

Abstract
目的
对一个Ⅰ型神经纤维瘤家系进行遗传学病因分析及产前诊断。
方法
应用目标捕获高通量测序和Sanger测序技术,对一个散发的Ⅰ型神经纤维瘤家系进行基因突变分析。提取先证者及其父母外周血淋巴细胞RNA,行RT-PCR及扩增产物测序分析。明确突变致病性后,抽取羊水标本对胎儿行产前基因诊断。
结果
先证者 NF1基因存在c.1260+4A>T杂合剪接突变,为新发突变。RNA剪接分析提示先证者 NF1基因发生转录时,11号外显子3'端发生相邻内含子区13个碱基的插入,理论上可导致蛋白编码提前终止,产生截短蛋白,影响蛋白功能。产前基因检测结果显示胎儿未携带该突变。
结论
NF1:c.1260+4A>T杂合剪接突变是该Ⅰ型神经纤维瘤患者的致病原因,本研究结果为该家系遗传咨询和产前诊断提供了理论依据。
Abstract
Objective
To identify pathogenic mutation for a family with neurofibromatosis type 1(NF1) and provide prenatal diagnosis for them.
Methods
Mutation analysis of the sporadic family with NF1 was performed with target captured next generation sequencing and Sanger sequencing. RNA samples were extracted from the lymphocytes of NF1 patient and her parents. RT-PCR and Sanger sequencing were performed to analyze the relative mRNA expression in the samples. Prenatal diagnosis of the pathogenic mutation was offered to the fetus.
Results
A novel splicing mutation c.1260+4A>T in the NF1 gene was found in the proband of the family, but was not found in her parents.cDNA sequencing showed that 13 bases inserted into the 3' end of exon 11 in the NF1 gene lead to a frameshift mutation. Prenatal diagnosis suggested that the fetus did not carried the mutant.
Conclusion
The NF1: c.1260+4A>T mutation found in the NF1 patient is considered to be pathogenic, which provides information for family genetic counseling and prenatal diagnosis.
Keywords: Neurofibroma/genetics; Genes, neurofibromatosis 1; Amniotic fluid; Prenatal diagnosis; Genetic testing; Pedigree; Mutation; Exons; Polymerase chain reaction
Ⅰ型神经纤维瘤(neurofibromatosis type Ⅰ,NF1, OMIM 162200)是一种常见的常染色体显性遗传病,发病率为1/2500~1/3000 [ 1] ,其主要临床特征为多发性的牛奶咖啡斑、腋窝和腹股沟雀斑、多发性皮肤神经纤维瘤、虹膜错构瘤和(或)特殊骨骼异常,如蝶骨嵴或胫骨发育不良等 [ 2] 。 NF1 基因(NM_000267.2)位于染色体17q11.2,基因总长度约350 Kb,包含60个外显子,其编码的神经纤维瘤蛋白包含2818个氨基酸,是RAS-MAPK信号通路的关键成分,可促进Ras-GTP失活 [ 3- 4] 。 NF1 基因突变导致神经纤维瘤蛋白功能丧失,从而引起下游细胞生长激活 [ 5- 6] 。至少78 %符合美国国立卫生研究院(NIH)诊断标准的患者可以发现 NF1 基因突变 [ 1] 。迄今,人类基因突变数据库(HGMD)报道的 NF1 基因突变超过3000种。 NF1 基因新发突变率较高,约50 %的NF1患者无家族史 [ 7] 。本研究中,我们应用目标捕获高通量测序技术对一例散发的NF1患儿进行基因检测,明确了其遗传学病因,为家系遗传咨询和产前诊断提供了理论依据。
1 对象与方法
1.1 对象
先证者,女,11岁,全身牛奶咖啡斑,腹部最多;椎骨神经纤维瘤,手术切除后臀部又出现纤维瘤;双下肢长度不一。根据NIH的诊断标准 [ 8] ,该患者明确诊断为NF1,其父母未见明显临床表型。其母亲就诊时已再次妊娠,孕16周 +,要求对胎儿行产前基因诊断。
