

Abstract
Background
CHILD syndrome is an X‐linked dominant disorder associated with pathogenic mutations in the NSDHL gene. The condition is predominantly found in females as it is lethal in males. Most cases present at birth with extensive unilateral ichthyosiform erythroderma involving the trunk and limbs. Milder and less extensive presentations have been reported, leading to misdiagnosis especially during early childhood.
Methods and Results
We report an adult female of Malay ancestry who presented with minimal skin and limb involvement. She was only diagnosed in adulthood when she presented with gastrointestinal symptoms and worsening of skin manifestations. The clinical diagnosis was suspected after a combination of clinical, pathological and immunohistochemistry correlation, and molecularly confirmed with the discovery of a frameshift variant in NSDHL. The novel variant was inherited from her mother who had some linear hypopigmented patches over the medial aspects of both her arms and right forearm.
Conclusion
We uncovered a novel frameshift variant associated with presentations that cast a new light on the clinical features of CHILD syndrome.
Keywords: CHILD syndrome, ichthyosis, NSDHL, xanthoma, X‐linked dominant disorder
A novel NSDHL frameshift variant in a patient with subtle skin features and almost no limb defects, with gastrointestinal symptoms and presence of multiple whitish plaques in the proximal duodenum, stomach and distal oesophagus.
1. INTRODUCTION
Congenital hemidysplasia with ichthyosiform erythroderma and limb defects (CHILD) syndrome (OMIM# 308050) is an ichthyosiform disorder associated with pathogenic mutations in the NAD(P)‐dependent steroid dehydrogenase‐like protein (NSDHL) gene (OMIM 300275). The almost 39 kb gene is located near the terminal of the q arm of the X chromosome and encodes an enzyme involved in cholesterol biosynthesis. As having a germline mutation is lethal for male foetuses, the condition is almost exclusively found in females.
Most patients with CHILD syndrome present at birth with extensive ichthyosiform erythroderma involving the trunk and limbs on one side of the body, either diffuse or follow lines of Blaschko or both, with a sharp midline demarcation. Yellowish‐waxy scales, ptychotropism (affinity for body folds) and the occurrence of verruciform xanthomas are additional distinguishing features (Happle et al., 1995). Ipsilateral skeletal abnormalities ranging from hypoplasia of some fingers to the complete absence of limbs on the same side are often also observed. Other organs below the areas of ichthyosis may also be affected (Ramphul et al., 2021). Milder and less extensive presentations of the condition may lead to misdiagnosis, especially during early childhood (Ormerod et al., 2019).
We report an adult female with features of CHILD syndrome but had minimal skin and limb involvement. She was only diagnosed in adulthood when she presented with gastrointestinal symptoms and worsening of skin manifestations. The clinical diagnosis was suspected after histopathological correlation and molecularly confirmed with the identification of a pathogenic variant in the NSDHL gene via next‐generation sequencing.
2. CLINICAL DESCRIPTION
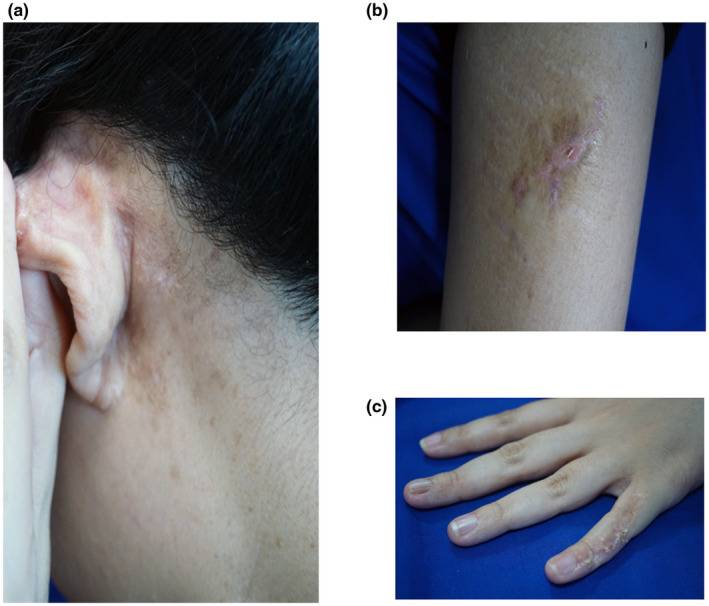
The patient of Malay ancestry presented at the age of 23 years to the dermatology department with an itchy and erosive rash affecting her left retro‐auricular area over a 2‐year period. On examination, there were multiple hyperpigmented, crusted and hyperkeratotic papules (Figure 1a). Dermoscopy of these lesions showed a brown reticulate pattern with yellowish scales (Figure 1b). In addition, there were yellow‐brown, scaly, hyperkeratotic plaques on her anterior left thigh and dorsum of her left little finger (Figure 1c,d) in a linear configuration, as well as a linear hypopigmented streak on the inner aspect of her left arm and left axilla (Figure 1e) and a depigmented macule on her lower back. She had previously been diagnosed with incontinentia pigmenti during infancy by physicians from a tertiary institution elsewhere and her parents provided a history that she was born with blisters affecting the aforementioned areas. These blisters subsequently resolved, resulting in the hyperkeratotic plaques and hypopigmented patches. Her mother's skin had similar linear hypopigmented patches over the medial aspects of both arms and the right forearm.
