

 This work is licensed under a
This work is licensed under a Abstract
There is a worldwide trend for women to have their first pregnancy later in life. However, as oocyte quality declines with maternal aging, this trend leads to an increase in subfertility. The cellular mechanisms underlying this decline in oocyte competence are poorly understood. Oocyte mitochondria are the subcellular organelles that supply the energy that drives early embryogenesis, and thus their quality is critical for successful conception. Mitochondria contain their own DNA (mtDNA) and mutations in mtDNA cause mitochondrial diseases with severe symptoms, such as neurodegeneration and heart disease. Since mitochondrial function declines in tissues as humans age accompanied by an accumulation of mtDNA mutations, mtDNA is implicated as a cause of declining oocyte quality in older mothers. While this mutation load could be caused by declining accuracy of the mitochondrial replisome, age-related decline in mitochondrial quality control likely contributes, however knowledge is lacking. Mitophagy, a cellular process which specifically targets and recycles damaged mitochondria may be involved, but studies are scarce. And although assisted reproductive technologies can help older mothers, how these techniques affect the mechanisms that regulate mitochondrial and oocyte quality have not been studied. With the long-term goal of understanding the molecular mechanisms that control mitochondrial quality in the oocyte, model systems including Drosophila and mouse as well as human oocytes have been used. In this review, we explore the contribution of mitophagy to oocyte quality and the need for further systematic investigation in oocytes during maternal aging using different systems.
Lay summary
Mitochondria are small parts of cells called organelles that generate the chemical energy needed for life. Hundreds of thousands of mitochondria in the developing eggs of the mother support the initial growth and development of the fertilized egg. However, due to increasingly diminished function over time, mitochondria generate less energy as we age, posing real problems for older women considering pregnancy. It is possible that this declining energy could be responsible for declining fertility as women age. Energy may decline because mitochondria fail and the cell’s way of keeping them healthy become less efficient as we age. This review summarizes what is known about mitochondrial quality control in developing eggs as they age. In the future, understanding how the best mitochondria are selected and maintained in the egg, and hence the future baby, may enable older women with or without mitochondrial problems, to have healthy children.
Keywords: mitophagy, mitochondria, ovary, oocyte, Drosophila, mouse, human, mtDNA, ARTs
Introduction
Our understanding of human reproduction and fertility has greatly increased in the last two decades. This is due to the knowledge gleaned from experimental model systems and through advances in assisted reproductive technologies (ART). Female fertility rates decline with age. Concurrently, women in developed countries are increasingly having children later in life (O’Brien & Wingfield 2019). However, many of the underlying intrinsic cellular conditions that potentially influence whether each fertilized egg will result in a healthy embryo are still not understood.
The mature egg is the largest cell in the body, and the cytoplasm contains everything required for early embryo development apart from the paternal DNA contributed by the sperm. Thus, at fertilization, all the energetic and nutritional requirements for development of the embryo prior to implantation are derived from the egg. Mature ovulated eggs are generated from primordial germ cells (PGCs) by processes named 'oocyte development' in mammals and 'oogenesis' in Drosophila (Fig. 1). The process is similar but not identical in these very different organisms: the oocytes develop from what is believed to be a finite pool in the majority of mammals, whereas in Drosophila, the oocytes arise from germline stem cells (GSCs) (Matova & Cooley 2001). The process of oocyte development is a complex and dynamic process, requiring intricate interactions between different cells within the gonad as well as appropriate endocrine support. The oocyte increases in size from just a few microns in diameter to around 100 mm in humans, around 70 µm in mouse, and 0.5 mm in Drosophila. The oocyte was believed to be a bystander in this process for many years. However, ablation of oocyte-specific growth and differentiation factor 9 (GDF9) in mouse in 1996 revealed the oocyte as the master conductor in driving the development of the follicle and hence its own destiny (Dong et al. 1996). Likewise, the Drosophila oocyte is responsible for signaling to the surrounding somatic follicle cells to establish the body pattern of the resulting embryo (Lehmann 1995).
Figure 1.

Timing and conserved aspects of oocyte development in humans, mouse, and Drosophila. Human, mouse, and Drosophila undergo female germ cell cyst formation, cyst breakdown, and oocyte maturation and development. Human and mouse germ cell cyst and follicle stages are indicated along the top. Birth occurs at ~week 40 of fetal development in humans (green) and embryonic (E) day 20 for mouse (blue). Drosophila (red) shares many cell biological similarities with human and mouse such as cyst breakdown and oocyte maturation. In Drosophila, germline stem cell division, cyst formation, and oocyte development occur throughout adulthood.
Studies on human ovarian tissue are clearly required to understand human ovarian function. However, experiments interrogating human ovarian function are inherently limited by tissue quantity, type (e.g. age), and source (e.g. patients in poor health or cadavers). In addition, experiments need to be carried out ex vivo. To enable us to explore ovarian function and development, model species have considerable advantages for elucidating the cellular processes underlying oocyte quality. Mice have been a model laboratory species for many years and undergone extensive investigation as they are small, versatile, and reproduce quickly, that is, fertilization to breeding in 9 weeks. Many transgenic mouse models have been created, made possible by advances in genetic modification, enabling targeted research into ovarian function as well as oocyte and embryo development (Barnett et al. 2006). Drosophila has been used as a model for over 100 years. Although Drosophila clearly differs in body plan compared to mice, deciphering the intricacies of cellular signaling pathways and molecular interactions has been advanced using Drosophila in ways not achievable with mouse models due to the speed of reproduction (7 days from germline stem cell to mature egg), the array of genetic tools available, and the accessibility of the ovaries.
Mitochondria are small subcellular organelles that provide cells with the chemical energy ATP. Growth and development of the oocyte demands highly functional mitochondria to support not only its own development, but also to ensure the final product, the mature egg, is commensurate to support early embryonic development. After fertilization, the zygote, as it is now known, embarks on a number of developmental stages resulting from multiple subsequent cell divisions. Critically, all energetics driven by the mitochondria are derived from the oocyte. Thus, in mammals and Drosophila, the generation and maintenance of high functioning mitochondria by the oocyte during development is crucial for early embryo development.
Mitochondrial numbers and dynamics change during oocyte development in mammals and Drosophila (see below). While there is some indication of decreased mitochondrial quality and function with age (Diot et al. 2016b), there is still much that we do not understand. There is a wide-spread appreciation of the requirement for ‘good’ mitochondria in oocytes, especially in the case of inherited mitochondrial disease (Poulton et al. 2010), and there are still many questions unanswered about the mechanisms that regulate this process within the oocyte and how this occurs during oocyte development and aging. One potential mechanism regulating mitochondrial quality during oocyte development, which may be affected by age, is mitophagy. Mitophagy is the process by which damaged mitochondria are targeted for destruction (Box 1). There are currently several known pathways regulating this process, however, new pathways are certain to emerge. This review summarizes the current knowledge of germ cell mitophagy using humans and two model species: mouse and Drosophila. The role of mitophagy during oocyte development and aging has not been extensively explored and yet is critical to understand. Knowledge derived from each model improves our ability to investigate mitochondrial function in human oocyte development.
Box 1 Summary of mitophagy pathways
Mitophagy shares much of the core machinery used by general autophagy. Mitophagy can be triggered by several different stimuli and there are likely pathways that have yet to be identified. Mitochondrial fragmentation often takes place before the organelle is degraded; thus, proteins controlling mitochondrial fission appear integral to the process and often interact with specific mitophagy components (Xian & Liou 2021). The descriptions are shown below (Fig. 2). The mitophagy pathway described originally involves PINK1 and PARKIN proteins (pathway 1) (reviewed in Pickles et al. 2018). Once a mitochondrion loses its mitochondrial membrane potential (Δψm), PINK1 (PTEN-induced kinase 1) is stabilized at the outer membrane, then phosphorylates the ubiquitins on various proteins. This signal recruits the cytoplasmic protein PARKIN (an E3 Ubiquitin ligase) to ubiquitinylate additional sites on proteins on the outer membrane. Subsequently, receptors are then recruited to the organelle and serve as a cue for degradation. The lipidated adaptor protein LC3 located on isolation membranes (also called phagophores) (green) binds to the receptors. This membrane grows and envelopes the damaged organelle, ultimately fusing with the lysosome for degradation. A second related pathway involves FUNDC1/NIX/BCL2 (pathway 2) (reviewed in Chen et al. 2020a). This pathway is triggered by loss of Δψm, as well as hypoxia (NIX/BLC2). NIX has been best characterized for mitochondrial clearance during red blood cell maturation (Sandoval et al. 2008). One difference is that FUNDC1, NIX, and BCL2 are outer membrane receptors that directly bind to LC3 and recruit an isolation membrane, thereby bypassing any need for ubiquitinylation (reviewed in Zachari & Ktistakis 2020). An additional pathway was recently identified that is ubiquitin-dependent but acts independently of PINK1/Parkin (pathway 3) (reviewed in Zachari & Ktistakis 2020). Exposing cells to the lactone ivermectin causes mitochondrial damage and fragmentation (Zachari et al. 2019). Once this happens, the E3 Ubiquitin ligases TIAF2, CIAP1, and CIAP2, conjugate ubiquitin on the mitochondrial membrane and ultimately recruit receptors (Optineurin, Atg13). The isolation membrane that subsequently binds to these receptors appears to originate from the endoplasmic reticulum (ER) (blue), unlike for pathways 1 and 2 (Zachari et al. 2019). An alternative non-canonical pathway that has been identified involves RAB9 positive vesicles or membranes (pathway 4) (reviewed in Zachari & Ktistakis 2020). This pathway does not appear to require LC3 lipidation, nor is it known whether ubiquitinylation or receptors are involved (reviewed in Zachari & Ktistakis 2020). In this pathway, phosphorylation of RAB9 stimulates mitochondrial fission (Saito et al. 2019). Subsequently, isolation membranes derived from the trans-Golgi network (yellow) engulf the damaged mitochondrion (Saito et al. 2019). In addition to specific mitophagy pathways, bulk autophagy also degrades mitochondria (pathway 5) (reviewed in Lahiri et al. 2019). Bulk autophagy and its effect on cell physiology and disease is a well-studied area. In response to stimuli such as starvation, an isolation membrane forms (green) and surrounds a part of the cytoplasm, thus scooping up multiple cellular components. Once membrane formation is complete, the autophagosome fuses with the lysosome in order to degrade and recycle the components for cellular use (Lahiri et al. 2019). A final pathway that does not involve whole organelle destruction involves mitochondrial-derived vesicles (MDVs) (pathway 6) (reviewed in Sugiura et al. 2014). This pathway appears to happen constitutively in the cell and may be important for normal mitochondrial homeostasis. Small MDVs containing oxidized mitochondrial proteins bud off individual mitochondria then fuse with endolysosomes for degradation in the lysosome (Soubannier et al. 2012a,b). This mechanism relies on PINK1 and PARKIN (McLelland et al. 2014).
Species comparison of mitochondrial changes during oocyte development
In human, mouse, and Drosophila, female germ cells undergo incomplete mitoses to form interconnected cysts which have been shown to have the capacity to share cellular contents, including mitochondria (de Cuevas et al. 1997, Pepling & Spradling 1998, Pepling et al. 1999). In human and mouse, cyst formation takes place during fetal development, then subsequently break down to form a pool of single primordial follicles with many undergoing apoptosis around birth. In Drosophila, cyst formation and breakdown occur continuously in adults, thus all stages can be viewed simultaneously. Mouse and human oocyte development have comparable developmental stages (Fig. 1). Mice reach puberty at around 4–5 weeks of age, whereas in humans, puberty occurs at approximately 12 years of age. After puberty, preantral follicles have the endocrine support required to enable growth and maturation into an ovulatory follicle; although many undergo apoptosis during the process of follicle development. As the follicle completes development, the oocyte resumes meiosis and undergoes two successive divisions that give rise to polar bodies. Follicle development and ovulation occur throughout a woman’s reproductive years terminating at menopause which occurs around 50 years of age on average.
