

Summary
Modulation of the neuronal K+/Cl− cotransporter 2 (KCC2) activity, which mediates Cl− export, is critical to neuronal function. Here, we demonstrate that KCC2 interacts with the SNARE protein synaptosome-associated protein 23, SNAP23, an essential component of membrane insertion machinery. Using KCC2 truncated mutants, we show that KCC2 C-terminal domain is essential for membrane targeting and SNAP23-dependent upregulation of KCC2 activity triggered by activation of the Zn2+-sensitive receptor mZnR/GPR39 in HEK293 cells. Expression of SNAP23 phosphorylation-insensitive mutants or inhibition of its upstream activator IκB kinase (IKK) prevents mZnR/GPR39 upregulation of KCC2 activity in mouse hippocampal neurons. We further find that SNAP23 interacts with Syntaxin 1A and KCC2, and that all three proteins exhibit increased membrane insertion following mZnR/GPR39 activation in neurons. Our results elucidate a G-protein-coupled receptor-dependent pathway for regulation of KCC activity, mediated via interaction with SNARE proteins.
Subject areas: Molecular physiology, Neuroscience, Cell biology
Graphical abstract
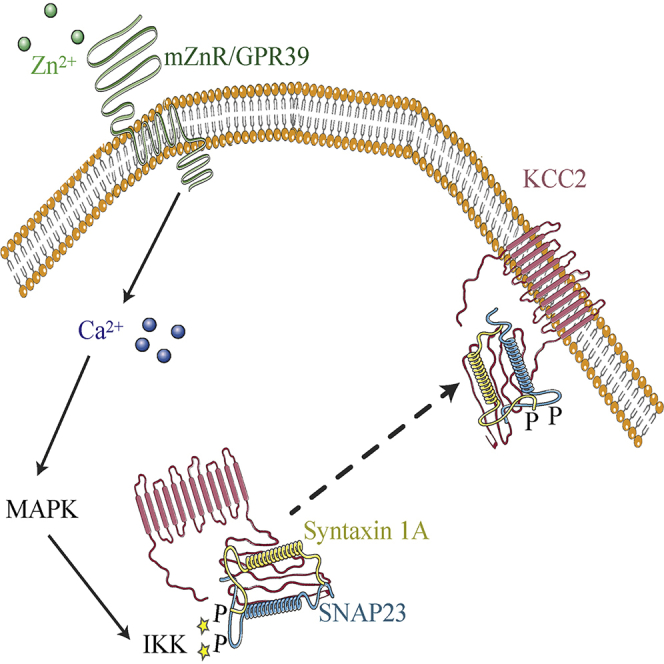
Highlights
-
•
Neuronal K+/Cl− cotransporter 2 (KCC2) is regulated via interaction with SNAP23
-
•
Zn2+ enhances interaction and membrane insertion of SNAP23, Syntaxin 1A, and KCC2
-
•
Zn2+-dependent mZnR/GPR39 regulation of KCC2 requires SNAP23 phosphorylation
-
•
Epithelial KCC3 regulation by ZnR/GPR39 also requires SNAP23
Molecular physiology; Neuroscience; Cell biology
Introduction
The K+/Cl− cotransporter KCC2 is responsible for Cl− extrusion in mature neurons and thus maintains the inward Cl− gradients essential for generation of hyperpolarizing GABAA and glycine responses (Kahle et al., 2016; Mapplebeck et al., 2019; Watanabe et al., 2019). Activity of KCC2 is highly regulated by synaptic signaling, thereby affecting Cl− extrusion capability and neuronal inhibitory tone (Mahadevan and Woodin, 2016; Spoljaric et al., 2019). Regulation of KCC2 is achieved by rapid changes in the phosphorylation status of multiple residues on KCC2, which, in turn, can directly modulate its activity (Cordshagen et al., 2018; Weber et al., 2014), or influence its presence on the plasma membrane (Côme et al., 2019a; Markkanen et al., 2017; Medina et al., 2014). Most prominent, phosphorylation of serine 940 (S940) by protein kinase C (PKC), a pathway activated by multiple G-protein coupled receptors, rapidly increases KCC2 surface stability and thereby Cl− transport (Lee et al., 2007; Leonzino et al., 2016). In contrast, surface expression of KCC2 is reduced by phosphorylation of threonine 1007 and 906, which results in a depolarized GABAA reversal potential (Friedel et al., 2015; Heubl et al., 2017). Phosphorylation of these threonine residues is mediated via activation of the with-no-lysine kinase (WNK) and its downstream target STE20/SPS1-related proline/alanine-rich kinase (SPAK) (Zhang et al., 2020). Other putative phosphorylation sites have been identified on both the N-and C-terminal domains of KCC2 and were shown to be selectively activated by various triggers in cells exogenously expressing the transporter (Cordshagen et al., 2018). In neurons, the membrane stability of KCC2 has been associated with modulation of the C-terminus while the N-terminus was suggested to control its delivery into the plasma membrane and is particularly important for its expression in distal dendrites (Friedel et al., 2017).
Zinc ions (Zn2+) that are synaptically released from neuronal presynaptic mossy fibers activate a postsynaptic metabotropic Gq-protein coupled receptor, mZnR/GPR39, triggering intracellular Ca2+ signaling (Besser et al., 2009). Subsequent activation of the mitogen-activated kinase (MAPK) pathway increases KCC2 surface expression in a soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE)-dependent manner (Chorin et al., 2011; Saadi et al., 2012). Further studies showed that mZnR/GPR39 knockout (KO) mice exhibit enhanced kainate-induced seizures, compared to wild-type mice, indicating that regulation of KCC2 activity by mZnR/GPR39 plays an important homeostatic role that can dampen epileptic activity (Gilad et al., 2015). The precise mechanism linking mZnR/GPR39 activation with KCC2 function, however, has not been fully delineated. Several other pathways that activate metabotropic neuronal signaling via G-protein coupled receptors also upregulate KCC2 activity (Mahadevan and Woodin, 2016). Among them, mGluR1 and mGluR5 (Banke and Gegelashvili, 2008), 5-HT2A serotonin receptors (Bos et al., 2013), the oxytocin receptor (Leonzino et al., 2016), and kainate receptors (Garand et al., 2019) all activate Ca2+-dependent PKC signaling that directly enhances KCC2 S940 phosphorylation and, subsequently KCC2 surface expression. We show here that the G-protein coupled receptor mZnR/GPR39 activates a distinct pathway that enhances interaction between the SNARE protein SNAP23 and KCC2 to regulate the membrane expression and activity of the transporter.
Results
KCC2 C-terminal domain is essential for regulation by mZnR/GPR39 signaling
We first asked if mZnR/GPR39 regulation of KCC2 activity is mediated via its N-terminal domain (NTD) or the C-terminal domain (CTD). For this purpose, we used HEK293 cells co-transfected with mZnR/GPR39 and the truncated isoforms ΔNTD-KCC2 or ΔCTD-KCC2, lacking, respectively, the NTD and CTD (Friedel et al., 2017). Transfection of HEK293 cells with the ΔNTD-KCC2 (deletion of amino acids 1–100) resulted in low, albeit measurable expression of the transporter (Figure 1A), while ΔCTD-KCC2 (deletion of amino acids 654–1114) yielded similar expression levels when compared to wild-type (WT) KCC2 (Figure 1A), in agreement with previous results (Li et al., 2007). To study KCC2-dependent transport, we determined rates of cellular acidification with BCECF fluorescence using established methodology (Chorin et al., 2011; Hershfinkel et al., 2009). Application of NH4Cl to the extracellular solution induces cellular alkalinization due to passive entry of NH3. This is followed by pronounced KCC2-dependent transport that acts in reverse mode and mediates influx of NH4+, a surrogate to K+, leading to intracellular acidification and markedly decreased BCECF fluorescence (Figure 1B). We monitored acidification rates in control cells treated with 200 μM Zn2+ for 2 min, a condition that activates mZnR/GPR39 without affecting intracellular Zn2+ levels (Besser et al., 2009). As previously noted (Chorin et al., 2011), cells treated with Zn2+ showed increased rates of NH4+ transport when compared to acidification rates in vehicle-treated controls (Figure 1B). Treatment of cells with the KCC2 inhibitor DIOA (100 μM) prevented Zn2+-dependent upregulation of transport activity, confirming that KCC2 mediates the NH4+-dependent acidification. Zn2+ treatment of HEK293 cells expressing KCC2 without mZnR/GPR39 did not influence the rates of NH4+-dependent acidification, indicating that mZnR/GPR39 is essential for enhancing KCC2-dependent transport by Zn2+ (Figure 1B). Next, we measured NH4+ transport rates in cells expressing mZnR/GPR39 together with the truncated ΔNTD-KCC2 or ΔCTD-KCC2 variants. We noted slower baseline rates of NH4+ transport activity in cells expressing ΔNTD-KCC2 (Figure 1C), when compared to the full-length KCC2 (Figure 1B), consistent with the lower expression levels of this variant. Nonetheless, NH4+ transport rates in cells expressing ΔNTD-KCC2 were still significantly enhanced following Zn2+ treatment (Figure 1C), suggesting that the N-terminus is not necessary for transporter regulation by mZnR/GPR39. In contrast, transport rates in ΔCTD-KCC2-expressing cells were completely insensitive to Zn2+ (Figure 1C), indicating that the C-terminal domain is required for mZnR/GPR39-mediated regulation of KCC2 activity.
Figure 1.

KCC2 C-terminal is essential for Zn2+-dependent upregulation of transport
(A) Immunoblot analysis of KCC2, ΔNTD-KCC2, or ΔCTD-KCC2 expression in HEK293 cells. Blots were exposed to a KCC2 antibody together with an anti-FLAG antibody, and normalized to actin as loading control. The KCC2 antibody, which is targeted to the C-terminus, works well with both the WT and ΔNTD KCC2. The FLAG antibody recognizes the WT KCC2 and ΔCTD truncated mutant but works poorly with the ΔNTD. White arrows on the blot point to the monomeric forms of each of the constructs at their expected molecular weight. Right panel shows quantification of expression level, and a schematic drawing of the KCC2 constructs used. (p = 0.0008, Welch’s t test).
