

Abstract
Background
The NADH:ubiquinone oxidoreductase complex assembly factor gene (NDUFAF5) has been linked to the occurrence of Leigh syndrome, but few causative mutations have been identified. Here we report a rare case of Leigh syndrome in an infant who died in the early postnatal period.
Methods
We performed whole‐exome sequencing (WES) and mutation analysis of NDUFAF5 to obtain genetic data on the patient and describe the clinical and genetic characteristics.
Results
The proband was a 2‐month‐old male infant who suffered from recurrent vomiting and persistent seizure and died at 2 months of age after early medical support and treatment. His parents reported the unexplained death of the infant's older brother at 1 year of age. WES of the patient's DNA revealed c.357C>G and c.611C>T compound heterozygous mutations in NDUFAF5; analysis with the MutationTaster application indicated that both were pathogenic (p = 0.99). Significant structural changes in the transport domain of the protein were predicted using SWISS‐MODEL. We estimated the stability of the mutant protein using a mutation cutoff scanning matrix and found reductions in Gibbs free energy (−0.623 kcal/mol for p.D119E and −0.813 kcal/mol for p.A204V), indicating that the mutations led to an unstable protein structure. We speculated that the patient died as a result of impaired mitochondrial function caused by the NDUFAF5 mutations, and made a diagnosis of Leigh syndrome.
Conclusion
Our results demonstrate that molecular genetic screening is useful for the diagnosis of mitochondrial diseases, especially in children with a positive family history. Leigh syndrome should be considered in the diagnosis of patients presenting with severe recurrent vomiting and feeding difficulties with persistent seizure. Our findings expand the mutation spectrum of the NDUFAF5 gene and contribute to the genotype–phenotype map of mitochondrial respiratory chain complex I deficiency.
Keywords: case report, genomic sequence, Leigh syndrome, mitochondrial deficiency, NDUFAF5
Our results demonstrate that molecular genetic screening is useful for the diagnosis of mitochondrial diseases, especially in children with a positive family history. Leigh syndrome should be considered in the diagnosis of patients presenting with severe recurrent vomiting and feeding difficulties with persistent seizure. Our findings expand the mutation spectrum of the NDUFAF5 gene and contribute to the genotype‐phenotype map of mitochondrial respiratory chain complex I deficiency.

1. INTRODUCTION
Mitochondria are essential for maintaining normal cellular function and sustaining life, and account for more than 50% of the total weight in several cell types. Mitochondrial components are encoded by more than 100 nuclear (n)DNA and mitochondrial (mt)DNA genes that are involved in mitochondrial biogenesis and function. Mitochondria drive cell proliferation and differentiation, control reactive oxygen species accumulation and calcium (Ca2+) release and provide energy to the cell. Defects in mitochondria usually have severe developmental consequences and can lead to early postnatal mortality—for example, from disorders of energy, which affect at least 1 in 10,000 live births.
The mitochondrial respiratory chain (RC) consists of four membrane‐bound, multimeric RC complexes (RCCs) that catalyze the oxidation of reducing equivalents (nicotinamide adenine dinucleotide [NADH]). RCC I (NADH‐Q reductase or NADH dehydrogenase) is the first and largest multisubunit complex in the RC and oxidative phosphorylation system and is responsible for transferring electrons to coenzyme Q10 and pumping protons to maintain the electrochemical gradient across the mitochondrial inner membrane, providing proton power for ATP synthesis (Fassone & Rahman, 2012; Formosa et al., 2018). Mammalian RCC I is composed of 45 subunits, of which 7 are encoded by mtDNA and 38 by nDNA (Distelmaier et al., 2009). These are assembled into mature holoenzymes in a coordinated process. RCC I deficiency is usually associated with neurologic disorders that have variable clinical presentation and disease onset. Mutations in factors that participate in RCC I assembly can cause a reduction in complex activity, with pathogenic consequences (Carilla‐Latorre et al., 2013).
Leigh syndrome (MIM#256000) is a subacute necrotizing encephalopathy and heterogeneous disorder associated with mutations in mtDNA and nDNA encoding mitochondria‐related factors. Leigh syndrome manifests in infancy or early childhood as nonspecific symptoms that include recurrent vomiting, hypotonia, nystagmus, and muscle weakness. The syndrome progresses rapidly soon after birth with a fatal outcome, although several atypical late‐onset and delayed progression cases have been reported. RRC I deficiency is the most common molecular finding detected by genetic analyses in Leigh syndrome patients. Of the 15 putative assembly factors for RCC I, NADH:ubiquinone oxidoreductase complex assembly factor (NDUFAF)2, NDUFAF3, NDUFAF4, NDUFAF5, NDUFAF6, NDUFAF8, and FOXRED1 have all been implicated in Leigh syndrome, with NDUFAF5 being one of the most important. Seven mutations have been reported in the NDUFAF5 gene including c.155A>C, c.327G>C, c.477C>T, c.562C>T, c.719T>C, c.749G>T, and c.836T>G (Gerards et al., 2010; Simon et al., 2019; Sugiana et al., 2008; Tong et al., 2018).
