

Abstract
Flow cytometry allows for the visualization of physical, functional, and/or biological properties of cells including antigens, cytokines, size, and complexity. With increasingly large flow cytometry panels able to analyze up to 50 parameters, there is a need to standardize flow cytometry protocols to achieve high quality data that can be input to analysis algorithms. Without this clean data, algorithms may incorrectly categorize the cell populations present in the samples. In this protocol, we outline a comprehensive methodology to prepare samples for polychromatic flow cytometry. The use of multiple washing steps and rigorous controls creates high-quality data with good separation between cell populations. Experimental data acquired using this protocol can be analyzed via computational algorithms that perform end-to-end analysis.
Keywords: Phenotyping, immunology, flow cytometry, sample preparation, controls
INTRODUCTION:
High-dimensional flow cytometry data, containing excess of 15 parameters, is difficult to analyze using conventional analysis methods such as manual gating of cells on 2-dimensional plots. In the past decade, researchers have worked to develop data analysis tools for flow cytometry. However, data input must be reliable for these tools to accurately analyze it. By executing this current protocol, it will be possible to acquire clean flow cytometry data that can be input into data analysis pipelines. With increasingly complex flow cytometry development, it is important to acquire data following a very strict and reproducible flow cytometry staining procedure to ensure high quality data prior to analysis. By the end of this protocol, the high quality flow cytometry data and controls are ready to be entered into and analyzed using a Cyto-feature engineering pipeline (Fox et al., Under Revision).
Basic Protocol 1 describes how to prepare single-cell suspension for flow cytometry after harvesting either lung or spleen from an animal. This protocol can further be modified to harvest cells from other organs. Basic Protocol 2 describes how to discriminate live and dead cells using a viability dye and stain cells with surface and intracellular markers.
STRATEGIC PLANNING
Prior to employing this protocol for an experiment, the flow cytometry panel should be optimized for the user’s flow cytometer to ensure there is no overlap between different fluorophores. Users should familiarize themselves with their flow cytometer configuration and the possible combination of fluorophores that can be used with that cytometer. Panel design will be based on this configuration and possible fluorophores. Excellent resources for panel design include the website FluoroFinder (FluoroFinder, 2020) and a recent publication in Current Protocols (Ferrer-Font et al., 2020).
BASIC PROTOCOL 1: Preparation of single-cell suspension for flow cytometry
In this protocol, tissues are dissociated into single-cell suspension in preparation for flow cytometry. Whole tissue is macerated through a cell strainer after which red blood cells are lysed. Note, this protocol should occur in a biosafety cabinet to prevent sample contamination and ensure technician safety.
Materials:
Reagents, Solutions, and test animals
Lung of mouse
Spleen of mouse
DMEM 1x with 4.5g/L glucose without L-glutamine, sodium pyruvate (Corning, cat# 15–017-CV)
Red Blood Cell (RBC) Lysing Buffer (Sigma, cat# R7757)
Complete media (see recipe)
Phosphate Buffered Saline (VWR, cat. No. 45000–446)
Hardware and Instruments
Biosafety cabinet (e.g., Labconco Type A2)
70 μm Cell Strainers (Corning, cat# 352350)
60 × 15 mm Petri Dishes (Thermo Fisher Scientific, cat# AS4051)
3 mL LuerLok Syringes (BD Biosciences, cat# 309657)
5 mL serological pipets (Thermo Fisher Scientific, cat# 170355)
Motorized Serological Pipette Filler (SCILOGEX, cat# 740200029999)
15 mL Conical Tubes (Thermo Fisher Scientific, cat# 12565269)
Tabletop Centrifuge (e.g., Beckman Coulter Allegra 6)
1 mL Pipettor (Sigma Aldrich, cat# EP3124000121)
1 mL Pipet Tips (VWR, cat# 83007–382)
10 mL serological pipets (Thermo Fisher Scientific, cat# 170356)
Flow cytometer (e.g., BD LSR-II)
200 μL pipettor (Sigma Aldrich, cat# EP3124000083)
200 μL pipet tips (VWR, cat# 53508–810)
96-well plates with V-bottom (Sigma Aldrich, cat# M9686)
Paper towels (Supply Works, cat# SCAHB9201)
Protocol steps—Step annotations:
-
Harvest spleen and lung into 1 mL of DMEM media in a 15 mL conical tube.
