

Abstract
Background
Premature ovarian insufficiency (POI) is a highly heterogeneous disease, and up to 25% of cases can be explained by genetic causes. The transcription factor WT1 has long been reported to play a crucial role in ovary function. Wt1‐mutated female mice exhibited POI‐like phenotypes.
Methods and Results
In this study, whole exome sequencing (WES) was applied to find the cause of POI in Han Chinese women. A nonsense variant in the WT1 gene: NM_024426.6:c.1387C>T(p.R463*) was identified in a non‐syndromic POI woman. The variant is a heterozygous de novo mutation that is very rare in the human population. The son of the patient inherited the mutation and developed Wilms’ tumor and urethral malformation at the age of 7. According to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) guidelines, the novel variant is categorized as pathogenic. Western blot analysis further demonstrated that the WT1 variant could produce a truncated WT1 isoform in vitro.
Conclusions
A rare heterozygous nonsense WT1 mutant is associated with non‐syndromic POI and Wilms’ tumor. Our finding characterized another pathogenic WT1 variant, providing insight into genetic counseling.
Keywords: premature ovarian insufficiency (POI), truncated protein, whole exome sequencing (WES), Wilms’ tumor, WT1
A heterozygous nonsense WT1 mutant is associated with non‐syndromic POI and Wilms’ tumor. The variant is a very rare mutation in the human population and is categorized as pathogenic. The nonsense variant caused a truncated protein product in vitro.
1. INTRODUCTION
Premature ovarian insufficiency (POI) is defined as absent menarche or premature depletion of ovarian follicles before the age of 40 years (Persani et al., 2009). POI is an extremely heterogeneous disorder with variable clinical presentations and multiple causes. It is estimated that genetic causes account for approximately 20%–25% of cases of POI (Jiao et al., 2018). In the past decade, multiple genetic analysis methods including whole exome sequencing (WES) have offered great opportunities to identify pathogenic variants in POI. POI‐associated causative genes fall within pathways critical for ovarian development and function, such as DNA damage repair, meiosis, recombination, gene transcription or translation, follicle development, steroidogenesis, etc. (Jiao et al., 2020; Rossetti et al., 2017).
The human WT1 gene (OMIM 607102), located at 11p13 (GRCh37), encodes a transcription factor involved in transcriptional regulation, self‐association, and RNA recognition (Kennedy et al., 1996; Moffett et al., 1995; Reddy et al., 1995; Rose et al., 1990). Initially, WT1 was found to be expressed at a high level in the glomeruli of the kidney and was first known as a tumor suppressor gene for Wilms’ tumor in the 1990s (Haber et al., 1990; Pelletier, Schalling, et al., 1991). WT1 protein contains a proline/glutamine‐rich domain at the N‐terminus and four zinc fingers in the C‐terminal region (Bardeesy & Pelletier, 1998). A repression domain is located within residues 84–179, and an activation domain with independent function is between residues 180 and 294 (Wang et al., 1993). WT1 binds to DNA helix through the four carboxyl‐terminal Cys2His2 zinc fingers, which have bidirectional activities of transcriptional regulation depending on the cellular or chromosomal context (Parenti et al., 2015; Ullmark et al., 2018).
To date, WT1 is found to be expressed and functional in many tissues, with essential roles in the regulation of ovarian cell proliferation, apoptosis, and steroidogenesis (Park et al., 2014; Pelletier, Bruening, et al., 1991; Wang et al., 2021). For instance, activation of WT1 through the regulation of the upstream activator Bax is necessary for the maintenance of granulosa cell survival during the early stage of follicles in rats (Park et al., 2014). Multiple steroidogenic enzyme‐encoding genes have also been reported to be putative targets of WT1. In mouse ovaries, the mRNA levels of P450scc, 3β‐HSD, Hsd17b1, Cyp17a1, Star, and Arx were significantly increased in Wt1‐deficient XX gonads compared with those in control ovaries (Chen et al., 2017). Moreover, variants in the Wt1 gene in animals are associated with ovarian insufficiency. Severe reproductive defects such as smaller ovaries and reduced number of follicles were observed in Wt1 +/R394W female mice (Gao et al., 2004).
Here, we identified a nonsense variant of WT1 in a non‐syndromic POI patient and her son from a non‐consanguineous Chinese family through WES data processing. Human genome variation databases were utilized to investigate the minor allele frequency, and bioinformatic tools were utilized to evaluate the pathogenicity. Sanger sequencing was performed on the patient and her family members to confirm their genotypes. The western blot assay suggested that the WT1 variant could encode a truncated protein, which might contribute to the development of POI.
2. MATERIALS AND METHODS
2.1. Study subject and clinical evaluations
POI patients were diagnosed at the Affiliated Obstetrics and Gynecology Hospital of Fudan University. The criteria for POI diagnosis follow the recommendations provided by the European Society for Human Reproduction and Embryology (2016) (European Society for Human Reproduction and Embryology (ESHRE) Guideline Group on POI et al., 2016). Women with ovarian surgery and radiotherapeutic or chemotherapeutic interventions were excluded. A detailed clinical query including environment, behavior, diet, and poison exposure was also performed. Familial history was ascertained. Written informed consent was obtained from participants or the parent of participants under the age of 18.
2.2. DNA extraction and assessment
Genomic DNA was extracted from peripheral blood using the QIAamp DNA Mini Kit (QIAGEN, Hilden, Germany) according to the manufacturer's instructions. Briefly, optimized buffers and enzymes were used to lyse the peripheral blood from the patient, stabilize nucleic acids, and enhance the genome DNA adsorption to the QIAamp membrane. Then, alcohol was added, and the whole lysates were loaded onto the QIAamp spin column. Afterward, wash buffers were used to remove impurities, and pure ready‐to‐use DNA was then eluted in water or a low‐salt buffer. Finally, the quality and quantity of DNA were assessed by agarose gel electrophoresis and SimpliNano (Harvard Bioscience).
