

Abstract
The immune checkpoint blockade therapy has profoundly revolutionized the field of cancer immunotherapy. However, despite great promise for a variety of cancers, the efficacy of immune checkpoint inhibitors is still low in colorectal cancer (CRC). This is mainly due to the immunosuppressive feature of the tumor microenvironment (TME). Emerging evidence reveals that certain chemotherapeutic drugs induce immunogenic cell death (ICD), demonstrating great potential for remodeling the immunosuppressive TME. In this study, the potential of ginsenoside Rg3 (Rg3) as an ICD inducer against CRC cells was confirmed using in vitro and in vivo experimental approaches. The ICD efficacy of Rg3 could be significantly enhanced by quercetin (QTN) that elicited reactive oxygen species (ROS). To ameliorate in vivo delivery barriers associated with chemotherapeutic drugs, a folate (FA)-targeted polyethylene glycol (PEG)-modified amphiphilic cyclodextrin nanoparticle (NP) was developed for co-encapsulation of Rg3 and QTN. The resultant nanoformulation (CD-PEG-FA.Rg3.QTN) significantly prolonged blood circulation and enhanced tumor targeting in an orthotopic CRC mouse model, resulting in the conversion of immunosuppressive TME. Furthermore, the CD-PEG-FA.Rg3.QTN achieved significantly longer survival of animals in combination with Anti-PD-L1. The study provides a promising strategy for the treatment of CRC.
KEY WORDS: Nano drug delivery system, Chemotherapy, Immunotherapy, Combination therapy, Immunogenic cell death, Reactive oxygen species, Tumor microenvironment, Colorectal cancer
Abbreviations: ATF6, activating transcription factor 6; ATP, adenosine triphosphate; CI, combination index; CXCL9, C-X-C motif chemokine 9; CXCL10, C-X-C motif chemokine 10; CTLA-4, cytotoxic T lymphocyte antigen 4; CRC, colorectal cancer; CRT, calreticulin; DAMPs, damage-associated molecular patterns; DCs, dendritic cells; ECL, enhanced chemiluminescence; EE, encapsulation efficiency; ER, endoplasmic reticulum; FA, folate; HMGB1, high-mobility group box 1; ICD, immunogenic cell death; IFN-γ, interferon-gamma; IL-4, interleukin-4; IL-6, interleukin-6; IL-10, interleukin-10; IL-12, interleukin-12; IRE1, inositol-requiring enzyme 1; LC, loading capacity; MDSCs, myeloid derived suppressor cells; MMR, mismatch repair; MR, molar ratio; NAC, N-acetyl-l-cysteine; NP, nanoparticle; p-IRE1, phosphorylation of IRE1; p-PERK, phosphorylation of PERK; PD-L1, programmed death-ligand 1; PEG, polyethylene glycol; PERK, PKR-like ER kinase; PFA, paraformaldehyde; PVDF, polyvinylidene fluoride; QTN, quercetin; ROS, reactive oxygen species; TAAs, tumor-associated antigens; TME, tumor microenvironment; UPR, unfolded protein response
Graphical abstract
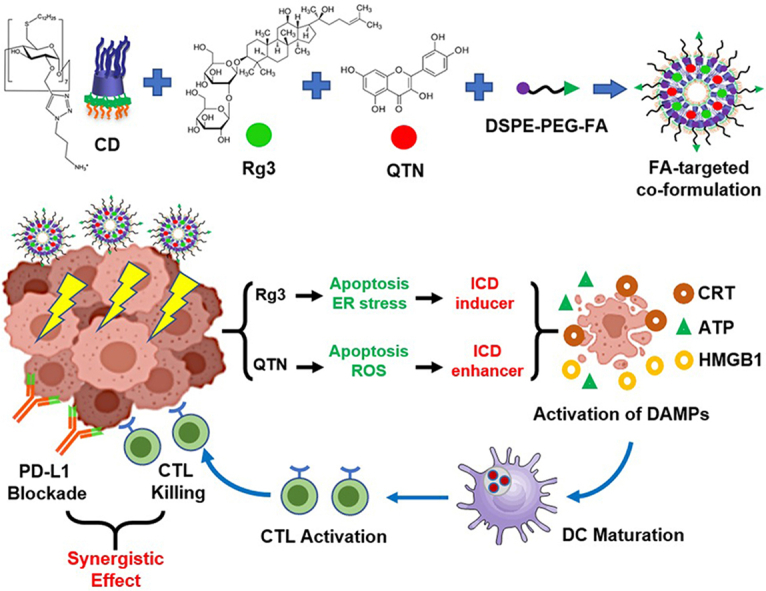
A folate-targeted PEGylated cyclodextrin-based nanoparticle was developed for co-delivery of Rg3 and QTN. The combination of the resultant co-formulation and anti-PD-L1 antibody achieved chemo-immunotherapy for colorectal cancer.
1. Introduction
As the third leading cause of cancer-related deaths worldwide (e.g., 881,000 deaths estimated in 2018)1, colorectal cancer (CRC, a cancer of the colon or rectum) is an obvious disease burden requiring effective, safe and widely-applicable treatments. Recent research in cancer immunology has led to the development of different immunotherapeutic strategies2, 3, 4. Among these, strategies that exert the blockade of immune checkpoint pathways [e.g., cytotoxic T lymphocyte antigen 4 (CTLA-4) and programmed death-ligand 1 (PD-L1)] have achieved favorable outcomes in a range of solid tumors5. However, it has been reported that only a minority of patients (up to 15% of the CRC population), who are identified with mismatch repair (MMR)-deficient CRC, respond positively to immune checkpoint blockade therapy6, while the response rate remains low in MMR-proficient CRC patients7. This failure is strongly attributed to the immunosuppressive feature of the tumor microenvironment (TME)8. Therefore, approaches designed to reprogram the TME may improve the therapeutic efficacy of immune checkpoint inhibitors9, potentially providing therapeutic benefit for the wide spectrum of CRC patients.
Immunogenic cell death (ICD) is characterized as immunogenic apoptosis that activates damage-associated molecular patterns (DAMPs) in dying or dead tumor cells in response to certain stimuli10. DAMPs as danger signals activate dendritic cells (DCs) for the presentation of tumor-associated antigens (TAAs), which subsequently induce T cell-mediated immunological responses against living tumor cells of the same kind10. The concept of ICD has revolutionized the traditional view of chemotherapeutic agents that are considered cytotoxic and poorly immunogenic. For example, chemotherapeutic drugs such as anthracyclines11, oxaliplatin12, bortezomib13 and cardiac glycosides14 have been identified as the putative ICD inducers. Recently, evaluation of chemotherapeutics as potential ICD inducers has gained increasing attention. In addition, ICD is often concomitant with the production of reactive oxygen species (ROS)10, and the efficacy of ICD may be enhanced by ROS-inducing strategies15, 16, 17. Therefore, it is hypothesized that a combination of ICD- and ROS-inducing strategies will mediate the remodeling of immunosuppressive TME and achieve synergistic immunotherapeutic efficacy with immune checkpoint blockade.
Ginsenosides are a group of naturally occurring chemicals within the extract of ginseng (a traditional medicine with a long history of human use18). One of the well-studied ginsenosides, ginsenoside Rg3 has demonstrated different pharmacological effects19, 20, 21, 22. The difference is the stereocenter on the C20 of Rg3 generates two epimers namely 20(S)- and 20(R)-Rg3. They both exhibit cardio-protective functions, promote antitumor effects, and mediate immunological responses, albeit to different extents23. In this study, the role of 20(S)-Rg3 (thereafter referred as Rg3, Fig. 1) as a potential ICD inducer was confirmed, to the best of our knowledge, for the first time using accepted and validated experimental approaches for the identification of ICD agents24. In addition, quercetin (thereafter referred as QTN, Fig. 1) is a natural pigment (flavonoid), and has obtained wide attention in cancer treatment and prevention25. In this study, ROS was effectively generated by QTN, which enhanced the activity of Rg3-mediated ICD. When Rg3 and QTN were co-formulated in a targeted amphiphilic cyclodextrin NP, they exerted chemo-immunotherapeutic effects in an orthotopic CRC mouse model, significantly improving the survival of animals when combined with Anti-PD-L1.
Figure 1.

A combination of folate-targeted Rg3/QTN cyclodextrin-based co-formulation and anti-PD-L1 antibody for chemo-immunotherapy in CRC.
2. Materials and methods
2.1. Materials
20(S)-Ginsenoside-Rg3 (Cat. No. DST180521-028) and quercetin (Cat. No. DST180130-011) were purchased from DESITE Biotech (Chengdu, China). DSPE-mPEG2000 (Cat. No. C12251) was obtained from Xi’an Biological Technology Co., Ltd., and DSPE-PEG2000-Folate (Cat. No. C09235) were obtained from Xi’an Biological Technology Co., Ltd. (Xi’an, China). The other chemicals and reagents were purchased from Sigma–Aldrich unless mentioned otherwise. In addition, the amphiphilic cationic cyclodextrin (Fig. 5A) was produced as previously reported in our laboratories26.
Figure 5.

Preparation and physicochemical characterization of FA-targeted co-formulation. (A) Formulation schematic. (B) The EE%, LC%, particle size and surface charge of targeted co-formulation. Data are presented as mean ± SD (n = 3). (C) TEM image of targeted co-formulation (scale bar = 100 nm). (D) The in vitro release of drugs from targeted co-formulation in 0.01 M PBS (pH = 5.5 and 7.4). Data are presented as mean ± SD (n = 4). (E) Particle size of targeted co-formulation following storage at 4 °C in aqueous solution. Data are presented as mean ± SD (n = 4). ∗P < 0.05 and ∗∗P < 0.01 relative to Day 0; NS, no significance. Non-targeted co-formulation also demonstrated similar physicochemical results observed by targeted counterpart.
2.2. Cell culture
CT26 (mouse CRC cell line) and HCT116 (human CRC cell line) were maintained within RPMI-1640 (Corning) with 10% fetal bovine serum (FBS; Corning) and 1% Penicillin-Streptomycin (TransGen Biotech, Beijing, China). CT26-Luc (mouse CRC cell line, stably expressing luciferase) was maintained within RPMI-1640 with 10% FBS, 1% Penicillin-Streptomycin, and 1 μg/mL puromycin (ThermoFisher). DC2.4 cells (mouse dendritic cell line) were maintained within RPMI-1640 with 10% FBS, 1 × nonessential amino acids, 1 × HEPES buffer, and 5.4 mmol/L β-mercaptoethanol. All cells were maintained at 37 °C with 5% CO2 and 95% relative humidity.
2.3. Animals
Six to eight-week old female BALB/c and nude mice were obtained from Changchun Institute of Biological Products, China. The experiments have been approved by the Animal Ethics Committee of Jilin University. Mice were allowed to acclimate for at least 7 days in the housing facility before the experiment. Animals were group-housed (5 animals per cage) in standard conditions (room temperature of 22 ± 2 °C under a 12 h light/dark cycle: lights on at 08:00 and lights off at 20:00). All mice received food and water ad libitum.
