

Abstract
B cells must extract antigens attached to the surface of antigen presenting cells to generate high-affinity antibodies. Antigen extraction requires force, and recent studies have implicated actomyosin-dependent pulling forces generated within the B cell as the major driver of antigen extraction. These actomyosin-dependent pulling forces also serve to test the affinity of the B cell antigen receptor for antigen prior to antigen extraction. Such affinity discrimination is central to the process of antibody affinity maturation. Here we review the evidence that actomyosin-dependent pulling forces generated within the B cell promote affinity discrimination and power antigen extraction. Our take on these critical B cell functions is influenced significantly by the recent identification of formin-generated, myosin-rich, concentric actin arcs in the medial portion of the T cell immune synapse, as B cells appear to contain a similar contractile actomyosin structure.
Introduction
B lymphocytes drive the humoral component of the immune response by generating antibodies to foreign substances (antigens), which serve to neutralize the antigen and enhance its clearance by innate immune cells like macrophages. To generate an antibody, the B cell must first recognize the antigen, which it accomplishes via its B cell receptor (BCR), itself a surface-exposed, plasma membrane-bound antibody. BCR: antigen recognition must then be followed by the internalization of the antigen if the B cell is to begin the long journey to eventually producing a high-affinity, secreted antibody [1–7]. While B cells do recognize and internalize soluble antigens, much stronger antibody responses occur when they recognize and internalize the same antigen presented on the surface of an antigen presenting cell (APC) like a macrophage or dendritic cell (DC) [8, 9]. Moreover, this latter route is used almost exclusively during the complex process of antibody maturation, wherein repeated rounds of somatic hypermutation (SHM) of the BCR, coupled with force-dependent testing of BCR: antigen affinity and force-dependent extraction of the antigen from the APC surface, culminate in the generation of high-affinity antibodies [6, 10, 11]. Importantly, recent studies indicate that B cells use pulling forces generated by the conventional non-muscle myosin, myosin 2, to test BCR: antigen affinity and to power the extraction of antigens from APC surfaces [12–14]. Our central focus here is to review these data and suggest approaches that might further clarify the mechanism by which actomyosin-dependent pulling forces drive antigen extraction. Secondarily, we review the roles played by actin (and possibly myosin 2) in two important steps that precede antigen extraction/internalization: the spreading of the B cell over the APC surface, and the subsequent contraction of the B cell. Our thoughts on the roles played by actin and myosin in driving all of these B cell phenomena are influenced considerably by the recent identification of actomyosin arcs in the medial portion of the T cell immune synapse (IS) [15–17], as there is growing evidence that the B cell IS contains a similar contractile structure [18, 19].
We begin with a brief description of B cell biology. We then review the organization and dynamics of the actin and actomyosin structures known to be present at the T cell IS and compare that to what we currently know about the actin and actomyosin structures that are formed at the B cell IS. We then discuss in more detail how these actin and actomyosin structures contribute to B cell spreading, B cell contraction, BCR: antigen affinity testing, and finally antigen extraction. We close with a discussion of microscopy-based approaches that might yield greater insight into how B cells harness actomyosin-dependent pulling forces to extract antigens from APCs.
The essential role of antigen extraction in B cell biology
B lymphocytes are an essential arm of the adaptive immune system that defend the body against infection by performing several important functions, the most well-known of which is their ability to produce antibodies. Antibodies act by binding to ligands derived from whole or parts of bacteria, viruses and allergens, which are collectively called antigens. Upon binding, antibodies can neutralize their targets (i.e. prevent toxins and pathogens from attaching to host cells), promote the phagocytosis of antibody-coated particles by scavenger cells, and initiate the direct killing of pathogens [20]. Naïve B cells, which have not “seen” antigen before, make only a membrane-bound version of an antibody that is expressed on their cell surface and that, together with a non-covalently bound Igα/βheterodimer (also known as the CD79 signaling subunit), is called the B cell receptor (BCR) [21]. The binding of antigen by the BCR induces the rapid phosphorylation of immunoreceptor tyrosine activation motifs on the Igα/βheterodimer by Src-family kinases (add references). This in turn leads to the recruitment of intracellular signaling molecules and adaptors that drive most aspects of B cell activation, including robust changes in actin assembly (for review, see). Only upon activation and differentiation into plasma cells do B cells switch from making membrane-bound BCRs to secreted antibodies [21]. Remarkably, each B cell bears a clonal BCR such that all of its BCRs have an identical antigen-binding site that binds only one or a few structurally-related antigens with sufficient binding affinity to induce BCR signaling [22]. Collectively, the vast diversity of BCR specificities provided by the millions of B cells in the body confers the ability to recognize an enormous number of different antigens.
To facilitate the rare encounter between a B cell and its cognate antigen, antigens throughout the body are collected and concentrated within secondary lymphoid organs (SLOs) like lymph nodes and the spleen via the blood and lymphatic systems (Figure 1A). The architecture of SLO tissues separates B cells into specific compartments called follicles wherein naïve B cells that enter from the blood scan for antigens. Although B cells can readily recognize soluble antigens, the most physiologically relevant form of antigen encountered by B cells is antigen presented on the surface of antigen presenting cells (APCs) [8]. APCs that are particularly important for presenting antigen to B cells include subcapsular sinus macrophages, DCs and follicular dendritic cells (FDCs) [8, 9] (Figure 1B).
Figure 1.
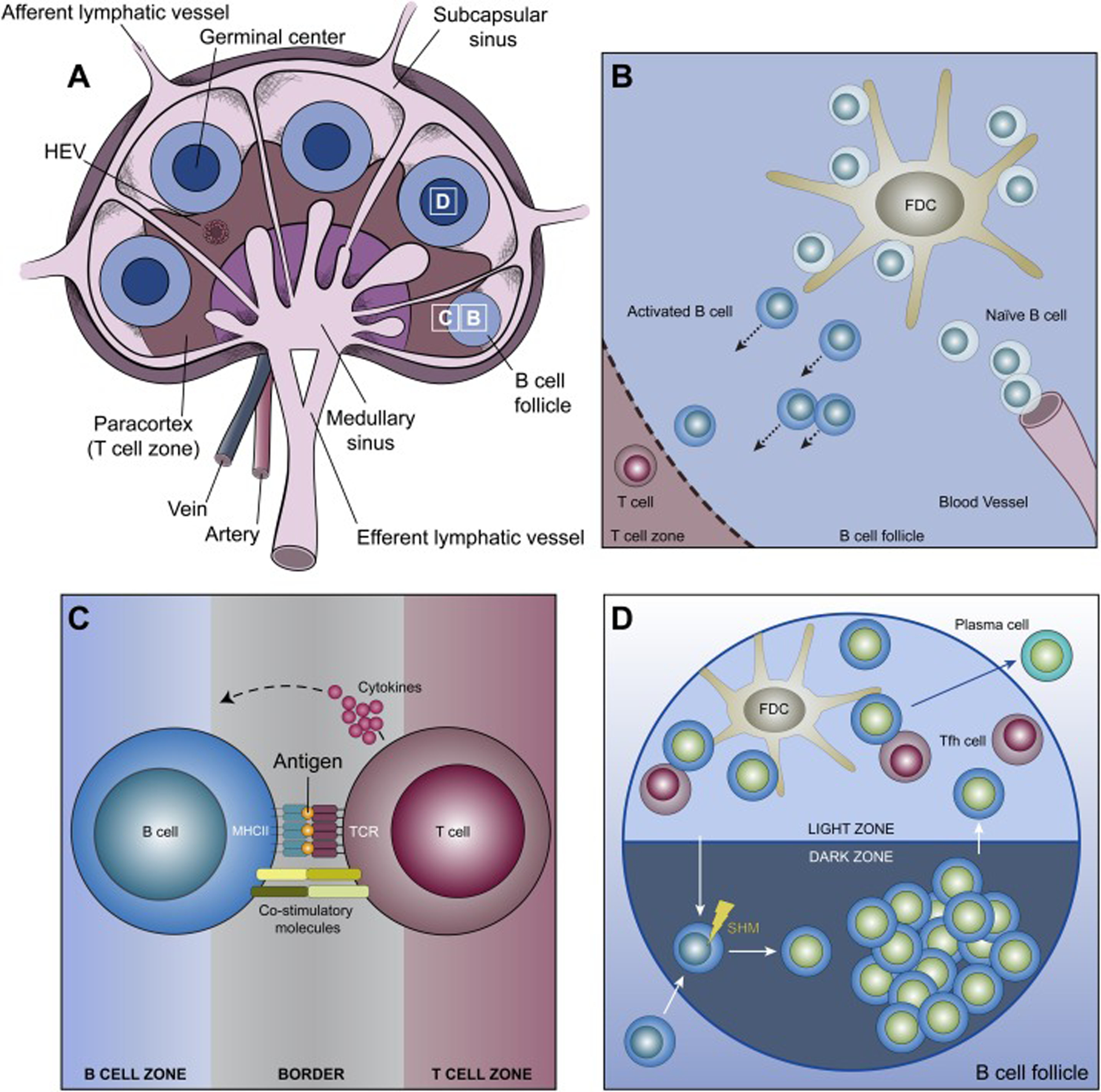
The basics of B cell biology. Shown are cartoons depicting the structure of the lymph node (note the presence of B cell follicles at different stages of maturation) (A), the initial interaction of B cells with antigen on APCs in the B cell follicle (B), the interaction of an activated B cell with a T cell at the border between the B and T cell zones that can result in the B cell receiving signals from the T cell to form a germinal center (C), and the organization within a B cell follicle of a germinal center, which is divided into dark and light zones where SHM of the BCR and BCR: antigen affinity testing occur, respectively (D). See the text for additional details. HEV, high endothelial venule; Tfh, T follicular helper.
B cell activation and differentiation proceed through a series of steps where each step, from initial encounter with antigen in the SLO to exit into the blood stream as a plasma cell, relies on the B cell’s ability to extract antigen from APCs. B cells that bind cognate antigen presented on APCs form a structure called the IS that facilitates BCR signaling and antigen uptake [23]. The mechanisms that drive IS formation will be discussed below. Upon IS formation, B cells extract antigen from APC membranes using their BCR and then internalize the extracted antigen [12, 23]. Following internalization, the antigen is trafficked to processing compartments, where it is partially degraded [5]. The antigen peptides thus produced are loaded onto molecules of MHC II and then trafficked back to the B cell surface [5]. B cells at this stage then migrate towards the T cell zone, which is adjacent to the B cell follicle [24]. Concomitantly, activated helper T cells migrate towards the B cell follicle, resulting in the two cell types meeting at the boundary between these two regions [25] (Figure 1C). A T cell bearing a T cell receptor (TCR) that recognizes the peptide antigen presented by the activated B cell at this boundary provides, in a process commonly referred to as “T cell help”, activation signals in the form of co-stimulatory molecules and secreted cytokines that direct this B cell to seed the formation of a germinal center (GC) [24]. This process involves rapid B cell proliferation and clustering with antigen-bearing FDCs in the B cell follicle, culminating in the formation of a GC containing dark and light zones [25] (Figure 1D). B cells in the dark zone undergo both clonal expansion and BCR editing, wherein point mutations are introduced into the BCR’s antigen-binding site by SHM. Importantly, this stochastic process can produce BCR variants that bind antigen more strongly. Dark zone B cells then migrate to the light zone, where the affinity of their mutated BCRs is tested by engaging a limited amount of antigen on FDCs [26]. GC B cell clones bearing mutated BCRs that bind antigen on FDCs with higher affinity outcompete clones with lower affinity BCRs because they can extract and internalize more antigen, resulting in additional help in the form of survival signals from T follicular helper (Tfh) cells following antigen processing and presentation [27]. It is in the light zone, therefore, that the process of antigen extraction and internalization is harnessed to fine-tune the antibody response by selecting for B cell clones bearing higher-affinity BCRs. After several rounds of receptor editing in the dark zone and affinity testing in the light zone, GC B cells bearing high-affinity BCRs differentiate into either memory B cells or plasma cells that exit the follicle to produce high-affinity, secreted antibodies with the same specificity/affinity as that B cell’s BCR prior to differentiation [11, 25]. Consistent with the critical importance of antigen extraction and internalization, B cells undergo apoptosis in the absence of antigen uptake due to the lack of T cell-derived survival signals [28]. In other words, antigen presentation to T cells during the affinity maturation process provides the essential signals for B cells to differentiate into high-affinity memory B cells and long-lived plasma cells.
Brief overview of the actin-dependent events leading up to actomyosin-dependent antigen extraction
When a B cell recognizes antigen on the surface of an APC, a series of signaling events downstream of this BCR: antigen interaction results in the formation of a mature B cell IS over a period of about 10 minutes [29]. The dominant feature of this mature synapse is the accumulation of BCR: antigen complexes at its center [23, 30] (although see below regarding GC B cells). This distribution sets the stage for the actomyosin force-dependent extraction of antigen from the APC surface. Two major actin-dependent events lead to the accumulation of BCR: antigen clusters at the center of the IS: the spreading of the B cell over the APC, which helps the B cell gather antigen, and the subsequent contraction of the B cell, which drives BCR: antigen clusters to the center of the IS [29]. A full understanding of the roles played by actin and myosin in antigen uptake requires, therefore, an understanding of their roles in B cell spreading and B cell contraction as well. Insight into how actin and myosin accomplish these tasks can come from understanding the organization and dynamics of actin and myosin at the B cell IS. Our first task is to review what is known in this regard. Before considering what is known (and what is likely), however, we will review what is known about the organization and dynamics of actin and myosin at the T cell IS. We do this in part because our understanding of these cytoskeletal proteins in T cells is in some respects more advanced than in B cells, and therefore may prove instructive for future studies in B cells. We also do this because of the growing sense that several key aspects of actomyosin dynamics and function might be shared between these two immune cell types despite the fact that their synapses serve different purposes (polarized secretion in the T cell and antigen uptake in the B cell). Of note, we will not review the important role that actin dynamics play in controlling BCR diffusion and clustering in resting and activated B cells, and we will not discuss in depth the signaling pathways leading to actin assembly. The reader is referred to several excellent reviews that cover these important aspects of B cell biology [4, 31–33].