本研究经浙江大学医学院附属妇产科医院伦理委员会审查通过[(2019)伦审科第(038)号],家系成员均签署了知情同意书。
1.2 试剂和仪器
QIAamp DNA Blood Mini Kit为德国QIAGEN公司产品;2×GoldStar MasterMix为北京康为世纪生物科技有限公司产品;RNAiso Blood试剂和PrimeScript TM Ⅱ 1st Strand cDNA Synthesis Kit为宝日医生物技术(北京)有限公司产品;AmpFLSTR TM Identifiler TM PCR Amplification Kit为美国Applied Biosystems公司产品。
NanoDrop 2000分光光度计为美国Thermo Fisher Scientific公司产品;BGISEQ-500高通量测序仪为深圳华大基因股份有限公司产品;ABI3730全自动测序仪为美国Applied Biosystems公司产品;PCR扩增仪为美国Bio-Rad公司产品。
1.3 外周血DNA、RNA提取及cDNA合成
采集外周血5 mL,置于EDTA二钾抗凝管中,-20 ℃冻存。取200 μL抗凝血,按照QIAamp DNA Blood Mini Kit说明书进行DNA提取。孕20周经腹超声引导下羊膜腔穿刺抽取羊水30 mL,其中20 mL用于胎儿染色体核型分析,10 mL用于胎儿基因检测。羊水培养2周后收集细胞,利用QIAamp DNA Blood Mini Kit提取基因组DNA,测定浓度后于-20 ℃保存备用。取先证者及其父母250 μL新鲜外周血,采用RNAiso Blood试剂提取外周血总RNA,定量后-70 ℃保存备用。以RNA为模板,采用PrimeScript TM Ⅱ 1st Strand cDNA Synthesis Kit合成第一链cDNA,-20 ℃保存。
1.4 高通量测序检测基因突变
构建DNA文库,通过芯片捕获先证者遗传性神经纤维瘤病相关基因外显子及其邻近±10 bp内含子区突变(包括点突变,20 bp以内的缺失插入突变),通过BGISEQ-500高通量测序仪进行测序,目标区覆盖度达100.00 %,平均深度633.88×,平均深度>30×位点所占比例达99.91 %。高通量测序由深圳华大临床检验中心完成。
1.5 Sanger测序验证基因突变
根据高通量测序结果,使用Primer 5.0软件设计引物。 NF1 基因参考转录本NM_000267。引物由生工生物工程(上海)股份有限公司合成。以先证者及其父母基因组DNA为模板,分别扩增突变位点所在的第11号外显子及邻近内含子片段,片段大小为250 bp,扩增引物序列:F1 5′-AC-TGCCTTGTTTCTTGCTTTCGTAT-3′,R1 5′-TATGG-TCCCTTCGGTCAAGACTTAA-3′。PCR总体积为50 μL:2×GoldStar MasterMix 25 μL、上下游引物(20 μmol/L)各1 μL、DNA模板2 μL、去离子水21 μL。PCR扩增条件:95 ℃预变性10 min,94 ℃变性30 s,60 ℃退火30 s,72 ℃延伸45 s,35个循环后,72 ℃ 10 min。PCR产物送上海华大医学检验所进行Sanger测序。
1.6 生物信息学软件分析基因突变和致病性
从NCBI( https://www.ncbi.nlm.nih.gov/)下载 NF1 基因参考序列,应用Mutation Surveyor软件分析测序结果。查询ClinVar和HGMD数据库有无该突变的致病性报道,应用Human Splicing Finder ()预测可能存在的隐匿剪接位点及基因突变对剪接位点的改变。通过ExAC browser ()和gnomAD browser()查询该突变在正常人群中的发生频率。
1.7 RT-PCR检测RNA剪接产物
以先证者及其父母的cDNA为模板,扩增 NF1 基因编码区,大小840 bp,PCR产物直接进行Sanger测序。扩增引物序列为:F2 5′-AAAGC-AGCAGTTTGGCCACTACA-3′,R2 5′-AAGAACCAG-CAGAGCCTCCATTG-3′。
1.8 根据基因突变位点对胎儿进行产前诊断
基因突变确定后,对胎儿基因组DNA进行针对性突变位点PCR扩增,并行Sanger测序,检测羊水标本是否存在致病突变。运用AmpFLSTR TM Identifiler TM PCR Amplification Kit对胎儿羊水细胞基因组DNA及其父母外周血基因组DNA进行扩增,对15个短串联重复序列位点进行分析,以排除母体DNA的污染。
1.9 妊娠结局及新生儿随访
对孕妇妊娠结局和新生儿出生1个月后的体格发育、皮肤情况进行电话随访。
2 结果
2.1 基因检测结果
高通量测序结果显示,先证者存在 NF1 :c.1260+4A>T杂合突变。Sanger测序结果提示,先证者存在 NF1 :c.1260+4A>T杂合突变,未见其父母存在该突变( 图 1)。
图1.