Here we report a rare case of Leigh syndrome presenting with severe recurrent vomiting and convulsions leading to malnutrition, severe growth retardation, and delayed neurologic development, ultimately resulting in the early death of the infant. We identified the compound heterozygous mutations c.357C>G (p.D119E) and c.611C>T (p.A204V) in NDUFAF5 by whole‐exome sequencing (WES), and made the diagnosis based on the genetic findings and clinical symptoms.
2. METHODS
2.1. Ethical compliance
The study was approved by the ethics committee of the West China Second Hospital of Sichuan University (approval no. 2014–034). We obtained written, informed consent from the patient's parents prior to performing WES and for the inclusion of the patient's clinical and imaging details in publications.
2.2. DNA extraction and WES analysis
A peripheral blood sample was obtained from the patient in an EDTA anticoagulant blood sample tube that was stored at 4°C for less than 6 h. DNA was extracted using the Blood Genome Column Medium Extraction Kit (Tiangen Biotech, Beijing, China) according to the manufacturer's instructions. Protein‐coding exome enrichment was performed using the xGen Exome Research Panel v1.0, comprising 429,826 individually synthesized and quality‐controlled probes targeting 49.11 Mb of protein‐coding region (>23,000 genes) of the human genome. WES was performed using the NovaSeq 6000 platform (Illumina, San Diego, CA, USA), and the raw data were processed using FastP to remove adapters and filter low‐quality reads. Paired‐end reads were aligned to the Ensembl GRCh38/hg38 reference genome using the Burrows–Wheeler Aligner. Variant annotation was performed in accordance with database‐sourced minor allele frequencies (MAFs) and practical guidelines on pathogenicity issued by the American College of Medical Genetics. The annotation of MAFs was performed based on the 1000 Genomes, dbSNP, ESP, ExAC, and Chigene in‐house MAF database, Provean, Sift, Polypen2_hdiv, and Polypen2_hvar databases using R software (R Foundation for Statistical Computing, Vienna, Austria).
2.3. Mutation analysis of NDUFAF5
To elucidate the molecular architecture of the human NDUFAF5 gene, we used MutationTaster with R software to predict the pathogenicity of NDUFAF5 c.357C>G and c.611C>T and assess the impact of these mutations on protein structure. We performed comparative modeling using the SWISS‐MODEL (https://swissmodel.expasy.org/) with the 2p35.1.A template. We estimated the change in the free energy of the model using the mutation cutoff scanning matrix (mCSM) method (http://biosig.unimelb.edu.au/mcsm/) and Site Directed Mutator (SDM; http://marid.bioc.cam.ac.uk/sdm2). To assess the impact of the mutations on the stability of NDUFAF5, we used the DUET server (http://biosig.unimelb.edu.au/duet/) to integrate the results obtained with mCSM and SDM and thereby improve the overall prediction accuracy of the mutations under consideration. The signature vector that was ultimately generated was used to train the predictive classification and regression model for calculating the change in Gibbs folding free energy (ΔΔG) induced by the mutations.
3. CLINICAL DESCRIPTION AND MOLECULAR RESULTS
3.1. History of illness and physical examination
This proband was a 2‐month‐old male infant who had suffered from recurrent vomiting, severe choking, extreme feeding difficulties, obvious abdominal distension, and significant hypotonia since birth. He was born full term via cesarean section and had required rescue breathing at birth. His family history was unremarkable. The first symptoms of choking on milk and vomiting were observed starting on postnatal day 3. The patient was admitted to the pediatric hospital for treatment. Mechanical ventilation support with antibiotics was provided for 2 weeks, and the patient was discharged from the hospital after partial recovery from his initial symptoms.
Once at home, the patient presented feeding difficulties and his body weight decreased from 3.71 kg (+1 SD) at birth to 3.24 kg (−4 SD) at 47 days after birth. He also experienced several seizures (>5 times in the 10 days after leaving the hospital) accompanied by recurrent and progressively worsening vomiting. The patient was readmitted to our hospital. A physical examination revealed dysphagia, multiple deformities on the face, fingers, and heart‐suggesting a complex congenital heart disease such as ventricular or atrial septal defect as well as central hypothyroidism. Unfortunately, the patients demonstrated a severe neurological development delay. And the motor movement had been identified as impaired and it was difficult to complete limbs movement intensively. At the time of hospital administration, the baby demonstrated significant dystonia, presenting obviously hypertonia. However, as the disease progressed, the baby presented severer hypotonia occasionally, which led to the involuntary muscle contractions. The patient had severe growth retardation and his body weight was −4 SD; he also had a small head circumference (34.5 cm), poor skin elasticity, severe malnutrition with the absence of subcutaneous fat, widened cranial sutures, narrowed left eye cleft, small mandible, high palate arch, heart murmur, bulging abdomen, deformity of the palms on both sides, abnormal carpal and finger joints. The parents reported the unexplained death of the infant's older brother at the age of 1 year (Figure 1b).