Actin and actomyosin structures at the T cell IS
The mature T cell IS is composed of three distinct actin networks that correspond in position to the three supramolecular activation clusters (SMACs) that comprise the T cell IS, i.e. the distal SMAC or dSMAC, the peripheral SMAC or pSMAC, and the central SMAC or cSMAC [34] (Figure 2A). Specifically, the distal portion of the synapse (i.e. the dSMAC) is composed of a lamellipodia-like branched actin network, the medial portion of the synapse (i.e. the pSMAC) is composed of a lamella-like actomyosin arc network, and the central portion of the synapse (i.e. the cSMAC) is composed of a hypodense actin network [34] (Figure 2B and inset). Importantly, these actin structures are evident in cells that are simply fixed and stained for F-actin and myosin 2, so they are not induced by imaging with dynamic actin and myosin reporters [15–17] (Figure 2C and 2D). Moreover, all three networks are present at synapses made by primary T cells as well as at synapses made by Jurkat T cells [15–17]. Finally, small actin foci that partially overlap with TCR microclusters (MCs) are also present in the dSMAC and pSMAC of primary T cells [35] (Figure 2B).
Figure 2.
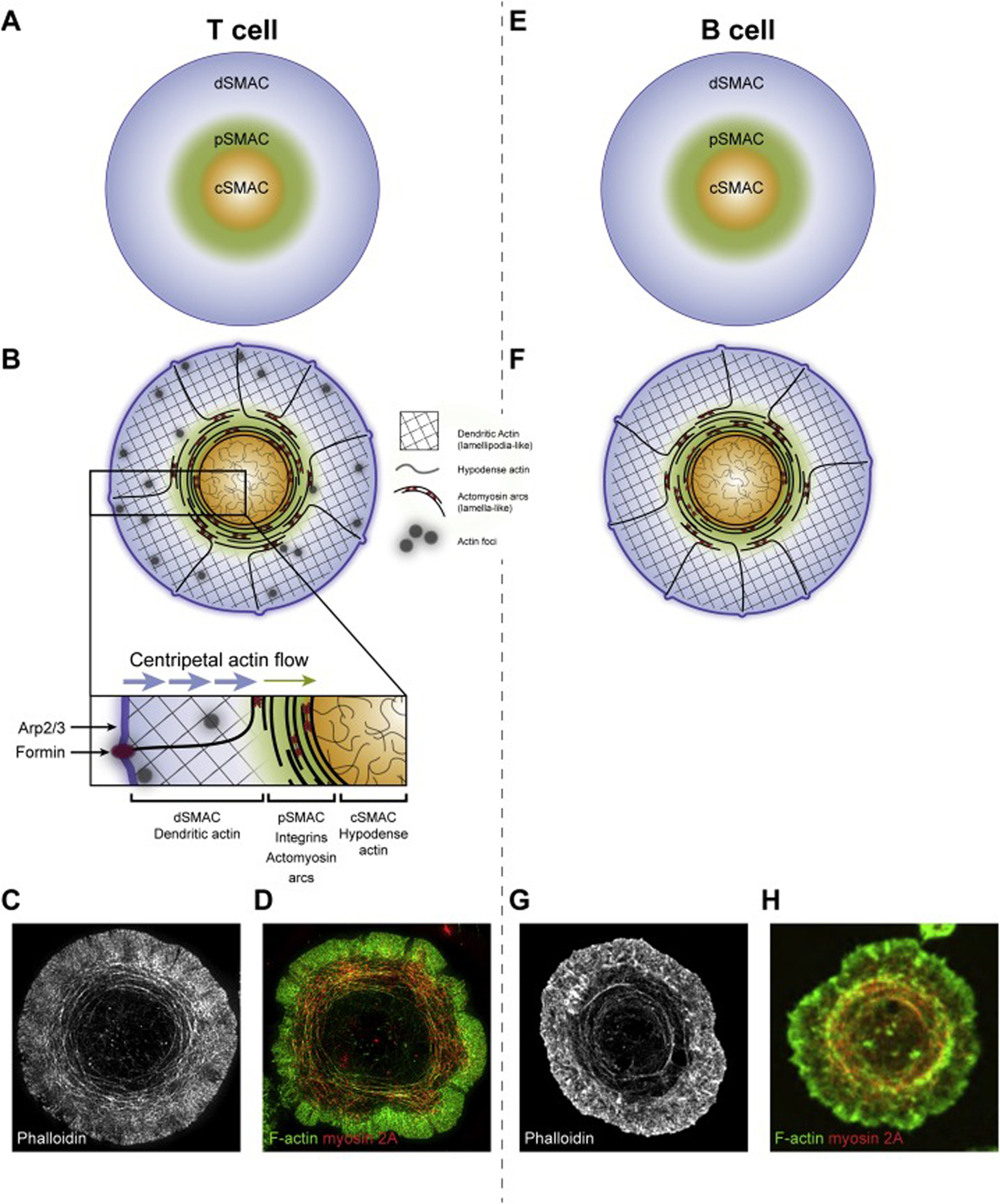
The organization of the SMACs, actin and actomyosin at immune synapses made by T cells and B cells. Shown are cartoons depicting the organization of the SMACs at T cell (A) and B cell (E) synapses, and the organization of actin and actomyosin at T cell (B) and B cell (F) synapses (the Panel B inset shows details discussed in the text). Panel C shows a representative TIRF-SIM image of a phalloidin-stained Jurkat T cell, revealing the branched F-actin network comprising the outer dSMAC, the actin arcs comprising the medial pSMAC, and the actin hypodense region comprising the central cSMAC (reproduced from [16]). Panel D shows a representative TIRF-SIM image of a Jurkat T cell stained with phalloidin to visualize F-actin (green) and an antibody against myosin 2A (red), showing that the pSMAC arcs are highly decorated with endogenous myosin 2A (reproduced from [16]). See also Movies 1 and 2. Panel G shows a representative STED image of a phalloidin-stained A20 B cell, which looks remarkedly similar to the TIRF-SIM image of the Jurkat T cell in Panel C, including the presence of actin arcs in the medial pSMAC portion of the B cell synapse (reproduced from [19]). Panel H shows a representative spinning disk confocal image of an A20 B cell expressing GFP-tagged myosin 2A (pseudo-colored red) and stained for F-actin (green) (reproduced from [19]). This image also closely resembles the image of the Jurkat T cell in Panel D because the pSMAC arcs in the B cell are also highly decorated with myosin 2A. Regarding the inward flows of the branched and arc actin networks depicted by the arrows in the Panel B inset, while these two flows have been shown to explain completely the centralization of TCR MCs [15, 16], one study implicated the dynein-dependent movement of TCR MCs on microtubules in the centralization process [123].
The branched actin network comprising the outer dSMAC, which can be seen without sophisticated microscopy [36], is created by Arp2/3 complex-dependent branched actin nucleation occurring just under the plasma membrane at the outer edge of the radially symmetric IS (Figure 2B and inset). The Arp2/3 complex is stimulated to generate this branched network by the nucleation promoting factor (NPF) WAVE2. This NPF is recruited to and activated at the plasma membrane by the WAVE Regulatory Complex (WRC) downstream of the recruitment of the WRC to the plasma membrane by active GTP-bound Rac1 and the phosphoinositide PIP3 [37, 38]. The assembly of this branched actin network initially drives the spreading of the T cell over the activating surface/APC. But once spreading is complete, continued assembly drives an inward or “retrograde flow” of this network at a rate of ~0.1 μm/s [15, 16, 36, 39] (Figure 2B and inset). TCR MCs form within this network and, when visualized using planar lipid bilayers (PLBs), move centripetally in lock step with actin retrograde flow [15, 16]. Blocking the formation of this network using the small molecule inhibitor of the Arp2/3 complex, CK666, dramatically attenuates TCR MC formation, TCR MC centralization, and the creation of a mature IS [16, 39]. Finally, this network occupies just the dSMAC portion of the IS, as it is largely disassembled at the dSMAC/pSMAC boundary based on imaging (it disappears rather abruptly at this boundary) (Figure 2B–2D; see also Movies 1 and 2) and on the rapid appearance of a bright actin ring at this boundary when actin filament disassembly is blocked using jasplakinolide [15, 39].
The lamella-like actomyosin arc network comprising the medial pSMAC, which requires super-resolution imaging modalities to see clearly [16, 17, 40], is created by the formin Dia1 acting just under the plasma membrane at the outer edge of the IS [16] (Figure 2B and inset). Dia1 is recruited to and activated at the plasma membrane by GTP-bound Rho A and PIP2 [41]. The linear actin filaments generated by Dia1 dive down through the branched actin network in the dSMAC and, upon exiting it, are organized by bipolar filaments of myosin 2A (the major isoform of myosin 2 in T cells) into concentric, contractile actomyosin arcs that permeate the pSMAC [16] (Figure 2B–2D). Of note, Rho A also activates myosin 2 by activating Rho Kinase (ROCK), which promotes myosin 2 filament assembly and mechanical activity by phosphorylating the regulatory light chain of myosin [42]. Indeed, the joint activation of myosin 2 and formin by Rho A is found in many examples of actomyosin function in biology (e.g. contractile ring formation and function in dividing cells) because it links the activation of this contractile motor with the linear, formin-generated actin filaments that it cooperates with to drive contractility [42, 43].
The T cell’s actomyosin arc network flows inward at about one third the rate of the branched network (~ 0.035 μm/s) [15, 16, 39] until it undergoes almost complete disassembly at the pSMAC/cSMAC boundary (Figure 2B–2D). When visualized using PLBs, TCR MCs move inward across the pSMAC at the same speed as the slower inward flow rate exhibited by these actomyosin arcs [15, 16, 39]. Moreover, super-resolution imaging reveals that the arcs propel inward TCR MC transport via a frictional coupling mechanism (like a broom sweeping dirt), although the possibility of very weak, transient interactions between TCR MCs and the arcs cannot be excluded [16, 44–46]. Consistent with the pSMAC being the zone in the mature IS where adhesion to the APC is most robust, the open, active form of the major T cell integrin, LFA-1, co-localizes extensively with the actomyosin arcs. Moreover, the contractility of the actomyosin arcs drives an enhanced accumulation of integrin clusters at the pSMAC/cSMAC boundary late in IS formation [15]. These observations argue that while actin retrograde flow plays a major role in the activation of LFA-1 [47, 48], the contractile structures comprising the pSMAC probably play a critical role in maintaining or further enhancing T cell adhesion to the APC (similar to how contractile actomyosin structures in mesenchymal cells maintain and enhance focal adhesions [49, 50]).
Blocking the formation of actomyosin arcs using the pan formin inhibitor SMIFH2 [51] or disrupting their concentric organization and contractility by dissociating myosin 2 from arc actin using Blebbistatin [52], shows that these structures are required for the centralization of TCR MCs at the cSMAC and for the formation of a normal adhesive ring in the pSMAC [15–17, 39]. These alterations result in significant defects in adhesion to APCs, in TCR proximal signaling, and in discriminating between antigens of different affinities [15–17, 39]. Interestingly, blocking Arp2/3-dependent nucleation using CK666 results in a large increase in the content of actin arcs at the IS most likely because the formin is no longer constrained by limiting levels of actin monomer [16, 53–55]. Finally, imaging actomyosin arcs is quite challenging, as it is best done on an activating surface using structured illumination microscopy (SIM) combined with total internal reflection (TIRF) illumination (Figure 2C; see also Movies 1 and 2). Moreover, it is critical that an appropriate dynamic probe for F-actin be used (e.g. F-Tractin). Importantly, GFP-tagged actin should not be used as it incorporates poorly into formin-generated structures [15–17, 56, 57] (of note, this is also a problem for Lifeact, albeit less so than for GFP-actin) [57]. When imaged properly, actomyosin arcs can be seen to form an even greater fraction of the synapse in primary T cells than in Jurkat T cells [16, 17].
The hypodense actin network comprising the cSMAC, which requires super-resolution imaging modalities like STED and TIRF-SIM to see clearly, is composed of a fine isotropic network containing small actin foci embedded within both straight and branched filaments/fibers [58–61] (Figure 2B–2D). While the origin of these filaments is not entirely clear, both the Arp2/3 complex and formin(s) may contribute to creating them [60]. Unlike the branched and arc networks in the dSMAC and pSMAC, respectively, this network is largely static in terms of directional flow. In naïve T cells and natural killer (NK) cells, the average pore size of this fine actin network is smaller than the diameter of typical lytic granules (LGs), suggesting that this network serves to limit the access of LGs to the plasma membrane machinery that drives exocytosis [59–61]. Upon activation, however, many of these pores increase in size as a result of local increases in actin dynamics and myosin 2-based contractions, thereby allowing LGs to access the plasma membrane to undergo exocytosis. Therefore, while largely static in terms of directional flow, the nanoscale dynamics of this cSMAC actin network appear to control LG secretion (for review, see [62]).