FIGURE 1.
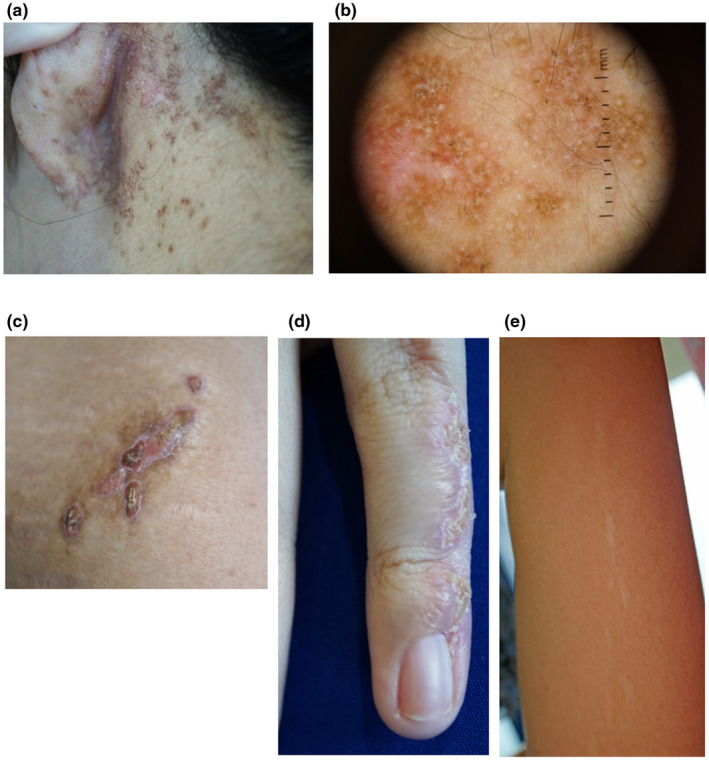
Images from the patient's skin lesions in various regions: (a) Multiple hyperpigmented, crusted and hyperkeratotic papules over the left retro‐auricular area. (b) Dermoscopic image of the papules over the left retro‐auricular area. (c) Yellow‐brown, scaly, hyperkeratotic, linear plaque on the anterior left thigh. (d) Yellow‐brown, scaly, hyperkeratotic linear plaque on the left little finger. (e) Linear hypopigmented patch on the inner aspect of the left arm
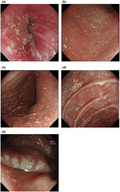
Biopsies of the plaques on the patient's left retro‐auricular region and thigh showed hyperkeratosis, columns of parakeratosis, acanthosis of the epidermis and multiple xanthoma cells within the papillary dermis (Figure 2a–d). At the same time, she presented to the gastroenterologist with complaints of recurrent dyspepsia and heartburn. An esophagogastroduodenoscopy revealed multiple whitish plaques in the proximal duodenum, stomach and distal oesophagus (Figure 3a–e). Biopsies of these areas showed similar xanthoma cells with finely vacuolated cytoplasm in the lamina propria (Figure 4a–c). These cells were positive for CD163 stain (Figure 4d), confirming their macrophage lineage and the diagnosis of gastrointestinal xanthomatosis.
FIGURE 2.