Germ cell mitochondria in mammals and Drosophila
Mitochondrial shape, numbers, activity, localization, and ultrastructure have been examined by transmission electron microscopy (TEM) and fluorescent microscopy in human, mouse, and Drosophila germ cells and oocytes. In Drosophila, mitochondria were originally described during oogenesis using TEM (reviewed in Mulligan 2003). Immunofluorescence offered a more comprehensive examination of numbers, shape, and location.
During cyst formation in the Drosophila germarium (Fig. 1), mitochondria remain fragmented as the germ cells undergo rapid mitoses to form the 16-cell cyst (Cox & Spradling 2003). Once cyst formation is complete, the mitochondria associate with a germline-specific ER-derived organelle called the fusome. As the oocyte is specified from one of the 16 germ cells in the cyst, a subset of mitochondria from the 15 connected nurse cells moves into the oocyte using microtubules and their associated motors to form the Balbiani body, an organelle-rich body found in most oocytes (Cox & Spradling 2006). As the oocyte continues to mature, the connected sister germ cells stop transferring mitochondria, in while the oocyte’s own mitochondria undergo biogenesis to replicate. Late in oogenesis, the nurse transport their cytoplasmic contents, including mitochondria, into the oocyte and subsequently undergo apoptosis (de Cuevas et al. 1997).
In mouse early embryos, TEM and northern dot blot analysis of mitochondrial rRNA and mRNA estimated that there is approximately one copy of mitochondrial DNA (mtDNA) per mitochondrion (Piko & Matsumoto 1976) (Piko & Taylor 1987). Increased sophistication of imaging techniques makes it possible to use fluorescence microscopy to get a full picture of entire oocytes. However, given the large size of the growing oocyte (~80 µm mice, ~100 µm human) and the heterogeneous mitochondrial distribution, acquiring accurate mitochondrial numbers can be challenging. In mouse and humans, the number of mitochondria reaches a plateau during oocyte development and the organelles do not resume biogenesis until after embryo implantation. Low level mtDNA turnover has however been documented in mice (McConnell & Petrie 2004), and increased copy number and transcription in cows (May-Panloup et al. 2005). There are approximately 100–200 mitochondria in PGCs in mouse and human. These mitochondria are spherical and have reduced cristae. During oocyte development, this number increases and reaches over 100,000 in the mature oocyte (Cao et al. 2007, Wai et al. 2008). In addition to a large increase in numbers, there are also general changes to mitochondrial localization and activity. In mouse and human PGCs and primary follicles, mitochondria associate with the ER and the Balbiani body (Motta et al. 2000, Pepling et al. 2007). In the developing oocyte, mitochondria are mostly homogeneously localized throughout the cytoplasm, however, the mitochondria located at the plasma membrane have higher membrane potential and thus may be more active (Van Blerkom 2009). In oocytes from women and mice of advanced maternal age, this membrane potential is greatly decreased and thus indicative of lower activity (Pasquariello et al. 2019).
The effect of oxidative stress on oocytes
As mitochondria provide important metabolites and energy, and contain their own error-prone DNA, understanding the cell biological mechanisms governing mitochondrial location and function in germ cells has been recognized as critical to understanding oocyte fitness and almost certainly affects oocyte quality during maternal aging. Pyruvate oxidation is essential for oocyte development in mouse (Johnson et al. 2007). In contrast, ovulated oocyte and preimplantation embryo metabolism is based on low levels of oxidation of pyruvate, lactate, and specific amino acids, followed by a dramatic increase in oxidative phosphorylation at blastocyst formation (Gardner & Wale 2013). Likewise, in Drosophila, during the majority of oogenesis, germ cells rely on oxidative phosphorylation (Sieber et al. 2016). The first half of embryogenesis is also heavily reliant on oxidative phosphorylation, after which a switch to glycolysis occurs in preparation for the exponential growth experienced during larval growth (Tennessen et al. 2011).
Oocytes in mouse and human primarily use oxidative phosphorylation for their energy demands, with oxygen consumption low in the zygote and first few cell divisions and increasing several folds at the blastocyst stage (Leese 2012). This burst requires a robustly functioning electron transport chain (Dumollard et al. 2007a,b, Dalton et al. 2014). However, electron transport is the main source of mitochondrial oxidative damage due to the inevitable production of reactive oxygen species (ROS). Oxidative stress is associated with maternal aging and supplementing aging mice with the antioxidant CoenzymeQ can preserve ovarian reserve (Ben-Meir et al. 2015). Since the resumption of meiosis is an energy-intensive process, it is perhaps not surprising that impaired mitochondrial function can lead to chromosomal aberrations. Aneuploidy greatly increases with age with up to 60% of oocytes exhibiting aneuploidy after age 40 (Angell 1994, Pellestor et al. 2003). Oocytes retrieved from women with advanced maternal age have decreased mitochondrial function associated with increased oxidative stress and spindle abnormalities (Pasquariello et al. 2019). In addition, there is evidence that treating mouse and human oocytes with compounds that ameliorate ROS and their subsequent damage can improve chromosome segregation (Pasquariello et al. 2019, Al-Zubaidi et al. 2021). Reduced numbers of mitochondria (Reynier et al. 2001) and mtDNA (Wai et al. 2010) may also play a role.
In Drosophila, females mutant for Superoxide Dismutase 2 (SOD2), the ROS scavenger in mitochondria, only live for <24 h, have greatly reduced ATP levels and increased oxidative damage (Sen et al. 2013). While this is insufficient time for egg laying to start, mitochondria in the developing follicles are abnormally localized and clumped, indicative of stress (Sheard et al. 2020). Together, these observations give strong support that maintaining undamaged and fully competent mitochondria during oocyte development is critical for healthy oocytes.
Germ cell mitochondrial changes during aging
The mechanisms underlying ageing have been discussed for decades. Advances in molecular and cell biology of the last three decades have identified multiple potential mechanisms that contribute to aging. It has become clear that mitochondrial function, which plays a major role in many cellular pathways including ATP production, nuclear gene expression and epigenetic alterations, declines with age. In Drosophila, mitochondrial fission increases with age in GSCs, and if fission is genetically increased, GSCs are lost more frequently from the stem cell niche (Amartuvshin et al. 2020). Conversely, they found increasing fusion makes GSCs more competitive for remaining in the niche. How mitochondrial dynamics changes with age has not been examined in more developed follicles in Drosophila.
In humans, declining oocyte potential is strongly associated with oocyte karyotypic instability in older women. Both chaotic mosaicism and morphologically abnormal meiotic apparatus are significantly associated with low mitochondrial membrane potential in preimplantation embryos (Wilding et al. 2003). Oocytes retrieved from women of increased maternal age who were being treated for idiopathic infertility showed structural and functional mitochondrial deficits (Muller-Hocker et al. 1996, Chan et al. 2005, Murakoshi et al. 2013). Mitophagy is one of the mechanisms for maintaining mitochondrial quality that appears to decline in aging. While the quality and the quantity of mtDNA are directly implicated in oocyte function, their importance in aging and the role of mitophagy remain unclear.
Mitophagy as a potential driver of oocyte quality
Mitophagy mechanisms
Superfluous or damaged mitochondria are recycled within the cell by either bulk-autophagy, or the more selective mitochondria specific process, mitophagy (Fig. 2 and Box 1, reviewed in Pickles et al. 2018, Zachari & Ktistakis 2020, Doblado et al. 2021). However, the developmental in vivo contexts for mitophagy are poorly understood. While the molecular details of some pathways, such as the best characterized PINK1/Parkin pathway (Fig. 2, pathway 1), are well understood (Narendra et al. 2010), others are still poorly defined. PINK1 and PARK2 (Parkin) are mutated in inherited forms of early onset Parkinson’s disease. This pathway causes ubiquitination of mitochondrial proteins, triggering formation of a specialized membrane called a phagophore (Fig. 2, green), which engulfs the mitochondrion forming an autophagosome (Narendra et al. 2010). In contrast, pathways mediated by receptors on the mitochondrial outer membrane, such as NIX, which functions specifically during red blood cell development (Fig. 2, pathway 2) trigger mitophagy independently of ubiquitination (Sandoval et al. 2008). These receptors bind to the adaptor protein LC3 that characterizes autophagosomes that then engulf their target. While the receptors and source of the phagophore membrane differ, the final downstream steps to degrade the organelles are often shared (Fig. 2). Two other pathways have been identified that are PINK1/PARK independent (Fig. 2, pathways 2 and 4). Once the autophagosome forms (pathways 1–5), it fuses with a lysosome and its contents are degraded at low pH. For cell biological studies, a key mitophagy readout is co-localization of mitochondria with autophagosomes or lysosomes. Finally, a unique quality control pathway that uses mitochondrial derived vesicles can recycle small portions of the mitochondrion for direct degradation by the lysosome (Fig. 2, pathway 6) (Sugiura et al. 2014). Please see other literature for more detail (Pickles et al. 2018, Zachari & Ktistakis 2020, Doblado et al. 2021) and for RAB9-dependent alternative/non-canonical mitophagy (Arakawa et al. 2017) and Table 1 (adapted from other publications (Dickinson et al. 2016, Zachari & Ktistakis 2020)) for essential genes.
Figure 2.

Mitochondrial quality control pathways. Mechanisms governing mitochondrial turnover can be subdivided into six major pathways (see Box 1 for further detail). Representative cellular cues that stimulate each pathway are at the top and examples of the process and the proteins required for each pathway are listed at the bottom. Pathways 1–5 ultimately result in the mitochondrion engulfed in an autophagosome membrane (green) that will fuse with the lysosome, destroying the organelle. The source of this membrane differs depending on the pathway (pathways 3 and 4) and in some instances has not yet been identified (pathways 1 and 2). Mitochondria are also turned over by non-selective autophagy (pathway 5). Mitochondrial derived vesicles turn over small portions of the mitochondrion (Sugiura et al. 2014) by transporting them to the lysosome, arguably distinct from mitophagy because autophagosomes are not involved (pathway 6). RAB9-dependent alternative/non-canonical mitophagy involves autophagosomes but not ATG5 or 7 (Arakawa et al. 2017). Figure created with BioRender.
Table 1.