Finally, synapses made by primary T cells on activating surfaces contain small actin foci spread throughout the dSMAC and pSMAC (Figure 2B and inset) [35]. These structures, which represent a minor fraction of total synaptic actin, co-localize significantly with TCR MCs and are created by the Arp2/3 complex downstream of the NPF WASp. Consistently, T cells that lack WASp also lack actin foci. Foci are, however, the only IS actin structure that disappears in T cells lacking WASp [37, 63–65], as the Arp2/3-dependent branched actin network comprising the dSMAC is driven by the NPF WAVE2 [37, 38], and the actin arc network comprising the pSMAC is formin-generated [16]. In other words, the appearance and organization of actin and actomyosin at synapses formed by WASp null T cells is largely normal. Foci nevertheless make an important contribution to PLCƴ1 recruitment, activation and subsequent calcium flux, consistent with the defect in calcium signaling exhibited by WASp null T cells [35, 66, 67].
The pathway leading to foci formation is the pathway most often cited regarding actin assembly at the T cell IS, in which proximal signaling downstream of TCR engagement leads to the recruitment of the adaptor protein Slp76 to LAT clusters at the plasma membrane. Slp76 then recruits Vav, which serves as a guanine nucleotide exchange factor for Cdc42, the Rho-related GTPase that recruits, unfolds and activates WASp. Finally, Slp76 also recruits the adaptor protein Nck, which helps bring WASp to the membrane and activate it [68–71]. Recent in vitro work by Case et al. [72] has shed new light on the nature of focal actin assembly downstream of WASp. Utilizing planar lipid bilayers containing the transmembrane adhesion protein Nephrin in complex with N-WASp and Nck, Case et al. showed that the ability of these proteins to promote focal, Arp2/3-dependent actin assembly is potentiated by liquid-liquid phase separation (LLPS) of the Nephrin-Nck-N-WASp complex [72]. The LLPS of these proteins, which is driven by their ability to undergo multivalent protein: protein interactions through SH2 and SH3 domain-dependent binding events, serves to increase the membrane dwell time of N-WASp and the Arp2/3 complex, resulting in an increase in focal actin assembly. This exciting finding parallels the recent observation that bilayer-bound LAT, in complex with Grb2 and SOS (a guanine nucleotide exchange factor for Ras), also undergoes LLPS driven by similar SH2 and SH3 domain-dependent multivalent interactions, and that the increase in dwell time caused by LLPS promotes the SOS-dependent activation of membrane-bound Ras [73]. Together, these two seminal studies suggest that the generation of TCR MCs, and the actin foci with which they associate, are both promoted by LLPS, and that these two LLPS phenomena may act in concert to promote the kinetic proofreading of multistep activation pathways common in receptor-mediated signaling.
Regarding TCR MC transport across the dSMAC, recent work combining in vitro reconstitution of the TCR and its signaling components with live cell imaging has provided evidence that TCR MCs within the branched dSMAC physically associate with this network as it flows inward by means of basic regions in WASp and Nck that bind to F-actin [74]. Importantly, as TCR MCs exit the dSMAC, they lose Nck (and presumably WASp), and therefore their direct connection to F-actin. At this point, the MCs are picked up and moved inward across the pSMAC by the sweeping action of the actomyosin arcs [16], as discussed above. In support of this model, over-expression in Jurkats of a chimeric adaptor that prevents the normal uncoupling of TCR MCs from F-actin at the dSMAC/pSMAC boundary caused MCs to undergo a more circumferential inward path in the pSMAC, presumably because they are now bound to the telescoping actomyosin arcs [74]. Together, these data argue that WASp and Nck act as a clutch to connect TCR MCs to actin, and that changes in TCR MC composition at the dSMAC/pSMAC boundary cause the mode by which MCs are transported inward to change from an actin-attached mode in the dSMAC to an actin-detached, sweeping mode in the pSMAC. Implicit in this model given that it is WASp centric is that the TCR MC-associated, WASp-dependent actin foci discussed above may play a role in coupling TCR MCs to the WAVE-dependent branched actin network undergoing retrograde flow in the dSMAC.
Actin and actomyosin structures at the B cell IS
Like the T cell IS, the B cell synapse contains dSMAC, pSMAC and cSMAC zones (Figure 2E) (please note that, unless stated otherwise, we are describing synapses made by naïve follicular B cells; near the end of the review we discuss GC B cells, which appear to make different synaptic structures). Regarding the actin structures that comprise these three B cell IS zones (Figure 2F), the outer lamellipodia-like structure that drives B cell spreading and antigen gathering clearly corresponds to the dSMAC portion of the T cell IS. Like the T cell dSMAC, the B cell dSMAC is a branched actin network created by the Arp2/3 complex operating at the outer edge of the radially symmetric B cell synapse [19, 75]. With regard to the NPF in B cells that activates Arp2/3 to create this structure, B cells that lack both WASp and N-WASp fail to spread on antigen-presenting surfaces (Liu et al. PLoS Biol 2013), arguing that these two NPFs cooperate to drive the formation of this outer branched actin network. If true, however, this would be a significant departure from the norm, as WAVE downstream of the WRC and its plasma membrane activators Rac-GTP and PIP3 is the NPF driving the Arp2/3-dependent formation of the dSMAC in T cells and the lamellipodium in virtually every other cell type [35, 36, 71–73]. One way to reconcile this apparent discrepancy would be if B cells that lack both WASp and N-WASp exhibit such a pronounced deficit in early signaling that subsequent activation of the Arp2/3 complex by the WAVE/WRC/Rac/PIP3 pathway does not occur. In this way, WASp and N-WASp would not be the actual NPFs driving dSMAC formation, but their activity would be a prerequisite for WAVE to accomplish this task (see also below regarding the possible relationship between WASp/N-WASp and actin foci in B cells).”Recent work has shown that, as in T cells, the branched actin network comprising the B cell dSMAC flows inward once cell spreading is complete, allowing it to contribute to the centralization of BCR: antigen clusters [19]. This and other aspects of the B cell dSMAC are covered in more detail below in the sections on B cell spreading and B cell contraction.
While there is consensus that a branched actin network comprises the B cell dSMAC, our inclusion in Figure 2F of a T cell-like actomyosin arc network comprising the medial pSMAC portion of the B cell synapse is not a widely held idea. We note, however, that integrins have been known for a long time to be concentrated in the pSMAC portion of the B cell IS just as they are in the T cell synapse [30, 79]. In a sense, then, the current situation in B cells resembles the situation in T cells before the discovery of the integrin-rich actomyosin arcs that populate their pSMAC. But beyond simply drawing this comparison, two recent papers have provided high-resolution and super-resolution images of B cell synapses that reveal the presence of actin arcs in the medial/pSMAC portion of the synapse [18, 19] (Figure 2G). Moreover, these arcs stain strongly for endogenous myosin 2A (Figure 2H). Finally, recent unpublished work from our lab using TIRF-SIM imaging of actin and myosin 2A in A20 B cells and primary B cells on activating surfaces strongly corroborates these observations. Clearly, though, many questions remain. For example, are these B cell actin arcs created by a formin? Is their concentric organization dependent on myosin 2A? Do they co-localize with open, active integrin? Do they sweep BCR: antigen clusters inward towards the cSMAC? Do they drive B cell contraction? Do they promote BCR signaling, B cell: APC adhesion, and B cell effector functions? Despite these open questions, it seems reasonable to assume at this point that the B cell pSMAC, like the T cell pSMAC, is populated by contractile actomyosin arcs. Importantly, we will assume that this is the case for the purpose of reviewing below the known and likely functions of actin and myosin in B cell spreading, B cell contraction, BCR: antigen affinity testing, and antigen extraction.
A second area of general agreement is that the B cell cSMAC, the site where BCR: antigen clusters accumulate late in IS formation, corresponds to an actin-hypodense zone [18, 80] (Figure 2F–2H). This parallels the T cell cSMAC, which is also actin-hypodense and the site where TCR MCs accumulate late in IS formation. That said, efforts to define the origin, organization and dynamics of actin at the B cell cSMAC have been rather limited, especially as compared to T cells and NK cells. Whether the sparse actin filaments present within the B cell cSMAC have a role in antigen extraction, by analogy with the role played by the fine actin network at the T cell/NK cell cSMAC in regulating lytic granule secretion [60], is unknown. Indeed, the fact that actomyosin-dependent antigen extraction appears to occur preferentially at the cSMAC, the region of the B cell IS with the least amount of actin, represents a seeming mismatch that we discuss in more detail below.
Finally, whether the B cell IS contains BCR-associated actin foci that are analogous to the TCR-associated actin foci seen in T cells is unclear. That said, actin foci have been observed during B cell IS formation [81, 82] suggesting that T cells and B cells may share this actin structure as well. As discussed above, these structures are created in T cells by the Arp2/3 complex downstream of the NPF WASp. Interestingly, B cells lacking both WASp and N-WASp show profound defects in overall actin assembly at the IS [83, 84]. While this could be taken as evidence that WASp/N-WASp are the NPFs driving dSMAC formation (as this structure represents the largest fraction of F-actin at the IS), we suggest that a tight association between WASp-dependent actin foci and signaling clusters early in IS formation might be a prerequisite for subsequent actin assembly through WAVE-Arp2/3 (to create the dSMAC) and formin (to create the pSMAC).
B cell spreading
Upon initial contact with antigen, the B cell rapidly and transiently inactivates the actin-membrane linker protein ezrin [85] and activates the F-actin severing protein cofilin [75]. These two responses act synergistically to transiently remodel the B cell’s actin cortex in such a way as to promote the coalescence of BCRs into BCR MCs, thereby initiating BCR signaling (Figure 3A; reviewed in depth in [4, 6, 31]). Once signaling is underway, the B cell undergoes a rapid spreading phase in which its plasma membrane extends outward over the surface of the APC (Figure 3B). This spreading phase, which typically lasts 2 to 5 minutes [29], serves to maximize the area of contact between the B cell and the APC. The functional significance of this spreading is to promote antigen gathering, wherein additional BCRs engage antigen, resulting in the formation of additional BCR MCs and more robust BCR signaling. Spreading operates, therefore, in a feed-forward fashion, with higher affinity and/or avidity antigen on the APC surface driving more BCR MC formation/signaling, leading to more spreading [29]. That said, spreading can also facilitate the uptake of low-affinity antigens or antigens that are presented at low densities.
Figure 3.
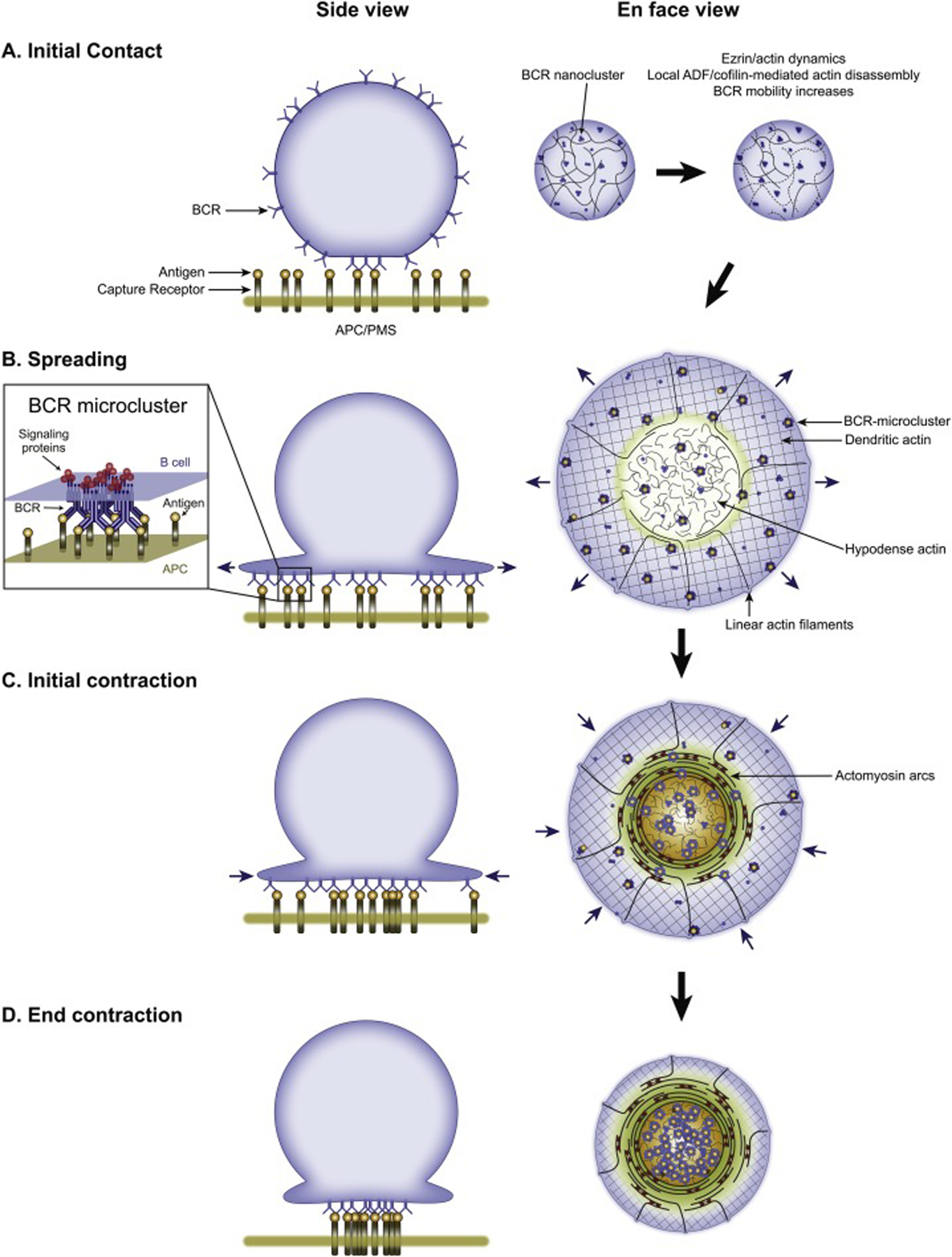
B cell events leading up to antigen extraction. Shown on the left are cartoons depicting the initial contact of the B cell with the APC/activating surface (A), the spreading of the B cell over this surface (B), and the subsequent contraction phase divided into initial contraction (C) and the end of contraction (D). The inset in (B) depicts an antigen-bound BCR microcluster (MC) with associated signaling molecules. On the right are cartoons depicting the organization and dynamics (blue arrows) of the B cell synaptic actin and actomyosin networks described in Figure 2 during these four phases. Please note that these latter cartoons are largely conjecture at this point (especially the possible role of the actomyosin arcs in the pSMAC portion of the B cell synapse in driving the contraction phase). Also of note, one study implicated the dynein-dependent movement of BCR MCs on microtubules in the BCR centralization process [124]. Finally, we note that while the steps shown in this figure and discussed in the corresponding text are presented for the sake of clarity as if they are always completely sequential and very well defined both temporally and spatially, in reality they often blend together to some extent due to variations in the kinetics of their formation and their spatial distribution. PMS, plasma membrane sheet; ADF, actin depolymerizing factor.
