

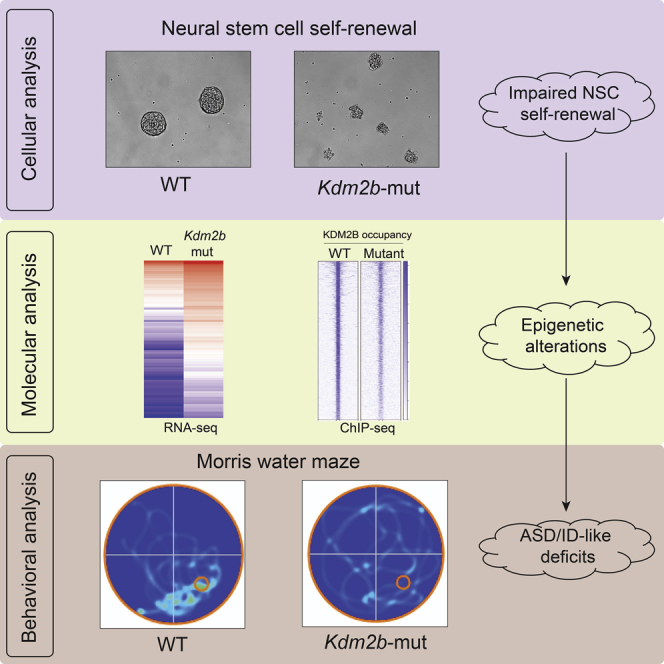
Summary
Recent clinical studies report that chromosomal 12q24.31 microdeletions are associated with autism spectrum disorder (ASD) and intellectual disability (ID). However, the causality and underlying mechanisms linking 12q24.31 microdeletions to ASD/ID remain undetermined. Here we show Kdm2b, one gene located in chromosomal 12q24.31, plays a critical role in maintaining neural stem cells (NSCs) in the mouse brain. Loss of the CxxC-ZF domain of KDM2B impairs its function in recruiting Polycomb repressive complex 1 (PRC1) to chromatin, resulting in de-repression of genes involved in cell apoptosis, cell-cycle arrest, NSC senescence, and loss of NSC populations in the brain. Of importance, the Kdm2b mutation is sufficient to induce ASD/ID-like behavioral and memory deficits. Thus, our study reveals a critical role of KDM2B in normal brain development, a causality between the Kdm2b mutation and ASD/ID-like phenotypes in mice, and potential molecular mechanisms linking the function of KDM2B-PRC1 in transcriptional regulation to the 12q24.31 microdeletion-associated ASD/ID.
Subject areas: Biological sciences, Molecular biology, Neuroscience
Graphical abstract
Highlights
-
•
Kdm2b mutation impairs neural stem cell self-renewal
-
•
Kdm2b mutation causes autistic-like behaviors and memory deficits
-
•
Kdm2b mutation derepresses genes related to impaired NSC self-renewal
-
•
Kdm2b mutation impairs PRC1 recruitment to chromatin
Biological sciences; Molecular biology; Neuroscience
Introduction
Autism spectrum disorder (ASD) is one of the most prevalent neurodevelopmental diseases (NDDs) associated with various genetic mutations (Lord et al., 2020). Recent genetic studies have revealed that human ASD risk genes are enriched with genes encoding chromatin modifying factors and transcription regulators (Grove et al., 2019; Lalli et al., 2020; Ruzzo et al., 2019; Satterstrom et al., 2020), highlighting the importance of epigenetic factors and their function in regulating gene expression in ASD pathogenesis. However, the molecular and cellular mechanisms underlying the mutations of specific epigenetic factors and NDD genesis are largely unknown.
Recent genetic and clinical studies report that chromosomal 12q24.31 microdeletions are associated with NDDs in human patients (Baple et al., 2010; Chouery et al., 2013; Labonne et al., 2016; Palumbo et al., 2015; Qiao et al., 2013). Clinically, the patients display intellectual disability (ID), autistic behaviors, epilepsy, craniofacial deformity, and developmental delay, suggesting the genes located in 12q24.31 potentially have important functions for normal brain development. Further genetic mapping analysis identified a 445-kb common region in the chromosomal 12q24.31 microdeletion variants, in which one of the genes, KDM2B, was found to have reduced expression in patients compared with their healthy siblings, suggesting KDM2B was a candidate ASD pathogenic gene and its loss-of-function mutations potentially contributed to the chromosomal 12q24.31 microdeletion-associated ASD/ID (Labonne et al., 2016).
KDM2B is a histone H3 lysine 36 di-methylation (H3K36me2)-specific demethylase (Tsukada et al., 2006). In addition to having enzymatic activity, KDM2B interacts with Polycomb repressive complex 1 (PRC1) and recruits the KDM2B-PRC1 complex to chromatin through its CxxC zinc finger (CxxC-ZF) domain-mediated DNA binding to non-methylated CpG islands (CGIs) and CGI-associated gene promoters in mammalian cells (Farcas et al., 2012; He et al., 2013; Wu et al., 2013). The recruitment of PRC1 to chromatin is essential for PRC1 to ubiquitylate histone H2A at gene promoters and repress gene expression in mammalian cells (Turberfield et al., 2019; Wang et al., 2004). Although the mechanisms underlying the KDM2B-PRC1 complex-mediated transcriptional regulation have been well studied, the molecular and cellular mechanisms linking the KDM2B loss in 12q24.31 microdeletions to ASD/ID pathogenesis remain unknown.
To investigate the function of KDM2B in normal brain development and ASD/ID genesis, we generated a Kdm2b conditional mutant mouse line in which a mutant KDM2B with loss of its CxxC-ZF domain was introduced in the developing mouse brain. The genetic analysis demonstrated that the heterozygous Kdm2b mutation in the developing mouse brain impaired self-renewal of neural stem cells (NSCs), induced NSC senescence, and reduced NSC populations in vitro and in vivo, which was accompanied by increased cell apoptosis, cell-cycle arrest, and precocious NSC differentiation. Behavioral studies showed that the heterozygous adult Kdm2b mutant mice displayed both ID-like memory deficits and core autistic-like behaviors, suggesting that the KDM2B mutation-induced NSC loss in developing brains was likely to be a causative factor leading to the pathogenesis of 12q24.31 microdeletion-associated ASD/ID. At the molecular level, the Kdm2b mutation in NSCs resulted in decreased occupancy of KDM2B-PRC1 at gene promoters and de-repression of multiple genes involved in cell apoptosis, cell proliferation blockade, and promoting NSC differentiation. Thus, our study reveals a critical role of KDM2B in maintaining NSCs in the developing mouse brain and provides the molecular and cellular mechanisms linking the KDM2B-PRC1 function in transcriptional regulation to NSC self-renewal and 12q24.31 microdeletion-associated ASD/ID.
Results
Loss of KDM2B CxxC-ZF domain impairs NSC self-renewal
Previous studies have shown that the CxxC-ZF domain of KDM2B is required for recruiting PRC1 to chromatin through its binding to non-methylated CpG-rich DNA sequences (Farcas et al., 2012; He et al., 2013; Wu et al., 2013). To examine the function of KDM2B-PRC1 in vivo, we generated a Kdm2b conditional mutant (Kdm2b-cMut) mouse line by inserting two LoxP elements into the exon 13-flanking sites at the Kdm2b gene locus (Kdm2b-cMut: Kdm2b+/2f). A CRE recombinase-mediated deletion of exon 13 resulted in altered splicing of mRNA encoding a mutant KDM2B protein with deletion of amino acids 552–626 encompassing the entire CxxC-ZF domain (Figures S1A and S1B). The mutant KDM2B protein contained all functional domains except its CxxC-ZF domain and thus lost its function in recruiting PRC1 to chromatin. To preclude mouse strain-specific effects on animal phenotypes, we backcrossed the wild-type Kdm2b+/2f founders with C57BL/6 mice for more than five generations to reach a pure genetic background.