(B) HEK293 cells co-transfected with GPR39 and KCC2 or KCC2 alone were loaded with the pH-sensitive fluorescent indicator BCECF (1 μM). Cells were washed with K+-free Ringer’s solution, baseline, and addition of NH4Cl (15 mM) resulted in increase in fluorescence due to NH3 diffusion. Subsequent NH4+ transport, used as surrogate by K+ transporter, resulted in acidification (Hershfinkel et al., 2009). Shown are representative fluorescent signal traces from cells treated with Zn2+ (200 μM, 2 min) compared to control cells or cells treated with the KCC inhibitor DIOA (100 μM). Right panel shows averaged rates of NH4+ transport, mediated by KCC2, obtained over initial 100 s from maximal signal.(∗∗∗p< 0.0001, t test).
(C) BCECF fluorescent signal traces from cells co-transfected with GPR39 and ΔNTD-KCC2 or ΔCTD-KCC2, treated with or without Zn2+. Averaged rates of NH4+ transport, representing KCC2 activity, are shown in right panel. (∗∗p = 0.0032, t test). Dotted line indicates the rate of transport in cells expressing GPR39 and WT KCC2 that were treated with Zn2+ as in (B).
(D) Biotinylation assay of KCC2 variants in HEK293 cells co-transfected with GPR39 and KCC2 or ΔNTD-KCC2 or ΔCTD-KCC2 (upper panel). Comparison of surface expression between cells treated with Zn2+ (200 μM, 2 min) and control cells, using KCC2 and FLAG antibodies (as in A). Loading controls for surface expression were the membrane protein transferrin receptor and cytoplasmic tubulin. Total protein levels (lower panel) determined in cell lysates were used for normalization, tubulin was used as loading control. The arrows mark the protein isoform, quantified in the right panel, at the expected molecular weight. Quantification of normalized membrane expression of KCC2 variants in Zn2+-treated cells versus control (∗∗p = 0.0064 KCC2 with and without Zn2+, ∗∗p = 0.0012 ΔNTD-KCC2 with and without Zn2+, Mann-Whitney test). All bar graphs values are averages ± SD, with all data points presented.
We previously reported that mZnR/GPR39 upregulates KCC2 activity in neurons by enhancing its surface expression (Chorin et al., 2011). We therefore tested whether the observed changes in KCC2 transport following Zn2+ treatment in HEK293 cells were similarly accompanied by increased membrane expression levels. Using a biotinylation assay, we noted that WT-KCC2 expression was increased in the membrane fraction of Zn2+-treated HEK293 cells (Figure 1D), suggesting that mZnR/GPR39 regulation of KCC2 is mediated by a similar pathway in both neurons and HEK293 cells. In cells expressing ΔNTD-KCC2, we also observed increased levels of the construct in the membrane fraction following Zn2+ treatment (Figure 1D). In contrast, Zn2+ treatment did not enrich ΔCTD-KCC2 expression level in the membrane (Figure 1D). These results indicate that KCC2 C-terminus is essential for mZnR/GPR39 upregulation of transport activity, likely as a result of the increased cell membrane insertion of the transporter.
mZnR/GPR39-dependent upregulation of KCC2 activity is not mediated through phosphorylation sites on the C-terminus of the transporter
The results presented thus far indicate that mZnR/GPR39-dependent regulation of KCC2 transport activity requires the C-terminus, which contains numerous regulatory phosphorylation sites (Côme et al., 2019b). As mentioned earlier, various Gq-coupled metabotropic receptors upregulate KCC2 activity via PKC phosphorylation of S940 (Banke and Gegelashvili, 2008; Lee et al., 2007; Leonzino et al., 2016; Mahadevan and Woodin, 2016). We thus asked whether phosphorylation of S940 (Figure 2A) is also required for mZnR/GPR39-dependent upregulation of KCC2. We mutated S940 into a phosphorylation-insensitive alanine residue (S940A), and determined NH4+ transport rate in HEK293 cells co-expressing this mutant and mZnR/GPR39 (Figure 2B). The S940A KCC2 mutant showed Zn2+-dependent upregulation of transport activity similar to the WT KCC2 (Figure 2B).Because other residues on KCC2 may be phosphorylation targets of PKC, we tested whether kinase inhibitors staurosporine (1 nM) and GO6983 (10 μM) could block the modulatory actions of Zn2+ on transporter function. However, cells expressing WT KCC2 and mZnR/GPR39 exhibited similar Zn2+-dependent upregulation of KCC2 activity in the presence of both inhibitors (Figure 2C). Taken together, these results suggest that mZnR/GPR39 upregulation of KCC2 activity does not involve PKC signaling or phosphorylation of KCC2 at S940.
Figure 2.

ZnR/GPR39-dependent upregulation of KCC2 activity is not mediated through its phosphorylation
(A) Scheme of residues that are putative phosphorylation sites on KCC2.
(B) BCECF fluorescent signal traces from HEK293 cells co-transfected with GPR39 and WT KCC2 or KCC2 mutant S940A, treated with Zn2+ (200 μM, 2 min) and compared to controls. Cells were treated with NH4Cl and rates of acidification following NH4+ transport were determined. Bar graph shows averaged rates of NH4+ transport, calculated over initial 100 s from maximal signal.(∗∗∗p<0.0001 Mann-Whitney test).
(C) BCECF fluorescent signal traces from HEK293 cells co-transfected with WT KCC2 and GPR39, pretreated with the PKC inhibitor staurosporine and then with Zn2+, compared to controls. Right panel shows averaged rates of acidification following NH4+ transport in control cells or cells treated with the PKC inhibitors staurosporine (1 nM) or GO6983 (10 μM). (∗∗∗p<0.0001 KCC2 with and without Zn2+, ∗∗p = 0.0022 staurosporine with and without Zn2+, ∗∗∗p = 0.0005 GO6983 with and without Zn2+, Mann-Whitney test).
(D and E) Averaged rates of acidification following NH4+ transport monitored in HEK293 cells co-transfected with GPR39 and the indicated KCC2 mutants. Cells were treated with Zn2+ or without it. (all comparisons are between control and Zn2+-treated mutants, panel D using t test: p = 0.0046 KCC2, p = 0.0212 T1007E, p = 0.0088 T906E; for panel E, t test was used: p<0.0001 KCC2, p = 0.0002 S988A, p = 0.0002 S986/988A, p = 0.0003 S986/988D, p<0.0001 T584A, p = 0.0002 S868A, p = 0.0274 SPVSS; and p = 0.0041 S986A using Mann-Whitney test). All bar graphs values are averages ± SD, with all data points presented.
We then explored the possible role of threonines 906 and 1007, previously shown to enhance KCC2 surface expression following dephosphorylation (Friedel et al., 2015). We transfected HEK293 cells with KCC2 mutants that mimic the constitutively phosphorylated state at these sites, T906E or T1007E (Figure 2A), and monitored Zn2+-dependent NH4+ transport rates. In cells expressing either T906E or T1007E, which could not be dephosphorylated to enhance KCC2 membrane expression levels, we still observed Zn2+-dependent upregulation of KCC2 activity (Figure 2D). This suggests that T906 or T1007 are also not involved in mZnR/GPR39 regulation of KCC2.
Previous results showed that similar to neuronal KCC2, the epithelial KCC family members are also activated by mZnR/GPR39 via Ca2+ signaling and activation of MAP kinase (Mero et al., 2019; Sunuwar et al., 2017). To determine if this kinase phosphorylated a previously unknown site on KCC2, we identified putative MAPK phosphorylation sites that are shared between KCC family members using the Group-based Prediction System (GPS) Website (Xue et al., 2011) (http://gps.biocuckoo.cn/online.php). Putative phosphorylation sites receiving a high score were mutated into alanine, namely S988A, S986A, T584, and S868A (Figure 2A). In addition, we deleted a region that is enriched with serine residues (SPVSS, residues 1045–1049) also suspected to be phosphorylated by MAPK. However, we found that all these mutants continued to show Zn2+-dependent upregulation of KCC2 transport activity (Figure 2E). Notably, we found that the S988A mutant showed an increase in baseline transport activity, suggesting that dephosphorylation of S988 may be involved in regulation of KCC2 that is independent of mZnR/GPR39, but mutation of both S988 and S986 to alanine reversed this baseline effect. Importantly, both mutants showed Zn2+-dependent upregulation of transport activity. Moreover, mutation of both S986 and 988 into negatively charged aspartate residues also yielded Zn2+-dependent upregulation of transport, further suggesting that the phosphorylation or dephosphorylation of these sites are not linked to mZnR/GPR39 regulation of KCC2. In addition, mutation of T584 to a phosphorylation-insensitive alanine resulted in an increased effect of Zn2+ on transport activity, suggesting that following Zn2+-dependent insertion of KCC2 into the membrane, dephosphorylation of this site may enhance membrane stability of the transporter, as shown for T906 and T1007 (Friedel et al., 2015). Thus, although MAPK inhibition attenuates mZnR/GPR39 activation of neuronal KCC2 (Chorin et al., 2011), we could not identify a direct phosphorylation site of KCC2 that is required for mZnR/GPR39 regulation of transport.
mZnR/GPR39 regulation of KCC2 is mediated by SNAP23
In light of the aforementioned results, we next sought other potential mechanisms that could be responsible for mediating mZnR/GPR39-dependent upregulation of KCC2. In a prior study, we reported that SNARE proteins are essential for regulation of neuronal KCC2 function following mZnR/GPR39 activation (Saadi et al., 2012). In a separate line of experiments, we found that mZnR/GPR39 regulates the epithelial KCC isoforms KCC1 in colon cells and KCC3 in breast cancer cells (Chakraborty et al., 2021; Mero et al., 2019; Sunuwar et al., 2017). As the epithelial SNARE protein SNAP23 regulates surface expression of major ion transporters, including the cystic fibrosis transmembrane conductance regulator (CFTR) and epithelial sodium channel (ENaC) (Cormet-Boyaka et al., 2002; Saxena et al., 2006), we asked if SNAP23 could mediate the enhanced neuronal KCC2 activity following mZnR/GPR39 activation. For this purpose, we used SHSY-5Y neuroblastoma cells, which endogenously express KCC2 and mZnR/GPR39 (Chorin et al., 2011), thus do not require overexpression that can promote promiscuous protein–protein interactions. We first confirmed that mZnR/GPR39 activation enhanced KCC-dependent transport in these cells using mZnR/GPR39-silencing (siGPR39) constructs. We found that Zn2+ treatment upregulated NH4+ transport in cells transfected with a scrambled siRNA (Figure 3A). In contrast, siGPR39 that effectively reduced mRNA expression of mZnR/GPR39 (inset, Figure 3A) abolished the stimulatory actions of Zn2+ (Figure 3A). To determine if KCC2 mediates the observed transport, we applied the KCC inhibitor DIOA or the KCC2 selective inhibitor VU0463271. Each of these inhibitors effectively diminished Zn2+-dependent upregulation of NH4+ transport (Figure S1). We noted, however, that inhibition of KCC2 by VU0463271 showed enhanced baseline transport activity, a potential compensatory mechanism mediated by the sodium/potassium/chloride cotransporter NKCC1. We therefore asked if NKCC may be regulated by Zn2+. But, the NKCC inhibitor bumetanide did not reverse the effect of Zn2+ (Figure 3B), indicating that NKCC is not upregulated by mZnR/GPR39 in SHSY-5Y cells. Finally, we found that silencing of KCC2 using siRNA constructs reduced endogenous KCC2 mRNA (inset, Figure 3B) and prevented mZnR/GPR39 upregulation of NH4+ transport activity (Figure 3B). These results suggest that SHSY-5Y cells can effectively serve as a model system to investigate the pathway for mZnR/GPR39 activation of endogenous KCC2.
