

Abstract
Objective
Early identification of chronic diseases is a pillar of precision medicine as it can lead to improved outcomes, reduction of disease burden, and lower healthcare costs. Predictions of a patient’s health trajectory have been improved through the application of machine learning approaches to electronic health records (EHRs). However, these methods have traditionally relied on “black box” algorithms that can process large amounts of data but are unable to incorporate domain knowledge, thus limiting their predictive and explanatory power. Here, we present a method for incorporating domain knowledge into clinical classifications by embedding individual patient data into a biomedical knowledge graph.
Materials and Methods
A modified version of the Page rank algorithm was implemented to embed millions of deidentified EHRs into a biomedical knowledge graph (SPOKE). This resulted in high-dimensional, knowledge-guided patient health signatures (ie, SPOKEsigs) that were subsequently used as features in a random forest environment to classify patients at risk of developing a chronic disease.
Results
Our model predicted disease status of 5752 subjects 3 years before being diagnosed with multiple sclerosis (MS) (AUC = 0.83). SPOKEsigs outperformed predictions using EHRs alone, and the biological drivers of the classifiers provided insight into the underpinnings of prodromal MS.
Conclusion
Using data from EHR as input, SPOKEsigs describe patients at both the clinical and biological levels. We provide a clinical use case for detecting MS up to 5 years prior to their documented diagnosis in the clinic and illustrate the biological features that distinguish the prodromal MS state.
Keywords: knowledge graph, electronic health records, multiple sclerosis, preventative medicine
INTRODUCTION
Efforts to move toward precision and preventative medicine have increased in the last decade and are now pervasive in most aspects of biomedicine.1 As a result, there has been a sharp increase in medical research studies that implement machine learning (ML) approaches using electronic health records (EHRs).2,3 ML approaches have been moderately successful and have substantially advanced tasks such as disease diagnosis and specimen classification.4 However, because they identify patterns in data without knowledge of the underlying clinical or biological meaning, their overall performance has been limited and interpretability of the results remains a black box.
Most chronic diseases lack a unique sign or symptom at presentation. On the contrary, patients may consult a specialist following a clinical event, but often acknowledge that symptoms presented months or even years prior. Early identification of individuals at risk for chronic diseases who are still healthy or have subclinical manifestations would be beneficial for both patients (to receive early treatment or close monitoring) and the health system as a whole (to help optimize across multiple visits and expensive testing).
In order to systematically assess the earliest symptoms (ie, prodromal period) and the biological changes underlying a chronic disease, clinical record standardization is critical in order to overcome the incompleteness in a patient’s biomedical history. The Observational Medical Outcomes Partnership (OMOP) format5 helps bridge the incompatibility of disparate EHR systems and facilitates the unification of patient records and timelines. Additionally, projects that incorporate basic science-level data (genomics, proteomics, etc.) into EHR research, such as Electronic Medical Records and Genomics, have furthered our understanding of disease pathogenesis and offered practical applications.6–9 A recently recognized need is the consideration of known general biological mechanisms in patient-specific health data analytics.10 This need can be addressed by knowledge graphs (KGs) which naturally bridge the gap between basic science research and medical practice.11 KGs connect information from multiple classes of biological and medical concepts, thus allowing to constraint the vast solution space faced by traditional ML methods.12–15 SPOKE is a KG that connects information from over 30 databases and contains more than 3 million nodes of 16 types and more than 16 million edges of 32 types.16,17 The subset of nodes and edges used here is listed in Tables 1 and 2.
Table 1.
SPOKE node statistics
| Node type | Count |
|---|---|
| Compound | 286 790 |
| Protein | 33 857 |
| Gene | 19 567 |
| Anatomy | 13 257 |
| Biological process | 13 156 |
| Disease | 9128 |
| Side effect | 3865 |
| Molecular function | 3407 |
| Pathway | 2428 |
| Pharmacologic class | 1748 |
| Cellular component | 1725 |
| Symptom | 369 |
Table 2.
SPOKE edge statistics
| Node type 1 | Edge | Node type 2 | Count |
|---|---|---|---|
| Disease | ASSOCIATES_DaG | Gene | 1 998 072 |
| Gene | PARTICIPATES_GpBP | Biological process | 1 480 742 |
| Protein | INTERACTS_PiP | Protein | 1 238 535 |
| Compound | BINDS_CbP | Protein | 1 098 776 |
| Anatomy | EXPRESSES_AeG | Gene | 1 052 814 |
| Gene | REGULATES_GrG | Gene | 531 344 |
| Gene | INTERACTS_GiG | Gene | 294 328 |
| Gene | PARTICIPATES_GpMF | Molecular function | 260 152 |
| Gene | PARTICIPATES_GpCC | Cellular component | 226 582 |
| Gene | PARTICIPATES_GpPW | Pathway | 221 080 |
| Anatomy | DOWNREGULATES_AdG | Gene | 204 480 |
| Anatomy | UPREGULATES_AuG | Gene | 195 696 |
| Disease | RESEMBLES_DrD | Disease | 128 000 |
| Gene | COVARIES_GcG | Gene | 123 380 |
| Compound | CAUSES_CcSE | Side effect | 86 400 |
| Disease | LOCALIZES_DlA | Anatomy | 79 010 |
| Protein | TRANSLATEDFROM_PtG | Gene | 67 332 |
| Compound | TREATS_CtD | Disease | 64 872 |
| Pharmacologic class | INCLUDES_PCiC | Compound | 62 952 |
| Disease | PRESENTS_DpS | Symptom | 47 606 |
| Compound | DOWNREGULATES_CdG | Gene | 42 204 |
| Compound | CONTRAINDICATES_CcD | Disease | 41 302 |
| Compound | UPREGULATES_CuG | Gene | 37512 |
| Anatomy | ISA_AiA | Anatomy | 37 304 |
| Anatomy | CONTAINS_AcA | Anatomy | 37 304 |
| Disease | ISA_DiD | Disease | 22 952 |
| Disease | CONTAINS_DcD | Disease | 22 952 |
| Anatomy | PARTOF_ApA | Anatomy | 19 502 |
| Disease | UPREGULATES_DuG | Gene | 15 462 |
| Disease | DOWNREGULATES_DdG | Gene | 15 246 |
| Compound | RESEMBLES_CrC | Compound | 12 972 |
| Compound | INTERACTS_CiP | Protein | 6390 |
| Compound | PALLIATES_CpD | Disease | 780 |
| Compound | AFFECTS_CamG | Gene | 718 |
Early detection of chronic diseases such as diabetes or hypertension has enabled their effective management to avoid or delay clinical complications.18,19 However, despite current efforts in quantifying genetic and environmental risk factors,20 accurate methods to predict diagnosis of multiple sclerosis (MS) do not yet exist. MS is a chronic, autoimmune disease of the central nervous system (CNS) with severe and life-long consequences. Early symptoms of MS, such as fatigue or depression, are often nonspecific, which can make it difficult for the general practitioner to identify and refer the patient to a neurologist. However, previous studies suggest that healthcare utilization by some patients increases even 10 years prior to their MS diagnosis.21 Since early treatment of MS is associated with improved long-term neurological outcomes,22 early recognition of a (sub)clinical presentation and understanding its biological basis could have a major impact on disease trajectories of individual patients. Here, we present a computational method to identify patients before they are diagnosed with MS using only the structured portion of their medical records and biological knowledge from a KG. This method for incorporating biological knowledge in health data analysis has broad applicability to other chronic conditions.