FIGURE 1.
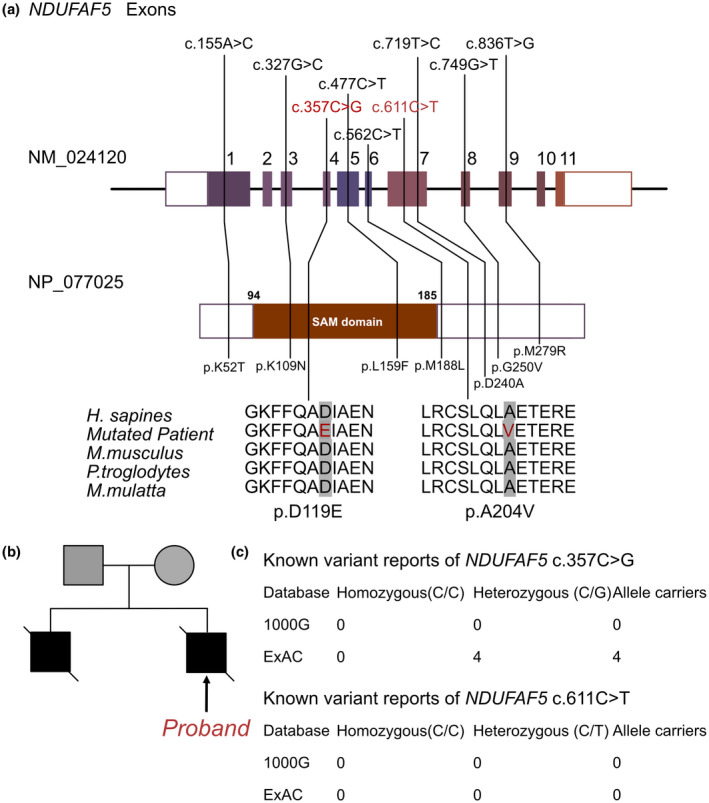
The information of NDUFAF5 mutations in this family. (a) Summary of current reports on the individuals of NDUFAF5. (b) Family pedigree reveals the maternal carrier of NDUFAF5 c.357C > G (p.D119E) and parental carrier of c.611C > T (p.A204V), and his elder brother demonstrated an early lethal without genomic sequencing. This proband presented infant vomiting and serious with compound heterozygous mutations of NDUFAF5 c.357C > G and c.611C > T. (c) The prevalence of NDUFAF5 mutations of c.357C > G and c.611C > T
3.2. Laboratory results
The infant's respiratory rate was elevated at 65 breaths/min, and pulse oximetry declined to 85% when nasal oxygen was administered. He responded poorly to external stimuli and was irritable. Scattered bilateral wet rales were evident in his lungs. Blood gas analysis revealed respiratory alkalosis combined with metabolic alkalosis (pH 7.718), along with electrolyte disturbance (in mmol/L: K+, 2.6; Na+, 132; Ca2+, 0.99; P, 1.26; Mg2+, 0.58). The minimum level of peripheral blood hemoglobin was 64 g/L. Blood biochemical examination showed decreased aspartate aminotransferase (83 U/L; normal value [n.v.] <43 U/L), total protein (34.4 g/L; n.v. >51 g/L), albumin (20.1 g/L; n.v. >38.g/L), and globulin (7.6 g/L; n.v. >23 g/L), while other parameters of kidney function and myocardial enzymes showed no obvious abnormalities. Thyroid function test results were as follows: triiodothyronine (T3), 0.62 nmol/L (n.v. >1.6 nmol/L); thyroxine (T4), 74.1 nmol/L (n.v. >93 nmol/L); thyroid‐stimulating hormone, 0.082 mIU/L (n.v. >1.7 mIU/L); free T3, 1.48 pmol/L (n.v. >4.3 pmol/L); and free T4, 11.49 pmol/L (n.v. >14 pmol/L). The decreased serum level of total 25‐OH vitamin D (22.1 ng/mL; n.v. 30–100 ng/mL) indicated vitamin D deficiency. Cellular immune function showed a reduction, as determined by the following lymphocyte subsets: cluster of differentiation (CD)3+, 2.11% (n.v. 2.3%–7.0%); CD3+CD4+, 1.47% (n.v. 1.7%–5.3%); and CD19+, 0.48% (n.v. 0.6%–1.9%). Escherichia coli was extracted in the bronchial secretions culture test. A series of tests ruled out infection with respiratory viruses including severe acute respiratory syndrome coronavirus 2, influenza A/B, adenoviruses, and respiratory syncytial virus as well as mycoplasma and chlamydia. Computed tomography (CT) scans revealed reduced light transmittance of both lung fields and extensive bilateral areas with diffuse infiltrates, indicating a parenchymal lung lesion. Additionally, severe intestinal edema was recorded by abdominal CT. Echocardiography revealed ventricular and atrial septal defects and patent ductus arteriosus with normal heart systolic and diastolic functions. Rectal pathologic biopsy revealed no ganglion cells in the muscle walls. The patient underwent a head magnetic resonance imaging (MRI) scan at 2 weeks after birth during his first hospitalization, which did not reveal any abnormalities in the basal ganglia. Because of the severity of his condition, the patient did not undergo further MRI scanning; therefore, we were unable to examine the brain for characteristic lesions.