Figure 3.

SNAP23 phosphorylation is required for ZnR/GPR39-dependent KCC3 and KCC2 upregulation
(A) SHSY-5Y cells were transfected with an siRNA sequence targeted to silence GPR39, or with a scrambled sequence (SCR) to determine the role of mZnR/GPR39 in upregulating transport in these cells. Insert shows mRNA expression level of GPR39 in SCR and siGPR39 cells. NH4+ transport was monitored in cells loaded with BCECF, and representative fluorescence signals in cells treated with Zn2+ (200 μM) are shown versus controls. Right panel shows averaged rates of NH4+ transport, calculated over initial 100 s from maximal signal.(∗∗∗p<0.0001 for SCR with and without Zn2+, t test).
(B) BCECF fluorescent signal traces from SHSY-5Y cells treated with the SCR constructs, or treated with the NKCC inhibitor bumetanide (5 μM) or transfected with siKCC2 and treated with bumetanide, which was added to attenuate compensatory NKCC-dependent baseline transport. Insert shows mRNA expression level of KCC2 in cells transfected with SCR or siKCC2. Right panel shows averaged rates of NH4+ transport. (∗∗p = 0.0024 for SCR, ∗∗p = 0.0067 for bumetanide, with and without Zn2+, t test).
(C) Fluorescent signal traces from SHSY-5Y cells transfected with the siSNAP23 construct or from cells treated with the IKK inhibitor BAY11-8075 (2 μM), which were treated with Zn2+ or without it. Scheme shows the mZnR/GPR39 pathway blocked by the above conditions. Note that SCR siRNA-transfected cells shows Zn2+-dependent upregulation of transport, marked by the dashed lines, taken from (A). Insert shows mRNA expression of SNAP23 in cells treated with the siSNAP23 or scrambled sequences. Right panel shows averaged rates of NH4+ transport. (non-significant differences between control and Zn2+ treatment, t test).
(D) SHSY-5Y cells were transfected with an siRNA sequence targeted against human SNAP23, as in (C). Note the schemes that describe the experimental conditions. Cells deficient of the human SNAP23 were then transfected with either WT-mouse SNAP23 (left panels) or with a phosphorylation-insensitive S95/120A mouse SNAP23 mutant (middle panels). Subsequently, NH4+ transport was monitored in cells loaded with BCECF. Representative fluorescence signals in cells treated with Zn2+ compared to controls are shown. Right panel shows averaged rates of NH4+ transport. (∗∗p = 0.027 between Zn2+-treated and control WT SNAP23-expressing cells, t test). All bar graphs values are averages ± SD, with all data points presented.
Next, we silenced SNAP23 in SHSY-5Y cells (mRNA expression shown in inset, Figure 3C) and monitored rates of neuronal KCC2-dependent transport activity. Remarkably, Zn2+ failed to enhance the rate of NH4+ transport in SHSY-5Y cells transfected with siSNAP23 (Figure 3C), while expression of a scrambled siRNA construct showed Zn2+-dependent increase in transport activity (Figure 3A). This suggests that SNAP23 is required for mZnR/GPR39-dependent modulation of endogenous KCC2 activity. Similar siSNAP23 silencing in HEK293 cells expressing mZnR/GPR39 and the epithelial KCC3, showed reversal of Zn2+-dependent upregulation of NH4+ transport (Figure S2). This finding suggests a general role for SNAP23 in mZnR/GPR39-dependent regulation of KCC transporters. In mast cells, phosphorylation of SNAP23 by IκB kinase (IKK) induces granule membrane fusion and exocytosis (Naskar and Puri, 2017; Suzuki and Verma, 2008). We therefore investigated whether Zn2+-dependent upregulation of NH4+ transport in SHSY-5Y cells is modulated by inhibition of IKK. Pretreatment of SHSY-5Y cells with the IKK inhibitor BAY11-7085 (2 μM) abolished mZnR/GPR39 enhancement of NH4+ transport (Figure 3C), suggesting that IKK phosphorylation of SNAP23 may be a critical component of KCC2 regulation by mZnR/GPR39. As IKK phosphorylates serine 95 and serine 120 of SNAP23 to regulate its activity (Hepp et al., 2005), we next tested if a phosphorylation-insensitive mutant of the SNARE protein, in which both serine 95 and serine 120 had been replaced by alanine (S95/120A), would diminish mZnR/GPR39 activation of KCC2. Because SHSY-5Y cells express an endogenous SNAP23, we used cells in which the endogenous human SNAP23 isoform was silenced using our siRNA construct (Figure 3C). We then transfected the cells with a mouse wild-type SNAP23 (WT-mSNAP23) or a mouse S95/120A mutant SNAP23, which are insensitive to the human isoform-directed siRNA. In SHSY-5Y cells silenced for the human SNAP23, but expressing the mouse SNAP23 isoform (WT-mSNAP23), we observed Zn2+-dependent upregulation of acidification rates (Figure 3D), suggesting that the mouse isoform of SNAP23 can rescue mZnR/GPR39 upregulation of KCC2. In contrast, the phosphorylation-insensitive mouse SNAP23 mutant (S95/120A) failed to rescue upregulation KCC2-dependent transport by Zn2+ (Figure 3D). These results indicate that in neuronal SHSY-5Y cells, Zn2+-dependent KCC2 upregulation requires IKK-dependent signaling and phosphorylation of SNAP23 at Ser95 and Ser120.
mZnR/GPR39 enhances SNAP23 binding to KCC2 to enhance its surface expression in hippocampal neurons
To assess the role of SNAP23 in the modulation of KCC2 in a more relevant model, mouse hippocampal neuronal cultures were used. In agreement with our previous results (Chorin et al., 2011; Saadi et al., 2012), Zn2+ enhanced NH4+ transport, monitored by cellular acidification of BCECF-loaded neurons (Figure 4A). In BAY11-7085 (5 μM)-treated neurons, however, Zn2+ did not enhance rates of cellular acidification (Figure 4A), suggesting that the pathway under study is also involved in mZnR/GPR39 regulation of KCC2 in this preparation. To specifically determine the role of SNAP23, hippocampal neurons were incubated with a cell-permeable blocking peptide that mimics the N-terminal domain of SNAP23 that is critical for activation of this SNARE protein (Cheng et al., 2013). We found significant Zn2+-dependent upregulation of KCC2 activity in cells incubated with a scrambled peptide, used as control (Figure 4B). In contrast, the SNAP23 blocking peptide completely abolished upregulation of KCC2 activity by Zn2+ (Figure 4B). Altogether, these results support a role for IKK and SNAP23 in mZnR/GPR39-mediated regulation of KCC2 in primary neurons.
Figure 4.

ZnR/GPR39 triggers SNAP23 interaction with KCC2 to enhance KCC2 membrane expression and activity in primary hippocampal neurons
(A)Mouse hippocampal neuronal cultures (DIV 15) loaded with the pH-sensitive fluorescent indicator BCECF (1 μM) were treated with Zn2+ (100 μM, 15 s) and rates of NH4+ transport (following application of 5 mM NH4Cl) were compared to control. To study the role of IKK phosphorylation of SNAP23, neurons were pretreated with the IKK inhibitor BAY11-7085 (5 μM), and rates of Zn2+-dependent NH4+ transport were determined. Right panel shows averaged rates of NH4+ transport, calculated over initial 100 s from maximal signal. (Box and Whisker plots, line indicates median, each dot represents the rate averaged over at least 10 neurons from an independent slide, ∗∗p<0.05 Independent-Samples Kruskal-Wallis Test).
(B) BCECF fluorescent signal traces from hippocampal neurons pretreated with a cell-permeable SNAP23-binding peptide or with a scrambled (SCR) control (3 μM). Cells were treated with Zn2+ or without, as in (A). Right panel shows averaged rates of NH4+ transport. (Box and Whisker plots, line indicates median, each dot represents the rate averaged over at least 10 neurons from an independent slide, ∗∗p<0.05 Independent-Samples Kruskal-Wallis Test).
(C) Representative confocal fluorescence microscope images (60x) following PLA between KCC2 and SNAP23, in mouse hippocampal neuronal cultures that were treated with Zn2+ (100 μM, 30 s) or control. Red puncta are PLA immunolabeling, DAPI fluorescence (blue) marks nuclei. (Scale bar is 50 μm). Quantification of PLA puncta is shown on right panel (dots are quantification of single images from three independent experiments, ∗∗p<0.05, t test).
(D) Hippocampal slices were treated with Zn2+ (200 μM, 1 min) or without it. Membrane fractions were immunoprecipitated with an antibody against SNAP23. Lysates were then separated on SDS-PAGE and subjected to immunoblotting with antibodies against KCC2, SNAP23, or Syntaxyin 1A to determine if these proteins were co-immunoprecipitated.
