHPPA only affects the acetylation of H3K9 histone but not the expression of histone H3 (Supporting Information Fig. S6A). We also monitored the activity of HDAC in macrophage isolated from HIRI mice livers, and we found that the inhibition of HDAC activity of HIRI liver macrophages by 3,4-DHPPA at ZT12 showed strong trend (P = 0.076) (Supporting Information Fig. S6B). These results demonstrated that 3,4-DHPPA inhibited HDAC activity in macrophages.
We then hypothesized that 3,4-DHPPA may directly bind to the active site of HDAC. To address this, we performed surface plasmon resonance (SPR) analysis to determine the binding activity of this metabolite. However, Fig. 5A and Supporting Information Fig. S7 showed that 3,4-DHPPA could not directly bind HDACs in a dose dependent manner, in which HDAC inhibitor SAHA was used as positive control. Taken together, these data demonstrated that 3,4-DHPPA may inhibit HDAC activity in an indirect manner. Previous studies have shown that HDAC activity was positively correlated with its phosphorylation and casein kinase 2 (CK2) is a major protein kinase for HDAC phosphorylation26, 27, 28. Studies have also shown that CK2 promotes phosphorylation of HDAC1 through phosphorylation sites (Ser393, Ser421, Ser423, Ser445, Ser460, and Ser465)29,30. Thus, we performed phosphoproteomics analysis to detect phosphorylated HDACs levels by LC–MS/MS. The results showed that 3,4-DHPPA and 4,5,6,7-tetrabromobenzotriazole (TBB, selective inhibitor of CK2) can both significantly decrease the level of phosphorylated Ser393 in HDAC1 but did not affect total level of HDAC1 (Fig. 5B–E). Additionally, 3,4-DHPPA did not further reduce the HDAC activity and inflammatory factor levels in macrophages co-treated with TBB (Fig. 5F and G), demonstrating that the anti-inflammation effects of 3,4-DHPPA may be dependent on HDAC phosphorylation. Following this, we further explored whether HDAC inhibition mediated the beneficial effects of 3,4-DHPPA on macrophage activation and HIRI development. Fig. 6A shows that HDAC inhibition [HDAC inhibitors: SAHA and trichostatin A (TSA)] markedly reduced expression of pro-inflammatory factors upon LPS challenge; however, 3,4-DHPPA administration did not have an additional effect. This supports the notion that the anti-inflammation effects of 3,4-DHPPA may be, at least in part, dependent on HDAC inhibition.
Figure 5.

The anti-inflammatory effect of 3,4-DHPPA was dependent on inhibition of HDAC1 phosphorylation. (A) The direct interactions between 3,4-DHPPA or SAHA and HDAC1 was determined by SPR. (B–E) The nucleoprotein of LPS-activated RAW264.7 cells were analyzed by phosphoproteomics LC–MS/MS. Full mass spectra of enriched phosphorpeptides (B) and MS/MS spectra (C) of a HDAC1 specific phosphorylated peptide [MLPHAPGVQMQAIPEDAIPEESGDEDEEDPDKR, Ser393, the point indicated by red arrow in (B)] in LPS-activated RAW264.7 cells for 1 h. Relative expression of total HDAC1 in nucleus (n = 3, D) and intensity of the specific HDAC1 phosphorylated peptide (Ser393) (n = 3, E) were shown. (F, G) BMDMs and RAW264.7 cells were activated by LPS for 6 h with 20 μmol/L 3,4-DHPPA and 20 μmol/L TBB. HDAC activity in BMDMs (n = 4, F) and the mRNA levels of key cytokines and chemokine in BMDMs and RAW264.7 (n = 4, G) were shown. Data were expressed as mean ± SEM. ∗P < 0.05; n.s. indicates nonsignificant.
Figure 6.

The protective effect of 3,4-DHPPA was dependent on HDAC inhibition. (A) BMDMs and RAW264.7 cells were activated by LPS for 6 h with 20 μmol/L 3,4-DHPPA, 5 μmol/L SAHA or 5 μmol/L trichostatin A (TSA). The mRNA levels of key cytokines and chemokines (n = 4) were shown. (B–D) Mice treated with 25 mg/kg SAHA and underwent hepatic I/R surgery at ZT0 or ZT12. Serum transaminase levels and hepatic MPO activity (n = 5–8, B), the mRNA levels of key cytokines and chemokine in HIRI mice livers (n = 5–8, C), and HE staining and Suzuki scores (n = 5, D) were shown. (E–G) Mice treated with 100 mg/kg 3,4-DHPPA by gavage once daily for 3 days, then 25 mg/kg SAHA was intraperitoneally injected before hepatic I/R surgery at ZT12. Serum transaminase levels and hepatic MPO activity (n = 5–12, E), the mRNA levels of key cytokines and chemokine in whole liver (n = 7, F), and HE staining and Suzuki scores (n = 5, G) were shown. Scale bar: 50 μm. Data were expressed as mean ± SEM. ∗P < 0.05; n.s. indicates nonsignificant.