2.4. Induction of immunogenic cell death by ginsenoside Rg3
The in vitro cytotoxicity of Rg3 was determined using MTT assay. CT26 and HCT116 cells (1 × 104 per well) were seeded within 96-well plates for one day, respectively. Subsequently, Rg3 ([c] = 1, 5, 10, 20, 40, 80, 100, 120 and 150 μmol/L) was added to cells for 24 h. Cells were then added with MTT reagent (5 mg/mL in PBS) at 37 °C for ~4 h, and the purple precipitate was dissolved by DMSO before measurement at 570 nm. IC50 was calculated using the GraphPad Prism software.
The in vitro apoptosis of Rg3 was assessed using flow cytometry (Becton Dickinson, FACSCalibur, NJ, USA). CT26 and HCT116 cells (2 × 105 per well) were seeded in 6-well plates for one day, respectively. After this, Rg3 ([c] = 30 μmol/L) was added was added to cells for 6, 12 and 24 h. Subsequently, apoptotic cells (%) were detected using Annexin V-FITC/propidium iodide assay (YEASEN Biotech, Shanghai, China) by BD FACSCalibur.
The activity of UPR signaling pathways was evaluated using Western blotting assay. CT26 and HCT116 cells (2 × 105 per well) were seeded in 6-well plates for one day, respectively. After this, Rg3 ([c] = 30 μmol/L) was added to cells for 6, 12 and 24 h. Subsequently, proteins were extracted from cells using ProteinExt® Mammalian Total Protein Extraction Kit (TransGen Biotech) and qualified using BCA assay (Beyotime Biotech, Beijing, China). Proteins (35–50 μg per sample) were loaded onto the SDS-polyacrylamide gel and run at 80–100 V for 30–60 min. Proteins were then transferred to polyvinylidene fluoride (PVDF) membrane (Invitrogen) at 90 V for 1 h. The membrane was incubated overnight with antibodies (Supporting Information Table S1) at 4 °C. The secondary antibody (Table S1) was added to the membrane for 1.5 h. Proteins were detected using the enhanced chemiluminescence (ECL) solution (GE Healthcare). Quantification of protein bands was performed using densitometry (ImageJ), and all results were normalized to β-Actin.
The exposure of CRT was detected using immunofluorescent staining assay. CT26 and HCT116 cells (1 × 105 per well) were seeded in 24-well plates with glass bottoms for one day, respectively. Rg3 ([c] = 30 μmol/L) was added to cells for 6 h. After this, cells were incubated with 0.25% paraformaldehyde (PFA) for 5 min. Cells were then washed with PBS, which were followed by anti-CRT antibody (Table S1) for 1 h. After PBS washes, FITC-conjugated secondary antibody (Table S1) was added for 30 min. Subsequently, cells were treated by 4% PFA for 20 min and stained using DAPI (Beyotime Biotech) for confocal imaging (OLYMPUS, Olympus FV3000, Tokyo, Japan).
The secretion of ATP and release of HMGB1 were assessed using bioluminescent and ELISA assays. CT26 and HCT116 cells (5 × 105 per well) were seeded in 6-well plates for one day. Rg3 ([c] = 30 μmol/L) was added to cells for 12 h. Subsequently, the level of ATP and HMGB1 in the supernatants was measured using ENLITEN® ATP Assay System Bioluminescence Detection Kit (Promega) and ELISA kit (LS-F11641 and LS-F11642, LifeSpan BioSciences), respectively.
The maturation of DC2.4 cells was determined using flow cytometry (Becton Dickinson). CT26 (5 × 105 per well) were seeded in 6-well plates for one day. After this, Rg3 ([c] = 30 μmol/L) was added to cells for one day. Separately, DC2.4 cells (2 × 105 per well) were seeded in 6-well plates. Following 24 h, the supernatants were collected from Rg3-treated CT26 cells and transferred to DC2.4 cells for another 24 h. DC2.4 cells were treated with fluorophore-conjugated antibodies (Table S1), and the expression (%) of CD11c and CD86 was assessed using by BD FACSCalibur (Becton Dickinson).
The in vivo vaccination assay was performed as previously described27. Briefly, 3 × 106 CT26 cells, either treated with DMSO, freeze-thawing three times on dry ice, or 30 μmol/L Rg3 for 12 h, were s.c. implanted into the right flank of BALB/c mice or nude mice. One week later, 3 × 105 CT26 cells were s.c. implanted into the left flank. Tumor development in left flank was monitored to determine tumor-free mice.
2.5. Generation of reactive oxygen species by quercetin
The in vitro cytotoxicity of QTN was determined using MTT assay. CT26 and HCT116 cells (1 × 104 per well) were seeded within 96-well plates for one day, respectively. Subsequently, QTN ([c] = 1, 5, 10, 20, 40, 80, 100, 120 and 150 μmol/L) was added to cells for 24 h. Cells were then added with MTT reagent at 37 °C for ~4 h, and the purple precipitate was dissolved by DMSO before measurement at 570 nm. IC50 was calculated using the GraphPad Prism software. In a separate study, following seeding, cells were treated with or without N-acetylcysteine (NAC; 5 mmol/L) for 4 h. Subsequently, QTN ([c] = 80 μmol/L) was added into cells for 24 h, and cytotoxicity was measured as mentioned above.
The in vitro apoptosis of QTN was assessed using flow cytometry (Becton Dickinson). CT26 and HCT116 cells (2 × 105 per well) were seeded in 6-well plates for one day, respectively. After this, cells were treated with or without NAC (5 mmol/L) for 4 h. Subsequently, QTN ([c] = 80 μmol/L) was added to cells for 24 h, and apoptotic cells (%) were detected as mentioned above.
The activity of BCL-2/BAX/caspase 9/caspase 3 signaling pathways was evaluated using Western blotting assay. CT26 and HCT116 cells (2 × 105 per well) were seeded in 6-well plates for one day, respectively. After this, QTN ([c] = 80 μmol/L) was added to cells for 6, 12 and 24 h. The Western blotting assay was performed as mentioned above. Quantification of protein bands was performed using densitometry (ImageJ), and all results were normalized to β-actin.
To measure the formation of ROS, CT26 and HCT116 cells (2 × 105 per well) were seeded in 6-well plates for one day, respectively. After this, QTN ([c] = 80 μmol/L) was added to cells for 6, 12 and 24 h. The ROS level in cells was detected using 2ʹ,7ʹ-dichlorodihydrofluorescein diacetate-based Reactive Oxygen Species Assay Kit (YEASEN Biotech) by BD FACSCalibur (Becton Dickinson) (488 nm/525 nm).
2.6. In vitro effects of ginsenoside Rg3 and quercetin
The in vitro cytotoxicity of “Rg3 + QTN” was determined using MTT assay. CT26 cells (1 × 104 per well) were seeded within 96-well plates for one day, respectively. Subsequently, “Rg3 + QTN” at different molar ratios (MR = 1:5, 1:2, 1:1, 2:1 and 5:1) was added to cells for 24 h, and IC50 was measured as mentioned above.
The in vitro apoptosis of “Rg3 + QTN” was assessed using flow cytometry (Becton Dickinson). CT26 (2 × 105 per well) were seeded in 6-well plates for one day, respectively. After this, cells were added with either single drugs or “Rg3 + QTN” ([c] = 12 and 12 μmol/L, respectively) for 24 h, and apoptotic cells (%) were detected as mentioned above.
The exposure of CRT was detected using immunofluorescent staining assay. CT26 (1 × 105 per well) were seeded in 24-well plates with glass bottoms for one day, respectively. Cells were then treated with or without NAC (5 mmol/L) for 4 h. Subsequently, cells were added with either single drugs or “Rg3 + QTN” ([c] = 12 and 12 μmol/L, respectively) for 6 h. The exposure of CRT was detected as mentioned above.
The secretion of ATP and release of HMGB1 were assessed using bioluminescent and ELISA assays. CT26 cells (5 × 105 per well) were seeded in 6-well plates for one day. Cells were then treated with or without NAC (5 mmol/L) for 4 h. Subsequently, cells were added with either single drugs or “Rg3 + QTN” ([c] = 12 and 12 μmol/L, respectively) for 12 h. The level of ATP and HMGB1 in the supernatants was measured as mentioned above.
The maturation of DC2.4 cells was determined using flow cytometry (Becton Dickinson). CT26 (5 × 105 per well) were seeded in 6-well plates for one day. After this, cells were then treated with or without NAC (5 mmol/L) for 4 h. Subsequently, “Rg3 + QTN” ([c] = 12 and 12 μmol/L, respectively) was added to cells for one day. Following 24 h, the supernatants were collected from Rg3-treated CT26 cells and transferred to DC2.4 cells for another 24 h. The expression (%) of CD11c and CD86 was assessed as mentioned above.
2.7. Preparation and characterization of co-formulations
The preparation has been optimized in terms of encapsulation efficiency (EE%) and loading capacity (LC%) as follows: 2 mg of CD, 0.5 mg of Rg3 and 0.2 mg of QTN were dissolved in 5 mL chloroform (CHCl3) in a round bottom flask and dried to form thin film using rotary evaporator. Subsequently, the thin film was rehydrated using 5 mL of ultrapure water, sonicated at 0 °C for 30 min, and incubated by slight shaking at 37 °C for 1–1.5 h, in order to obtain the CD.Rg3.QTN complex. In addition, a solution of DSPE-mPEG2000 and DSPE-mPEG2000-Folate (MR, ~4:1, 0.5 mg/mL) was prepared in 20 mmol/L HEPES buffer (pH = 7.4) at 60 °C with shaking for 15 min. As previously described28,29, the “postinsertion” of DSPE-mPEG2000/DSPE-mPEG2000-folate into preformed CD. Rg3.QTN complex was carried out at 60 °C with shaking for 1 h. As a result, ~1.5% (mol/mol) of FA on the outer surface per formulation was achieved. Non-targeted co-formulation was produced as mentioned above without the use of DSPE-mPEG2000-folate. In addition, the rhodamine-labelled formulations were prepared as described above containing ~0.02% (w/w) of rhodamine.