(E) Hippocampal tissue slices were treated with Zn2+ (200 μM, 1 min) or controls. Representative immunoblots of membrane-enriched fractions and total lysates that were exposed to antibodies against KCC2, SNAP23, or Syntaxin 1A. Right panel shows boxplot quantification (line at mean) of membrane expression of KCC2, SNAP23, or Syntaxin 1A following Zn2+ treatment, normalized to total protein in the same fractions, dashed line notes the membrane expression level of the proteins in non-treated slices. (n = 3 independent experiments, # <0.05 Independent-Samples Kruskal-Wallis test compared to non-treated).Data are presented as box and whisker (min. to max.) plots with median marked by the line, or in bar graphs as mean ± SD, all data points are presented.
We then asked if Zn2+ treatment could promote interaction between SNAP23 and KCC2 in neurons. We first studied the interaction between KCC2 and SNAP23 using a proximity ligation assay (PLA) (Krall et al., 2020; Söderberg et al., 2006), which allows detection of fluorescent puncta when target proteins are at least within 40 nm from each other. We found that in hippocampal neurons in culture Zn2+ treatment, which activated mZnR/GPR39 (Chorin et al., 2011), led to a significant increase in fluorescent PLA puncta (Figure 4C). The Zn2+-triggered enhanced proximity likely resulted from increased interaction between KCC2 and SNAP23. The interaction between these two proteins was further tested by immunoblot analysis of membrane protein fractions from hippocampal tissue slices immunoprecipitated with SNAP23 antibodies. Indeed, KCC2 immunoreactivity was observed in control and Zn2+-pretreated cells (Figure 4D), indicating that SNAP23 and KCC2 are found in a protein complex. We also found that syntaxin 1A, previously shown to interact with SNAP23 (Cormet-Boyaka et al., 2002; Saxena et al., 2006), was present in the SNAP23-immunoprecipitated fraction that contained KCC2 (Figure 4D), suggesting all three of these proteins are found within the complex. While co-IP cannot provide an accurate quantitative measure of expression levels, due to differences in antibody pull-down efficiency, we did note that in both control and Zn2+-treated tissue, expression of KCC2 and syntaxin 1A is seen in the SNAP23-immunoprecipitated fractions (Figure 4D). To determine if Zn2+-pretreatment enhances membrane insertion of SNAP23 and syntaxin 1A, as we have shown previously for KCC2 (Chorin et al., 2011), we studied the expression level of these proteins in membrane-enriched fractions. Immunoblot analysis of KCC2, SNAP23, and syntaxin 1A in non-immunoprecipitated membrane-enriched fractions from hippocampal tissue slices indicated that expression of all three proteins was indeed increased in Zn2+-treated tissues compared to control (Figure 4E). Thus, our results indicate that in neurons, Zn2+ treatment enhances the interaction between SNAP23, syntaxin 1A, and KCC2, as well as their expression in membrane-enriched fractions.
Discussion
Mechanisms for modulation of KCC2 transport activity, directly or via its trafficking to the cell membrane, have been the focus of numerous studies due to the major role of this transporter in influencing neuronal excitability (Mahadevan and Woodin, 2016). Phosphorylation of KCC2 is considered a fundamental feature of the function of this transporter, required for rapid regulation of its membrane insertion and stability (Côme et al., 2019a). In the current study, we identify a previously unrecognized molecular component that regulates KCC2 membrane insertion via a transporter phosphorylation-independent pathway, namely SNAP23. We show that the interaction of KCC2 with the SNARE proteins SNAP23 and syntaxin 1A is essential for the mZnR/GPR39-dependent upregulation of KCC2 activity.
Modulation of neuronal inhibitory tone by the Gq-coupled mZnR/GPR39 is mediated via increased membrane expression of KCC2 (Chorin et al., 2011; Gilad et al., 2015). Signaling activated by mZnR/GPR39 involves release of Ca2+ from intracellular stores, similar to the cellular signaling cascade activated by the metabotropic glutamate receptor (mGluR1) (Banke and Gegelashvili, 2008). Previous studies demonstrated that metabotropic Gq-coupled receptors, among them mGLUR1, mGLUR5, or 5-hydroxytryptamine type 2A serotonin receptors (5-HT2Rs), regulate KCC2 activity via PKC-dependent phosphorylation of serine 940 (S940) (Banke and Gegelashvili, 2008; Bos et al., 2013; Garand et al., 2019; Lee et al., 2007; Mahadevan and Woodin, 2016). Therefore, it seemed likely that mZnR/GPR39 would activate a similar pathway to regulate KCC2 activity (Mahadevan and Woodin, 2016). Surprisingly, mZnR/GPR39-dependent upregulation of transport activity was maintained in the presence of the phosphorylation-insensitive S940A KCC2 mutant or PKC inhibitors. Moreover, other major phosphorylation sites on KCC2 were similarly not involved in mZnR/GPR39 regulation of KCC2 transport. In contrast, we find that mZnR/GPR39 activation of IKK and SNAP23 are required for increased transport activity. Thus, despite shared Ca2+ signaling by these Gq-coupled receptors, mZnR/GPR39 and the mGluRs activate distinct, possibly context-dependent signaling pathways that can regulate KCC2 activity via distinct phosphorylation-independent and dependent components. It thus would be interesting to determine if these pathways can synergistically enhance KCC2 activity, especially since Zn2+ and glutamate co-exist in synaptic vesicles in a large number of excitatory synapses (Krall et al., 2021; McAllister and Dyck, 2017).
In a previous study, we demonstrated that mZnR/GPR39 upregulation of KCC2 activity in neurons is prevented by a botulinum toxin C1-expressing plasmid (Saadi et al., 2012). This toxin cleaves SNARE proteins SNAP25 and syntaxin 1A, but not SNAP23 (Gu et al., 2016; Pantano and Montecucco, 2014; Saadi et al., 2012). Our present results show that KCC2 interacts with SNAP23 and syntaxin 1A in a protein complex, suggesting that our previous observations can be explained by the cleavage for syntaxin 1A, preventing from complexing with SNAP23 and KCC2. In neurons, expression of SNAP25 might be more prominent in presynaptic membranes, where it regulates synaptic vesicle trafficking and membrane fusion to induce neurotransmitter release (Rizo and Südhof, 2012; Tao-Cheng et al., 2000; Zamponi, 2003). In contrast, SNAP23 is found in postsynaptic dendritic spines where it regulates surface expression of NMDA (Suh et al., 2010; Washbourne et al., 2004) and GABAA receptors (Gu et al., 2016), via its interaction with receptor-containing vesicles. Interestingly, SNAP23 was also shown to directly interact with mGluR1 and regulate its membrane expression (Raynaud et al., 2018). Our results point to another role for SNAP23, namely, regulation of KCC2 surface expression following activation of mZnR/GPR39, likely by recruiting Syntaxin 1A. Moreover, our results also show that mZnR/GPR39 signaling activates SNAP23-dependent upregulation of transport mediated by the epithelial KCC3 isoform. Thus, we suggest that SNAP23 interaction with KCC transporters may be a ubiquitous pathway for regulation of this family of transporters.
Various signaling pathways induce SNAP23-dependent vesicle release, mediated by the phosphorylation of the SNARE protein at distinct residues. For example, LPS-dependent cytokines release from mast cells is triggered by Toll-like TLR4 receptor via intracellular Ca2+ rise and IκB kinase (IKK) activation, which, in turn, induces SNAP23 phosphorylation at serines 120 and 95 (Suzuki and Verma, 2008). Astroglia signaling via a Ca2+-dependent pathway induced a similar pattern of SNAP23 phosphorylation important for exocytosis (Yasuda et al., 2011). Our results show that mZnR/GPR39 signaling, via IKK pathway and phosphorylation of serine 120 and serine 95 of SNAP23, enhances KCC2 membrane expression. In contrast, the histamine H1 receptor in HeLa cells regulates exosome secretion via release of Ca2+ and activation of PKC, leading to phosphorylation of SNAP23 serine 110 (Verweij et al., 2018). Exosome release from cancer cells is triggered by pyruvate kinase type M2 (PKM2) phosphorylation of serine 95 on SNAP23, enhancing metastasis and invasion of the cancer cells through the extracellular matrix (Wei et al., 2017). Hence, distinct upstream events may differentially regulate SNAP23 phosphorylation on sites that induce unique protein–protein interactions and control exocytosis of vesicles or specific proteins.
Previous studies suggested that KCC2 membrane insertion is determined by the N-terminal domain under baseline expression conditions and that mutants lacking the C-terminus are more rapidly internalized (Friedel et al., 2017). While the surface expression of the N-terminal truncated protein was previously shown to be ambiguous in HEK-293 cells (Friedel et al., 2017; Li et al., 2007), our results show that mZnR/GPR39 regulation of KCC2 is maintained in the absence of the N-terminus. Although baseline activity of the ΔNTD-KCC2 mutant was low, activation of mZnR/GPR39 increased its membrane localization and facilitated an increase in transport activity. In contrast, we show that the C-terminal is necessary for SNAP23-mediated membrane insertion. Thus, in addition to the known phosphorylation sites that regulate KCC2 membrane stability (Friedel et al., 2017), the C-terminus of KCC2 likely also has a binding site for SNAP23 that controls its membrane insertion. Interestingly, previous results have revealed an interaction of the KCC2 C-terminus with the actin cytoskeleton, leading to enhanced neuronal differentiation and migration (Horn et al., 2010).
Taken together, our results indicate that SNAP23 interacts with KCC2 to enhance its surface expression, a pathway critical for modulating inhibitory postsynaptic responses in hippocampal neurons.
Limitations of the study
We found that the C-terminal domain of KCC2 is essential for membrane insertion of the transporter by SNAP23. Future studies should identify the precise interaction domains on KCC2 (Krall et al., 2020; Raynaud et al., 2018), which will enable selective targeting of this interaction to control neuronal inhibitory tone. In addition, previous studies found that KCC2 regulation is mediated by phosphorylation, which can be triggered by Gq-coupled receptors signaling. It would be interesting to determine the role of SNAP23 activation by other GPCRs.