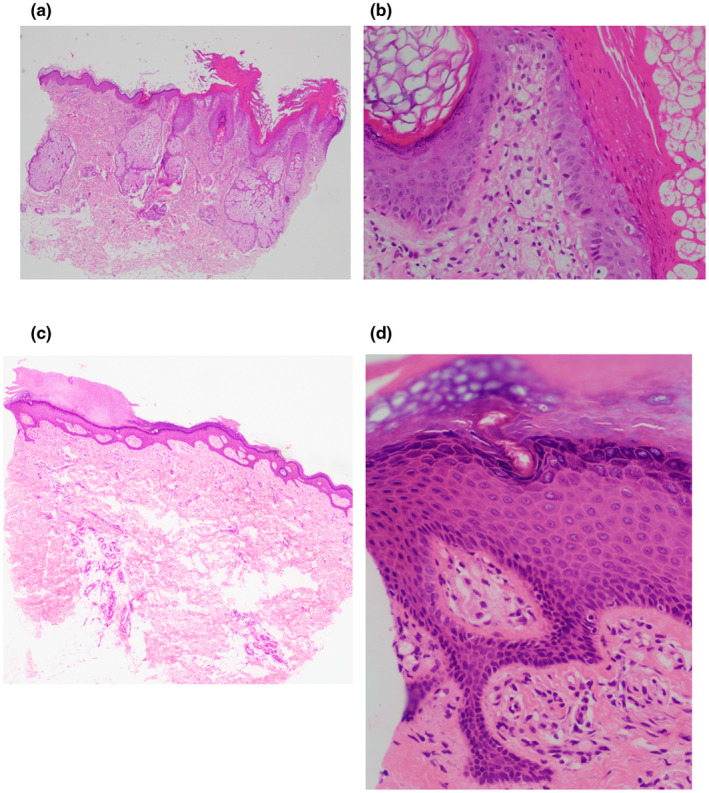
Images from biopsy samples of the patient's skin from various regions stained with hematoxylin and eosin at 400× magnification: (a) From the retro‐auricular area showing hyperkeratosis, columns of parakeratosis, acanthosis of the epidermis and multiple xanthoma cells within the papillary dermis. (b) From the retro‐auricular area showing multiple xanthoma cells within the papillary dermis. (c) From the thigh showing hyperkeratosis and acanthosis of the epidermis. (d) From the thigh showing hyperkeratosis, acanthosis of the epidermis and multiple xanthoma cells within the papillary dermis
FIGURE 3.
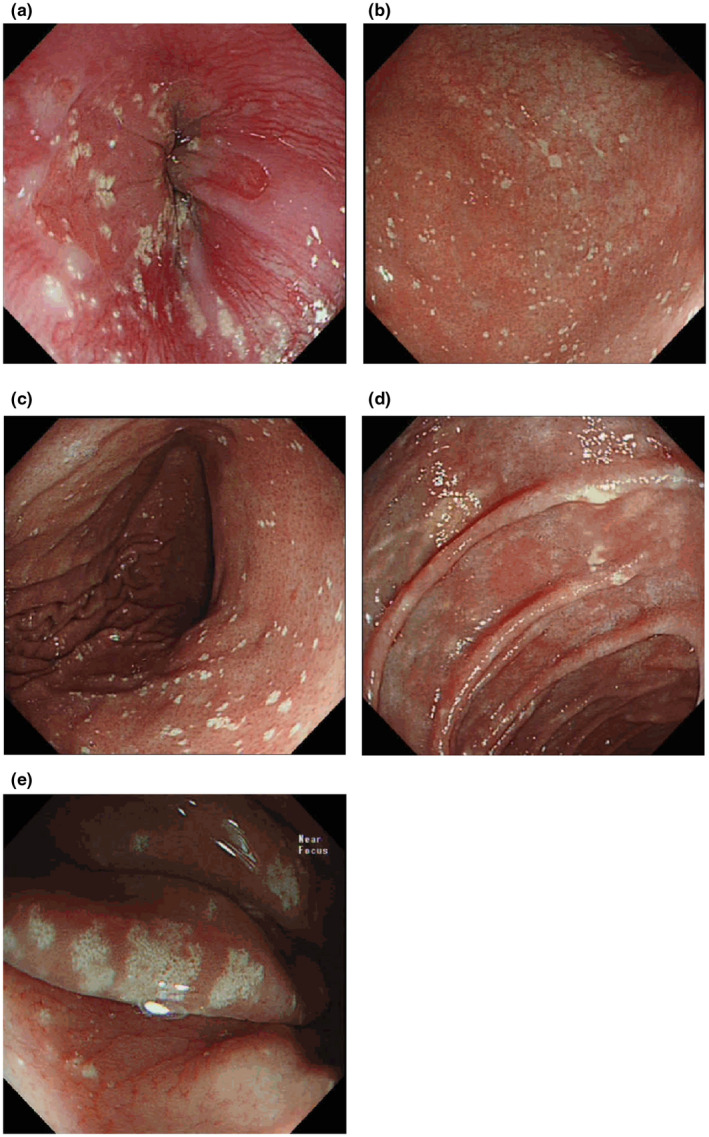
Images from the patient's upper gastrointestinal tract endoscopy showing the presence of multiple whitish plaque‐like lesions in various regions: (a) Z‐line; (b) antrum; (c) corpus; (d) duodenum; (e) Near focus magnified view of the gastric xanthelasma at the gastric corpus greater curve
FIGURE 4.