To examine the function of Kdm2b in central nervous system development, we crossed the Kdm2b-cMut mice with a neural progenitor cell (NPC)-specific Cre (Nestin-Cre) mouse line to generate neural Kdm2b conditional mutant mice (Kdm2b-Nes-cMut: Kdm2b2f/+;Nestin-Cre+/-). The following mating (Kdm2b2f/2f x Kdm2b2f/+;Nestin-Cre+/-) only produced wild-type (Kdm2b-Nes-WT: Kdm2b2f/2f;Nestin-Cre−/− or Kdm2b2f/+;Nestin-Cre−/−) and heterozygous (Kdm2b-Nes-Het: Kdm2b2f/+;Nestin-Cre+/-) mutant pups (Figure S1C), suggesting the homozygous Kdm2b (Kdm2b2f/2f;Nestin-Cre+/-) mutation in the developing mouse brain was embryonic lethal. The Kdm2b-Nes-Het mice did not display any gross abnormalities and had similar postnatal growth as their wild-type littermates.
To investigate the function of KDM2B and its CxxC-ZF domain-mediated PRC1 recruitment in neural stem cells (NSCs) in the developing mouse brain, we isolated NSCs from the subventricular zone (SVZ) at embryonic day 16 (E16) and examined their self-renewal by serial neurosphere formation assays. Compared with the wild-type NSCs, the Kdm2b-Nes-Het mutant NSCs formed primary neurospheres with smaller sizes in the first round of plating. After splitting into single cells, the wild-type NSCs formed secondary spheres with sizes comparable with those of the primary spheres, indicating the wild-type NSCs could maintain their self-renewing capacity under the experimental condition. In contrast, the Kdm2b-mutant cells failed to form secondary spheres in the second round of plating (Figures 1A and 1B), suggesting that the mutant NSCs were unable to maintain the stem cell property of self-renewal. Further senescence-associated beta-galactosidase (β-gal) staining assays showed that, compared with the wild-type NSCs, the Kdm2b-Nes-Het mutant NSCs displayed a high percentage of β-gal+ cells at early passages, indicating that the Kdm2b mutation induced premature NSC senescence (Figures 1C and 1D). Taken together, these results suggested that heterozygous Kdm2b mutation with loss of its CxxC-ZF domain impaired NSC self-renewal and induced premature cellular senescence in vitro.
Figure 1.

Kdm2b mutation impairs NSC self-renewal
(A) Representative photos showing the size of primary and secondary neurospheres formed by wild-type and Kdm2b mutant NSCs in suspension culture; scale bar, 100 μm.
(B) The dot plot showing the diameters of primary and secondary neurospheres formed by wild-type and Kdm2b mutant NSCs in suspension culture. p Values were calculated using a two-way ANOVA; error bars represent mean ± SEM; ∗∗∗p <0.001.
(C) Representative photos showing the senescence-associated galactosidase-positive wild-type and Kdm2b mutant NSCs in the monolayer culture; scale bar, 50 μm.
(D) The bar plot showing the percentage of senescence-associated galactosidase-positive cells in each field. p Values were calculated using a two-tailed t test; error bars represent mean ± SEM; ∗∗∗p <0.001.
Kdm2b mutation induces cellular changes leading to impaired NSC self-renewal
Next, we examined the cellular changes that potentially could cause the NSC self-renewing defect observed in the Kdm2b-Nes-Het mutant cells, including cell proliferation, apoptosis, and precocious NSC differentiation. Bromodeoxyuridine (BrdU) incorporation assays showed that Kdm2b-mutant NSCs had significantly lower BrdU+ populations compared with wild-type cells, indicating that the Kdm2b mutation induced cell-cycle arrest and blocked NSC proliferation (Figures 2A and 2B). Moreover, terminal deoxynucleotidyl transferase dUTP nick end label (TUNEL) assays showed that the Kdm2b-mutant NSCs exhibited a higher rate of apoptosis compared with the wild-type NSCs (Figures 2C and 2D), suggesting that loss of the CxxC-ZF domain of KDM2B induced cell apoptosis. To examine whether the Kdm2b mutation caused abnormal NSC differentiation, we stained the primary NSCs maintained in the NSC culture medium for the early differentiation marker GFAP. The results showed that the Kdm2b-Nes-Het mutant cells had a higher percentage of GFAP+ cells compared with the wild-type cells (Figures 2E and 2F), suggesting the Kdm2b mutation induced precocious NSC differentiation in vitro.
Figure 2.

Kdm2b mutation induces cellular changes leading to impaired NSC self-renewal
(A and B) Representative photos (A) and bar plots (B) showing the percentage of BrdU+ cells in the Kdm2b-Nes-WT and Kdm2b-Nes-Het NSCs cultured in vitro; scale bar, 20 μm.
(C and D) Representative photos (C) and bar plots (D) showing the percentage of TUNEL+ apoptotic cells in the Kdm2b-Nes-WT and Kdm2b-Nes-Het NSCs cultured in vitro; scale bar, 20 μm.
(E and F) Representative photos (E) and bar plots (F) showing the GFAP+ cells/field in the Kdm2b-Nes-WT and Kdm2b-Nes-Het NSCs cultured in vitro; scale bar, 20 μm.
(G–I) Representative photos (G) and bar plots (H, I) showing the BrdU+ cells in the SVZ and DG of Kdm2b-Nes-WT and Kdm2b-Nes-Het mouse brains; scale bar, 100 μm.
(J–L) Representative photos (J) and bar plots (K, L) showing the SOX2+ cells in the SVZ and DG of Kdm2b-Nes-WT and Kdm2b-Nes-Het mouse brains; scale bar, 100 μm. For B, D, F, H, I, K, and L, four to six biological replicates per genotype were included in each experiment. Arrows (J): SOX2+ cells. p Values were calculated using a two-tailed t test; error bars represent mean ± SEM; ∗∗p <0.01, ∗∗∗∗p <0.0001.
To examine whether the Kdm2b mutation compromised NSC populations in vivo, we injected BrdU into adult mice and examined the BrdU-incorporated proliferating cells in the brain. The results showed that, compared with the wild-type littermates, the Kdm2b-Nes-Het mice had significantly fewer BrdU-incorporated cells in the SVZ and dentate gyrus (DG) of the hippocampus (Figures 2G–2I), which was consistent with lower numbers of SOX2+ NSCs in both regions (Figures 2J–2L). Grossly, although the mutant brain had normal morphology and similar weight to that of the wild type, the hippocampus isolated from the mutant mice had ∼20% less weight than the wild-type littermates (Figures S2A–S2C). Taken together, these results suggested that the Kdm2b mutation in the developing mouse brain induced cell apoptosis, repressed proliferation, and promoted precocious NSC differentiation, which subsequently led to the impaired NSC self-renewal and loss of NSC populations in the developing brain.
Kdm2b mutation in the developing brain causes ASD/ID-like behavioral and memory deficits
Since the Kdm2b mutant mice had reduced NSC populations in the DG and smaller hippocampi, we further asked whether the Kdm2b mutation in the developing mouse brain could lead to hippocampus-related memory deficits in adult mice. To this end, we performed the novel object recognition (NOR) test to examine object recognition memory. Twenty-four hours after initial familiarization with two identical objects in an arena, the mice were allowed to explore the same arena in the presence of a familiar object and a novel object. The results showed that, compared with wild-type mice, both male and female Kdm2b-Nes-Het mice had reduced discrimination indexes (t = 2.252, df = 28, p = 0.032) (Figures 3A, S3A and S3B), indicating a deficit in long-term object recognition memory. The impaired memory was unlikely to be caused by hyperactivity or increased anxiety-like behaviors since the open field tests showed the mutant mice had comparable locomotor activity and exploring time in the center of the arena as their wild-type littermates (Figures S3C and S3D). To further confirm the memory deficit observed in the mutant mice, we used the Morris water maze to compare spatial memory between wild-type and Kdm2b-Nes-Het mice. Although the mutant mice had a similar swimming speed as the wild-type controls (Figure S3E), both male and female mutant mice showed significantly delayed spatial learning during the first 5-day acquisition training phase (F1,188 = 8.067, p = 0.005) (Figures 3B, S3F and S3G), and they spent less time in the platform quadrant during the post-training probe tests (t = 2.732, df = 38, p = 0.0098) (Figures 3C, 3D, S3H and S3I), suggesting that the Kdm2b-Nes-Het mice had impaired spatial learning and memory.