We also examined HIRI in SAHA pretreated mice. Interestingly, SAHA pretreatment significantly mitigated liver injury at ZT12, which diminished the diurnal variation of HIRI (Fig. 6B–D). These data suggest that HDAC inhibition is a significant factor in mediating the diurnal variation of HIRI in the animal model. Furthermore, 3,4-DHPPA administration did not influence liver injury in HDAC inhibitor-pretreated animals (Fig. 6E–G), suggesting that the protective effects of 3,4-DHPPA may be, at least in part, dependent on HDAC inhibition.
4. Discussion
Recently amoxicillin pretreatment was shown to mitigate hepatic injury after liver transplant, which supports the possible contribution of gut microbiota to HIRI31. However, the detailed connection between gut microbe and host response during HIRI is still largely unknown. In the present study, by focusing on the intestinal microbiota, we explored the mechanism of the diurnal variation of HIRI in mice. We found that gut microbial metabolite, 3,4-DHPPA, was a novel protective compound against HIRI. The underlying mechanism was related to inhibition of HDAC activity in macrophages, which suppressed inflammation. Our current study revealed several important findings that potentially have clinical translational significance.
As an important peripheral organ, a variety of hepatic pathological and physiological processes exhibit rhythmic oscillations. These diurnal variations help the liver to adapt to exogenous environmental changes32, 33, 34, 35. Understanding these alterations has the potential to improve the treatment of patients with liver diseases. Our current study revealed that HIRI was more severe in the evening when compared with the morning. Based upon this key observation, we pursued the explanation for this interesting finding. In recent years, the circadian rhythm is attracting tremendous attention due to its remarkable effects on the development of various diseases. In particular, convincing clinical evidence has indicated that circadian rhythms have effects on surgical outcomes of liver transplantation for patients36. A retrospective analysis of 147 patients indicated that compared with that at daytime, liver transplantation performed at night had higher incidence of intraoperative complications and postoperative abdominal infection. Moreover, patients which received liver transplantation at night needed longer time to restore hepatic function to normal. Thus, not only our preclinical data, but also the clinical evidence from previous studies, makes it clear that diurnal variation has an important effect on the prognosis of patients who underwent liver surgery. However, the mechanism by which circadian rhythm influences the complications and prognosis of liver transplantation remains unclear. In our research, we found the circadian rhythm in HIRI of mice is possibly related to the gut microbiota. Our data revealed that 3,4-DHPPA modulates HIRI progression. 3,4-DHPPA can be generated by bacterial of reduction of caffeic acid37. It is a well-established bacterial metabolite that could be found in both human and mice, and exhibits many protective effects on host cells38,39. In an ischemia-induced brain injury model, 3,4-DHPPA suppressed matrix metalloproteinase-2 (MMP-2) and MMP-9 activity and protected against neuronal damage40. Furthermore, 3,4-DHPPA reduced ROS level and mitigated oxidative stress in different cell lines41,42. In the present study, we expanded the investigation of 3,4-DHPPA to the activation of macrophages, which are key mediators of inflammation, and found this metabolite exerted anti-inflammatory effects. In the current work, we confirmed that the level of 3,4-DHPPA was higher in ZT0 feces than ZT12 feces under non-surgical conditions. We further found basal DHPPA level was comparable between the mice livers under non-surgical conditions at ZT0 and ZT12 (data not shown). However, after I/R surgery, 3,4-DHPPA accumulated in the liver at ZT0 but not at ZT12. We speculate that the normal liver efficiently metabolizes or excretes 3,4-DHPPA at both ZT0 and ZT12. However, upon I/R challenge, the hepatic metabolism of 3,4-DHPPA between ZT0 and ZT12 may be different, leading to the accumulation of 3,4-DHPPA in ZT0 compared with that at ZT12. More gut derived 3,4-DHPPA accumulates in the liver and exerts protective effects at ZT0. It is important to point out that based on metabolomics analysis, additional beneficial metabolites generated from bacteria may contribute, and future studies should assess the potential protective effects of other compounds.