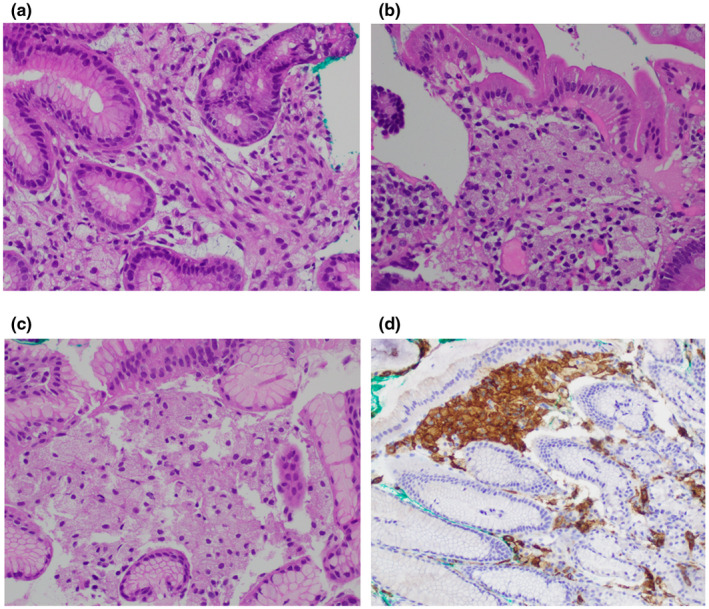
Haematoxylin and eosin staining of gut sections at 400× magnification: (a) Gastric antrum showing multiple xanthoma cells within the lamina propria. (b) Duodenum showing multiple xanthoma cells within the lamina propria. (c) Z‐line mucosa showing multiple xanthoma cells within the lamina propria. (d) CD163 (200×) staining of a section from Z‐line mucosa showing multiple xanthoma cells in the lamina propria
After clinical‐pathological correlation, a diagnosis of CHILD syndrome was suspected, and this was confirmed on molecular testing. The patient was commenced on a compounded combination cream containing 1% simvastatin and 2% cholesterol, with a good response after 3 months of treatment (Figure 5a–c). Her gastrointestinal symptoms improved on treatment with a proton pump inhibitor and antacids.
FIGURE 5.
Images from the patient's skin after 3 months of treatment with compounded combination cream containing 1% simvastatin and 2% cholesterol over the (a) left retro‐auricular area, (b) anterior left thigh and (c) left little finger
3. MOLECULAR ANALYSIS
Genetic investigation under the research setting was obtained from the SingHealth Centralised Institutional Review Board which has oversight of all research activities in the hospital. Punch biopsy specimens were obtained from the lesional skin of the left retro‐auricular region and left thigh after written informed consent. Venous blood was collected from the patient and both her parents. DNA was extracted using DNeasy Blood and Tissue kit (Qiagen Inc., USA) for skin tissues and PureGene Blood Kit (Qiagen Inc., USA) for venous blood. Targeted panel sequencing using Illumina's TruSight One kit with the DNA extracted from the thigh tissue uncovered a duplication of a G nucleotide in exon 3 of the NSDHL gene which would lead to a frameshift starting from an amino acid change in the next codon (NM_015922.3: c.131dupG: p. Gly45Trpfs*24). The variant was confirmed by targeted Sanger sequencing for DNA from both skin tissues and blood. Testing of parental DNA indicated maternal inheritance, with her father testing negative for the variant.