Known female germ cell phenotypes for autophagy/mitophagy genes.
| Process/proteina |
Species |
Mouse fertilityb
|
Fly fertility |
Mouse viabilityb
|
|
|---|---|---|---|---|---|
| Turning on the pathway | |||||
| mTORC1 | Many | H/M/D | Reduced fertility1 | Tor RNAi-sterile | |
| TFEB | Transcription factor | H/M/D | ND | ND | Lethal2 |
| RHEB | Small GTP-binding proteins, Ras superfamily | H/M/D | Fertile3 | Female sterile (viable mutation)4 | Embryonic lethal5 |
| Initiation of autophagosome biogenesis | |||||
| ULK complex | |||||
| ULK1, 2 | Ser/Thr kinase catalytic subunit of ULK | H/M/D | ND | (Atg1) Female sterile (RNAi)6, 7 | DKO for ULK1/2 Embryonic lethal8 |
| ATG13 | Regulatory subunit of ULK | H/M/D | ND | Fertile | lethal9 |
| ATG101 | Subunit of ULK | H/M/D | ND | ND | ND |
| FIP200 | Subunit of ULK | H/M/D | ND | (Atg17) ND | Embryonic lethal10 |
| AMPK (α, β, γ) | Ser/Thr kinase | H/M/D | (α1) Decreased litter size, abnormal mitochondrial physiology11 | Female sterile (RNAi)12 | Viable (α1, 2)13 Viable (γ)13 |
| ATG9 | Transmembrane protein | H/M/D | ND | Sterile (null mutant)14 | Lethal neonatal15 |
| VPS34-I | |||||
| VPS34 | Class III PI-3 kinase subunit of VPS34-1 | H/M/D | ND | ND | Embryonic lethal16 |
| VPS15 | Subunit of VPS34-1 | H/M/D | ND | Semi-sterile (RNAi)12 | Embryonic lethal17 |
| BECN1 | Regulatory subunit of VPS34-1 | H/M/D | Infertile18 | (Atg6) ND | Embryonic lethal19 |
| ATG14L | Subunit of VPS34-1 | H/M/D | ND | ND | ND |
| AMBRA | Regulator of BECN1 | H/M | ND | – | Lethal20 |
| MAPKAP2,3 | Ser/Thr kinases | H/M/D | Fertile21 | Semi-sterile (RNAi)12 | Viable21 |
| DFCP1 | PI(3)P-binding protein | H/M | ND | – | Viable22 |
| WIPI1,2 | PI(3)P-binding protein | H/M/D | ND | ND | ND |
| Building the autophagosome | |||||
| Lipidation complex | |||||
| ATG12 | Part of ubiquitin ligation-like (E3) complex | H/M/D | ND | ND | Lethal neonatal23 |
| ATG3 | Part of ubiquitin ligation-like (E3) complex | H/M/D | ND | ND | Lethal neonatal24 |
| ATG5 | Part of ubiquitin ligation-like (E3) complex | H/M/D | Oocyte development normal, early embryonic lethal25 | Fertile | Lethal neonatal26 |
| ATG7 | Part of ubiquitin ligation-like (E3) complex | H/M/D | Subfertile27 | ND | Lethal neonatal28 |
| ATG16L | Part of ubiquitin ligation-like (E3) complex | H/M/D | ND | Fertile | Lethal neonatal29 |
| LC3A, B, C | Ubiquitin-like proteins | H/M/D | LC3B Fertile (JAX) | (Atg8a, Atg8b) ND | LC3B viable30 |
| GABARAP, L1, L2 | Ubiquitin-like proteins | H/M/D | ND | (Atg8a, Atg8b) ND | GABARAP viable31 L1 viable32
L2 lethal33 |
| ATG2b | Phospholipid binding/transfer protein | H/M/D | ND | ND | Viable (IMPC) |
| VMP1 | ER-resident protein | H/M/D | ND | ND | Lethal34 |
| Fusing autophagosome with lysosome | |||||
| STX17 | SNARE protein | H/M/D | ND | (Syx17) fertile | ND |
| RAB7 | Small GTP-binding proteins, Ras superfamily | H/M/D | ND | ND | s |
| EPG5 | RAB7 effector protein | H/M/D | ND | ND | Viable, reduced survival 36 |
| HOPS (VPS11, VPS16, VPS18, VPS33A) | Tethering complex | H/M/D | ND | (Car) ND (Cm) ND |
VPS33A, 16, viable, impaired motor function37 |
| PLEKHM1 | HOPS-interacting protein | H/M/D | Fertile 38 | ND | Viable38 |
| Ubiquitin-mediated mitophagy | |||||
| PINK1 | Kinase | H/M/D | Fertile (JAX) | Sterile39 | Viable40 |
| OPTN | Mitophagy receptor | H/M/D | Fertile41 | (Nemo) Semi-sterile 7 | Viable41 |
| PARK2 | E3 ubiquitin ligase | H/M/D | Fertile42 | Semi-sterile 12, 43 | Viable42 |
| P62 (SQSTM1) | Mitophagy receptor | H/M/D | ND | (Ref(2)P) Fertile | Viable44 |
| TAX1BP1 | Mitophagy receptor | H/M | ND | – | Viable45 |
| NDP52 | Mitophagy receptor | H/M | ND | – | Viable (IMPC) |
| NBR1 | Mitophagy receptor | H/M | ND | – | Viable (IMPC) |
| OMM mitophagy receptors | – | ||||
| BNIP3 | Mitophagy receptor | H/M | Fertile46 | – | Viable46 |
| BNIP3L (NIX) | Mitophagy receptor | H/M | ND | – | Viable47 |
| BCL2L13 | Mitophagy receptor | H/M | Reduced fertility 48 | – | Viable48 |
| FUNDC1 | Mitophagy receptor | H/M | Fertile49 | – | Male lethal (IMPC) |
| Lysosomal | |||||
| LAMP1 | H/M/D | Fertile50 | ND | Viable50 | |
aProtein functions listed are those that are related to mitophagy/autophagy. Other important cellular functions may have been ascribed to individual proteins. bFertility and viability were assessed from literature, Jackson Laboratory (JAX) breeding information, Mouse Genome Informatics (MGI), and the International Mouse Phenotyping Consortium (IMPC). Fertility information may indicate that homozygotes can breed or produce offspring however this does not necessarily mean oocyte development in normal. In some cases, while a strain is viable it may have abnormalities and it is not clear if it is fertile (ND). Viability was assessed for available information on whole body knockout. Phenotypic description of whole body knockouts may not be included in the original study generating the knockout strain. DKO, double knockout; H/M/D, human/mouse/Drosophila; ND, no data; OMM, outer mitochondrial membrane. 1Guo and Yu (2019); 2Steingrimsson et al. (1998); 3Baker et al. (2014); 4Stocker et al. (2003); 5Goorden et al. (2011); 6Lieber et al. (2019); 7Kuhn et al. (2015); 8Cheong et al. (2014); 9Kaizuka and Mizushima (2016); 10Gan et al. (2006); 11Bertoldo et al. (2015); 12Sopko et al. (2014); 13Jorgensen et al. (2004); 14Wen et al. (2017); 15Saitoh et al. (2009); 16Zhou et al. (2011); 17Nemazanyy et al. (2013); 18Gawriluk et al. (2011); 19Yue et al. (2003); 20Fimia et al. (2007); 21Ronkina et al. (2007); 22Zhong et al. (2020); 23Malhotra et al. (2015); 24Sou et al. (2008); 25Tsukamoto et al. (2008b); 26Kuma et al. (2004); 27Song et al. (2015); 28Komatsu et al. (2005); 29Saitoh et al. (2008); 30Cann et al. (2008); 31O’Sullivan et al. (2005); 32Sasai et al. (2017); 33Skarnes et al. (2011); 34Morishita et al. (2019); 35Kawamura et al. (2012); 36Zhao et al. (2013); 37Zhen and Li (2015); 38Fujiwara et al. (2016); 39Politi et al. (2014); 40Kitada et al. (2007); 41Slowicka et al. (2016); 42Itier et al. (2003); 43Cox and Spradling (2009); 44Wada et al. (2006); 45Iha et al. (2008); 46Diwan et al. (2007); 47Yuan et al. (2017); 48D’Alonzo and Hong (2017); 49Zhang et al. (2016); 50Andrejewski et al. (1999).
The effect of mitophagy gene loss on oocyte development and fertility
Mitophagy has the potential to remove mtDNA mutants during oogenesis and oocyte development due to its specificity removing damaged mitochondria. While there are several pathways known to regulate mitophagy, which ones play a role during oogenesis and oocyte development has not been fully investigated.
The Drosophila genome has excellent homology to the human genome, with over 75% of known human diseases having a homolog in flies (Bier 2005). Thus, mutants or RNAi knockdown have been analyzed for many of the known genes involved in mitophagy and autophagy (Table 1). If mutations in a gene are non-viable, it is straight-forward to use germ cell specific RNAi to identify fertility defects. Of those genes that have been examined, many cause female sterility or semi-sterility when lost in the germline (Table 1). However, most of the genes involved in autophagy and mitophagy have not been examined for their effect on mitochondrial quality and turnover in Drosophila female germ cells. The PINK1/Parkin pathway is required for fertility in Drosophila and mutants in either gene have clumped, mislocalized germ cell mitochondria that are hyperfused. However, Parkin and PINK1 do not appear to alter mtDNA inheritance (see below).
Autophagy is essential for normal preimplantation development in mice and components of the autophagy pathway have been knocked out in mouse models. In general, the resulting phenotypes indicate mice lacking the proteins required for the pre-conjugation system die very early in embryogenesis and whereas the mice lacking the proteins required for conjugation die either at birth or surviving to adulthood (reviewed in Kuma et al. 2017). A germline-specific knockout of Atg5 is fertile but the resulting embryos do not develop past the embryonic four to eight cell stages (Tsukamoto et al. 2008b). Using LC3 to identify autophagosomes, Meng et al. showed that autophagy is a key process in preantral follicular atresia, while antral follicles degenerate mostly through apoptosis (Meng et al. 2018).
Global knockout of several key autophagy and mitochondrial dynamics genes, including Beclin1, Atg7, Opa1, Mfn1, and Mfn2 are embryonic lethal. BECLIN1 is an essential autophagy protein involved in autophagosome formation. Mice that are homozygous for Beclin1 knockout die as embryos (Qu et al. 2007) and heterozygotes have a shortened lifespan and increased risk of mouse mammary cancer (Vega-Rubin-de-Celis 2019). When targeted to mouse ovary, deficiency of Beclin1 results in 56% fewer primordial follicles at postnatal day 1 (Gawriluk 2014). Constitutively, active Beclin1 demonstrates the central importance of mitophagy, because by increasing autophagy it can rescue a model of mouse aging that has impaired fertility and mitochondrial function (Murata et al. 2009, Fernandez et al. 2018). Oocyte-specific deletion of Mfn1, but not Mfn2, results in a complete loss of oocyte growth and ovulation due to a block in folliculogenesis at the preantral-to-antral follicle transition (Carvalho et al. 2020). In addition, Mfn1 knockdown mice exhibit abnormal mitochondrial clustering and enlarged mitochondria with disrupted cristae. Together, this work demonstrates that the balanced expression of modulators of mitochondrial dynamics is critical for proper oocyte development.
With aging, Pink1 mutant mice have increased mitochondrial neuronal deficits (Gispert et al. 2009). However, it is well established that mice mutant for Pink1 and Parkin do not normally display characteristic Parkinson’s disease neuronal loss, nor are they infertile (Goldberg et al. 2003, Perez & Palmiter 2005, Kitada et al. 2007). Recent literature has demonstrated Parkinson’s disease phenotypes, including loss of dopaminergic neurons, after stress induced by exhaustive exercise or intestinal infection (Sliter et al. 2018, Matheoud et al. 2019). Neither fertility nor oocyte development were examined under these conditions and thus need to be explored.