Figure 3.

Kdm2b mutation in the developing brain causes ASD/ID-like behavioral and memory deficits
(A) Quantitative discrimination ratio of NOR tests. The discrimination ratio was calculated as (time spent on the novel object-time spent on the familiar object)/total time. n = 15 for each genotype; p values were calculated using a two-tailed t test; error bars represent mean ± SEM; ∗p <0.05.
(B) The plot showing the escape latency during the 5-day training phase. n = 20 for each genotype; p values were calculated using two-way ANOVA; error bars represent mean ± SEM
(C) The time in the target zones during the post-training probe tests. n = 20 for each genotype; p values were calculated using a two-tailed t test; error bars represent mean ± SEM; ∗∗p <0.01.
(D) Representative heatmap showing the time in the target zone during the post-training probe test. The red circle represents the location of platform.
(E) Quantitative results showing the time wild-type and Kdm2b-Nes-Het mice spent in the chamber containing a social partner in the three-chamber sociability tests. n = 15 for each genotype; p values were calculated using two-way ANOVA; error bars represent mean ± SEM; ∗∗∗p <0.001, ns, not significant.
(F) Time spent in the chamber containing a novel animal in the three-chamber social novelty tests. n = 15 for each genotype; p values were calculated using two-way ANOVA; error bars represent mean ± SEM; ∗∗∗p <0.001, ns, not significant.
To examine whether the Kdm2b mutant mice developed autistic-like behaviors, we performed a three-chamber test to examine voluntary exploration of a social versus a non-social stimulus (sociability) and the voluntary exploration of a familiar versus a novel social stimulus (social novelty/social memory) (Kaidanovich-Beilin et al., 2011; Moy et al., 2004). In the sociability portion of the test, the results showed main effects of stimulus type (F1,56 = 13.48, p = 0.0005). Planned comparisons revealed that the wild-type controls spent more time with the social stimulus than with the object (t = 3.713, df = 56, p = 0.0009), representing normal sociability. In contrast, both male and female Kdm2b-Nes-Het mice showed a reduced preference for the social stimulus, suggesting impaired sociability (Figures 3E, S3J and S3K). In the social novelty portion of the test, there was an effect of genotype by stimulus interaction (F1,68 = 7.929, p = 0.006). Planned comparisons showed that the wild-type mice preferred a novel over a familiar animal (p = 0.0001) (Figure 3F), indicating preference for social novelty. In contrast, both male and female Kdm2b-Nes-Het mice showed no preference (Figures 3F, S3L and S3M), indicating loss of social interest or reduced social memory.
Thus, the collective behavioral tests revealed that the Kdm2b-Nes-Het mice with loss of KDM2B CxxC-ZF domain in the developing mouse brain displayed ASD/ID-like behavioral deficits, including impaired cognitive and spatial memory, reduced sociability, and impaired social memory.
Kdm2b mutation derepresses genes related to impaired NSC self-renewal
Next, we set out to investigate the molecular mechanisms underlying the phenotypic changes in the Kdm2b mutant NSCs by examining transcriptome changes. Consistent with the function of KDM2B-PRC1 in transcriptional repression, RNA-seq analysis revealed that the Kdm2b-Nes-Het mutant NSCs had more upregulated (n = 302) than downregulated (n = 191) genes (cutoff: RPKM ≥3, fold changes ≥2, FDR <0.05) (Figures S4A). The gene ontology (GO) enrichment analysis showed that the upregulated genes could be divided into three main functional groups (cutoff: FDR <0.05): (1) inducing cell apoptosis, (2) repressing cell proliferation, and (3) inducing neural development (Figures 4A and Table S1), which was consistent with the increased apoptosis, reduced cell proliferation, and precocious NSC differentiation of Kdm2b-Nes-Het mutant NSCs (Figures 2A–2F). This suggested that the genes aberrantly upregulated in the Kdm2b-Nes-Het mutant NSCs were likely to play a major role in inducing the phenotypic changes observed in the Kdm2b mutant cells. In contrast, the GO enrichment analysis showed that the downregulated genes were involved in initiation of DNA replication (Figure 4B and Table S2), suggesting that the decreased NSC proliferation could also be caused by reduced genes involved in mitotic division.
Figure 4.

Kdm2b mutation derepresses genes related to impaired NSC self-renewal
(A) The upregulated genes in the Kdm2b-Nes-Het NSCs and their related gene functions. The gene expression was measured by log2FPKM (fragments per kilobase of transcript per million mapped reads).
(B) The downregulated genes in the Kdm2b-Nes-Het NSCs and their related gene functions. The gene expression was measured by log2FPKM.
(C) KEGG pathway enrichment analysis showing the functional pathways related to the up- and downregulated genes in the Kdm2b-Nes-Het NSCs.
(D) Heatmap showing the expression of Cdkn1a, Cdkn2a, and Cdkn2b in the Kdm2b-Nes-WT and Kdm2b-Nes-Het NSCs. The gene expression was measured by log2 FPKM.
Further Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis showed the upregulated genes in the Kdm2b-Nes-Het mutant were involved in the activation of p53 and FOXO signaling pathways that induce both cell apoptosis and cell-cycle arrest (Burgering and Kops, 2002; Harris and Levine, 2005; Zhang et al., 2011), whereas the downregulated genes were involved in regulating cell cycle (Figure 4C, Tables S3 and S4). Of note, Cdkn2a and Cdkn2b in the Ink4a-Arf locus and Cdkn1a, which play a central role in inducing both RB-mediated cell-cycle arrest and p53-mediated apoptosis (Sharpless, 2005; Sherr, 2001), were highly upregulated in the Kdm2b-Nes-Het mutant NSCs (Figures 4D and S4B), consistent with previous findings that Polycomb-mediated epigenetic modification was critical for repressing the Ink4a-Arf locus to maintain NSCs in the developing mouse brain (Bracken et al., 2007; Bruggeman et al., 2005; He et al., 2008; Molofsky et al., 2005).
Thus, the collective results suggested that, at the molecular level, the KDM2B mutation with loss of its CxxC-ZF domain derepressed the genes involved in apoptosis, cell proliferation blockade, and precocious NSC differentiation, which led to impaired NSC self-renewal, NSC senescence, and loss of NSC populations in the developing brain.
Kdm2b mutation impairs the PRC1 recruitment to chromatin
To examine whether the derepressed gene expression in the Kdm2b-Nes-Het mutant NSCs was caused by impaired KDM2B-mediated PRC1 recruitment to chromatin, we performed chromatin immunoprecipitation coupled with next-generation sequencing (ChIP-seq) assays to examine the occupancy of KDM2B and PRC1 core component RING1B in wild-type and Kdm2b-Nes-Het mutant NSCs. The results showed that genome-wide KDM2B occupancy at the 7125 KDM2B-binding sites was reduced to ∼50% in the Kdm2b-Nes-Het mutant NPCs compared with that in the wild-type cells (Figure 5A), confirming that loss of the KDM2B CxxC-ZF domain impaired its chromatin binding. To examine whether the mutant KDM2B reduced the occupancy of the PRC1 core component RING1B at gene promoters, we compared the total RING1B occupancy at the promoters of differentially expressed gene in wild-type and Kdm2b-Nes-Het mutant NSCs (Figures S4A). The results showed that RING1B occupancy was reduced for ∼25% at the promoters of upregulated genes in the Kdm2b-Nes-Het mutant NSCs, whereas its occupancy at the downregulated gene promoters had a slight increase (Figures 5B and S5A). The ChIP-seq results were further confirmed by ChIP-qPCR analysis at the representative Cdkn2a/2b, Bmp7, and Wnt7b gene loci (Figures S5B–S5D). Specifically, both KDM2B and RING1B occupancy was reduced at the Ink4a-Arf locus (Figures 5C and S5B), which was consistent with the upregulated expression of Cdkn2a and Cdkn2b in the Kdm2b-Nes-Het mutant NSCs (Figures 4d and S4B). Taken together, these results suggested that aberrant gene expression in the Kdm2b-Nes-Het mutant NSCs was a direct outcome of reduced KDM2B-mediated PRC1 recruitment and impaired PRC1-mediated transcriptional repression.