3.3. Molecular results
As previously noted, the patient's family history included the death of a 1.5‐year‐old male sibling from an unknown cause. Therefore, a genetic disorder was strongly suspected. We performed WES using the Illumina NovaSeq 6000 platform and identified the c.357C>G and c.611C>T compound heterozygous mutations in the NDUFAF5 gene. The patient's mother and father were carriers of the c.357C>G and c.611C>T mutations, respectively. According to the American College of Medical Genetics, these variants have uncertain pathogenicity (PM2+ and PP3). Furthermore, c.611C>T has not been reported in any populations; this is the first report of this variant (Figure 1c). Meanwhile, c.357C>G has been reported in 4 heterozygous patients in the ExAC database. An analysis performed with MutationTaster revealed that both mutations are pathogenic due to amino acid sequence and splice site changes (p = 0.99 for both c.357C>G and c.611C>T). The PolyPhen score of 1.000 predicted that p.D119E (c.357C>G) (sensitivity = 0.85, specificity = 0.92) and p.A204V (c.611C>T) (sensitivity = 0.00, specificity = 1.00) are damaging mutations.
We used the SWISS‐MODEL to predict the variant's wild‐type and mutated protein crystal structures (Figure 2a). Significant structural changes were observed in the transport domain (Figure 2a,b). The DUET, mCSM, and SDM tools were used to predict protein stability; the results showed decreases in Gibbs free energy (mCSM for −0.623 kcal/mol for p.D119E and −0.813 kcal/mol for p.A204V), indicating an unstable protein structure associated with these mutations (Figure 2c,d).
FIGURE 2.
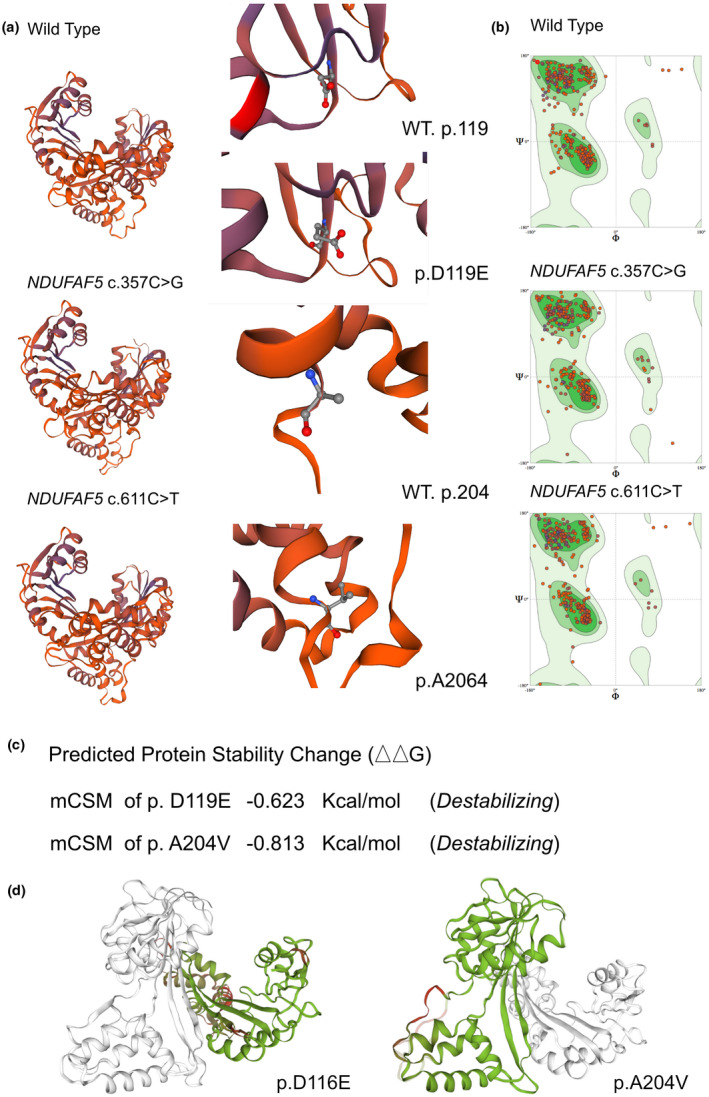
The impacts of NDUFAF5 c.357C > G and c.611C > T mutations on the molecular structure of protein. (a) SWISS‐MODEL to predict the variant's wild type and mutated protein crystal structure. And changes of the structure have been identified in its transporter part. (b) Ramachandran plots of NDUFAF5 with or without p.D119E and p.A204V mutations respectively. (c and d) mCSM tool used to predict protein stability, revealed a decline in Gibbs free energy, indicating an unstable protein structure associated with this mutation
3.4. Treatment and clinical outcome
Timely and continuous gastrointestinal decompression was performed. Attempts were made to adjust alkalosis, accompanied by intravenous nutrition and other supportive treatments. Mechanical ventilation and antibiotics were administrated. Sedatives and antiepileptic drugs were used to reduce the onset of convulsions. However, the patient's clinical symptoms worsened, with continuous vomiting and intestinal dysfunction and more frequent convulsions that progressed to a persistent state of seizure. The parents ultimately rejected any further medical treatment and the patient died shortly thereafter.
4. DISCUSSION AND CONCLUSION