We also explored the level of 3,4-DHPPA in fasting mice at ZT0 and ZT12 which showed that 3,4-DHPPA was significantly reduced in the feces of mice after fasting at both ZT0 and ZT12 (Supporting Information Fig. S2A). In fact, 3,4-DHPPA is abundantly found in various foods and usually present as simple esters with quinic acid, glucose, polysaccharides, or other carboxylic acids, and 3,4-DHPPA is also present in blood and in urine as a metabolite of various polyphenols found in food, beverages, medicinal plants, or extracts43. Thus, we propose the rhythmic oscillation of fecal 3,4-DHPPA was dependent on food intake. Interestingly, we found the degree of hepatic I/R injury was significantly reduced after fasting of mice (Supporting Information Fig. S2B), which is consistent with recent research which showed that short-term fasting can effectively reduce hepatic I/R injury. It was proposed that this was related to the promotion of autophagy in hepatocytes and the up-regulated expression of antioxidant genes in fasting mice44,45. These results indicated that physiology of mice livers is greatly changed in various pathways after fasting such as autophagy and oxidative stress. Therefore, 3,4-DHPPA might not be recognized as a key factor to prevent HIRI in fasting mice. However, our data still fully support the application of 3,4-DHPPA to serve as a potential substance for protecting HIRI.
HDAC is a key modulator of innate immune activation. Inhibition of HDAC enzymatic activity shifts macrophages to an M2-like phenotype and reduces inflammation in the context of disease development46, 47, 48, 49. Thus HDAC inhibition is a well-established target to reduce pro-inflammatory responses. HDAC activity is tightly regulated by its phosphorylation50,51. For example, Ser421 and Ser423 phosphorylation of HDAC1 is required for its enzymatic activity52. Due to technical limitations, we only observed the difference of Ser393 phosphorylation of HDAC1 between 3,4-DHPPA treated and untreated macrophages by phosphoproteomics analysis. However, we cannot rule out the possibility that this metabolite may inhibit phosphorylation of other sites on HDAC1 and other HDAC family members, and contribute together to suppress HDAC activity during inflammatory challenge in macrophages in HIRI. Although we observed 3,4-DHPPA had similar effects as TBB, the selective inhibitor of protein kinase CK2, we did not verify if 3,4-DHPPA directly targets protein kinase CK2, or other potential HDAC's kinases. Future work is needed to specify which kinase was directly modulated by 3,4-DHPPA. It is also possible that DHPPA may regulate HDAC activity through indirect mechanisms. For example, 3,4-DHPPA may affect the key type of miRNA and in turn influence the HDAC activity53. 3,4-DHPPA may also coordinate HDAC activity by inhibiting the secretion of inflammatory cytokines such as interleukin 10 (IL-10) and thereby suppressing inflammatory response54. In addition, considering the off-target effect of HDAC inhibitors, other host pathway or molecular may be also modulated by 3,4-DHPPA, for example, 3,4-DHPPA may regulate DNA methylation in immune cells to influence inflammatory activity55. We therefore cannot exclude the possibility that 3,4-DHPPA also inhibits inflammatory response and improves hepatic ischemia reperfusion injury through other pathways that are independent of HDAC activity.
In the current research, we provide evidence that HDAC inhibition as a potential therapeutic target for HIRI. However, the association between HDAC and HIRI is controversial. It has been reported that butyrate inhibits HDAC activity and alleviates liver ischemia reperfusion injury56. We also confirmed the protective effects of butyrate against HIRI (data not shown). However, Ruess et al.57 have reported that HDAC inhibition by valproic acid (VPA) and SAHA did not protect against liver injury after ischemia reperfusion challenge in the rat. In fact, they found the HDAC inhibitor augmented liver injury at approximately 24 h post reperfusion. Different phenotypes observed in our study and the results of Ruess et al.57 may be due to the following reasons: we used a different animal model (mice vs. rats), which may reflect different phenotypes; and we administered SAHA or 3,4-DHPPA once compared with multiple treatments in Ruess’ study. Therefore, continuous HDAC inhibitor treatment may lead to adverse effects when compared with a single administration. In addition, the previous study revealed that HDAC inhibition augmented acetylated HMGB1 release from hepatocytes during ischemia condition58. However, we found that 3,4-DHPPA or SAHA administration had limited beneficial effects on hepatocytes (Supporting Information Figs. S4 and S8); therefore, continuous HDAC inhibitor administration may cause harmful effects on hepatocytes and mask the protective effects. Indeed, we also assessed the effects of 3,4-DHPPA and SAHA on HIRI at multiple time points (12, 24, and 48 h post reperfusion) after single administration. Figure 2, Figure 6B–D and Supporting Information Fig. S9 showed that both 3,4-DHPPA and SAHA can effectively protect against liver injury in the early stage of hepatic I/R (6 and 12 h). However, 3,4-DHPPA and SAHA showed no significant improvement on the ALT levels of the HIRI mice (but also did not augmented HIRI) at the time points after 24 h. We proposed that early phase (6–12 h) is the most important injury stage during HIRI progression59. After 24 h, liver rapidly recovers and such improvement could conceal the protective effects of these compounds. Thus, 3,4-DHPPA was able to reduce liver injury at both 6 and 12 h, the peak injury state during HIRI. In the model we used, though mice fully recover, a more severe liver injury which is lethal or with more prolonged recovery might reveal beneficial effects from early treatment on the ultimate outcome. Furthermore, more rapid recovery in humans may decrease susceptibility to other complications or shorten intensive care unit (ICU) stay. In addition, we cannot ignore the possibility that other diversified gut microbiota or gut microbial metabolites may also exert potential beneficial effects on hepatic I/R injury. The effect of other gut microbial metabolites on hepatic I/R injury requires further investigation in the future.