Figure 5.

Kdm2b mutation impairs PRC1 recruitment to chromatin
(A) Heatmap showing KDM2B occupancy at KDM2B binding sites and their 10-kb flanking sites in the Kdm2b-Nes-WT and Kdm2b-Nes-Het NSCs.
(B) Heatmaps and plots showing the 302 upregulated genes in the Kdm2b-Nes-Het NSCs (left panel) and RING1B occupancy at the gene code regions and their 10-kb flanking regions (right panel). TSS, transcriptional starting sites; TES, transcriptional ending sites.
(C) IGV genome browser view of KDM2B occupancy, RING1B occupancy, and expression levels of Cdkn2a/2b, Bmp7, and Wnt7b genes.
Discussion
In this study, we used a Kdm2b conditional mutant mouse model to investigate two questions related to chromosomal 12q24.31 microdeletion-associated ASD/ID: (1) Is the loss-of-function mutation of KDM2B a causative driver leading to ASD/ID-like behavioral and memory deficits? (2) What are the molecular mechanisms linking Kdm2b mutations to ASD/ID pathogenesis?
At the cellular level, the Kdm2b mutant NSCs displayed impaired self-renewal and underwent premature cellular senescence (Figure 1), which was accompanied with increased cell apoptosis, reduced cell proliferation, and premature NSC differentiation. Correspondingly, the mutant adult mice had markedly reduced SOX2+ and BrdU+ proliferating NSCs in the SVZ and DG (Figure 2), suggesting that the CxxC-ZF domain of KDM2B was required for maintaining the neural stem cell populations in the developing mouse brain through preventing apoptosis, cell-cycle arrest, and premature NSC differentiation.
Previous studies have shown that the CxxC-ZF domain of KDM2B mediates the recruitment of PRC1 to chromatin through its binding to unmethylated CpG-rich DNA sequences (Farcas et al., 2012; He et al., 2013; Wu et al., 2013). After binding to chromatin, PRC1 catalyzes the mono-ubiquitination of histone H2A at gene promoters and mediates transcriptional silencing (Blackledge et al., 2020; Wang et al., 2004). Consistent with these findings, transcriptome analyses showed that loss of the CxxC-ZF domain of KDM2B in NSCs derepressed genes involved in cell apoptosis, cell proliferation blockade, and NSC premature differentiation. Specifically, Cdkn2a and Cdkn2b in the Ink4a-Arf locus, which are located at the apex of signaling pathways to activate RB and p53-mediated cell-cycle arrest and apoptosis (Sharpless, 2005; Sherr, 2001), were highly upregulated in the Kdm2b mutant NSCs (Figure 4). Furthermore, ChIP-seq analysis showed that the occupancy of KDM2B and RIGN1B was reduced at the promoters of activated genes (Figure 5), suggesting that de-repression of genes in the Kdm2b mutant NSCs was a direct outcome of reduced KDM2B-mediated PRC1 recruitment to chromatin. Taken together, these results suggest that the KDM2B mutation with loss of its CxxC-ZF domain impaired the KDM2B-mediated PRC1 recruitment to chromatin, compromised the function of PRC1 in transcriptional silencing, and led to de-repression of genes. The overall consequences of these molecular changes were the defective NSC self-renewal and loss of NSC populations in the Kdm2b mutant brain.
Further behavioral studies showed that the KDM2B mutation with loss of its CxxC-ZF domain in the developing mouse brain was sufficient to cause ID-like cognitive and spatial memory deficits as well as autistic-like deficits in social behaviors (Figure 3), suggesting that loss-of-function mutation of KDM2B in the developing mouse brain was likely to be a causative factor leading to 12q24.31 microdeletion-associated ASD/ID pathogenesis. Although currently the roles of NSCs in ASD/ID pathogenesis remain undetermined, earlier studies have shown that loss of NSCs in young postnatal mice causes progressive memory impairment (Zhu et al., 2010), which is consistent with our findings that the Kdm2b mutation-induced loss of NSC populations in the developing brain results in ID-like cognitive and spatial memory deficits. Based on these results, we propose that KDM2B is critical for the maintenance of NSC populations in the developing mouse brain by recruiting PRC1 to chromatin to silence the genes involved in cell apoptosis, cell cycle blockade, and precocious NSC differentiation. The mutant KDM2B with loss of its CxxC-ZF domain causes aberrant gene expression, impaired NSC self-renewal, and loss of NSC populations in the developing brain, which subsequently causes the ASD/ID-like behavioral and memory deficits in adult mice (Figure 6).
Figure 6.

Proposed model for the impaired function of KDM2B-PRC1 in ASD/ID pathogenesis
KDM2B is critical for the maintenance of NSC populations in the developing mouse brain by recruiting PRC1 to chromatin to silence the genes involved in cell apoptosis, cell-cycle blockade, and precocious NSC differentiation. The KDM2B mutation with loss of its CxxC-ZF domain derepresses genes involved in apoptosis, cell-cycle arrest, and premature NSC differentiation, which leads to NSC self-renewing defects and ASD/ID pathogenesis.
Our current study is exciting but has some limitations: (1) unlike the loss of the entire protein in the chromosomal 12q24.31 microdeletions, the heterozygous Kdm2b mutant mice used in this study generate both wild-type and mutant KDM2B proteins in the brain, in which the mutant protein- might have a dominant-negative effect to exacerbate the phenotypes; (ii) although the Kdm2b mutation-induced NSC self-renewing defects and loss of NSC populations in the brain are likely to play a major role in causing the ASD/ID-like abnormalities in this mouse model, our current study does not exclude other mechanisms, such as aberrant neural lineage differentiation caused by de-repression of lineage-specific transcription factors, that might also be involved in the genesis of ASD/ID-like phenotypes.
Finally, our mouse model has shown that the impaired KDM2B-mediated recruitment of PRC1 to chromatin is sufficient to impair NSC self-renewal and deplete NSC populations in the brain, which provides a useful genetic tool to study the functions of NSCs in adult neurogenesis, brain aging, and brain degenerative diseases by introducing the KDM2B mutation to deplete adult NSC populations in the brain.
Limitations of the study
Although this study indicates that the Kdm2b mutation-induced defective NSC self-renewal and loss of NSC populations in the brain are likely to play a major role in causing the ASD/ID-like abnormalities in this mouse model, our current study does not exclude other mechanisms, such as aberrant neural lineage differentiation or functional abnormalities of differentiated neurons, that might also be involved in the genesis of ASD/ID-like phenotypes. Induced Kdm2b mutation in different neural lineages will help to elucidate the functions of Kdm2b in individual neural lineages and their roles in ASD/ID pathogenesis.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti Sox2 | Cell signaling technology | Cat#:3728, lot 4; RRID: AB_2194037 |
| Rabbit monoclonal anti-GFAP | Cell signaling technology | Cat#: 12389, lot 5; RRID: AB_2631098 |
| Rat monoclonal anti-BrdU | Bio-rad | MCA2060; RRID: AB_323427 |
| Rabbit polyclonal anti-KDM2B | Dr. Robert Klose (Oxford University) | N/A |
| Rabbit monoclonal anti-RING1B | Cell signaling technology | Cat#: 5694, lot 1; RRID: AB_10705604 |
| Rabbit polyclonal anti-TUBULIN alpha | Proteintech | Cat#: 11224-1-AP; RRID: AB_2210206 |
| Critical commercial assays | ||
| Senescence β-Galactosidase Staining Kit | Cell signaling technology | 9860 |
| In Situ Cell Death Detection Kit | Sigma-aldrich | 11684795910 |
| Deposited data | ||
| Raw and processed data | This paper | GEO: GSE190807 |
| Experimental models: Organisms/strains | ||
| Kdm2b conditional mutant mouse line | This paper | N/A |
| B6.Cg-Tg (Nes-cre) 1Kln/J | The Jackson laboratory | JAX: 003771 |
| C57BL/6J | The Jackson laboratory | JAX: 000664 |
| Oligonucleotides | ||
| Primers for qRT-PCR, see Table S5 | This paper | N/A |
| Primers for ChIP-qPCR, see Table S5 | This paper | N/A |
| Primers for genotyping, see Table S5 | This paper | N/A |
| Software and algorithms | ||
| Bowtie2 | Langmead and Salzberg (2012) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| MACS | Feng et al. (2012) | https://github.com/macs3-project/MACS |
| Tophat2 | Kim et al. (2013) | https://ccb.jhu.edu/software/tophat/index.shtml |
| Cufflink | Trapnell et al. (2013) | http://cole-trapnell-lab.github.io/cufflinks/ |
| deepTools | Ramirez et al. (2016) | https://deeptools.readthedocs.io/en/develop/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jin He (hejin1@msu.edu).