The EE% and LC% were assessed using HPLC (Shimadzu, SPD-20A, Kyoto, Japan) [C18 column; UV at 203 nm for Rg3, mobile phase = acetonitrile and water (0.05% phosphoric acid), 50:50; UV at 370 nm for QTN, mobile phase = methanol and water (0.2% phosphoric acid), 70:30]. As EE% was >95% for both drugs, co-formulations were used for in vitro and in vivo experiments without further purification as shown in Eqs. (1), (2).
| (1) |
| (2) |
The particle size and zeta potential were measured using Malvern Nano-ZS as described previously30. The morphology of NPs was observed using TEM (Jeol, JEOL JEM1230, Tokyo, Japan) as described previously31. Briefly, ~5 μL of samples were added to a 400-mesh carbon-filmed copper grid and stained using 2% (w/w) uranyl acetate before TEM analysis. Moreover, co-formulations containing 250 μg and 100 μg QTN (MR = 1:1) in 0.01 mol/L PBS (pH = 5.5 and 7.4) was placed within the dialysis bag (MWCO = 2 kDa; Solarbio®, Beijing, China). The dialysis bag was incubated at 37 °C within the release medium (0.5% Tween-80 0.01 mol/L PBS solution). Samples were collected at different time points (the release medium with the same volume was supplemented), and the concentration of free drugs was determined using HPLC (Shimadzu) as mentioned above.
2.8. In vitro studies of co-formulations
The cellular uptake of co-formulations (containing 0.05% Rhodamine, w/w) was assessed using confocal microscopy (Olympus) and flow cytometry (Becton Dickinson). CT26 and HCT116 cells (1 × 105 per well) were seeded in 24-well plates with glass bottoms for one day, respectively. Rhodamine-containing co-formulations (12 μmol/L Rg3 and 12 μmol/L QTN) were added to cells for 6 h. After this, cells were treated by 4% PFA for 20 min and stained using DAPI (Beyotime Biotech) for confocal imaging (Olympus). In addition, CT26 and HCT116 cells (1 × 105 per well) were seeded in 24-well plates for one day, respectively. Rhodamine-containing co-formulations (12 μmol/L Rg3 and 12 μmol/L QTN) were added to cells for 4 h. Subsequently, rhodamine-positive cells (%) were detected using BD FACSCalibur.
The in vitro cytotoxic, antiproliferative and antimetastatic activities of co-formulations were determined using MTT, scratch and colony formation assays, respectively. CT26 and HCT116 cells (1 × 104 per well) were seeded within 96-well plates for one day, respectively. Subsequently, co-formulations (12 μmol/L Rg3 and 12 μmol/L QTN) were added to cells for 24 h, and IC50 was measured as mentioned above. In addition, the in vitro scratch assay was carried out as previously described32. Briefly, when CT26 and HCT116 cells reached confluence, the cell monolayer was washed thoroughly with PBS, scraped with a p200 pipette tip to create a “scratch”, and washed again with PBS. Cells were replaced with serum-free growth medium and added with co-formulations (12 μmol/L Rg3 and 12 μmol/L QTN) for 12 h. The cell-free areas before and after the incubation of co-formulations were imaged under the microscope and measured using ImageJ. Furthermore, the in vitro colony formation assay was performed as previously described33. Briefly, CT26 and HCT116 cells seeded in 6-well plates with 30%–50% confluence were treated with co-formulations (12 μmol/L Rg3 and 12 μmol/L QTN) for 4 weeks. The colonies were stained with 0.2% crystal violet and counted under the microscope (OLYMPUS, Olympus CK2, Tokyo, Japan).
2.9. In vivo toxicity, pharmacokinetics and biodistribution of co-formulations
Healthy BALB/c mice were treated with co-formulations as described in Fig. 7A (n = 5). Body weight was regularly recorded. In addition, major organs, the whole blood and the serum were obtained on Day 30 to analyze histopathology, myelosuppression, and hepatic/renal functions as previously described (Fig. 7B‒D)33,34.
Figure 7.

In vivo toxicity, pharmacokinetics and biodistribution of targeted co-formulation. (A) The body weight over a 30-day period following i.v. treatment of PBS and targeted co-formulation on Days 1, 3, 5, and 7. Data are presented as mean ± SD (n = 5). (B) Major organs were collected on Day 30 and assessed using H&E staining assay. No significant toxic sign was found in targeted co-formulation as compared to PBS (scale bar = 50 μm) (C) Hematological analysis including red blood cells (RBCs), white blood cells (WBCs), platelets (PLTs) and hemoglobin (HGB) was carried out on Day 30. Data are presented as mean ± SD (n = 4). (D) The liver/kidney functions including alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood urea nitrogen (BUN) and creatinine (CRE) were determined on Day 30. Data are presented as mean ± SD (n = 4). (E) The concentration of drugs in the plasma was plotted at different time points. Data are presented as mean ± SD (n = 4). (F) Biodistribution of DiD-labeled co-formulations was detected (640 nm/670 nm) using IVIS® In Vivo Optical System. Data are presented as mean ± SD (n = 4). ∗P < 0.05 in orthotopic CRC mouse model.
Following the procedures as previously described34, the cecum wall of BALB/c mice was injected with 5 × 105 CT26-Luc cells to establish orthotopic CRC mouse model. After the inoculation (Day 0), 100 μL of luciferin (10 mg/mL; Pierce) were intraperitoneally (i.p.) administrated to animals, and tumor progression was monitored using IVIS® In Vivo Optical System (Perkin Elmer). When tumor was developed to ~5 to 10 × 108 p/s/cm2/sr, mice were used for pharmacokinetics and tissue distribution: 1) Co-formulations containing 10 mg/kg of Rg3 and 4 mg/kg of QTN were i.v. administrated, and the blood (~50 μL) was collected at different time points (Fig. 7E, n = 4). Drugs in the plasma were extracted with ethyl acetate, dried with nitrogen, and reconstituted in the mobile phase for HPLC (Shimadzu). Half-life was evaluated using DAS 2.0 software. 2) Co-formulations containing ~0.05% (w/w) of DiD (ThermoFisher), 10 mg/kg of Rg3 and 4 mg/kg of QTN were i.v. injected to animals, and biodistribution was detected (640 nm/670 nm) using IVIS® In Vivo Optical System (PerkinElmer, IVIS Kinetic, MA, USA) (n = 4).
2.10. Combination of targeted co-formulation and anti-PD-L1 for CRC therapy
When tumor was developed to ~5 to 10 × 108 p/s/cm2/sr, as described in Fig. 8A, mice (n = 5) were treated with either Anti-PD-L1 (Bioxcell, clone 10F.9G2, 100 μg per mouse, i.p.), targeted co-formulation (10 mg/kg of Rg3 and 4 mg/kg of QTN per mouse, i.v.), or the combination. Tumor progression was monitored using IVIS® In Vivo Optical System (PerkinElmer).
Figure 8.

Combination therapy of targeted co-formulation and Anti-PD-L1 for CRC. (A) Treatment schedule and IVIS images. (B) The CRC progression over a 35-day period. Data are presented as mean ± SD (n = 5). ∗P < 0.05 and ∗∗P < 0.01; NS, no significance. (C) Animal survival (median survival: PBS ~38 days, Anti-PD-L1 ~40 days, targeted co-formulation ~62 days, and combination ≈ 96 days). Data are presented as mean ± SD (n = 5). ∗∗P < 0.01 and ∗∗∗P < 0.001. (D) Immunofluorescent staining assay (green = DNA fragments and blue = nuclei) on Day 20 to assess apoptosis in the tumor (scale bar = 50 μm). Data are presented as mean ± SD (n = 3). ∗P < 0.05 and ∗∗P < 0.01, relative to PBS. (E) Level of immune cells in the tumor on Day 20 was analyzed using flow cytometry (BD). Data are presented as mean ± SD (n = 4). ∗P < 0.05 and ∗∗P < 0.01; NS, no significance. (F) The mRNA expression of cytokines and chemokines in the tumor on Day 20 was analyzed using real time RT-PCR. Data are presented as mean ± SD (n = 4). ∗P < 0.05 and ∗∗P < 0.01; NS, no significance. (G) Orthotopic CRC mice treated with targeted co-formulation following the removal of CD4+ or CD8+ T cells. Data are presented as mean ± SD (n = 4). ∗P < 0.05 and ∗∗P < 0.01; NS, no significance.
Separately, 2 days after two injections (Day 20; the timepoint chosen to analyze immunological effects was generally within one week following treatment)35, 36, 37, 38, 39, tumors were collected for following studies: 1) Apoptosis. Tumors (n = 3) were fixed with 4% PFA, conducted on paraffin-embedded slides, and permeabilized33,34. DNA fragments were detected using the TransDetect®Fluorescein TUNEL Cell Apoptosis Kit (TransGen Biotech), and nuclei were stained with DAPI (Beyotime Biotech), for confocal microscopic analysis (Olympus); 2) Measurement of immune cells. Single cells from tumors (n = 4) were generated using collagenase A (1 mg/mL; Sigma) and DNAse (200 μg/mL; Invitrogen)33,34. After removal of red blood cells using the ACK buffer (Gibco), cells were incubated with fluorophore-labeled antibodies (Table S1), fixed using 4% PFA, and analyzed using BD FACSCalibur. 3) Measurement of cytokines and chemokines. Tumors (n = 4) were homogenized within TriZol Up reagent (TransGen Biotech) using the tissue grinder (Scientz, Zhengjiang, China). The homogenates collect the supernatant for RT-PCR. First-strand cDNA was generated using the TransScript® First-Strand cDNA Synthesis SuperMix kit (TransGen Biotech). Quantitative real-time RT-PCR was performed using TransStart® Top Green qPCR SuperMix kit (TransGen Biotech) by StepOnePlus™ Real-Time PCR System (ThermoFisher, QuantStudio 3, MA, USA). The reaction was carried out: 94 °C for 30 s, 45 cycles of 5 s at 94 °C, and 30 s at 60 °C. The primers were listed in Supporting Information Table S2.
The depletion study of T cells was carried out as previously reported33,34. Briefly, 100 μg of either anti-CD4 (clone GK1.5, Bioxcell), anti-CD8 (clone 53-6.72, Bioxcell) or IgG (polyclonal, Bioxcell) antibodies were i.p. given per mouse (Fig. 8H) before the treatment of targeted co-formulation (10 mg/kg of Rg3 and 4 mg/kg of QTN). Tumor progression was monitored using IVIS® In Vivo Optical System (PerkinElmer) (n = 4).
2.11. Statistical analysis
GraphPad prism software was applied for statistical analysis. Results were exhibited as mean ± standard deviation (SD). The significance between two groups was assessed using unpaired Student’s t-test. The significance between three or more groups was assessed using the one-way ANOVA (Bonferroni’s correction). Kaplan–Meier survival analysis with log-rank Mantel–Cox test was used to determine the overall survival rate and tumor free rate. The combination index (CI) was determined as previously described40, CI < 1 suggests the synergistic effect. In this work, P < 0.05 was considered statistically significant.
3. Results
3.1. Ginsenoside Rg3 induces immunogenic cell death in CRC cells
The cytotoxicity (IC50) of Rg3 was assessed in mouse CT26 and human HCT116 CRC cell lines using the MTT assay (Fig. 2A). The antiproliferative effect of Rg3 was similar in two cell lines, with IC50 ~32 μmol/L for CT26 (24 h incubation) and IC50 ~30 μmol/L for HCT116 (24 h incubation) (Fig. 2A). Flow cytometry (Becton Dickinson) results show that Rg3 induced apoptosis in a time-dependent manner, causing ~40% and ~35% apoptosis (24 h incubation) in CT26 and HCT116 cell lines, respectively (Fig. 2B). These results indicate that Rg3-elicited apoptosis is the mechanism for inhibition of CRC cell growth.