Control of mtDNA mutation load during oogenesis and oocyte aging: implications for mitochondrial disease
Importance of the developmental genetic bottleneck in mtDNA transmission
Individual animals contain overwhelmingly WT mtDNA in their mitochondria, which is called homoplasmy. WT mtDNA mixed with mtDNA containing mutations, such as in a diseased state, or single nucleotide polymorphisms, such as in an experimental state, is called heteroplasmy. In addition to maternal inheritance, mitochondrial inheritance is driven by a developmental genetic bottleneck (Fig. 3) (Olivo et al. 1983). The general principle behind the genetic bottleneck is that heteroplasmic mothers produce offspring with ratios of WT to mutant mtDNA that are not the same as the mother. This rapid transgenerational segregation suggests that the number of segregating units is smaller than the 100,000 or so mtDNAs in the mammalian oocyte. Using allele-specific PCR, shifts in heteroplasmy have been used as a read out for pinpointing the developmental time points for the bottleneck (Johnston et al. 2015). In mouse and fly, this time point occurs very early in oogenesis and coincides in mouse with, but is not identical with, a reduction in mtDNA germ cell content (Cao et al. 2007, Wai et al. 2008) (Fig. 3). For mathematical modeling and cross-generational analysis, advances in mtDNA deep sequencing have been very useful to study the bottleneck (Arbeithuber et al. 2020). These types of analyses have been done for humans as well (Boucret et al. 2017).
Figure 3.
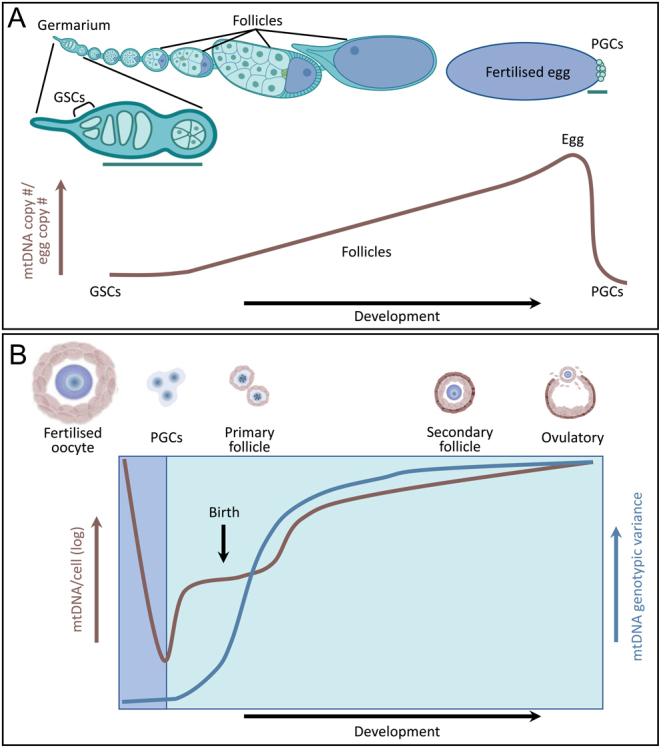
The mitochondrial bottleneck. (A) Mitochondrial DNA (mtDNA) copy number throughout Drosophila oogenesis. Cartoon of the ovariole at the top which contains developing follicles including the oocyte (blue) with a newly developing fertilized egg on the right. Germline stem cells (GSCs) are at the anterior of a specialized structure called the germarium. GSCs are present throughout the adult lifespan and continuously give rise to the germline. Primordial germ cells (PGCs) are the first cells formed at the posterior of the newly fertilized and deposited egg. The graph represents mtDNA copy number relative to the egg measured using quantitative mtDNA fluorescence in situ hybridization (FISH) and verified with qPCR for accessible stages (eggs, PGCs) (Hurd et al. 2016). mtDNA copy number greatly increases during follicle development then decreases when PGCs form. The green lines represent developmental time points that have been implicated in the genetic bottleneck due to decreased mtDNA copy number, mitochondrial dynamics, and mtDNA replication. (B) mtDNA copy number and genotypic variance throughout mammalian follicle development. Germ cell stages are indicated at the top. The profound drop in mtDNA copy number in PGCs followed by ~500-fold increase in copy number to mature oocyte enables clonal proliferation of mtDNA as well as passive selection of the best oocyte at the cellular level, the so-called 'ovarian bottleneck' (Wolf et al. 2017). Following fertilization, the oocyte divides and forms the inner cell mass of the blastocyst with little mtDNA replication (McConnell & Petrie 2004), where ~3 cells will develop into the embryo. In this 'postfertilization bottleneck' there may be active selection at the mtDNA level as well as passive compartmentalization. Evidence to date suggests that the major component of the variance in germline development arises prenatally during oogenesis (Li et al. 2016) and postnatally during folliculogenesis (Johnston et al. 2015).
In Drosophila, allelic-specific PCR and mtDNA Single Molecular Fluorescent In Situ Hybridization showed a steady increase in mtDNA copy number during oogenesis from GSCs to egg formation with a drastic decrease once the PGCs are formed at the beginning of embryogenesis (Fig. 3B) (Hurd et al. 2016). Examining mtDNA inheritance, studies identifying and analyzing mtDNA size variants indicated the larger size was preferentially transmitted as the heteroplasmic mothers aged (Solignac et al. 1984, 1987, Kann et al. 1998). A breakthrough occurred when researchers were able to create deleterious mtDNA mutations using mitochondria-targeted endonucleases (Srivastava & Moraes 2001, Xu et al. 2008). Using a temperature-sensitive mtDNA mutation and sequencing, Ma et al. found evidence for a genetic bottleneck early in oogenesis (Ma et al. 2014) (Fig. 3A). Using a different temperature-sensitive mtDNA mutation and imaging experiments to identify replicating mtDNA, Hill et al. found evidence supporting preferential replication of WT over mutant mtDNA could be responsible for the bottleneck (Hill et al. 2014). Recently, disruptions to fission and fusion have been shown to influence the bottleneck and inheritance, suggesting reducing organelle size and mtDNA content could be a mechanism supporting mtDNA inheritance and the genetic bottleneck (Lieber et al. 2019, Chen et al. 2020b). Interestingly, knocking down atg1 and BNIP3 disrupted mtDNA selection, whereas parkin knockdown had no effect (Ma et al. 2014, Lieber et al. 2019). How maternal aging affects the bottleneck or mtDNA transmission in these models has not yet been investigated.
We and others demonstrated purifying selection against detrimental mtDNA mutants in mouse (Marchington et al. 1999), a process that may have evolved to maintain germline homoplasmy. However, purifying selection fails in human families with maternally inherited heteroplasmic mtDNA diseases and 'selfish' transmission of detrimental mtDNA is well established in flies (Klucnika & Ma 2019). The risks of transmitting disease to future children are poorly understood so reproductive decisions of potential parents are fraught with uncertainty. This is largely because of the mtDNA bottleneck. Indeed, high profile mtDNA replacement therapy has been developed in the UK as a reproductive option that aims to reduce this uncertainty (Poulton et al. 2019). While the time course of the mtDNA bottleneck is now emerging, the mechanisms are not clear, however, mitophagy is one of the processes that could contribute (Diot et al. 2016a). During transmission of mtDNA from mother to child, significant fluctuations in heteroplasmy are already apparent in mature oocytes (Marchington et al. 1997, Marchington et al. 1998) and this highlights the importance of understanding mitophagy in oocyte development.
Studies of artificially generated heteroplasmic mice suggest that mtDNA segregation occurs during oocyte development (Jenuth et al. 1996) and in preimplantation embryos between late morula and early gastrula (Latorre-Pellicer et al. 2019). Interactions with nuclear genes and metabolic factors can drive segregation, suggesting that mitochondrial fitness plays a role in the bottlenecking process (Lechuga-Vieco et al. 2020). Massive expansion in mtDNA content per cell occurs during development of PGCs to mature oocytes and provides an opportunity for mtDNA subpopulations to proliferate (Fig. 3B). Some authors have suggested that the segregation of pathogenic mtDNA mutations is non-random (Otten et al. 2018). Furthermore, the huge excess of oocytes in the ovary at birth compared with the numbers that are ovulated is a second opportunity for selection against poor mitochondrial subpopulations. However, the cellular mechanisms of purifying selection have barely been elucidated.
MtDNA as a measure of mitochondrial quality in aging oocytes
A threshold level for mtDNA content has been associated with successful fertilization in mice (Wai et al. 2010) pigs (El Shourbagy et al. 2006) and cows (Lamas-Toranzo et al. 2018). However, whether a reduction in mtDNA content underlies the reduced success in oocytes from aged females remains unclear (Reynier et al. 2001, Chan et al. 2005, Murakoshi et al. 2013, Cree et al. 2015). Measurement of mtDNA copy number in single blastomeres from cleavage stage human embryos suggested that increased mtDNA copy number was associated with poor outcome and with oocytes from older mothers (Fragouli et al. 2013). However, this is controversial because data from other investigators does not support it (Murakoshi et al. 2013, Kim & Seli 2019). If so, the increased mtDNA copy number could be a compensation for a decline in mitochondrial function driven by aging.
Thousands of identical copies of mtDNA are present in most types of cells in normal individuals (homoplasmy), but in heteroplasmic mtDNA disease, normal and mutant mtDNA co-exist in the same cells. These findings soon suggested an mtDNA-based theory of aging in which an increase in heteroplasmic mtDNA point mutations and deletions (Cortopassi & Arnheim 1990) may underlie the decline in energy in aging individuals. The quality of mtDNA was directly implicated in a study in which multiple mtDNA mutations were generated in mice with deficient in the enzyme POLG (necessary for mtDNA synthesis and accurate replication) (Trifunovic et al. 2004). When maternally transmitted in mice, with a WT nuclear genome, such mtDNA mutations can induce mild aging phenotypes including impaired fertility (both litter size and number of litters) and shortened lifespan (Ross et al. 2013). Furthermore, accumulation of maternally transmitted mtDNA mutations down generations exacerbates this reduced fertility. Because oocytes contain a significant load of mutant mtDNA relative to controls in this model, mtDNA was explored in oocytes from aging women. However, the links between aging and oocyte mtDNA quality are by no means clear. Early investigators reported that they could detect mtDNA deletions in oocytes from older women using PCR (Chan et al. 2005), and findings were similar in cows (Hammond et al. 2016). However, these methods do not readily distinguish between deleted mtDNAs and other types of rearrangement such as mtDNA duplications. Once next-generation sequencing became available (Arbeithuber et al. 2020), germline heteroplasmy could be tested in more depth, but it is still not yet clear whether heteroplasmic point mutations increase in oocytes from aging women (Boucret et al. 2017) or cows (Hammond et al. 2016). Again, PCR was used to investigate arrested human embryos, and this suggested that mtDNA deletions were common. However, healthy embryos were not sampled so it remains unresolved as to whether mtDNA deletions are associated with poor outcome.
Other groups have investigated mtDNA heteroplasmy in normal human oocytes at different stages. Human PGCs can only be sampled from ovaries prenatally, so the data are scanty. At this stage, the mtDNA content may be as low as 200–2000 mtDNAs per cell and five mtDNAs per mitochondrion (Floros et al. 2018). The data are consistent with selection against non-synonymous mtDNA mutations prior to clonal expansion during oogenesis. In the later stages of oocyte development, cytoplasm adherent to the two polar bodies mitochondria can be sampled sequentially (De Fanti et al. 2017). While their data would be consistent with purifying selection between extrusion of the two polar bodies, the polar cytoplasm may be enriched in 'cellular garbage' and hence reflect the whole oocyte or zygote rather poorly.
In summary, while the mtDNA bottleneck appears to have evolved to improve mitochondrial quality in the offspring, it fails to eliminate mtDNA disease in humans and selfish mtDNA in flies. However, the physiological basis of the bottleneck is not clear and mitophagy could be important. There is little solid data demonstrating a decline in mitochondrial quality in oocytes from aging women and almost no published data on mitophagy during oocyte development.
