

Abstract
Hippocampal atrophy is endemic in ‘normal aging’ but it is unclear what factors drive age-related changes in medial temporal lobe (MTL) structural measures. We investigated cross-sectional (n=191) and longitudinal (n=164) MTL atrophy patterns in cognitively normal older adults from ADNI-GO/2 with no to low cerebral β-amyloid and assessed whether white matter hyperintensities (WMHs) and cerebrospinal fluid (CSF) phospho tau (p-tau) levels can explain age-related changes in the MTL. Age was significantly associated with hippocampal volumes and Brodmann Area (BA) 35 thickness, regions affected early by neurofibrillary tangle pathology, in the cross-sectional analysis and with anterior/posterior hippocampus, entorhinal cortex and BA35 in the longitudinal analysis. CSF p-tau was significantly associated with hippocampal volumes and atrophy rates. Mediation analyses showed that CSF p-tau levels partially mediated age effects on hippocampal atrophy rates. No significant associations were observed for WMHs. These findings point towards a role of tau pathology, potentially reflecting Primary Age-Related Tauopathy, in age-related MTL structural changes and suggests a potential role for tau-targeted interventions in age-associated neurodegeneration and memory decline.
Introduction
Both hippocampal atrophy and memory problems are endemic to ‘normal aging’ (Barnes et al., 2009, Fjell and Walhovd, 2010, Hoyer and Verhaeghen, 2006, Rönnlund et al., 2005). While age effects on the whole hippocampus are well established, studies investigating age-effects on more granular medial temporal lobe (MTL) regions, including adjacent cortical structures, report inconsistent results (i.e. entorhinal cortex (ERC) (Du, A. T. et al., 2003, Insausti et al., 1998, Knoops et al., 2012, Price, J. L. et al., 2001, Raz, N. et al., 2004, Wisse et al., 2014)) and are sparse with regard to the regions represented, as studies on age-effects often do not include MTL regions such as the perirhinal and parahippocampal cortex (PRC, PHC) or anterior and posterior subsections of the hippocampus (but note (Chen et al., 2010, Dickerson et al., 2009, Fjell et al., 2009, Insausti et al., 1998, Jack et al., 1997, Malykhin et al., 2008, Raz, Naftali et al., 1997)). As different disease processes differentially involve MTL subregions, a better characterization of age-related changes in the MTL is of great importance to gain more insight in age-related cognitive decline and to be able to separate “normal” age-related changes in the MTL from the “pathological” changes of preclinical stages of dementia. Moreover, it is still unclear what ‘normal aging’ entails and what drives aging effects on the MTL.
There have been several barriers to gaining a more precise understanding of the effects of age on the MTL. First, there are several methodological challenges when assessing the MTL. MTL subregions are a difficult to quantify accurately due to anatomical variants, mostly tied to the depth of the collateral sulcus (Ding and Van Hoesen, 2010). The location of ERC and adjacent PRC subregions differ in these anatomical variants and not taking this into account will decrease the validity of the measurements. Moreover, MTL cortical regions, such as ERC, can be confounded by dura mater which has similar intensity as cortex on T1-weighted magnetic resonance imaging (MRI) scans and can therefore lead to segmentation errors (Xie, L. et al., 2016, Xie, Long et al., 2019). We have recently developed a pipeline to segment MTL subregions on T1-weighted MRI overcoming these challenges and provided granular MTL subregional structural measures. Importantly, subregions of the PRC, Brodmann Area (BA) 35 and 36, but also anterior and posterior subregions of the hippocampus, are incorporated in this method. These granular measures are important to assess, as, for example, BA35 approximates the transentorhinal region which is the first supratentorial brain region with neurofibrillary tangle (NFT) pathology (Braak, H. and Braak, 1991).
A second barrier with most extant studies is the use of cross-sectional rather than longitudinal designs (but note (Du, A. T. et al., 2003, Du, An-Tao et al., 2006, Raz, N. et al., 2005)). Longitudinal measurement may be preferable as it is not confounded by, for example, developmental factors, and longitudinal measures of structural change are more closely linked to active neurodegenerative processes.
A third major factor which may have confounded the previous literature’s precision for measuring age-specific effects on the MTL is a lack of accounting for the presence of preclinical Alzheimer’s Disease (AD) in cognitively normal older adults. Between ~25-35% of cognitively normal older adults harbor a significant amount of β-amyloid pathology (Jansen et al., 2015), reflecting preclinical AD, which likely influences the observed ‘age-related’ changes in extant studies. Indeed, cognitively normal adults with β-amyloid pathology have been shown to have more atrophy in regions typically affected by NFT pathology (Wolk et al., 2017, Xie, Long et al., 2020), compared to those with no or low levels of β-amyloid pathology. Individuals with high levels of β-amyloid pathology should therefore be eliminated as a factor when studying ‘normal aging’ effects on the brain.
Outside of preclinical AD, there are at least four major factors commonly considered potential contributors to observed age-related changes in the MTL: cerebrovascular disease (CVD), Primary Age-related Tauopathy (PART) (Crary et al., 2014), TAR DNA-binding Protein 43 (TDP-43) pathology in the form of the recently codified limbic-predominant age-related TDP-43 encephalopathy (LATE) (Nelson, Peter et al., 2019), and non-specific age effects. While it could be argued that CVD, PART and LATE do not reflect ‘normal aging’ but are rather forms of pathological aging, we still focus on these processes as they may reflect changes that have traditionally been considered normal aging. Additionally, some of these factors are so common, e.g. PART, that it would be difficult to disentangle them from normal aging. CVD is thought to specifically affect the cornu ammonis (CA) 1 subfield of the hippocampus and lead to neuron loss, potentially through mechanisms of hypoxia and ischemia (Schmidt-Kastner and Freund, 1991, Zola-Morgan et al., 1992). PART is also thought to specifically affect the MTL, by targeting the transentorhinal cortex (approximately the same as BA35) first, then ERC followed by the CA1 subfield of the hippocampus (Braak, H. and Braak, 1995, Crary et al., 2014, Price, Joseph L. and Morris, 1999), although there is evidence that the pattern of MTL tau pathology is slightly different in PART than AD ((Jellinger, 2018), but note (Zhang et al., 2020)). Tau pathology in PART has been found to lead to neuron loss and memory impairments (Crary et al., 2014, Jefferson-George et al., 2017, Josephs et al., 2017, Quintas-Neves et al., 2019), albeit less severe than in the presence of β-amyloid (Bell et al., 2019, Besser et al., 2019). Third, LATE (Nelson, Peter T. et al., 2019) may also drive age-related changes in the MTL, particularly involving anterior MTL regions, producing an amnestic syndrome with increasing prevalence in adults generally over 80 years (Josephs et al., 2014, Nag et al., 2017). Finally, there are non-specific age effects, such as impaired neurogenesis (Apple et al., 2017, Galvan and Jin, 2007, Isaev et al., 2019), impaired synaptic plasticity (Yankner et al., 2008), downstream effects of inflammation (Wyss-Coray, 2016), glucocorticoids (McEwen et al., 1999, Nichols et al., 2001), and potential other mechanisms. While there are biomarkers that provide measures of CVD (e.g. white matter hyperintensities (WMHs)) and potentially PART [e.g. cerebrospinal fluid (CSF) phosphor-tau181 (p-tau)], TDP-43 and these other non-specific factors are more difficult to assess. MTL atrophy pattern may therefore also provide clues as to what may be the primary drivers of age-associated atrophy.
In this study, we first investigate age effects on MTL subregional structural measures obtained from structural MRI in older adults with no to low β-amyloid levels (often referred to as β-amyloid negative; from here referred to as low β-amyloid) in ADNI-GO/2, both in a cross-sectional and longitudinal manner. Second, we will investigate the role of p-tau in CSF, as proxy for PART, and WMHs, as a proxy for CVD, as potential drivers of aging effects. Given the known regional distribution of early NFTs in PART (Braak, H. and Braak, 1995, Crary et al., 2014, Price, Joseph L. and Morris, 1999), we hypothesize that if PART is a driver of age-associated effects, BA35 and ERC would likely be most affected followed by hippocampal measures. We would also expect an association of CSF p-tau with these regions. Alternatively, if CVD is a primary driver, we would anticipate hippocampal volumes to be the most saliently affected region of the MTL (Schmidt-Kastner and Freund, 1991, Zola-Morgan et al., 1992) and would expect an association with WMHs. If TDP-43 is driving age-associated effects, we would expect atrophy in anterior MTL regions, particularly the anterior hippocampus and ERC (de Flores et al., 2020, Josephs et al., 2014, Nag et al., 2017) with age.