MATERIALS AND METHODS
Patient encounter snapshots
The initial cohort consisted of deidentified EHR from 2 180 882 patients who visited UCSF between 2011 and 2018. Available “snapshots” from the medical history of 5752 patients with a confirmed diagnosis of MS were taken using only past encounters 1–7 years prior to their first MS diagnosis code (t0; Figure 1A). These snapshots represent everything a doctor knows about a patient (through their EHRs), up to a given point in time (ie, snapshot at year −1 contains data up to 1 year before MS diagnosis). These snapshots represent the de facto prodromal period of MS.
Figure 1.

Patient timeline aligning and filtering. (A) Timepoint 0 (t0) is the point of alignment for the multiple sclerosis (MS) and non-MS timelines. For MS patients, t0 was the first visit in which a patient received a diagnosis code for MS. The duration of time a patient has been diagnosed with MS is represented by a red line between the first and last visits with an MS diagnosis code. For Non-MS patients, t0 was set to 6 months (purple line) prior to their most recent visit (hexagon). Left of t0 are the patient snapshots that encompass all of the information (electronic health record data) a doctor has on a patient up to a given point of time. The snapshot at year −1 (blue line) contains all data between the first visit (triangle) and −1 year from t0. The remaining snapshots (years −3, −5, and −7) become smaller as their endpoints move farther from t0. (B) Two patient encounter groups were followed throughout the workflow: All-Visit (left) and Primary Care Physician only (PCP-Only) (right). The All-Visit analysis uses all possible encounters at UCSF, whereas the PCP-Only analysis only includes patient encounters at primary (or emergency) care visits. The number of MS or non-MS patients at each year goes from t0 (top) to −7 years (bottom) is shown.
A control group (non-MS, n = 2 175 130) was selected among individuals who never received an MS diagnosis during the observational period. For the non-MS group, t0 was set at 6 months prior to their most recent visit to UCSF. This aligned MS and non-MS snapshots and ensured that the control population had a follow-up period without MS equal to the minimum amount of observation time available for MS patients after diagnosis.
Parallel analyses were conducted to simulate 2 possible scenarios: patients who visited multiple specialists all visit types (All-Visits) and patients with only primary or emergency care providers (PCP-Only) visits. A patient could potentially be in both simulations if they received both primary and specialist care at UCSF, but only data collected during primary care type visits were used for the PCP-Only analysis. Figure 1B depicts the number of MS and non-MS patients included in the All-Visits (left) and PCP-Only (right) groups for each snapshot (years −1 to −7).
Embedding EHRs into SPOKE
The EHRs used for this analysis were translated into the OMOP Common Data Model. We first created Propagated SPOKE Entry Vectors (PSEVs), machine-readable embeddings that quantify the significance of each node in SPOKE for a given cohort of patients.23 To create PSEVs, SPOKE Entry Points (SEPs) were first identified by finding all concepts that are present in both the EHRs and SPOKE. For this work, we identified 7535 SEPs, defined as the EHR concepts from the primary tables “condition_occurrence,” “drug_exposure,” and “measurement” that directly corresponded to nodes in SPOKE. Then, for a given concept (eg, carbamazepine), a connection was made between a patient’s SEPs in the EHRs and SPOKE. A modified version of topic-sensitive Page Rank24 was then used to generate PSEVs for each SEP (Figure 2A and B). Specifically, a random walker was placed onto a node in SPOKE and allowed it to randomly traverse edges within the network until the walker is forced to restart (P = .1) at one of the input patients (that was prescribed carbamazepine in this example). This process continues until the amount of time (importance) the walker spends on each node becomes stable. The resulting PSEV holds weights for each node in SPOKE based on how important a node is for the corresponding patient population.
Figure 2.

Embedding individual patients in SPOKE. (A) Example embedding the electronic health record (EHR) concept for the drug carbamazepine into SPOKE. First, SPOKE Entry Points (SEPs) are created by finding all concepts that are present in both the EHRs and SPOKE. Then each patient that was prescribed carbamazepine is connected to SPOKE through the SEPs in their EHRs. A random walker is then placed onto a node in SPOKE and randomly traverses edges within the network until the walker is restarted at 1 of the patients that was prescribed carbamazepine (probability of restart = .1). (B) This process continues until the amount of time the walker spends on each node becomes stable. The nodes are then ranked such that the most important nodes are given the highest rank (dark teal) and the least important nodes are given the lowest rank (white). Here the medically or biologically important nodes for carbamazepine are darker teal. Meanwhile, heartburn, which is not related to carbamazepine, is white. (C) A SPOKEsig is produced for a patient at a given snapshot by summing the PSEVs associated with the SEPs in their EHRs during that time period. During this example snapshot, Patient X had 3 SEPs: carbamazepine, epilepsy, and constipation. Therefore, the PSEVs for carbamazepine, epilepsy, and constipation are summed to create this snapshot for Patient X. Just as the elements in the PSEVs, each element in the SPOKEsig corresponds to a single node in SPOKE.