The actin cytoskeletal structure driving spreading is the Arp2/3-dependent branched actin network comprising the dSMAC (Figure 3B). As discussed briefly above, this network is created by Arp2/3 complex-dependent branched actin nucleation occurring just under the plasma membrane at the outer edge of the synapse. Newly-formed branched actin powers the radial extension of the “leading-edge” via a Brownian ratchet mechanism in which spaces caused by thermal fluctuations in the plasma membrane are rapidly filled by the expanding branched network [86]. This process can continue until the cell becomes maximally spread, at which point continued branched actin assembly drives an inward or “retrograde flow” of this network until it is disassembled at the dSMAC/pSMAC boundary [18, 19]. As discussed above, the formation of the B cell dSMAC is most likely dependent on the NPF WAVE downstream of the WRC and its plasma membrane recruiters/activators Rac-GTP and PIP3. Relevant to this, the B cell membrane protein CD19, which associates with the BCR during MC formation and is required for B cell spreading, mediates the recruitment and activation of PI3K and Vav at the synapse [9]. Although not directly proven, it is likely that CD19-dependent signaling promotes B cell dSMAC formation by promoting the PIK3-dependent production of PIP3 and the Vav-dependent activation of Rac, as these two plasma membrane signals recruit the WRC to activate WAVE-dependent Arp2/3 activity. In summary, B cell spreading is harnessed to (i) increase BCR signaling output, which serves to overcome the signaling threshold required for B cell activation, and (ii) to maximize the amount of antigen bound to BCRs, which determines the amount of antigen taken up at the mature IS and subsequently presented to T cells.
B cell contraction/IS formation
Once the B cell has finished spreading across the APC, it reverses direction and begins to shrink its area of contact with the APC in a process known as B cell contraction [29] (Figure 3C and 3D). This contraction phase, which typically lasts for 5 to 10 minutes, serves to concentrate BCR: antigen clusters in the center of the IS for eventual uptake (although see below regarding GC B cells). While B cell contraction is widely considered to be the major driver of BCR MC centralization and B cell IS maturation [7, 32, 87], how the cortical actin cytoskeleton switches from robust spreading to robust contraction is largely unknown. Also unknown are the contractile elements within the B cell that power contraction, and whether retrograde flows of actin and actomyosin also contribute to the centralization of BCR: antigen clusters. We next consider these and other open questions in light of recent results regarding the maturation of the T cell IS.
As mentioned above, the consensus in the B cell field is that B cell contraction is the main driver of BCR: antigen cluster centralization and IS maturation. This view stands in sharp contrast to the consensus in the T cell field because T cell contraction, when it does occur, usually happens after a mature IS has formed. Indeed, recent T cell studies have focused almost exclusively on the role of inward flows of cortical actin and actomyosin at synapses of constant size as the main drivers of TCR MC centralization and IS maturation (reviewed above). These studies raise an obvious question for B cells: do similar inward flows of cortical actin and actomyosin at the B cell synapse contribute, along with B cell contraction, to the centralization of BCR MCs and IS maturation? With regard to the branched actin network comprising the B cell dSMAC, this same structure in T cells flows inward once cell spreading is complete, carrying TCR MCs with it. Importantly, recent results indicate that the B cell dSMAC also undergoes retrograde flow at ~0.06 μm/s following the spreading phase, and that this inward actin flow propels BCR MCs towards the center of the IS [19] (Figure 3C). Whether the contractile actomyosin arcs that populate the B cell pSMAC also flow inward and propel BCR MCs towards the cSMAC in the process remains to be seen. As with T cells, this question could be addressed using live TIRF-SIM imaging of F-Tractin-expressing B cells engaged with fluorescent antigen-containing PLBs. If the actomyosin arcs do in fact propel BCR MCs towards the cSMAC, then BCR MC centralization/B cell IS maturation will parallel TCR MC centralization/T cell IS maturation, with the one significant difference being that BCR MC centralization is also promoted by the overall contraction of the B cell’s footprint on the APC.
What contractile element drives the contraction of the B cell? We suggest that the actomyosin arcs that populate the B cell pSMAC are probably responsible, as they are contractile in nature and their radial symmetric organization should promote the symmetric shrinkage of the IS footprint. If true, then this myosin-based contractile structure might well be driving BCR MC centralization and B cell IS maturation by powering two distinct centering forces: an inward flow of actin arcs across the pSMAC, and the overall contraction of the B cell IS. Tools and approaches to address this and other open questions, such as whether these actomyosin structures also power antigen extraction once a mature IS has formed, are discussed in the final section of this review.
Antigen extraction and internalization
As discussed above, the humoral response to an antigen presented on an APC membrane is typically much stronger than the response to the same antigen presented in soluble form [8, 23, 88]. Moreover, the process within GCs of selecting B cell clones possessing a mutated BCR with a higher affinity for antigen involves almost exclusively the uptake of antigen from the surface of an APC (typically a FDC) [26]. Such uptake is thought to require the generation within the B cell of actomyosin-dependent pulling forces that are exerted on the BCR to extract the attached antigen from the APC surface [6]. Once the antigen is extracted, the BCR: antigen complex is then internalized via clathrin-mediated endocytosis (CME) to complete the two-step antigen uptake process [6, 32]. Below we review the evidence that actomyosin-dependent pulling forces generated within the B cell are harnessed to test the affinity of the BCR for its antigen, and to extract those antigens whose interaction with the BCR survives the pulling force.
As summarized in Figure 4, the B cell and the APC are connected during antigen uptake via a linear series of interactions, each with its own characteristic features and affinity. First, the connection between the BCR and the B cell membrane (Figure 4, Box 1), as well as the connection between the antigen capture receptor (i.e. an Fc receptor (FcR) or a complement receptor) and the APC membrane (Figure 4, Box 5), involve transmembrane domains, which makes them essentially unbreakable. Second, the connection between the antigen capture receptor on the APC and its opsonizing partner (i.e. between the FcR and the constant region of IgG on the antibody-opsonized antigen (Figure 4, Box 4A), or between the complement receptor and complement on the complement-opsonized antigen (Figure 4, Box 4B)), while breakable, are fairly high-affinity. For FcR: IgG interactions, KD values range from ~0.5 μM to ~5 μM depending on the specific FcR and IgG subtype [89]. For complement receptor: complement interactions, values range from ~0.1 μM to ~1 μM depending on the specific complement receptor and complement factor [90–92]. Notably, the affinity of the capture receptor for its opsonizing partner may remain fairly constant throughout the affinity maturation process, suggesting that it may set an approximate upper threshold for the force-dependent testing of the BCR’s affinity for antigen. Third, the connection between the opsonizing antibody and the antigen (Figure 4, Box 3A) is breakable and may be subject to increasing strength as opsonizing antibodies of increasing affinity for the antigen are generated during the affinity maturation process (this may help select for BCRs with increased affinity for antigen- see the legend to Figure 4 for more details). On the other hand, the connection between the opsonizing complement factor and the antigen (Figure 4, Box 3B) is essentially unbreakable as it involves a cysteine-based covalent bond [93]. Finally, and most importantly, the connection between the BCR and the antigen (Figure 4, Box 2) is breakable and subject to large increases in affinity as BCRs with improved affinity for antigen are generated by SHM within GCs.
Figure 4.
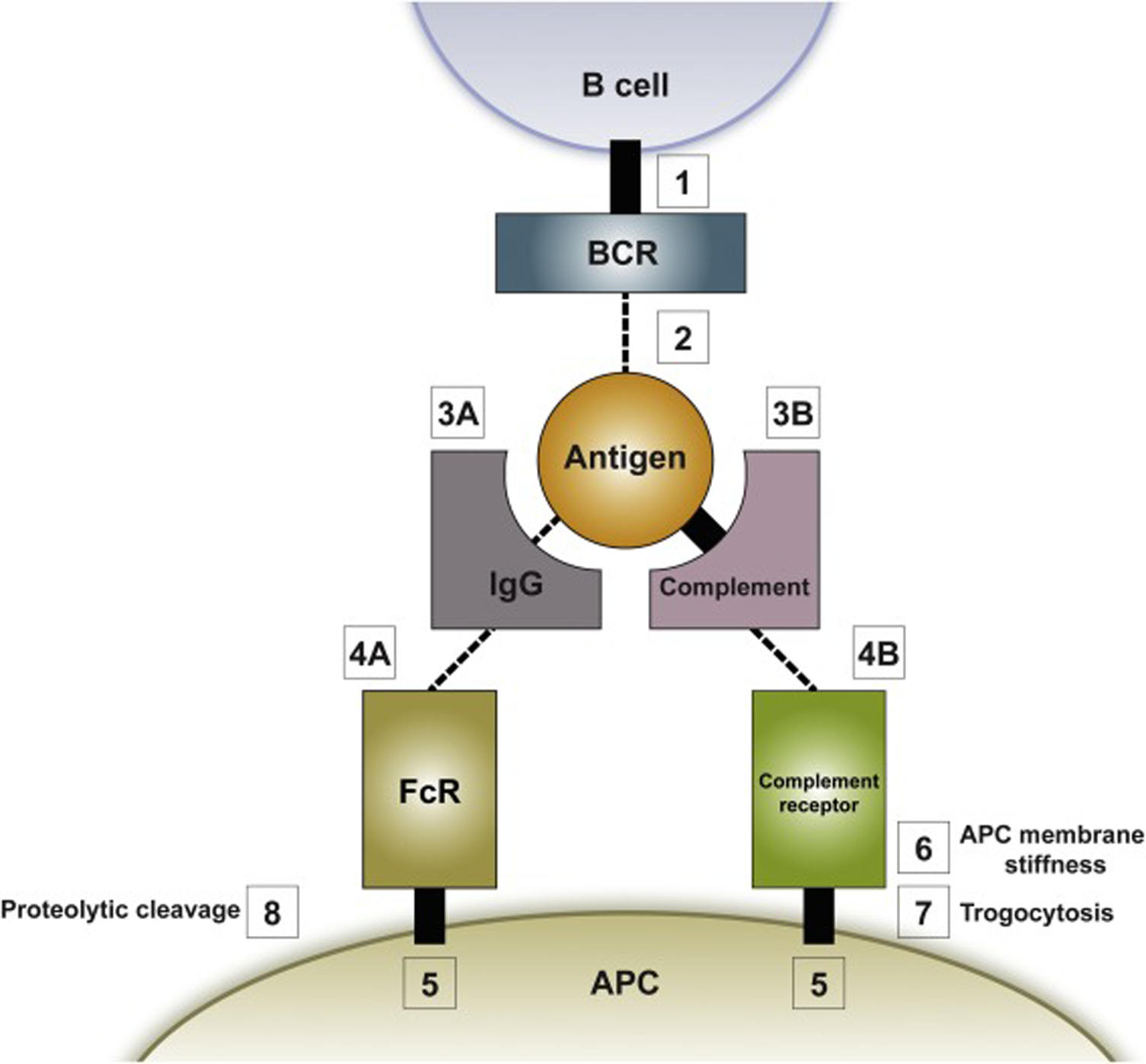
The connections linking the B cell to the APC that dictate the outcome of force-dependent BCR: antigen affinity testing and antigen extraction efforts. Shown is a cartoon summarizing the series of connections that link in linear fashion the BCR to antigen presented on the surface of the APC. As discussed in the text, the relative strengths of connections 1 through 5 determine which one breaks first under load placed on the BCR by actomyosin-dependent pulling forces generated inside the B cell. This mechanism forms the basis by which force is used to discriminate between BCR: antigen pairs of different affinities, and it dictates the fate of the antigen during extraction efforts. See the text for additional details. Note that the cartoon does not include details regarding how the trogocytosis (box 7) and proteolytic cleavage (box 8) pathways operate.