Figure 2.

Ginsenoside Rg3 induced immunogenic cell death in CRC cells. (A) IC50 of Rg3 for CT26 and HCT116 cells at 24 h. Data are presented as mean ± SD (n = 3). (B) Apoptosis (%) in CT26 and HCT116 cells following treatment of Rg3 at 6, 12 and 24 h. Data are presented as mean ± SD (n = 3). ∗P < 0.05 and ∗∗P < 0.01 relative to DMSO. (C) The activity of UPR signaling pathways following treatment of Rg3 ([c] = 30 μmol/L) at 6, 12 and 24 h. The quantification was demonstrated in Fig. S1. (D) The characterization of ICD in CRC cells following treatment of Rg3 ([c] = 30 μmol/L), including CRT exposure (6 h), ATP secretion (12 h) and HMGB1 release (12 h). Data are presented as mean ± SD (n = 3). ∗P < 0.05 relative to DMSO; scale bar = 20 μm. (E) The expression of CD11c and CD86 in DCs stimulated with the supernatant from Rg3 ([c] = 30 μmol/L) -treated cells (24 h). Data are presented as mean ± SD (n = 3). ∗∗P < 0.01 relative to DMSO. (F) The in vivo vaccination assay using BALB/C and nude mice. Data are presented as mean ± SD (n = 4). ∗∗∗P < 0.001.
The potential of Rg3 as the ICD inducer was assessed in CT26 and HCT116 cell lines. The induction of ICD is critically linked to two types of stress, namely endoplasmic reticulum (ER) stress and autophagy41,42. When cancer cells undergo the ER stress, the unfolded protein response (UPR) signaling pathways including inositol-requiring enzyme 1 (IRE1), PKR-like ER kinase (PERK) and activating transcription factor 6 (ATF6) are activated43. As shown in Fig. 2C, the phosphorylation of IRE1 protein (p-IRE1) was upregulated by Rg3, and the expression of p-IRE1 relative to total IRE1 protein was significantly increased (P < 0.001) in two cell lines (Supporting Information Fig. S1). Rg3 also mediated two downstream effects associated with the IRE1 signaling pathway, namely the downregulation of BCL-2 protein and the upregulation of BAX protein (Fig. 2C and Fig. S1). In addition, the phosphorylation of PERK protein (p-PERK) was induced by Rg3, and the expression of p-PERK relative to total PERK protein was significantly enhanced (P < 0.05 and P < 0.01) in CT26 and HCT116 cells (Fig. 2C and Fig. S1). Accordingly, the cleavage of caspase 9 and caspase 3 (c-caspase 9 and c-caspase 3, two downstream proteins of the PERK signaling pathway) was significantly upregulated (P < 0.01 and P < 0.001; Fig. 2C and Fig. S1). Furthermore, the expression of ATF6 protein and the cleavage of caspase 4 (c-caspase 4, one downstream protein of the ATF6 signaling pathway) were significantly (P < 0.05, P < 0.01 and P < 0.001) enhanced by Rg3 in CT26 and HCT116 cells (Fig. 2C and Fig. S1). When autophagy (or autophagocytosis, a natural process for removal of cytoplasmic components44) proceeds in cancer cells, autophagosomes, a double-membrane vesicle, transport unnecessary or dysfunctional components into lysosomes for degradation45. The microtubule-associated protein light chain 3 (LC3) is an essential protein associated with autophagosome biogenesis, and has been confirmed as the most widely used marker for the autophagy pathway46. The formation of autophagosomes is evident with the transformation of LC3-I (a cytosolic form of LC3) into LC3-II (a membrane-bound form of LC3)47. As shown in Fig. 2C and Fig. S1, the ratio of LC3-II/LC3-I was significantly (P < 0.05) increased by Rg3. Therefore, results in Fig. 2C indicate that ER stress and autophagy, two types of stress necessary for the ICD induction, were effectively induced by Rg3 in CRC cells.
Endogenous DAMPs are activated in cancer cells during ICD in response to ER stress and autophagy. The exposure of calreticulin (CRT, a protein mainly residing in the endoplasmic reticulum), secretion of adenosine triphosphate (ATP) and release of high-mobility group box 1 (HMGB1, a nuclear protein) are three essential hallmarks that can be used to accurately predict the ICD potential of candidate drugs48. In response to the ER stress, CRT, a protein located inside the ER lumen, is translocated onto the plasma membrane of pre-apoptotic cancer cells49. During apoptosis, autophagy is required for ATP secretion from dying cells into the extracellular milieu42. At the late stage of apoptosis, HMGB1, a non-histone chromatin protein, is released from the nucleus into the extracellular environment50. The capacity of Rg3 to activate these ICD hallmarks was assessed in CT26 and HCT116 cells (Fig. 2D). Results indicate that the exposure of CRT, secretion of ATP and release of HMGB1 were significantly (P < 0.05) activated following treatment with Rg3 (Fig. 2D), suggesting that Rg3 could elicit the apoptosis, ER stress and autophagy.
DAMPs can activate the transition of immature DCs to a mature phenotype51. When DC2.4 cells (mouse DCs) were challenged with PBS and the growth medium used for DMSO-treated or Rg3-treated CT26 cells, the number of CD11c+ and CD86+ population (defined as mature DCs52, 53, 54) was measured using flow cytometry (Becton Dickinson). Results show that no significant difference in the number of CD11c+ and CD86+ cells was found between the DMSO-treated group (Fig. 2E) and the PBS group (data not shown). In contrast, DC maturation was significantly (P < 0.01) increased in the Rg3-treated group (Fig. 2E), indicating that Rg3-induced cell death was immunogenic, and could promote the mature status of DCs.
Tumor cells undergoing ICD in vitro can be used as the vaccine to induce T cell-mediated antitumor immunity against living tumor cells of the same kind in vivo24. Thus, the in vivo vaccination assay was performed to further identify the ICD potential of Rg3 (Fig. 2F). CT26 cells were pre-treated with either DMSO, freeze-thawing or Rg3. Subsequently, these cells were subcutaneously (s.c.) injected into the right flank of immunocompetent mice. The same animals were s.c. injected with living CT26 cells at the left flank one week later. Consequently, mice injected with DMSO-treated or freeze-thawed cells succumbed to the rechallenging of living CT26 cells, and tumor growth was found in all animals within 15 days (Fig. 2F). In contrast, inoculation of mice with CT26 cells treated by Rg3 significantly prevented subsequent growth of living CT26 cells (P < 0.001) (Fig. 2F). As a confirmatory study, nude mice (immunodeficient), which are characterized with a lack of functional T cells, were used for the vaccination assay. As a result, the antitumor effect arising from Rg3-treated CT26 cells was significantly abolished in nude mice (Fig. 2F). These results suggest that Rg3 was able to convert the CRC cells into the endogenous vaccine, which may mediate the maturation of DCs to activate T cells against live cells of the same type. Taken together, results in Fig. 2 confirmed the potential of Rg3 as the ICD inducer against CRC.
3.2. Quercetin causes reactive oxygen species in CRC cells
The cytotoxicity (IC50) of QTN was assessed in CT26 and HCT116 cell lines using the MTT assay (Fig. 3A). QTN led to similar antiproliferative effect in the two cell lines, with IC50 ~80 μmol/L for CT26 (24 h incubation) and IC50 ~81 μmol/L for HCT116 (24 h incubation) (Fig. 3A). QTN also caused ~70% and ~50% apoptosis (24 h incubation) in CT26 and HCT116 cells, respectively (Fig. 3B). The apoptotic effects of QTN were confirmed by the downregulation of BCL-2 (an apoptosis-suppressing protein) and the upregulation of BAX (an apoptosis-regulating protein), caspase 9, and caspase 3 (Fig. 3C and Supporting Information Fig. S2). Thus, QTN-induced apoptosis is the main mechanism for the inhibition of CRC cell growth.
Figure 3.

Quercetin caused reactive oxygen species in CRC cells. (A) IC50 of QTN for CT26 and HCT116 cells at 24 h. Data are presented as mean ± SD (n = 3). (B) Apoptosis (%) in CT26 and HCT116 cells following treatment of QTN ([c] = 80 μmol/L) at 24 h. Data are presented as mean ± SD (n = 3). ∗∗P < 0.01 relative to DMSO. (C) The activity of Bcl-2/BAX/caspase 9/caspase 3 signaling pathways following treatment of QTN ([c] = 80 μmol/L) at 6, 12 and 24 h. The quantification was demonstrated in Fig. S2. (D) The ROS level in CT26 and HCT116 cells following treatment of QTN ([c] = 80 μmol/L) at 6, 12 and 24 h. Data are presented as mean ± SD (n = 3). ∗∗P < 0.01 and ∗∗∗P < 0.001 relative to DMSO. (E) Cell viability (%) of CT26 and HCT116 cells with or without NAC prior to treatment of QTN ([c] = 80 μmol/L) (24 h). Data are presented as mean ± SD (n = 3). ∗P < 0.05 and ∗∗P < 0.01 relative to untreated control. (F) Apoptosis (%) in CT26 and HCT116 cells with or without NAC prior to treatment of QTN ([c] = 80 μmol/L) (24 h). Data are presented as mean ± SD (n = 3). ∗P < 0.05 and ∗∗P < 0.01 relative to untreated control.
The BCL-2/BAX/caspase 9/caspase 3 signaling pathways are closely linked with the formation of ROS (one of apoptotic stimuli)55. The level of ROS in two CRC cell lines was measured using a bioluminescent assay, showing that QTN generated ROS in a time-dependent manner (Fig. 3D). To investigate the role of QTN-mediated ROS in antitumor effects, CRC cells were treated with N-acetyl-l-cysteine (NAC, a chemical used for the neutralization of ROS56,57) prior to the treatment of QTN (Fig. 3E). Results show that QTN-mediated cytotoxicity and apoptosis were significantly (P < 0.05 and P < 0.01) impeded by the pretreatment of NAC (Fig. 3E and F), suggesting that QTN could induce the formation of ROS for antiproliferative and apoptotic effects in CRC cells.