2.3. WES and data processing
Approximately 1.5 μg of genomic DNA was used to prepare a captured library using an Agilent SureSelectXT Human All Exon V6 kit and then sequenced on a HiSeq X Ten platform (Illumina). Raw data were aligned to the human reference genome sequence (UCSC Genome Browser hg19) with the Burrows‐Wheeler Alignment tool (http://bio‐bwa.sourceforge.net/). Variant calling was accomplished using the Genome Analysis Toolkit (https://www.broadinstitute.org/gatk/) (McKenna et al., 2010) and ANNOVAR software was used to annotate all variants.
The raw data collected from WES were subjected to analysis as previously described (Yang et al., 2019). Briefly, genetic variants in the exonic and splicing regions were chosen. Variant filtering was performed based on a minor allele frequency (MAF) ≤0.1% in the 1000 Genomes Project (1KG Project; http://browser.1000genomes.org), Genome Aggregation Database (gnomAD; http://gnomad‐old.broadinstitute.org/), and Exome Aggregation Consortium (ExAC; http://exac.broadinstitute.org). Predictions of deleterious nonsynonymous variants were performed using four bioinformatics tools: SIFT (http://sift.jcvi.org), PolyPhen‐2 (http://genetics.bwh.harvard.edu/pph2/), MutationTaster (http://www.mutationtaster.org), and CADD (http://cadd.gs.washington.edu).
2.4. Variant confirmation
Sanger sequencing was performed to confirm the potential causative variants in the family. Genomic DNA was used for variant confirmation. Primers for the WT1 (NM_024426.6) variant were designed using the “Primer‐BLAST” program (https://www.ncbi.nlm.nih.gov/tools/primer‐blast/). Primer specificity was checked using the alignment search tool BLAST (https://www.ncbi.nlm.nih.gov/blast).
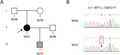
Primer sequences were as follows: forward, 5′–GGAA ACAGTAGGGACCTGGC‐3′; reverse, 5′–CAGATGCAGAC ATTGCAGGC‐3′. The results of Sanger sequencing were analyzed using SnapGene 4.2.4 software (Figure 1).
FIGURE 1.
Identification of a WT1 mutation in a Chinese family. (a) A heterozygous WT1 variant (M) was identified in a non‐consanguineous family. The black arrow in the pedigree plot indicates the proband. (b) Sanger sequencing confirmed heterozygous WT1 mutations in the proband (W/M). Both of the proband's father and mother are wild type (W/W). The red arrow indicates the mutation sites
2.5. Plasmid construction and mutagenesis
Full‐length human WT1 cDNA was synthesized (Weizhen, Jinan, China) and constructed into the pCMV‐FLAG vector (Takara). Site‐directed mutagenesis was performed to generate the null variant (c.1387C>T) of WT1 according to the instructions of the KOD‐Plus‐Mutagenesis Kit (Toyobo). The relevant primers were as follows: forward, 5′‐TGAAAGTTCTCCCGGTCCGACCACC‐3′; reverse, 5′‐CTGACAAGTTTTACACTGGAATGGTTTCACACCTGT‐3′. The recombinant plasmids were verified by direct Sanger sequencing prior to functional studies.
2.6. Cell culture and transfection
Human embryonic kidney 293T (HEK293T) cells were purchased from the Cell Bank of the Chinese Academy of Sciences. HEK293T cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (FBS) (Gibco) and 1% penicillin–streptomycin–neomycin (PSN) antibiotic mixture (Gibco) at 37℃ with 5% CO2. HEK293T cells were transfected with the wild‐type or mutated WT1 plasmids using Lipofectamine 3000 (Invitrogen) according to the manufacturer's instructions.
2.7. Western blotting
Whole cell lysates were separated by SDS‐PAGE and transferred onto PVDF membranes. After being blocked with nonfat milk, each membrane was incubated with specific antibodies against different proteins at 4℃ overnight, followed by incubation with an HRP‐conjugated secondary antibody. Membranes were visualized using an enhanced chemiluminescence kit (GE Healthcare Life Science). The images acquired were representative of three independent experiments with consistent results. β‐actin was used as a loading control. The related antibodies included anti‐FLAG (cat. no. F3165, Sigma‐Aldrich), anti‐GFP (cat. no. G6539, Sigma‐Aldrich), HRP‐labeled anti‐β‐actin (cat. no. HRP–60008, Proteintech), HRP‐labeled goat anti‐mouse IgG (cat. no. I‐0031, DingGuo Changsheng Biotech), and HRP‐labeled goat anti‐rabbit IgG (cat. no. IH‐0011, DingGuo Changsheng Biotech).
3. RESULT
3.1. Clinical findings
The diagnosis of POI is based on the presence of menstrual disturbance and biochemical confirmation, in brief: (i) oligo/amenorrhea for at least 4 months; (ii) an elevated FSH level >25 mIU/ml on two occasions >4 weeks aside; (iii) no fallopian tube abnormalities; (iv) no radioactive, surgical, or chemotherapeutic injury; (v) no inflammation or autoimmune response of the pelvic cavity or reproductive system; and (vi) no karyotypic abnormality.