The insertion of a duplicated G nucleotide results in a frameshift that leads to premature termination 24 codons downstream of the duplication site. This variant has not been previously reported in any patient or population databases. However, a de novo single nucleotide substitution leading to a missense in the same codon (c.130G>A:p. Gly44Ser) has been reported in a Filipino girl with CHILD syndrome. It was classified as “likely pathogenic” by the investigators (Maceda et al., 2020).
4. DISCUSSION
We report a rare case of CHILD syndrome diagnosed only in adulthood, and only after a combination of clinical, pathological, and molecular correlation and confirmation. Her skin lesions were subtle and present from birth, becoming more prominent only in adulthood. A diagnosis of CHILD syndrome was not considered initially with these early clinical features. Instead, she was diagnosed with incontinentia pigmenti. On current presentation, the main differential diagnosis of her skin lesions was mosaic Darier's disease. However, no variants were identified for both ATP2A2 and IKBKG, excluding Darier's disease and incontinentia pigmenti. Our case illustrates that the clinical spectrum of CHILD syndrome can include patients with subtle skin features and almost no limb defects, necessitating the requirement for skin biopsies to confirm the presence of multiple xanthoma cells in the dermis associated with other classical features of hyperkeratosis, orthokeratosis or epidermal acanthosis (Gantner et al., 2014).
Our patient also presented with gastrointestinal symptoms which are rare in cases of CHILD syndrome but may also provide clues to the diagnosis, with the presence of similar xanthoma cells in biopsies of the stomach and duodenum (Ryan et al., 2013). This extensive distribution of xanthomas in the gastrointestinal tract is postulated to be reactive secondary to mucosal inflammation, damage or trauma, and may be related to deposition of toxic lipid metabolites due to mutations in the NSDHL gene (Mavilia & Wu, 2018). Xanthomas in the gastrointestinal tract have also been reported in association with carcinoma, lymphoma and cytomegalovirus colitis. Our case adds CHILD syndrome to the differential diagnoses of multiple xanthomas in the gastrointestinal tract.
Ever since mutations in the NSDHL gene were identified to be the underlying cause of CHILD syndrome (König et al., 2000), no other gene has been linked to this disorder. The other NSDHL‐associated disorder is CK syndrome (OMIM# 300831) which is characterized by variable cognitive impairment, seizures, microcephaly, cerebral cortical malformations and dysmorphic facial features (McLarren et al., 2010; du Souich et al., 2008). CK syndrome is an X‐linked recessive disorder affecting mostly males. In contrast, CHILD syndrome affects females almost exclusively as it is X‐linked dominant. Only two cases of affected males have been reported (Happle et al., 1996; Zellweger & Uehlinger, 1948), and they are most likely due to postzygotic and not germline mutations. Another related disorder, X‐linked chondrodysplasia punctata 2 (CDPX2, OMIM# 302960) can exhibit clinical features similar to CHILD syndrome and has been attributed to mutations in the EBP (Emopamil‐binding protein) gene, which encodes a protein that functions downstream of NSDHL in a common biosynthetic pathway (Herman, 2003). In addition to ichthyosiform lesions distributed in a linear or whorled pattern and shortening of bones of the upper arms and thighs, these patients also exhibit radiological features of stippling near the ends of bones and in cartilage.
The NSDHL gene codes for 3β‐hydroxysteroid dehydrogenase, an enzyme involved in the biosynthesis of cholesterol. As cholesterol is one of the key components of the lamellar structures found in the stratum corneum, its insufficiency can result in the disruption of these lamellar structures (Feingold & Elias, 2014). Accumulation of sterol intermediates may also result in abnormal Hedgehog signalling pathways, giving rise to skeletal deformities as seen in CHILD syndrome patients (Cooper et al., 2003). The success of the use of combination topicals containing cholesterol and statins (e.g. 2% lovastatin / 2% cholesterol) to treat the cutaneous lesions in CHILD syndrome imply that the skin lesions are not only due to a deficiency of cholesterol alone but to accumulation of toxic metabolites (Christiansen et al., 2015; Paller et al., 2011; Sandoval et al., 2019). The improvement seen in our patient adds further evidence of the efficacy of this combination in the treatment of cutaneous lesions of CHILD syndrome.