Unanswered questions and future directions
In conclusion, there are many gaps in our knowledge of how mitochondrial quality is maintained during oocyte development. While mitophagy is a cellular process that may well be involved, there is little if any hard data demonstrating its effects in oogenesis, oocyte development, and preimplantation development. Autophagy is clearly an essential process in early embryos in which transcription initiation must be accompanied by turnover of maternally inherited RNAs (Tsukamoto et al. 2008a) and autophagy-mediated apoptosis protects against aneuploidy (Singla et al. 2020). However, a role for mitophagy can only be inferred (Diot et al. 2016a). A group studying developmental programming were unable to detect mitophagy in oocytes, but their study was not exhaustive (Boudoures et al. 2017). Given that autophagy is required for maintaining healthy stem cells (Phadwal et al. 2013) and declines with aging, it is likely that the same is true of mitophagy. If mitophagy is required to maintain oocyte quality, it may also impact on our understanding of inherited mtDNA diseases and have implications for potential interventions such as mitochondrial replacement therapy. However, any connections between mitophagy and the decline in oocyte developmental potential in aging women remain conjectural.
Finally, the use of ARTs and cryopreservation for fertility preservation are being used ever more frequently, however, knowledge regarding the effect of these different modalities and processes on mitophagy is severely lacking. To address these gaps in our knowledge, in addition to studies using human reproductive tissues, which can be limited in supply, we should fully exploit valuable model organisms such as Drosophila and mouse. By adopting a multi-faceted strategy and using all available tools, we will move toward understanding the basic cell and molecular mechanisms governing mitophagy in aging germ cells and elucidate how ARTs influence mitophagy.
Declaration of interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results. Suzannah Williams is a Lay Editor of Reproduction and Fertility. Suzannah Williams was not involved in the review or editorial process for this paper, on which she is listed as an author.
Funding
Salary support for J P was from the NHS Highly Specialised services ‘Rare Mitochondrial Disorders of Adults and Children Diagnostic Service’. The views expressed are those of the authors and not necessarily those of the NHS.
Author contribution statement
Conceptualization and writing were carried out by R T C, J P, and S A W. All authors have read and agreed to the published version of the manuscript.
Acknowledgements
The authors would like to thank Dr Eszter Dombi and Dr Aditya Sen for critically reading the manuscript.
References
- Al-Zubaidi U, Adhikari D, Cinar O, Zhang QH, Yuen WS, Murphy MP, Rombauts L, Robker RL, Carroll J.2021Mitochondria-targeted therapeutics, MitoQ and BGP-15, reverse aging-associated meiotic spindle defects in mouse and human oocytes. Human Reproduction 36771–784. ( 10.1093/humrep/deaa300) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amartuvshin O, Lin CH, Hsu SC, Kao SH, Chen A, Tang WC, Chou HL, Chang DL, Hsu YY, Hsiao BSet al. 2020Aging shifts mitochondrial dynamics toward fission to promote germline stem cell loss. Aging Cell 19 e13191. ( 10.1111/acel.13191) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrejewski N, Punnonen EL, Guhde G, Tanaka Y, Lullmann-Rauch R, Hartmann D, von Figura K, Saftig P.1999Normal lysosomal morphology and function in LAMP-1-deficient mice. Journal of Biological Chemistry 27412692–12701. ( 10.1074/jbc.274.18.12692) [DOI] [PubMed] [Google Scholar]
- Angell RR.1994Aneuploidy in older women. Higher rates of aneuploidy in oocytes from older women. Human Reproduction 91199–1200. ( 10.1093/oxfordjournals.humrep.a138675) [DOI] [PubMed] [Google Scholar]
- Arakawa S, Honda S, Yamaguchi H, Shimizu S.2017Molecular mechanisms and physiological roles of Atg5/Atg7-independent alternative autophagy. Proceedings of the Japan Academy: Series B, Physical and Biological Sciences 93378–385. ( 10.2183/pjab.93.023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbeithuber B, Hester J, Cremona MA, Stoler N, Zaidi A, Higgins B, Anthony K, Chiaromonte F, Diaz FJ, Makova KD.2020Age-related accumulation of de novo mitochondrial mutations in mammalian oocytes and somatic tissues. PLoS Biology 18 e3000745. ( 10.1371/journal.pbio.3000745) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker MD, Ezzati M, Aloisio GM, Tarnawa ED, Cuevas I, Nakada Y, Castrillon DH.2014The small GTPase Rheb is required for spermatogenesis but not oogenesis. Reproduction 147615–625. ( 10.1530/REP-13-0304) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett KR, Schilling C, Greenfeld CR, Tomic D, Flaws JA.2006Ovarian follicle development and transgenic mouse models. Human Reproduction Update 12537–555. ( 10.1093/humupd/dml022) [DOI] [PubMed] [Google Scholar]
- Ben-Meir A, Burstein E, Borrego-Alvarez A, Chong J, Wong E, Yavorska T, Naranian T, Chi M, Wang Y, Bentov Yet al. 2015Coenzyme Q10 restores oocyte mitochondrial function and fertility during reproductive aging. Aging Cell 14887–895. ( 10.1111/acel.12368) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoldo MJ, Guibert E, Faure M, Rame C, Foretz M, Viollet B, Dupont J, Froment P.2015Specific deletion of AMP-activated protein kinase (alpha1AMPK) in murine oocytes alters junctional protein expression and mitochondrial physiology. PLoS ONE 10 e0119680. ( 10.1371/journal.pone.0119680) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bier E.2005Drosophila, the golden bug, emerges as a tool for human genetics. Nature Reviews: Genetics 69–23. ( 10.1038/nrg1503) [DOI] [PubMed] [Google Scholar]
- Boucret L, Bris C, Seegers V, Goudenege D, Desquiret-Dumas V, Domin-Bernhard M, Ferre-L’Hotellier V, Bouet PE, Descamps P, Reynier Pet al. 2017Deep sequencing shows that oocytes are not prone to accumulate mtDNA heteroplasmic mutations during ovarian ageing. Human Reproduction 322101–2109. ( 10.1093/humrep/dex268) [DOI] [PubMed] [Google Scholar]
- Boudoures AL, Saben J, Drury A, Scheaffer S, Modi Z, Zhang W, Moley KH.2017Obesity-exposed oocytes accumulate and transmit damaged mitochondria due to an inability to activate mitophagy. Developmental Biology 426126–138. ( 10.1016/j.ydbio.2017.04.005) [DOI] [PubMed] [Google Scholar]
- Cann GM, Guignabert C, Ying L, Deshpande N, Bekker JM, Wang L, Zhou B, Rabinovitch M.2008Developmental expression of LC3alpha and beta: absence of fibronectin or autophagy phenotype in LC3beta knockout mice. Developmental Dynamics 237187–195. ( 10.1002/dvdy.21392) [DOI] [PubMed] [Google Scholar]
- Cao L, Shitara H, Horii T, Nagao Y, Imai H, Abe K, Hara T, Hayashi J, Yonekawa H.2007The mitochondrial bottleneck occurs without reduction of mtDNA content in female mouse germ cells. Nature Genetics 39386–390. ( 10.1038/ng1970) [DOI] [PubMed] [Google Scholar]
- Carvalho KF, Machado TS, Garcia BM, Zangirolamo AF, Macabelli CH, Sugiyama FHC, Grejo MP, Augusto Neto JD, Tostes K, Ribeiro FKSet al. 2020Mitofusin 1 is required for oocyte growth and communication with follicular somatic cells. FASEB Journal 347644–7660. ( 10.1096/fj.201901761R) [DOI] [PubMed] [Google Scholar]
- Chan CC, Liu VW, Lau EY, Yeung WS, Ng EH, Ho PC.2005Mitochondrial DNA content and 4977 bp deletion in unfertilized oocytes. Molecular Human Reproduction 11843–846. ( 10.1093/molehr/gah243) [DOI] [PubMed] [Google Scholar]
- Chen G, Kroemer G, Kepp O.2020aMitophagy: an emerging role in aging and age-associated diseases. Frontiers in Cell and Developmental Biology 8 200. ( 10.3389/fcell.2020.00200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Wang ZH, Zhang G, Bleck CKE, Chung DJ, Madison GP, Lindberg E, Combs C, Balaban RS, Xu H.2020bMitochondrial DNA segregation and replication restrict the transmission of detrimental mutation. Journal of Cell Biology 219 e201905160. ( 10.1083/jcb.201905160) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong H, Wu J, Gonzales LK, Guttentag SH, Thompson CB, Lindsten T.2014Analysis of a lung defect in autophagy-deficient mouse strains. Autophagy 1045–56. ( 10.4161/auto.26505) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortopassi GA, Arnheim N.1990Detection of a specific mitochondrial DNA deletion in tissues of older humans. Nucleic Acids Research 186927–6933. ( 10.1093/nar/18.23.6927) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox RT, Spradling AC.2003A Balbiani body and the fusome mediate mitochondrial inheritance during Drosophila oogenesis. Development 1301579–1590. ( 10.1242/dev.00365) [DOI] [PubMed] [Google Scholar]
- Cox RT, Spradling AC.2006Milton controls the early acquisition of mitochondria by Drosophila oocytes. Development 1333371–3377. ( 10.1242/dev.02514) [DOI] [PubMed] [Google Scholar]
- Cox RT, Spradling AC.2009Clueless, a conserved Drosophila gene required for mitochondrial subcellular localization, interacts genetically with parkin. Disease Models and Mechanisms 2490–499. ( 10.1242/dmm.002378) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cree LM, Hammond ER, Shelling AN, Berg MC, Peek JC, Green MP.2015Maternal age and ovarian stimulation independently affect oocyte mtDNA copy number and cumulus cell gene expression in bovine clones. Human Reproduction 301410–1420. ( 10.1093/humrep/dev066) [DOI] [PubMed] [Google Scholar]
- D’Alonzo D, Hong Z.2017Apoptosis in Bcl2l13 epididymal cells of mice. Cell and Developmental Biology 6 2. ( 10.4172/2168-9296.1000187) [DOI] [Google Scholar]
- Dalton CM, Szabadkai G, Carroll J.2014Measurement of ATP in single oocytes: impact of maturation and cumulus cells on levels and consumption. Journal of Cellular Physiology 229353–361. ( 10.1002/jcp.24457) [DOI] [PubMed] [Google Scholar]
- de Cuevas M, Lilly MA, Spradling AC.1997Germline cyst formation in Drosophila. Annual Review of Genetics 31405–428. ( 10.1146/annurev.genet.31.1.405) [DOI] [PubMed] [Google Scholar]