Note: Organs may be required for multiple experimental tests. In this case, we take ½ spleen and two of the lung lobes for flow cytometry. The remaining lung lobes and spleen are utilized for enumerating colony forming units or other downstream applications. However, this could be modified according to the experimental design and goal. For lungs, follow Supplementary Protocol 1 prior to step 2 below.
Add organ and liquid into 70 μm cell strainer placed inside a petri dish.
-
Macerate organs using 3 mL plunger in order to pass cells through the strainer mesh.
Note: Remove the 3 mL plunger from the 3 mL syringe and use only the non-rubber side of the plunger.
Flush cell strainer mesh with 5 mL of DMEM media and harvest cells from the Petri Dish after resuspending. Transfer cells back into the 15 ml conical tube.
-
Centrifuge cells at 380 G-force for 10 min at 4°C.
The RPM (revolutions per minute) for specific centrifuges can be calculated with Equation 1, where g is the g-force and Radius is the radius of the rotor in centimeters.Equation 1 -
Discard supernatant and resuspend cell pellet in 1 mL RBC lysing buffer – incubate for 1 minute at room temperature.
Note: During the incubation time, cells should be mixed by running the 15 mL conical tubes against the grate of the biosafety cabinet, disrupting the pellet.
Add 6 mL of complete media to dilute RBC lysing buffer.
Centrifuge cells at 380 G-force for 10 min at 4°C.
Discard supernatant by gently flipping tube upside down.
-
Resuspend cells in PBS and keep at 4°C while counting.
Note: Resuspend spleens in 800 μl of PBS and lungs in 400 μL of PBS.
-
Count cells in each tube using a flow cytometer (See Supplementary Protocol 2).
Note: Alternatively, cells could be counted with a hemocytometer or cell counter, but this method is more time consuming.
Adjust cell suspension to 2 × 107 cells/mL for spleen and 5 × 106 cells/mL for lung.
-
Add 200 μL of lung cells (1 × 106 total cells) or 100 μl of spleen cells (2 × 106 total cells) to each well of a 96-well plate (Figure 1).
Note: There should be a well for every stained sample, a well for every Fluorescence Minus One control (FMO), a single well that will not be stained (Unstained), and a single well that will only be stained with live-dead (Zombie-NIR) stain. Single-color controls will also need to be stained, but beads can be used for these controls (see support protocol 3). Due to different levels of autofluorescence and viability, each organ requires the following controls: FMOs, unstained cells, and live-dead staining. To run these controls for each organ, cells can be pooled from different experimental groups or biological replicates within a group.
-
Centrifuge plates at 380 G-force for 10 min at 4°C.
Note: At this point, it should be possible to see a pellet of cells at the bottom of each well.
Remove supernatant by gently tapping the liquid out of the wells onto a paper towel.
-
Wash cells by adding 100 μL of PBS to each well and centrifuge at 380 G-force for 10 minutes at 4°C.
Note: This wash minimizes any residual protein in the sample, which can quench the Zombie NIR viability stain.
Remove supernatant by gently tapping the liquid out of the wells onto a paper towel.
Figure 1. Cell plate layout with controls.