5. Conclusions
We report a novel pathophysiologic feature of HIRI in murine model: HIRI was more severe at ZT12 compared with ZT0. Furthermore, the metabolite of gut microbiota, 3,4-DHPPA, attenuated macrophage activation and inflammation during HIRI, at least in part via suppression of HDAC activation (Fig. 7). Our present study provides new insights into the “gut–liver” axis in the context of HIRI. More clinical translational research regarding the diurnal variation of HIRI, protective effects of 3,4-DHPPA, and HDAC inhibition against HIRI are required in the future.
Figure 7.

Working model: HIRI exhibited diurnal variation which is related to the changes of gut microbiota. Gut microbiota produced and accumulated 3,4-DHPPA at ZT0. 3,4-DHPPA could inhibit the pro-inflammatory activity of macrophages and improve hepatic ischemia–reperfusion injury of mice by reducing the level of HDAC phosphorylation and inhibiting the activity of HDAC.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (81873926, 32071124); Natural Science Funds for Distinguished Young Scholar of Guangdong province grant (2016A030306043, China) to Peng Chen; Grants from the NSFC-Guangdong Joint Foundation of China (U1601225), Natural Science Foundation of China (81971895), and Special Support Plan for Outstanding Talents of Guangdong Province (2019JC05Y340, China) to Yong Jiang.
Author contributions
Rui Li, Li Xie, Lei Li, Xiaojiao Chen, Tong Yao, Cui Li, and Yifan Li performed the experiments and analyzed the data; Yuanxin Tian participated in the molecular binding analysis; Qingping Li and Kai Wang provided assistant to hepatic ischemia/reperfusion model; Chenyang Huang was responsible for proteomics analysis; Hongwei Zhou was responsible for the analysis of bacterial sequencing data; Neil Kaplowitz interpreted the data and edited the manuscript; Yong Jiang and Peng Chen designed the study, interpreted the data, draft and edit the manuscript, and supervised the study.
Conflicts of interest
The authors declare no conflicts of interest.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2021.05.029.
Contributor Information
Yong Jiang, Email: jiang48231@163.com.
Peng Chen, Email: perchen@smu.edu.cn.