As shown in Figure 1a, a 26‐year‐old woman (II–1) diagnosed with POI from a Chinese Han non‐consanguineous family was ascertained in this study (Figure 1a). The proband had normal puberty, and menarche occurred at 16 years of age. Her menses became irregular at 23 years of age and completely stopped at 26 years of age. Other probable histories including ovarian operation, chemotherapy, radiotherapy, or immune disease were all excluded. Physical examination showed a normal body mass index. No other known urologic diseases (Table S1; Figure 2), endocrinopathies, or autoimmune disorders (Table S2) were observed for the proband. Transvaginal ultrasonography revealed a normal uterus but small ovaries with few antral follicles (Figure 3). Consecutive hormonal measurements revealed elevated FSH levels. Clinical information regarding the POI subjects is summarized in Table 1.
FIGURE 2.
Transabdominal ultrasound image of the POI subject. Ultrasound of the right kidney (a) and left kidney (b) from the proband showed normal size, structure and position of kidneys and ureters
FIGURE 3.
Transvaginal ultrasound image of the POI subject. (a) The thickness of endometrium. (b) Size of the right ovary. (c and d) Size of the left ovary
TABLE 1.
Clinical characteristics of the POI patient affected by WT1 variant
| Characteristic | Proband |
|---|---|
| First menses (y old) | 16 |
| Age of POI (y old) | 26 |
| Weight (kg) | 53 |
| Height (cm) | 158 |
| FSH (mIU/ml) | 67.84 |
| LH (mIU/ml) | 49.4 |
| PRL (ng/ml) | 11.44 |
| E2 (pg/ml) | 71 |
| P (pg/ml) | 0.5 |
| T (ng/ml) | 0.43 |
| Size of ovary (right/left) (mm) | 16 × 13 × 10/25 × 23 × 17 |
| Size of follicle (right/left) (mm) | Not detected/19 × 16 × 14 |
Abbreviations: E2, estradiol; FSH, follicle‐stimulating hormone; LH, luteinizing hormone; P, progesterone; PRL, prolactin; T, testosterone.
3.2. Identification of a rare WT1 variant by WES
WES was performed on peripheral blood DNA from the patient. Analysis of WES data was performed as previously described (Yang et al., 2019). Filtering steps and variants identified in each step are shown in Table 2. Among all variants called by WES, 11,633 variants of high calling quality and sited in exonic and splicing regions were reserved. Variants with a MAF of more than 0.1% were then excluded according to three public human genome variation databases (1KG Project, ExAC, and gnomAD). Then, 195 synonymous variants were further excluded. Among the 502 remaining variants, 321 were missense, which was subjected to functional prediction using in silico tools. Relevance to phenotype was considered based on previous reports and animal studies. Finally, a heterozygous variant of WT1, NM_024426.6:c.1387C>T (p.R463*; rs121907909) was identified. This was confirmed by Sanger sequencing (Figure 1). As shown in Table 3, the allele frequency of WT1 c.1387C>T in total population is 0.000006583 (1/151,896), and the only case is a European male. It is predicted to be pathogenic by DANN, MutationTaster, and CADD.
TABLE 2.
Filtering steps and variants identified in each step
| Step | Number of variants |
|---|---|
| All variants called by WES | 43362 |
| High calling quality | 38783 |
| In exonic and splicing regions | 11633 |
| Allele frequencies ≤0.001 in databases a | 697 |
| After elimination of synonymous SNVs | 502 |
| Nonsense, frameshift, non‐frameshift indel, splicing site, or deleterious missense variants b | 243 |
| Known pathogenic genes of POI | 1 |
Allele frequencies were estimated according to 1KG Project, ExAC, and gnomAD databases.
All missense variants were assessed using the SIFT, PolyPhen‐2, MutationTaster, and CADD tools. From those, deleterious variants were selected.
TABLE 3.
In silico analysis of identified variant in WT1 gene
| Gene | Mutation type | cDNA Change a | Protein change | Minor allele frequency b | Functional prediction c | ||||
|---|---|---|---|---|---|---|---|---|---|
| 1KG | ExAC | gnomAD | DANN | MutationTaster | CADD | ||||
| WT1 | Heterozygous | c. C1387T | p.R463* | 0 | 0 | 0.000006583 | Damaging | Damaging | 14.003 |
The GenBank accession number of WT1 is NM_024426.4.
Allele frequencies were estimated according to the 1KG Project, ExAC, and gnomAD databases.
Mutation assessment using MutationTaster and CADD tools. High CADD scores suggest that a variant is likely to have deleterious effects. The CADD cutoff is usually set at 4.
3.3. Family follow‐up and genetical analysis
The proband's parents were both healthy without any other diseases. Her mother (I–2) was now 51 years old and still experiencing a regular period. She also denied a history of any reproductive and urological diseases. Sanger sequencing revealed that both parents of the proband were wild type. Therefore, WT1 c.1387C>T is a de novo variant for the proband. Classification of the variant was then performed according to the ACMG/AMP guidelines and this novel variant was classified as “pathogenic.”
Additionally, the proband has one son (III–1) and he has been diagnosed with Wilms’ tumor and urethral malformation at 7 years of age. Sanger sequencing demonstrated that he inherited the mutant WT1 variant from his mother and a wild‐type WT1 allele from his father, so his genotype was the same as his mother.
3.4. In vitro functional characteristics of the WT1 variants
WT1 c.1387C>T variant was located in the ninth exon of WT1, and it introduced a premature stop codon in the second zinc finger of WT1 (NP_077744.4) (Figure 4a,b). The putative impact of the nonsense variant on WT1 was further investigated in vitro. Recombinant plasmids of full‐length wild‐type and mutated human WT1 were introduced into HEK293T cells, respectively. The western blotting analysis revealed that a truncated protein of approximately 51 kDa in cells overexpressing the mutated WT1 (Figure 4c), which was consistent with our prediction.