Mitochondrial diseases are hereditary and can occur at any age and affect any organ or tissue (usually those with high energy requirements). They are characterized by mitochondrial dysfunction caused by mutations in mtDNA and nDNA genes and are associated with high morbidity and mortality (Gorman et al., 2016). The prevalence of all forms of childhood‐onset (<16 years of age) mitochondrial diseases is estimated to range from 5 to 15 cases per 100,000 individuals (Gorman et al., 2016). While in adults, the prevalence of mitochondrial diseases caused by mutations in mtDNA and nDNA is 9.6 and 2.9 cases per 100,000 individuals, respectively (Gorman et al., 2015).
The mitochondrial oxidative phosphorylation pathway is typically affected in mitochondrial diseases (Finsterer, 2020). The electron transport chain or RC consists of four protein complexes (I–IV) located in the inner mitochondrial membrane. RRC I, as the largest and most complex of these proteins, plays an important role in the development of mitochondrial diseases. RCC I in humans has 45 subunits that are assembled into the mature holoenzyme by assembly factors. Mutations in these factors or in the subunits can lead to mitochondrial diseases (Formosa et al., 2018). NDUFAF5, also known as C20orf7, is an assembly factor of RCC I that regulates the translation of the mitochondrial factor NADH‐ubiquinone oxidoreductase chain (ND)1 and its insertion into the membrane, or promotes ND1 assembly into an early membrane arm intermediate (McKenzie & Ryan, 2010; Nouws et al., 2012; Sugiana et al., 2008). NDUFAF5 mutations that affect the function of the highly conserved S‐adenosylmethionine‐dependent methyltransferase domain may contribute to Leigh syndrome (Gerards et al., 2010). They may also affect the translation of ND1 protein, thereby reducing the activity of RCC I and giving rise to clinical symptoms (Carilla‐Latorre et al., 2013). It was reported that NDUFAF5 has arginine hydroxylase activity and adds a hydroxyl group to Arg73 of the RCC I core subunit NDUFS7, demonstrating how the assembly factor plays an important role in regulating the early stages of RCC I formation (Formosa et al., 2018). The mitochondrial function is critical for cellular function. And impairment of mitochondria would lead to fetal lethal. RCC I contains 44 subunits, and there are many potential targets for pathogenic mutations, both on the nuclear and mitochondrial genomes. Generally, the neurological and cardiovascular are mostly rely on energy supplementary, indicating that these two systems should be mainly affected by RCC I deficiency. The defects of single subunit of RCC I might lead to cardiac hypertrophy and stroke in cardiovascular disease(Forte et al., 2019), and lead to bilateral striatal necrosis, progressive dystonia, optic atrophy, leukoencephalomyelopathy, and Leigh syndrome(Baide‐Mairena et al., 2019; Ortigoza‐Escobar et al., 2016; Scheffler, 2015). However, based on literature review, there is no significant difference among individual mutations in single subunit in RCC I. So that, such group of disorder could be affected by RCC I deficiency in total (Fiedorczuk & Sazanov, 2018).