Materials availability
The Kdm2b mutant mouse line is available upon request. This study did not generate other unique reagents.
Experimental model and subject details
Mice
The Kdm2b conditional mutant mice were generated in Dr. Yi Zhang lab. The Kdm2b conditional mutant target construct was generated by modifying the BAC clone (RP23-214I6) based on the Recombneering method (Thomason et al., 2014). Two LoxP elements were inserted into the exon 13-flanking sites. The targeting construct was electroporated into the C57BL/6:129 hybrid murine ES cells. Homologous recombinant ES cell clones were identified by PCR-based genotyping and injected into blastocysts. The genetically modified ES cells were micro-injected to blastocysts and transferred to the uterus of CD-1 pseudo-pregnant females to generate chimeric founder mice. The chimeric founder mice were crossed to an FLP recombinase mouse line to remove the FRT-flanked selection cassette. All mice were backcrossed to C57BL/6 mice for at least five generations to reach a pure C57BL/6 background before further mating to specific Cre lineages. Mice were housed under standard conditions (12h light: 12h dark cycles) with food and water ad libitum. Neural stem cells were isolated from E16 mouse brains. 12-week-old mice were used for behavioral studies. The data obtained from all embryos were pooled for analysis without discrimination of sexes. All behavioral tests were performed on both male and female littermate wild-type and heterozygous Kdm2b mutant mice. The mice were labeled by ear-tags. All the behavioral tests were carried out by researchers blinded to the genotypes of animals during the behavioral tests. All mouse experiments were performed with the approval of the Michigan State University Institutional Animal Care & Use Committee.
Mouse breeding strategy
All mice were backcrossed to C57BL/6 mice for at least five generations to reach a pure C57BL/6 background. (1) Generating Kdm2b-Nestin-cMut mice: The Kdm2b neural conditional mutant mice were generated by mating Kdm2b floxed mice with Nestin-cre mice (B6.Cg-Tg [Nes-cre] 1Kln/J, The Jackson Laboratory). The wild-type (Kdm2b2f/2f;Nestin-Cre−/−) and heterozygous (Kdm2b2f/+;Nestin-Cre+/-) mice were generated by Kdm2b2f/2f;Nestin-Cre−/− (female) x Kdm2b2f/+;Nestin-Cre+/- (male) mating.
Method details
Isolation, culture, and neurosphere formation of neural stem cells
The neural tissues were isolated from the subventricular zone of E16 brains and dissociated with 0.05% trypsin-EDTA (Life Technologies) at 37°C for 15 min. The reaction was stopped by trypsin inhibitor (10 mg/mL, Worthington Biochemical Corporation). The dissociated cells were washed with cold PBS for 3 times and plated onto the non-coated Petri dishes in the proliferation medium (Neurobasal medium [Life Technologies] supplemented with 1× B27 supplement [Gibco], 1x GlutaMAX [Life Technologies], 20 ng/mL murine epithelial growth factor [PeproTech], 20 ng/mL basic fibroblast growth factor [PeproTech], and 100U/mL penicillin/streptomycin [Life Technologies]). For the first round of neurosphere formation assays, ∼5000 single cells isolated from SVZ were cultured in the ultra-low binding plates (Corning). The neurospheres formed after 7-day culture were the primary neurospheres. The second round of neurosphere formation assays were carried out by dissociating the primary neurospheres into single cells and replating around 1000 single cells in the ultra-low binding plates. The cells were further cultured for another 7 days to form secondary neurospheres. The diameters of neurospheres were measured using a Zeiss Axio Imager microscope (Carl Zeiss GmbH, Oberkochen, Germany).
NSC proliferation and differentiation assays
To study cell proliferation, we dissociated NSCs with trypsin and plated them on poly-L-ornithin/laminin-coated glass coverslips at a density of 50,000 cells/well in proliferation medium (see above). At 20 h post-plating, 5 μM 5-bromo-2′-deoxyuridine (BrdU, Sigma-Aldrich) was added into the culture medium for 10 h. NSCs were then washed with PBS and fixed with 4% paraformaldehyde for 30 min at room temperature, followed by immunohistochemical analysis. To detect BrdU incorporation, fixed cells were pretreated with 1.0 M HCl for 30 min at 37°C, and then washed with borate buffer (pH 8.5) for 30 min. To induce differentiation of neural progenitor cell, the cultured neurospheres were collected and re-plated onto the Poly-L-Ornithine (R&D systems) and Laminin (Corning)-coated coverslips to form a monolayer culture. The neural progenitor cells were passaged at a 1:5 ratio every 3 days. The NPC monolayer cells were gently washed with PBS 3 times and cultured under differentiation medium (Neurobasal medium supplemented with 1× N2 supplement [Giboco], 1x GlutaMAX [Life Technologies]). Before immunocytochemistry staining, monolayer cultured cells were washed with DPBS and fixed with 4% PFA for 10 min. After pre-treatment, cells were pre-blocked with DPBS containing 5% normal donkey serum and 0.2% Triton X-100 for 30 min, followed by overnight incubation with primary antibody. The antibodies were rat anti-BrdU (1:500, MCA2060, Bio-Rad) or rabbit anti-GFAP (1:300, 12389, Cell Signaling technology). After washing with DPBS, cells were incubated with secondary antibodies R-Phycoerythrin AffiniPure F (ab’) two Fragment Donkey anti-rat IgG (1:300; 712-116-153; Jackson ImmunoResearch) or Alexa Fluor 568 donkey anti-rabbit IgG (1:300, Life Science technology) for 1 hour and mounted using Vectorshield mounting media with DAPI (H1200, Vector Laboratories). Images were captured using a Zeiss Axio Imager microscope (Carl Zeiss GmbH, Oberkochen, Germany) and an AxioCam HRc camera (Carl Zeiss GmbH) with image acquisition via Zeiss Zen Pro software (v.2.3; Carl Zeiss GmbH).
Genotyping
Genomic DNA was extracted from mouse tails with a lysis buffer of 0.01 M NaOH. After neutralizing with Tris-HCl (PH 7.6), the extracted genomic DNA was used for genotyping PCR assays. Primers used for genotyping are listed in Table S5.
TUNEL assay
In Situ Cell Death Detection Kit (11684795910, Sigma-aldrich) was used to evaluate cell death in monolayer cultured neural progenitor cells according to the manufacturer’s instruction. Fluorescence images were captured by Zeiss Axio Imager microscope (Carl Zeiss GmbH, Oberkochen, Germany).
Senescence-associated β-galactosidase staining assay
Senescence β-Galactosidase Staining Kit (9860, Cell Signaling technology) was applied to measure the senescent monolayer cultured neural progenitor cells according to the manufacturer’s instruction. Images were captured by Zeiss Axio Imager microscope (Carl Zeiss GmbH, Oberkochen, Germany).