STAR★Methods
Key resources table
| REAGENT | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti SNAP23 | Synaptic System | 111–213; RRID:AB_10805651 |
| Anti SNAP23 | Synaptic System | 111–202; RRID:AB_887788 |
| Anti KCC2 (D1R2R) | Cell Signaling Technology | 94725; RRID:AB_2800232 |
| Anti-KCC2 [N1/12] | Abcam | Cat# ab134300 |
| Anti Syntaxin 1A | Synaptic Systems | 110–111; RRID:AB_887848 |
| Anti FLAG | Genscript | A 00187; RRID:AB_1720813 |
| Anti Transferrin receptor | Thermofisher scientific | 13–6800; RRID:AB_2533029 |
| β-Actin | MP Biomedicals | Cat# 0869100 |
| β-Tubulin | Santa Cruz Biotechnology | sc5274; RRID:AB_2288090 |
| Chemicals, Peptides and recombinant proteins | ||
| Dulbecco's Modified Eagle's Medium (DMEM) | Biological Industries | 01-055-1A |
| Fetal Calf Serum | Biological Industries | 04-001-1A |
| pen-strep | Biological Industries | 03-031-1B |
| L-Glutamine | Biological Industries | 03-020-1B |
| Zinc sulfate heptahydrate | Sigma- Aldrich | z4750 |
| BCECF-AM | Tef Labs | Not available |
| BSA | MP Biomedicals | 216006980 |
| TransIT-X2 | mirusbio | MIR 6000 |
| DIOA 2-[(2-butyl-6,7-dichloro-2-cyclopentyl-1-oxo-3H-inden-5-yl)oxy]acetic acid |
Sigma-Aldrich | D129 |
| Bumetanide 3-(butylamino)-4-phenoxy-5-sulfamoylbenzoic acid |
Sigma-Aldrich | B3023 |
| Staurosporine | Santa Cruz Biotechnology | sc3510A |
| Go6983 3-[1-[3-(dimethylamino)propyl]-5-methoxyindol-3-yl]-4-(1H-indol-3-yl)pyrrole-2,5-dione |
Sigma-Aldrich | G1918 |
| BAY11-7085 (E)-3-(4-tert-butylphenyl)sulfonylprop-2-enenitrile | APExBio | B3033 |
| VU0463271 N-cyclopropyl-N-(4-methyl-1,3-thiazol-2-yl)-2-(6-phenylpyridazin-3-yl)sulfanylacetamide |
Tocris | 4719 |
| Iodoacetamide 2-iodoacetamide |
Sigma-Aldrich | I6125 |
| Protease inhibitor cocktail | A2S | K1007 |
| Papain 2-[[2-[[2-[(2-aminoacetyl)amino]acetyl]amino] -3-(4-hydroxyphenyl)propanoyl]amino]-5-(diaminomethylideneamino)pentanoic acid |
Worthington | LS033126 |
| Hanks' balanced salts solution | Biological Industries | 02-015-1B |
| HEPES 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid |
Biological Industries | 03-025-1C |
| poly-D-lysine | Sigma-Aldrich | p0899 |
| Neurobasal medium | GIBCO | 21103049 |
| Fetal Bovine Serum | HyClone | sh30070.03 |
| B-27 | GIBCO | 17504044 |
| Glutamax-100X | GIBCO | 35050038 |
| Gentamicin | Biological Industries | 03-035-1C |
| Veriblot IP detection reagent | Abcam | ab131366; RRID:AB_2892718 |
| protein A magbeads | GenScript | L00273 |
| qPCR bio probe blue mix | PCR biosystem | pb20.25-05 |
| Cytosine β-D-arabinofuranoside (AraC) | Sigma- Aldrich | c6645 |
| SNAP23 blocking peptide | GRKKRRQRRRPQMDDLSPEEIQLRAHQVTDSEQ | GenScript USA Inc |
| SCR blocking peptide | GRKKRRQRRRPQGFAESLFQSIEKESGFSCG | GenScript USA Inc |
| Critical commercial assays | ||
| Bio-Rad protein assay | Bio-Rad | 5000006 |
| Q5 Site-Directed Mutagenesis Kit | New England Biolabs | E0554S |
| EZ-Link Sulfo-NHS-Biotinylation Kit | Pierce Biotechnology, Thermoscientific | PIR-89881 |
| Duolink in situ red starter kit | SigmaAldrich | DU092101 |
| PureLink™ RNA Mini Kit | Invitrogene | 12183018a |
| cDNA synthesis kit | Quanta bio | 95047 |
| Experimental models: Cell lines | ||
| HEK293 | ATCC | CRL-1573 |
| SHSY-5Y | ATCC | CRL-2266 |
| Experimental models: Organisms/strains | ||
| C57BL/6JRccHsd mice | Envigo | 043 |
| Oligonucleotides | ||
| siGPR39 | 5′-CCAUGGAGUUCUACAGCAU-3′ | Sigma- Aldrich |
| siSNAP23 | 5′-CCAACAGAGAUCGUAUUGA-3′ | Integrated DNA Technologies, IDT |
| siKCC2 | 5′-CAAACAGAUGCAUUUAACCAAGAAT-3′ | Integrated DNA Technologies, IDT |
| SCR | 5'-GCCCAGAUCCCUGUACGU-3′ | Sigma- Aldrich |
| KCC2 S940A | 5′-GTCACGAGGCGCAATCCGGAG-3′ | 5′-TCATCTGTGATACTCTGGATCTC-3′ |
| KCC2 S988A | 5′-CAGCTCCCCGGCCCCAGGGGA-3′ | 5′-CTGGGGCAGCTGGGAGCACTC-3′ |
| KCC2 S986A | 5′-CCCCAGCAGCGCCCCGTCCCC-3′ | 5′-CAGCTGGGAGCACTCTGATCGTGG-3′ |
| KCC2 T584A | 5′-GCTGCTGAGGGCACCCAACTG-3′ | 5′-GTCTGCACTGCACAGGCC-3′ |
| KCC2 T906E | 5′-CTATGAGAAGGAGTTGGTGATGGAG-3′ | 5′-GTGTAAGCTGAGATGTCG-3′ |
| KCC2 T1007E | 5′-GGTGCATCTCGAGTGGACCAAGGACAAG-3′ | 5′-TTCTCCGGATCTGTCTCC-3′ |
| KCC2 S868A | 5′-GGATGACAATGCCATCCAGATGAAGAAG-3′ | 5′-ATCTGGGCCACAGTGAAG-3′ |
| SPVSS | 5′-GAGGGCATCAAGGACTTC-3′ | 5′-GGGGCCCTTATTCTTCTC-3′ |
| ΔNCD-KCC2 | 5′-ATGGGCGTGTACCTGCCG-3′ | 5′-TTTATCATCATCATCTTTATAA TCCATAGCCGAATTC-3′ |
| ΔCTD-KCC2 | 5′-TGCTCTAGAGCATCATATC CCATCGCCCCACTCC-3′ |
|
| SNAP23 S95/120A | 5′-GAACTTTGAGGCTGGAAAGAAC-3′ | 5′-TTTGTCCTATTACAAGGG-3′ |
| 5′-TAAGCAACCGGCCCGGATTACAAATG-3′ | 5′-GATACTACATTGCTAGGTG-3′ | |
| Recombinant DNA | ||
| human KCC2 cDNA | Prof. Gamba | |
| mouse SNAP23 | OriGene | MR202271 |
| Human GPR39 | OriGene- with mcherry addition and deletion of FLAG-MYC | www.origene.com/catalog/cdna-clones/expression-plasmids/rc219212/gpr39-nm_001508-human-tagged-orf-clone |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact: Michal Hershfinkel (hmichal@bgu.ac.il)
Materials availability
Reagents and plasmids generated in this study are available from the lead contact upon request.
Experimental model and subjectdetails
Cell cultures
HEK293 and SHSY-5Y cell lines, purchased from ATCC and not authenticated. Cells were grown in Dulbecco's Modified Eagle's Medium (DMEM, Biological Industries, Israel) supplemented with 10% (v/v) fetal calf serum (Biological Industries, Israel), 1% pen-strep, 1% L-Glutamine solution. All cells were grown in 5% CO2 humidified atmosphere at 37°C.
Primary mouse neuronal cultures
Primary hippocampal neurons were prepared from postnatal day 0 C57BL/6JRccHsd mouse neonates of both sex, using a protocol approved by the Institutional Animal Care and Use Committee of the Ben-Gurion University of the Negev. Hippocampi were treated with papain (Worthington, Lakewood, NJ) and dissociated by mechanic pipetting in Hanks' balanced salts solution (HBSS) supplied with 20mM HEPES (pH 7.4). Cells were seeded on 50 μg poly-D-lysin-coated coverslips (13 mm) in 24-well plates at the density of 5 × 105 cells/mL (coverslip). During initial 24h cells were cultured in neurobasal medium (GIBCO), supplemented with 5% fetal bovine serum (FBS), 2% B-27, 1% Glutamax-100X, and 1 μg/mL Gentamicin, after which the medium was changed to serum-free medium. To suppress glial growth, 5 μM Cytosine β-D-arabinofuranoside (AraC) was added to the medium on DIV5. All cells were cultured in a humidified atmosphere of 5% CO2 at 37°C.
Mouse tissues for co-IP
C57BL/6JRccHsd male mice of both sex (18 days) were sacrificed, hippocampal slices were sectioned (Chorin et al., 2011), and incubated at 37°C in O2 bubbled ACSF buffer (124 mM NaCl, 26 mM NaHCO3, 1.25 mM NaH2PO4, 2 mM MgSO4, 2 mM CaCl2, 3 mM KCl, and 10 mM glucose, at pH 7.4), for 30 min.
Method details
Cell culture preparation and transfection
For imaging experiments, cells were seeded at 1 × 106 cells/mL on 10 mm glass coverslips, and used after 24–48 h. For siGPR39, siKCC2, siSNAP23 silencing, HEK293 or SHSY-5Y cells were seeded in 60 mm plates, 24 hprior to transfection. Cells were transfected with siRNA constructs (Sigma- Aldrich, Israel, and Integrated DNA Technologies, IDT, IOWA, USA) using the CaPO4 precipitation protocol or Mirus transit-X2, according to the manufacturer’s protocol. Sequences of siRNA used for GPR39 silencing were: 5′-CCAUGGAGUUCUACAGCAU-3′, for SNAP23: 5′-CCAACAGAGAUCGUAUUGA-3′, for KCC2 5′-CAAACAGAUGCAUUUAACCAAGAAT-3′ and the control siRNA (scrambled) sequence: 5′-GCCCAGAUCCCUGUACGU-3′. At 48 h after transfection, cells were used for imaging experiments or immunoblots.