Appendix ASupplementary data
The following is the Supplementary data to this article:
References
- 1.Casillas-Ramirez A., Mosbah I.B., Ramalho F., Roselló-Catafau J., Peralta C. Past and future approaches to ischemia–reperfusion lesion associated with liver transplantation. Life Sci. 2006;79:1881–1894. doi: 10.1016/j.lfs.2006.06.024. [DOI] [PubMed] [Google Scholar]
- 2.Duffy J.P., Kao K., Ko C.Y., Farmer D.G., McDiarmid S.V., Hong J.C., et al. Long-term patient outcome and quality of life after liver transplantation: analysis of 20-year survivors. Ann Surg. 2010;252:652–661. doi: 10.1097/SLA.0b013e3181f5f23a. [DOI] [PubMed] [Google Scholar]
- 3.Konishi T., Lentsch A.B. Hepatic ischemia/reperfusion: mechanisms of tissue injury, repair, and regeneration. Gene Expr. 2017;17:277–287. doi: 10.3727/105221617X15042750874156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Motiño O., Francés D.E., Casanova N., Fuertes-Agudo M., Cucarella C., Flores J.M., et al. Protective role of hepatocyte cyclooxygenase-2 expression against liver ischemia–reperfusion injury in mice. Hepatology. 2019;70:650–665. doi: 10.1002/hep.30241. [DOI] [PubMed] [Google Scholar]
- 5.Katwal G., Baral D., Fan X., Weiyang H., Zhang X., Ling L., et al. SIRT3 a major player in attenuation of hepatic ischemia–reperfusion injury by reducing ROS via its downstream mediators: SOD2, CYP-D, and HIF-1α. Oxid Med Cell Longev. 2018;2018:2976957. doi: 10.1155/2018/2976957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vardanian A.J., Busuttil R.W., Kupiec-Weglinski J.W. Molecular mediators of liver ischemia and reperfusion injury: a brief review. Mol Med. 2008;14:337–345. doi: 10.2119/2007-00134.Vardanian. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ji H., Liu Y., Zhang Y., Shen X.D., Gao F., Busuttil R.W., et al. T-cell immunoglobulin and mucin domain 4 (TIM-4) signaling in innate immune-mediated liver ischemia-reperfusion injury. Hepatology. 2014;60:2052–2064. doi: 10.1002/hep.27334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakamura K., Zhang M., Kageyama S., Ke B., Fujii T., Sosa R.A., et al. Macrophage heme oxygenase-1–SIRT1–p53 axis regulates sterile inflammation in liver ischemia–reperfusion injury. J Hepatol. 2017;67:1232–1242. doi: 10.1016/j.jhep.2017.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yue S., Zhu J., Zhang M., Li C., Zhou X., Zhou M., et al. The myeloid heat shock transcription factor 1/beta-catenin axis regulates NLR family, pyrin domain-containing 3 inflammasome activation in mouse liver ischemia/reperfusion injury. Hepatology. 2016;64:1683–1698. doi: 10.1002/hep.28739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu B., Guo Y., Garbacz W.G., Jiang M., Xu M., Huang H., et al. Fatty acid binding protein-4 (FABP4) is a hypoxia inducible gene that sensitizes mice to liver ischemia/reperfusion injury. J Hepatol. 2015;63:855–862. doi: 10.1016/j.jhep.2015.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson B.P., Walisser J.A., Liu Y., Shen A.L., McDearmon E.L., Moran S.M., et al. Hepatocyte circadian clock controls acetaminophen bioactivation through NADPH-cytochrome P450 oxidoreductase. Proc Natl Acad Sci U S A. 2014;111:18757–18762. doi: 10.1073/pnas.1421708111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gälman C., Angelin B., Rudling M. Bile acid synthesis in humans has a rapid diurnal variation that is asynchronous with cholesterol synthesis. Gastroenterology. 2005;129:1445–1453. doi: 10.1053/j.gastro.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 13.Zhang F., Duan Y., Xi L., Wei M., Shi A., Zhou Y., et al. The influences of cholecystectomy on the circadian rhythms of bile acids as well as the enterohepatic transporters and enzymes systems in mice. Chronobiol Int. 2018;35:673–690. doi: 10.1080/07420528.2018.1426596. [DOI] [PubMed] [Google Scholar]
- 14.Nobs S.P., Tuganbaev T., Elinav E. Microbiome diurnal rhythmicity and its impact on host physiology and disease risk. EMBO Rep. 2019;20 doi: 10.15252/embr.201847129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thaiss C.A., Levy M., Korem T., Dohnalová L., Shapiro H., Jaitin D.A., et al. Microbiota diurnal rhythmicity programs host transcriptome oscillations. Cell. 2016;167:1495–1510. doi: 10.1016/j.cell.2016.11.003. [DOI] [PubMed] [Google Scholar]