Once population-level embeddings (PSEVs) were created for all matching EHR concepts, they were aggregated to create vectors for the individual patient snapshots. Similar to other machine-learning algorithms,25,26 we applied vector/matrix arithmetic to produce the Patient-Specific SPOKE Profile Vectors (SPOKEsigs, see Supplementary Methods). Following this principle, SPOKEsigs were computed for each patient, at each snapshot (Figure 2C). The resulting vectors represent the importance of each node in SPOKE for each patient at that time point.
Building a classifier for early detection of MS
Random forest classifiers were used to determine if SPOKEsigs could predict prodromal MS. Random forest was chosen based on its combination of interpretability and performance.27 To measure the importance of the knowledge network in the prediction, we also created a classifier using only the binary vector corresponding to the patient’s SEP. Since SEPs are simply the EHR input variables used to derive the SPOKEsigs, comparing the performance between the 2 classifiers allowed us to gauge the predictive performance gained by using SPOKE.
In order to build a classifier that could be used to compute risk of MS in the general population, the classifier was tested using the prevalence of MS at UCSF, which approached ∼1:1000 for all groups (comparable to the prevalence of MS in the United States).28,29 The classifiers (using either SPOKEsigs or SEPs) were run from snapshots at years −5, −3, and −1 from diagnosis for both the All-Visits and PCP-Only groups.
RESULTS
MS-related nodes increase in significance as time of diagnosis approaches
In order to measure the flow of information from thousands of subjects through the “MS” node in SPOKE before diagnosis, we generated SPOKEsigs without using the PSEV corresponding to the concept MS (as MS is naturally the top-ranked node within the MS PSEV).23 Of interest, nodes related to the physiopathology of MS were found to be highly ranked likely due to the biologically meaningful connections within SPOKE. To investigate the importance of the MS node in our subject population, the rank distribution of MS was compared for years −7 to −1 relative to MS diagnosis in the index group. Figure 3A shows that MS increases in significance as time to diagnosis approaches for both the All-Visits and PCP-Only groups (r2 = 0.93; P < .037 PCP-Only and r2 = 0.96; P < .018 All-Visits). Furthermore, when compared with all other diseases in SPOKE, MS remains within the top 1% in the All-Visits group and (and within 2% for PCP-Only visits), during years −7 to −1. Further, the importance of MS is statistically significant (t test) for both groups between years −5 (5.5e-6 PCP-Only; 1.6e-26 All-Visits) to −1 (6.4e-62 PCP-Only; 3.4e-147 All-Visits). Note that this cannot be explained by prescriptions of MS-specific disease-modifying medications (DMTs), as these individuals have not been yet diagnosed with MS. There is a noticeable gap between the P-values for the All-Visits and the PCP-Only groups, suggesting a substantial increase in information related to MS being recorded during specialist visits. Though this increase in significance (overtime as well as the difference between PCP-Only and All-Visits groups) can partially be attributed to the increased sample size, the average P-value at any time point is not significant. Further, the slope for the MS node compared with the slope of the average P-value over time is 215× and 127× higher (All-Visits and PCP-Only, respectively), suggesting that only a small portion of the increase in significance can be attributed to increased sample size.
Figure 3.
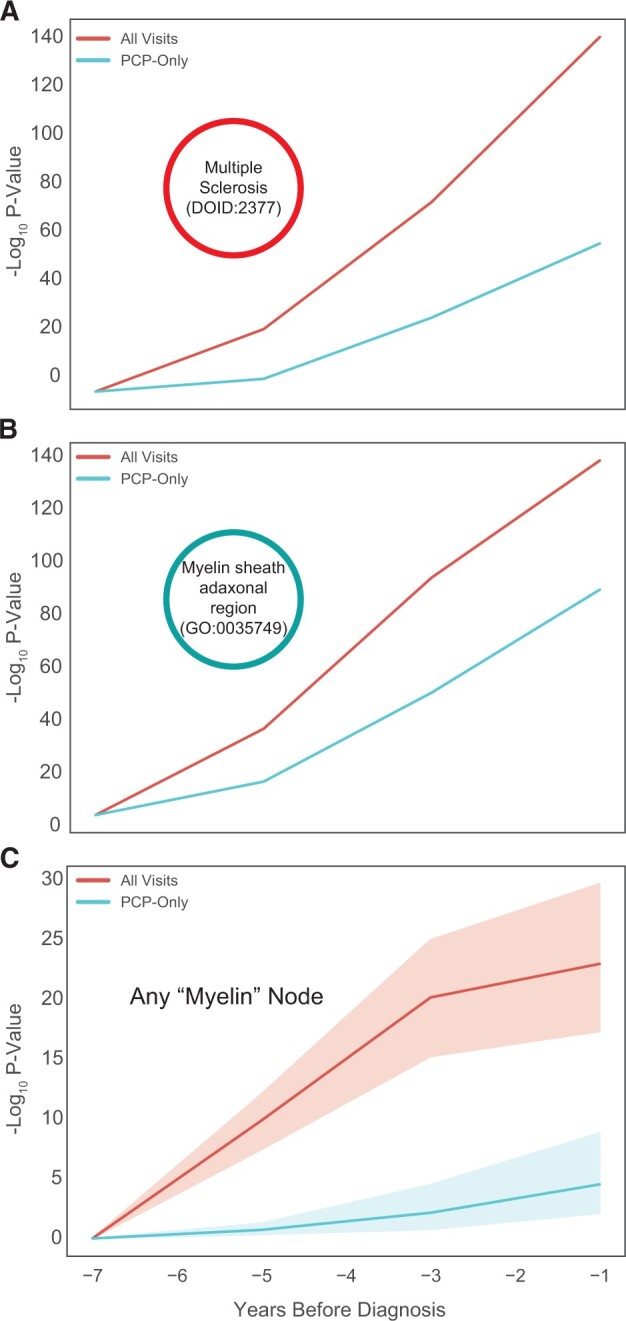
Multiple sclerosis (MS) biology nodes become more significant with time to diagnosis. (A–C) For each node, the t test was used to compare the distribution of ranks between the MS and non-MS patients. Here the −log10 P-value from the t test is plotted against time to MS diagnosis (or lack thereof) for the (A) MS node (DOID:2377), (B) Myelin sheath adaxonal region (GO:0035749), and (C) group of nodes with “myelin” in the name.