Inhibitors
DIOA, #D129 SigmaAldrich.Bumetanide, #B3023 Sigma Aldrich.Staurosporine, #sc3510A Santa Cruz Biotechnology.Go6983, #G1918 SigmaAldrich.BAY11-7085, #B3033 APExBio.VU0463271, #4719 Tocris. All inhibitors were applied in Ringer’s solution at concentrations and incubation times shown in the relevant figure legend.
Molecular tools
The human KCC2 cDNA coding plasmid was a kind gift from Prof. Gamba (Song et al., 2002). The mouse SNAP23 plasmid was purchased from OriGene (OriGene Technologies Inc. MR202271). All site-directed mutagenesis was performed using Q5 Site-Directed Mutagenesis Kit, according to the manufacturer’s protocols (New England Biolabs). Primers used for generation of mutations were as follows: KCC2 point mutations: Ser940 to Ala (S940A; 5′-GTCACGAGGCGCAATCCG GAG-3′, 5′-TCATCTGTGATA CTCTGGATCTC-3′); Ser988 to Ala (S988A; 5′-CAGCTCCCCGGCCCCAGGGGA-3′, 5′-CTGGGGCAGCTGGGAGCACTC-3′); Ser986 to Ala (S986A; 5′-CCCCAGCAGCGCCCCGTCCCC-3′, 5′-CAGCTGGGAGCACTCTGATCGTGG-3′); Thr584 to Ala (T584A; 5′-GCTGCTGAGGGCACCCAACTG-3′, 5′-GTCTGCACTGCACAGGCC-3′); Thr906 to Glu (T906E; 5′-CTATGAGAAGGAGTTGGTGATGGAG-3′, 5′-GTGTAAGCTGAGATGTCG-3′); Thr1007 to Glu (T1007E; 5′-GGTGCATCTCGAGTGGACCAAGGACAAG-3′, 5′-TTCTCCGGATCTGTCTCC-3′); Ser868 to Ala (S868A; 5′-GGATGACAATGCCATCCAGATGAAGAAG-3′, 5′-ATCTGGGCCACAGTGAAG-3′), SPVSS deletion (SPVSS; 5′-GAGGGCATCAAGGACTTC-3′; 5′-GGGGCCCTTATTCTTCTC-3′).Deletion of 1–100 amino acids ΔNCD-KCC2; 5′-ATGGGCGTGTACCTGCCG-3′, 5′-TTTATCATCATCATCTTTATAATCCATAGCCGAATTC-3′ (Friedel et al., 2017). Deletion of 654–1114 amino acids was created using PCR and insertion of stop codon and XbaI restriction enzyme site at the C-terminal domain (ΔCTD-KCC2; 5′-TGCTCTAGAGCATCATATCCCATCGCCCCACTCC-3′) (Friedel et al., 2017). All KCC2 plasmids contained FLAG at the N-terminal of the protein. For SNAP23 point mutation: Ser95 to Ala and Ser120 to Ala (S95/120A; 5′-GAACTTTGAGGCTGGAAAGAAC-3′, 5′-TTTGTCCTATTACAAGGG-3′, 5′-TAAGCAACCGGCCCGGATTACAAATG-3′, 5′-GATACTACATTGCTAGGTG-3′). All mutations were verified using sequence analysis.
qPCR assay
Silencing efficiency of SNAP, GPR39 or KCC2 in SHSY-5Y cells was quantified using qPCR. Cells were seeded on 60 mm plates, 24 hprior to transfection. Cells were transfected with siRNA constructs, and after 48 h the total RNA was purified using RNeasy Mini Kit (Invitrogen) as described by the manufacturer. 1 μg RNA was converted to cDNA using cDNA synthesis kit as described by the manufacturer (Quanta bio). cDNA was diluted 1:10 (as per calibration) with ultrapure water and subjected to PCR cycles (BIOER). The following sequences were used for the primers and probes (IDT): KCC2: forward primer- TGCATCTGTTTGAGGATCTGG, reverse primer- CTG TATCATTTACGCATCACTGC probe CTGCTCCATCACCAACGTCTTC TCA. GPR39: forward primer- CTCGTCCAG TCGTGCTTG, reverse primer- CAGGAGGCAGACCATCATC. probe TCGGAGGATCAT GGCTGCGG. SNAP23: forward primer- GCCACAGCATTTGTTGAGTTC reverse primer- GCAGGAATC AAGACCATCACT probe ACCGCATAGAAGAAGGCTTGGACC primer, Actin: forward primer ACAGAGCCTCGCCTTTG, reverse primer CCTTGCACATGC CGGAG, probe TCATCCATGGTGAGCTGGCGG. qPCR assay was performed using qPCR bio probe blue mix (pb20.25–05, PCR biosystem).
Fluorescent imaging
For measurement of KCC-dependent transport, we used the NH4Cl paradigm (Chorin et al., 2011) to cells loaded with a pH-sensitive dye, 2',7′-Bis-(2-Carboxyethyl)-5-(and-6)-Carboxyfluorescein (BCECF). Cells grown on coverslips were loaded for 15minat room temperature with 1 μM BCECF-AM (Tef Labs) in Ringer’s solution containing 0.1% BSA and were subsequently incubated in Ringer’s solution containing 0.1% BSA for additional 15minat room temperature. Inhibitors were added in some experiments to the incubating solution, as described in figure legends. For SNAP23 inhibition, a cell permeant blocking peptide for the N-terminus (SNAP23: GRKKRRQRRRPQMDDLSPEEIQLRA HQVTDSEQ or SCR: GRKKRRQRRRPQGFAESLFQSIEKESGFSCG) was designed to include a trans-activator of transcription (TAT) cell-penetrating peptide (YGRKKRRQRRR) (Cheng et al., 2013; Krall et al., 2020). Primary cultured neurons were incubated with the peptide, or the scrambled control, for 6 h prior to BCECF loading for imaging. Treatment with extracellular Zn2+ for mZnR/GPR39 activation was done by addition of 200 μM Zn2+ for 2 minprior to imaging. Coverslips were then mounted in a perfusion chamber and BCECF was excited at 440 and 470 nm and imaged with a 510-nm long-pass filter. Briefly, the NH4Cl paradigm consists of perfusion of the cells with K+-free Ringer's solution containing NH4Cl (15 mM, HEK293 or 5 mM, SHSY-5Y and Hippocampal neurons primary culture). This triggers initial alkalization that is induced by NH3 diffusion, resulting in an increase in fluorescence signal. Subsequently, activation of ion transport systems induces transport of NH4+, which is a surrogate to K+, and results in cellular acidification that is representing NH4+ efflux rate. The rate of acidification was determined using a linear fit obtained during a 100s period following the peak fluorescent signal.
The imaging system consisted of an Axiovert 100 inverted microscope using a 20 objective (Zeiss, Germany), Polychrome V monochromator (TILL Photonics, Germany) and a SensiCam cooled charge-coupled device (PCO, Germany). Fluorescent imaging measurements were acquired with Imaging Workbench 5 (Indec, USA).
Protein expression and co-Immunoprecipitation
To monitor changes in KCC2 membrane expression HEK293 cells were seeded in 10 cm plates and co-transfected with 1 μg of the GPR39-mcherry and 8 μg of the either the WT-KCC2, ΔCTD-KCC2 or ΔNTD-KCC2 plasmids using the TransIT-X2 Transfection Reagent (Mirus). Cell surface biotinylation assay was performed using the EZ-Link Sulfo-NHS-Biotinylation Kit (Pierce Biotechnology, Thermoscientific). Cells were treated with 200 μM Zn2+ for 2 min then incubated with sulfo-NHS-Biotin for 30minat 4°C, cells were then lysed (50 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 10% glycerol, 1% Triton X-, 100 mM MgCl2, 10 mM iodoacetamide and protease inhibitor cocktail, A2S) 250 μg lysates were then incubated with Neutravidin Gel (Pierce) overnight at 4°C. Sample were resolved on 8–15% gradient SDS-PAGE followed by immunoblot analysis.
Membrane expression and interaction of KCC2, SNAP23 and Syntaxin-1A were studied using membrane fractions of hippocampal brain section. C57BL/6JRccHsd male mice (18 days) were sacrificed, hippocampal slices were sectioned as described (Chorin et al., 2011), and incubated at 37°C in O2 bubbled ACSF buffer (124 mM NaCl, 26 mM NaHCO3, 1.25 mM NaH2PO4, 2 mM MgSO4, 2 mM CaCl2, 3 mM KCl, and 10 mM glucose, at pH 7.4), for 30 min. Slices were then washed with phosphate-free ACSF, followed by 200 μM Zn2+ treatment for 1 min. and hippocampi were isolated and lysed in fractionation buffer (20 mM HEPES, pH 7.4, 10 mM KCl, 2 mM MgCl2, 1 mM EDTA, 1 mM EGTA with protease inhibitors), lysates were centrifuged at 720 g for 5 min. Supernatant, containing total cell lysates, was used to measure the total cell protein. Protein concentrations were determined using Bio-Rad protein assay only for the total cell protein lysate. The supernatant was further ultracentrifuged at 140,000 g for 1 h, the pellet that contains membrane proteins was resuspended in fractionation buffer and used to evaluate membrane protein levels with immunoblots. Briefly, SDS sample buffer was added, and samples were heated to 40°C for 5 min. Proteins were separated on gradient gel 8–15% SDS-PAGE and blotted onto nitrocellulose membranes.
In addition, the membrane-enriched fractions of the hippocampal slices, described above, wasfurther used for co-immunoprecipitation. Membrane fractions were incubated overnight at 4°C with an antibody against the SNAP23 (111–213, Synaptic System), then subjected to protein A magbeads (L00273, Genescript) for 1hat room temperature. Beads were washed and proteins were isolated from the beads with sample buffer containing beta-mercapthoethanol. Proteins were separated on gradient gel 8–15% SDS-PAGE and blotted onto nitrocellulose membranes.