Methods
Participants
Data from all cognitively normal subjects with available biomarkers of cerebral amyloidosis (β-amyloid positron emission tomography (PET)) and neurodegeneration (baseline structural T1 magnetic resonance images (MRI)) from the Alzheimer’s NeuroImaging Initiative (ADNI)-GO and ADNI-2 cohorts were included.
The MRI at baseline as well as all follow up scans within 1.2 to 4.5 years were selected, to maximize the number of subjects included in the longitudinal analyses. Additional information, including in- and exclusion criteria for cognitively normal older adults, are provided in the Supplementary Methods.
Demographics, medical and neuropsychological information
Standard demographics information was obtained, including race. Self-reported history of head injury, smoking, hypertension, stroke and cardiovascular disease was obtained as well as the family history of dementia. Note that certain factors related to cardiovascular disease were reason for exclusion, such as presence of pacemakers, aneurysm clips, artificial heart valves, multiple lacunes or lacunes in a critical memory structure, a Hachinski score (Rosen et al., 1980) less than or equal to 4 and multi-infarct dementia (see Supplementary Methods). History of head injury was not assessed in a standardized way. We therefore searched the self-reported medical history and included terms as traumatic brain injury, head injury, concussion and loss of consciousness. Note that history of significant head trauma followed by persistent neurologic defaults or known structural brain abnormalities was an exclusion criterion (see Supplementary Methods). Additionally, measurements of mean arterial pressure (MAP) and body mass index (BMI) were obtained. APOE-ε4 carrier status was obtained via standard methods (Saykin et al., 2010). Positive APOE-ε4 status was defined as having at least one APOE-ε4 allele. Finally, the Mini Mental State Examination (MMSE) (Folstein et al., 1975) and Clinical Dementia Rating (Morris, 1993) were obtained.
Phospho-tau and β-amyloid PET
Both the CSF and PET measures came from publicly available, processed data on the ADNI website. CSF levels of phospho-tau181 (p-tau) were measured using the Elecsys phosphotau (181P) immunoassays on a cobas e 601 analyzer according to the preliminary kit manufacturer’s instructions, as previously described (Bittner et al., 2016, Hansson et al., 2018). For both measures data from the baseline visit was used. A composite, standardized uptake value ratio (SUVR) for the florbetapir images was calculated by taking the mean SUVR of a set of regions typically associated with increased uptake in AD (lateral and medial frontal, anterior and posterior cingulate, lateral parietal and lateral temporal regions), using gray matter of the cerebellum as reference region (Landau et al., 2012). β-amyloid status was defined by a florbetapir SUVR value of 1.11 (Landau et al., 2012). Only cognitively normal older adults with low β-amyloid levels were included.
Imaging protocol and image processing
T1-weighted MRI scans were acquired from different scanners at multiple sites. Up-to-date information about MRI imaging protocols can be found at adni.loni.usc.edu/methods/mri-tool/mri-analysis. The resolution of the scans ranged from 0.86x0.86x1.00 to 1.02x1.02x1.20 mm3. The MRI at baseline as well as all follow up scans within 1.2 to 4.5 years were selected.
MTL structural measures
The anterior and posterior hippocampus, ERC, BA35 and BA36 and the PHC were automatically segmented using the Automated Segmentation of Hippocampal Subfields (ASHS) package for T1-weighted MRI segmentation (ASHS-T1) (Xie, Long et al., 2019). Intracranial volume (ICV) was also measured using ASHS-T1. For the MTL cortical subregions (ERC, BA35, BA36 and PHC), a graph-based multi-template thickness analysis pipeline (Xie, Long et al., 2018) was applied to the automatic segmentation to derive the thickness of each subregion. See Supplementary Figure 1 for an example segmentation.
Symmetric diffeomorphic registration (Avants et al., 2008) was performed between the baseline and each of the follow-up MRI scans, as implemented in the Automatic Longitudinal Hippocampal Atrophy (ALOHA) software (Das, S. R. et al., 2012), to obtain unbiased estimates of the volume of each subregion of all subsequent MRI scans. Additional information can be found in the supplements. It should be noted that between 11.7% and 33.7% of all included subjects had a positive atrophy rate for the different MTL subregions. This is a common phenomenon, reported for different algorithms and these percentages fall well within the range of what is reported in the literature (Sankar et al., 2017). The percentage of cases with positive atrophy rates is partly due to the selection of low β-amyloid cognitively normal older adults. For example, the percentages are notably lower in the high β-amyloid Mild Cognitive Impairment (MCI) group (between 8.0-12.8%). It is unclear if the positive atrophy rates reflect a measurement error or a true biological phenomenon. We have therefore decided to keep all subjects with a positive atrophy rate in our analyses, especially since scan-rescan reliability can likely explain part of the positive atrophy rate and only between 2.5% and 11.7% of low β-amyloid cognitively normal older adults have a positive atrophy rate higher than 0.5% for the different MTL subregions. Supplementary Figure 2 shows the distribution of the atrophy rates for each of the six subregions.
Bilateral measurements of each subregion were averaged for both the cross-sectional and the longitudinal data.
White matter hyperintensities
WMHs were characterized using a deep learning-based segmentation method (Doshi et al., 2019). The proposed method is built upon the UNet architecture (Ronneberger et al., 2015) with the convolutional layers in the network replaced by an Inception ResNet architecture (Szegedy et al., 2016), which was previously shown to outperform traditional convolutional network architectures while also achieving dramatically improved training speed. The deep-learning model used inhomogeneity corrected and co-registered Fluid-attenuated inversion recovery (FLAIR) and T1-weighted images for segmentation. The model was trained using a separate training set with human-validated segmentation of WMH. The model was applied to participants to calculate binary WMH masks and the lesion volumes in different regions of interest. WMHs were obtained from the baseline FLAIR and T1-weighted images.
Statistical analyses
Structural MTL measures of left and right hemisphere were averaged. Sex and ICV were regressed out for hippocampal volumes and sex was regressed out for thickness measures and atrophy rates. All continuous variables were assessed for normality. CSF p-tau and WMH volume (also corrected for ICV) measures were log-transformed for normality. Model assumptions were checked and met for all analyses. All analyses were performed in IBM SPSS Statistics Version 26.0 (Armonk, NY, IBM Corp.) unless otherwise specified.
Linear regression models were used to assess the cross-sectional association of age. For longitudinal atrophy rates, first the atrophy rates were compared to 0 with a one-sample t-test. Second, the atrophy rates of the different MTL subregions were compared with each other using paired sample t-tests. Third, a linear regression was used to assess the association of age with atrophy rates.
Pearson correlations were performed to assess the association of p-tau and WMHs with MTL cross-sectional and longitudinal measures. For WMHs in the different lobes log transformations did not approach normality. Spearman correlations were therefore performed using the ppcor package in R Studio (www.r-project.org).
Finally, mediation analyses were performed using the Lavaan package in R Studio to assess whether p-tau or WMH volumes mediate the effect of age on MTL structural measures. In the p-tau mediation analyses we additionally corrected for continuous β-amyloid florbetapir levels to ensure that any effects of p-tau levels are not due to subthreshold β-amyloid levels.
We corrected for multiple comparisons using a Holm-Bonferroni correction per analysis.
Results
Demographics
Table 1 displays the demographics of full dataset of older adults with low β-amyloid levels (n=191) and the subset with longitudinal data (n=164). Age, sex, education, MAP, BMI, APOE-ε4 carrier status, possible history of head injury, history of smoking, hypertension, stroke, cardiovascular disease and family history of dementia were not different between participants with and without longitudinal data. However, those without longitudinal data had lower MMSE scores (28.3±1.7 vs 29.1±1.2; p=0.01), as well as a trend towards a higher percentage of history of hypertension (62.1% versus 43.7%, p=0.07) and a slightly different distribution of race (see Supplementary Table 1, p=0.09).
Table 1.