To ensure that these results were MS-specific and not simply the outcome of visiting a neurologist (in the All-Visits group), a similar analysis was conducted using snapshots from patients diagnosed with amyotrophic lateral sclerosis (ALS). Similarly, ALS was the most important disease (P < 3.17e-9) in the ALS snapshots at year −1. In contrast, the MS node was not differentially ranked compared with the control population (P > .9). This indicates that although both MS and ALS patients can see neurologists during the prodromal period, each prodromal disease has a distinct signal in SPOKE.
Considering that a first demyelinating event must occur prior to the diagnosis of MS,30,31 we speculated that SPOKE nodes related to myelin might also increase in significance as time to diagnosis approached. Figure 3B illustrates the increased significance of the concept Myelin sheath adaxonal region (GO:0035749). Furthermore, the same trend is observed for any node with “myelin” in its name (Figure 3C). These results suggest that the biological underpinnings of the disease might be detectable during the prodromal period using only information from the EHR.
Predicting prodromal MS
After confirming that SPOKEsigs contained meaningful information related to MS, a predictive model was built using patient-specific SPOKEsigs as inputs to a random forest classifier. The average area under the receiver operating characteristic (ROC) curve (AUC) for the SPOKEsig All-Visit (AV) classifier was 0.76 at −7 years, and progressively increased to 0.84 for year −1. This same trend was observed for all 4 classifier types (AUCSPOKE AV: 0.76–0.84, AUCSPOKE PCP: 0.6–0.78, AUCSEP AV: 0.7–0.83, and AUCSEP PCP: 0.53–0.75; Figure 4). As expected, the classifier that used all encounters outperformed the classifier that used PCP-Only encounters (Avg. ΔAUCSPOKE Years −1 to −5: 0.11 and Avg. ΔAUCSEP Years −1 to −5: 0.15; Avg. ΔAUC = Avg. AUC All-Visits—Avg. AUC PCP-Only). In all cases of information loss, either from smaller time windows (time from diagnosis) or missing specialist visits (PCP-Only), the enhancement of EHRs with SPOKE drove classifier performance. The greatest improvement was seen at 3 years prior to diagnosis using PCP-Only encounters (ΔAUCSPOKE-SEP: 0.12). Altogether, these results demonstrate that embeddings of patients’ clinical data from the structured portion of the EHR onto a KG contain relevant information about their health status. Furthermore, adding structured knowledge to EHR data through SPOKE can compensate for missing and incomplete EHR data.
Figure 4.

Integrating SPOKE enhances classifier AUC. ROC curves for predicting MS diagnosis at year(s) −1, −3, and −5 (A–C accordingly) with a random forest classifier. The classifiers that used encounters from All-Visits are in blue (SPOKEsig input vector) and green (SPOKE Entry Point [SEP] input vector). The classifiers that only used encounters from primary care provider (PCP) visits are shown in orange (SPOKEsig input vector) and red (SEP input vector). In all instances the SPOKEsig input vectors out preformed the corresponding SEP input vector. The largest gain in AUC was for the PCP encounter classifier 3 years prior to diagnosis.
More SEPs will likely improve classifier performance
We recognize SEPs themselves are incomplete because they currently do not map every EHR concept to SPOKE (88% of conditions, 79% of medications, and 47% of measurements for All-Visits at year −1). To estimate how much SPOKEsigs could improve if each EHR concept was mapped to SPOKE, the same classifiers were run using the full set of EHR concepts. Interestingly, the average difference in AUC between full OMOP and SPOKEsigs was the same as that between SPOKEsigs and SEPs (ΔAUC: 0.053). The majority of OMOP concepts that drove the full OMOP classifiers were measurements that were not mapped to SPOKE (Supplementary Tables S1 and S2). These results suggest that if more EHR concepts were mapped to SPOKE, a significant improvement in the classifier could be achieved.
Biological drivers of the classifier
Our previous results suggest that the improved performance of classifiers using SPOKEsigs over those using only SEPs (ie, straight from the EHR) is due to biologically relevant information from SPOKE being utilized in the computation (ie, because the network connects these variables). To understand how the incorporation of biological knowledge increased the AUC, we extracted the scores of each biological node using the average feature value across all years for both the All-Visits and PCP-Only groups. Next, the top 20 nodes from each biological node type (Gene, Protein, Biological Process, Molecular Function, Cellular Component, and Pathway) were selected and split into MS or non-MS significant groups according to the sign of the t-statistic (Figure 5A and B, respectively). To further interpret how each group of top nodes was connected to one another, additional SPOKE nodes were added if they had direct edges to at least 2 top biological nodes (Figure 5A and B). Remarkably, the highest-ranked nodes in the MS groups corresponded to myelin biology (myelin sheath adaxonal region, MAG, glial cell differentiation, etc.), neurophysiological functions (axonogenesis, ceramide binding, etc.), and adaptive immunity (CD4+T cells and B cell-specific pathways, CCR5, etc.; Figure 5A and Supplementary Table S3). Also significant were nodes related to the CNS, muscle behavior, the extracellular matrix (eg, matrix metalloproteinases, collagen, NCAM, Basigin interactions, etc.), and genes associated with other neurological diseases such as spastic paraplegia (MPV17L2), ataxia (RNF170) Alzheimer’s disease (APBA3), and lysosomal storage disease (NAGLU). Together, these nodes illustrate how the classifier detected the importance of neurological and immunological processes in MS patients several years before their diagnoses. In contrast, the highest-ranked nodes within the non-MS group were related to Th2 cell differentiation (eosinophil migration, prostaglandins, CCR3 chemokine receptor binding, etc.), an immune subset associated with protection against inflammatory diseases like MS (Figure 5B).32–35
Figure 5.