Given the connections shown in Figure 4, it stands to reason that the application of an actomyosin-dependent pulling force on the BCR will result in a break in the connection between the B cell and the APC at the weakest link. So how is this basic fact utilized to initially select for a BCR: antigen interaction of reasonable affinity, and then to select for mutated versions of this BCR generated during the affinity maturation process that bind the antigen with even higher affinity? In its simplest guise, an initial BCR: antigen interaction of very low affinity will likely represent the weakest link in the overall connection between the B cell and the APC. As a result, the antigen’s connection to the BCR will be the first connection to break under force, thereby abrogating its uptake. This B cell clone will then undergo apoptosis due to the lack of survival signals. Conversely, an initial BCR: antigen interaction of reasonable affinity may not be the weakest link in the overall connection between the B cell and the APC. As a result, this antigen’s connection to the BCR will not be the first link to break under load, allowing its uptake. If this happens, this B cell clone will receive T cell help, enter the GC, and begin the affinity maturation process. Once SHM of this BCR is underway, the actomyosin-dependent pulling force can then be used to select for those mutated versions of this BCR that exhibit increased affinity for the antigen, as those BCR: antigen interactions will become increasingly the strongest of the breakable connections in the linkage between the B cell and the APC. In this way, then, actomyosin-dependent pulling forces can drive B cell clonal selection, leading eventually to a B cell clone/plasma cell that produces high-affinity, secreted antibodies.
While the simplified scenario just described encompasses the essence of actomyosin force-based affinity discrimination, many other factors likely make important contributions. One such factor is valency. For example, multivalent BCR: antigen interactions favored by larger BCR MCs will serve to reduce the likelihood that the BCR: antigen linkage will break under load by sharing that load among many BCR: antigen pairs [6, 14]. For the same reason, load sharing should also enhance the extraction of antigens that are bound by BCRs with lower affinity [14]. On the APC side, while the affinity of capture receptors for their opsonizing partner may be relatively constant during affinity maturation, the strength of this connection can be enhanced by multivalency (e.g. multiple capture receptors bound to one antigen molecule; enhanced clustering of capture receptors could also strengthen the connection) [14]. A second modifying factor is the relative stiffness of the APC membrane (Figure 4, Box 6), as a stiffer/less compliant APC membrane will raise the threshold for affinity discrimination, leading to the preferential uptake of higher-affinity BCR: antigen pairs [94]. Consistently, the plasma membranes of FDCs, which drive affinity maturation in GCs, are much stiffer than the plasma membranes of non-follicular DCs employed earlier in the humoral response [94]. A third factor may be mechanotransduction related, as some data suggests that the BCR, like the TCR [95, 96], may switch from being a slip bond to a much stronger catch bond when bound strongly to an antigen and then placed under load [97, 98]. In the case of complement receptors in the APC membrane, two of them (CR3 and CR4) are β2 integrins, suggesting that their interaction with complement proteins might also be potentiated by load [99]. A fourth factor that may potentiate affinity discrimination in conjunction with applied actomyosin force revolves around the advantage gained from increasing the dwell time of interactions between BCR-proximal signaling proteins and adaptors, which likely happens with stronger BCR: antigen interactions. This concept is best exemplified by the recent work from the Groves lab [73] (see also above), which demonstrates that increases in interaction dwell time between proteins participating in multistep activation pathways are created by their ability to undergo LLPS driven by multivalent SH2 domain- and SH3 domain-dependent binding events. Current thinking is that multistep activation pathways common in receptor-mediated signaling pathways may often be promoted by the LLPS of key signaling molecules. Whether this is also the case for the BCR and its signaling partners like CD19 and BLNK remains an exciting possibility worth exploring as its contribution to BCR proximal signaling could promote many B cell functions including antigen extraction.
Once extracted, BCR: antigen complexes appear to be internalized via the canonical CME pathway [12, 32]. Briefly, tyrosine-based sorting motifs present in the cytoplasmic tail of the BCR are recognized by the clathrin adaptor AP2, the primary clathrin adaptor driving CME in most if not all cell types [100]. AP2 then recruits clathrin to create a nascent clathrin-coated pit (CCP) in which the BCR with bound antigen becomes concentrated. Conversion of the initially planar clathrin array into a three-dimensional, spherical structure, together with other factors (e.g. proteins that recognize/promote membrane curvature, local actin assembly), drives a 50–100 nm invagination of the plasma membrane [101, 102]. Endosome formation is then completed by the dynamin-dependent severing of the narrow neck connecting the invaginated CCP with the plasma membrane, followed by the uncoating of the nascent clathrin-coated vesicle [103]. Consistent with these statements, clathrin co-localizes with BCRs undergoing internalization, and BCR internalization is inhibited by the RNAi-mediated knockdown of clathrin or AP2, by mutation of one specific tyrosine-based sorting motif in the BCR’s cytoplasmic tail, and by the knockdown or chemical inhibition of dynamin [12, 100, 104]. Finally, while CME drives the efficient internalization of plasma membrane receptors with attached cargo (in this case, a BCR: antigen complex), it is not a mechanism that can generate any significant force on the APC (e.g. it is probably incapable of pinching off APC membrane - see below).
When the strength of every connection in Figure 4 is very strong, the B cell may extract the antigen along with a patch of the surrounding APC membrane in a poorly understood process known as trogocytosis [105–108] (Figure 4, Box 7). Exactly how this occurs is not yet clear, but may involve the tearing of the APC membrane under strong load, although other mechanisms have been suggested (see more below). Once extracted, the membrane patch with associated antigen must be internalized by the B cell to allow processing and presentation. This pathway may have been at play when Batista and colleagues mixed BCR-transgenic B cells with an APC expressing the BCR’s high-affinity cognate antigen that was artificially attached to the APC surface via a non-breakable transmembrane domain [23]. That said, B cells, as well as other types of immune cells, have been observed in vivo to possess fragments of APC membranes [109], indicating that trogocytosis can occur under physiological conditions.
By far the best evidence to date that actomyosin-dependent pulling forces promote affinity discrimination and drive antigen extraction comes from the seminal study of Tolar and colleagues published in 2013 [12]. To image antigen extraction and define its functional requirements, these authors engaged B cells with fluid, flexible, plasma membrane sheets (PMSs) created by sonication of glass-adhered HEK293 cells, which leaves the ventral membrane of the HEK293 cell attached to the glass. When PMSs were coated with antigen (initially a stimulatory antibody against the BCR, but later cognate antigen) via a biotin-streptavidin linkage, they served as surrogate APCs, as B cells spread rapidly across them and then clustered the antigen at the center of their contact with the PMS. Using PMSs marked with the fluorescent membrane dye DiI, Tolar and colleagues observed PMS membranes with associated antigen undergoing invagination into the B cell [12] (Figure 5A). Dynamic imaging of F-actin using Lifeact and myosin 2A using a GFP-tagged version of its regulatory light chain showed that actomyosin accumulated at sites of invagination. Moreover, inhibition of myosin 2A using Blebbistatin, or reducing its level of activation by inhibiting the myosin light chain kinase ROCK, blocked the formation of invaginations. Together, these observations argued that actomyosin pulling forces generated within the B cell and exerted on the BCR were responsible for pulling antigen-associated PMS membranes upward to form the invaginations (Figure 5A).
Figure 5.

Antigen extraction by naïve and GC B cells. Shown are cartoons depicting the site of antigen uptake as well as the antigen extraction and CME-dependent internalization steps that together drive antigen uptake in both naïve B cells (A) and GC B cells (B). Note that the distributions of the actomyosin structures that power antigen extraction in both B cells types (indicated in red) are largely hypothetical. See the text for additional details.
When BCR-transgenic B cells were engaged with PMSs coated with a low-affinity version of their cognate antigen, the antigen clusters that formed were small and most antigen-associated invaginations retreated abruptly after about 5 seconds. The authors interpreted these events as ruptures under load of the bond between the BCR and the weak antigen, thereby abrogating the uptake of this low-affinity antigen [12]. Consistent with this interpretation, engaging these B cells with PMSs coated with the high-affinity version of their cognate antigen resulted in larger antigen clusters that were associated with longer-lived invaginations. Moreover, these longer-lived invaginations often culminated in the apparent internalization of the antigen, based on the disappearance of the antigen and DiI signals from the TIRF field, and on the DiI signal remaining with the antigen signal presumably inside the B cell. Consistently, clathrin was seen to accumulate at these longer-lived invaginations, and the knockdown of the clathrin adaptor AP2, as well as the knockdown or inhibition of dynamin 2, reduced the amount of labeled antigen inside B cells, presumably by inhibiting the CME of the extracted antigen. Of note, CME inhibition did not block the appearance of invaginations, indicating that their formation depends solely on actin and myosin. Together, these results argued that actomyosin-dependent pulling forces generated inside the B cell drive the extraction of antigens presented by an APC, and, in collaboration with CME, promote the uptake of BCR: antigen complexes of higher affinity and higher valency by mechanically testing the overall strength of the BCR-antigen connection [12] (Figure 5A).
One point of ambiguity in the observations made by Tolar and colleagues revolves around how the PMS membrane ends up inside the B cell upon antigen uptake. If this is correct (ideally the B cell plasma membrane would have been labeled with a dye to confirm that the PMS membrane was internalized), then the uptake observed using PMSs likely represents a version of trogocytosis. Consistent with this interpretation, the connection between the antigen and the PMS (biotin- streptavidin), and the connection between the antigen and the BCR (an antibody to a BCR subunit or a strong cognate antigen), were both very high affinity. This suggests that the weakest link in the overall connection between the B cell and the PMS under these conditions might have been the integrity of the PMS membrane, resulting in it tearing upon force exertion. The authors proposed, on the other hand, that the B cell internalized the antigen by “pinching off” the flexible, invaginated PMS membrane [12]. How this would occur is unclear, however, as CME is probably incapable of pinching off membranes (other than the endocytosing cell’s own plasma membrane), and actomyosin-based internalization mechanisms like phagocytosis generally do not bite through/pinch off portions of the substrate. It is also unclear how membrane fragments could be incorporated effectively into 50–100 nm diameter clathrin-coated vesicles. Finally, trogocytosis in T cells was reported to be clathrin-independent [105, 107]. Future imaging-based studies of antigen extraction should seek to clarify when and how APC/PMS membranes get internalized along with antigen.
One final, fascinating observation made by Tolar and colleagues was that adding the myosin 2 inhibitor Blebbistatin at a low concentration in an effort to reduce the magnitude of the actomyosin-dependent pulling force without eliminating it resulted in a reduction in the uptake of the high-affinity antigen, but an increase in the uptake of the low-affinity antigen (presumably by reducing the rupture of the weaker BCR: antigen bond) [12]. This result suggests that while the B cell signaling machinery must activate actomyosin contractility to drive antigen extraction (possibly by promoting the formation of contractile actomyosin arcs through the RhoA-dependent activation a formin and myosin 2 - see below), it might also regulate the extent of actomyosin contractility to influence the threshold for affinity discrimination.
In a follow-up study, Tolar and colleagues also addressed the role of myosin 2 in B cell function in vivo by crossing a myosin 2A conditional knockout mouse with two cre driver mice specific for the B cell lineage [110]. Early deletion of myosin 2A (Mb1-cre) broadly suppressed B cell development, consistent with the central role played by myosin 2 in cytokinesis, while deletion in mature B cells (CD23-cre) interfered with the development/maintenance of several major B cell subtypes in the periphery, most notably GC B cells. Importantly, the ability of surviving myosin 2-deficient B cells to extract antigen from PMS membranes in vitro was reduced by ~50%. Moreover, mature HEL-BCR transgenic B cells in which myosin 2A had been deleted using CD23-cre exhibited a modest but significant decrease in the uptake of HEL-containing immune complexes in vivo. Finally, myosin 2A KO mice exhibited reduced circulating levels of IgG and IgM and, when immunized, mounted only weak IgM responses and did not class switch to IgG antibodies. This inability to mount an efficient antibody response probably stems in large part from the fact that the surviving B cells have difficulty acquiring membrane-bound antigen, leading to the loss of GC B cells. While these in vivo experiments provide additional support for the idea that B cells use myosin 2-dependent forces to extract membrane-bound antigens, they also highlight the challenges of studying B cells that lack myosin 2 constitutively (see more below), and they indicate that significant antigen extraction/uptake can occur in the absence of myosin 2-dependent pulling forces [110].
Antigen extraction and internalization by GC B cells
Two recent studies addressing the mechanism of antigen uptake by GC B cells have revealed several striking differences between this critical B cell subtype and naïve follicular B cells [13, 111] (Figure 5B). Like the latter, GC B cells spread on contact with an antigen-presenting surface to collect antigen. This event is not followed, however, by the accumulation of BCR: antigen complexes into large clusters at the center of the synapse/cSMAC as in naïve B cells. Instead GC B cells retain their BCR: antigen complexes as small clusters at the periphery of their synapse [13, 111]. Moreover, these small, peripheral BCR: antigen clusters co-localize with actin- and ezrin-rich plasma membrane surface projections that the authors have termed “pods” [111] (Figure 5B). The application of internal reflection microscopy to identify sites of surface attachment showed that GC B cells attach almost exclusively via these peripheral pods. This is in sharp contrast to naïve B cells, which showed robust surface attachment across much of their flatter synaptic interface. Perhaps consistent with all these observations, the distribution of F-actin at the GC synapse is restricted largely to the peripheral pods, while naïve B cells exhibit F-actin across much of their synapse (Figure 2F–H). Together, these observations argue that the differentiation of naïve B cells into GC B cells is accompanied by major changes in IS organization, and most likely in the actin cytoskeletal dynamics that drive IS organization.