3.3. Combination of quercetin and ginsenoside Rg3 shows a synergistic effect in CRC cells
The concomitant activation of ER stress and ROS production is critical for the activity of DAMPs58, and the efficacy of ICD may be effectively improved by ROS-inducing strategies15, 16, 17. MTT results show that combining the two drugs achieved a synergistic antiproliferative effect (Fig. 4A). When the molar ratio (MR) of the two drugs was optimized at 1:1 (Fig. 4A and Supporting Information Fig. S3), the IC50 of the combined QTN and Rg3 regimen was reduced to ~12 μmol/L and (Fig. 4A), which was significantly (P < 0.05) lower compared to results following exposure to the individual drugs (IC50 of QTN ~80 μmol/L and IC50 of Rg3 ~32 μmol/L) (Figure 2, Figure 3A). The combined regimen also significantly (P < 0.01) enhanced the apoptotic effects (~80%, 24 h incubation) in CT26 cells as compared to either QTN (~20%) or Rg3 (~30%) (Fig. 4B). Similar cytotoxic and apoptotic effects in HCT116 cells, were also recorded in the CT26 cells. These results indicate that a synergistic antitumor effect was achieved by the combination of QTN and Rg3.
Figure 4.

Synergistic effects of Rg3 and QTN in CT26 cells. (A) IC50 of drug combination at 24 h. Data are presented as mean ± SD (n = 3). CI values at IC50 were shown in Fig. S3. (B) Apoptosis (%) caused by drug combination at 24 h. Data are presented as mean ± SD (n = 3). ∗P < 0.05 and ∗∗P < 0.01 relative to DMSO. (C) The CRT exposure with or without NAC before treatment of drug combination (6 h). Data are presented as mean ± SD (n = 3). ∗P < 0.05 and ∗∗P < 0.01 relative to DMSO; scale bar = 20 μm. (D) The ATP secretion with or without NAC before treatment of drug combination at 12 h. Data are presented as mean ± SD (n = 3).∗P < 0.05 and ∗∗P < 0.01, between NAC and No NAC. (E) The HMGB1 release with or without NAC before treatment of drug combination at 12 h. Data are presented as mean ± SD (n = 3). ∗P < 0.05 and ∗∗P < 0.01, between NAC and No NAC. (F) The expression of CD11c and CD86 in DCs stimulated (24 h) by the supernatant from Rg3-treated cells with or without pretreatment of NAC. Data are presented as mean ± SD (n = 3). ∗P < 0.05, ∗∗P < 0.01 and ∗∗∗P < 0.001, relative to DMSO.
The synergistic ICD effects of QTN and Rg3 were assessed in CT26 cells, as this cell line was chosen to establish the tumor-bearing mouse model for analysis of the in vivo immunological response. As shown in Fig. 4C, the exposure of CRT onto the cell membrane was not achieved by treatment with either DMSO or QTN, in contrast Rg3 significantly (P < 0.05) triggered the translocation of CRT. Notably, the combination further (P < 0.01) activated the CRT exposure (Fig. 4C). In addition, the combined regimen significantly (P < 0.01 and P < 0.001) elicited the secretion of ATP and the release of HMGB1 in CT26 cells (Fig. 4D and E) and triggered the maturation (CD11c+ and CD86+) of DCs (Fig. 4F) relative to single drugs. It is worth noting that the pretreatment of NAC significantly (P < 0.05 and P < 0.01) dampened the activity of ICD hallmarks and the maturation of DCs (Fig. 4C‒F), suggesting that the ICD efficacy of Rg3 in the CRC cells was significantly enhanced by QTN and that this synergy was, at least, in part due to the production of ROS.
3.4. Preparation and physicochemical characterization of co-formulations
The in vivo application of chemotherapeutic agents is seriously impeded by low solubility, poor pharmacokinetics, and non-specific tissue distribution. Recently, development of nano delivery systems has achieved high bioavailability, controlled drug release, prolonged systematic circulation, and improved tumor distribution for chemotherapeutic agents33,34,59,60. In addition, Rg3 and QTN possess distinctive physicochemical characteristics (e.g., the logP of Rg3 and QTN ≈ 2.63 and 1.81, and the molecular weight of Rg3 and QTN = 785 and 302), therefore, a nano delivery system is desirable for co-delivery of two drugs in order to achieve synergistic effects. However, to our best knowledge, no study has been reported for co-encapsulation of Rg3 and QTN in a nanoformulation for CRC.
Previously, a range of cyclodextrin-based NPs have been generated and characterized using a variety of in vitro and in vivo models28,61, 62, 63, 64, 65. Among these functionalized cyclodextrins, an amphiphilic cationic β-cyclodextrin (thereafter referred as CD, Fig. 5A) has demonstrated high gene delivery efficacy28,63, 64, 65 and low levels of cytotoxicity and immunotoxicity66. In this study, a FA-targeted PEG-modified CD-based nanoformulation was developed for co-encapsulation of Rg3 and QTN (Fig. 5A). The FA-targeted co-formulation (CD-PEG-FA.Rg3.QTN) demonstrated loading capacity (LC%, ~12% (w/w) for Rg3 and ~6% (w/w) for QTN, MR of Rg3 and QTN ~ 1:1), a low particular size (~110 nm) and a near neutral zeta potential (Fig. 5B), which were similar to those achieved by the non-targeted counterpart (CD-PEG.Rg3.QTN). In addition, the targeted co-formulation displayed a spherical structure (Fig. 5C), which was similar to that observed by non-targeted counterpart.
As shown in Fig. 5D, ~30% of both drugs were released from the targeted co-formulation at 8 h in neutral PBS (pH 7.4), while drug release was remarkably increased (~70%) at 8 h in acidic PBS (pH 5.5). At 48 h, ~70% and ~90% of drug release was observed in the targeted co-formulation at the neutral and acidic PBS, respectively. This data indicates that the CD formulation may achieve a higher drug release at acidic pH environment, which is also similar to other amine-functionalized NPs that can also facilitate pH-sensitive drug release67,68. The mechanism underlying this phenomenon is likely due to the protonation of amine groups in response to the external acidic conditions (see review in Ref. 69). It is worth noting that the release of both Rg3 and QTN from the targeted co-formulation was similar at either pH, suggesting that two drugs may be simultaneously delivered inside the bloodstream and released at the tumor site. Following storage at 4 °C the targeted co-formulation remained stable for up to one week when no significant aggregation of was detected (Fig. 5E). In addition, the non-targeted co-formulation demonstrated similar drug release and stability to that observed for the targeted counterpart.
3.5. In vitro anticancer effects of co-formulations
To confirm the active targeting delivery, cellular uptake of non-targeted and targeted co-formulations containing rhodamine was assessed using confocal microscopy (Olympus) (Fig. 6A) and flow cytometry (Becton Dickinson) (Fig. 6B) in CT26 and HCT116 cells (they both express the folate receptor70,71). Results show that targeted co-formulation achieved significantly higher uptake of rhodamine (P < 0.05 and P < 0.01) than non-targeted counterpart in two cell lines (Fig. 6A and B, and Supporting Information Fig. S4), confirming the FA-mediated delivery effect.
Figure 6.

In vitro studies of co-formulations. (A) Cellular uptake of co-formulations (NT co-formulation = non-targeted co-formulation; co-formulation = targeted co-formulation) containing Rhodamine was assessed at 6 h using confocal microscopy (scale bar = 5 μm). The quantification was shown in Fig. S4. (B) Cellular uptake of co-formulations containing Rhodamine was assessed at 4 h using flow cytometry (BD). Data are presented as mean ± SD (n = 3). ∗P < 0.05 and ∗∗P < 0.01, relative to PBS. (C) Cell viability (%) of co-formulations at 24 h. Data are presented as mean ± SD (n = 3). ∗P < 0.05 and ∗∗P < 0.01, relative to free drugs. (D) Cell-free areas before and after treatment of co-formulations (12 h) were imaged and measured for the relative scratch area (%) (scale bar = 50 μm). Data are presented as mean ± SD (n = 3). ∗P < 0.05 and ∗∗P < 0.01, relative to free drugs. (E) The colony formation following treatment of co-formulations (4 weeks) (scale bar = 50 μm). Data are presented as mean ± SD (n = 3). ∗P < 0.05 and ∗∗P < 0.01, relative to free drugs.
As shown in Fig. 6C, targeted co-formulation significantly reduced cell viability (P < 0.05, ~25% at 24 h incubation) relative to non-targeted counterpart (~40% at 24 h incubation) and combination of free drugs (~55% at 24 h incubation) in CT26 and HCT116 cells. Notably, antiproliferative effect achieved by co-formulations was not due to nanotoxicity, as blank co-formulations could not inhibit cell growth (Supporting Information Fig. S5). Furthermore, antimetastatic activity of targeted co-formulation was assessed using scratch assay (Fig. 6D) and colony formation assay (Fig. 6E). Results show that targeted co-formulation significantly (P < 0.01) slowed down the migration of cancer cells (Fig. 6D, ~35%) and reduced the number of colonies (Fig. 6E, ~5) as compared to non-targeted counterpart (~20% and ~12) and combination of free drugs (~5% and ~20). Results in Fig. 6 indicate that targeted co-formulation achieved FA-mediated antiproliferative and antimetastatic effects in CRC cells.
3.6. In vivo toxicity, pharmacokinetics and biodistribution of co-formulation
The in vivo toxicity of free drugs and co-formulations was assessed in healthy mice (Fig. 7A and Supporting Information Fig. S6). As shown in Fig. S6, no significant body weight loss was found in animals intravenously (i.v.) injected with free Rg3 up to 50 mg/kg or free QTN up to 25 mg/kg relative to PBS. In addition, the body weight was not significantly reduced in animals i.v. injected with targeted co-formulation containing two drugs at different doses (Rg3 up to 50 mg/kg and QTN up to 20 mg/kg, MR ~1:1) as compared to PBS (Fig. 7A). The H&E staining results show that no significant histological changes were found in major organs of mice following i.v. injection of targeted co-formulation (Rg3 = 50 mg/kg and QTN = 20 mg/kg) as compared to PBS (Fig. 7B). Moreover, no significant hematological toxicity (Fig. 7C) and liver/kidney injuries (Fig. 7D) were caused by targeted co-formulation (Rg3 = 50 mg/kg and QTN = 20 mg/kg) as compared to PBS. These indicate that no systemic toxicity was caused by targeted co-formulation under the doses tested.
In general, following systemic administration chemotherapeutic drugs display short half-lives and rapid clearance from the body, which will significantly lessen therapeutic efficacy. It is well established that modification with PEG sterically prevents NPs from non-specific absorption of serum proteins, improving the blood circulation of NPs72. In this study, the half-lives of the free drugs and drugs encapsulated within the targeted co-formulation was evaluated using an orthotopic CT26-Luc derived CRC mouse model (Fig. 7E). Results showed that the concentration of two drugs in the plasma decreased quickly, and the minimum level was detected at 8 h post injection (t1/2–20 min for QTN and 10 min for Rg3; Fig. 7E). By contrast, the two drugs in targeted co-formulation were significantly more slowly removed from the plasma (t1/2–1.4 h for QTN and 1.3 h for Rg3; Fig. 7E), indicating prolonged blood circulation of the drugs. Of note, free drugs demonstrated distinct half-life; in contrast, they presented similar half-life when co-delivered by targeted co-formulation. The data indicate that the two drugs may be simultaneously delivered into the blood. In addition, non-targeted formulation demonstrated similar half-life observed by targeted counterpart.