Demographics table of the full cross-sectional dataset and the subset with longitudinal data of older adults with low β-amyloid levels
| Cross-sectional dataset | Longitudinal dataset | |
|---|---|---|
| Number | 191 | 164 |
| Age (years) (range) | 71.7±6.0 (56-89) | 71.5±6.1 (56-89) |
| Sex (% female) | 47.6 | 46.3 |
| Race (%) | ||
| America Indian or Alaskan native | 0.5 | 0.6 |
| Asian | 2.1 | 2.4 |
| Black or African American | 4.7 | 3.0 |
| White | 90.1 | 90.9 |
| More than one race | 2.6 | 3.0 |
| APOE-ε4 carrier (%)* | 20.4 | 22.0 |
| Education (years) | 16.9±2.4 | 16.9±2.4 |
| BMI (kg/m2) | 27.9±5.1 | 27.9±5.0 |
| MAP (mm Hg) | 93.0±9.7 | 93.3±9.6 |
| History of smoking (%) | 36.6 | 36.6 |
| History of hypertension (%) | 46.6 | 46.3 |
| History of stroke (%) | 1.0 | 1.2 |
| History of cardiovascular disease | 63.4 | 62.2 |
| Possible history of head injury (%) | 3.1 | 3.7 |
| Family history of dementia (%) | 51.9 | 52.8 |
| MMSE | 29.0±1.3 | 29.1±1.2 |
| Follow up time (years) | - | 3.3±1.0 |
One or two alleles.
BMI=body mass index, kg=kilogram, m=meter, MAP=mean arterial pressure, mm Hg=millimeters of mercury, MMSE=Mini Mental State Examination
MTL atrophy patterns related to aging
We first investigated how MTL structural measures are related to age to better characterize age-related MTL atrophy patterns. Cross-sectional associations were found between age and anterior and posterior hippocampal volumes and BA35 thickness (Table 2, Figure 1a, Supplementary Figure 3), although anterior hippocampal volume was no longer significant when correcting for multiple comparisons. No significant associations were observed when a quadratic term for age was included in the model.
Table 2.
Relationship of age with cross-sectional and longitudinal MTL structural measures in cognitively normal older adults with low β-amyloid levels
| Cross-sectional dataset | Longitudinal dataset | |||
|---|---|---|---|---|
|
| ||||
| Standardized beta | p-value | Standardized beta | p-value | |
| AH | −0.17 | 0.02 | −0.23 | 0.004 |
| PH | −0.24 | 0.001 | −0.27 | 0.001 |
| ERC | 0.07 | 0.34 | −0.22 | 0.004 |
| BA35 | −0.24 | 0.001 | −0.30 | <0.001 |
| BA36 | −0.09 | 0.20 | −0.17 | 0.03 |
| PHC | −0.12 | 0.09 | −0.12 | 0.12 |
Bolded p-values meet significant after correction for multiple comparisons using the Holm-Bonferroni correction. Linear regression models were performed without covariates. Sex and intracranial volume are regressed out for hippocampal volumes, sex is regressed out for MTL cortical thickness measures and MTL atrophy rates.
AH=anterior hippocampus, BA=Brodmann Area, ERC=entorhinal cortex, MTL=medial temporal lobe, PH=posterior hippocampus, PHC=parahippocampal cortex.
Figure 1.
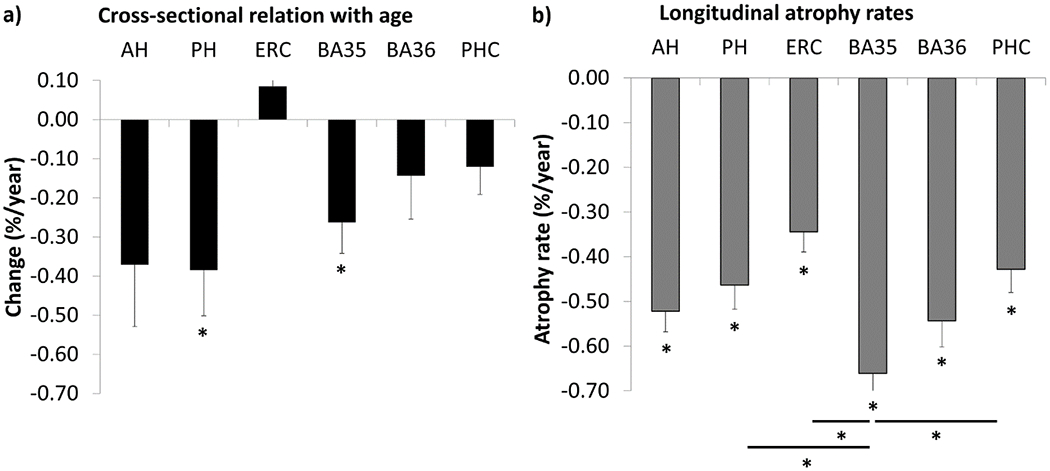
The association of age and cross-sectional structural MTL measures and longitudinal atrophy rates in cognitively normal older adults with low β-amyloid levels. a) The association of cross-sectional MTL measures (volume for hippocampus, thickness for cortical regions) with age is displayed as percentage volume loss per year. b) Longitudinal atrophy rates of MTL structural measures are displayed.
*Significant after correction for multiple comparisons using the Holm-Bonferroni correction. Significant results before multiple comparison correction are not shown in this figure. For the cross-sectional data, % volume loss/year was calculated using the regression coefficients. Sex and intracranial volume are regressed out for hippocampal volumes, sex is regressed out for MTL cortical thickness measures and MTL atrophy rates. The error bars represent standard errors. Negative values on the y-axis represent volume or thickness loss. AH=anterior hippocampus, BA=Brodmann Area, ERC=entorhinal cortex, MTL=medial temporal lobe, PH=posterior hippocampus, PHC=parahippocampal cortex.
Longitudinal atrophy rates of each region are displayed in Figure 1b and show a roughly similar pattern as the cross-sectional measures, although the estimated atrophy rates are slightly lower cross-sectionally and the relative degree of change between the regions is different. Atrophy rates in all MTL subregions were significantly different from 0 after multiple comparison correction, indicating that each region shows significant atrophy over the average follow up time of 3.3 years. BA35 atrophy rates were significantly larger than all other MTL regions, and still reached significance for the posterior hippocampus, ERC, and PHC after multiple comparison correction (see Supplementary Table 2). Additionally, the anterior hippocampus had significantly larger atrophy rates than ERC and BA36 than PHC, but not after multiple comparison correction. As it is not entirely certain whether %/year or mm3/year is a better reflection of burden in a given region, we also report the absolute annualized change in volumes in Supplementary Table 3.
Age was significantly associated with atrophy rates in the anterior and posterior hippocampus, ERC, BA35 and BA36, and remained significant after multiple comparison correction for all regions except BA36 (Table 2, Supplementary Figure 4). No significant associations were observed for quadratic models.
In general, the atrophy pattern associated with age had most pronounced atrophy in BA35 and the hippocampus, and to a lesser extent ERC, which mostly coincides with the hypothesized atrophy pattern in PART. The lack of a clear greater anterior-to-posterior gradient in these age-effects argues against the role of LATE as a factor. In the next sections we investigate whether MTL structural changes are associated with CSF p-tau levels as a proxy for PART and WMH volume, as a proxy for CVD.
Role of WMHs as contributor to age-related changes in the MTL
All analyses were run initially without correcting for age, as age is associated with increased WMHs and is therefore not necessarily a confounder but may be part of the causal pathway. In secondary analyses, we corrected for age.
The cross-sectional analyses did not reveal a significant association of WMH volumes with MTL structural measures, except for posterior hippocampal volume, which survived multiple comparison and age correction (Table 3). Note that this was positive correlation where larger WMH volumes were associated with larger hippocampal volumes. This was likely driven by the few subjects with low WMH volumes (Supplementary Figure 5a). For the longitudinal analyses, significant associations were found for BA35, BA36 and PHC (Supplementary Figure 5b–d), although not after multiple comparison correction. Correction for age weakened the association for all regions (BA35: r=−0.12, p=0.12; BA36: r=−0.14, p=0.08; PHC: r=−0.15, p=0.06).
Table 3.