Regarding the site of antigen extraction by GC B cells, consistent with the observations discussed above, GC B cells extract antigen at the periphery of their synapse rather that at the IS center as in naïve B cells [13, 111]. Moreover, these studies argue that GC B cells extract antigen specifically at their peripheral BCR-, actin- and ezrin-rich pods via pod-associated myosin 2 [13, 111] (Figure 5B). Importantly, elegant experiments employing DNA-based force probes in combination with internal reflection microscopy indicated that the actomyosin-dependent pulling forces generated by GC B cells at their peripheral pods are stronger and more persistent than the actomyosin-dependent pulling forces generated by naïve B cells at the center of their IS [13]. This fact, together with the lower density of BCRs on GC B cells relative to naive B cells (which will reduce the degree of load sharing), should allow GC B cells to extend the upper limit of force-dependent affinity discrimination to manage the higher-affinity BCR: antigen bonds arising from the SHM of their BCRs. This upper limit may be further extended by the increased membrane stiffness exhibited by the APCs that typically present antigen to GC B cells (i.e. FDCs; [94]), and by the increasing affinity of opsonizing antibodies for the antigen as affinity maturation progresses. Consistent with all these ideas, experiments designed to compare the responses of GC and naïve B cells to strong and weak antigens showed that GC B cells exhibit a higher affinity threshold for BCR signaling and antigen extraction than naïve B cells [111]. A final major difference between GC B cells and naïve B cells is the site where CME internalizes extracted antigen, as this happens along the sides of the GC B cell instead of at the center of the IS as in naïve B cells (Figure 5B). In other words, antigen extraction and CME-dependent antigen internalization are not spatially coupled in GC B cells as they are in naïve B cells.
Antigen uptake by proteolysis
BCR: antigen pairs that fail to be extracted mechanically by myosin-generated forces may be internalized via a proteolytic cleavage mechanism [94, 112, 113] (Figure 4, #8). In this pathway, unknown signals that sense the inability to mechanically extract the antigen instruct the B cell to translocate its lysosomes to the IS, where they undergo exocytosis [94, 112]. The released lysosomal hydrolases are then thought to proteolytically cleave the antigen from the presenting surface, allowing it to be internalized along with its BCR via CME. Importantly, this event would take advantage of the fact that B cell: APC contacts, like T cell: APC contacts, cause the MTOC and attached interphase microtubule array to move to a position immediately adjacent to the APC, thereby focusing the microtubule-based secretory machinery of these cells in the direction of the APC. Indeed, the polarized secretion of lysosomes by B cells could be seen as a variation of the polarized secretion of lytic granules (secretory lysosomes) by T cells. That said, many questions remain regarding this pathway in B cells. For example, what are the signals that instruct lysosomes to move to the IS and become secreted when the mechanical extraction of antigen fails (which, it is argued, occurs preferentially when the presenting surface is very stiff [94])? What specific machinery controls the secretion of lysosomes (while the V-SNARE Vamp 7 has been implicated [114], a role for a specific Rab GTPase, akin to the role played by Rab27a in regulating lytic granule secretion in T cells [115, 116], has not been identified)? Is the synaptic cleft sufficiently acidified following lysosome secretion to permit acid hydrolase function? Is there any specificity as regards where the released lysosomal enzymes cut the connection between the B cell and the APC (e.g. is the BCR protected in some way)? Does this mechanism allow for any form of affinity testing? Does this pathway explain why myosin 2A-deficient B cells retain some capacity to acquire membrane-bound antigens [110]? Can GC B cells, which typically do not reposition their MTOC to the IS [13, 111], use this mechanism? Future studies should seek to answer these and other open questions regarding this alternate pathway of antigen uptake.
The future of antigen extraction
While the evidence that B cells use actomyosin pulling forces to extract antigens from APC surfaces is quite strong, exactly how actin and myosin are organized inside the B cell to accomplish this task is not at all clear. In the case of naïve B cells, it is tempting to speculate that the contractile actomyosin arcs populating the medial, pSMAC portion of their synapse create the pulling forces that drive antigen extraction. For this to occur, however, significant intermixing of the arcs with the BCR: antigen clusters accumulated at the center of the IS (i.e. at the cSMAC) would likely be required. Assuming the actomyosin arcs in B cells are heavily decorated with active integrin as they are in T cells, then it is interesting to note that integrin engagement has long been known to increase B cell responses to low affinity/low avidity antigens [30, 79]. This suggests that the actomyosin arcs populating the B cell’s pSMAC may play a role in the mechanism by which integrins serve as “co-receptors” for the BCR. Finally, it is likely that active RhoA promotes both the formin-dependent synthesis of arc actin, and the ROCK-dependent activation of myosin 2 required for organizing arc actin into a concentric, contractile structure. If true, then the activation of RhoA would drive a coordinated activation pathway downstream of BCR ligation that leads to the formation of a force-generating actomyosin machine to power antigen extraction. Even if this turns out to be the case, many questions would remain. For example, actomyosin contraction would lead to APC membrane invagination only if the contractile network within the B cell is anchored to an internal structure that is more resistant to displacement than the APC membrane. Perhaps the nucleus serves this role. How the ideas discussed above relate to antigen extraction by GC B cells is another open question given that GC B cells appear to organize their actin and myosin quite differently than naïve B cells [13, 111].
Perhaps the best approach to defining the mechanism by which actomyosin contractility extracts antigens from APC surfaces is to image the antigen extraction process as it happens. The degree to which this approach is successful will depend heavily, however, on the specifics of how it is done. First, the extraction process should be imaged using primary B cells interacting with real APCs, as this would be the most physiological approach (short of imaging in vivo). Second, antigen should be presented by the APC in the most physiologically relevant fashion (ideally opsonized and presented via FcRs or complement receptors). This can be done in conjunction with the use of B cells isolated from a BCR transgenic mouse so that each B cell imaged has the same affinity for the antigen, and extraction efforts in the presence of strong and weak versions of the antigen can be readily compared. Third, the antigen, the plasma membrane of the APC, and the plasma membrane of the B cell should be tagged with different fluorescent labels so that one can know exactly where the antigen is in 3D space at all times (e.g. on the surface of the B cell or inside the B cell) and to follow possible trogocytosis events. Fourth, F-actin and myosin 2A inside the B cell should be followed in 3D over time to define their location and dynamics during the antigen extraction process. Ideally this imaging should be performed using a microscope that provides high resolution in x, y and z. One imaging modality that can accomplish this is 3D SIM, as it provides ~100 nm resolution in x and y and ~300 nm resolution in z (of note, this resolution allows one to see individual, ~300 nm-long myosin 2 bipolar filaments [117]). Importantly, TIRF imaging should probably be avoided because the rapid loss of fluorescent signal with increased z-depth may preclude a full understanding of how actomyosin generated pulling forces arranged perpendicular to the plasma membrane of the B cell drive APC membrane invagination and antigen extraction. Of note, these experiments should be performed using myosin 2 tagged on its heavy chain rather than its regulatory light chain because the latter is present on several other classes of myosin [118]. Additionally, F-Tractin should be used to follow F-actin dynamics instead of Lifeact (and especially instead of GFP-actin) because it labels formin-generated actin filaments much more robustly [57]. Hopefully the application of these approaches in various combinations (or even approximations of these approaches) will yield additional insight into how actomyosin is organized in the B cell to generate the pulling forces that power antigen extraction.
A major benefit of addressing the mechanism of antigen extraction using imaging is that this approach is non-intrusive; one is simply observing the event. The inhibition of specific players in the process like myosin 2, formins, and RhoA can, however, provide further insight into the underlying molecular mechanism of antigen extraction. Ideally, one has available a highly-specific, membrane-permeant, fast-acting, small-molecule inhibitor of the target protein, as this allows the protein’s function to be abrogated as close in time as possible to the actual antigen extraction event. This temporal control of inhibition is important because players in the extraction process may also have important roles in steps that precede extraction and are a prerequisite for extraction. For example, if myosin 2 is required for B cell contraction as well as antigen extraction, then its premature inhibition may preclude a full characterization of its role in antigen extraction. Alternatively, the premature inhibition of a protein suspected of playing a role in antigen extraction that in reality only functions earlier in IS formation may result in this protein being incorrectly assigned a role in antigen extraction. Given these considerations, approaches like RNAi and especially the use of B cells from KO mice where the protein is constitutively absent should be applied with caution. Fortunately, there are effective small-molecule inhibitors of several known or suspected players in antigen extraction, including formins (SMIFH2) [51], myosin 2 (Blebbistatin) [52], and the Arp2/3 complex (CK666) [119]. That said, some of these inhibitors require short preincubation times to be effective. Moreover, care should be taken in their use. For example. the original version of Blebbistatin is phototoxic when imaged with blue light [120]. Fortunately, newer versions do not have this problem [121, 122].
Our primary goal in writing this review was to focus on the emerging evidence that the synapses formed by T cells and B cells share may characteristics, including the presence of contractile actomyosin arcs. That said, there will almost certainly be interesting differences between their synapses, although these differences won’t become clear until the details of actomyosin arc formation in B cells are presented in future publications. The most interesting difference between T cells and B cells is already clear, however: how do they harness essentially the same contractile structure (i.e. actomyosin arcs) to drive very different outcomes- target cell killing in the case of cytotoxic T cells and antigen extraction in the case of B cells. More specifically, how precisely is this force harnessed in T cells to strain the target cell membrane so as to enhance target cell killing (Huse), and in B cells to pull antigen off the surface of an APC? Resolution of this fascinating question will require imaging both events in physiological contexts with high temporal and spatial resolution.
In conclusion, previous studies that focused on the role of actomyosin-dependent force generation in driving BCR: antigen affinity discrimination and antigen extraction have set the stage for intensive, microcopy-based approaches to define more precisely how actomyosin contractility accomplishes these tasks. The application of state-of-the-art, super-resolution imaging modalities together with the temporally-controlled inhibition of known and new players in these processes should provide further mechanistic insight into how myosin 2 drives the biology of both naïve and GC B cells.
Supplementary Material
Acknowledgments
Funding was provided by the Intramural Program of the NHLBI, NIH (1ZIAHL006121-04).