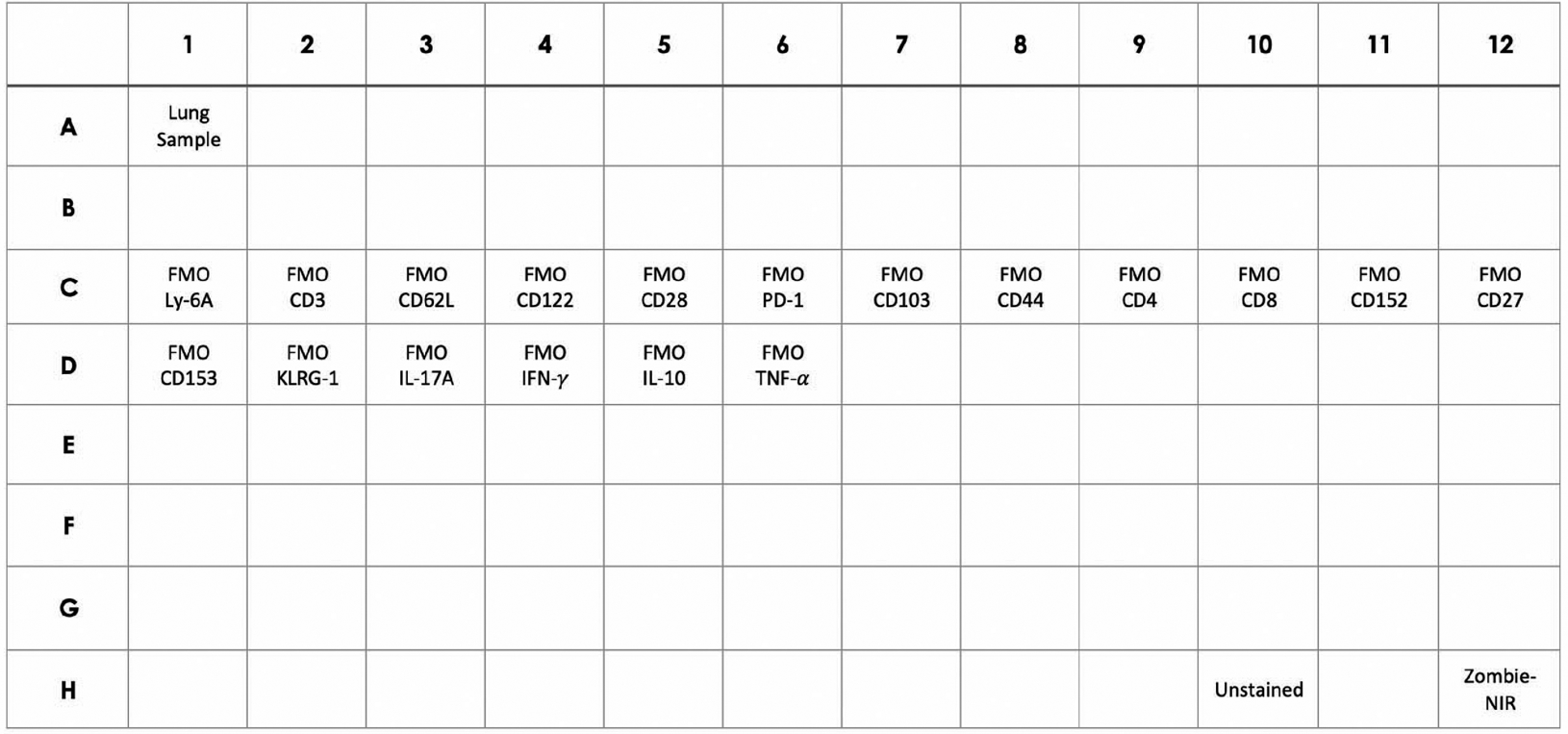
Example of the required cell samples for flow cytometry. Note that there will be a second plate with beads used for single-color controls.
SUPPORT PROTOCOL 1: Lung Preparation
To maximize cell yield and viability, lungs must first be enzymatically digested before dissociation and cell suspension. Collagen in the lungs’ extracellular matrix will be digested to improve cell dissociation when macerating through a 70 μm cell strainer. Inevitably, some cells lyse and release DNA during this procedure. DNase is included in the dissociation buffer to digest released DNA and minimize the formation of a viscous material trapping the cells. If this protocol is conducted properly, single-cell suspensions with a cell viability >70–80% will be obtained from the lungs.