Few cases of RCC I deficiency caused by mutations in NDUFAF5 have been described to date, and all the reported mutation sites had been listed in Figure 1a. Among the reported eight mutations, three of them were located in sterile alpha motif (SAM) domain. SAM domain had been proved to be involved in the development regulation. A series of genes had been identified to contain SAM domain. However, there was no significant difference in clinical manifestation of Leigh syndrome between NDUFAF5 mutations located in and out SAM domain. And we could not suspect the SAM domain mutations would lead to a more severe disease condition. Besides, considering the importance of SAM domain, we speculated that the mutation outside SAM domain would also alter the interaction of SAM domain and impairing the biological function of this protein. As NDUFAF5 is a structural and functional protein in mitochondrial RCC. The main function of this protein is to help produce ATP and proton, which the interaction of other domains to work as a regulator was not key biological function of NDUFAF5. So that, the clinical manifestation remains still when mutations are located inside or outside SAM domain. The first report was of a child from an Egyptian family who had intrauterine growth retardation and multiple malformations (heart, bones, etc.), and died from cardiac and respiratory arrest caused by progressive lactic acidosis on postnatal day 7; homozygous mapping revealed the missense mutation c.719T>C in exon 7 of NDUFAF5 (Sugiana et al., 2008). In another study of five affected children in two families (Saada et al., 2012). All of the patients showed developmental delay and regression and seizures, and two died at the ages of 5 years and 10 months. Mapping revealed that the homozygous mutation c.749G>T in exon 7 of NDUFAF5 resulted in the p.G250V substitution. An 8‐month‐old girl with recurrent seizures and difficulties in swallowing showed rapid disease progression and ultimately died of cardiovascular failure at the age of 2 years. WES showed a compound heterozygous c.836T>G (p.M279R) and c.145C>G (p.R49G) mutations in the NDUFAF5 gene (Tong et al., 2018). In another recently reported case, three patients were diagnosed with Leigh syndrome with gastroesophageal reflux and seizure attacks caused by NDUFAF5 mutations at c.327G>C and C.223‐907A>C (Simon et al., 2019). In one patient, the seizures persisted and he was intubated because of dyspnea at 8 months; his family decided not to continue aggressive care, and the patient was extubated and died shortly thereafter. WES revealed the potentially pathogenic variants c.155A>C, c.327G>C, and C.836T>G in exon 3 of NDUFAF5. In our patient, feeding difficulties, recurrent severe vomiting, facial abnormalities, multiple deformities, seizures, protein–energy malnutrition, delayed psychomotor development, and increased blood lactic acid was observed and the patient died in early infancy, similar to previously reported cases. We made a diagnosis of Leigh syndrome based on the presence of the heterozygous NDUFAF5 mutations c.357C>G (p.D119E) and c.611C>T (p.A204V) detected by WES. Although there is no clear evidence that these mutations are pathogenic, we speculate that they alter NDUFAF5 protein structure, which in turn affects the assembly and activity of RCC I, resulting in impaired electron transport, mitochondrial dysfunction, and Leigh syndrome.
RCC I deficiency leads to severe functional decline in multiple systems and has a poor prognosis with no cure. The main treatment strategy is symptom relief. This can improve quality of life and extend life expectancy, but cannot cure or even delay the progression of the disease (Nouws et al., 2012). The 2015 Mitochondrial Medicine Association's consensus statement on the diagnosis and treatment of mitochondrial diseases recommends coenzyme Q10 (ubiquinone) for patients with mitochondrial diseases along with α‐lipoic acid and riboflavin; for patients with central nervous system symptoms, folic acid should be considered; and l‐carnitine should be administered in the case of deficiency (Parikh et al., 2015). Idebenone was recently approved for the treatment of adolescents and adults with Leber's hereditary optic neuropathy. Nonetheless, managing mitochondrial diseases—especially Leigh syndrome‐remains clinically challenging. Gene therapy is an alternative treatment that combines an adeno‐associated virus delivery system with clustered regularly interspaced short palindromic repeats (CRISPR)‐CRISPR‐associated protein (Cas)9/13 or base editing techniques for isogenetic correction.
In conclusion, there are few known cases of Leigh syndrome, a mitochondrial disease caused by NDUFAF5 mutation. Whether in infants or children, when multiple seemingly unrelated organs are clinically affected mitochondrial disease should be considered as a possible diagnosis. Leigh syndrome should be suspected in patients who exhibit severe recurrent vomiting and feeding difficulties with persistent seizure. Molecular genetic screening is helpful for the diagnosis of mitochondrial diseases, especially in children with a positive family history. Our report expands the mutation spectrum of the NDUFAF5 gene and contributes to the genotype–phenotype map of mitochondrial RCC I deficiency.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
ETHICAL APPROVAL STATEMENT
This study was approved by the Ethics Committee of West China Second Hospital of Sichuan University (2014–034). And informed consent from the patient's parents prior to conducting the WES had been obtained, including the patient's clinical and imaging details in the manuscript for the purpose of publication.