FIGURE 4.
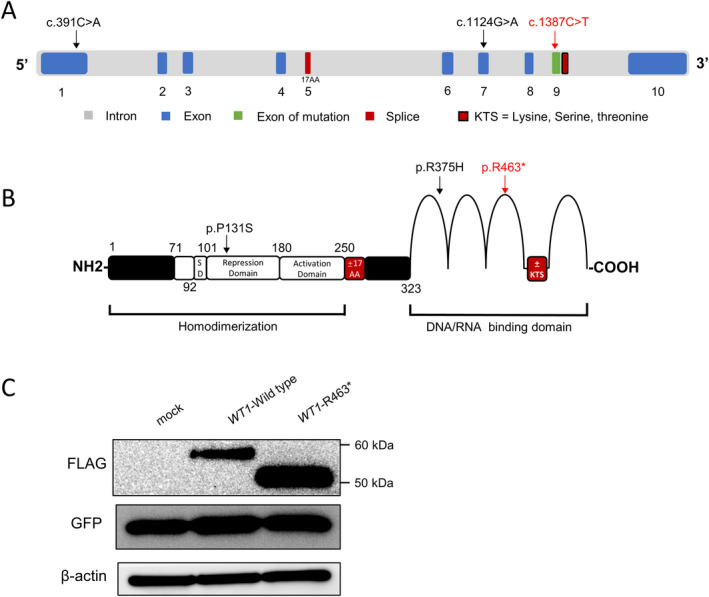
Schematic representation of the WT1 gene and protein. (a) WT1 is comprised of 10 exons. Two alternative splicing sites are indicated by red boxes. The black arrows indicate two mutations related to POI from previous report, while the red arrow indicates the variant we found. (b) Known functional domains of WT1 protein include the homodimerization domain and DNA/RNA‐binding domain (zinc finger domain). The inclusion of exon 5 leads to the insertion of 17 amino acid residues into the regulatory domain of WT1, which is indicated as “±17AA” in the red box. Four arcs represent the zinc finger domain. The alternative splicing at the end of exon 9 produced the tripeptide KTS, which is inserted between zinc fingers III and IV and indicated by a red box. The variant reported by us is marked by a red arrow, and two mutations from previous report are marked by black arrows. (c) A truncated WT1 protein with approximately 51 kDa caused by the WT1 variant. Western blotting analysis of the WT1 protein expression in HEK293T cells transfected with equal amounts of indicated WT1 constructs. GFP was used to evaluate the transfection efficiency and β‐actin was used as a loading control
4. DISCUSSION
WT1 is a vital factor in maintaining female gonad development (Kreidberg et al., 1993). To date, a few studies have focused on how WT1 functions in regulating gonad development and female fertility using genetically modified animals. Herein, we summarized the female reproductive phenotypes from representative mouse models carrying different Wt1 variants (Table 4). The targeted total deletion of the Wt1 gene produced mice displaying hermaphroditism or gonadal dysgenesis, while heterozygous loss induced similar but much milder gonadal developmental defects, irrespective of the strain. Furthermore, mice with different mutation types differ in manifestations. Some showed masculinization with normal fertility while others had POI‐like phenotypes, indicating that characterization of different WT1 variants is important in genetic analysis of females with ovarian dysfunction.
TABLE 4.
Representative studies of Wt1 genetically modified mice with ovarian defects
| Index | Strain | Type | Phenotype of ovary | Ref |
|---|---|---|---|---|
| 1 | C57BL/6 | Wt1−/− | Complete agenesis of the gonads. | Kreidberg et al. (1993) |
| Wt1+/− | Normal in gonad | |||
| 2 | C57BL/6×129/Sv | Wt1+/− | Smaller ovaries with fewer ova; normal appearance and maintenance of corpus lutea; no implanted embryos | Kreidberg et al. (1999) |
| 3 | 129S7/SvEvBrd×C57BL/6J | Wt1tm1Asc/tm1Asc | Germ cells are fewer and abnormally organized; Gonads of XY mice are ovarian‐like and cryptorchid | Hammes et al. (2001) |
| Wt1+/tm1Asc | Same as homozygous | |||
| 4 | 129S7/SvEvBrd×C57BL/6J | Wt1tm2Asc/tm2Asc | Streak gonad found in both XX and XY genotypes and obvious by E12.5; abnormal internal genital duct development | Hammes et al. (2001) |
| 5 | 129P2/OlaHsd×C57BL/6 | Wt1tm1Mlh/tm1Mlh | Agonadal (ovary absent in all E13.5 embryos) in embryos | Patek et al. (2008) |
| Wt1+/tm1Mlh | Infertile | |||
| 6 | B6/129 | Wt1+/R394W | Subfertile; ovulation rate significantly decreased; ovaries significantly smaller; total number of developing follicles significantly reduced | Gao et al. (2014) |
| 7 | NA | Wt1+/− | Ectopic development of 3β‐HSD‐positive steroidogenic cells; aberrant differentiation of somatic cells in Wt1‐deficient gonads; SF1 expression was dramatically upregulated in Wt1‐deficient XX gonads | Chen et al. (2017) |
| 8 | C57BL/6 | Wt1+/− | Loss of sex‐specific gene expression pattern; reduced proliferating cells in XX gonad/mesonephroi explants | Rudigier et al. (2017) |
| 9 | 129/SvEv×C57BL/6 | Wt1+/− | Aberrant ovary development; pre‐granulosa cells to steroidogenic cells transformation; delayed meiosis progression in germ cells; abnormal degeneration of wolffian duct in Wt1‐deficient female embryos | Cen et al. (2020) |
| 10 | NA | Wt1+/R495G | Normal and fully fertile | Eozenou et al. (2020) |
| Wt1R495G/R495G | Distinct signs of masculinization |
Genetic variation is one of the main causes of POI (Jiao et al., 2018; Persani et al., 2010; Veitia, 2020). In our study, we identified a de novo nonsense variant of the WT1 gene in a non‐syndromic POI patient through WES. Wang et al. have previously identified two novel missense mutations and four intronic variants of WT1 in 384 Chinese POI women (Wang et al., 2015). Mutations in WT1 can cause many different diseases including non‐syndromic POI and syndromic POI such as Denys–Drash syndrome (Wang et al., 2018), WAGR syndrome (Huynh et al., 2017), and Frasier syndrome (Barbaux et al., 1997; Klamt et al., 1998). For our proband, the heterozygous WT1 c.1387C>T variant caused non‐syndromic POI. As to the son, the renal cells might receive a second hit in its remaining functional copy of WT1, leading to the development of Wilms’ tumor (Cresswell et al., 2016).