Relationship of WMHs (logtransformed) with cross-sectional and longitudinal MTL structural measures in cognitively normal older adults with low β-amyloid levels
| Cross-sectional dataset | Longitudinal dataset | |||
|---|---|---|---|---|
|
| ||||
| Spearman correlation | p-value | Spearman correlation | p-value | |
| AH | 0.13 | 0.09 | −0.10 | 0.24 |
| PH | 0.22 | 0.003 | −0.11 | 0.19 |
| ERC | −0.03 | 0.68 | −0.10 | 0.20 |
| BA35 | −0.09 | 0.23 | −0.19 | 0.01 |
| BA36 | −0.12 | 0.10 | −0.18 | 0.02 |
| PHC | −0.04 | 0.57 | −0.18 | 0.02 |
Bolded p-values meet significant after correction for multiple comparisons using the Holm-Bonferroni correction. Sex and intracranial volume are regressed out for hippocampal volumes, sex is regressed out for MTL cortical thickness measures and MTL atrophy rates.
AH=anterior hippocampus, BA=Brodmann Area, ERC=entorhinal cortex, MTL=medial temporal lobe, PH=posterior hippocampus, PHC=parahippocampal cortex, WMHs=white matter hyperintensities.
Mediation analyses with WMH volume as mediator for age effects on BA35, BA36 or PHC atrophy rates did not reach significance.
Exploratory analyses with regional WMHs revealed significant associations of BA35 and PHC atrophy rates with frontal WMHs and of BA35 and hippocampal atrophy rates with parietal WMHs after multiple comparison correction (Supplementary Table 4; Supplementary Figure 6). These associations survived correction for age.
Role of CSF p-tau as contributor to age-related changes in the MTL
Similar as in the previous section, all analyses were run initially without correcting for age, as age is associated with increased CSF p-tau levels and is therefore not necessarily a confounder but may be part of the causal pathway. In secondary analyses, we corrected for age.
CSF p-tau levels were associated with cross-sectional anterior hippocampal volume and at a trend level with posterior hippocampal volume (Table 4, Supplementary Figure 7a–b). CSF p-tau levels were also significantly associated with anterior and posterior hippocampal, ERC and BA35 atrophy rates (Table 4, Supplementary Figure 7c–f). After correction for multiple comparisons, only the association of CSF p-tau levels with anterior hippocampal volumes and anterior and posterior hippocampal atrophy rates still reached significance. Correcting for age did not notably change the results.
Table 4.
Relationship of CSF p-tau (log-transformed) with cross-sectional and longitudinal MTL structural measures in cognitively normal older adults with low β-amyloid levels
| Cross-sectional dataset | Longitudinal dataset | |||
|---|---|---|---|---|
|
| ||||
| Pearson correlation | p-value | Pearson correlation | p-value | |
| AH | −0.22 | 0.006 | −0.31 | <0.001 |
| PH | −0.15 | 0.06 | −0.28 | 0.001 |
| ERC | 0.10 | 0.19 | −0.08 | 0.32 |
| BA35 | −0.09 | 0.25 | −0.22 | 0.007 |
| BA36 | −0.09 | 0.25 | −0.19 | 0.02 |
| PHC | 0.05 | 0.54 | −0.13 | 0.14 |
Bolded p-values meet significant after correction for multiple comparisons using the Holm-Bonferroni correction. Sex and intracranial volume are regressed out for hippocampal volumes, sex is regressed out for MTL cortical thickness measures and MTL atrophy rates.
AH=anterior hippocampus, BA=Brodmann Area, CSF=cerebrospinal fluid, ERC=entorhinal cortex, MTL=medial temporal lobe, p-tau=phospho tau, PH=posterior hippocampus, PHC=parahippocampal cortex.
As subthreshold β-amyloid levels could potentially influence the results we also performed exploratory associations of β-amyloid PET levels with MTL structural measures. For the cross-sectional data, none of the associations reached significance, although a trend was found for the ERC (r=−0.12, p=0.09). Adjusting the association between cross-sectional hippocampal volumes and p-tau levels for β-amyloid PET levels did not notably change the results. In the longitudinal dataset, no significant associations were found between β-amyloid PET levels and MTL structural measures, except for a positive association between posterior hippocampal atrophy rates and β-amyloid PET levels (r=0.21, p=0.012) and a trend for ERC (r=0.14, p=0.06), although this did not survive multiple comparison correction. Adjusting the association between longitudinal hippocampal atrophy rates and p-tau levels for β-amyloid PET levels did not notably change the results.
Given that the associations of both age and CSF p-tau levels with anterior and posterior hippocampal measures were roughly similar, we combined the two in total hippocampal volume/atrophy rates for the mediation analyses. The cross-sectional mediation analysis indicated that CSF p-tau partially mediates the association of age with total hippocampal volume, although at a trend level, while correcting for β-amyloid PET levels (Figure 2). For the longitudinal data a similar result was found for hippocampal atrophy rates whereby the indirect effect was significant. The indirect effect in the mediation model for BA35 atrophy rates reached a trend level, but the model for BA36 atrophy rates was not significant. Again, all models were corrected for β-amyloid PET levels.
Figure 2.
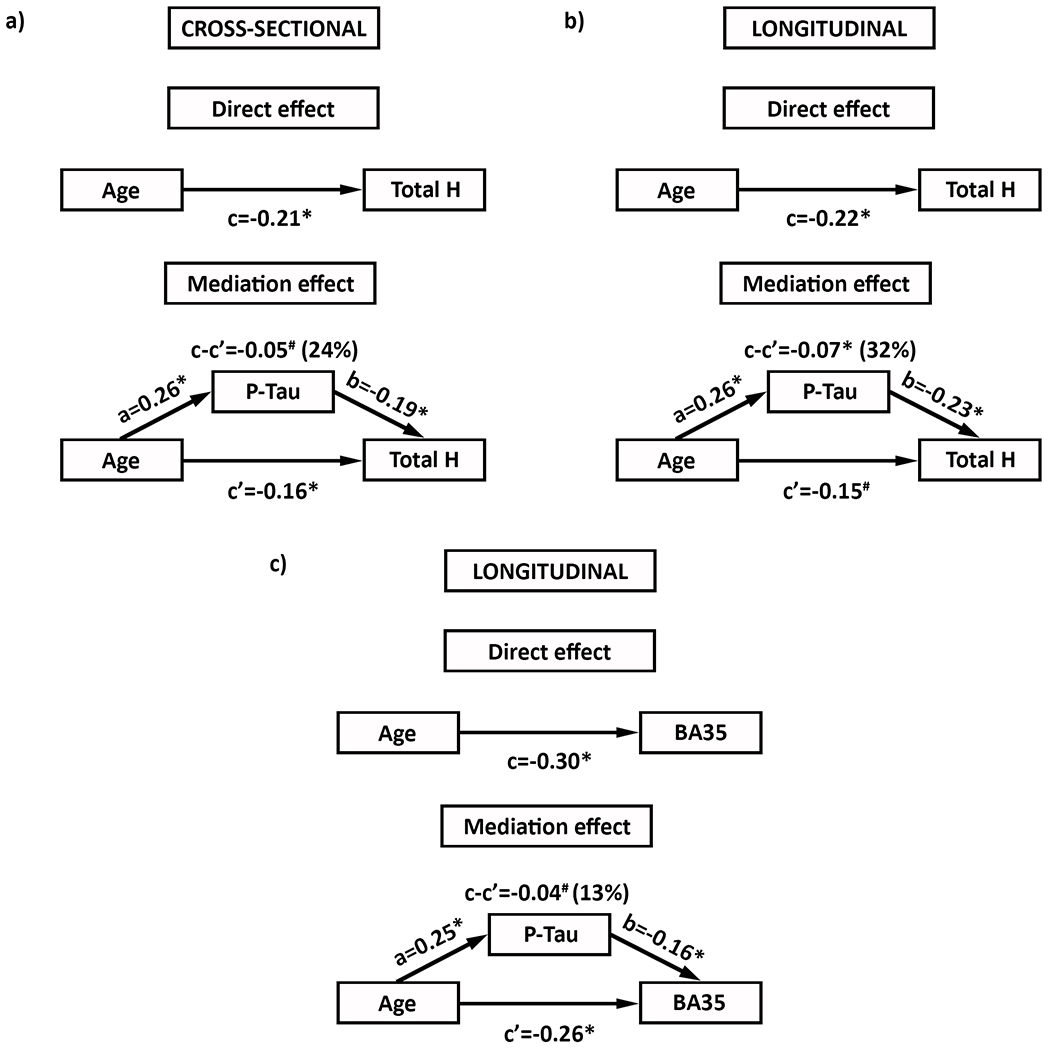
Mediation model indicating that CSF P-Tau levels partially mediate the effect of age on hippocampal volume (a, trend), hippocampal atrophy rates (b) and BA35 atrophy rates (c, trend), while correcting for PET β-amyloid levels, in cognitively normal older adults with low β-amyloid levels
#p<0.10; *p<0.05. % indicates the % of the model explained by the mediation. BA=Brodmann Area, CSF=cerebrospinal fluid, p-tau=phospho tau, T Hippo=total hippocampus.