Th1/Th2 balance and neurological nodes drive biological increase in AUC. (A, B) Networks of significant biological nodes for random forest classifier. Red nodes were higher ranked in the MS population (A), whereas blue nodes were higher ranked higher in the non-multiple sclerosis (MS) population (B) (color gradient based on t-statistic). The shape (diamond or oval) of the node denotes whether or not the node is in the top 20 of a given node type. If it is an oval, it must connect to ≥ 2 nodes in the top 20. Highlighted in the network are some of the nodes that correspond to Th1/Th2 balance or neurology. (C) Illustration of how a prescription for carbamazepine can send information to the Myelin sheath adaxonal region node (GO:0007404, GO:0043360; replaced by: GO:0010001). (D) Depiction of how asthma (a Th2-mediated disease that is more prevalent in the non-MS UCSF population) pushes information downstream to the Eicosanoid ligand-binding receptors node (Reactome R-HSA-391903).
Medications and common laboratory tests drive information flow to neurological nodes
The difficulty in identifying MS at an early stage is due to the combination of the EHRs being sparse and MS symptoms being vague and common in the general population. Often this results in OMOP codes only being associated with one or a small number of MS patients (Supplementary Figure S2) which does not contribute to the classifier. However, after mapping an OMOP concept to a SEP it is transformed into a multidimensional SPOKEsig that represents the importance of each node in SPOKE for that OMOP concept/SEP. Therefore, 2 distinct OMOP concepts could “push” information to the same downstream nodes.
To identify which OMOP concepts were responsible for “pushing” information downstream to each of the MS-significant biological nodes, network paths were traced back to the originating SEPs (see Materials and Methods). For most of the top MS nodes, the SEPs that were essential for the high rank of the MS-significant nodes were mapped from medication orders and common laboratory tests (note that MS DMTs are not SEPs, as none of these individuals had been diagnosed with MS at the time of analysis). Though these SEPs may not have been significant in the MS population as a whole, their propagation through SPOKE led to increased information flow to the MS top nodes. For example, while Carbamazepine and Lithium are not significant as distinct SEPs, they both direct information flow to the GO concept “Myelin sheath adaxonal region” (GO:0035749, a highly ranked MS-relevant node) in a representative patient shown in Figure 5C. For this patient, information flows from Carbamazepine to a set of Disease nodes (either through “treated by” or “contraindicated for” edges) and then (either directly or through an additional Disease or Gene node) to the genes CNP, MAG, or PTEN which are all components of “Myelin sheath adaxonal region.” Interestingly, Carbamazepine or Lithium can be used to treat symptoms and comorbidities of MS such as trigeminal and glossopharyngeal neuralgia or depression, respectively, which are common symptoms experienced by MS patients. This further demonstrates that distinct clinical presentations can lead to similar SPOKE representations of MS patients.
Similarly, the paths between the laboratory test for Aspartate aminotransferase travel through aspartic acid (Compound) and then traverse 1–2 edge(s) before reaching MAG and PTEN (Genes) (Supplementary Figure S3). Despite the different paths of entry into SPOKE, data are repetitively sent through nodes such as MAG and PTEN, which then converge at the “Myelin sheath adaxonal region” node. Similar patterns were observed for multiple other neurological nodes.
Th2-mediated diseases drive information to non-MS biological nodes
The same method for abstracting the pertinent OMOP concepts information flow was then applied to the top non-MS biological nodes. After retracing several paths, we found that the OMOP concepts that facilitated the flow of information to nodes related to eosinophils, eicosanoids, and T cells were driven by Th2-mediated diseases such as asthma and allergies which are more prevalent in the non-MS population (–log2 odds ratio of −2.46 and −1.97, accordingly). Figure 5D provides an example of how these diseases transfer information to the (non-MS significant) biological node Eicosanoid ligand-binding receptors. In this representative patient, data start at the node for asthma and then either directly connect to or are 1 neighbor apart from genes that participate in Eicosanoid ligand-binding receptor (Pathway). In the latter case, the information first flows through diseases similar to asthma or its associated genes. These straightforward routes from Th2-mediated diseases to their associated genes are what power the Th2 signal in the non-MS significant biological nodes.
Taken together, our results show that SPOKE nodes useful for the classifier include nodes with both strongly positive (highly ranked in MS) and negative (highly ranked in controls) associations with MS. In both cases, the biological interpretation of those nodes is consistent with the known pathogenesis of MS.
DISCUSSION
The purported prodromal period of MS is often described in terms of healthcare utilization.36,37 MS patients in the prodromal stage are, by definition, months or even years away from a recorded diagnosis code for MS. During this period, however, they are not just standing idly—in fact, their healthcare use both within and beyond the primary care setting, steadily increases until time to diagnosis.36 Previous research revealed that MS patients have more encounters with psychiatrists and urologists, as well as higher proportions of musculoskeletal, genito-urinary, or hormonal-related prescriptions.38These findings hint that underlying biological signals must be present months or even years before diagnosis and the information from these specialist visits could be pivotal in uncovering those differences.
Although patients often pay multiple visits to a specialist before receiving an MS diagnosis, the process of obtaining an appointment with a specialist can itself be prolonged, usually requiring a referral and insurance coverage. As a result, a patient’s initial interface with a health system is often through primary or emergency care. Appreciating the different roles primary care and specialist clinicians play in the diagnosis process, we ran 2 analyses in parallel using data from either PCP-Only or All-Visits. Though it is possible for symptoms to be recorded in the structured portion of EHRs, this typically only occurs if it is necessary for billing. Additional patient data can be extracted from the patient notes using natural language processing (NLP). However, NLP methods to date generate rather sparse data, and need further validation in healthcare settings; thus their incorporation is out of scope for this work.