WT1 c.1387C>T has been reported in several patients with Wilms’ tumor. As shown in Table S3, among all patients carrying WT1 c.1387C>T, 18 cases are male and the other 4 cases are female with highly variable presentations in clinic. Particularly, all female patients showed unilateral or bilateral Wilms’ tumor. Additionally, one woman showed ovarian dysgenesis and another had cysts in ovaries, indicating ovaries of 50% of the female patients were affected. By contrast, our proband did not exhibit any clinical features of Wilms’ tumor. And her POI symptoms were much milder compared to ovarian dysgenesis. So we reported an isolated POI patient carrying WT1 c.1387C>T for the first time, suggesting that this pathogenic variant may only affect the function of ovaries during reproductive aging process.
Variable clinical features were also found in the male patients. Among the 18 cases, 2 (No. 21, 22) were diagnosed as Denys–Drash syndrome, 1 (No. 23) was diagnosed as Frasier syndrome, and 3 (No. 18, 19, 23) showed disorders of sex development without developing tumor. And the remaining 12 cases exhibited unilateral/bilateral Wilms’ tumor with or without nephrotic phenotypes/urinary tract malformations. It was hypothesized that the tumor tissues probably suffered a second hit during the gamete or embryonic stage due to specific factors, resulting in homozygous mutations or 11p13 loss of heterozygosity in the tumor (Cardoso et al., 2013). As to our male patient, the son of the proband, he was diagnosed with Wilms’ tumor and urethral malformation at 7 years of age without other symptoms, which was similar to several reported patients (No. 1, 2, 8, 9). Additionally, the variable clinical characteristics of patients carrying the same WT1 variants might be related to gender, genetic background, environmental factors, and the mechanisms of WT1 mutation, etc. All these findings are important for genetic counseling in clinic. And construction of Wt1 knockout or knock‐in mice would be beneficial for determining the underlying pathogenic mechanism of WT1 variants in POI.
Abbas et al. have found that mutant WT1 mRNA transcripts that carry premature termination codons were sensitive to nonsense‐mediated RNA decay (NMD) in primary acute myeloid leukemia. According to the “50 bp rule”, WT1 c.1387C>T, which is 61 bp upstream of the last exon–exon junction, may be likely to escape from NMD (Abbas et al., 2010). However, western blotting results using HEK293T cells further demonstrated that the c.1387C>T (p.R463*) variant could produce a truncated protein of WT1. The role of the truncated WT1 protein in ovarian development is of great value to be addressed in future work. WT1 has been found to regulate apoptosis and proliferation of immature granulosa cells through regulation of the Wnt/β‐catenin signaling pathway (Y. Wang et al., 2019). These results would probably provide some insight into the subsequent specific functional assays to investigate the harmfulness of p.R463* altered WT1 protein.
Collectively, we report for the first time that a heterozygous c.1387C>T variant of WT1 was associated with non–syndromic POI and Wilms’ tumor in a Chinese family. All our findings provide novel insight into the molecular mechanism of WT1 and genetic counseling for women with POI.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
Xiaojin Zhang, Yanhua Wu, and Yingchen Wang designed the study. Yingchen Wang and Xiaojin Zhang performed clinical assessments. Qing Chen performed WES and data processing. Yingchen Wang, Xi Yang, Lingyue Shang, Yuncheng Pan, and Shuting Ren conducted Sanger sequencing, molecular cloning, and western blotting. Yingchen Wang, Xiaojin Zhang, Yanhua Wu, Qing Chen, Feng Zhang, Xi Yang, Zixue Zhou, Guoqing Li, Yunzheng Fang, and Li Jin analyzed the data. Yingchen Wang wrote the manuscript. Xiaojin Zhang and Yanhua Wu edited the manuscript. All authors confirmed the manuscript.
5. ETHICS STATEMENTS
The study was approved by the review boards of the Affiliated Obstetrics and Gynecology Hospital of Fudan University (Grant nos. 2017–19). Written informed consent was obtained from the patients and their families through interviews.
Supporting information
Table S1‐S3‐Fig S1
ACKNOWLEDGMENTS
The authors gratefully acknowledge the patients for participating and supporting this study.
Wang, Y. , Chen, Q. , Zhang, F. , Yang, X. , Shang, L. , Ren, S. , Pan, Y. , Zhou, Z. , Li, G. , Fang, Y. , Jin, L. , Wu, Y. , & Zhang, X. (2022). Whole exome sequencing identified a rare WT1 loss‐of‐function variant in a non‐syndromic POI patient. Molecular Genetics & Genomic Medicine, 10, e1820. 10.1002/mgg3.1820
Yingchen Wang and Qing Chen contributed equally to this work.