ACKNOWLEDGMENT
This work was supported by grants from the National Natural Science Foundation of China (No. 81700360), Technology Project of Sichuan Province of China (2020YFS0102).
Wen, Y. , Lu, G. , Qiao, L. , & Li, Y. (2022). A Leigh syndrome caused by compound heterozygous mutations on NDUFAF5 induce early infant death: A case report. Molecular Genetics & Genomic Medicine, 10, e1852. 10.1002/mgg3.1852
Yan Wen and Guoyan Lu contributed equally to this article.
Contributor Information
Lina Qiao, Email: iaqiao@163.com.
Yifei Li, Email: liyfwcsh@scu.edu.cn.
DATA AVAILABILITY STATEMENT
Data sets used in this study are available from the corresponding author upon reasonable request.
REFERENCES
- Baide‐Mairena, H. , Gaudó, P. , Marti‐Sánchez, L. , Emperador, S. , Sánchez‐Montanez, A. , Alonso‐Luengo, O. , Correa, M. , Grau, A. M. , Ortigoza‐Escobar, J. D. , Artuch, R. , Vázquez, E. , Del Toro, M. , Garrido‐Pérez, N. , Ruiz‐Pesini, E. , Montoya, J. , Bayona‐Bafaluy, M. P. , & Pérez‐Dueñas, B. (2019). Mutations in the mitochondrial complex I assembly factor NDUFAF6 cause isolated bilateral striatal necrosis and progressive dystonia in childhood. Molecular Genetics and Metabolism, 126(3), 250–258. 10.1016/j.ymgme.2019.01.001 [DOI] [PubMed] [Google Scholar]
- Carilla‐Latorre, S. , Annesley, S. J. , Munoz‐Braceras, S. , Fisher, P. R. , & Escalante, R. (2013). Ndufaf5 deficiency in the Dictyostelium model: new roles in autophagy and development. Molecular Biology of the Cell, 24(10), 1519–1528. 10.1091/mbc.E12-11-0796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Distelmaier, F. , Koopman, W. J. , van den Heuvel, L. P. , Rodenburg, R. J. , Mayatepek, E. , Willems, P. H. , & Smeitink, J. A. (2009). Mitochondrial complex I deficiency: from organelle dysfunction to clinical disease. Brain, 132(Pt 4), 833–842. 10.1093/brain/awp058 [DOI] [PubMed] [Google Scholar]
- Fassone, E. , & Rahman, S. (2012). Complex I deficiency: Clinical features, biochemistry and molecular genetics. Journal of Medical Genetics, 49(9), 578–590. 10.1136/jmedgenet-2012-101159 [DOI] [PubMed] [Google Scholar]
- Fiedorczuk, K. , & Sazanov, L. A. (2018). Mammalian mitochondrial complex I Structure And Disease‐Causing Mutations. Trends in Cell Biology, 28(10), 835–867. 10.1016/j.tcb.2018.06.006 [DOI] [PubMed] [Google Scholar]
- Finsterer, J. (2020). Secondary manifestations of mitochondrial disorders. Journal of Zhejiang University Science B, 21(7), 590–592. 10.1631/jzus.B2000010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formosa, L. E. , Dibley, M. G. , Stroud, D. A. , & Ryan, M. T. (2018). Building a complex complex: Assembly of mitochondrial respiratory chain complex I. Seminars in Cell & Developmental Biology, 76, 154–162. 10.1016/j.semcdb.2017.08.011 [DOI] [PubMed] [Google Scholar]
- Forte, M. , Palmerio, S. , Bianchi, F. , Volpe, M. , & Rubattu, S. (2019). Mitochondrial complex I deficiency and cardiovascular diseases: Current evidence and future directions. Journal of Molecular Medicine (Berlin), 97(5), 579–591. 10.1007/s00109-019-01771-3 [DOI] [PubMed] [Google Scholar]
- Gerards, M. , Sluiter, W. , van den Bosch, B. J. C. , de Wit, L. E. A. , Calis, C. M. H. , Frentzen, M. , Akbari, H. , Schoonderwoerd, K. , Scholte, H. R. , Jongbloed, R. J. , Hendrickx, A. T. M. , de Coo, I. F. M. , & Smeets, H. J. M. (2010). Defective complex I assembly due to C20orf7 mutations as a new cause of Leigh syndrome. Journal of Medical Genetics, 47(8), 507–512. 10.1136/jmg.2009.067553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman, G. S. , Chinnery, P. F. , DiMauro, S. , Hirano, M. , Koga, Y. , McFarland, R. , Suomalainen, A. , Thorburn, D. R. , Zeviani, M. , & Turnbull, D. M. (2016). Mitochondrial diseases. Nature Reviews Disease Primers, 2, 16080. 10.1038/nrdp.2016.80 [DOI] [PubMed] [Google Scholar]