The generation of PSEVs is comparable with word2vec, another machine-learning vector embedding method.25,26 Similar to how word2vec learns the embedding of a word by using the words around it as context, PSEVs utilize patient cohorts to give context to the nodes in SPOKE. PSEVs are then added together to produce the Patient-Specific SPOKE Profile Vectors (SPOKEsigs) that describe a patient in terms of node weights in SPOKE. The main difference between these 2 embedding techniques is that PSEVs (and therefore SPOKEsigs) are based on a “clear box” algorithm that constructs machine-readable vectors while maintaining human interpretability. This means each element in the vector corresponds to a node in SPOKE and it is possible to trace back how information travels from sparse EHRs to downstream nodes. The diffusion of EHRs through SPOKE enabled the prioritization of the MS Disease node in the SPOKEsigs of MS patients compared with controls. Additionally, the significance of this differential prioritization increases as the time to diagnosis decreases. Further, we have shown that the known biological underpinnings of MS could be abstracted using these sparse clinical data. This is evident by the prioritization of myelin-related nodes within the SPOKEsigs of MS patients—whose disease is characterized by demyelination in the CNS—compared with controls up to 7 years prior to MS diagnosis.
We hypothesized that SPOKEsigs contained deeper information about a patient than the equivalent EHR vectors (SEPs). Remarkably, SPOKEsigs outperformed SEPs (ie, EHR-only information) at all time points for both the All-Visits and PCP-Only analyses. The All-Visit AUCs were always higher than the PCP-Only AUCs due to the greater power of the All-Visit group in both the number of patients and encounters. This difference was minimized by the addition of SPOKE, which enabled the use of PCP-Only data to achieve results closer to using All-Visit data using the SEPs alone. This enhancement of EHRs using SPOKE was particularly striking for the PCP-Only analysis performed 3 years before diagnosis, which showed a 12% improvement in AUC (over SEPs alone). These results hint at a future where, after adequate validation including consideration of possible biases, SPOKE could be used at the point of care to support or target supplementary evaluation for primary care providers.
The top biological drivers of the classifier were split into 2 groups (MS significant or non-MS significant) according to whether they were ranked higher in the MS or Control SPOKEsigs. Notably, neurophysiological functions, CNS, and muscle behavior nodes were among the top MS-significant nodes. In contrast, there were many Th2-related nodes (indicating immunoregulatory activity) dominating the non-MS significant nodes. Interestingly, phospholipase C activity, which was high in the MS group, is known to play a role or interact within both the MS and non-MS top immune features. Moreover, phospholipase C39 was recently implicated in female-specific neuropathic pain induce a myelin basic protein peptide (MBP84–104) in mice. This study showed that after MBP exposure, T cells attack the DRG and spinal cord in females but remain localized in males.40 Notably, multiple top nodes from both the MS and non-MS groups participate together in this pathway in a way that is consistent with both this observed sexual dimorphism as well as the increased prevalence of MS among women. This connection between top immune nodes within MS and non-MS groups further supports the hypothesis that MS (and others like RA) results from an imbalance between proinflammatory (Th1 or Th17) and immunoregulatory Th2 responses.41 In contrast, asthma and allergies are mediated by Th2 responses, which presumably protect against Th1/Th17-driven diseases.42,43
PSEVs represent a new class of clear (as opposed to a black) box algorithms. This property allowed us to trace back how key biological nodes became significant. The propagation of information to nodes that were ranked higher in non-MS patients mostly originated from Th2-mediated diseases such as allergies and asthma, which were more prevalent in the non-MS population. In contrast, a heterogeneous set EHRs mainly from commonly ordered laboratory tests or treatments for comorbidities facilitated information to move to the MS significant nodes. These results demonstrate that clinical presentation and biological changes are inherently linked and the intersection can be uncovered using EHRs during the MS prodromal period.
To move toward the delivery of precision medicine, disease biology and clinical manifestations must be investigated side by side. Increasing amounts of data are being obtained for individual patients, and knowledge networks will play a key role in bridging the gap between biological knowledge derived from basic science research, and medical knowledge. As more measurements (genomics, proteomics, microbiome) become available, we hypothesize the SPOKEsigs will become even more informative. Further, the transition from curative to preventative medicine can only be possible through a better understanding of the prodromal biology of a disease. It is our hope that such methods will be used for a variety of diseases to advance both precision and preventative medicine.
CONCLUSIONS
This work presents a strategy to embed EHR data onto a knowledge graph (SPOKE) to obtain high-dimensional health status profiles (SPOKEsigs). SPOKEsigs were computed for hundreds of thousands of individuals and a random-forest classifier was trained to identify individuals at risk of MS. This approach was able to detect MS up to 5 years prior to their documented diagnosis in the clinic. SPOKEsigs represent a new kind of “clear box” explainable predictable models with broad applicability to other chronic medical conditions where early diagnosis can benefit patients.
FUNDING
This work was supported in part by a grant from the US National Science Foundation (Convergence Accelerator NSF_1937160). Partial support also comes from the Bakar Family Foundation and the Bakar Computational Health Sciences Institute. SEB holds the Heidrich Family and Friends endowed chair in Neurology at UCSF. SEB holds the Professorship in Neurology I at UCSF.
AUTHOR CONTRIBUTIONS
CAN: developed methods for analysis, gathered data, and performed analysis. Created Figures, and drafted article. RB: analyzed clinical data, ensured medical accuracy, and edited article. AJB: contributed to the study design, methods development and article revision, and editing. SEB: study conception, design, and supervision. Data analysis and Figure design. Drafted and edited article. Accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
SUPPLEMENTARY MATERIAL
Supplementary material is available at Journal of the American Medical Informatics Association online.