Sensitivity analyses
Follow up time:
As the atrophy rates were obtained using linear models (see Supplementary Methods), we believe it is unnecessary to correct our analyses for follow up time. However, out of caution we performed all our main analyses including follow up time and this did not change the results.
APOE-ε4-carrier status:
As low levels of β-amyloid can still be present in the selected cognitively normal older adults, APOE-ε4 carrier status might be an important factor to consider. APOE-ε4 carrier status was not associated with cross-sectional hippocampal volumes or parahippocampal cortical thickness measures, nor with longitudinal MTL atrophy rates (data not shown). We were only able to look at the association between presence of an APOE-ε4 allele and structural measures in a binarized manner as only 3 subjects in both the cross-sectional and longitudinal dataset had 2 APOE-ε4 alleles.
Cognitive change over time:
Of the 164 participants with longitudinal data, 138 had information on diagnosis at the time of their last scan. For 26 participants this was missing. For those participants, we determined cognitive decline at the time of the follow up scan by a CDR of 0.5 or higher, a CDR Memory Box score of 0.5 or higher or an MMSE score of 24 or lower as per the ADNI criteria for cognitively normal older adults listed in the Supplementary Methods section. In total 17 developed MCI, dementia or cognitive decline during their follow up time, leaving 147 without cognitive decline. Excluding all subjects with cognitive decline, all longitudinal analyses are repeated in this smaller subset to explore if these subjects were driving the observed results. The results were largely similar, see Supplementary Tables 5–7 and Supplementary Figures 8–9.
Cerebrospinal levels of β-amyloid:
CSF β-amyloid might potentially reflect earlier pathological changes than PET (Palmqvist et al., 2016), and by using PET to determine β-amyloid status we may have inadvertently included subjects who have positive β-amyloid levels according to CSF. We therefore repeated the mediation analyses for p-tau using CSF β-amyloid 1-42 instead of PET β-amyloid, where both β-amyloid status was determined by the CSF marker (980 pg/mL (Hansson et al., 2018)) and the continuous CSF β-amyloid measure was included in the mediation model. We obtained relatively similar, even slightly stronger, results for the longitudinal data, where the indirect effects were significant for hippocampal and BA35 atrophy rates. The cross-sectional analyses no longer reached a trend (Supplementary Figure 10).
Additionally, removing 26 subjects who had low β-amyloid levels or were β-amyloid negative according to the CSF measures, but were positive for β-amyloid (i.e. they had significant β-amyloid levels) according to the PET cut off, did not notably change the results (Supplementary Figure 11).
Discussion
In this study we aimed to obtain a comprehensive overview of age effects on MTL structural measures, avoiding some of the pitfalls of the extant literature by, uniquely, including only older adults with no to low levels of β-amyloid, analyzing both cross-sectional and longitudinal data, and assessing potential drivers of observed age effects.
Both the cross-sectional and longitudinal analyses indicated most pronounced atrophy in the hippocampus and BA35, with atrophy rates increasing with age in these regions, potentially reflecting accumulating pathologies and other detrimental processes with increasing age. The association of age with BA35, the region that in absolute terms displayed the highest percent rate of atrophy in this older adult population, is consistent with the role of PART as a driver of age-related MTL changes, whereas the relationship with both anterior and posterior hippocampus is less specific. That said, the associations of hippocampal and BA35 structural measures (before multiple comparison correction) with CSF p-tau and the mediation effect of p-tau on the age effect of the hippocampus (trend for the cross-sectional data and significant for the longitudinal data; a trend for BA35 atrophy rates) is further supportive of PART explaining, at least partially, the age-related effects on the MTL. Indeed, NFTs in low β-amyloid individuals have been found to be related to neuron loss (Josephs et al., 2017, Price, Joseph L. and Morris, 1999) and atrophy in the MTL measured on MRI (Josephs et al., 2017, Quintas-Neves et al., 2019). As a controversy remains around PART, including whether it is a benign condition or if it is linked to neurodegeneration, this study advances the field by providing important novel information that PART is potentially already linked to neurodegeneration in cognitively normal individuals and may therefore potentially also be implicated in age-related memory impairments.
The above demonstrated some relationship between p-tau and BA35. However, as neurofibrillary tangles first target BA35 and ERC (Braak, H. and Braak, 1995, Crary et al., 2014, Price, Joseph L. and Morris, 1999), we initially expected even stronger associations of structural measures of these regions with CSF p-tau levels. However, the reliability of ERC and BA35 segmentation is lower than that of the hippocampal regions (Xie, Long et al., 2019), which may have limited the detection of an association with these regions given the subtlety of the effects.
Notably, the mediation model was corrected for β-amyloid PET levels, indicating that subthreshold β-amyloid levels likely do not explain the observed association. However, it is possible that even though the included individuals in this study had no to low β-amyloid levels, they still have Thal stage 1 or 2 which would mean that would fall into the ‘possible PART’ category rather than “definite PART” and the degree to which this influences atrophy or biomarker measures of tau remains unclear. Additionally, CSF β-amyloid might potentially reflect earlier pathological changes than PET (Palmqvist et al., 2016), and by using PET to determine β-amyloid status we may have inadvertently included subjects who have high β-amyloid levels (i.e. β-amyloid positive) according to CSF. However, we chose to do our primary analyses using the PET data, as a larger set in the ADNI database has PET than CSF measures. When repeating the analyses with CSF instead of PET β-amyloid levels, a mediation effect was found for the longitudinal but not cross-sectional data. This finding even held when only including individuals with low β-amyloid levels (i.e. β-amyloid negative) according to both CSF and PET measures. The lack of a significant effect in the cross-sectional data may be due to the fact that cross-sectional data is generally confounded by other factors not associated with neurodegeneration.
In general, these findings support a mediating effect of tau on age-related MTL atrophy and that this effect is independent of subthreshold β-amyloid levels. While our findings indicate that CSF p-tau levels might be sensitive to PART, there is currently still controversy on this topic and future work needs to elucidate this. It is possible that other age-related tauopathies contributed to the signal in CSF and MTL atrophy (e.g. Argyrophilic Grain Disease), which may be difficult to differentiate from PART, which requires neuropathological confirmation and cannot currently be definitely determined in vivo. An additional point of controversy to keep in mind is whether PART is actually a distinct entity from AD and whether tau pathology in the presence of low levels of β-amyloid really reflects possible PART and not low AD neuropathologic change (Braak, Heiko and Del Tredici, 2014, Duyckaerts et al., 2015).
It should be noted though that we only observed a partial mediation with between 32-46%, dependent on the group selection, of the age-structure relation being explained by CSF p-tau levels. It is possible that we only observed a partial mediation as CSF p-tau is not only reflective of MTL tau which would most likely drive MTL neurodegeneration. However, it is also likely tau only explains part of the age-related changes in MTL structure and that other processes likely play a role in this. CVD is also a prime candidate for driving age-related processes and has been mostly linked to hippocampal atrophy ((Den Heijer et al., 2005, O’BRIEN et al., 1997, van der Flier, Wiesje M et al., 2005), but note (Du, An-Tao et al., 2006)) because of CA1 neurons’ sensitivity to ischemia/hypoxia (Schmidt-Kastner and Freund, 1991, Zola-Morgan et al., 1992). Surprisingly, no relationship of WMHs with hippocampal atrophy was found (except for a likely spurious positive association with hippocampal volumes), but rather with extrahippocampal regions, BA35, BA36 and PHC, although these did not survive multiple comparison correction. While some studies have reported an association of WMHs (Wang et al., 2020) or cardiovascular risk factors with MTL cortices (Cox et al., 2019, de Toledo Ferraz Alves, Tania Correa et al., 2011, Gourley et al., 2020), this has generally not been well researched and required further study and replication. Further, mediation models were not significant. While this is potentially due to insufficient power, this might indicate less support for CVD driving the observed age-related structural changes in the MTL.