Tissue distribution of targeted co-formulation was also determined using an orthotopic CRC mouse model. Twelve h following i.v. injection of DiD-labeled co-formulations, tumors and major organs were ex vivo imaged using the IVIS® In Vivo Imaging System (Fig. 7F). Results show that targeted co-formulation achieved significantly higher tumor accumulation (~3.5 folds; P < 0.05) but significantly less liver accumulation (~1.5 folds; P < 0.05) than non-targeted counterpart (Fig. 7F). The results indicate that the targeted co-formulation significantly improved tumor retention and reduced non-specific tissue distribution.
It is known that chemotherapeutic drugs, due to poor pharmacokinetics and non-specific tumor delivery, have to be used in large doses for therapeutic outcome, but such excessive treatment will cause serious side effects. In this study, the targeted co-formulation significantly extended the circulation time and enhanced delivery to the tumor (Fig. 7E and F), suggesting that the targeted co-formulation potentially provides a low-dosage strategy that is sufficient for treating CRC as compared with free unformulated drugs. The chemo-immunotherapeutic efficacy of the targeted co-formulation in combination with anti-PD-L1 was investigated in the following in vivo studies.
3.7. Combination therapy of targeted co-formulation and anti-PD-L1 in an orthotopic CRC model
Chemo-immunotherapeutic efficacy was assessed using an orthotopic CT26-Luc derived CRC mouse model. To investigate the potential of a low-dosage strategy, co-formulations containing Rg3 (10 mg/kg) and QTN (4 mg/kg) (MR ~1:1) were chosen in this study. Results of Supporting Information Fig. S7 show that no significant tumor growth was achieved by the combination of free drugs at higher doses (25 mg/kg Rg3 and 10 mg/kg QTN; toxic signs were observed when doses of Rg3 and QTN of 50 and 20 mg/kg, respectively, were used as compared to PBS. In contrast, tumor growth was significantly (P < 0.05) slowed down by the non-targeted co-formulation relative to the combination of free drugs, while therapeutic efficacy was further (P < 0.05) improved by targeted co-formulation (Fig. S7). These results confirm that the targeted co-formulation achieved FA-mediated delivery effect, and could significantly improve the therapeutic efficacy at lower doses as compared to the free drugs at higher doses. Based on these results, targeted co-formulation containing Rg3 (10 mg/kg) and QTN (4 mg/kg) was chosen for combination therapy with anti-PD-L1.
The response rate of immune checkpoint blockade remains low in patients diagnosed with MMR-proficient CRC7. Indeed, anti-PD-L1 could not generate antitumor efficacy compared to PBS in an orthotopic CRC mouse model (established with CT26 cells, characterized as an MSS CRC cell line73,74 (Fig. 8A and B), which was similar to results previously observed75. In contrast, the targeted co-formulation achieved significantly (P < 0.01) better antitumor efficacy than Anti-PD-L1 alone (Fig. 8A and B). Notably, a combination of the “targeted co-formulation + anti-PD-L1” further (P < 0.05) enhanced therapeutic outcome relative to targeted co-formulation alone (Fig. 8A and B). Consequently, the combined strategy significantly prolonged the survival of diseased mice (median survival ~96 days) as compared to either PBS (~38 days), anti-PD-L1 (~40 days) or targeted co-formulation (~62 days) (Fig. 8C).
Immunofluorescent staining results showed that no difference was detected in apoptotic tumor cells between anti-PD-L1 and PBS, while the targeted co-formulation significantly induced the apoptosis in tumor cells (P < 0.05; ~11%) (Fig. 8D). The combined strategy further (P < 0.05) enhanced the apoptosis in tumor cells (~27%) (Fig. 8D).
In addition, the TME was significantly reprogrammed by the combined strategy as compared to PBS, anti-PD-L1 or the targeted co-formulation alone (Fig. 8E and F). Immunostimulatory cells such as CD8+ T cells, CD4+ T cells and activated DCs were significantly (P < 0.05) upregulated within the tumor by the combined strategy (Fig. 8E), which were accompanied by the increment of IFN-γ, IL-12, CXCL9 and CXCL10 (IFN-γ and IL-12 are responsible for the activation of antitumor immunity76), and CXCL9 and CXCL10 function positively for T cell infiltration at the tumor site77 (Fig. 8F). In addition, the immunosuppressive cells such as regulatory T cells (Tregs), myeloid derived suppressor cells (MDSCs) and tumor-associated macrophages (M2) were significantly downregulated within the tumor by the combined strategy (Fig. 8E), and were accompanied by the reduction of IL-4, IL-6 and IL-10 (they promote the activation of immune suppressive cells78) (Fig. 8F).
It has been reported that the ICD-mediated antitumor immunity relies on the activation of effector T cells10. When orthotopic CRC mice were injected with targeted co-formulation following the removal of CD4+ or CD8+ T cells, the anticancer outcome was significantly abolished (Fig. 8G). In contrast, the therapeutic efficacy of targeted co-formulation was not affected by pretreatment with the isotype IgG antibody (Fig. 8G), confirming the role of targeted co-formulation in the induction of T cell-mediated immunological responses against CRC.
Taken together, results in Fig. 8 indicate that the targeted co-formulation was able to induce ICD (T cell-mediated immunogenic responses) for reprogramming the immunosuppressive TME, which significantly improved the therapeutic efficacy when combined with PD-L1, providing a promising strategy for CRC patients.
4. Discussion
Increasing evidence demonstrates that certain chemotherapeutic drugs can induce ICD, which profoundly modulate the immunosuppressive TME, remodeling “cold” (non-T cell-inflamed) tumors into “hot” (T cell-inflamed) ones10. The combination of ICD-inducing chemotherapeutics with other immunotherapies has demonstrated great promise for improving the survival of cancer patients79. Therefore, investigation of chemotherapeutic drugs (considered cytotoxic and poorly immunogenic) as potential ICD agents has received increasing attention. In this study, the potential of Rg3 [20(S)-Rg3] as an ICD inducer against CRC cells was confirmed for the first time using in vitro and in vivo experimental approaches (Fig. 2) that have been accepted and validated for the identification of ICD agents24. When Rg3 was combined with QTN (a ROS inducer, Fig. 3), the ICD efficacy was significantly improved (Fig. 4).
Recent developments of nano delivery systems have significantly facilitated in vivo delivery of chemotherapeutic agents for cancer therapy35, 36, 37, 38, 39,80, 81, 82, 83, 84. However, to the best of our knowledge, no study has previously reported the co-encapsulation of Rg3 and QTN in a nanoformulation for CRC. In this study, a FA-targeted PEGylated amphiphilic cyclodextrin NP was developed for co-encapsulation of Rg3 and QTN at an optimal molar ratio (1:1) with favorable physicochemical properties (Fig. 5). The targeted co-formulation (CD-PEG-FA.Rg3.QTN) achieved synergistic in vitro anticancer effects (Fig. 6), and improved the pharmacokinetics and biodistribution in an orthotopic CRC mouse model (Fig. 7). Consequently, the CD-PEG-FA.Rg3.QTN altered the immunosuppressive nature of the TME, significantly prolonging the survival of orthotopic CRC mice when combined with anti-PD-L1 (Fig. 8). It is known that only patients with MMR-deficient CRC respond to anti-Anti-PD-L1 as monotherapy6, while the response rate is poor in MMR-proficient CRC7. Therefore, our combined strategy potentially provides therapeutic benefit for a wider spectrum of CRC patients.
It must be borne in mind that the identified features of ICD are only “thin end of the wedge”, therefore, future investigation of ICD hallmarks is of critical importance for progress in the validation of emerging ICD inducers. In addition, the hepatic metastasis is known as the commonest form of distant metastasis in CRC, therefore, therapeutic efficacy of “targeted co-formulation + PD-L1 blockade” strategy will be assessed using mice with experimental CRC liver metastases85, in order to confirm the potential for the treatment of CRC liver metastasis.
5. Conclusions
Rg3 is demonstrate to be an inducer of ICD, and QTN enhances chemo-immunotherapeutic effects in an orthotopic CRC mouse model by increasing ROS. Therefore, identification of emerging ICD inducers has received increasing attention, which will advance ICD-based cancer immunotherapy and synergize with immune checkpoint blockade therapy.
Acknowledgments
Jianfeng Guo acknowledges financial support from the Department of Education of Jilin Province, China (JJKH20190099KJ), the Outstanding Youth Foundation from the Department of Science and Technology of Jilin Province, China (20170520046JH), Health Commission of Jilin Province, China (2020Q012), Fundamental Research Funds for the Central Universities (China), and Talents Cultivation Program of Jilin University. Zhuo Yu acknowledges financial support from National Natural Science Foundation of China (81774240, 82074154) and Siming Scholar from Shanghai Shuguang Hospital (SGXZ-201904, China). Caitriona M O'Driscoll acknowledges financial support from Science Foundation Ireland co-funded under the European Regional Development: Centre for Research in Medical Devices, CURAM (13/RC/2073, Ireland); Synthesis and Solid State Cluster, SSPC (12/RC/2275, Ireland), and Centre for Advanced Materials and BioEngineering Research, AMBER (12/RC/2275, Ireland).
Author contributions
Jianfeng Guo supervised the project. Zhuo Yu and Jianfeng Guo designed the research. Dandan Sun carried out the experiments and performed data analysis. Yifang Zou, Liu Song, Shulan Han, Hao Yang and Di Chu participated part of the experiments. Yun Dai, Jie Ma, Caitriona M O'Driscoll, Zhuo Yu and Jianfeng Guo provided resources. Dandan Sun, Zhuo Yu and Jianfeng Guo wrote the manuscript. Yun Dai, Jie Ma, Caitriona M O'Driscoll and Jianfeng Guo revised the manuscript. All authors have read and approved the final manuscript.
Conflicts of interest
The authors have no conflicts of interest to declare.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2021.06.005.
Contributor Information
Zhuo Yu, Email: zhuoyu@shutcm.edu.cn.
Jianfeng Guo, Email: jguo@jlu.edu.cn.