CONFLICT OF INTEREST STATEMENT
RB is funded by the National Multiple Sclerosis Society Harry Weaver Award, the National Science Foundation and the National Institutes of Health National Library Medicine. She has received research grant funding from Biogen, Novartis, and Roche Genentech. She has received consulting honoraria from Alexion, Biogen, EMD Serono, Genzyme Sanofi, Novartis, and Roche Genentech. SEB is cofounder and holds shares in MATE Bioservices, a company that commercializes uses of SPOKE knowledge graph. C.A.N holds shares of MATE Bioservices. AB is a cofounder and consultant to Personalis and NuMedii; consultant to Samsung, Mango Tree Corporation, and in the recent past, 10× Genomics, Helix, Pathway Genomics, and Verinata (Illumina); has served on paid advisory panels or boards for Geisinger Health, Regenstrief Institute, Gerson Lehman Group, AlphaSights, Covance, Novartis, Genentech, Merck, and Roche; is a shareholder in Personalis and NuMedii; is a minor shareholder in Apple, Facebook, Alphabet (Google), Microsoft, Amazon, Snap, Snowflake, 10x Genomics, Illumina, Nuna Health, Assay Depot (Scientist.com), Vet24seven, Regeneron, Sanofi, Royalty Pharma, Pfizer, BioNTech, AstraZeneca, Moderna, Biogen, Twist Bioscience, Pacific Biosciences, Editas Medicine, Invitae, Doximity, and Sutro, and several other non-health-related companies and mutual funds; and has received honoraria and travel reimbursement for invited talks from Johnson and Johnson, Roche, Genentech, Pfizer, Merck, Lilly, Takeda, Varian, Mars, Siemens, Optum, Abbott, Celgene, AstraZeneca, AbbVie, Westat, several investment and venture capital firms, and many academic institutions, medical- or disease-specific foundations and associations, and health systems. AB receives royalty payments through Stanford University, for several patents and other disclosures licensed to NuMedii and Personalis. AB’s research has been funded by NIH, Northrup Grumman (as the prime on an NIH contract), Genentech, Johnson and Johnson, FDA, Robert Wood Johnson Foundation, Leon Lowenstein Foundation, Intervalien Foundation, Priscilla Chan and Mark Zuckerberg, the Barbara and Gerson Bakar Foundation, and in the recent past, the March of Dimes, Juvenile Diabetes Research Foundation, California Governor’s Office of Planning and Research, California Institute for Regenerative Medicine, L’Oreal, and Progenity.
DATA AVAILABILITY
Due to the sensitive nature of EHR, we are not able to share patient data, even in deidentified form. To facilitate the reproducibility and advancement of this research, we have created an API for generating SPOKEsigs alongside a jupyter notebook with instructions on how to use it, which can be accessed at https://github.com/BaranziniLab/SPOKEsigs. Anyone with access to EHRs can now create SPOKEsigs for their own patient populations and test the concepts presented in this work. SPOKE can be accessed at https://spoke.rbvi.ucsf.edu/neighborhood.html.
Supplementary Material
Contributor Information
Charlotte A Nelson, Integrated Program in Quantitative Biology, University of California San Francisco, San Francisco, California, USA; Bakar Computational Health Sciences Institute, University of California San Francisco, San Francisco, California, USA.
Riley Bove, Department of Neurology, UCSF Weill Institute for Neurosciences, University of California San Francisco, San Francisco, California, USA.
Atul J Butte, Bakar Computational Health Sciences Institute, University of California San Francisco, San Francisco, California, USA; Department of Pediatrics, University of California San Francisco, San Francisco, California, USA.
Sergio E Baranzini, Integrated Program in Quantitative Biology, University of California San Francisco, San Francisco, California, USA; Bakar Computational Health Sciences Institute, University of California San Francisco, San Francisco, California, USA; Department of Neurology, UCSF Weill Institute for Neurosciences, University of California San Francisco, San Francisco, California, USA.
REFERENCES
- 1. Auffray C, Charron D, Hood L. Predictive, preventive, personalized and participatory medicine: back to the future. Genome Med 2010; 2 (8): 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tate AR, Beloff N, Al-Radwan B, et al. Exploiting the potential of large databases of electronic health records for research using rapid search algorithms and an intuitive query interface. J Am Med Inform Assoc 2014; 21 (2): 292–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jensen PB, Jensen LJ, Brunak S. Mining electronic health records: towards better research applications and clinical care. Nat Rev Genet 2012; 13 (6): 395–405. [DOI] [PubMed] [Google Scholar]
- 4. Shickel B, Tighe PJ, Bihorac A, Rashidi P. Deep EHR: a survey of recent advances in deep learning techniques for electronic health record (EHR) analysis. IEEE J Biomed Health Inform 2018; 22 (5): 1589–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stang PE, Ryan PB, Racoosin JA, et al. Advancing the science for active surveillance: rationale and design for the Observational Medical Outcomes Partnership. Ann Intern Med 2010; 153 (9): 600–6. [DOI] [PubMed] [Google Scholar]
- 6. Denny JC, Ritchie MD, Basford MA, et al. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics 2010; 26 (9): 1205–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ritchie MD, Denny JC, Crawford DC, et al. Robust replication of genotype-phenotype associations across multiple diseases in an electronic medical record. Am J Hum Genet 2010; 86 (4): 560–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kho AN, Pacheco JA, Peissig PL, et al. Electronic medical records for genetic research: results of the eMERGE consortium. Sci Transl Med 2011; 3 (79): 79re1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.eMERGE Consortium. Harmonizing clinical sequencing and interpretation for the eMERGE III Network. Am J Hum Genet 2019; 105 (3): 588–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gustafsson M, Nestor CE, Zhang H, et al. Modules, networks and systems medicine for understanding disease and aiding diagnosis. Genome Med 2014; 6 (10): 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nicholson DN, Greene CS. Constructing knowledge graphs and their biomedical applications. Comput Struct Biotechnol J 2020; 18: 1414–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Barabasi AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet 2011; 12 (1): 56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Goh K-I, Cusick ME, Valle D, Childs B, Vidal M, Barabási A-L. The human disease network. Proc Natl Acad Sci U S A 2007; 104 (21): 8685–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang L, Himmelstein DS, Santaniello A, Parvin M, Baranzini SE. iCTNet2: integrating heterogeneous biological interactions to understand complex traits. F1000Res 2015; 4: 485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhou X, Menche J, Barabasi AL, Sharma A. Human symptoms-disease network. Nat Commun 2014; 5: 4212. [DOI] [PubMed] [Google Scholar]
- 16. Himmelstein DS, Baranzini SE. Heterogeneous network edge prediction: a data integration approach to prioritize disease-associated genes. PLoS Comput Biol 2015; 11 (7): e1004259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Woolhandler S, Himmelstein DU. Single-payer reform. Ann Intern Med 2017; 167 (7): 527. [DOI] [PubMed] [Google Scholar]
- 18. O’Connor PJ, Sperl-Hillen JM, Rush WA, et al. Impact of electronic health record clinical decision support on diabetes care: a randomized trial. Ann Fam Med 2011; 9 (1): 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ye C, Fu T, Hao S, et al. Prediction of incident hypertension within the next year: prospective study using statewide electronic health records and machine learning. J Med Internet Res 2018; 20 (1): e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xia Z, Steele SU, Bakshi A, et al. Assessment of early evidence of multiple sclerosis in a prospective study of asymptomatic high-risk family members. JAMA Neurol 2017; 74 (3): 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Disanto G, Zecca C, MacLachlan S, et al. Prodromal symptoms of multiple sclerosis in primary care. Ann Neurol 2018; 83 (6): 1162–73. [DOI] [PubMed] [Google Scholar]
- 22. Kappos L, Edan G, Freedman MS, et al. ; BENEFIT Study Group. The 11-year long-term follow-up study from the randomized BENEFIT CIS trial. Neurology 2016; 87 (10): 978–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nelson CA, Butte AJ, Baranzini SE. Integrating biomedical research and electronic health records to create knowledge-based biologically meaningful machine-readable embeddings. Nat Commun 2019; 10 (1): 3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haveliwala TH. Topic-sensitive pagerank. In: WWW '02: proceedings of the 11th International Conference on World Wide Web; New York, NY: Association for Computing Machinery; May 2002: 517–26. https://doi.org/10.1145/511446.511513
- 25. Mikolov T, Chen K, Corrado G. J. D. Efficient estimation of word representations in vector space. 2013; (arXiv:1301.3781v3).