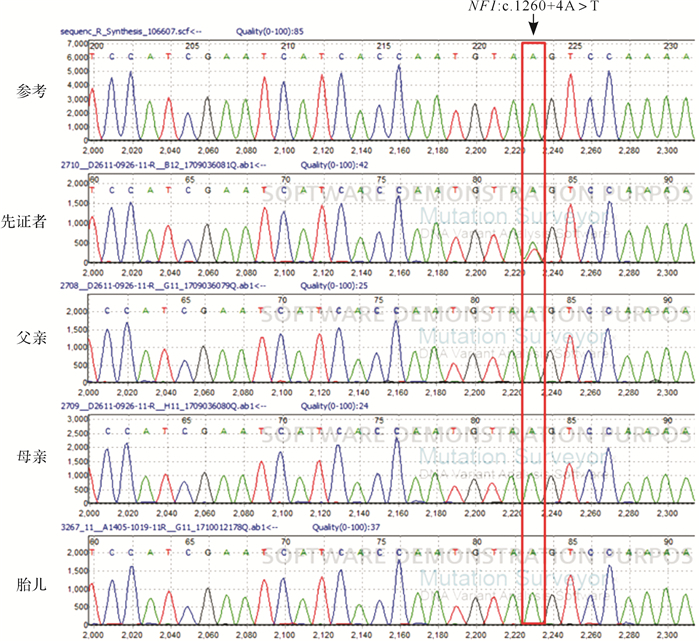
本组Ⅰ型神经纤维瘤家系 NF1 基因测序结果
2.2 :c.1260+4A>T突变对 基因表达剪接产物的影响
Human Splicing Finder预测 NF1 :c.1260+4A>T突变可能影响正常剪接,因此本文应用RT-PCR及Sanger测序进行验证。先证者的cDNA测序结果提示杂合,结果分析显示存在两种剪接产物,一种与参考序列一致,另一种则由于该突变产生新的剪接位点,引起11号外显子3′端发生相邻内含子区13个碱基的插入,导致421位后的编码氨基酸发生移码,并在第432位引入终止密码,使 NF1 编码的全长2818个氨基酸组成的蛋白质缩减至由432个氨基酸组成的截短型蛋白( 图 2),健康对照(父母)仅有一个剪接产物。
图2.
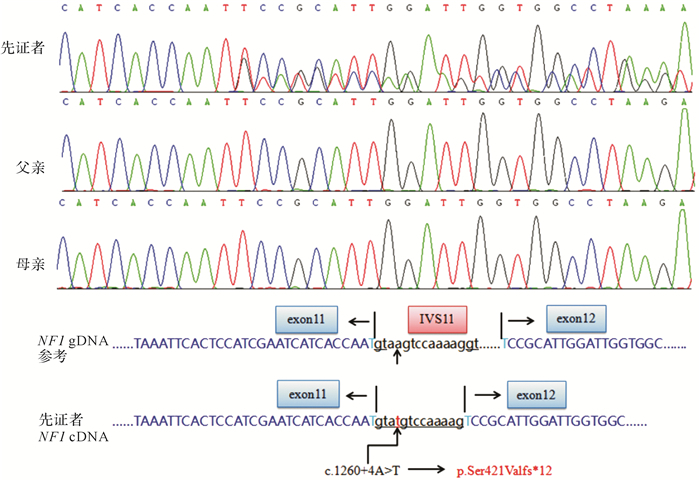
本组Ⅰ型神经纤维瘤家系cDNA测序结果和剪接分析
2.3 突变位点致病性分析预测结果
先证者存在 NF1 :c.1260+4A>T杂合突变,ClinVar和HGMD数据中未见报道,且未见该突变致病性的相关文献报道,因此根据美国医学遗传学和基因组学学会(ACMG)2015年指南 [ 9] 对该突变进行分析:先证者的新发突变,无家族史[PS2:患者的新发突变,且无家族史(经双亲验证)];RNA剪接分析提示产生新的剪接位点,可能影响蛋白功能(PS3:功能实验已明确会导致基因功能受损);ExAC和gnomAD数据库中未见该突变在健康人群中的发生频率,为较强的致病证据(PM2:数据库中健康对照人群中未发现的突变);该突变导致患者表型高度符合NF1(PP4:突变携带者的表型或家族史高度符合某种单基因遗传疾病)。根据ACMG(2015)指南,将该突变评级为“致病”。
2.4 胎儿产前诊断结果
对羊水基因组DNA行测序分析,结果显示胎儿未携带与先证者相同的突变( 图 1D)。短串联重复序列位点检测结果排除羊水标本母源污染。
2.5 新生儿随访情况
先证者母亲妊娠足月后自然分娩一女活婴,皮肤未见异常;产后1个月再次电话随访,婴儿体格发育正常,皮肤未见牛奶咖啡斑。
3 讨论
NF1是由于生殖细胞系 NF1 基因失活导致的一种肿瘤易感性综合征 [ 4] ,主要临床表现为牛奶咖啡色斑、腋窝或腹股沟雀斑、神经纤维瘤、视神经胶质瘤、虹膜结节和(或)特异性骨质缺陷,如蝶骨发育不良或长骨骨质偏薄。根据NIH的诊断标准,只要存在两个及以上的主要临床症状(或一个临床症状伴一级亲属为NF1患者)就可以临床诊断为NF1 [ 10] 。NF1虽然外显率达100 %,但其临床异质性大,在无亲缘关系患者之间存在明显的表现性差异,即使同一家系患者间甚至同一患者不同年龄段也会有不同的表现 [ 11- 12] 。
NF1 基因是人体中突变率最高的基因之一,比一般的基因突变率高出十倍左右,因此大约一半的病例由新发突变引起 [ 13- 14] 。对于生育过NF1患者的夫妻,尽管无家族病史,但不能排除夫妻双方可能存在生殖腺细胞嵌合突变,故再次妊娠时仍存在生育NF1患儿的风险,建议行产前诊断。HGMD报道的 NF1 突变目前已超过3000种,突变类型包含错义突变、无义突变、剪接突变、缺失/插入突变、复杂重排等,尚未发现明确的突变热点。大约2 %的深部内含子突变可产生新的受体/供体剪接位点,使转录mRNA包含一个新的隐匿外显子,导致异常的神经纤维瘤蛋白产生 [ 15] 。此外大约30 %的 NF1 基因突变预测可引起异常剪接,因此基因筛查时分析患者的RNA和DNA十分必要 [ 16] 。
本研究搜集到一个散发的NF1家系,通过目标区域捕获高通量测序和Sanger测序家系验证分析发现,先证者 NF1 基因存在一个新的杂合剪接突变c.1260+4A>T,其父母不存在该突变。推测先证者 NF1 :c.1260+4A>T突变为新发突变的可能性大,但不能排除其父母存在生殖腺嵌合或体细胞低比例嵌合的可能。RNA剪接分析提示该突变导致11号外显子后插入13个碱基,使突变mRNA阅读框移码,理论上造成氨基酸编码提前终止,产生截短蛋白,从而影响蛋白功能。根据ACMG(2015)指南综合分析,该突变可评级为致病突变。突变致病性的明确为产前诊断提供了理论依据。产前诊断结果提示胎儿不存在该突变,从基因水平推测胎儿发生NF1的可能性不大。
综上所述,本研究在一个散发的NF1家系中发现 NF1 基因新的剪接突变c.1260+4A>T,且从RNA水平明确该突变可导致剪接异常。这一发现不仅丰富了 NF1 基因引起NF1的突变谱,同时为该家系的遗传咨询和产前诊断提供了理论依据。
Funding Statement
浙江省重点研发计划(2019C03025);浙江省自然科学基金(LQ19H090019);浙江省医药卫生科技计划(2018PY022)
References