Another potential pathology that may influence MTL structural integrity in aging is TDP-43 (Nelson, Peter T. et al., 2019) which increases in prevalence after the age of 80. Prior work has suggested that TDP-43 may particularly affect anterior hippocampus and ERC (de Flores et al., 2020, Josephs et al., 2014, Nag et al., 2017). Given that there was no clear anterior-to-posterior gradient of atrophy in relation to aging in this study, it does not appear likely that TDP-43 is a significant driver of atrophy in this cohort. However, this may be due in part to the fact that only a small portion of our study population was older than 80 years (only 21 subjects). It is worth noting that there also may be non-specific age effects, such as for example decreased neurogenesis (Galvan and Jin, 2007), or downstream effects of inflammation (Wyss-Coray, 2016) or glucocorticoids (Nichols et al., 2001), that may also contribute to MTL atrophy, but it is not possible to parse this out from the current findings.
For this study, we included all individuals who were cognitively normal at baseline, irrespective of future changes in their cognition. While ADNI has longer follow up data for many of the participants, the rationale not to exclude individuals with impaired cognition at a later timepoints is that we aimed to study ‘normal aging’ rather than ‘super aging’. Moreover, given that tau pathology is very common in older age (Braak, Heiko et al., 1996) and PART leads to cognitive decline (Crary et al., 2014, Jefferson-George et al., 2017), removal of subjects who progress to MCI or dementia may obscure effects of PART on MTL structure. Moreover, dementia is considered by some as continuous with aging and an acceleration of aging (Von Dras and Blumenthal, 1992, Wyss-Coray, 2016) and the definition of ‘normal aging’ is still under debate. For example, if PART is associated with cognitive decline to MCI or dementia level impairment, it is unclear whether to consider this normal or pathological aging. On the other hand, one could argue that some of the age-related changes we observed are not actually reflective of ‘normal aging’ but are rather precursors to later occurring dementia. As there is no clear answer to this question, we decided to repeat the analyses in a subgroup that does not convert to MCI or dementia or show cognitive decline at their latest follow up scan and report largely similar findings. Finally, we note that normal aging was assessed at a group level and that heterogeneity in normal aging was not taken into account, where other factors such as diet, exercise and psychosocial factors could also have influenced brain health and potential resilience to different pathologies (Rowe and Kahn, 1987).
In conclusion, we performed a comprehensive characterization of aging effects on MTL structural measures in cognitively normal older adults with low levels of β-amyloid and showed age-related changes in all MTL regions, but most pronounced in BA35 and the hippocampus. Combined with the fact that age-related hippocampal atrophy was partly mediated by CSF p-tau levels, these findings are suggestive of a role of PART in age-related structural changes in the MTL. This finding is consistent with other work in individuals with low β-amyloid levels demonstrating correlations of MTL tau, measured using tau PET, with both atrophy in early Braak regions and memory (Das, Sandhitsu R. et al., 2019), but provides a major advance on previous literature by establishing a relationship between a marker of tau pathology and neurodegeneration already in cognitively normal older adults. In general, this study indicates the importance of assessing age-related changes in a well-characterized cohort where the separate contributions of different pathologies can be quantified. Further, if the present results are replicated, it also suggests a potential role for tau-targeted interventions in age-associated memory decline.
Supplementary Material
Acknowledgements
This work was supported by NIH grants R01-AG056014, R01-AG040271, P30-AG010124, R01-EB017255, R01-AG055005, RF1AG054409, the donors of Alzheimer’s Disease Research, a program of the BrightFocus Foundation, Alzheimer’s Association (AARF-19-615258), Penn Institute on Aging, Fondation Philippe Chatrier.
Disclosures
D.A.W has received grant support from Merck, Biogen, and Eli Lilly/Avid and consultation fees from Neuronix and is on the DSMB for a clinical trial run by Functional Neuromodulation. L.X. received personal consultation fees from Galileo CDS, Inc. None of the other authors had any disclosures.
References
- Apple DM, Solano-Fonseca R, Kokovay E, 2017. Neurogenesis in the aging brain. Biochem.Pharmacol 141, 77–85. [DOI] [PubMed] [Google Scholar]
- Avants BB, Epstein CL, Grossman M, Gee JC, 2008. Symmetric diffeomorphic image registration with cross-correlation: evaluating automated labeling of elderly and neurodegenerative brain. Med.Image Anal 12, 26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes J, Bartlett JW, van de Pol LA, Loy CT, Scahill RI, Frost C, Thompson P, Fox NC, 2009. A meta-analysis of hippocampal atrophy rates in Alzheimer’s disease. Neurobiol.Aging 30, 1711–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell WR, An Y, Kageyama Y, English C, Rudow GL, Pletnikova O, Thambisetty M, O’Brien R, Moghekar AR, Albert MS, 2019. Neuropathologic, genetic, and longitudinal cognitive profiles in primary age-related tauopathy (PART) and Alzheimer’s disease. 15, 8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besser LM, Mock C, Teylan MA, Hassenstab J, Kukull WA, Crary JF, 2019. Differences in cognitive impairment in primary age-related tauopathy versus Alzheimer disease. 78, 219–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittner T, Zetterberg H, Teunissen CE, Ostlund RE Jr, Militello M, Andreasson U, Hubeek I, Gibson D, Chu DC, Eichenlaub U, 2016. Technical performance of a novel, fully automated electrochemiluminescence immunoassay for the quantitation of β-amyloid (1–42) in human cerebrospinal fluid. 12, 517–526. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E, 1995. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol.Aging 16, 271–278. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E, 1991. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E, Bohl J, Reintjes R, 1996. Age, neurofibrillary changes, Aβ-amyloid and the onset of Alzheimer’s disease. Neurosci.Lett 210, 87–90. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, 2014. Are cases with tau pathology occurring in the absence of Aβ deposits part of the AD-related pathological process?. Acta Neuropathol. 128, 767–772. [DOI] [PubMed] [Google Scholar]
- Chen KH, Chuah LY, Sim SK, Chee MW, 2010. Hippocampal region-specific contributions to memory performance in normal elderly. Brain Cogn. 72, 400–407. [DOI] [PubMed] [Google Scholar]