Appendix ASupplementary data
The following is the Supplementary data to this article:
References
- 1.Rawla P., Sunkara T., Barsouk A. Epidemiology of colorectal cancer: incidence, mortality, survival, and risk factors. Przeglad Gastroenterol. 2019;14:89–103. doi: 10.5114/pg.2018.81072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sahin U., Türeci Ö. Personalized vaccines for cancer immunotherapy. Science. 2018;359:1355–1360. doi: 10.1126/science.aar7112. [DOI] [PubMed] [Google Scholar]
- 3.Weiner L.M., Surana R., Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol. 2010;10:317–327. doi: 10.1038/nri2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu X., Li T., Shen S., Wang J., Abdou P., Gu Z., et al. Advances in engineering cells for cancer immunotherapy. Theranostics. 2019;9:7889–7908. doi: 10.7150/thno.38583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wei S.C., Duffy C.R., Allison J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069–1086. doi: 10.1158/2159-8290.CD-18-0367. [DOI] [PubMed] [Google Scholar]
- 6.Ruiz-Banobre J., Goel A. DNA mismatch repair deficiency and immune checkpoint inhibitors in gastrointestinal cancers. Gastroenterology. 2019;156:890–903. doi: 10.1053/j.gastro.2018.11.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sinicrope F.A., Sargent D.J. Molecular pathways: microsatellite instability in colorectal cancer: prognostic, predictive, and therapeutic implications. Clin Cancer Res. 2012;18:1506–1512. doi: 10.1158/1078-0432.CCR-11-1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herbst R.S., Soria J.C., Kowanetz M., Fine G.D., Hamid O., Gordon M.S., et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patel S.A., Minn A.J. Combination cancer therapy with immune checkpoint blockade: mechanisms and strategies. Immunity. 2018;48:417–433. doi: 10.1016/j.immuni.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kroemer G., Galluzzi L., Kepp O., Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51–72. doi: 10.1146/annurev-immunol-032712-100008. [DOI] [PubMed] [Google Scholar]
- 11.Casares N., Pequignot M.O., Tesniere A., Ghiringhelli F., Roux S., Chaput N., et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. 2005;202:1691–1701. doi: 10.1084/jem.20050915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tesniere A., Schlemmer F., Boige V., Kepp O., Martins I., Ghiringhelli F., et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene. 2010;29:482–491. doi: 10.1038/onc.2009.356. [DOI] [PubMed] [Google Scholar]
- 13.Spisek R., Charalambous A., Mazumder A., Vesole D.H., Jagannath S., Dhodapkar M.V. Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: therapeutic implications. Blood. 2007;109:4839–4845. doi: 10.1182/blood-2006-10-054221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Menger L., Vacchelli E., Adjemian S., Martins I., Ma Y., Shen S., et al. Cardiac glycosides exert anticancer effects by inducing immunogenic cell death. Sci Transl Med. 2012;4:143ra99. doi: 10.1126/scitranslmed.3003807. [DOI] [PubMed] [Google Scholar]
- 15.Duan X., Chan C., Han W., Guo N., Weichselbaum R.R., Lin W. Immunostimulatory nanomedicines synergize with checkpoint blockade immunotherapy to eradicate colorectal tumors. Nat Commun. 2019;10:1–15. doi: 10.1038/s41467-019-09221-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Q., Chen J., Yang Z., Xu J., Xu L., Liang C., et al. Nanoparticle-enhanced radiotherapy to trigger robust cancer immunotherapy. Adv Mater. 2019;31:1802228. doi: 10.1002/adma.201802228. [DOI] [PubMed] [Google Scholar]
- 17.Wang D., Wang T., Yu H., Feng B., Zhou L., Zhou F., et al. Engineering nanoparticles to locally activate T cells in the tumor microenvironment. Sci Immunol. 2019;4 doi: 10.1126/sciimmunol.aau6584. [DOI] [PubMed] [Google Scholar]
- 18.Park H.J., Kim D.H., Park S.J., Kim J.M., Ryu J.H. Ginseng in traditional herbal prescriptions. J Ginseng Res. 2012;36:225–241. doi: 10.5142/jgr.2012.36.3.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei X., Chen J., Su F., Su X., Hu T., Hu S. Stereospecificity of ginsenoside Rg3 in promotion of the immune response to ovalbumin in mice. Int Immunol. 2012;24:465–471. doi: 10.1093/intimm/dxs043. [DOI] [PubMed] [Google Scholar]
- 20.Wu K., Li N., Sun H., Xu T., Jin F., Nie J. Endoplasmic reticulum stress activation mediates ginseng Rg3-induced anti-gallbladder cancer cell activity. Biochem Biophys Res Commun. 2015;466:369–375. doi: 10.1016/j.bbrc.2015.09.030. [DOI] [PubMed] [Google Scholar]
- 21.Xie Q., Wen H., Zhang Q., Zhou W., Lin X., Xie D., et al. Inhibiting PI3K-AKt signaling pathway is involved in antitumor effects of ginsenoside Rg3 in lung cancer cell. Biomed Pharmacother. 2017;85:16–21. doi: 10.1016/j.biopha.2016.11.096. [DOI] [PubMed] [Google Scholar]
- 22.Pan X.Y., Guo H., Han J., Hao F., An Y., Xu Y., et al. Ginsenoside Rg3 attenuates cell migration via inhibition of aquaporin 1 expression in PC-3M prostate cancer cells. Eur J Pharmacol. 2012;683:27–34. doi: 10.1016/j.ejphar.2012.02.040. [DOI] [PubMed] [Google Scholar]
- 23.Mohanan P., Subramaniyam S., Mathiyalagan R., Yang D.C. Molecular signaling of ginsenosides Rb1, Rg1, and Rg3 and their mode of actions. J Ginseng Res. 2018;42:123–132. doi: 10.1016/j.jgr.2017.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Humeau J., Levesque S., Kroemer G., Pol J.G. Gold standard assessment of immunogenic cell death in oncological mouse models. Methods Mol Biol. 2019;1884:297–315. doi: 10.1007/978-1-4939-8885-3_21. [DOI] [PubMed] [Google Scholar]
- 25.Tang S.M., Deng X.T., Zhou J., Li Q.P., Ge X.X., Miao L. Pharmacological basis and new insights of quercetin action in respect to its anti-cancer effects. Biomed Pharmacother. 2020;121:109604. doi: 10.1016/j.biopha.2019.109604. [DOI] [PubMed] [Google Scholar]
- 26.O'Mahony A.M., Godinho B.M., Ogier J., Devocelle M., Darcy R., Cryan J.F., et al. Click-modified cyclodextrins as nonviral vectors for neuronal siRNA delivery. ACS Chem Neurosci. 2012;3:744–752. doi: 10.1021/cn3000372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hossain D.M.S., Javaid S., Cai M., Zhang C., Sawant A., Hinton M., et al. Dinaciclib induces immunogenic cell death and enhances anti-PD1-mediated tumor suppression. J Clin Invest. 2018;128:644–654. doi: 10.1172/JCI94586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo J., Russell E.G., Darcy R., Cotter T.G., McKenna S.L., Cahill M.R., et al. Antibody-targeted cyclodextrin-based nanoparticles for siRNA delivery in the treatment of acute myeloid leukemia: physicochemical characteristics, in vitro mechanistic studies, and ex vivo patient derived therapeutic efficacy. Mol Pharm. 2017;14:940–952. doi: 10.1021/acs.molpharmaceut.6b01150. [DOI] [PubMed] [Google Scholar]
- 29.Evans J.C., Malhotra M., Guo J., O'Shea J.P., Hanrahan K., O'Neill A., et al. Folate-targeted amphiphilic cyclodextrin.siRNA nanoparticles for prostate cancer therapy exhibit PSMA mediated uptake, therapeutic gene silencing in vitro and prolonged circulation in vivo. Nanomedicine. 2016;12:2341–2351. doi: 10.1016/j.nano.2016.06.014. [DOI] [PubMed] [Google Scholar]
- 30.Luan X., Rahme K., Cong Z., Wang L., Zou Y., He Y., et al. Anisamide-targeted PEGylated gold nanoparticles designed to target prostate cancer mediate: enhanced systemic exposure of siRNA, tumour growth suppression and a synergistic therapeutic response in combination with paclitaxel in mice. Eur J Pharm Biopharm. 2019;137:56–67. doi: 10.1016/j.ejpb.2019.02.013. [DOI] [PubMed] [Google Scholar]
- 31.Guo J., O'Driscoll C.M., Holmes J.D., Rahme K. Bioconjugated gold nanoparticles enhance cellular uptake: a proof of concept study for siRNA delivery in prostate cancer cells. Int J Pharm. 2016;509:16–27. doi: 10.1016/j.ijpharm.2016.05.027. [DOI] [PubMed] [Google Scholar]
- 32.Liang C.C., Park A.Y., Guan J.L. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2:329–333. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- 33.Yu Z., Guo J., Hu M., Gao Y., Huang L. Icaritin exacerbates mitophagy and synergizes with doxorubicin to induce immunogenic cell death in hepatocellular carcinoma. ACS Nano. 2020;14:4816–4828. doi: 10.1021/acsnano.0c00708. [DOI] [PubMed] [Google Scholar]
- 34.Guo J., Yu Z., Das M., Huang L. Nano codelivery of oxaliplatin and folinic acid achieves synergistic chemo-immunotherapy with 5-fluorouracil for colorectal cancer and liver metastasis. ACS Nano. 2020;14:5075–5089. doi: 10.1021/acsnano.0c01676. [DOI] [PubMed] [Google Scholar]
- 35.Zhao X., Yang K., Zhao R., Ji T., Wang X., Yang X., et al. Inducing enhanced immunogenic cell death with nanocarrier-based drug delivery systems for pancreatic cancer therapy. Biomaterials. 2016;102:187–197. doi: 10.1016/j.biomaterials.2016.06.032. [DOI] [PubMed] [Google Scholar]
- 36.Feng B., Zhou F., Hou B., Wang D., Wang T., Fu Y., et al. Binary cooperative prodrug nanoparticles improve immunotherapy by synergistically modulating immune tumor microenvironment. Adv Mater. 2018;30:e1803001–e1803010. doi: 10.1002/adma.201803001. [DOI] [PubMed] [Google Scholar]
- 37.Chen Q., Liu L., Lu Y., Chen X., Zhang Y., Zhou W., et al. Tumor microenvironment-triggered aggregated magnetic nanoparticles for reinforced image-guided immunogenic chemotherapy. Adv Sci (Weinh) 2019;6:1802134. doi: 10.1002/advs.201802134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Q., Zhang D., Zhang J., Jiang Y., Song A., Li Z., et al. A three-in-one immunotherapy nanoweapon via cascade-amplifying cancer-immunity cycle against tumor metastasis, relapse, and postsurgical regrowth. Nano Lett. 2019;19:6647–6657. doi: 10.1021/acs.nanolett.9b02923. [DOI] [PubMed] [Google Scholar]
- 39.Huang H., Jiang C., Shen S., Liu A., Gan Y., Tong Q., et al. Nanoenabled reversal of Ido1-mediated immunosuppression synergizes with immunogenic chemotherapy for improved cancer therapy. Nano Lett. 2019;19:5356–5365. doi: 10.1021/acs.nanolett.9b01807. [DOI] [PubMed] [Google Scholar]
- 40.Chou T.C. Drug combination studies and their synergy quantification using the chou-talalay method. Cancer Res. 2010;70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 41.Garg A.D., Krysko D.V., Verfaillie T., Kaczmarek A., Ferreira G.B., Marysael T., et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012;31:1062–1079. doi: 10.1038/emboj.2011.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Michaud M., Martins I., Sukkurwala A.Q., Adjemian S., Ma Y., Pellegatti P., et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334:1573–1577. doi: 10.1126/science.1208347. [DOI] [PubMed] [Google Scholar]