References
- [1].Lanzavecchia A, Antigen-specific interaction between T and B cells, Nature 314(6011) (1985) 537–9. [DOI] [PubMed] [Google Scholar]
- [2].Kuokkanen E, Sustar V, Mattila PK, Molecular control of B cell activation and immunological synapse formation, Traffic 16(4) (2015) 311–26. [DOI] [PubMed] [Google Scholar]
- [3].Song W, Liu C, Upadhyaya A, The pivotal position of the actin cytoskeleton in the initiation and regulation of B cell receptor activation, Biochim Biophys Acta 1838(2) (2013) 569–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Mattila PK, Batista FD, Treanor B, Dynamics of the actin cytoskeleton mediates receptor cross talk: An emerging concept in tuning receptor signaling, J Cell Biol 212(3) (2016) 267–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Yuseff MI, Pierobon P, Reversat A, Lennon-Dumenil AM, How B cells capture, process and present antigens: a crucial role for cell polarity, Nature reviews. Immunology 13(7) (2013) 475–86. [DOI] [PubMed] [Google Scholar]
- [6].Tolar P, Cytoskeletal control of B cell responses to antigens, Nature reviews. Immunology 17(10) (2017) 621–634. [DOI] [PubMed] [Google Scholar]
- [7].Harwood NE, Batista FD, The cytoskeleton coordinates the early events of B-cell activation, Cold Spring Harb Perspect Biol 3(2) (2011) a002360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Carrasco YR, Batista FD, B cell recognition of membrane-bound antigen: an exquisite way of sensing ligands, Current opinion in immunology 18(3) (2006) 286–91. [DOI] [PubMed] [Google Scholar]
- [9].Depoil D, Fleire S, Treanor BL, Weber M, Harwood NE, Marchbank KL, Tybulewicz VL, Batista FD, CD19 is essential for B cell activation by promoting B cell receptor-antigen microcluster formation in response to membrane-bound ligand, Nature immunology 9(1) (2008) 63–72. [DOI] [PubMed] [Google Scholar]
- [10].Hauser AE, Junt T, Mempel TR, Sneddon MW, Kleinstein SH, Henrickson SE, von Andrian UH, Shlomchik MJ, Haberman AM, Definition of germinal-center B cell migration in vivo reveals predominant intrazonal circulation patterns, Immunity 26(5) (2007) 655–67. [DOI] [PubMed] [Google Scholar]
- [11].Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, Nussenzweig MC, Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter, Cell 143(4) (2010) 592–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Natkanski E, Lee WY, Mistry B, Casal A, Molloy JE, Tolar P, B cells use mechanical energy to discriminate antigen affinities, Science (New York, N.Y.) 340(6140) (2013) 1587–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nowosad CR, Spillane KM, Tolar P, Germinal center B cells recognize antigen through a specialized immune synapse architecture, Nature immunology 17(7) (2016) 870–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Spillane KM, Tolar P, Mechanics of antigen extraction in the B cell synapse, Mol Immunol 101 (2018) 319–328. [DOI] [PubMed] [Google Scholar]
- [15].Yi J, Wu XS, Crites T, Hammer JA 3rd, Actin retrograde flow and actomyosin II arc contraction drive receptor cluster dynamics at the immunological synapse in Jurkat T cells, Molecular biology of the cell 23(5) (2012) 834–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Murugesan S, Hong J, Yi J, Li D, Beach JR, Shao L, Meinhardt J, Madison G, Wu X, Betzig E, Hammer JA, Formin-generated actomyosin arcs propel T cell receptor microcluster movement at the immune synapse, J Cell Biol 215(3) (2016) 383–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hong J, Murugesan S, Betzig E, Hammer JA, Contractile actomyosin arcs promote the activation of primary mouse T cells in a ligand-dependent manner, PLoS One 12(8) (2017) e0183174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wang JC, Lee JY, Christian S, Dang-Lawson M, Pritchard C, Freeman SA, Gold MR, The Rap1-cofilin-1 pathway coordinates actin reorganization and MTOC polarization at the B cell immune synapse, J Cell Sci 130(6) (2017) 1094–1109. [DOI] [PubMed] [Google Scholar]
- [19].Bolger-Munro M, Choi K, Scurll JM, Abraham L, Chappell RS, Sheen D, Dang-Lawson M, Wu X, Priatel JJ, Coombs D, Hammer JA, Gold MR, Arp2/3 complex-driven spatial patterning of the BCR enhances immune synapse formation, BCR signaling and cell activation, Elife 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Forthal DN, Functions of Antibodies, Microbiol Spectr 2(4) (2014) 1–17. [PMC free article] [PubMed] [Google Scholar]
- [21].Murphy KM, Weaver C, Mowat A, Berg L, Chaplin D, Janeway CA, Travers P, Walport M, Janeway’s immunobiology, 2017. [Google Scholar]
- [22].Batista FD, Neuberger MS, Affinity dependence of the B cell response to antigen: a threshold, a ceiling, and the importance of off-rate, Immunity 8(6) (1998) 751–9. [DOI] [PubMed] [Google Scholar]
- [23].Batista FD, Iber D, Neuberger MS, B cells acquire antigen from target cells after synapse formation, Nature 411(6836) (2001) 489–94. [DOI] [PubMed] [Google Scholar]
- [24].Qi H, T follicular helper cells in space-time, Nature reviews. Immunology 16(10) (2016) 612–25. [DOI] [PubMed] [Google Scholar]
- [25].Mesin L, Ersching J, Victora GD, Germinal Center B Cell Dynamics, Immunity 45(3) (2016) 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Heesters BA, Myers RC, Carroll MC, Follicular dendritic cells: dynamic antigen libraries, Nature reviews. Immunology 14(7) (2014) 495–504. [DOI] [PubMed] [Google Scholar]
- [27].Crotty S, A brief history of T cell help to B cells, Nature reviews. Immunology 15(3) (2015) 185–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mayer CT, Gazumyan A, Kara EE, Gitlin AD, Golijanin J, Viant C, Pai J, Oliveira TY, Wang Q, Escolano A, Medina-Ramirez M, Sanders RW, Nussenzweig MC, The microanatomic segregation of selection by apoptosis in the germinal center, Science (New York, N.Y.) 358(6360) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Fleire SJ, Goldman JP, Carrasco YR, Weber M, Bray D, Batista FD, B cell ligand discrimination through a spreading and contraction response, Science (New York, N.Y.) 312(5774) (2006) 738–41. [DOI] [PubMed] [Google Scholar]
- [30].Carrasco YR, Fleire SJ, Cameron T, Dustin ML, Batista FD, LFA-1/ICAM-1 interaction lowers the threshold of B cell activation by facilitating B cell adhesion and synapse formation, Immunity 20(5) (2004) 589–99. [DOI] [PubMed] [Google Scholar]
- [31].Li J, Yin W, Jing Y, Kang D, Yang L, Cheng J, Yu Z, Peng Z, Li X, Wen Y, Sun X, Ren B, Liu C, The Coordination Between B Cell Receptor Signaling and the Actin Cytoskeleton During B Cell Activation, Frontiers in immunology 9 (2018) 3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Song W, Liu C, Upadhyaya A, The pivotal position of the actin cytoskeleton in the initiation and regulation of B cell receptor activation, Biochim Biophys Acta 1838(2) (2014) 569–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Abraham L, Wang JC, Bolger-Munro M, Gold MR, Structure, function, and spatial organization of the B cell receptor, in: Ratcliffe MJH (Ed.), Encyclopedia of Immunobiology, Elsevier; 2016, pp. 40–54. [Google Scholar]
- [34].Hammer JA, Wang JC, Saeed M, Pedrosa AT, Origin, Organization, Dynamics, and Function of Actin and Actomyosin Networks at the T Cell Immunological Synapse, Annual review of immunology 37 (2019) 201–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kumari S, Depoil D, Martinelli R, Judokusumo E, Carmona G, Gertler FB, Kam LC, Carman CV, Burkhardt JK, Irvine DJ, Dustin ML, Actin foci facilitate activation of the phospholipase C-gamma in primary T lymphocytes via the WASP pathway, Elife 4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kaizuka Y, Douglass AD, Varma R, Dustin ML, Vale RD, Mechanisms for segregating T cell receptor and adhesion molecules during immunological synapse formation in Jurkat T cells, Proceedings of the National Academy of Sciences of the United States of America 104(51) (2007) 20296–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Nolz JC, Gomez TS, Zhu P, Li S, Medeiros RB, Shimizu Y, Burkhardt JK, Freedman BD, Billadeau DD, The WAVE2 complex regulates actin cytoskeletal reorganization and CRAC-mediated calcium entry during T cell activation, Current biology : CB 16(1) (2006) 24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zipfel PA, Bunnell SC, Witherow DS, Gu JJ, Chislock EM, Ring C, Pendergast AM, Role for the Abi/wave protein complex in T cell receptor-mediated proliferation and cytoskeletal remodeling, Current biology : CB 16(1) (2006) 35–46. [DOI] [PubMed] [Google Scholar]
- [39].Babich A, Li S, O’Connor RS, Milone MC, Freedman BD, Burkhardt JK, F-actin polymerization and retrograde flow drive sustained PLCgamma1 signaling during T cell activation, J Cell Biol 197(6) (2012) 775–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Fritzsche M, Fernandes RA, Chang VT, Colin-York H, Clausen MP, Felce JH, Galiani S, Erlenkamper C, Santos AM, Heddleston JM, Pedroza-Pacheco I, Waithe D, de la Serna JB, Lagerholm BC, Liu TL, Chew TL, Betzig E, Davis SJ, Eggeling C, Cytoskeletal actin dynamics shape a ramifying actin network underpinning immunological synapse formation, Sci Adv 3(6) (2017) e1603032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kuhn S, Geyer M, Formins as effector proteins of Rho GTPases, Small GTPases 5 (2014) e29513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Beach JR, Hammer JA 3rd, Myosin II isoform co-assembly and differential regulation in mammalian systems, Exp Cell Res 334(1) (2015) 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Cheffings TH, Burroughs NJ, Balasubramanian MK, Actomyosin Ring Formation and Tension Generation in Eukaryotic Cytokinesis, Current biology : CB 26(15) (2016) R719–R737. [DOI] [PubMed] [Google Scholar]
- [44].DeMond AL, Mossman KD, Starr T, Dustin ML, Groves JT, T cell receptor microcluster transport through molecular mazes reveals mechanism of translocation, Biophys J 94(8) (2008) 3286–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Yu CH, Wu HJ, Kaizuka Y, Vale RD, Groves JT, Altered actin centripetal retrograde flow in physically restricted immunological synapses, PLoS One 5(7) (2010) e11878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Smoligovets AA, Smith AW, Wu HJ, Petit RS, Groves JT, Characterization of dynamic actin associations with T-cell receptor microclusters in primary T cells, J Cell Sci 125(Pt 3) (2012) 735–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Comrie WA, Li S, Boyle S, Burkhardt JK, The dendritic cell cytoskeleton promotes T cell adhesion and activation by constraining ICAM-1 mobility, J Cell Biol 208(4) (2015) 457–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Comrie WA, Babich A, Burkhardt JK, F-actin flow drives affinity maturation and spatial organization of LFA-1 at the immunological synapse, J Cell Biol 208(4) (2015) 475–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Gardel ML, Schneider IC, Aratyn-Schaus Y, Waterman CM, Mechanical integration of actin and adhesion dynamics in cell migration, Annual review of cell and developmental biology 26 (2010) 315–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Case LB, Waterman CM, Integration of actin dynamics and cell adhesion by a three-dimensional, mechanosensitive molecular clutch, Nature cell biology 17(8) (2015) 955–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Rizvi SA, Neidt EM, Cui J, Feiger Z, Skau CT, Gardel ML, Kozmin SA, Kovar DR, Identification and characterization of a small molecule inhibitor of formin-mediated actin assembly, Chem Biol 16(11) (2009) 1158–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Straight AF, Cheung A, Limouze J, Chen I, Westwood NJ, Sellers JR, Mitchison TJ, Dissecting temporal and spatial control of cytokinesis with a myosin II Inhibitor, Science (New York, N.Y.) 299(5613) (2003) 1743–7. [DOI] [PubMed] [Google Scholar]
- [53].Burke TA, Christensen JR, Barone E, Suarez C, Sirotkin V, Kovar DR, Homeostatic actin cytoskeleton networks are regulated by assembly factor competition for monomers, Current biology : CB 24(5) (2014) 579–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Rotty JD, Wu C, Haynes EM, Suarez C, Winkelman JD, Johnson HE, Haugh JM, Kovar DR, Bear JE, Profilin-1 serves as a gatekeeper for actin assembly by Arp2/3-dependent and -independent pathways, Dev Cell 32(1) (2015) 54–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Lomakin AJ, Lee KC, Han SJ, Bui DA, Davidson M, Mogilner A, Danuser G, Competition for actin between two distinct F-actin networks defines a bistable switch for cell polarization, Nature cell biology 17(11) (2015) 1435–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Chen Q, Nag S, Pollard TD, Formins filter modified actin subunits during processive elongation, J Struct Biol 177(1) (2012) 32–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Belin BJ, Goins LM, Mullins RD, Comparative analysis of tools for live cell imaging of actin network architecture, Bioarchitecture 4(6) (2014) 189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Parsey MV, Lewis GK, Actin polymerization and pseudopod reorganization accompany anti-CD3-induced growth arrest in Jurkat T cells, Journal of immunology (Baltimore, Md. : 1950) 151(4) (1993) 1881–93. [PubMed] [Google Scholar]
- [59].Rak GD, Mace EM, Banerjee PP, Svitkina T, Orange JS, Natural killer cell lytic granule secretion occurs through a pervasive actin network at the immune synapse, PLoS Biol 9(9) (2011) e1001151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Carisey AF, Mace EM, Saeed MB, Davis DM, Orange JS, Nanoscale Dynamism of Actin Enables Secretory Function in Cytolytic Cells, Current biology : CB 28(4) (2018) 489–502 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Brown AC, Oddos S, Dobbie IM, Alakoskela JM, Parton RM, Eissmann P, Neil MA, Dunsby C, French PM, Davis I, Davis DM, Remodelling of cortical actin where lytic granules dock at natural killer cell immune synapses revealed by super-resolution microscopy, PLoS Biol 9(9) (2011) e1001152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Hammer JA, Immunology: Is Actin at the Lytic Synapse a Friend or a Foe?, Current biology : CB 28(4) (2018) R155–R157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Huang Y, Burkhardt JK, T-cell-receptor-dependent actin regulatory mechanisms, J Cell Sci 120(Pt 5) (2007) 723–30. [DOI] [PubMed] [Google Scholar]