Proteins were detected using antibodies raised against KCC2 (94725, Cell Signaling Technology), SNAP23 (111–202, Synaptic Systems), Syntaxin 1A (110–111, Synaptic Systems) and FLAG (A00187, Genscript), transferrin receptor (13–6800, Thermofisher scientific), β-Actin (0869100, MP), β-Tubulin (sc5274, Santa Cruz Biotechnology). For quantification of KCC2 expression levels the monomeric form of the protein was quantified from immunoblots and normalized as mentioned in the figure legends. Note that KCC2 oligomers may be associated with other proteins and in ectopic expression system their levels may depend on the preparation (Medina et al., 2014). To minimized detection of light and heavy chains of the IgG we used the Veriblot IP detection reagent (ab131366, Abcam).
Proximity ligation assay
PLA assay was performed using on PFA fixed neuronal cultures that were treated with Zn2+ (100 μM, 30 s) or without it as control, and the Duolink in situ red starter kit (DU092101, SigmaAldrich) was applied as per the manufacturer protocol, using antibodies against KCC2 (ab134300, Abcam) and against SNAP23 (111–213, Synaptic Systems). Coverslips seeded with cells were treated with Zn2+ to activate mZnR/GPR39, and then PFA 4% for 10 min, rinsed in PBS (PBS), and then permeabilized with 0.1% Triton X- in PBS. Coverslips were incubated in Duolink secondary antibodies (anti-rabbit SNAP23 and anti-mouse KCC2), which were conjugated with complementary oligonucleotides and PLA probes, and fluorescently labeled. Fluorescent images were acquired with appropriate filters using Nikon Eclipse Ti confocal microscope using an X60 objective. PLA puncta were quantified by counting the fluorescent particles using ImageJ software module in each field of view. PLA experiments were performed on three independent cultures, and n refers to the number of images used for quantification.
Quantification and statistical analysis
All imaging traces were generated and fitted, using a linear curve fit, in Microsoft Excel. Rates of KCC2 transport were determined by the decrease in fluorescence signal during a 100s period following the peak fluorescent signal, all acquired rates are presented in bar graphs with mean ± SD (SD). Primary neurons imaging data are presented as boxplots, with min and max, showing median values and all data points. All imaging experiments were done on at least three independent cultures. For western blots, quantification was done using ImageJ module for blot analysis (Gels) and data from all gels are presented in bar graphs with mean ± SD or boxplots, min to max, with median value. PLA puncta were quantified by counting the fluorescent particles using ImageJ software module in each field of view. PLA experiments were performed on three independent cultures, and n refers to the number of images used for quantification.
All data are expressed as means ± SD in graphs generates by Microsoft Excel. Statistical significance between the groups was determined following Shapiro-Wilk normality test (GraphPad Prism 8.4.2), using the Student's t test or ANOVA with multiple comparisons, as appropriate (SPSS). In addition, Mann-Whitney or Independent-Samples Kruskal-Wallis Test, as appropriate, were used for analysis of non-parametric data (GraphPad Prism 7.4.2). The sample sizes for each experiment (n) is shown in all graphs and extent of significance is included in figure legends. ∗p<0.05; ∗∗, p<0.01; ∗∗∗, p<0.001.
Acknowledgments
We thank Dr. Igor Medina for critical reading of the manuscript and his advice and support throughout this project. This work was funded by a joint National Science Foundation-US-Israel Binational Science Foundation grant NSF-IOS-BSF 1655480 (MH, EA). MH is also funded by Israel Science Foundation grant 812/20.
Author contributions
H.A and M.H designed experiments, H. A., N.G., and M.B. performed experiments and analyzed data, H.A., I.S., E. A., and M.H. interpreted the data, wrote and edited the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: February 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.103751.
Supplementalinformation
Data and code availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the supplemental information. All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
References
- Banke T.G., Gegelashvili G. Tonic activation of group I mGluRs modulates inhibitory synaptic strength by regulating KCC2 activity. J. Physiol. 2008;586:4925–4934. doi: 10.1113/jphysiol.2008.157024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besser L., Chorin E., Sekler I., Silverman W.F., Atkin S., Russell J.T., Hershfinkel M. Synaptically released zinc triggers metabotropic signaling via a zinc-sensing receptor in the hippocampus. J. Neurosci. 2009;29:2890–2901. doi: 10.1523/jneurosci.5093-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos R., Sadlaoud K., Boulenguez P., Buttigieg D., Liabeuf S., Brocard C., Haase G., Bras H., Vinay L. Activation of 5-HT2A receptors upregulates the function of the neuronal K-Cl cotransporter KCC2. Proc. Natl. Acad. Sci. U S A. 2013;110:348–353. doi: 10.1073/pnas.1213680110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty M., Asraf H., Sekler I., Hershfinkel M. ZnR/GPR39 controls cell migration by orchestrating recruitment of KCC3 into protrusions, re-organization of actin and activation of MMP. Cell Calcium. 2021;94:102330. doi: 10.1016/j.ceca.2020.102330. [DOI] [PubMed] [Google Scholar]
- Cheng J., Liu W., Duffney L.J., Yan Z. SNARE proteins are essential in the potentiation of NMDA receptors by group II metabotropic glutamate receptors. J. Physiol. 2013;591:3935–3947. doi: 10.1113/jphysiol.2013.255075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chorin E., Vinograd O., Fleidervish I., Gilad D., Herrmann S., Sekler I., Aizenman E., Hershfinkel M. Upregulation of KCC2 activity by zinc-mediated neurotransmission via the mZnR/GPR39 receptor. J. Neurosci. 2011;31:12916–12926. doi: 10.1523/jneurosci.2205-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Côme E., Heubl M., Schwartz E.J., Poncer J.C., Lévi S. Reciprocal regulation of KCC2 trafficking and synaptic activity. Front Cell Neurosci. 2019;13:48. doi: 10.3389/fncel.2019.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Côme E., Marques X., Poncer J.C., Lévi S. KCC2 membrane diffusion tunes neuronal chloride homeostasis. Neuropharmacology. 2019;169:107571. doi: 10.1016/j.neuropharm.2019.03.014. [DOI] [PubMed] [Google Scholar]
- Cordshagen A., Busch W., Winklhofer M., Nothwang H.G., Hartmann A.M. Phosphoregulation of the intracellular termini of K(+)-Cl(-) cotransporter 2 (KCC2) enables flexible control of its activity. J. Biol. Chem. 2018;293:16984–16993. doi: 10.1074/jbc.RA118.004349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormet-Boyaka E., Di A., Chang S.Y., Naren A.P., Tousson A., Nelson D.J., Kirk K.L. CFTR chloride channels are regulated by a SNAP-23/syntaxin 1A complex. Proc. Natl. Acad. Sci. U S A. 2002;99:12477–12482. doi: 10.1073/pnas.192203899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedel P., Kahle K.T., Zhang J., Hertz N., Pisella L.I., Buhler E., Schaller F., Duan J., Khanna A.R., Bishop P.N., et al. WNK1-regulated inhibitory phosphorylation of the KCC2 cotransporter maintains the depolarizing action of GABA in immature neurons. Sci.Signal. 2015;8:ra65. doi: 10.1126/scisignal.aaa0354. [DOI] [PubMed] [Google Scholar]
- Friedel P., Ludwig A., Pellegrino C., Agez M., Jawhari A., Rivera C., Medina I. A novel view on the role of intracellular tails in surface delivery of the potassium-chloride cotransporter KCC2. eNeuro. 2017;4 doi: 10.1523/eneuro.0055-17.2017. ENEURO.0055-0017.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garand D., Mahadevan V., Woodin M.A. Ionotropic and metabotropic kainate receptor signalling regulates Cl(-) homeostasis and GABAergic inhibition. J. Physiol. 2019;597:1677–1690. doi: 10.1113/jp276901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad D., Shorer S., Ketzef M., Friedman A., Sekler I., Aizenman E., Hershfinkel M. Homeostatic regulation of KCC2 activity by the zinc receptor mZnR/GPR39 during seizures. Neurobiol. Dis. 2015;81:4–13. doi: 10.1016/j.nbd.2014.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y., Chiu S.L., Liu B., Wu P.H., Delannoy M., Lin D.T., Wirtz D., Huganir R.L. Differential vesicular sorting of AMPA and GABAA receptors. Proc. Natl. Acad. Sci. U S A. 2016;113:E922–E931. doi: 10.1073/pnas.1525726113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepp R., Puri N., Hohenstein A.C., Crawford G.L., Whiteheart S.W., Roche P.A. Phosphorylation of SNAP-23 regulates exocytosis from mast cells. J. Biol. Chem. 2005;280:6610–6620. doi: 10.1074/jbc.M412126200. [DOI] [PubMed] [Google Scholar]
- Hershfinkel M., Kandler K., Knoch M.E., Dagan-Rabin M., Aras M.A., Abramovitch-Dahan C., Sekler I., Aizenman E. Intracellular zinc inhibits KCC2 transporter activity. Nat. Neurosci. 2009;12:725–727. doi: 10.1038/nn.2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heubl M., Zhang J., Pressey J.C., Al Awabdh S., Renner M., Gomez-Castro F., Moutkine I., Eugène E., Russeau M., Kahle K.T., et al. GABA(A) receptor dependent synaptic inhibition rapidly tunes KCC2 activity via the Cl(-)-sensitive WNK1 kinase. Nat. Commun. 2017;8:1776. doi: 10.1038/s41467-017-01749-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn Z., Ringstedt T., Blaesse P., Kaila K., Herlenius E. Premature expression of KCC2 in embryonic mice perturbs neural development by an ion transport-independent mechanism. Eur. J. Neurosci. 2010;31:2142–2155. doi: 10.1111/j.1460-9568.2010.07258.x. [DOI] [PubMed] [Google Scholar]