- De Fanti S, Vicario S, Lang M, Simone D, Magli C, Luiselli D, Gianaroli L, Romeo G.2017Intra-individual purifying selection on mitochondrial DNA variants during human oogenesis. Human Reproduction 321100–1107. ( 10.1093/humrep/dex051) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson ME, Flenniken AM, Ji X, Teboul L, Wong MD, White JK, Meehan TF, Weninger WJ, Westerberg H, Adissu Het al. 2016High-throughput discovery of novel developmental phenotypes. Nature 537508–514. ( 10.1038/nature19356) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diot A, Dombi E, Lodge T, Liao C, Morten K, Carver J, Wells D, Child T, Johnston IG, Williams Set al. 2016aModulating mitochondrial quality in disease transmission: towards enabling mitochondrial DNA disease carriers to have healthy children. Biochemical Society Transactions 441091–1100. ( 10.1042/BST20160095) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diot A, Morten K, Poulton J.2016bMitophagy plays a central role in mitochondrial ageing. Mammalian Genome 27381–395. ( 10.1007/s00335-016-9651-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diwan A, Krenz M, Syed FM, Wansapura J, Ren X, Koesters AG, Li H, Kirshenbaum LA, Hahn HS, Robbins Jet al. 2007Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. Journal of Clinical Investigation 1172825–2833. ( 10.1172/JCI32490) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doblado L, Lueck C, Rey C, Samhan-Arias AK, Prieto I, Stacchiotti A, Monsalve M.2021Mitophagy in human diseases. International Journal of Molecular Sciences 22 3903. ( 10.3390/ijms22083903) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Albertini DF, Nishimori K, Kumar TR, Lu N, Matzuk MM.1996Growth differentiation factor-9 is required during early ovarian folliculogenesis. Nature 383531–535. ( 10.1038/383531a0) [DOI] [PubMed] [Google Scholar]
- Dumollard R, Duchen M, Carroll J.2007aThe role of mitochondrial function in the oocyte and embryo. Current Topics in Developmental Biology 7721–49. ( 10.1016/S0070-2153(06)77002-8) [DOI] [PubMed] [Google Scholar]
- Dumollard R, Ward Z, Carroll J, Duchen MR.2007bRegulation of redox metabolism in the mouse oocyte and embryo. Development 134455–465. ( 10.1242/dev.02744) [DOI] [PubMed] [Google Scholar]
- El Shourbagy SH, Spikings EC, Freitas M, St John JC.2006Mitochondria directly influence fertilisation outcome in the pig. Reproduction 131233–245. ( 10.1530/rep.1.00551) [DOI] [PubMed] [Google Scholar]
- Fernandez ÁF, Sebti S, Wei Y, Zou Z, Shi M, Mcmillan KL, He C, Ting T, Liu Y, Chiang WCet al. 2018Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature 558136–140. ( 10.1038/s41586-018-0162-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz Pet al. 2007Ambra1 regulates autophagy and development of the nervous system. Nature 4471121–1125. ( 10.1038/nature05925) [DOI] [PubMed] [Google Scholar]
- Floros VI, Pyle A, Dietmann S, Wei W, Tang WCW, Irie N, Payne B, Capalbo A, Noli L, Coxhead Jet al. 2018Segregation of mitochondrial DNA heteroplasmy through a developmental genetic bottleneck in human embryos. Nature Cell Biology 20144–151. ( 10.1038/s41556-017-0017-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragouli E, Spath K, Alfarawati S, Wells D.2013Quantification of mitochondrial DNA predicts the implantation potential of chromosomally normal embryos. Fertility and Sterility 100 (Supplement 1). ( 10.1016/j.fertnstert.2013.07.125) [DOI] [Google Scholar]
- Fujiwara T, Ye S, Castro-Gomes T, Winchell CG, Andrews NW, Voth DE, Varughese KI, Mackintosh SG, Feng Y, Pavlos Net al. 2016PLEKHM1/DEF8/RAB7 complex regulates lysosome positioning and bone homeostasis. JCI Insight 1 e86330. ( 10.1172/jci.insight.86330) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan B, Peng X, Nagy T, Alcaraz A, Gu H, Guan JL.2006Role of FIP200 in cardiac and liver development and its regulation of TNFalpha and TSC-mTOR signaling pathways. Journal of Cell Biology 175121–133. ( 10.1083/jcb.200604129) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner DK, Wale PL.2013Analysis of metabolism to select viable human embryos for transfer. Fertility and Sterility 991062–1072. ( 10.1016/j.fertnstert.2012.12.004) [DOI] [PubMed] [Google Scholar]
- Gawriluk T.2014Targeted knockout of Beclin-1 reveals an essential function in ovary and testis. PhD thesis. USA: University of Kentucky. [Google Scholar]
- Gawriluk TR, Hale AN, Flaws JA, Dillon CP, Green DR, Rucker 3rd EB.2011Autophagy is a cell survival program for female germ cells in the murine ovary. Reproduction 141759–765. ( 10.1530/REP-10-0489) [DOI] [PubMed] [Google Scholar]
- Gispert S, Ricciardi F, Kurz A, Azizov M, Hoepken HH, Becker D, Voos W, Leuner K, Muller WE, Kudin APet al. 2009Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLoS ONE 4 e5777. ( 10.1371/journal.pone.0005777) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJet al. 2003Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. Journal of Biological Chemistry 27843628–43635. ( 10.1074/jbc.M308947200) [DOI] [PubMed] [Google Scholar]
- Goorden SM, Hoogeveen-Westerveld M, Cheng C, van Woerden GM, Mozaffari M, Post L, Duckers HJ, Nellist M, Elgersma Y.2011Rheb is essential for murine development. Molecular and Cellular Biology 311672–1678. ( 10.1128/MCB.00985-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Yu Q.2019Role of mTOR signaling in female reproduction. Frontiers in Endocrinology 10 692. ( 10.3389/fendo.2019.00692) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond ER, Green MP, Shelling AN, Berg MC, Peek JC, Cree LM.2016Oocyte mitochondrial deletions and heteroplasmy in a bovine model of ageing and ovarian stimulation. Molecular Human Reproduction 22261–271. ( 10.1093/molehr/gaw003) [DOI] [PubMed] [Google Scholar]
- Hill JH, Chen Z, Xu H.2014Selective propagation of functional mitochondrial DNA during oogenesis restricts the transmission of a deleterious mitochondrial variant. Nature Genetics 46389–392. ( 10.1038/ng.2920) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurd TR, Herrmann B, Sauerwald J, Sanny J, Grosch M, Lehmann R.2016Long Oskar controls mitochondrial inheritance in Drosophila melanogaster. Developmental Cell 39560–571. ( 10.1016/j.devcel.2016.11.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iha H, Peloponese JM, Verstrepen L, Zapart G, Ikeda F, Smith CD, Starost MF, Yedavalli V, Heyninck K, Dikic Iet al. 2008Inflammatory cardiac valvulitis in TAX1BP1-deficient mice through selective NF-kappaB activation. EMBO Journal 27629–641. ( 10.1038/emboj.2008.5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itier JM, Ibanez P, Mena MA, Abbas N, Cohen-Salmon C, Bohme GA, Laville M, Pratt J, Corti O, Pradier Let al. 2003Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Human Molecular Genetics 122277–2291. ( 10.1093/hmg/ddg239) [DOI] [PubMed] [Google Scholar]
- Jenuth JP, Peterson AC, Fu K, Shoubridge EA.1996Random genetic drift in the female germline explains the rapid segregation of mammalian mitochondrial DNA. Nature Genetics 14146–151. ( 10.1038/ng1096-146) [DOI] [PubMed] [Google Scholar]
- Johnson MT, Freeman EA, Gardner DK, Hunt PA.2007Oxidative metabolism of pyruvate is required for meiotic maturation of murine oocytes in vivo. Biology of Reproduction 772–8. ( 10.1095/biolreprod.106.059899) [DOI] [PubMed] [Google Scholar]
- Johnston IG, Burgstaller JP, Havlicek V, Kolbe T, Ruelicke T, Brem G, Poulton J, Jones NS.2015Stochastic modelling, Bayesian inference, and new in vivo measurements elucidate the debated mtDNA bottleneck mechanism. eLife 4 e07464. ( 10.7554/eLife.07464) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen SB, Viollet B, Andreelli F, Frosig C, Birk JB, Schjerling P, Vaulont S, Richter EA, Wojtaszewski JF.2004Knockout of the alpha2 but not alpha1 5′-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. Journal of Biological Chemistry 2791070–1079. ( 10.1074/jbc.M306205200) [DOI] [PubMed] [Google Scholar]
- Kaizuka T, Mizushima N.2016Atg13 is essential for autophagy and cardiac development in mice. Molecular and Cellular Biology 36585–595. ( 10.1128/MCB.01005-15) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kann LM, Rosenblum EB, Rand DM.1998Aging, mating, and the evolution of mtDNA heteroplasmy in Drosophila melanogaster. PNAS 952372–2377. ( 10.1073/pnas.95.5.2372) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura N, Sun-Wada GH, Aoyama M, Harada A, Takasuga S, Sasaki T, Wada Y.2012Delivery of endosomes to lysosomes via microautophagy in the visceral endoderm of mouse embryos. Nature Communications 3 1071. ( 10.1038/ncomms2069) [DOI] [PubMed] [Google Scholar]
- Kim J, Seli E.2019Mitochondria as a biomarker for IVF outcome. Reproduction 157R235–R242. ( 10.1530/REP-18-0580) [DOI] [PubMed] [Google Scholar]
- Kitada T, Pisani A, Porter DR, Yamaguchi H, Tscherter A, Martella G, Bonsi P, Zhang C, Pothos EN, Shen J.2007Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. PNAS 10411441–11446. ( 10.1073/pnas.0702717104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klucnika A, Ma H.2019A battle for transmission: the cooperative and selfish animal mitochondrial genomes. Open Biology 9180267. ( 10.1098/rsob.180267) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Yet al. 2005Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. Journal of Cell Biology 169425–434. ( 10.1083/jcb.200412022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn H, Sopko R, Coughlin M, Perrimon N, Mitchison T.2015The Atg1-Tor pathway regulates yolk catabolism in Drosophila embryos. Development 1423869–3878. ( 10.1242/dev.125419) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N.2004The role of autophagy during the early neonatal starvation period. Nature 4321032–1036. ( 10.1038/nature03029) [DOI] [PubMed] [Google Scholar]
- Kuma A, Komatsu M, Mizushima N.2017Autophagy-monitoring and autophagy-deficient mice. Autophagy 131619–1628. ( 10.1080/15548627.2017.1343770) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri V, Hawkins WD, Klionsky DJ.2019Watch what you (self-) eat: autophagic mechanisms that modulate metabolism. Cell Metabolism 29803–826. ( 10.1016/j.cmet.2019.03.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamas-Toranzo I, Pericuesta E, Bermejo-Alvarez P.2018Mitochondrial and metabolic adjustments during the final phase of follicular development prior to IVM of bovine oocytes. Theriogenology 119156–162. ( 10.1016/j.theriogenology.2018.07.007) [DOI] [PubMed] [Google Scholar]
- Latorre-Pellicer A, Lechuga-Vieco AV, Johnston IG, Hamalainen RH, Pellico J, Justo-Mendez R, Fernandez-Toro JM, Claveria C, Guaras A, Sierra Ret al. 2019Regulation of mother-to-offspring transmission of mtDNA heteroplasmy. Cell Metabolism 301120, .e5–1130.e5. ( 10.1016/j.cmet.2019.09.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechuga-Vieco AV, Latorre-Pellicer A, Johnston IG, Prota G, Gileadi U, Justo-Mendez R, Acin-Perez R, Martinez-De-Mena R, Fernandez-Toro JM, Jimenez-Blasco Det al. 2020Cell identity and nucleo-mitochondrial genetic context modulate OXPHOS performance and determine somatic heteroplasmy dynamics. Science Advances 6 eaba5345. ( 10.1126/sciadv.aba5345) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leese HJ.2012Metabolism of the preimplantation embryo: 40 years on. Reproduction 143417–427. ( 10.1530/REP-11-0484) [DOI] [PubMed] [Google Scholar]
- Lehmann R.1995Cell-cell signaling, microtubules, and the loss of symmetry in the Drosophila oocyte. Cell 83353–356. ( 10.1016/0092-8674(95)90111-6) [DOI] [PubMed] [Google Scholar]