- Gorman, G. S. , Schaefer, A. M. , Ng, Y. , Gomez, N. , Blakely, E. L. , Alston, C. L. , & McFarland, R. (2015). Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Annals of Neurology, 77(5), 753–759. 10.1002/ana.24362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie, M. , & Ryan, M. T. (2010). Assembly factors of human mitochondrial complex I and their defects in disease. IUBMB Life, 62(7), 497–502. 10.1002/iub.335 [DOI] [PubMed] [Google Scholar]
- Nouws, J. , Nijtmans, L. G. , Smeitink, J. A. , & Vogel, R. O. (2012). Assembly factors as a new class of disease genes for mitochondrial complex I deficiency: Cause, pathology and treatment options. Brain, 135(Pt 1), 12–22. 10.1093/brain/awr261 [DOI] [PubMed] [Google Scholar]
- Ortigoza‐Escobar, J. D. , Oyarzabal, A. , Montero, R. , Artuch, R. , Jou, C. , Jiménez, C. , Gort, L. , Briones, P. , Muchart, J. , López‐Gallardo, E. , Emperador, S. , Pesini, E. R. , Montoya, J. , Pérez, B. , Rodríguez‐Pombo, P. , & Pérez‐Dueñas, B. (2016). Ndufs4 related Leigh syndrome: A case report and review of the literature. Mitochondrion, 28, 73–78. 10.1016/j.mito.2016.04.001 [DOI] [PubMed] [Google Scholar]
- Parikh, S. , Goldstein, A. , Koenig, M. K. , Scaglia, F. , Enns, G. M. , Saneto, R. , Anselm, I. , Cohen, B. H. , Falk, M. J. , Greene, C. , Gropman, A. L. , Haas, R. , Hirano, M. , Morgan, P. , Sims, K. , Tarnopolsky, M. , Van Hove, J. L. K. , Wolfe, L. , & DiMauro, S. (2015). Diagnosis and management of mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Genetics in Medicine, 17(9), 689–701. 10.1038/gim.2014.177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saada, A. , Edvardson, S. , Shaag, A. , Chung, W. K. , Segel, R. , Miller, C. , Jalas, C. , & Elpeleg, O. (2012). Combined OXPHOS complex I and IV defect, due to mutated complex I assembly factor C20ORF7. Journal of Inherited Metabolic Disease, 35(1), 125–131. 10.1007/s10545-011-9348-y [DOI] [PubMed] [Google Scholar]
- Scheffler, I. E. (2015). Mitochondrial disease associated with complex I (NADH‐CoQ oxidoreductase) deficiency. Journal of Inherited Metabolic Disease, 38(3), 405–415. 10.1007/s10545-014-9768-6 [DOI] [PubMed] [Google Scholar]
- Simon, M. T. , Eftekharian, S. S. , Stover, A. E. , Osborne, A. F. , Braffman, B. H. , Chang, R. C. , Wang, R. Y. , Steenari, M. R. , Tang, S. , Hwu, P.‐L. , Taft, R. J. , Benke, P. J. , & Abdenur, J. E. (2019). Novel mutations in the mitochondrial complex I assembly gene NDUFAF5 reveal heterogeneous phenotypes. Molecular Genetics and Metabolism, 126(1), 53–63. 10.1016/j.ymgme.2018.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiana, C. , Pagliarini, D. J. , McKenzie, M. , Kirby, D. M. , Salemi, R. , Abu‐Amero, K. K. , Dahl, H.‐H. , Hutchison, W. M. , Vascotto, K. A. , Smith, S. M. , Newbold, R. F. , Christodoulou, J. , Calvo, S. , Mootha, V. K. , Ryan, M. T. , & Thorburn, D. R. (2008). Mutation of C20orf7 disrupts complex I assembly and causes lethal neonatal mitochondrial disease. American Journal of Human Genetics, 83(4), 468–478. 10.1016/j.ajhg.2008.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong, W. , Wang, Y. , Lu, Y. , Ye, T. , Song, C. , Xu, Y. , Li, M. , Ding, J. , Duan, Y. , Zhang, L. E. , Gu, W. , Zhao, X. , Yang, X.‐A. , & Jin, D. (2018). Whole‐exome sequencing helps the diagnosis and treatment in children with neurodevelopmental delay accompanied unexplained dyspnea. Scientific Reports, 8(1), 5214. 10.1038/s41598-018-23503-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sets used in this study are available from the corresponding author upon reasonable request.