Funding information
This work was supported by Natural Science Foundation of Shanghai (20ZR1407000), National Key Research and Development Program of China (2017YFC1001100), and National Natural Science Foundation of China (31625015, 31521003, and 81373869).
Contributor Information
Yanhua Wu, Email: yanhuawu@fudan.edu.cn.
Xiaojin Zhang, Email: zxjgcp@sina.com.
REFERENCES
- Abbas, S. , Erpelinck‐Verschueren, C. A. , Goudswaard, C. S. , Lowenberg, B. , & Valk, P. J. (2010). Mutant Wilms’ tumor 1 (WT1) mRNA with premature termination codons in acute myeloid leukemia (AML) is sensitive to nonsense‐mediated RNA decay (NMD). Leukemia, 24(3), 660–663. 10.1038/leu.2009.265 [DOI] [PubMed] [Google Scholar]
- Barbaux, S. , Niaudet, P. , Gubler, M.‐C. , Grünfeld, J.‐P. , Jaubert, F. , Kuttenn, F. , Fékété, C. N. , Souleyreau‐Therville, N. , Thibaud, E. , Fellous, M. , & McElreavey, K. (1997). Donor splice‐site mutations in WT1 are responsible for Frasier syndrome. Nature Genetics, 17(4), 467–470. 10.1038/ng1297-467 [DOI] [PubMed] [Google Scholar]
- Bardeesy, N. , & Pelletier, J. (1998). Overlapping RNA and DNA binding domains of the wt1 tumor suppressor gene product. Nucleic Acids Research, 26(7), 1784–1792. 10.1093/nar/26.7.1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso, L. C. , De souza, K. R. , De o. reis, A. H. , Andrade, R. C. , Britto, A. C. , De lima, M. A. , Santos, A. C. D. , De faria, P. S. , Ferman, S. , Seuánez, H. N. , & Vargas, F. R. (2013). WT1, WTX and CTNNB1 mutation analysis in 43 patients with sporadic Wilms’ tumor. Oncology Reports, 29(1), 315–320. 10.3892/or.2012.2096 [DOI] [PubMed] [Google Scholar]
- Cen, C. , Chen, M. , Zhou, J. , Zhang, L. , Duo, S. , Jiang, L. , Hou, X. , & Gao, F. (2020). Inactivation of Wt1 causes pre‐granulosa cell to steroidogenic cell transformation and defect of ovary developmentdagger. Biology of Reproduction, 103(1), 60–69. 10.1093/biolre/ioaa042 [DOI] [PubMed] [Google Scholar]
- Chen, M. , Zhang, L. , Cui, X. , Lin, X. , Li, Y. , Wang, Y. , Wang, Y. , Qin, Y. , Chen, D. , Han, C. , Zhou, B. , Huff, V. , & Gao, F. (2017). Wt1 directs the lineage specification of sertoli and granulosa cells by repressing Sf1 expression. Development, 144(1), 44–53. 10.1242/dev.144105 [DOI] [PubMed] [Google Scholar]
- Cresswell, G. D. , Apps, J. R. , Chagtai, T. , Mifsud, B. , Bentley, C. C. , Maschietto, M. , Popov, S. D. , Weeks, M. E. , Olsen, Ø. E. , Sebire, N. J. , Pritchard‐Jones, K. , Luscombe, N. M. , Williams, R. D. , & Mifsud, W. (2016). Intra‐tumor genetic heterogeneity in Wilms tumor: Clonal evolution and clinical implications. EBioMedicine, 9, 120–129. 10.1016/j.ebiom.2016.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eozenou, C. , Gonen, N. , Touzon, M. S. , Jorgensen, A. , Yatsenko, S. A. , Fusee, L. , Kamel, A. K. , Gellen, B. , Guercio, G. , Singh, P. , Witchel, S. , Berman, A. J. , Mainpal, R. , Totonchi, M. , Meybodi, A. M. , Askari, M. , Merel‐Chali, T. , Bignon‐Topalovic, J. , Migale, R. , … Bashamboo, A. (2020). Testis formation in XX individuals resulting from novel pathogenic variants in Wilms’ tumor 1 (WT1) gene. Proceedings of the National Academy of Sciences of the United States of America, 117(24), 13680–13688. 10.1073/pnas.1921676117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- European Society for Human Reproduction and Embryology (ESHRE) Guideline Group on POI , Webber, L. , Davies, M. , Anderson, R. , Bartlett, J. , Braat, D. , Cartwright, B. , Cifkova, R. , de Muinck Keizer‐Schrama, S. , Hogervorst, E. , Janse, F. , Liao, L. , Vlaisavljevic, V. , Zillikens, C. , & Vermeulen, N. (2016). ESHRE guideline: Management of women with premature ovarian insufficiency. Human Reproduction, 31(5), 926–937. 10.1093/humrep/dew027 [DOI] [PubMed] [Google Scholar]
- Gao, F. , Maiti, S. , Sun, G. , Ordonez, N. G. , Udtha, M. , Deng, J. M. , Behringer, R. R. , & Huff, V. (2004). The Wt1+/R394W mouse displays glomerulosclerosis and early‐onset renal failure characteristic of human Denys‐Drash syndrome. Molecular and Cellular Biology, 24(22), 9899–9910. 10.1128/MCB.24.22.9899-9910.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, F. , Zhang, J. , Wang, X. , Yang, J. , Chen, D. , Huff, V. , & Liu, Y. X. (2014). Wt1 functions in ovarian follicle development by regulating granulosa cell differentiation. Human Molecular Genetics, 23(2), 333–341. 10.1093/hmg/ddt423 [DOI] [PubMed] [Google Scholar]