- 26. Mikolov T, Sutskever I, Chen K, Corrado G, Dean J. Distributed representations of words and phrases and their compositionality. In: Proceedings of the 26th International Conference on Neural Information Processing Systems. Volume 2. Lake Tahoe, Nevada: Curran Associates Inc.; 2013: 3111–9. [Google Scholar]
- 27. Couronne R, Probst P, Boulesteix AL. Random forest versus logistic regression: a large-scale benchmark experiment. BMC Bioinformatics 2018; 19 (1): 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hirtz D, Thurman DJ, Gwinn-Hardy K, Mohamed M, Chaudhuri AR, Zalutsky R. How common are the “common” neurologic disorders? Neurology 2007; 68 (5): 326–37. [DOI] [PubMed] [Google Scholar]
- 29. Wallin MT, Culpepper WJ, Campbell JD, et al. ; US Multiple Sclerosis Prevalence Workgroup The prevalence of MS in the United States. Neurology 2019; 92 (10): e1029–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Okuda DT, Mowry EM, Beheshtian A, et al. Incidental MRI anomalies suggestive of multiple sclerosis the radiologically isolated syndrome. Neurology 2009; 72 (9): 800–5. [DOI] [PubMed] [Google Scholar]
- 31. Okuda DT, Siva A, Kantarci O, et al. ; on behalf of the Radiologically Isolated Syndrome Consortium (RISC) and Club Francophone de la Sclérose en Plaques (CFSEP). Radiologically isolated syndrome: 5-year risk for an initial clinical event. PLoS One 2014; 9 (3): e90509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Raphael I, Nalawade S, Eagar TN, Forsthuber TG. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015; 74 (1): 5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hirahara K, Nakayama T. CD4+T-cell subsets in inflammatory diseases: beyond the Th1/Th2 paradigm. Int Immunol 2016; 28 (4): 163–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bomprezzi R. Dimethyl fumarate in the treatment of relapsing–remitting multiple sclerosis: an overview. Adv Neurol Disord 2015; 8 (1): 20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Crane IJ, Forrester JV. Th1 and Th2 lymphocytes in autoimmune disease. Crit Rev Immunol 2005; 25 (2): 75–102. [DOI] [PubMed] [Google Scholar]
- 36. Wijnands JM, Kingwell E, Zhu F, et al. Infection-related health care utilization among people with and without multiple sclerosis. Mult Scler 2017; 23 (11): 1506–16. [DOI] [PubMed] [Google Scholar]
- 37. Wijnands JMA, Kingwell E, Zhu F, et al. Health-care use before a first demyelinating event suggestive of a multiple sclerosis prodrome: a matched cohort study. Lancet Neurol 2017; 16 (6): 445–51. [DOI] [PubMed] [Google Scholar]
- 38. Wijnands JMA, Zhu F, Kingwell E, et al. Five years before multiple sclerosis onset: Phenotyping the prodrome. Mult Scler 2019; 25 (8): 1092–101. [DOI] [PubMed] [Google Scholar]
- 39. Bush WS, McCauley JL, DeJager PL, et al. ; International Multiple Sclerosis Genetics Consortium. A knowledge-driven interaction analysis reveals potential neurodegenerative mechanism of multiple sclerosis susceptibility. Genes Immun 2011; 12 (5): 335–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chernov AV, Hullugundi SK, Eddinger KA, et al. A myelin basic protein fragment induces sexually dimorphic transcriptome signatures of neuropathic pain in mice. J Biol Chem 2020; 295 (31): 10807–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bar-Or A. The immunology of multiple sclerosis. Semin Neurol 2008; 28 (1): 29– 45. [DOI] [PubMed] [Google Scholar]
- 42. Tremlett HL, Evans J, Wiles CM, Luscombe DK. Asthma and multiple sclerosis: an inverse association in a case-control general practice population. QJM 2002; 95 (11): 753–6. [DOI] [PubMed] [Google Scholar]
- 43. Eagar TN, Miller SD. Helper T-cell subsets and control of the inflammatory response. In: Rich RR, Fleisher TA, Shearer WT, Schroeder HW, Frew AJ, Weyand CM, eds. Clinical Immunology , 5th ed. London: Elseiver; 2019: 235–45.e1. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Due to the sensitive nature of EHR, we are not able to share patient data, even in deidentified form. To facilitate the reproducibility and advancement of this research, we have created an API for generating SPOKEsigs alongside a jupyter notebook with instructions on how to use it, which can be accessed at https://github.com/BaranziniLab/SPOKEsigs. Anyone with access to EHRs can now create SPOKEsigs for their own patient populations and test the concepts presented in this work. SPOKE can be accessed at https://spoke.rbvi.ucsf.edu/neighborhood.html.