In vivo BrdU incorporation assay
4–5-week-old mice were injected with 100 mg/kg BrdU (Sigma-Aldrich) intraperitoneally once every 3 days for six total doses. Seven days after the final injection, mice were anesthetized via ketamine (80 mg/kg) and xylazine (6 mg/kg) intraperitoneally, perfused intracardially with ice-cold solutions of 10 mL of DPBS followed by 10 mL of 4% PFA in PBS. Brains were extracted and fixed in 4% PFA for 24 h at 4°C, then embedded in paraffin. Coronal sections (5 μm) were harvested using a microtome.
Immunohistology assays
Mouse tissues were fixed in 4% PFA in PBS overnight at 4°C and embedded in paraffin. For immunofluorescence, tissue sections of 5 μm were cut, dewaxed, and rehydrated. Antigen retrieval was performed by microwaving the sections in 0.01 M sodium citrate buffer (pH 6.0) for 4 min. Tissue sections were blocked in 5% normal donkey serum (NDS) for 30 min after washing with PBS. Tissue sections were then incubated with rabbit anti-SOX2 (1:200, 3728, Cell Signaling technology) diluted in 5% NDS overnight at 4°C. After washing with PBS, sections were incubated with Alexa Fluor 568 donkey anti-rabbit IgG (1:300, Life Science technology) for 1 hour and mounted using Vectorshield mounting media with DAPI (H1200, Vector Laboratories). Alternately, when IHC was required for BrdU visualization, sections were sequentially subjected to HCl treatment, incubated in ice-cold 1 N HCl for 10 min, in 2 N HCl for 10 min at room temperature, and in 2 N HCl for 40 min at 37°C. After finishing the above steps, 1x PBS was used to wash the sections. Sections were then processed as described in neural progenitor cell proliferation assay.
Western blot analysis
Total proteins were extracted from mouse cortices and separated by electrophoresis by 8% PAGE gel. The protein was transferred to the nitrocellulose membrane and blotted with rabbit polyclonal anti-KDM2B (gift from Dr. Robert Klose, Oxford University), rabbit polyclonal anti-TUBULIN (Proteintech, 11224-1-AP), and IRDye 680 donkey anti-rabbit second antibody (1: 10000, Li-Cor). The images were developed by Odyssey Li-Cor Imager (Li-Cor).
Behavioral tests
The mice were labeled by ear-tags. All the behavioral tests were carried out by researchers blinded to the genotypes of animals during the behavioral tests.
Open field test
The open-field apparatus consisted of a custom-made, square white polyvinylchloride foam box (38 × 38 × 35 cm). Mouse behavior was recorded for the first 30 min of habituation to measure time spent in open field and distance traveled with a digital CCD camera connected to a computer running an automated video tracking software package (ANYMaze; Stoelting).
Novel object recognition test (NOR)
NOR was assessed using a 3-day paradigm that included habituation, training, and testing as described previously (Benice and Raber, 2008; Ennaceur et al., 1989; Ennaceur and Delacour, 1988; Ennaceur and Meliani, 1992). Each day, mice were acclimated for 60 min to the behavioral testing room before assessment. All tests were performed under red lights, and behaviors were video recorded and automatically scored using Clever Sys. During habituation (day1), mice were placed into the open field apparatus, a square white polyvinylchloride foam 38 × 38 × 35 cm box, for 60 min while video recorded. For training (day 2), two identical objects consisting of miniature wheels, knobs, spark plugs, or Lego blocks were placed in opposite corners of the open field apparatus, and the animals were allowed to explore the objects for 60 min. The object pairs used were counterbalanced across treatments. For testing (day 3), mice were placed in the same apparatus, but this time one object of the pair was replaced with another dissimilar object (novel object), and mice were allowed to freely explore for 5 min. Their behavior was recorded, and the time the mice spent with their nose oriented toward the object within 3.5 cm of the object edge was considered exploration time. Throughout testing, objects and apparatus were cleaned with 70% ethanol between trials. Discrimination index was calculated as:
Sociability and preference for social novelty test
This test was adapted from Crawley's sociability and preference for social novelty protocol, which consists of three phases (Kaidanovich-Beilin et al., 2011; Moy et al., 2004). Mice were acclimated for 60 min to the behavioral testing room under red lights before testing. The behaviors during all three phases were video recorded and automatically scored using Clever Sys. In phase I (habituation), the experimental mouse was placed in the center of a three-chamber apparatus (polyvinylchloride, 60 × 40 × 22 cm) and allowed to freely explore for 5 min. During this time, the mouse had free access to all three chambers, which are connected by small openings at the bottom of the dividers. In phase two (sociability), two identical, wire cup-like containers were placed one in each of the side chambers. In this phase, an unfamiliar same-sex mouse was placed in one of the containers (“social stimulus”), while the other remained empty (“object”). The experimental mouse was allowed to freely explore the three chambers again for 5 min. In phase three (social memory), the container with the mouse (now “known”) was moved to the opposite chamber, and a new same-sex mouse (“unknown”) was placed in the other container. The experimental mouse was allowed to freely explore the three chambers for 5 min. Throughout testing, objects and apparatus were cleaned with 70% ethanol between trials. For analysis, the time with total body spent in each of the three chambers was recorded.
Morris water maze
Animal activity was measured by a video-based tracking system (ANYMaze; Stoelting). The pool was filled with opaque water (by adding washable white paint) and surrounded by extra maze cues. During the visible platform training, the escape platform (10 cm in diameter) was placed in the center of a designated quadrant with its top positioned 1 cm above the water surface, and the platform was marked by a flag. Mice were trained by four trials per day for 2 days. The four trials were performed with 2 min interval between the trials (intertrial interval [ITI]). Mice were allowed to navigate in the circular pool for up to 60 s until they found the platform. Mice were allowed to stay on the platform for 60 s. If mice failed to find and land on the platform within 60 s, they were manually guided to the platform. The visible platform was randomly placed in different locations for each trial. After the visible platform training, the mice were further trained by the hidden platform paradigm, during which the platform was placed 1 cm beneath the opaque water. Mice were trained by four trials (with 2 min ITI) per day for 5 days. For each trial, mice were dropped into the pool randomly from four different designated start points. The time each mouse spent to land on the platform was scored as escape latency. Probe trials were performed 1 day after the hidden platform training. With the escape platform removed, the mice were allowed to swim in the pool for 60 s. The time spent in target quadrant, number of crossings of the target quadrant, and the location of the hidden platform were recorded.
RT-qPCR assays
RNA was extracted and purified from cells using QI shredder (Qiagen) and RNeasy (Qiagen) spin columns. Total RNA (1 μg) was subjected to reverse transcription using Iscript reverse transcription supermix (1708840, Bio-Rad). cDNA levels were assayed by real-time PCR using iTaq universal SYBR green supermix (1725120, Bio-Rad) and detected by CFX386 Touch Real-Time PCR detection system (Bio-Rad). Primer sequences for qPCR are listed in Table S5.
ChIP-seq sample preparation
For KDM2B and RING1B ChIP, primary wild-type and Kdm2b1f/+ NSCs were fixed with 2 mM ethylene glycol bis(succinimidylsuccinate) (Thermo Scientific) for 1 h, followed by 10 min in 1% formaldehyde and 5 min in 0.125 M glycine to sequence the reaction. Cells were lysed in 1% SDS, 10 mM EDTA, and 50 mM Tris-HCl (pH 8.0), and the DNA was fragmented to approximately 200–400 bp by sonication (Branson Sonifier 450). Immunoprecipitation was performed with 2 μg rabbit polyclonal anti-KDM2B (gift from Dr. Robert Klose, Oxford University) and rabbit anti-RING1B antibody (1:100, 5694, Cell Signaling Technology) overnight at 4°C. Antibody bound DNA-protein complexes were isolated by protein G plus/protein A agarose beads (EMD Millipore), washed and eluted, and reverse cross-linked DNA was extracted by phenol/chloroform and precipitated.