- Haber, D. A. , Buckler, A. J. , Glaser, T. , Call, K. M. , Pelletier, J. , Sohn, R. L. , Douglass, E. C. , & Housman, D. E. (1990). An internal deletion within an 11p13 zinc finger gene contributes to the development of Wilms’ tumor. Cell, 61(7), 1257–1269. 10.1016/0092-8674(90)90690-g [DOI] [PubMed] [Google Scholar]
- Hammes, A. , Guo, J.‐K. , Lutsch, G. , Leheste, J.‐R. , Landrock, D. , Ziegler, U. , Gubler, M.‐C. , & Schedl, A. (2001). Two splice variants of the Wilms’ tumor 1 gene have distinct functions during sex determination and nephron formation. Cell, 106(3), 319–329. 10.1016/s0092-8674(01)00453-6 [DOI] [PubMed] [Google Scholar]
- Huynh, M. T. , Boudry‐Labis, E. , Duban, B. , Andrieux, J. , Tran, C. T. , Tampere, H. , Ceraso, D. , Manouvrier, S. , Tachdjian, G. , Roche‐Lestienne, C. , & Vincent‐Delorme, C. (2017). WAGR syndrome and congenital hypothyroidism in a child with a Mosaic 11p13 deletion. American Journal of Medical Genetics. Part A, 173(6), 1690–1693. 10.1002/ajmg.a.38206 [DOI] [PubMed] [Google Scholar]
- Jiao, S. Y. , Yang, Y. H. , & Chen, S. R. (2020). Molecular genetics of infertility: loss‐of‐function mutations in humans and corresponding knockout/mutated mice. Human Reproduction Update, 27(1), 154–189. 10.1093/humupd/dmaa034 [DOI] [PubMed] [Google Scholar]
- Jiao, X. , Ke, H. , Qin, Y. , & Chen, Z. J. (2018). Molecular genetics of premature ovarian insufficiency. Trends in Endocrinology and Metabolism, 29(11), 795–807. 10.1016/j.tem.2018.07.002 [DOI] [PubMed] [Google Scholar]
- Kennedy, D. , Ramsdale, T. , Mattick, J. , & Little, M. (1996). An RNA recognition motif in Wilms’ tumour protein (WT1) revealed by structural modelling. Nature Genetics, 12(3), 329–331. 10.1038/ng0396-329 [DOI] [PubMed] [Google Scholar]
- Klamt, B. , Koziell, A. , Poulat, F. , Wieacker, P. , Scambler, P. , Berta, P. , & Gessler, M. (1998). Frasier syndrome is caused by defective alternative splicing of WT1 leading to an altered ratio of WT1 +/‐KTS splice isoforms. Human Molecular Genetics, 7(4), 709–714. 10.1093/hmg/7.4.709 [DOI] [PubMed] [Google Scholar]
- Kreidberg, J. A. , Natoli, T. A. , McGinnis, L. , Donovan, M. , Biggers, J. D. , & Amstutz, A. (1999). Coordinate action of Wt1 and a modifier gene supports embryonic survival in the oviduct. Molecular Reproduction and Development, 52(4), 366–375. [DOI] [PubMed] [Google Scholar]
- Kreidberg, J. A. , Sariola, H. , Loring, J. M. , Maeda, M. , Pelletier, J. , Housman, D. , & Jaenisch, R. (1993). WT‐1 is required for early kidney development. Cell, 74(4), 679–691. 10.1016/0092-8674(93)90515-r [DOI] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , Garimella, K. , Altshuler, D. , Gabriel, S. , Daly, M. , & DePristo, M. A. (2010). The Genome Analysis Toolkit: A MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Research, 20(9), 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffett, P. , Bruening, W. , Nakagama, H. , Bardeesy, N. , Housman, D. , Housman, D. E. , & Pelletier, J. (1995). Antagonism of WT1 activity by protein self‐association. Proceedings of the National Academy of Sciences of the United States of America, 92(24), 11105–11109. 10.1073/pnas.92.24.11105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parenti, R. , Salvatorelli, L. , Musumeci, G. , Parenti, C. , Giorlandino, A. , Motta, F. , & Magro, G. (2015). Wilms’ tumor 1 (WT1) protein expression in human developing tissues. Acta Histochemica, 117(4–5), 386–396. 10.1016/j.acthis.2015.03.009 [DOI] [PubMed] [Google Scholar]
- Park, M. , Choi, Y. , Choi, H. , & Roh, J. (2014). Wilms’ tumor suppressor gene (WT1) suppresses apoptosis by transcriptionally downregulating BAX expression in immature rat granulosa cells. J Ovarian Res, 7, 118. 10.1186/s13048-014-0118-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patek, C. E. , Brownstein, D. G. , Fleming, S. , Wroe, C. , Rose, L. , Webb, A. , Berry, R. L. , Devenney, P. S. , Walker, M. , Maddocks, O. D. K. , Lawrence, N. J. , Harrison, D. J. , Wood, K. M. , Miles, C. G. , & Hooper, M. L. (2008). Effects on kidney disease, fertility and development in mice inheriting a protein‐truncating Denys‐Drash syndrome allele (Wt1tmT396). Transgenic Research, 17(3), 459–475. 10.1007/s11248-007-9157-0 [DOI] [PubMed] [Google Scholar]
- Pelletier, J. , Bruening, W. , Li, F. P. , Haber, D. A. , Glaser, T. , & Housman, D. E. (1991). WT1 mutations contribute to abnormal genital system development and hereditary Wilms’ tumour. Nature, 353(6343), 431–434. 10.1038/353431a0 [DOI] [PubMed] [Google Scholar]