- 1.XU W, YANG X, HU X, et al. Fifty-four novel mutations in the NF1 gene and integrated analyses of the mutations that modulate splicing. Int J Mol Med. 2014;34(1):53–60. doi: 10.3892/ijmm.2014.1756. [XU W, YANG X, HU X, et al. Fifty-four novel mutations in the NF1 gene and integrated analyses of the mutations that modulate splicing[J]. Int J Mol Med, 2014, 34(1):53-60.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.KEHRER-SAWATZKI H, MAUTNER V F, COOPERD N. Emerging genotype-phenotype relation-ships in patients with large NF1 deletions. Hum Genet. 2017;136(4):349–376. doi: 10.1007/s00439-017-1766-y. doi: 10.1007/s00439-017-1766-y. [KEHRER-SAWATZKI H, MAUTNER V F, COOPERD N. Emerging genotype-phenotype relation-ships in patients with large NF1 deletions[J]. Hum Genet, 2017, 136(4):349-376.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.HINMAN M N, SHARMA A, LUO G, et al. Neurofibromatosis type 1 alternative splicing is a key regulator of Ras signaling in neurons. Mol Cell Biol. 2014;34(12):2188–2197. doi: 10.1128/MCB.00019-14. [HINMAN M N, SHARMA A, LUO G, et al. Neurofibromatosis type 1 alternative splicing is a key regulator of Ras signaling in neurons[J]. Mol Cell Biol, 2014, 34(12):2188-2197.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.CUI Y, MORRISON H. Construction of cloning-friendly minigenes for mammalian expression of full-length human NF1 isoforms. Hum Mutat. 2019;40(2):187–192. doi: 10.1002/humu.23681. [CUI Y, MORRISON H. Construction of cloning-friendly minigenes for mammalian expression of full-length human NF1 isoforms[J]. Hum Mutat, 2019, 40(2):187-192.] [DOI] [PubMed] [Google Scholar]
- 5.PELTONEN S, KALLIONPÄÄ R A, PELTONEN J. Neurofibromatosis type 1(NF1) gene:Beyond café au lait spots and dermal neurofibromas. Exp Dermatol. 2017;26(7):645–648. doi: 10.1111/exd.13212. [PELTONEN S, KALLIONPÄÄ R A, PELTONEN J. Neurofibromatosis type 1(NF1) gene:Beyond café au lait spots and dermal neurofibromas[J]. Exp Dermatol, 2017, 26(7):645-648.] [DOI] [PubMed] [Google Scholar]