- Li M, Rothwell R, Vermaat M, Wachsmuth M, Schröder R, Laros JF, Van Oven M, De Bakker PI, Bovenberg JA, Van Duijn CMet al. 2016Transmission of human mtDNA heteroplasmy in the genome of the Netherlands families: support for a variable-size bottleneck. Genome Research 26417–426. ( 10.1101/gr.203216.115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber T, Jeedigunta SP, Palozzi JM, Lehmann R, Hurd TR.2019Mitochondrial fragmentation drives selective removal of deleterious mtDNA in the germline. Nature 570380–384. ( 10.1038/s41586-019-1213-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Xu H, O’Farrell PH.2014Transmission of mitochondrial mutations and action of purifying selection in Drosophila melanogaster. Nature Genetics 46393–397. ( 10.1038/ng.2919) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra R, Warne JP, Salas E, Xu AW, Debnath J.2015Loss of Atg12, but not Atg5, in pro-opiomelanocortin neurons exacerbates diet-induced obesity. Autophagy 11145–154. ( 10.1080/15548627.2014.998917) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchington DR, Hartshorne GM, Barlow D, Poulton J.1997Homopolymeric tract heteroplasmy in mtDNA from tissues and single oocytes: support for a genetic bottleneck. American Journal of Human Genetics 60408–416. [PMC free article] [PubMed] [Google Scholar]
- Marchington DR, Macaulay V, Hartshorne GM, Barlow D, Poulton J.1998Evidence from human oocytes for a genetic bottleneck in an mtDNA disease. American Journal of Human Genetics 63769–775. ( 10.1086/302009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchington DR, Barlow D, Poulton J.1999Transmitochondrial mice carrying resistance to chloramphenicol on mitochondrial DNA: developing the first mouse model of mitochondrial DNA disease. Nature Medicine 5957–960. ( 10.1038/11403) [DOI] [PubMed] [Google Scholar]
- Matheoud D, Cannon T, Voisin A, Penttinen AM, Ramet L, Fahmy AM, Ducrot C, Laplante A, Bourque MJ, Zhu Let al. 2019Intestinal infection triggers Parkinson’s disease-like symptoms in Pink1(-/-) mice. Nature 571565–569. ( 10.1038/s41586-019-1405-y) [DOI] [PubMed] [Google Scholar]
- Matova N, Cooley L.2001Comparative aspects of animal oogenesis. Developmental Biology 231291–320. ( 10.1006/dbio.2000.0120) [DOI] [PubMed] [Google Scholar]
- May-Panloup P, Vignon X, Chretien MF, Heyman Y, Tamassia M, Malthiery Y, Reynier P.2005Increase of mitochondrial DNA content and transcripts in early bovine embryogenesis associated with upregulation of mtTFA and NRF1 transcription factors. Reproductive Biology and Endocrinology 3 65. ( 10.1186/1477-7827-3-65) [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell JM, Petrie L.2004Mitochondrial DNA turnover occurs during preimplantation development and can be modulated by environmental factors. Reproductive Biomedicine Online 9418–424. ( 10.1016/s1472-6483(10)61277-1) [DOI] [PubMed] [Google Scholar]
- McLelland GL, Soubannier V, Chen CX, Mcbride HM, Fon EA.2014Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO Journal 33282–295. ( 10.1002/embj.201385902) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng L, Jan SZ, Hamer G, Van Pelt AM, Van Der Stelt I, Keijer J, Teerds KJ.2018Preantral follicular atresia occurs mainly through autophagy, while antral follicles degenerate mostly through apoptosis. Biology of Reproduction 99853–863. ( 10.1093/biolre/ioy116) [DOI] [PubMed] [Google Scholar]
- Morishita H, Zhao YG, Tamura N, Nishimura T, Kanda Y, Sakamaki Y, Okazaki M, Li D, Mizushima N.2019A critical role of VMP1 in lipoprotein secretion. eLife 8 e48834. ( 10.7554/eLife.48834) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motta PM, Nottola SA, Makabe S, Heyn R.2000Mitochondrial morphology in human fetal and adult female germ cells. Human Reproduction 15 (Supplement 2) 129–147. ( 10.1093/humrep/15.suppl_2.129) [DOI] [PubMed] [Google Scholar]
- Muller-Hocker J, Schafer S, Weis S, Munscher C, Strowitzki T.1996Morphological-cytochemical and molecular genetic analyses of mitochondria in isolated human oocytes in the reproductive age. Molecular Human Reproduction 2951–958. ( 10.1093/molehr/2.12.951) [DOI] [PubMed] [Google Scholar]
- Mulligan PK.2003Robert C. King: an appreciation of his work. Genetics 165939–943. ( 10.1093/genetics/165.3.939) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakoshi Y, Sueoka K, Takahashi K, Sato S, Sakurai T, Tajima H, Yoshimura Y.2013Embryo developmental capability and pregnancy outcome are related to the mitochondrial DNA copy number and ooplasmic volume. Journal of Assisted Reproduction and Genetics 301367–1375. ( 10.1007/s10815-013-0062-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata M, Miwa Y, Sato I.2009Expression of respiratory chain enzyme mRNA and the morphological properties of mitochondria in the masseter muscles of klotho mutant mice. Okajimas Folia Anatomica Japonica 8693–103. ( 10.2535/ofaj.86.93) [DOI] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ.2010PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biology 8 e1000298. ( 10.1371/journal.pbio.1000298) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemazanyy I, Blaauw B, Paolini C, Caillaud C, Protasi F, Mueller A, Proikas-Cezanne T, Russell RC, Guan KL, Nishino Iet al. 2013Defects of Vps15 in skeletal muscles lead to autophagic vacuolar myopathy and lysosomal disease. EMBO Molecular Medicine 5870–890. ( 10.1002/emmm.201202057) [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien Y, Wingfield MB.2019Reproductive ageing-turning back the clock? Irish Journal of Medical Science 188161–167. ( 10.1007/s11845-018-1769-2) [DOI] [PubMed] [Google Scholar]
- Olivo PD, Van De Walle MJ, Laipis PJ, Hauswirth WW.1983Nucleotide sequence evidence for rapid genotypic shifts in the bovine mitochondrial DNA D-loop. Nature 306400–402. ( 10.1038/306400a0) [DOI] [PubMed] [Google Scholar]
- O’Sullivan GA, Kneussel M, Elazar Z, Betz H.2005GABARAP is not essential for GABA receptor targeting to the synapse. European Journal of Neuroscience 222644–2648. ( 10.1111/j.1460-9568.2005.04448.x) [DOI] [PubMed] [Google Scholar]
- Otten ABC, Sallevelt SCEH, Carling PJ, Dreesen JCFM, Drusedau M, Spierts S, Paulussen ADC, De Die-Smulders CEM, Herbert M, Chinnery PFet al. 2018Mutation-specific effects in germline transmission of pathogenic mtDNA variants. Human Reproduction 331331–1341. ( 10.1093/humrep/dey114) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquariello R, Ermisch AF, Silva E, Mccormick S, Logsdon D, Barfield JP, Schoolcraft WB, Krisher RL.2019Alterations in oocyte mitochondrial number and function are related to spindle defects and occur with maternal aging in mice and humansdagger. Biology of Reproduction 100971–981. ( 10.1093/biolre/ioy248) [DOI] [PubMed] [Google Scholar]
- Pellestor F, Andreo B, Arnal F, Humeau C, Demaille J.2003Maternal aging and chromosomal abnormalities: new data drawn from in vitro unfertilized human oocytes. Human Genetics 112195–203. ( 10.1007/s00439-002-0852-x) [DOI] [PubMed] [Google Scholar]
- Pepling ME, Spradling AC.1998Female mouse germ cells form synchronously dividing cysts. Development 1253323–3328. ( 10.1242/dev.125.17.3323) [DOI] [PubMed] [Google Scholar]
- Pepling ME, De Cuevas M, Spradling AC.1999Germline cysts: a conserved phase of germ cell development? Trends in Cell Biology 9257–262. ( 10.1016/s0962-8924(99)01594-9) [DOI] [PubMed] [Google Scholar]
- Pepling ME, Wilhelm JE, O’Hara AL, Gephardt GW, Spradling AC.2007Mouse oocytes within germ cell cysts and primordial follicles contain a Balbiani body. PNAS 104187–192. ( 10.1073/pnas.0609923104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez FA, Palmiter RD.2005Parkin-deficient mice are not a robust model of parkinsonism. PNAS 1022174–2179. ( 10.1073/pnas.0409598102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phadwal K, Watson AS, Simon AK.2013Tightrope act: autophagy in stem cell renewal, differentiation, proliferation, and aging. Cellular and Molecular Life Sciences 7089–103. ( 10.1007/s00018-012-1032-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickles S, Vigie P, Youle RJ.2018Mitophagy and quality control mechanisms in mitochondrial maintenance. Current Biology 28R170–R185. ( 10.1016/j.cub.2018.01.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piko L, Matsumoto L.1976Number of mitochondria and some properties of mitochondrial DNA in the mouse egg. Developmental Biology 491–10. ( 10.1016/0012-1606(76)90253-0) [DOI] [PubMed] [Google Scholar]
- Piko L, Taylor KD.1987Amounts of mitochondrial DNA and abundance of some mitochondrial gene transcripts in early mouse embryos. Developmental Biology 123364–374. ( 10.1016/0012-1606(87)90395-2) [DOI] [PubMed] [Google Scholar]
- Politi Y, Gal L, Kalifa Y, Ravid L, Elazar Z, Arama E.2014Paternal mitochondrial destruction after fertilization is mediated by a common endocytic and autophagic pathway in Drosophila. Developmental Cell 29305–320. ( 10.1016/j.devcel.2014.04.005) [DOI] [PubMed] [Google Scholar]
- Poulton J, Chiaratti MR, Meirelles FV, Kennedy S, Wells D, Holt I.2010Transmission of mitochondrial DNA diseases and ways to prevent them. PLoS Genetics 8 e1001066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulton J, Steffann J, Burgstaller J, McFarland R. & workshop participants 2019243rd ENMC International Workshop: developing guidelines for management of reproductive options for families with maternally inherited mtDNA disease, Amsterdam, the Netherlands. Neuromuscular Disorders 29725–733. ( 10.1016/j.nmd.2019.08.004) [DOI] [PubMed] [Google Scholar]
- Qu X, Zou Z, Sun Q, Luby-Phelps K, Cheng P, Hogan RN, Gilpin C, Levine B.2007Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell 128931–946. ( 10.1016/j.cell.2006.12.044) [DOI] [PubMed] [Google Scholar]
- Reynier P, May-Panloup P, Chretien MF, Morgan CJ, Jean M, Savagner F, Barriere P, Malthiery Y.2001Mitochondrial DNA content affects the fertilizability of human oocytes. Molecular Human Reproduction 7425–429. ( 10.1093/molehr/7.5.425) [DOI] [PubMed] [Google Scholar]
- Ronkina N, Kotlyarov A, Dittrich-Breiholz O, Kracht M, Hitti E, Milarski K, Askew R, Marusic S, Lin LL, Gaestel Met al. 2007The mitogen-activated protein kinase (MAPK)-activated protein kinases MK2 and MK3 cooperate in stimulation of tumor necrosis factor biosynthesis and stabilization of p38 MAPK. Molecular and Cellular Biology 27170–181. ( 10.1128/MCB.01456-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross JM, Stewart JB, Hagstrom E, Brene S, Mourier A, Coppotelli G, Freyer C, Lagouge M, Hoffer BJ, Olson Let al. 2013Germline mitochondrial DNA mutations aggravate ageing and can impair brain development. Nature 501412–415. ( 10.1038/nature12474) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Nah J, Oka SI, Mukai R, Monden Y, Maejima Y, Ikeda Y, Sciarretta S, Liu T, Li Het al. 2019An alternative mitophagy pathway mediated by Rab9 protects the heart against ischemia. Journal of Clinical Investigation 129802–819. ( 10.1172/JCI122035) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu Met al. 2008Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 456264–268. ( 10.1038/nature07383) [DOI] [PubMed] [Google Scholar]
- Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, Matsunaga K, Kageyama S, Omori H, Noda Tet al. 2009Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. PNAS 10620842–20846. ( 10.1073/pnas.0911267106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J.2008Essential role for Nix in autophagic maturation of erythroid cells. Nature 454232–235. ( 10.1038/nature07006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasai M, Sakaguchi N, Ma JS, Nakamura S, Kawabata T, Bando H, Lee Y, Saitoh T, Akira S, Iwasaki Aet al. 2017Essential role for GABARAP autophagy proteins in interferon-inducible GTPase-mediated host defense. Nature Immunology 18899–910. ( 10.1038/ni.3767) [DOI] [PubMed] [Google Scholar]
- Sen A, Damm VT, Cox RT.2013Drosophila clueless is highly expressed in larval neuroblasts, affects mitochondrial localization and suppresses mitochondrial oxidative damage. PLoS ONE 8 e54283. ( 10.1371/journal.pone.0054283) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheard KM, Thibault-Sennett SA, Sen A, Shewmaker F, Cox RT.2020Clueless forms dynamic, insulin-responsive bliss particles sensitive to stress. Developmental Biology 459149–160. ( 10.1016/j.ydbio.2019.12.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieber MH, Thomsen MB, Spradling AC.2016Electron transport chain remodeling by GSK3 during oogenesis connects nutrient state to reproduction. Cell 164420–432. ( 10.1016/j.cell.2015.12.020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singla S, Iwamoto-Stohl LK, Zhu M, Zernicka-Goetz M.2020Autophagy-mediated apoptosis eliminates aneuploid cells in a mouse model of chromosome mosaicism. Nature Communications 11 2958. ( 10.1038/s41467-020-16796-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, Mujica AO, Thomas M, Harrow J, Cox Tet al. 2011A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474337–342. ( 10.1038/nature10163) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, Burman JL, Li Y, Zhang Z, Narendra DPet al. 2018Parkin and PINK1 mitigate STING-induced inflammation. Nature 561258–262. ( 10.1038/s41586-018-0448-9) [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Slowicka K, Vereecke L, Mc Guire C, Sze M, Maelfait J, Kolpe A, Saelens X, Beyaert R, van Loo G.2016Optineurin deficiency in mice is associated with increased sensitivity to Salmonella but does not affect proinflammatory NF-kappaB signaling. European Journal of Immunology 46971–980. ( 10.1002/eji.201545863) [DOI] [PubMed] [Google Scholar]
- Solignac M, Génermont J, Monnerot M, Mounolou JC.1984Genetics of mitochondria in Drosophila: mtDNA inheritance in heteroplasmic strains of D. mauritiana. Molecular and General Genetics 197183–188. ( 10.1007/BF00330961) [DOI] [Google Scholar]
- Solignac M, Genermont J, Monnerot M, Mounolou JC.1987Drosophila mitochondrial genetics: evolution of heteroplasmy through germ line cell divisions. Genetics 117687–696. ( 10.1093/genetics/117.4.687) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song ZH, Yu HY, Wang P, Mao GK, Liu WX, Li MN, Wang HN, Shang YL, Liu C, Xu ZLet al. 2015Germ cell-specific Atg7 knockout results in primary ovarian insufficiency in female mice. Cell Death and Disease 6 e1589. ( 10.1038/cddis.2014.559) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sopko R, Foos M, Vinayagam A, Zhai B, Binari R, Hu Y, Randklev S, Perkins LA, Gygi SP, Perrimon N.2014Combining genetic perturbations and proteomics to examine kinase-phosphatase networks in Drosophila embryos. Developmental Cell 31114–127. ( 10.1016/j.devcel.2014.07.027) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sou YS, Waguri S, Iwata J, Ueno T, Fujimura T, Hara T, Sawada N, Yamada A, Mizushima N, Uchiyama Yet al. 2008The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Molecular Biology of the Cell 194762–4775. ( 10.1091/mbc.e08-03-0309) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubannier V, Mclelland GL, Zunino R, Braschi E, Rippstein P, Fon EA, Mcbride HM.2012aA vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Current Biology 22135–141. ( 10.1016/j.cub.2011.11.057) [DOI] [PubMed] [Google Scholar]
- Soubannier V, Rippstein P, Kaufman BA, Shoubridge EA, Mcbride HM.2012bReconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS ONE 7 e52830. ( 10.1371/journal.pone.0052830) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Moraes CT.2001Manipulating mitochondrial DNA heteroplasmy by a mitochondrially targeted restriction endonuclease. Human Molecular Genetics 103093–3099. ( 10.1093/hmg/10.26.3093) [DOI] [PubMed] [Google Scholar]
- Steingrimsson E, Tessarollo L, Reid SW, Jenkins NA, Copeland NG.1998The bHLH-Zip transcription factor Tfeb is essential for placental vascularization. Development 1254607–4616. ( 10.1242/dev.125.23.4607) [DOI] [PubMed] [Google Scholar]
- Stocker H, Radimerski T, Schindelholz B, Wittwer F, Belawat P, Daram P, Breuer S, Thomas G, Hafen E.2003Rheb is an essential regulator of S6K in controlling cell growth in Drosophila. Nature Cell Biology 5559–565. ( 10.1038/ncb995) [DOI] [PubMed] [Google Scholar]
- Sugiura A, Mclelland GL, Fon EA, Mcbride HM.2014A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO Journal 332142–2156. ( 10.15252/embj.201488104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennessen JM, Baker KD, Lam G, Evans J, Thummel CS.2011The Drosophila estrogen-related receptor directs a metabolic switch that supports developmental growth. Cell Metabolism 13139–148. ( 10.1016/j.cmet.2011.01.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly-Y M, Gidlof S, Oldfors A, Wibom Ret al. 2004Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429417–423. ( 10.1038/nature02517) [DOI] [PubMed] [Google Scholar]
- Tsukamoto S, Kuma A, Mizushima N.2008aThe role of autophagy during the oocyte-to-embryo transition. Autophagy 41076–1078. ( 10.4161/auto.7065) [DOI] [PubMed] [Google Scholar]
- Tsukamoto S, Kuma A, Murakami M, Kishi C, Yamamoto A, Mizushima N.2008bAutophagy is essential for preimplantation development of mouse embryos. Science 321117–120. ( 10.1126/science.1154822) [DOI] [PubMed] [Google Scholar]
- Van Blerkom J.2009Mitochondria in early mammalian development. Seminars in Cell and Developmental Biology 20354–364. ( 10.1016/j.semcdb.2008.12.005) [DOI] [PubMed] [Google Scholar]
- Vega-Rubin-de-Celis S.2019The role of beclin 1-dependent autophagy in cancer. Biology 9 4. ( 10.3390/biology9010004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada T, Nakashima T, Hiroshi N, Penninger JM.2006RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends in Molecular Medicine 1217–25. ( 10.1016/j.molmed.2005.11.007) [DOI] [PubMed] [Google Scholar]
- Wai T, Teoli D, Shoubridge EA.2008The mitochondrial DNA genetic bottleneck results from replication of a subpopulation of genomes. Nature Genetics 401484–1488. ( 10.1038/ng.258) [DOI] [PubMed] [Google Scholar]
- Wai T, Ao A, Zhang X, Cyr D, Dufort D, Shoubridge EA.2010The role of mitochondrial DNA copy number in mammalian fertility. Biology of Reproduction 8352–62. ( 10.1095/biolreprod.109.080887) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen JK, Wang YT, Chan CC, Hsieh CW, Liao HM, Hung CC, Chen GC.2017Atg9 antagonizes TOR signaling to regulate intestinal cell growth and epithelial homeostasis in Drosophila. eLife 6 e29338. ( 10.7554/eLife.29338) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilding M, De Placido G, De Matteo L, Marino M, Alviggi C, Dale B.2003Chaotic mosaicism in human preimplantation embryos is correlated with a low mitochondrial membrane potential. Fertility and Sterility 79340–346. ( 10.1016/s0015-0282(02)04678-2) [DOI] [PubMed] [Google Scholar]
- Wolf DP, Hayama T, Mitalipov S.2017Mitochondrial genome inheritance and replacement in the human germline. EMBO Journal 362177–2181. ( 10.15252/embj.201797606) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xian H, Liou YC.2021Functions of outer mitochondrial membrane proteins: mediating the crosstalk between mitochondrial dynamics and mitophagy. Cell Death and Differentiation 28827–842. ( 10.1038/s41418-020-00657-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Deluca SZ, O’Farrell PH.2008Manipulating the metazoan mitochondrial genome with targeted restriction enzymes. Science 321575–577. ( 10.1126/science.1160226) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Zheng Y, Zhang X, Chen Y, Wu X, Wu J, Shen Z, Jiang L, Wang L, Yang Wet al. 2017BNIP3L/NIX-mediated mitophagy protects against ischemic brain injury independent of PARK2. Autophagy 131754–1766. ( 10.1080/15548627.2017.1357792) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Z, Jin S, Yang C, Levine AJ, Heintz N.2003Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. PNAS 10015077–15082. ( 10.1073/pnas.2436255100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachari M, Ktistakis NT.2020Mammalian mitophagosome formation: a focus on the early signals and steps. Frontiers in Cell and Developmental Biology 8 171. ( 10.3389/fcell.2020.00171) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachari M, Gudmundsson SR, Li Z, Manifava M, Cugliandolo F, Shah R, Smith M, Stronge J, Karanasios E, Piunti Cet al. 2019Selective autophagy of mitochondria on a ubiquitin-endoplasmic-reticulum platform. Developmental Cell 50 627.e5–643.e5. ( 10.1016/j.devcel.2019.06.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Ren H, Xu C, Zhu C, Wu H, Liu D, Wang J, Liu L, Li W, Ma Qet al. 2016Hypoxic mitophagy regulates mitochondrial quality and platelet activation and determines severity of I/R heart injury. eLife 5 e21407. ( 10.7554/eLife.21407) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Zhao YG, Wang X, Xu L, Miao L, Feng D, Chen Q, Kovacs AL, Fan D, Zhang H.2013Mice deficient in Epg5 exhibit selective neuronal vulnerability to degeneration. Journal of Cell Biology 200731–741. ( 10.1083/jcb.201211014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen Y, Li W.2015Impairment of autophagosome-lysosome fusion in the buff mutant mice with the VPS33A(D251E) mutation. Autophagy 111608–1622. ( 10.1080/15548627.2015.1072669) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong X, Feng L, Zang R, Lei CQ, Yang Q, Shu HB.2020ZFYVE1 negatively regulates MDA5- but not RIG-I-mediated innate antiviral response. PLoS Pathogens 16 e1008457. ( 10.1371/journal.ppat.1008457) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Takatoh J, Wang F.2011The mammalian class 3 PI3K (PIK3C3) is required for early embryogenesis and cell proliferation. PLoS ONE 6 e16358. ( 10.1371/journal.pone.0016358) [DOI] [PMC free article] [PubMed] [Google Scholar]