- Pelletier, J. , Schalling, M. , Buckler, A. J. , Rogers, A. , Haber, D. A. , & Housman, D. (1991). Expression of the Wilms’ tumor gene WT1 in the murine urogenital system. Genes & Development, 5(8), 1345–1356. 10.1101/gad.5.8.1345 [DOI] [PubMed] [Google Scholar]
- Persani, L. , Rossetti, R. , & Cacciatore, C. (2010). Genes involved in human premature ovarian failure. Journal of Molecular Endocrinology, 45(5), 257–279. 10.1677/JME-10-0070 [DOI] [PubMed] [Google Scholar]
- Persani, L. , Rossetti, R. , Cacciatore, C. , & Bonomi, M. (2009). Primary Ovarian Insufficiency: X chromosome defects and autoimmunity. Journal of Autoimmunity, 33(1), 35–41. 10.1016/j.jaut.2009.03.004 [DOI] [PubMed] [Google Scholar]
- Reddy, J. C. , Morris, J. C. , Wang, J. , English, M. A. , Haber, D. A. , Shi, Y. , & Licht, J. D. (1995). WT1‐mediated transcriptional activation is inhibited by dominant negative mutant proteins. Journal of Biological Chemistry, 270(18), 10878–10884. 10.1074/jbc.270.18.10878 [DOI] [PubMed] [Google Scholar]
- Rose, E. A. , Glaser, T. , Jones, C. , Smith, C. L. , Lewis, W. H. , Call, K. M. , Minden, M. , Champagne, E. , Bonetta, L. , Yeger, H. , & Housman, D. E. (1990). Complete physical map of the WAGR region of 11p13 localizes a candidate Wilms’ tumor gene. Cell, 60(3), 495–508. 10.1016/0092-8674(90)90600-j [DOI] [PubMed] [Google Scholar]
- Rossetti, R. , Ferrari, I. , Bonomi, M. , Persani, L. (2017). Genetics of primary ovarian insufficiency. Clinical Genetics, 91(2), 183–198. 10.1111/cge.12921 [DOI] [PubMed] [Google Scholar]
- Rudigier, L. J. , Dame, C. , Scholz, H. , & Kirschner, K. M. (2017). Ex vivo cultures combined with vivo‐morpholino induced gene knockdown provide a system to assess the role of WT1 and GATA4 during gonad differentiation. PLoS One, 12(4), e0176296. 10.1371/journal.pone.0176296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullmark, T. , Montano, G. , & Gullberg, U. (2018). DNA and RNA binding by the Wilms’ tumour gene 1 (WT1) protein +KTS and ‐KTS isoforms‐From initial observations to recent global genomic analyses. European Journal of Haematology, 100(3), 229–240. 10.1111/ejh.13010 [DOI] [PubMed] [Google Scholar]
- Veitia, R. A. (2020). Primary ovarian insufficiency, meiosis and DNA repair. Biomedical Journal, 43(2), 115–123. 10.1016/j.bj.2020.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, D. , Horton, J. R. , Zheng, Y. , Blumenthal, R. M. , Zhang, X. , & Cheng, X. (2018). Role for first zinc finger of WT1 in DNA sequence specificity: Denys‐Drash syndrome‐associated WT1 mutant in ZF1 enhances affinity for a subset of WT1 binding sites. Nucleic Acids Research, 46(8), 3864–3877. 10.1093/nar/gkx1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H. , Li, G. , Zhang, J. , Gao, F. , Li, W. , Qin, Y. , & Chen, Z. J. (2015). Novel WT1 missense mutations in Han Chinese women with premature ovarian failure. Scientific Reports, 5, 13983. 10.1038/srep13983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Meng, K. , Wang, Y. , Zhao, Y. , Lin, F. , Liu, X. , Zhang, Y. , & Quan, F. (2021). Wilms’ tumor (WT1) (+/‐KTS) variants decreases the progesterone secretion of bovine ovarian theca cells. Domestic Animal Endocrinology, 74, 106521. 10.1016/j.domaniend.2020.106521 [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Lu, E. , Bao, R. , Xu, P. , Feng, F. , Wen, W. , Dong, Q. , Hu, C. , Xiao, L. I. , Tang, M. , Li, G. , Wang, J. , & Zhang, C. (2019). Notch signalling regulates steroidogenesis in mouse ovarian granulosa cells. Reproduction, Fertility, and Development, 31(6), 1091–1103. 10.1071/RD18281 [DOI] [PubMed] [Google Scholar]
- Wang, Z. Y. , Qiu, Q. Q. , & Deuel, T. F. (1993). The Wilms’ tumor gene product WT1 activates or suppresses transcription through separate functional domains. Journal of Biological Chemistry, 268(13), 9172–9175. 10.1016/S0021-9258(18)98329-8 [DOI] [PubMed] [Google Scholar]
- Yang, X. I. , Zhang, X. , Jiao, J. , Zhang, F. , Pan, Y. , Wang, Q. , Chen, Q. , Cai, B. , Tang, S. , Zhou, Z. , Chen, S. , Yin, H. , Fu, W. , Luo, Y. , Li, D. A. , Li, G. , Shang, L. , Yang, J. , Jin, L. I. , … Wu, Y. (2019). Rare variants in FANCA induce premature ovarian insufficiency. Human Genetics, 138(11–12), 1227–1236. 10.1007/s00439-019-02059-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1‐S3‐Fig S1