- Cox SR, Lyall DM, Ritchie SJ, Bastin ME, Harris MA, Buchanan CR, Fawns-Ritchie C, Barbu MC, De Nooij L, Reus LM, 2019. Associations between vascular risk factors and brain MRI indices in UK Biobank. Eur.Heart J 40, 2290–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, Arnold SE, Attems J, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Gearing M, Grinberg LT, Hof PR, Hyman BT, Jellinger K, Jicha GA, Kovacs GG, Knopman DS, Kofler J, Kukull WA, Mackenzie IR, Masliah E, McKee A, Montine TJ, Murray ME, Neltner JH, Santa-Maria I, Seeley WW, Serrano-Pozo A, Shelanski ML, Stein T, Takao M, Thal DR, Toledo JB, Troncoso JC, Vonsattel JP, White CL 3rd, Wisniewski T, Woltjer RL, Yamada M, Nelson PT, 2014. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 128, 755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das SR, Avants BB, Pluta J, Wang H, Suh JW, Weiner MW, Mueller SG, Yushkevich PA, 2012. Measuring longitudinal change in the hippocampal formation from in vivo high-resolution T2-weighted MRI. Neuroimage. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das SR, Xie L, Wisse LE, Vergnet N, Ittyerah R, Cui S, Yushkevich PA, Wolk DA, Alzheimer’s Disease Neuroimaging Initiative, 2019. In vivo measures of tau burden are associated with atrophy in early Braak stage medial temporal lobe regions in amyloid-negative individuals. 15, 1286–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Flores R, Wisse LE, Das SR, Xie L, McMillan CT, Trojanowski JQ, Robinson JL, Grossman M, Lee E, Irwin DJ, 2020. Contribution of mixed pathology to medial temporal lobe atrophy in Alzheimer’s disease. 16, 843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Toledo Ferraz Alves, Tania Correa, Scazufca M, Squarzoni P, de Souza Duran, Fabio Luiz, Tamashiro-Duran JH, Vallada HP, Andrei A, Wajngarten M, Menezes PR, Busatto GF, 2011. Subtle gray matter changes in temporo-parietal cortex associated with cardiovascular risk factors. J. Alzheimer’s Dis 27, 575–589. [DOI] [PubMed] [Google Scholar]
- Den Heijer T, Launer LJ, Prins ND, Van Dijk EJ, Vermeer SE, Hofman A, Koudstaal PJ, Breteler M, 2005. Association between blood pressure, white matter lesions, and atrophy of the medial temporal lobe. 64, 263–267. [DOI] [PubMed] [Google Scholar]
- Dickerson BC, Feczko E, Augustinack JC, Pacheco J, Morris JC, Fischl B, Buckner RL, 2009. Differential effects of aging and Alzheimer’s disease on medial temporal lobe cortical thickness and surface area. Neurobiol.Aging 30, 432–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding SL, Van Hoesen GW, 2010. Borders, extent, and topography of human perirhinal cortex as revealed using multiple modern neuroanatomical and pathological markers. Hum.Brain Mapp 31, 1359–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doshi J, Erus G, Habes M, Davatzikos C, 2019. DeepMRSeg: A convolutional deep neural network for anatomy and abnormality segmentation on MR images. [Google Scholar]
- Du AT, Schuff N, Zhu XP, Jagust WJ, Miller BL, Reed BR, Kramer JH, Mungas D, Yaffe K, Chui HC, Weiner MW, 2003. Atrophy rates of entorhinal cortex in AD and normal aging. 60, 481–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du A, Schuff N, Chao LL, Kornak J, Jagust WJ, Kramer JH, Reed BR, Miller BL, Norman D, Chui HC, 2006. Age effects on atrophy rates of entorhinal cortex and hippocampus. Neurobiol.Aging 27, 733–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyckaerts C, Braak H, Brion J, Buée L, Del Tredici K, Goedert M, Halliday G, Neumann M, Spillantini MG, Tolnay M, 2015. PART is part of Alzheimer disease. Acta Neuropathol. 129, 749–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, Walhovd KB, 2010. Structural brain changes in aging: courses, causes and cognitive consequences. Rev.Neurosci 21, 187–221. [DOI] [PubMed] [Google Scholar]
- Fjell AM, Westlye LT, Amlien I, Espeseth T, Reinvang I, Raz N, Agartz I, Salat DH, Greve DN, Fischl B, 2009. High consistency of regional cortical thinning in aging across multiple samples. 19, 2001–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR, 1975. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J.Psychiatr.Res 12, 189–198. [DOI] [PubMed] [Google Scholar]
- Galvan V, Jin K, 2007. Neurogenesis in the aging brain. 2, 605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourley D, Pasha EP, Kaur SS, Haley AP, Tanaka H, 2020. Association of dementia and vascular risk scores with cortical thickness and cognition in low-risk middle-aged adults. 34, 313–317. [DOI] [PubMed] [Google Scholar]
- Hansson O, Seibyl J, Stomrud E, Zetterberg H, Trojanowski JQ, Bittner T, Lifke V, Corradini V, Eichenlaub U, Batrla R, 2018. CSF biomarkers of Alzheimer’s disease concord with amyloid-β PET and predict clinical progression: A study of fully automated immunoassays in BioFINDER and ADNI cohorts. 14, 1470–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer WJ, Verhaeghen P, 2006. Memory aging, in: Anonymous Handbook of the psychology of aging. Elsevier, pp. 209–232. [Google Scholar]
- Insausti R, Insausti AM, Sobreviela MT, Salinas A, Martinez-Penuela JM, 1998. Human medial temporal lobe in aging: anatomical basis of memory preservation. Microsc.Res.Tech 43, 8–15. [DOI] [PubMed] [Google Scholar]
- Isaev NK, Stelmashook EV, Genrikhs EE, 2019. Neurogenesis and brain aging. Rev.Neurosci 30, 573–580. [DOI] [PubMed] [Google Scholar]
- Jack CR Jr, Petersen RC, Xu YC, Waring SC, O’Brien PC, Tangalos EG, Smith GE, Ivnik RJ, Kokmen E, 1997. Medial temporal atrophy on MRI in normal aging and very mild Alzheimer’s disease. 49, 786–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen WJ, Ossenkoppele R, Knol DL, Tijms BM, Scheltens P, Verhey FR, Visser PJ, Amyloid Biomarker Study Group, Aalten P, Aarsland D, Alcolea D, Alexander M, Almdahl IS, Arnold SE, Baldeiras I, Barthel H, van Berckel BN, Bibeau K, Blennow K, Brooks DJ, van Buchem MA, Camus V, Cavedo E, Chen K, Chetelat G, Cohen AD, Drzezga A, Engelborghs S, Fagan AM, Fladby T, Fleisher AS, van der Flier WM, Ford L, Forster S, Fortea J, Foskett N, Frederiksen KS, Freund-Levi Y, Frisoni GB, Froelich L, Gabryelewicz T, Gill KD, Gkatzima O, Gomez-Tortosa E, Gordon MF, Grimmer T, Hampel H, Hausner L, Hellwig S, Herukka SK, Hildebrandt H, Ishihara L, Ivanoiu A, Jagust WJ, Johannsen P, Kandimalla R, Kapaki E, Klimkowicz-Mrowiec A, Klunk WE, Kohler S, Koglin N, Kornhuber J, Kramberger MG, Van Laere K, Landau SM, Lee DY, de Leon M, Lisetti V, Lleo A, Madsen K, Maier W, Marcusson J, Mattsson N, de Mendonca A, Meulenbroek O, Meyer PT, Mintun MA, Mok V, Molinuevo JL, Mollergard HM, Morris JC, Mroczko B, Van der Mussele S, Na DL, Newberg A, Nordberg A, Nordlund A, Novak GP, Paraskevas GP, Parnetti L, Perera G, Peters O, Popp J, Prabhakar S, Rabinovici GD, Ramakers IH, Rami L, Resende de Oliveira C, Rinne JO, Rodrigue KM, Rodriguez-Rodriguez E, Roe CM, Rot U, Rowe CC, Ruther E, Sabri O, Sanchez-Juan P, Santana I, Sarazin M, Schroder J, Schutte C, Seo SW, Soetewey F, Soininen H, Spiru L, Struyfs H, Teunissen CE, Tsolaki M, Vandenberghe R, Verbeek MM, Villemagne VL, Vos SJ, van Waalwijk van Doorn LJ, Waldemar G, Wallin A, Wallin AK, Wiltfang J, Wolk DA, Zboch M, Zetterberg H, 2015. Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. JAMA. 313, 1924–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferson-George KS, Wolk DA, Lee EB, McMillan CT, 2017. Cognitive decline associated with pathological burden in primary age-related tauopathy. 13, 1048–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA, 2018. Different patterns of hippocampal tau pathology in Alzheimer’s disease and PART. Acta Neuropathol. 136, 811–813. [DOI] [PubMed] [Google Scholar]
- Josephs KA, Murray ME, Tosakulwong N, Whitwell JL, Knopman DS, Machulda MM, Weigand SD, Boeve BF, Kantarci K, Petrucelli L, 2017. Tau aggregation influences cognition and hippocampal atrophy in the absence of beta-amyloid: a clinico-imaging-pathological study of primary age-related tauopathy (PART). Acta Neuropathol. 133, 705–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Murray ME, Whitwell JL, Parisi JE, Petrucelli L, Jack CR, Petersen RC, Dickson DW, 2014. Staging TDP-43 pathology in Alzheimer’s disease. Acta Neuropathol. 127, 441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoops AJ, Gerritsen L, van der Graaf Y, Mali WP, Geerlings MI, 2012. Loss of entorhinal cortex and hippocampal volumes compared to whole brain volume in normal aging: the SMART-Medea study. 203, 31–37. [DOI] [PubMed] [Google Scholar]
- Landau SM, Mintun MA, Joshi AD, Koeppe RA, Petersen RC, Aisen PS, Weiner MW, Jagust WJ, Alzheimer’s Disease Neuroimaging Initiative, 2012. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Ann.Neurol 72, 578–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malykhin NV, Bouchard TP, Camicioli R, Coupland NJ, 2008. Aging hippocampus and amygdala. 19, 543–547. [DOI] [PubMed] [Google Scholar]