- 16.Gong S., Lan T., Zeng L., Luo H., Yang X., Li N., et al. Gut microbiota mediates diurnal variation of acetaminophen induced acute liver injury in mice. J Hepatol. 2018;69:51–59. doi: 10.1016/j.jhep.2018.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leone V., Gibbons S.M., Martinez K., Hutchison A.L., Huang E.Y., Cham C.M., et al. Effects of diurnal variation of gut microbes and high-fat feeding on host circadian clock function and metabolism. Cell Host Microbe. 2015;17:681–689. doi: 10.1016/j.chom.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mazzoccoli G., De Cosmo S., Mazza T. The biological clock: a pivotal hub in non-alcoholic fatty liver disease pathogenesis. Front Physiol. 2018;9:193. doi: 10.3389/fphys.2018.00193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun L., Pang Y., Wang X., Wu Q., Liu H., Liu B., et al. Ablation of gut microbiota alleviates obesity-induced hepatic steatosis and glucose intolerance by modulating bile acid metabolism in hamsters. Acta Pharm Sin B. 2019;9:702–710. doi: 10.1016/j.apsb.2019.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gong S., Yan Z., Liu Z., Niu M., Fang H., Li N., et al. Intestinal microbiota mediates the susceptibility to polymicrobial sepsis-induced liver injury by granisetron generation in mice. Hepatology. 2019;69:1751–1767. doi: 10.1002/hep.30361. [DOI] [PubMed] [Google Scholar]
- 21.Khader A., Yang W.L., Godwin A., Prince J.M., Nicastro J.M., Coppa G.F., et al. Sirtuin 1 stimulation attenuates ischemic liver injury and enhances mitochondrial recovery and autophagy. Crit Care Med. 2016;44:e651–e663. doi: 10.1097/CCM.0000000000001637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu J., Luo H., Wang J., Tang W., Lu J., Wu S., et al. Enteric dysbiosis-linked gut barrier disruption triggers early renal injury induced by chronic high salt feeding in mice. Exp Mol Med. 2017;49 doi: 10.1038/emm.2017.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang F., Gong S., Wang T., Li L., Luo H., Wang J., et al. Soyasaponin II protects against acute liver failure through diminishing YB-1 phosphorylation and Nlrp3-inflammasome priming in mice. Theranostics. 2020;10:2714–2726. doi: 10.7150/thno.40128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang X.J., Cheng X., Yan Z.Z., Fang J., Wang X., Wang W., et al. An ALOX12–12-HETE–GPR31 signaling axis is a key mediator of hepatic ischemia–reperfusion injury. Nat Med. 2018;24:73–83. doi: 10.1038/nm.4451. [DOI] [PubMed] [Google Scholar]
- 25.Liu Y., Ji H., Zhang Y., Shen X., Gao F., He X., et al. Recipient T cell TIM-3 and hepatocyte galectin-9 signalling protects mouse liver transplants against ischemia–reperfusion injury. J Hepatol. 2015;62:563–572. doi: 10.1016/j.jhep.2014.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang H., Song C., Ding Y., Pan X., Ge Z., Tan B.H., et al. Transcriptional regulation of JARID1B/KDM5B histone demethylase by Ikaros, histone deacetylase 1 (HDAC1), and casein kinase 2 (CK2) in B-cell acute lymphoblastic leukemia. J Biol Chem. 2016;291:4004–4018. doi: 10.1074/jbc.M115.679332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eom G.H., Cho Y.K., Ko J.H., Shin S., Choe N., Kim Y., et al. Casein kinase-2alpha1 induces hypertrophic response by phosphorylation of histone deacetylase 2 S394 and its activation in the heart. Circulation. 2011;123:2392–2403. doi: 10.1161/CIRCULATIONAHA.110.003665. [DOI] [PubMed] [Google Scholar]
- 28.Khan D.H., He S., Yu J., Winter S., Cao W., Seiser C., et al. Protein kinase CK2 regulates the dimerization of histone deacetylase 1 (HDAC1) and HDAC2 during mitosis. J Biol Chem. 2013;288:16518–16528. doi: 10.1074/jbc.M112.440446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsai S.C., Seto E. Regulation of histone deacetylase 2 by protein kinase CK2. J Biol Chem. 2002;277:31826–31833. doi: 10.1074/jbc.M204149200. [DOI] [PubMed] [Google Scholar]
- 30.Pflum M.K., Tong J.K., Lane W.S., Schreiber S.L., et al. Histone deacetylase 1 phosphorylation promotes enzymatic activity and complex formation. J Biol Chem. 2001;276:47733–47741. doi: 10.1074/jbc.M105590200. [DOI] [PubMed] [Google Scholar]