- 6.BANERJEE S, LEI D, LIANG S, et al. Novel phenotypes of NF1 patients from unrelated Chinese families with tibial pseudarthrosis and anemia. http://cn.bing.com/academic/profile?id=638f14fb742771edcdbefada16f77a5a&encoded=0&v=paper_preview&mkt=zh-cn. Oncotarget. 2017;8(24):39695–39702. doi: 10.18632/oncotarget.13932. [BANERJEE S, LEI D, LIANG S, et al. Novel phenotypes of NF1 patients from unrelated Chinese families with tibial pseudarthrosis and anemia[J]. Oncotarget, 2017, 8(24):39695-39702.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.GUTMANN D H, FERNER R E, LISTERNICKR H, et al. Neurofibromatosis type 1. Nat Rev Dis Primers. 2017;3:17004. doi: 10.1038/nrdp.2017.4. [GUTMANN D H, FERNER R E, LISTERNICKR H, et al. Neurofibromatosis type 1[J]. Nat Rev Dis Primers, 2017, 3:17004.] [DOI] [PubMed] [Google Scholar]
- 8.LISTED N A. Nationalinstitutes of health consensus development conference statement on neurofibromatosis. Arch Neurol. 1988;45:575–578. doi: 10.1001/archneur.1988.00520290115023. [LISTED N A. Nationalinstitutes of health consensus development conference statement on neurofibromatosis[J]. Arch Neurol, 1988, 45:575-578.] [DOI] [PubMed] [Google Scholar]
- 9.RICHARDS S, AZIZ N, BALE S, et al. Standards and guidelines for the interpretation of sequence variants:a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [RICHARDS S, AZIZ N, BALE S, et al. Standards and guidelines for the interpretation of sequence variants:a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology[J]. Genet Med, 2015, 17(5):405-424.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.JETT K, FRIEDMAN J M. Clinical and genetic aspects of neurofibromatosis 1. Genet Med. 2010;12(1):1–11. doi: 10.1097/GIM.0b013e3181bf15e3. [JETT K, FRIEDMAN J M. Clinical and genetic aspects of neurofibromatosis 1[J]. Genet Med, 2010, 12(1):1-11.] [DOI] [PubMed] [Google Scholar]
- 11.TSIPI M, POULOU M, FYLAKTOU I, et al. Phenotypic expression of a spectrum of neurofibromatosis type 1( NF1) mutations identified through NGS and MLPA . J Neurol Sci. 2018;395:95–105. doi: 10.1016/j.jns.2018.10.006. [TSIPI M, POULOU M, FYLAKTOU I, et al. Phenotypic expression of a spectrum of neurofibromatosis type 1( NF1) mutations identified through NGS and MLPA[J]. J Neurol Sci, 2018, 395:95-105. ] [DOI] [PubMed] [Google Scholar]