- [64].Burkhardt JK, Carrizosa E, Shaffer MH, The actin cytoskeleton in T cell activation, Annual review of immunology 26 (2008) 233–59. [DOI] [PubMed] [Google Scholar]
- [65].Cannon JL, Burkhardt JK, Differential roles for Wiskott-Aldrich syndrome protein in immune synapse formation and IL-2 production, Journal of immunology (Baltimore, Md. : 1950) 173(3) (2004) 1658–62. [DOI] [PubMed] [Google Scholar]
- [66].Cianferoni A, Massaad M, Feske S, de la Fuente MA, Gallego L, Ramesh N, Geha RS, Defective nuclear translocation of nuclear factor of activated T cells and extracellular signal-regulated kinase underlies deficient IL-2 gene expression in Wiskott-Aldrich syndrome, J Allergy Clin Immunol 116(6) (2005) 1364–71. [DOI] [PubMed] [Google Scholar]
- [67].Calvez R, Lafouresse F, De Meester J, Galy A, Valitutti S, Dupre L, The Wiskott-Aldrich syndrome protein permits assembly of a focused immunological synapse enabling sustained T-cell receptor signaling, Haematologica 96(10) (2011) 1415–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Fuller CL, Braciale VL, Samelson LE, All roads lead to actin: the intimate relationship between TCR signaling and the cytoskeleton, Immunol Rev 191 (2003) 220–36. [DOI] [PubMed] [Google Scholar]
- [69].Courtney AH, Lo WL, Weiss A, TCR Signaling: Mechanisms of Initiation and Propagation, Trends Biochem Sci 43(2) (2018) 108–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Tybulewicz VL, Vav-family proteins in T-cell signalling, Current opinion in immunology 17(3) (2005) 267–74. [DOI] [PubMed] [Google Scholar]
- [71].Razidlo GL, Schroeder B, Chen J, Billadeau DD, McNiven MA, Vav1 as a central regulator of invadopodia assembly, Current biology : CB 24(1) (2014) 86–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Case LB, Zhang X, Ditlev JA, Rosen MK, Stoichiometry controls activity of phase-separated clusters of actin signaling proteins, Science (New York, N.Y.) 363(6431) (2019) 1093–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Huang WYC, Alvarez S, Kondo Y, Lee YK, Chung JK, Lam HYM, Biswas KH, Kuriyan J, Groves JT, A molecular assembly phase transition and kinetic proofreading modulate Ras activation by SOS, Science (New York, N.Y.) 363(6431) (2019) 1098–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Ditlev JA, Vega AR, Köster DV, Su X, Lakoduk AM, Vale RD, Mayor S, Jaqaman K, Rosen MK, A Composition-Dependent Molecular Clutch Between T Cell Signaling Clusters and Actin, bioRxiv (2018) 316414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Freeman SA, Lei V, Dang-Lawson M, Mizuno K, Roskelley CD, Gold MR, Cofilin-mediated F-actin severing is regulated by the Rap GTPase and controls the cytoskeletal dynamics that drive lymphocyte spreading and BCR microcluster formation, Journal of immunology (Baltimore, Md. : 1950) 187(11) (2011) 5887–900. [DOI] [PubMed] [Google Scholar]
- [76].Welch MD, Mullins RD, Cellular control of actin nucleation, Annual review of cell and developmental biology 18 (2002) 247–88. [DOI] [PubMed] [Google Scholar]
- [77].Pollard TD, Regulation of actin filament assembly by Arp2/3 complex and formins, Annu Rev Biophys Biomol Struct 36 (2007) 451–77. [DOI] [PubMed] [Google Scholar]
- [78].Firat-Karalar EN, Welch MD, New mechanisms and functions of actin nucleation, Curr Opin Cell Biol 23(1) (2011) 4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Carrasco YR, Batista FD, B-cell activation by membrane-bound antigens is facilitated by the interaction of VLA-4 with VCAM-1, EMBO J 25(4) (2006) 889–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Wang JC, Bolger-Munro M, Gold MR, Visualizing the Actin and Microtubule Cytoskeletons at the B-cell Immune Synapse Using Stimulated Emission Depletion (STED) Microscopy, J Vis Exp (134) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Keppler SJ, Burbage M, Gasparrini F, Hartjes L, Aggarwal S, Massaad MJ, Geha RS, Bruckbauer A, Batista FD, The Lack of WIP Binding to Actin Results in Impaired B Cell Migration and Altered Humoral Immune Responses, Cell Rep 24(3) (2018) 619–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Liu C, Fallen MK, Miller H, Upadhyaya A, Song W, The actin cytoskeleton coordinates the signal transduction and antigen processing functions of the B cell antigen receptor, Front Biol (Beijing) 8(5) (2013) 475–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Volpi S, Santori E, Abernethy K, Mizui M, Dahlberg CI, Recher M, Capuder K, Csizmadia E, Ryan D, Mathew D, Tsokos GC, Snapper S, Westerberg LS, Thrasher AJ, Candotti F, Notarangelo LD, N-WASP is required for B-cell-mediated autoimmunity in Wiskott-Aldrich syndrome, Blood 127(2) (2016) 216–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Liu C, Bai X, Wu J, Sharma S, Upadhyaya A, Dahlberg CI, Westerberg LS, Snapper SB, Zhao X, Song W, N-wasp is essential for the negative regulation of B cell receptor signaling, PLoS Biol 11(11) (2013) e1001704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Treanor B, Depoil D, Bruckbauer A, Batista FD, Dynamic cortical actin remodeling by ERM proteins controls BCR microcluster organization and integrity, J Exp Med 208(5) (2011) 1055–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Peskin CS, Odell GM, Oster GF, Cellular motions and thermal fluctuations: the Brownian ratchet, Biophys J 65(1) (1993) 316–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Song W, Liu C, Seeley-Fallen MK, Miller H, Ketchum C, Upadhyaya A, Actin-mediated feedback loops in B-cell receptor signaling, Immunol Rev 256(1) (2013) 177–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Batista FD, Neuberger MS, B cells extract and present immobilized antigen: implications for affinity discrimination, EMBO J 19(4) (2000) 513–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, Daeron M, Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses, Blood 113(16) (2009) 3716–25. [DOI] [PubMed] [Google Scholar]
- [90].Sarrias MR, Franchini S, Canziani G, Argyropoulos E, Moore WT, Sahu A, Lambris JD, Kinetic analysis of the interactions of complement receptor 2 (CR2, CD21) with its ligands C3d, iC3b, and the EBV glycoprotein gp350/220, Journal of immunology (Baltimore, Md. : 1950) 167(3) (2001) 1490–9. [DOI] [PubMed] [Google Scholar]
- [91].Dustin ML, Complement Receptors in Myeloid Cell Adhesion and Phagocytosis, Microbiol Spectr 4(6) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Bajic G, Yatime L, Sim RB, Vorup-Jensen T, Andersen GR, Structural insight on the recognition of surface-bound opsonins by the integrin I domain of complement receptor 3, Proceedings of the National Academy of Sciences of the United States of America 110(41) (2013) 16426–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Gadjeva M, Dodds AW, Taniguchi-Sidle A, Willis AC, Isenman DE, Law SK, The covalent binding reaction of complement component C3, Journal of immunology (Baltimore, Md. : 1950) 161(2) (1998) 985–90. [PubMed] [Google Scholar]
- [94].Spillane KM, Tolar P, B cell antigen extraction is regulated by physical properties of antigen-presenting cells, J Cell Biol 216(1) (2017) 217–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Liu B, Chen W, Evavold BD, Zhu C, Accumulation of dynamic catch bonds between TCR and agonist peptide-MHC triggers T cell signaling, Cell 157(2) (2014) 357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Hong J, Ge C, Jothikumar P, Yuan Z, Liu B, Bai K, Li K, Rittase W, Shinzawa M, Zhang Y, Palin A, Love P, Yu X, Salaita K, Evavold BD, Singer A, Zhu C, A TCR mechanotransduction signaling loop induces negative selection in the thymus, Nature immunology 19(12) (2018) 1379–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Wan Z, Chen X, Chen H, Ji Q, Chen Y, Wang J, Cao Y, Wang F, Lou J, Tang Z, Liu W, The activation of IgM- or isotype-switched IgG- and IgE-BCR exhibits distinct mechanical force sensitivity and threshold, Elife 4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Wan Z, Zhang S, Fan Y, Liu K, Du F, Davey AM, Zhang H, Han W, Xiong C, Liu W, B cell activation is regulated by the stiffness properties of the substrate presenting the antigens, Journal of immunology (Baltimore, Md. : 1950) 190(9) (2013) 4661–75. [DOI] [PubMed] [Google Scholar]
- [99].Sun Z, Guo SS, Fassler R, Integrin-mediated mechanotransduction, J Cell Biol 215(4) (2016) 445–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Busman-Sahay K, Drake L, Sitaram A, Marks M, Drake JR, Cis and trans regulatory mechanisms control AP2-mediated B cell receptor endocytosis via select tyrosine-based motifs, PLoS One 8(1) (2013) e54938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Bellve KD, Leonard D, Standley C, Lifshitz LM, Tuft RA, Hayakawa A, Corvera S, Fogarty KE, Plasma membrane domains specialized for clathrin-mediated endocytosis in primary cells, The Journal of biological chemistry 281(23) (2006) 16139–46. [DOI] [PubMed] [Google Scholar]
- [102].Sochacki KA, Taraska JW, From Flat to Curved Clathrin: Controlling a Plastic Ratchet, Trends Cell Biol 29(3) (2019) 241–256. [DOI] [PubMed] [Google Scholar]
- [103].Ferguson SM, De Camilli P, Dynamin, a membrane-remodelling GTPase, Nature reviews. Molecular cell biology 13(2) (2012) 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Stoddart A, Dykstra ML, Brown BK, Song W, Pierce SK, Brodsky FM, Lipid rafts unite signaling cascades with clathrin to regulate BCR internalization, Immunity 17(4) (2002) 451–62. [DOI] [PubMed] [Google Scholar]
- [105].Martinez-Martin N, Fernandez-Arenas E, Cemerski S, Delgado P, Turner M, Heuser J, Irvine DJ, Huang B, Bustelo XR, Shaw A, Alarcon B, T cell receptor internalization from the immunological synapse is mediated by TC21 and RhoG GTPase-dependent phagocytosis, Immunity 35(2) (2011) 208–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Aucher A, Magdeleine E, Joly E, Hudrisier D, Capture of plasma membrane fragments from target cells by trogocytosis requires signaling in T cells but not in B cells, Blood 111(12) (2008) 5621–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Dopfer EP, Minguet S, Schamel WW, A new vampire saga: the molecular mechanism of T cell trogocytosis, Immunity 35(2) (2011) 151–3. [DOI] [PubMed] [Google Scholar]
- [108].Puaux AL, Campanaud J, Salles A, Preville X, Timmerman B, Joly E, Hudrisier D, A very rapid and simple assay based on trogocytosis to detect and measure specific T and B cell reactivity by flow cytometry, Eur J Immunol 36(3) (2006) 779–88. [DOI] [PubMed] [Google Scholar]
- [109].Suzuki K, Grigorova I, Phan TG, Kelly LM, Cyster JG, Visualizing B cell capture of cognate antigen from follicular dendritic cells, J Exp Med 206(7) (2009) 1485–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Hoogeboom R, Natkanski EM, Nowosad CR, Malinova D, Menon RP, Casal A, Tolar P, Myosin IIa Promotes Antibody Responses by Regulating B Cell Activation, Acquisition of Antigen, and Proliferation, Cell Rep 23(8) (2018) 2342–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Kwak K, Quizon N, Sohn H, Saniee A, Manzella-Lapeira J, Holla P, Brzostowski J, Lu J, Xie H, Xu C, Spillane KM, Tolar P, Pierce SK, Intrinsic properties of human germinal center B cells set antigen affinity thresholds, Sci Immunol 3(29) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Yuseff M-I, Reversat A, Lankar D, Diaz J, Fanget I, Pierobon P, Randrian V, Larochette N, Vascotto F, Desdouets C, Jauffred B, Bellaiche Y, Gasman S, Darchen F, Desnos C, Lennon-Duménil A-M, Polarized Secretion of Lysosomes at the B Cell Synapse Couples Antigen Extraction to Processing and Presentation, Immunity 35(3) (2011) 361–374. [DOI] [PubMed] [Google Scholar]
- [113].Pierobon P, Lennon-Dumenil AM, To use or not to use the force: How B lymphocytes extract surface-tethered antigens, J Cell Biol 216(1) (2017) 17–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Obino D, Diaz J, Saez JJ, Ibanez-Vega J, Saez PJ, Alamo M, Lankar D, Yuseff MI, Vamp-7-dependent secretion at the immune synapse regulates antigen extraction and presentation in B-lymphocytes, Molecular biology of the cell 28(7) (2017) 890–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Haddad EK, Wu X, Hammer JA 3rd, Henkart PA, Defective granule exocytosis in Rab27a-deficient lymphocytes from Ashen mice, J Cell Biol 152(4) (2001) 835–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Stinchcombe JC, Barral DC, Mules EH, Booth S, Hume AN, Machesky LM, Seabra MC, Griffiths GM, Rab27a is required for regulated secretion in cytotoxic T lymphocytes, J Cell Biol 152(4) (2001) 825–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Beach JR, Shao L, Remmert K, Li D, Betzig E, Hammer JA 3rd, Nonmuscle myosin II isoforms coassemble in living cells, Current biology : CB 24(10) (2014) 1160–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Heissler SM, Sellers JR, Myosin light chains: Teaching old dogs new tricks, Bioarchitecture 4(6) (2014) 169–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Hetrick B, Han MS, Helgeson LA, Nolen BJ, Small molecules CK-666 and CK-869 inhibit actin-related protein 2/3 complex by blocking an activating conformational change, Chem Biol 20(5) (2013) 701–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Sakamoto T, Limouze J, Combs CA, Straight AF, Sellers JR, Blebbistatin, a myosin II inhibitor, is photoinactivated by blue light, Biochemistry 44(2) (2005) 584–8. [DOI] [PubMed] [Google Scholar]
- [121].Kepiro M, Varkuti BH, Vegner L, Voros G, Hegyi G, Varga M, Malnasi-Csizmadia A, para-Nitroblebbistatin, the non-cytotoxic and photostable myosin II inhibitor, Angew Chem Int Ed Engl 53(31) (2014) 8211–5. [DOI] [PubMed] [Google Scholar]
- [122].Varkuti BH, Kepiro M, Horvath IA, Vegner L, Rati S, Zsigmond A, Hegyi G, Lenkei Z, Varga M, Malnasi-Csizmadia A, A highly soluble, non-phototoxic, non-fluorescent blebbistatin derivative, Sci Rep 6 (2016) 26141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Hashimoto-Tane A, Yokosuka T, Sakata-Sogawa K, Sakuma M, Ishihara C, Tokunaga M, Saito T, Dynein-driven transport of T cell receptor microclusters regulates immune synapse formation and T cell activation, Immunity 34(6) (2011) 919–31. [DOI] [PubMed] [Google Scholar]
- [124].Schnyder T, Castello A, Feest C, Harwood NE, Oellerich T, Urlaub H, Engelke M, Wienands J, Bruckbauer A, Batista FD, B cell receptor-mediated antigen gathering requires ubiquitin ligase Cbl and adaptors Grb2 and Dok-3 to recruit dynein to the signaling microcluster, Immunity 34(6) (2011) 905–18. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.