- 31.Nakamura K., Kageyama S., Ito T., Hirao H., Kadono K., Aziz A., et al. Antibiotic pretreatment alleviates liver transplant damage in mice and humans. J Clin Invest. 2019;129:3420–3434. doi: 10.1172/JCI127550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guan D., Xiong Y., Borck P.C., Jang C., Doulias P.T., Papazyan R., et al. Diet-induced circadian enhancer remodeling synchronizes opposing hepatic lipid metabolic processes. Cell. 2018;174:831–842. doi: 10.1016/j.cell.2018.06.031. e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feng D., Liu T., Sun Z., Bugge A., Mullican S.E., Alenghat T., et al. A circadian rhythm orchestrated by histone deacetylase 3 controls hepatic lipid metabolism. Science. 2011;331:1315–1319. doi: 10.1126/science.1198125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sato S., Solanas G., Peixoto F.O., Bee L., Symeonidi A., Schmidt M.S., et al. Circadian reprogramming in the liver identifies metabolic pathways of aging. Cell. 2017;170:664–677. doi: 10.1016/j.cell.2017.07.042. e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang E.E., Liu Y., Dentin R., Pongsawakul P.Y., Liu A.C., Hirota T., et al. Cryptochrome mediates circadian regulation of cAMP signaling and hepatic gluconeogenesis. Nat Med. 2010;16:1152–1156. doi: 10.1038/nm.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ren S.S., Xu L.L., Wang P., Li L., Hu Y.T., Xu M.Q., et al. Circadian rhythms have effects on surgical outcomes of liver transplantation for patients with hepatocellular carcinoma: a retrospective analysis of 147 cases in a single center. Transplant Proc. 2019;51:1913–1919. doi: 10.1016/j.transproceed.2019.03.033. [DOI] [PubMed] [Google Scholar]
- 37.Fumeaux R., Menozzi-Smarrito C., Stalmach A., Munari C., Kraehenbuehl K., Steiling H., et al. First synthesis, characterization, and evidence for the presence of hydroxycinnamic acid sulfate and glucuronide conjugates in human biological fluids as a result of coffee consumption. Org Biomol Chem. 2010;8:5199–5211. doi: 10.1039/c0ob00137f. [DOI] [PubMed] [Google Scholar]
- 38.Huang J., de Paulis T., May J.M. Antioxidant effects of dihydrocaffeic acid in human EA.hy926 endothelial cells. J Nutr Biochem. 2004;15:722–729. doi: 10.1016/j.jnutbio.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 39.Monagas M., Khan N., Andres-Lacueva C., Urpí-Sardá M., Vázquez-Agell M., Lamuela-Raventós R.M., et al. Dihydroxylated phenolic acids derived from microbial metabolism reduce lipopolysaccharide-stimulated cytokine secretion by human peripheral blood mononuclear cells. Br J Nutr. 2009;102:201–206. doi: 10.1017/S0007114508162110. [DOI] [PubMed] [Google Scholar]
- 40.Lee K., Lee B.J., Bu Y. Protective effects of dihydrocaffeic acid, a coffee component metabolite, on a focal cerebral ischemia rat model. Molecules. 2015;20:11930–11940. doi: 10.3390/molecules200711930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.González-Sarrías A., Núñez-Sánchez M.A., Tomás-Barberán F.A., Espín J.C. Neuroprotective effects of bioavailable polyphenol-derived metabolites against oxidative stress-induced cytotoxicity in human neuroblastoma SH-SY5Y cells. J Agric Food Chem. 2017;65:752–758. doi: 10.1021/acs.jafc.6b04538. [DOI] [PubMed] [Google Scholar]
- 42.Lekse J.M., Xia L., Stark J., Morrow J.D., May J.M. Plant catechols prevent lipid peroxidation in human plasma and erythrocytes. Mol Cell Biochem. 2001;226:89–95. doi: 10.1023/a:1012741903996. [DOI] [PubMed] [Google Scholar]
- 43.Poquet L., Clifford M.N., Williamson G. Investigation of the metabolic fate of dihydrocaffeic acid. Biochem Pharmacol. 2008;75:1218–1229. doi: 10.1016/j.bcp.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 44.Verweij M., van Ginhoven T.M., Mitchell J.R., Sluiter W., van den Engel S., Roest H.P., et al. Preoperative fasting protects mice against hepatic ischemia/reperfusion injury: mechanisms and effects on liver regeneration. Liver Transplant. 2011;17:695–704. doi: 10.1002/lt.22243. [DOI] [PubMed] [Google Scholar]
- 45.van Ginhoven T.M., Mitchell J.R., Verweij M., Hoeijmakers J.H., Ijzermans J.N., de Bruin R.W. The use of preoperative nutritional interventions to protect against hepatic ischemia–reperfusion injury. Liver Transplant. 2009;15:1183–1191. doi: 10.1002/lt.21871. [DOI] [PubMed] [Google Scholar]