- 12.PALMA MILLA C, LEZANA ROSALES J M, LÓPEZ MONTIEL J, et al. Neurofibromatosis type Ⅰ:mutation spectrum of NF1 in spanish patients. Ann Hum Genet. 2018;82(6):425–436. doi: 10.1111/ahg.12272. [PALMA MILLA C, LEZANA ROSALES J M, LÓPEZ MONTIEL J, et al. Neurofibromatosis type Ⅰ:mutation spectrum of NF1 in spanish patients[J]. Ann Hum Genet, 2018, 82(6):425-436.] [DOI] [PubMed] [Google Scholar]
- 13.KOCZKOWSKA M, CHEN Y, CALLENS T, et al. Genotype-phenotype correlation in NF1:evidence for a more severe phenotype associated with missense mutations affecting NF1 codons 844-848. https://www.cell.com/ajhg/pdfExtended/S0002-9297(17)30490-1. Am J Hum Genet. 2018;102(1):69–87. doi: 10.1016/j.ajhg.2017.12.001. [KOCZKOWSKA M, CHEN Y, CALLENS T, et al. Genotype-phenotype correlation in NF1:evidence for a more severe phenotype associated with missense mutations affecting NF1 codons 844-848[J]. Am J Hum Genet, 2018, 102(1):69-87.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.UPADHYAYA M, KLUWE L, SPURLOCK G, et al. Germline and somatic NF1 gene mutation spectrum in NF1-associated malignant peripheral nerve sheath tumors (MPNSTs) http://d.old.wanfangdata.com.cn/NSTLQK/10.1002-humu.20601/ Hum Mutat. 2008;29(1):74–82. doi: 10.1002/humu.20601. [UPADHYAYA M, KLUWE L, SPURLOCK G, et al. Germline and somatic NF1 gene mutation spectrum in NF1-associated malignant peripheral nerve sheath tumors (MPNSTs)[J]. Hum Mutat, 2008, 29(1):74-82.] [DOI] [PubMed] [Google Scholar]
- 15.PROS E, GÓMEZ C, MARTÍN T, et al. Nature and mRNA effect of 282 different NF1 point mutations:focus on splicing alterations. Hum Mutat. 2008;29(9):E173–E193. doi: 10.1002/humu.20826. [PROS E, GÓMEZ C, MARTÍN T, et al. Nature and mRNA effect of 282 different NF1 point mutations:focus on splicing alterations[J/OL]. Hum Mutat, 2008, 29(9):E173-E193.] [DOI] [PubMed] [Google Scholar]
- 16.PHILPOTT C, TOVELL H, FRAYLING I M, et al. The NF1 somatic mutational landscape in sporadic human cancers. http://cn.bing.com/academic/profile?id=239720da30e7a94df45ef3d41bbc091e&encoded=0&v=paper_preview&mkt=zh-cn. Hum Genomics. 2017;11(1):13. doi: 10.1186/s40246-017-0109-3. [PHILPOTT C, TOVELL H, FRAYLING I M, et al. The NF1 somatic mutational landscape in sporadic human cancers[J]. Hum Genomics, 2017, 11(1):13.] [DOI] [PMC free article] [PubMed] [Google Scholar]