ChIP-qPCR analysis
Immunoprecipitated genomic DNA was analyzed by qPCR, and data are presented as the percentage of input as determined with CFX manager 3.1 software (Bio-rad). The amplicons were designed to locate at ∼ 0.5 kb upstream of transcriptional starting sites (TSS) and introns of Cdkn2a/2b, Bmp7, and Wnt7b genes. The primers for the ChIP-qPCR analysis were listed in the Table S5.
ChIP DNA preparation for NovaSeq6000 sequencing
ChIP DNA libraries were constructed for HiSeq4000 (Illumina) sequencing using NEBNext UltraII DNA library Prep Kit for Illumina (New England BioLabs, Inc) according to the manufacturer’s instructions. Adapter-ligated DNA was amplified by PCR for 12–14 cycles, followed by size selection using agarose gel electrophoresis. The DNA was purified using QIAquick gel extraction kits (Qiagen) and quantified both with an Agilent Bioanalyzer and Invitrogen Qubit. The DNA was diluted to a working concentration of 20 nM prior to sequencing. Sequencing on a NovaSeq6000 instrument was carried out by the Genomics Core Facility at Michigan State University.
ChIP-seq data analysis
For the ChIP-Seq data analysis, all sequencing reads were mapped to NCBI build 37 (mm9) of the mouse genome using Bowtie2 (Langmead and Salzberg, 2012). Mapped reads were analyzed using the MACS program and bound-regions (peaks) were determined using sequencing reads from input DNA as negative controls (Feng et al., 2012). When multiple reads mapped to the same genomic position, a maximum of two reads were retained. The statistical cutoff used for peak calling was p-value < 10−8 and >5-fold enrichment over the control. The mapped sequencing reads were normalized as Counts Per Million Reads (CPM). The normalized reads were binned into 50 bp windows along the genome using the bamCoverage of deepTools program and visualized in the IGV genome browser. The datasets of CpG islands and Refseq genes of mm9 mouse reference genome were retrieved from the UCSC table bowser. The heatmap and plot of ChIP-seq reads in the 10 kb windows flanking KDM2B binding center or Refseq genes were generated using plotHeatmap and plotProfile in the deepTools program (Ramirez et al., 2016).
RNA-seq sample preparation for HiSeq4000 sequencing
RNA was extracted and purified from wild-type (n = 3) and Kdm2b mutant (n = 3) NSCs using QI shredder (Qiagen) and RNeasy (Qiagen) spin columns. Total RNA (1 μg) was used to generate RNA-seq libraries using the NEBNext Ultra Directional RNA library Prep Kit for Illumina (New England BioLabs, Inc) according to the manufacturer’s instructions. Adapter-ligated cDNA was amplified by PCR and followed by size selection using agarose gel electrophoresis. The DNA was purified using the Qiaquick gel extraction kit (Qiagen) and quantified both with an Agilent Bioanalyzer and Invitrogen Qubit. The libraries were diluted to a working concentration of 10 nM prior to sequencing. Sequencing on an Illumina HiSeq4000 instrument was carried out by the Genomics Core Facility at Michigan State University.
RNA-seq data analysis
RNA-Seq data analysis was performed essentially as described previously (Aljazi et al., 2020). 18–20 million reads per sample were obtained by sequencing. The mean read length range from 45 to 50 nt. All sequencing reads were mapped mm9 of the mouse genome using Tophat2 (Kim et al., 2013). The ratios of mapped reads range from 97.8% to 98.4%. The mapped reads were normalized as Reads Per Kilobase of transcript per Million mapped reads (RPKM). The differential gene expression was calculated by the Cuffdiff program and the statistical cutoff for identification of differential gene expression was q <0.05 and 2-fold RPKM change between samples (Trapnell et al., 2013). The normalized mapped reads (RPKM) of each RNA-seq experiments were binned into 50bp windows along the genome using the bamCoverage of deepTools program and visualized in the IGV genome browser. The heatmap and plot of gene expression were generated using plotHeatmap and plotProfile in the deepTools program (Ramirez et al., 2016). The differentially expressed gene lists were then used as input for the DAVID Functional Annotation Bioinformatics Microarray Analysis for KEGG pathway and gene ontology enrichment analyses (https://david.ncifcrf.gov/).
Quantification and statistical analysis
All statistical analyses were performed using GraphPad Prism 8 (GraphPad Software). Parametric data were analyzed by a two-tailed t test or two-way ANOVA test for comparisons of multiple samples. p values <0.05 were considered statistically significant. Data are presented as mean ± SEM.
Acknowledgments
We thank MSU genomics core facility for processing the next-generation sequencing. This work was supported by the National Institutes of Health NIH grant R01GM127431.
Author contributions
J.H. conceived the project. The mouse model was generated in the Y.Z. lab. N.D.-W. and M.B.A. performed the experiments. J.H. and G.I.M. performed the sequencing data analysis. J.H., A.J.M., A.J.R., and Y.Z. discussed the results. Y.G. and J.H. wrote the manuscript.
Declaration of interests
Authors declare that they have no conflict of interests.
Inclusion and diversity
We worked to ensure sex balance in the selection of non-human subjects.
Published: February 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.103742.
Contributor Information
Yi Zhang, Email: yzhang@genetics.med.harvard.edu.
Jin He, Email: hejin1@msu.edu.
Supplemental information
Data and code availability
All data supporting the findings of this study are available within the paper and its supplemental information files. All RNA-seq and ChIP-seq reported in this paper have been deposited to NCBI Gene Expression Omnibus (GEO), GEO accession: GSE190807. This paper does not report original code.
References
- Aljazi M.B., Gao Y., Wu Y., Mias G.I., He J. Cell signaling coordinates global PRC2 recruitment and developmental gene expression in murine embryonic stem cells. iScience. 2020;23 doi: 10.1016/j.isci.2020.101646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baple E., Palmer R., Hennekam R.C. A microdeletion at 12q24.31 can mimic beckwith-wiedemann syndrome neonatally. Mol. Syndromol. 2010;1:42–45. doi: 10.1159/000275671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benice T.S., Raber J. Object recognition analysis in mice using nose-point digital video tracking. J. Neurosci. Methods. 2008;168:422–430. doi: 10.1016/j.jneumeth.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Blackledge N.P., Fursova N.A., Kelley J.R., Huseyin M.K., Feldmann A., Klose R.J. PRC1 catalytic activity is central to polycomb system function. Mol. Cell. 2020;77:857–874 e859. doi: 10.1016/j.molcel.2019.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken A.P., Kleine-Kohlbrecher D., Dietrich N., Pasini D., Gargiulo G., Beekman C., Theilgaard-Monch K., Minucci S., Porse B.T., Marine J.C., et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007;21:525–530. doi: 10.1101/gad.415507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruggeman S.W., Valk-Lingbeek M.E., van der Stoop P.P., Jacobs J.J., Kieboom K., Tanger E., Hulsman D., Leung C., Arsenijevic Y., Marino S., et al. Ink4a and Arf differentially affect cell proliferation and neural stem cell self-renewal in Bmi1-deficient mice. Genes Dev. 2005;19:1438–1443. doi: 10.1101/gad.1299305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgering B.M., Kops G.J. Cell cycle and death control: long live Forkheads. Trends Biochem. Sci. 2002;27:352–360. doi: 10.1016/s0968-0004(02)02113-8. [DOI] [PubMed] [Google Scholar]
- Chouery E., Choucair N., Abou Ghoch J., El Sabbagh S., Corbani S., Megarbane A. Report on a patient with a 12q24.31 microdeletion inherited from an insulin-dependent diabetes mellitus father. Mol. Syndromol. 2013;4:136–142. doi: 10.1159/000346473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennaceur A., Cavoy A., Costa J.C., Delacour J. A new one-trial test for neurobiological studies of memory in rats. II: effects of piracetam and pramiracetam. Behav. Brain Res. 1989;33:197–207. doi: 10.1016/s0166-4328(89)80051-8. [DOI] [PubMed] [Google Scholar]