- 43.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 44.Kroemer G., Marino G., Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoshii S.R., Mizushima N. Monitoring and measuring autophagy. Int J Mol Sci. 2017;18:1865. doi: 10.3390/ijms18091865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klionsky D.J., Abdelmohsen K., Abe A., Abedin M.J., Abeliovich H., Acevedo Arozena A., et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy. 2016;12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tanida I., Ueno T., Kominami E. LC3 and autophagy. Methods Mol Biol. 2008;445:77–88. doi: 10.1007/978-1-59745-157-4_4. [DOI] [PubMed] [Google Scholar]
- 48.Zhou J., Wang G., Chen Y., Wang H., Hua Y., Cai Z. Immunogenic cell death in cancer therapy: present and emerging inducers. J Cell Mol Med. 2019;23:4854–4865. doi: 10.1111/jcmm.14356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Panaretakis T., Kepp O., Brockmeier U., Tesniere A., Bjorklund A.C., Chapman D.C., et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009;28:578–590. doi: 10.1038/emboj.2009.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Galluzzi L., Buque A., Kepp O., Zitvogel L., Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17:97–111. doi: 10.1038/nri.2016.107. [DOI] [PubMed] [Google Scholar]
- 51.Garg A.D., Martin S., Golab J., Agostinis P. Danger signalling during cancer cell death: origins, plasticity and regulation. Cell Death Differ. 2014;21:26–38. doi: 10.1038/cdd.2013.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sousa R.E., Caetano Dendritic cells in a mature age. Nat Rev Immunol. 2006;6:476–483. doi: 10.1038/nri1845. [DOI] [PubMed] [Google Scholar]
- 53.Schlecht G., Mouriès J., Poitrasson-Rivière M., Leclerc C., Dadaglio G. Purification of splenic dendritic cells induces maturation and capacity to stimulate Th1 response in vivo. Int Immunol. 2006;18:445–452. doi: 10.1093/intimm/dxh384. [DOI] [PubMed] [Google Scholar]
- 54.Min K.K., Kim J. Properties of immature and mature dendritic cells: phenotype, morphology, phagocytosis, and migration. RSC Adv. 2019;9:11230–11238. doi: 10.1039/c9ra00818g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Circu M.L., Aw T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med. 2010;48:749–762. doi: 10.1016/j.freeradbiomed.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chytil M., Pekar M. Effect of new hydrophobic modification of hyaluronan on its solution properties: evaluation of self-aggregation. Carbohydr Polym. 2009;76:339–443. [Google Scholar]
- 57.Zafarullah M., Li W., Sylvester J., Ahmad M. Molecular mechanisms of N-acetylcysteine actions. Cell Mol Life Sci. 2003;60:6–20. doi: 10.1007/s000180300001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Garg A.D., Krysko D.V., Vandenabeele P., Agostinis P. The emergence of phox-ER stress induced immunogenic apoptosis. OncoImmunology. 2012;1:786–788. doi: 10.4161/onci.19750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Han X., Li Y., Xu Y., Zhao X., Zhang Y., Yang X., et al. Reversal of pancreatic desmoplasia by re-educating stellate cells with a tumour microenvironment-activated nanosystem. Nat Commun. 2018;9:1–18. doi: 10.1038/s41467-018-05906-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou F., Feng B., Yu H., Wang D., Wang T., Ma Y., et al. Tumor microenvironment-activatable prodrug vesicles for nanoenabled cancer chemoimmunotherapy combining immunogenic cell death induction and CD47 blockade. Adv Mater. 2019;31:1805888. doi: 10.1002/adma.201805888. [DOI] [PubMed] [Google Scholar]
- 61.Guo J., Ogier J.R., Desgranges S., Darcy R., O'Driscoll C. Anisamide-targeted cyclodextrin nanoparticles for siRNA delivery to prostate tumours in mice. Biomaterials. 2012;33:7775–7784. doi: 10.1016/j.biomaterials.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 62.McMahon A., O'Neill M.J., Gomez E., Donohue R., Forde D., Darcy R., et al. Targeted gene delivery to hepatocytes with galactosylated amphiphilic cyclodextrins. J Pharm Pharmacol. 2012;64:1063–1073. doi: 10.1111/j.2042-7158.2012.01497.x. [DOI] [PubMed] [Google Scholar]
- 63.McCarthy J., O'neill M., Bourre L., Walsh D., Quinlan A., Hurley G., et al. Gene silencing of TNF-alpha in a murine model of acute colitis using a modified cyclodextrin delivery system. J Control Release. 2013;168:28–34. doi: 10.1016/j.jconrel.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 64.Godinho B.M., Ogier J.R., Darcy R., O'Driscoll C.M., Cryan J.F. Self-assembling modified β-cyclodextrin nanoparticles as neuronal siRNA delivery vectors: focus on Huntington's disease. Mol Pharm. 2013;10:640–649. doi: 10.1021/mp3003946. [DOI] [PubMed] [Google Scholar]
- 65.Fitzgerald K.A., Guo J., Tierney E.G., Curtin C.M., Malhotra M., Darcy R., et al. The use of collagen-based scaffolds to simulate prostate cancer bone metastases with potential for evaluating delivery of nanoparticulate gene therapeutics. Biomaterials. 2015;66:53–66. doi: 10.1016/j.biomaterials.2015.07.019. [DOI] [PubMed] [Google Scholar]
- 66.Godinho B.M., McCarthy D.J., Torres-Fuentes C., Beltrán C.J., McCarthy J., Quinlan A., et al. Differential nanotoxicological and neuroinflammatory liabilities of non-viral vectors for RNA interference in the central nervous system. Biomaterials. 2014;35:489–499. doi: 10.1016/j.biomaterials.2013.09.068. [DOI] [PubMed] [Google Scholar]
- 67.Gao W., Chan J.M., Farokhzad O.C. pH-responsive nanoparticles for drug delivery. Mol Pharm. 2010;7:1913–1920. doi: 10.1021/mp100253e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.He Y., Luo L., Liang S., Long M., Xu H. Amino-functionalized mesoporous silica nanoparticles as efficient carriers for anticancer drug delivery. J Biomater Appl. 2017;32:524–532. doi: 10.1177/0885328217724638. [DOI] [PubMed] [Google Scholar]
- 69.Zhuo S., Zhang F., Yu J., Zhang X., Yang G., Liu X. pH-sensitive biomaterials for drug delivery. Molecules. 2020;25:5649. doi: 10.3390/molecules25235649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fani M., Tamma M.L., Nicolas G.P., Lasri E., Medina C., Raynal I., et al. In vivo imaging of folate receptor positive tumor xenografts using novel 68Ga-NODAGA-folate conjugates. Mol Pharm. 2012;9:1136–1145. doi: 10.1021/mp200418f. [DOI] [PubMed] [Google Scholar]
- 71.Soe Z.C., Poudel B.K., Nguyen H.T., Thapa R.K., Ou W., Gautam M., et al. Folate-targeted nanostructured chitosan/chondroitin sulfate complex carriers for enhanced delivery of bortezomib to colorectal cancer cells. Asian J Pharm Sci. 2019;14:40–51. doi: 10.1016/j.ajps.2018.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Suk J.S., Xu Q., Kim N., Hanes J., Ensign L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Deliv Rev. 2016;99:28–51. doi: 10.1016/j.addr.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Castle J.C., Loewer M., Boegel S., de Graaf J., Bender C., Tadmor A.D., et al. Immunomic, genomic and transcriptomic characterization of CT26 colorectal carcinoma. BMC Genom. 2014;15:1–12. doi: 10.1186/1471-2164-15-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Germano G., Lamba S., Rospo G., Barault L., Magrì A., Maione F., et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature. 2017;552:116–120. doi: 10.1038/nature24673. [DOI] [PubMed] [Google Scholar]
- 75.Song W., Shen L., Wang Y., Liu Q., Goodwin T.J., Li J., et al. Synergistic and low adverse effect cancer immunotherapy by immunogenic chemotherapy and locally expressed PD-L1 trap. Nat Commun. 2018;9:2237. doi: 10.1038/s41467-018-04605-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Showalter A., Limaye A., Oyer J.L., Igarashi R., Kittipatarin C., Copik A.J., et al. Cytokines in immunogenic cell death: applications for cancer immunotherapy. Cytokine. 2017;97:123–132. doi: 10.1016/j.cyto.2017.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bronger H., Singer J., Windmüller C., Reuning U., Zech D., Delbridge C., et al. CXCL9 and CXCL10 predict survival and are regulated by cyclooxygenase inhibition in advanced serous ovarian cancer. Br J Cancer. 2016;115:553–563. doi: 10.1038/bjc.2016.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu Y., Guo J., Huang L. Modulation of tumor microenvironment for immunotherapy: focus on nanomaterial-based strategies. Theranostics. 2020;10:3099. doi: 10.7150/thno.42998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang Y.J., Fletcher R., Yu J., Zhang L. Immunogenic effects of chemotherapy-induced tumor cell death. Genes Dis. 2018;5:194–203. doi: 10.1016/j.gendis.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guo J.F., Huang L. Membrane-core nanoparticles for cancer nanomedicine. Adv Drug Deliv Rev. 2020;156:23–39. doi: 10.1016/j.addr.2020.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sun D., Zhang J., Wang L., Yu Z., O'Driscoll C.M., Guo J. Nanodelivery of immunogenic cell death-inducers for cancer immunotherapy. Drug Discov Today. 2021:651–662. doi: 10.1016/j.drudis.2020.11.029. [DOI] [PubMed] [Google Scholar]
- 82.Gao J., Wang W., Pei Q., Lord M.S., Yu H. Engineering nanomedicines through boosting immunogenic cell death for improved cancer immunotherapy. Acta Pharmacol Sin. 2020;41:986–994. doi: 10.1038/s41401-020-0400-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gao A., Chen B., Gao J., Zhou F., Saeed M., Hou B., et al. Sheddable prodrug vesicles combating adaptive immune resistance for improved photodynamic immunotherapy of cancer. Nano Lett. 2019;20:353–362. doi: 10.1021/acs.nanolett.9b04012. [DOI] [PubMed] [Google Scholar]
- 84.Hu X., Hou B., Xu Z., Saeed M., Sun F., Gao Z., et al. Supramolecular prodrug nanovectors for active tumor targeting and combination immunotherapy of colorectal cancer. Adv Sci. 2020;7:1903332. doi: 10.1002/advs.201903332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Guo J., Yu Z., Sun D., Zou Y., Huang L. Two nanoformulations induce reactive oxygen species and immunogenetic cell death for synergistic chemo-immunotherapy eradicating colorectal cancer and hepatocellular carcinoma. Mol Cancer. 2021;20:10. doi: 10.1186/s12943-020-01297-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.