- 46.Sanchez S., Lemmens S., Baeten P., Sommer D., Dooley D., Hendrix S., et al. HDAC3 inhibition promotes alternative activation of macrophages but does not affect functional recovery after spinal cord injury. Exp Neurobiol. 2018;27:437–452. doi: 10.5607/en.2018.27.5.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kimbrough D., Wang S.H., Wright L.H., Mani S.K., Kasiganesan H., LaRue A.C., et al. HDAC inhibition helps post-MI healing by modulating macrophage polarization. J Mol Cell Cardiol. 2018;119:51–63. doi: 10.1016/j.yjmcc.2018.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang G., Shi Y., Jiang X., Leak R.K., Hu X., Wu Y., et al. HDAC inhibition prevents white matter injury by modulating microglia/macrophage polarization through the GSK3beta/PTEN/Akt axis. Proc Natl Acad Sci U S A. 2015;112:2853–2858. doi: 10.1073/pnas.1501441112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Han Z., Zhao H., Tao Z., Wang R., Fan Z., Luo Y., et al. TOPK promotes microglia/macrophage polarization towards M2 phenotype via inhibition of HDAC1 and HDAC2 activity after transient cerebral ischemia. Aging Dis. 2018;9:235–248. doi: 10.14336/AD.2017.0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McKinsey T.A., Zhang C.L., Lu J., Olson E.N. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature. 2000;408:106–111. doi: 10.1038/35040593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pluemsampant S., Safronova O.S., Nakahama K., Morita I. Protein kinase CK2 is a key activator of histone deacetylase in hypoxia-associated tumors. Int J Cancer. 2008;122:333–341. doi: 10.1002/ijc.23094. [DOI] [PubMed] [Google Scholar]
- 52.Karwowska-Desaulniers P., Ketko A., Kamath N., Pflum M.K. Histone deacetylase 1 phosphorylation at S421 and S423 is constitutive in vivo, but dispensable in vitro. Biochem Biophys Res Commun. 2007;361:349–355. doi: 10.1016/j.bbrc.2007.06.167. [DOI] [PubMed] [Google Scholar]
- 53.Xing T., Zhu J., Xian J., Li A., Wang X., Wang W., et al. miRNA-548ah promotes the replication and expression of hepatitis B virus by targeting histone deacetylase 4. Life Sci. 2019;219:199–208. doi: 10.1016/j.lfs.2018.12.057. [DOI] [PubMed] [Google Scholar]
- 54.Stanfield B.A., Purves T., Palmer S., Sullenger B., Welty-Wolf K., Haines K., et al. IL-10 and class 1 histone deacetylases act synergistically and independently on the secretion of proinflammatory mediators in alveolar macrophages. PLoS One. 2021;16 doi: 10.1371/journal.pone.0245169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang J., Hodes G.E., Zhang H., Zhang S., Zhao W., Golden S.A., et al. Epigenetic modulation of inflammation and synaptic plasticity promotes resilience against stress in mice. Nat Commun. 2018;9:477. doi: 10.1038/s41467-017-02794-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun J., Wu Q., Sun H., Qiao Y. Inhibition of histone deacetylase by butyrate protects rat liver from ischemic reperfusion injury. Int J Mol Sci. 2014;15:21069–21079. doi: 10.3390/ijms151121069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ruess D.A., Probst M., Marjanovic G., Wittel U.A., Hopt U.T., Keck T., et al. HDACi valproic acid (VPA) and suberoylanilide hydroxamic acid (SAHA) delay but fail to protect against warm hepatic ischemia–reperfusion injury. PLoS One. 2016;11 doi: 10.1371/journal.pone.0161233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Evankovich J., Cho S.W., Zhang R., Cardinal J., Dhupar R., Zhang L., et al. High mobility group box 1 release from hepatocytes during ischemia and reperfusion injury is mediated by decreased histone deacetylase activity. J Biol Chem. 2010;285:39888–39897. doi: 10.1074/jbc.M110.128348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abe Y., Hines I.N., Zibari G., Pavlick K., Gray L., Kitagawa Y., et al. Mouse model of liver ischemia and reperfusion injury: method for studying reactive oxygen and nitrogen metabolites in vivo. Free Radic Biol Med. 2009;46:1–7. doi: 10.1016/j.freeradbiomed.2008.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequencing data associated with microbial diversity analysis is accessible at https://www.ncbi.nlm.nih.gov/bioproject and the accession numbers: PRJNA659591; Raw sequencing data associated with transcriptome analysis is accessible at https://www.ncbi.nlm.nih.gov/bioproject and the accession numbers: PRJNA659337; The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the iProX database (https://iprox.org/) and is available via ProteomeXchange with identifier PXD021195.