- Kahle K.T., Khanna A.R., Duan J., Staley K.J., Delpire E., Poduri A. The KCC2 cotransporter and human epilepsy: getting excited about inhibition. Neuroscientist. 2016;22:555–562. doi: 10.1177/1073858416645087. [DOI] [PubMed] [Google Scholar]
- Krall R.F., Moutal A., Phillips M.B., Asraf H., Johnson J.W., Khanna R., Hershfinkel M., Aizenman E., Tzounopoulos T. Synaptic zinc inhibition of NMDA receptors depends on the association of GluN2A with the zinc transporter ZnT1. Sci. Adv. 2020;6 doi: 10.1126/sciadv.abb1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krall R.F., Tzounopoulos T., Aizenman E. The function and regulation of zinc in the brain. Neuroscience. 2021;457:235–258. doi: 10.1016/j.neuroscience.2021.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.H.C., Walker J.A., Williams J.R., Goodier R.J., Payne J.A., Moss S.J. Direct protein kinase C-dependent phosphorylation regulates the cell surface stability and activity of the potassium chloride cotransporter KCC2. J. Biol. Chem. 2007;282:29777–29784. doi: 10.1074/jbc.M705053200. [DOI] [PubMed] [Google Scholar]
- Leonzino M., Busnelli M., Antonucci F., Verderio C., Mazzanti M., Chini B. The timing of the excitatory-to-inhibitory GABA switch is regulated by the oxytocin receptor via KCC2. Cell Rep. 2016;15:96–103. doi: 10.1016/j.celrep.2016.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Khirug S., Cai C., Ludwig A., Blaesse P., Kolikova J., Afzalov R., Coleman S.K., Lauri S., Airaksinen M.S., et al. KCC2 interacts with the dendritic cytoskeleton to promote spine development. Neuron. 2007;56:1019–1033. doi: 10.1016/j.neuron.2007.10.039. [DOI] [PubMed] [Google Scholar]
- Mahadevan V., Woodin M.A. Regulation of neuronal chloride homeostasis by neuromodulators. J. Physiol. 2016;594:2593–2605. doi: 10.1113/jp271593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mapplebeck J.C.S., Lorenzo L.E., Lee K.Y., Gauthier C., Muley M.M., De Koninck Y., Prescott S.A., Salter M.W. Chloride dysregulation through downregulation of KCC2 mediates neuropathic pain in both sexes. Cell Rep. 2019;28:590–596 e594. doi: 10.1016/j.celrep.2019.06.059. [DOI] [PubMed] [Google Scholar]
- Markkanen M., Ludwig A., Khirug S., Pryazhnikov E., Soni S., Khiroug L., Delpire E., Rivera C., Airaksinen M.S., Uvarov P. Implications of the N-terminal heterogeneity for the neuronal K-Cl cotransporter KCC2 function. Brain Res. 2017;1675:87–101. doi: 10.1016/j.brainres.2017.08.034. [DOI] [PubMed] [Google Scholar]
- McAllister B.B., Dyck R.H. Zinc transporter 3 (ZnT3) and vesicular zinc in central nervous system function. Neurosci. Biobehav. Rev. 2017;80:329–350. doi: 10.1016/j.neubiorev.2017.06.006. [DOI] [PubMed] [Google Scholar]
- Medina I., Friedel P., Rivera C., Kahle K.T., Kourdougli N., Uvarov P., Pellegrino C. Current view on the functional regulation of the neuronal K(+)-Cl(-) cotransporter KCC2. Front. Cell. Neurosci. 2014;8:27. doi: 10.3389/fncel.2014.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mero M., Asraf H., Sekler I., Taylor K.M., Hershfinkel M. ZnR/GPR39 upregulation of K(+)/Cl(-)-cotransporter 3 in tamoxifen resistant breast cancer cells. Cell Calcium. 2019;81:12–20. doi: 10.1016/j.ceca.2019.05.005. [DOI] [PubMed] [Google Scholar]
- Naskar P., Puri N. Phosphorylation of SNAP-23 regulates its dynamic membrane association during mast cell exocytosis. Biol. Open. 2017;6:1257–1269. doi: 10.1242/bio.025791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantano S., Montecucco C. The blockade of the neurotransmitter release apparatus by botulinum neurotoxins. Cell. Mol. Life Sci. 2014;71:793–811. doi: 10.1007/s00018-013-1380-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynaud F., Homburger V., Seveno M., Vigy O., Moutin E., Fagni L., Perroy J. SNAP23-Kif5 complex controls mGlu1 receptor trafficking. J. Mol.Cell. Biol. 2018;10:423–436. doi: 10.1093/jmcb/mjy031. [DOI] [PubMed] [Google Scholar]
- Rizo J., Südhof T.C. The membrane fusion enigma: SNAREs, Sec1/Munc18 proteins, and their accomplices--guilty as charged? Annu. Rev.Cell Dev Biol. 2012;28:279–308. doi: 10.1146/annurev-cellbio-101011-155818. [DOI] [PubMed] [Google Scholar]
- Saadi R.A., He K., Hartnett K.A., Kandler K., Hershfinkel M., Aizenman E. SNARE-dependent upregulation of potassium chloride co-transporter 2 activity after metabotropic zinc receptor activation in rat cortical neurons in vitro. Neuroscience. 2012;210:38–46. doi: 10.1016/j.neuroscience.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena S.K., George C.M., Pinskiy V., McConnell B. Epithelial sodium channel is regulated by SNAP-23/syntaxin 1A interplay. Biochem.Biophys. Res. Commun. 2006;343:1279–1285. doi: 10.1016/j.bbrc.2006.03.093. [DOI] [PubMed] [Google Scholar]
- Söderberg O., Gullberg M., Jarvius M., Ridderstråle K., Leuchowius K.J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L.G., et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods. 2006;3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- Song L., Mercado A., Vázquez N., Xie Q., Desai R., George A.L., Jr., Gamba G., Mount D.B. Molecular, functional, and genomic characterization of human KCC2, the neuronal K-Cl cotransporter. Brain Res. Mol. Brain Res. 2002;103:91–105. doi: 10.1016/s0169-328x(02)00190-0. [DOI] [PubMed] [Google Scholar]
- Spoljaric I., Spoljaric A., Mavrovic M., Seja P., Puskarjov M., Kaila K. KCC2-Mediated Cl(-) extrusion modulates spontaneous hippocampal network events in perinatal rats and mice. Cell Rep. 2019;26:1073–1081 e1073. doi: 10.1016/j.celrep.2019.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh Y.H., Terashima A., Petralia R.S., Wenthold R.J., Isaac J.T., Roche K.W., Roche P.A. A neuronal role for SNAP-23 in postsynaptic glutamate receptor trafficking. Nat. Neurosci. 2010;13:338–343. doi: 10.1038/nn.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunuwar L., Asraf H., Donowitz M., Sekler I., Hershfinkel M. The Zn(2+)-sensing receptor, ZnR/GPR39, upregulates colonocytic Cl(-) absorption, via basolateral KCC1, and reduces fluid loss. Biochim.Biophys. Acta Mol. Basis Dis. 2017;1863:947–960. doi: 10.1016/j.bbadis.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K., Verma I.M. Phosphorylation of SNAP-23 by IkappaB kinase 2 regulates mast cell degranulation. Cell. 2008;134:485–495. doi: 10.1016/j.cell.2008.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao-Cheng J.H., Du J., McBain C.J. Snap-25 is polarized to axons and abundant along the axolemma: an immunogold study of intact neurons. J. Neurocytol. 2000;29:67–77. doi: 10.1023/a:1007168231323. [DOI] [PubMed] [Google Scholar]
- Verweij F.J., Bebelman M.P., Jimenez C.R., Garcia-Vallejo J.J., Janssen H., Neefjes J., Knol J.C., de Goeij-de Haas R., Piersma S.R., Baglio S.R., et al. Quantifying exosome secretion from single cells reveals a modulatory role for GPCR signaling. J.Cell Biol. 2018;217:1129–1142. doi: 10.1083/jcb.201703206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washbourne P., Liu X.-B., Jones E.G., McAllister A.K. Cycling of NMDA receptors during trafficking in neurons before synapse formation. J. Neurosci. 2004;24:8253–8264. doi: 10.1523/jneurosci.2555-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M., Zhang J., Mansuri M.S., Duan J., Karimy J.K., Delpire E., Alper S.L., Lifton R.P., Fukuda A., Kahle K.T. Developmentally regulated KCC2 phosphorylation is essential for dynamic GABA-mediated inhibition and survival. Sci. Signal. 2019;12:eaaw9315. doi: 10.1126/scisignal.aaw9315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M., Hartmann A.M., Beyer T., Ripperger A., Nothwang H.G. A novel regulatory locus of phosphorylation in the C terminus of the potassium chloride cotransporter KCC2 that interferes with N-ethylmaleimide or staurosporine-mediated activation. J. Biol. Chem. 2014;289:18668–18679. doi: 10.1074/jbc.M114.567834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y., Wang D., Jin F., Bian Z., Li L., Liang H., Li M., Shi L., Pan C., Zhu D., et al. Pyruvate kinase type M2 promotes tumour cell exosome release via phosphorylating synaptosome-associated protein 23. Nat. Commun. 2017;8:14041. doi: 10.1038/ncomms14041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y., Liu Z., Cao J., Ma Q., Gao X., Wang Q., Jin C., Zhou Y., Wen L., Ren J. GPS 2.1: enhanced prediction of kinase-specific phosphorylation sites with an algorithm of motif length selection. Protein Eng. Des. Sel. 2011;24:255–260. doi: 10.1093/protein/gzq094. [DOI] [PubMed] [Google Scholar]
- Yasuda K., Itakura M., Aoyagi K., Sugaya T., Nagata E., Ihara H., Takahashi M. PKC-dependent inhibition of CA2+-dependent exocytosis from astrocytes. Glia. 2011;59:143–151. doi: 10.1002/glia.21083. [DOI] [PubMed] [Google Scholar]
- Zamponi G.W. Regulation of presynaptic calcium channels by synaptic proteins. J. Pharmacol. Sci. 2003;92:79–83. doi: 10.1254/jphs.92.79. [DOI] [PubMed] [Google Scholar]
- Zhang J., Cordshagen A., Medina I., Nothwang H.G., Wisniewski J.R., Winklhofer M., Hartmann A.M. Staurosporine and NEM mainly impair WNK-SPAK/OSR1 mediated phosphorylation of KCC2 and NKCC1. PLoS One. 2020;15:e0232967. doi: 10.1371/journal.pone.0232967. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper and/or the supplemental information. All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.