To date, more than 30 different pathogenic variants (including insertions and deletions) spanning the NSDHL gene have been identified in patients with CHILD syndrome. Most are single nucleotide substitutions of which there are 13 missense, seven nonsense, two splice sites and one substitution in the 3’ untranslated region documented in the Human Gene Mutation Database. Multi‐exon deletions and a small insertion have also been described. The majority of patients in the reports were of European descent, with a smaller proportion of East Asians (Chinese and Japanese), and solitary cases from elsewhere in the world (Hettiarachchi et al., 2020; Tang et al., 2021). Our patient might be the first CHILD syndrome patient of Malay ethnicity (defined as descendants of the natives of the Southeast Asian region including Southern Thailand, the Malay Peninsula and Indonesian islands). Another unusual feature of this case is the maternal transmission when most cases are sporadic with de novo variants.
Although skin manifestations of CHILD syndrome are known to be restricted to one side of the body, bilateral presentations have been reported (Estapé et al., 2015; Fink‐Puches et al., 1997; Xu et al., 2020). Approximately two‐thirds of patients exhibited right‐sided involvement, with left‐sided involvement associated with a more severe phenotype or early death (Ramphul et al., 2021) (Hummel et al., 2003). Our patient had left‐sided involvement but her phenotype was so mild that mosaicism was initially suspected. The variation in clinical severity is marked when compared with two other patients with frameshift variants, who displayed more extensive phenotypes including skeletal abnormalities and areas of persistent erythroderma (Bornholdt et al., 2005; Estapé et al., 2015). Another patient carrying a missense variant in the same codon as our patient also showed classical symptoms of CHILD syndrome (Maceda et al., 2020). The variability in the presentation of the syndrome suggests the influence of epigenetic and environmental factors on phenotypic presentation.
As the NSDHL gene is located on the X chromosome, another plausible explanation of the variable severity in phenotype is skewed X inactivation which could range from 34 and 68% for each of the two X chromosomes (Migeon, 2020). However, that is less likely as the NSDHL protein appears to be essential for cell survival: Nsdhl‐null fibroblasts did not survive past 48 hours when cultured in vitro (Cunningham et al., 2005), and there is also evidence of negative selection against NSDHL‐deficient cells in various mouse tissues (Cunningham et al., 2009).
5. CONCLUSIONS
Our results add a novel pathogenic variant found in a mother‐child pair and widen the phenotypic spectrum of CHILD syndrome. Our case illustrates that the clinical spectrum of CHILD syndrome can include subtle skin features and almost no limb defects, and she was only diagnosed in adulthood when she presented with gastrointestinal symptoms and worsening of her skin manifestations. This case also emphasizes that the condition can present with a mild phenotype, with the presence of multiple xanthomas in both skin and gastrointestinal tissues providing clues to the diagnosis.|
CONFLICT OF INTEREST
All authors declare no conflict of interest.
ETHICAL COMPLIANCE
All samples were collected after the patient and her family had given their written informed consent, and the study was approved by the SingHealth Centralised Institutional Review Board.
ACKNOWLEDGEMENT
This work was supported by project no. NMRC/CG/M003/2017 from the Singapore Ministry of Health’s National Medical Research Council.
Tan, E.‐C. , Chia, S. Y. , Rafi’ee, K. , Lee, S. X. , Kwek, A. B. E. , Tan, S. H. , Ng, V. W. L. , Wei, H. , Koo, S. , Koh, A. L. , & Koh, M. J.‐A. (2022). A novel NSDHL variant in CHILD syndrome with gastrointestinal manifestations and localized skin involvement. Molecular Genetics & Genomic Medicine, 10, e1848. 10.1002/mgg3.1848
Contributor Information
Ene‐Choo Tan, Email: tan.ene.choo@kkh.com.sg.
Shan Xian Lee, Email: lee.shan.xian@singhealth.com.sg.
DATA AVAILABILITY STATEMENT
The data associated with this article are available upon request from the authors.