- Ennaceur A., Delacour J. A new one-trial test for neurobiological studies of memory in rats. 1: behavioral data. Behav. Brain Res. 1988;31:47–59. doi: 10.1016/0166-4328(88)90157-x. [DOI] [PubMed] [Google Scholar]
- Ennaceur A., Meliani K. A new one-trial test for neurobiological studies of memory in rats. III. Spatial vs. non-spatial working memory. Behav. Brain Res. 1992;51:83–92. doi: 10.1016/s0166-4328(05)80315-8. [DOI] [PubMed] [Google Scholar]
- Farcas A.M., Blackledge N.P., Sudbery I., Long H.K., McGouran J.F., Rose N.R., Lee S., Sims D., Cerase A., Sheahan T.W., et al. KDM2B links the polycomb repressive complex 1 (PRC1) to recognition of CpG islands. Elife. 2012;1 doi: 10.7554/eLife.00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J., Liu T., Qin B., Zhang Y., Liu X.S. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 2012;7:1728–1740. doi: 10.1038/nprot.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove J., Ripke S., Als T.D., Mattheisen M., Walters R.K., Won H., Pallesen J., Agerbo E., Andreassen O.A., Anney R., et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019;51:431–444. doi: 10.1038/s41588-019-0344-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris S.L., Levine A.J. The p53 pathway: positive and negative feedback loops. Oncogene. 2005;24:2899–2908. doi: 10.1038/sj.onc.1208615. [DOI] [PubMed] [Google Scholar]
- He J., Kallin E.M., Tsukada Y., Zhang Y. The H3K36 demethylase Jhdm1b/Kdm2b regulates cell proliferation and senescence through p15(Ink4b) Nat. Struct. Mol. Biol. 2008;15:1169–1175. doi: 10.1038/nsmb.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J., Shen L., Wan M., Taranova O., Wu H., Zhang Y. Kdm2b maintains murine embryonic stem cell status by recruiting PRC1 complex to CpG islands of developmental genes. Nat. Cell Biol. 2013;15:373–384. doi: 10.1038/ncb2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaidanovich-Beilin O., Lipina T., Vukobradovic I., Roder J., Woodgett J.R. Assessment of social interaction behaviors. J. Vis. Exp. 2011:2473. doi: 10.3791/2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labonne J.D., Lee K.H., Iwase S., Kong I.K., Diamond M.P., Layman L.C., Kim C.H., Kim H.G. An atypical 12q24.31 microdeletion implicates six genes including a histone demethylase KDM2B and a histone methyltransferase SETD1B in syndromic intellectual disability. Hum. Genet. 2016;135:757–771. doi: 10.1007/s00439-016-1668-4. [DOI] [PubMed] [Google Scholar]
- Lalli M.A., Avey D., Dougherty J.D., Milbrandt J., Mitra R.D. High-throughput single-cell functional elucidation of neurodevelopmental disease-associated genes reveals convergent mechanisms altering neuronal differentiation. Genome Res. 2020;30:1317–1331. doi: 10.1101/gr.262295.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord C., Brugha T.S., Charman T., Cusack J., Dumas G., Frazier T., Jones E.J.H., Jones R.M., Pickles A., State M.W., et al. Autism spectrum disorder. Nat. Rev. Dis. Primers. 2020;6:5. doi: 10.1038/s41572-019-0138-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molofsky A.V., He S., Bydon M., Morrison S.J., Pardal R. Bmi-1 promotes neural stem cell self-renewal and neural development but not mouse growth and survival by repressing the p16Ink4a and p19Arf senescence pathways. Genes Dev. 2005;19:1432–1437. doi: 10.1101/gad.1299505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy S.S., Nadler J.J., Perez A., Barbaro R.P., Johns J.M., Magnuson T.R., Piven J., Crawley J.N. Sociability and preference for social novelty in five inbred strains: an approach to assess autistic-like behavior in mice. Genes Brain Behav. 2004;3:287–302. doi: 10.1111/j.1601-1848.2004.00076.x. [DOI] [PubMed] [Google Scholar]
- Palumbo O., Palumbo P., Delvecchio M., Palladino T., Stallone R., Crisetti M., Zelante L., Carella M. Microdeletion of 12q24.31: report of a girl with intellectual disability, stereotypies, seizures and facial dysmorphisms. Am. J. Med. Genet. A. 2015;167A:438–444. doi: 10.1002/ajmg.a.36872. [DOI] [PubMed] [Google Scholar]
- Qiao Y., Tyson C., Hrynchak M., Lopez-Rangel E., Hildebrand J., Martell S., Fawcett C., Kasmara L., Calli K., Harvard C., et al. Clinical application of 2.7M Cytogenetics array for CNV detection in subjects with idiopathic autism and/or intellectual disability. Clin. Genet. 2013;83:145–154. doi: 10.1111/j.1399-0004.2012.01860.x. [DOI] [PubMed] [Google Scholar]
- Ramirez F., Ryan D.P., Gruning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dundar F., Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–W165. doi: 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzzo E.K., Perez-Cano L., Jung J.Y., Wang L.K., Kashef-Haghighi D., Hartl C., Singh C., Xu J., Hoekstra J.N., Leventhal O., et al. Inherited and de novo genetic risk for autism impacts shared networks. Cell. 2019;178:850–866 e826. doi: 10.1016/j.cell.2019.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satterstrom F.K., Kosmicki J.A., Wang J., Breen M.S., De Rubeis S., An J.Y., Peng M., Collins R., Grove J., Klei L., et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell. 2020;180:568–584 e523. doi: 10.1016/j.cell.2019.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpless N.E. INK4a/ARF: a multifunctional tumor suppressor locus. Mutat. Res. 2005;576:22–38. doi: 10.1016/j.mrfmmm.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Sherr C.J. The INK4a/ARF network in tumour suppression. Nat. Rev. Mol. Cell Biol. 2001;2:731–737. doi: 10.1038/35096061. [DOI] [PubMed] [Google Scholar]
- Thomason L.C., Sawitzke J.A., Li X., Costantino N., Court D.L. Recombineering: genetic engineering in bacteria using homologous recombination. Curr. Protoc. Mol. Biol. 2014;106:1 16 11–39. doi: 10.1002/0471142727.mb0116s106. [DOI] [PubMed] [Google Scholar]
- Trapnell C., Hendrickson D.G., Sauvageau M., Goff L., Rinn J.L., Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada Y., Fang J., Erdjument-Bromage H., Warren M.E., Borchers C.H., Tempst P., Zhang Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- Turberfield A.H., Kondo T., Nakayama M., Koseki Y., King H.W., Koseki H., Klose R.J. KDM2 proteins constrain transcription from CpG island gene promoters independently of their histone demethylase activity. Nucleic Acids Res. 2019;47:9005–9023. doi: 10.1093/nar/gkz607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Wang L., Erdjument-Bromage H., Vidal M., Tempst P., Jones R.S., Zhang Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature. 2004;431:873–878. doi: 10.1038/nature02985. [DOI] [PubMed] [Google Scholar]
- Wu X., Johansen J.V., Helin K. Fbxl10/Kdm2b recruits polycomb repressive complex 1 to CpG islands and regulates H2A ubiquitylation. Mol. Cell. 2013;49:1134–1146. doi: 10.1016/j.molcel.2013.01.016. [DOI] [PubMed] [Google Scholar]
- Zhang X., Tang N., Hadden T.J., Rishi A.K. Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta. 2011;1813:1978–1986. doi: 10.1016/j.bbamcr.2011.03.010. [DOI] [PubMed] [Google Scholar]
- Zhu C., Gao J., Karlsson N., Li Q., Zhang Y., Huang Z., Li H., Kuhn H.G., Blomgren K. Isoflurane anesthesia induced persistent, progressive memory impairment, caused a loss of neural stem cells, and reduced neurogenesis in young, but not adult, rodents. J. Cereb. Blood Flow Metab. 2010;30:1017–1030. doi: 10.1038/jcbfm.2009.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this study are available within the paper and its supplemental information files. All RNA-seq and ChIP-seq reported in this paper have been deposited to NCBI Gene Expression Omnibus (GEO), GEO accession: GSE190807. This paper does not report original code.