- McEwen BS, de Leon MJ, Lupien SJ, Meaney MJ, 1999. Corticosteroids, the aging brain and cognition. 10, 92–96. [DOI] [PubMed] [Google Scholar]
- Morris JC, 1993. The Clinical Dementia Rating (CDR): current version and scoring rules. 43, 2412–2414. [DOI] [PubMed] [Google Scholar]
- Nag S, Yu L, Wilson RS, Chen E, Bennett DA, Schneider JA, 2017. TDP-43 pathology and memory impairment in elders without pathologic diagnoses of AD or FTLD. 88, 653–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, Rademakers R, Alafuzoff I, Attems J, Brayne C, 2019. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. 142, 1503–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson P, Dickson D, Trokanowski J, Jack C Jr, Boyle P, Arfanakis K, Rademakers R, Alafuzoff I, Attems J, Brayne C, 2019. Limbic-predominant Age-related TDP-43 Encephalopathy (LATE): Consensus Working Group Report. 142, 1503–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols NR, Zieba M, Bye N, 2001. Do glucocorticoids contribute to brain aging?. Brain Res.Rev 37, 273–286. [DOI] [PubMed] [Google Scholar]
- O’BRIEN JT, Desmond P, Ames D, Schweitzer I, Tress B, 1997. Magnetic resonance imaging correlates of memory impairment in the healthy elderly: association with medial temporal lobe atrophy but not white matter lesions. Int.J.Geriatr.Psychiatry 12, 369–374. [PubMed] [Google Scholar]
- Palmqvist S, Mattsson N, Hansson O, Alzheimer’s Disease Neuroimaging Initiative, 2016. Cerebrospinal fluid analysis detects cerebral amyloid-β accumulation earlier than positron emission tomography. 139, 1226–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JL, Ko AI, Wade MJ, Tsou SK, McKeel DW, Morris JC, 2001. Neuron number in the entorhinal cortex and CA1 in preclinical Alzheimer disease. Arch.Neurol 58, 1395–1402. [DOI] [PubMed] [Google Scholar]
- Price JL, Morris JC, 1999. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. 45, 358–368. [DOI] [PubMed] [Google Scholar]
- Quintas-Neves M, Teylan MA, Besser L, Soares-Fernandes J, Mock CN, Kukull WA, Crary JF, Oliveira TG, 2019. Magnetic resonance imaging brain atrophy assessment in primary age-related tauopathy (PART). 7, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz N, Lindenberger U, Rodrigue KM, Kennedy KM, Head D, Williamson A, Dahle C, Gerstorf D, Acker JD, 2005. Regional brain changes in aging healthy adults: general trends, individual differences and modifiers. Cereb.Cortex 15, 1676–1689. [DOI] [PubMed] [Google Scholar]
- Raz N, Rodrigue KM, Head D, Kennedy KM, Acker JD, 2004. Differential aging of the medial temporal lobe: a study of a five-year change. 62, 433–438. [DOI] [PubMed] [Google Scholar]
- Raz N, Gunning FM, Head D, Dupuis JH, McQuain J, Briggs SD, Loken WJ, Thornton AE, Acker JD, 1997. Selective aging of the human cerebral cortex observed in vivo: differential vulnerability of the prefrontal gray matter. 7, 268–282. [DOI] [PubMed] [Google Scholar]
- Ronneberger O, Fischer P, Brox T, 2015. U-net: Convolutional networks for biomedical image segmentation, 234–241. [Google Scholar]
- Rönnlund M, Nyberg L, Bäckman L, Nilsson L, 2005. Stability, growth, and decline in adult life span development of declarative memory: cross-sectional and longitudinal data from a population-based study. Psychol. Aging 20, 3. [DOI] [PubMed] [Google Scholar]
- Rosen WG, Terry RD, Fuld PA, Katzman R, Peck A, 1980. Pathological verification of ischemic score in differentiation of dementias. 7, 486–488. [DOI] [PubMed] [Google Scholar]
- Rowe JW, Kahn RL, 1987. Human aging: usual and successful. 237, 143–149. [DOI] [PubMed] [Google Scholar]
- Sankar T, Park MTM, Jawa T, Patel R, Bhagwat N, Voineskos AN, Lozano AM, Chakravarty MM, Alzheimer’s Disease Neuroimaging Initiative, 2017. Your algorithm might think the hippocampus grows in Alzheimer’s disease: Caveats of longitudinal automated hippocampal volumetry. Hum.Brain Mapp 38, 2875–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saykin AJ, Shen L, Foroud TM, Potkin SG, Swaminathan S, Kim S, Risacher SL, Nho K, Huentelman MJ, Craig DW, Thompson PM, Stein JL, Moore JH, Farrer LA, Green RC, Bertram L, Jack CR Jr, Weiner MW, Alzheimer’s Disease Neuroimaging Initiative, 2010. Alzheimer’s Disease Neuroimaging Initiative biomarkers as quantitative phenotypes: Genetics core aims, progress, and plans. Alzheimers Dement. 6, 265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Kastner R, Freund TF, 1991. Selective vulnerability of the hippocampus in brain ischemia. 40, 599–636. [DOI] [PubMed] [Google Scholar]
- Szegedy C, Ioffe S, Vanhoucke V, Alemi A, 2016. Inception-v4, inception-resnet and the impact of residual connections on learning. [Google Scholar]
- van der Flier, Wiesje M, van Straaten EC, Barkhof F, Ferro JM, Pantoni L, Basile A, Inzitari D, Erkinjuntti T, Wahlund LO, Rostrup E, 2005. Medial temporal lobe atrophy and white matter hyperintensities are associated with mild cognitive deficits in non-disabled elderly people: the LADIS study. 76, 1497–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Dras DD, Blumenthal HT, 1992. Dementia of the aged: disease or atypical-accelerated aging? Biopathological and psychological perspectives. J.Am.Geriatr.Soc [DOI] [PubMed] [Google Scholar]
- Wang Y, Yang Y, Wang T, Nie S, Yin H, Liu J, 2020. Correlation between white matter hyperintensities related gray matter volume and cognition in cerebral small vessel disease. 29, 105275. [DOI] [PubMed] [Google Scholar]
- Wisse LE, Biessels GJ, Heringa SM, Kuijf HJ, Koek DH, Luijten PR, Geerlings MI, Utrecht Vascular Cognitive Impairment (VCI) Study Group, 2014. Hippocampal subfield volumes at 7T in early Alzheimer’s disease and normal aging. Neurobiol.Aging 35, 2039–2045. [DOI] [PubMed] [Google Scholar]
- Wolk DA, Das SR, Mueller SG, Weiner MW, Yushkevich PA, Alzheimer’s Disease Neuroimaging Initiative, 2017. Medial temporal lobe subregional morphometry using high resolution MRI in Alzheimer’s disease. Neurobiol.Aging 49, 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, 2016. Ageing, neurodegeneration and brain rejuvenation. 539, 180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Wisse LEM, Das SR, Wang H, Wolk DA, Manjon JV, Yushkevich PA, 2016. Accounting for the Confound of Meninges in Segmenting Entorhinal and Perirhinal Cortices in T1-Weighted MRI. Med.Image Comput.Comput.Assist.Interv 9901, 564–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Wisse LE, Das SR, Ittyerah R, Wang J, Wolk DA, Yushkevich PA, Alzheimer’s Disease Neuroimaging Initiative, 2018. Characterizing Anatomical Variability and Alzheimer’s Disease Related Cortical Thinning in the Medial Temporal Lobe Using Graph-Based Groupwise Registration and Point Set Geodesic Shooting, 28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Wisse LE, Das SR, Vergnet N, Dong M, Ittyerah R, de Flores R, Yushkevich PA, Wolk DA, Alzheimer’s Disease Neuroimaging Initiative, 2020. Longitudinal atrophy in early Braak regions in preclinical Alzheimer’s disease. Hum.Brain Mapp 41, 4704–4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Wisse LE, Pluta J, de Flores R, Piskin V, Manjón JV, Wang H, Das SR, Ding S, Wolk DA, 2019. Automated segmentation of medial temporal lobe subregions on in vivo T1-weighted MRI in early stages of Alzheimer’s disease. Hum.Brain Mapp 40, 3431–3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankner BA, Lu T, Loerch P, 2008. The aging brain. 3, 41–66. [DOI] [PubMed] [Google Scholar]
- Zhang L, Jiang Y, Zhu J, Liang H, He X, Qian J, Lin H, Tao Y, Zhu K, 2020. Quantitative Assessment of Hippocampal Tau Pathology in AD and PART. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zola-Morgan S, Squire LR, Rempel NL, Clower RP, Amaral DG, 1992. Enduring memory impairment in monkeys after ischemic damage to the hippocampus. J.Neurosci 12, 2582–2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.