REFERENCES
- Bornholdt, D. , König, A. , Happle, R. , Leveleki, L. , Bittar, M. , Danarti, R. , Vahlquist, A. , Tilgen, W. , Reinhold, U. , Poiares Baptista, A. et al (2005). Mutational spectrum of NSDHL in CHILD syndrome. Journal of Medical Genetics, 42, e17. 10.1136/jmg.2004.024448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen, A. G. , Koppelhus, U. , & Sommerlund, M. (2015). Skin abnormalities in CHILD syndrome successfully treated with pathogenesis‐based therapy. Acta Dermato Venereologica, 95, 752–753. 10.2340/00015555-2044 [DOI] [PubMed] [Google Scholar]
- Cooper, M. K. , Wassif, C. A. , Krakowiak, P. A. , Taipale, J. , Gong, R. , Kelley, R. I. , Porter, F. D. , & Beachy, P. A. (2003). A defective response to Hedgehog signaling in disorders of cholesterol biosynthesis. Nature Genetics, 33, 508–513. 10.1038/ng1134 [DOI] [PubMed] [Google Scholar]
- Cunningham, D. , Spychala, K. , McLarren, K. W. , Garza, L. A. , Boerkoel, C. F. , & Herman, G. E. (2009). Developmental expression pattern of the cholesterogenic enzyme NSDHL and negative selection of NSDHL‐deficient cells in the heterozygous Bpa(1H)/+ mouse. Molecular Genetics and Metabolism, 98, 356–366. 10.1016/j.ymgme.2009.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham, D. , Swartzlander, D. , Liyanarachchi, S. , Davuluri, R. V. , & Herman, G. E. (2005). Changes in gene expression associated with loss of function of the NSDHL sterol dehydrogenase in mouse embryonic fibroblasts. Journal of Lipid Research, 46, 1150–1162. 10.1194/jlr.M400462-JLR200 [DOI] [PubMed] [Google Scholar]
- du Souich, C. , Raymond, F. L. , Grzeschik, K. H. , & Boerkoel, C. F. (2008). NSDHL‐related disorders. In Adam M. P., Ardinger H. H., Pagon R. A., Wallace S. E., Bean L. J. H., Mirzaa G., & Amemiya A. (Eds), GeneReviews(®). University of Washington, Seattle. https://pubmed.ncbi.nlm.nih.gov/21290788/ [Google Scholar]
- Estapé, A. , Josifova, D. , Rampling, D. , Glover, M. , & Kinsler, V. A. (2015). Congenital hemidysplasia with ichthyosiform naevus and limb defects (CHILD) syndrome without hemidysplasia. British Journal of Dermatology, 173, 304–307. 10.1111/bjd.13636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feingold, K. R. , & Elias, P. M. (2014). Role of lipids in the formation and maintenance of the cutaneous permeability barrier. Biochimica Et Biophysica Acta, 1841, 280–294. 10.1016/j.bbalip.2013.11.007 [DOI] [PubMed] [Google Scholar]
- Fink‐Puches, R. , Soyer, H. P. , Pierer, G. , Kerl, H. , & Happle, R. (1997). Systematized inflammatory epidermal nevus with symmetrical involvement: An unusual case of CHILD syndrome? Journal of the American Academy of Dermatology, 36, 823–826. 10.1016/S0190-9622(97)70031-8 [DOI] [PubMed] [Google Scholar]
- Gantner, S. , Rütten, A. , Requena, L. , Gassenmaier, G. , Landthaler, M. , & Hafner, C. (2014). CHILD syndrome with mild skin lesions: Histopathologic clues for the diagnosis. Journal of Cutaneous Pathology, 41, 787–790. 10.1111/cup.12377 [DOI] [PubMed] [Google Scholar]
- Happle, R. , Effendy, I. , Megahed, M. , Orlow, S. J. , & Küster, W. (1996). CHILD syndrome in a boy. American Journal of Medical Genetics, 62, 192–194. [DOI] [PubMed] [Google Scholar]
- Happle, R. , Mittag, H. , & Küster, W. (1995). The CHILD nevus: A distinct skin disorder. Dermatology, 191, 210–216. 10.1159/000246548 [DOI] [PubMed] [Google Scholar]
- Herman, G. E. (2003). Disorders of cholesterol biosynthesis: Prototypic metabolic malformation syndromes. Human Molecular Genetics, 12(90001), R75–88. 10.1093/hmg/ddg072 [DOI] [PubMed] [Google Scholar]
- Hettiarachchi, D. , Panchal, H. , Lai, P. S. , & Dissanayake, V. H. W. (2020). Novel variant in NSDHL gene associated with CHILD syndrome and syndactyly‐ a case report. BMC Medical Genetics, 21, 164. 10.1186/s12881-020-01094-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummel, M. , Cunningham, D. , Mullett, C. J. , Kelley, R. I. , & Herman, G. E. (2003). Left‐sided CHILD syndrome caused by a nonsense mutation in the NSDHL gene. American Journal of Medical Genetics. Part A, 122a, 246–251. 10.1002/ajmg.a.20248 [DOI] [PubMed] [Google Scholar]
- König, A. , Happle, R. , Bornholdt, D. , Engel, H. , & Grzeschik, K. H. (2000). Mutations in the NSDHL gene, encoding a 3beta‐hydroxysteroid dehydrogenase, cause CHILD syndrome. American Journal of Medical Genetics, 90, 339–346. [PubMed] [Google Scholar]
- Maceda, E. B. G. , Kratz, L. E. , Ramos, V. M. E. , & Abacan, M. A. R. (2020). Novel NSDHL gene variant for congenital hemidysplasia with ichthyosiform erythroderma and limb defects (CHILD) syndrome. BMJ Case Reports, 13. https://pubmed.ncbi.nlm.nih.gov/33139364/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavilia, M. G. , & Wu, G. Y. (2018). The pathogenesis and clinical significance of gastrointestinal xanthelasma. A case report and review of literature. Clinical Medical Reviews and Case Reports, 5. https://clinmedjournals.org/articles/cmrcr/clinical‐medical‐reviews‐and‐case‐reports‐cmrcr‐5‐243.php?jid=cmrcr [Google Scholar]
- McLarren, K. W. , Severson, T. M. , du Souich, C. , Stockton, D. W. , Kratz, L. E. , Cunningham, D. , Hendson, G. , Morin, R. D. , Wu, D. , Paul, J. E. , An, J. , Nelson, T. N. , Chou, A. , DeBarber, A. E. , Merkens, L. S. , Michaud, J. L. , Waters, P. J. , Yin, J. , McGillivray, B. , … Boerkoel, C. F. (2010). Hypomorphic temperature‐sensitive alleles of NSDHL cause CK syndrome. American Journal of Human Genetics, 87, 905–914. 10.1016/j.ajhg.2010.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migeon, B. R. (2020). X‐linked diseases: Susceptible females. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 22, 1156–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormerod, E. , Bownass, L. , Smithson, S. , Zhang, Y. , & Dunnill, M. G. S. (2019). Mild clinical presentation of a patient with a mutation in the NSDHL gene. Clinical and Experimental Dermatology, 44, 456–458. [DOI] [PubMed] [Google Scholar]
- Paller, A. S. , van Steensel, M. A. , Rodriguez‐Martín, M. , Sorrell, J. , Heath, C. , Crumrine, D. , van Geel, M. , Cabrera, A. N. , & Elias, P. M. (2011). Pathogenesis‐based therapy reverses cutaneous abnormalities in an inherited disorder of distal cholesterol metabolism. The Journal of Investigative Dermatology, 131, 2242–2248. 10.1038/jid.2011.189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramphul, K. , Kota, V. , & Mejias, S. G. (2021). Child syndrome. In StatPearls. StatPearls Publishing. [PubMed] [Google Scholar]
- Ryan, C. , Quinn, S. , & McDermott, M. (2013). Small intestinal mucosal xanthoma in a patient with CHILD syndrome. Journal of Clinical Pathology, 66, 1094–1095. 10.1136/jclinpath-2013-201770 [DOI] [PubMed] [Google Scholar]
- Sandoval, K. R. , Machado, M. C. R. , Oliveira, Z. N. P. , & Nico, M. M. S. (2019). CHILD syndrome: Successful treatment of skin lesions with topical lovastatin and cholesterol lotion. Anais Brasileiros De Dermatologia, 94, 341–343. 10.1590/abd1806-4841.20198789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, M. M. , Tan, W. C. , Surana, U. , Leong, K. F. , & Pramano, Z. A. D. (2021). CHILD syndrome in a Malaysian adult with identification of a novel heterozygous missense mutation NSDHL c.602A>G. International Journal of Dermatology, 60, e154–e156. [DOI] [PubMed] [Google Scholar]
- Xu, X. G. , Zheng, S. , Di, Z. , Wan, Y. , & Gao, X. H. (2020). Congenital hemidysplasia with ichthyosiform nevus and limb defects syndrome: A rare case without hemidysplasia and limb defects. International Journal of Dermatology, 59, e272–e274. 10.1111/ijd.14889 [DOI] [PubMed] [Google Scholar]
- Zellweger, H. , & Uehlinger, E. (1948). A case of half‐sided bone enchondromatosis (Ollier) with ichthyosiform nevus. Helvetica Paediatrica Acta, 3, 153–163. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data associated with this article are available upon request from the authors.