Materials:
Reagents, Solutions, and test animals
Lung sample
70% ethanol
DMEM 1x with 4.5g/L glucose without L-glutamine, sodium pyruvate (Corning, cat# 15–017-CV)
2x DNase/Liberase (see recipe)
Hardware and Instruments
Biosafety cabinet (e.g., Labconco Type A2)
60 × 15 mm Drosophila Supplies Small Petri Dishes (Thermo Fisher Scientific, cat# AS4051)
Razor blades (VWR, cat # 55411–050)
37°C incubator
Protocol steps—Step annotations:
-
Make superficial cuts in the lung in a petri dish using 2 razors (rinse razors with 70% ethanol and then DMEM).
Note: Use one razor to hold tissue in place and the other to chop. Move tissue and chop in a crosshatch pattern. Be careful not to chop tissue too deep—the tissue needs to be kept whole.
Transfer lungs and liquid back into the 15 mL conical tubes.
-
Add 1 mL of 2x DNase/Liberase to each tube and incubate at 37°C for 30 minutes.
Note: The final concentration per sample of Liberase is 0.25 mg/mL and the concentration per sample of DNase is 0.125 mg/mL.
Continue with Step 2 in Basic Protocol 1.
SUPPORT PROTOCOL 2: Counting Cells on Flow Cytometer
To calculate the total number of cells obtained from an organ, the cells must be counted. Unless cell death is a readout, dead cells are usually excluded from this analysis as dead cells tend to be more autofluorescent and bind antibodies nonspecifically. Dead cells are stained with a viability dye such as 7-AAD, which binds to DNA in dead cells with compromised cell membranes. Therefore, live cells are those in which there is no 7-AAD present. Fluorescent counting beads of known concentration are added to the samples to determine the ratio of beads to live cells. For convenience, two different viability dyes (7-AAD and Zombie-NIR) were used in this protocol: 7-AAD to count cells on the cytometer and Zombie-NIR to analyze samples after surface/intracellular staining. This staining approach will be required when counting cells using equipment that does not have a laser to detect Zombie-NIR. One viability dye could, however, be used for both procedures upon further optimization. One advantage of using 7-AAD for the counting procedure is that it allows for quicker counting analysis as it can directly be added to flow tubes without washing steps. If this protocol is conducted properly, the concentration of live leukocytes in each sample can be calculated.
Materials:
FACS Staining Buffer (see recipe)
7-AAD Viability Staining (Thermo Fisher, cat# 00-6993-50)
CountBright Absolute Counting beads (Thermo Fisher, cat# C36950)
Hardware and Instruments
Biosafety cabinet (e.g., Labconco Type A2)
5 mL Polypropylene round bottom flow tubes (VWR, cat# 60819–794)
Flow cytometer (e.g., LSR II)
Protocol steps—Step annotations:
For each sample, add 200 μL of FACS Staining Buffer with 0.025 μg of 7-AAD to a flow tube.
Add 25 μL of counting beads and 25 μL of cells to each tube from step 1.
-
Collect 50,000 events on the flow cytometer.
Note: Ensure gains (voltages) for FSC and SSC allow for both the beads and cells to be seen on the plots (See Figure 2). This can be visualized by setting the SSC-A axis to log-scale.
Place a gate around the beads and count the number of beads as shown in Figure 2.
Gate the cells based on live leukocytes as shown in Figure 2.
- Calculate the volume of PBS to add to each sample based on the desired cell concentration using the following calculations.
- Sample volume: Volume in which the cells are suspended (e.g., 400 μL of PBS for lungs or 800 μL of PBS for spleen)
- Bead concentration/μL: Concentration of beads per μL (this information is written on the vial of beads. Note that bead concentration may vary between different lots and should be modified accordingly.)
- # Beads: Number of beads based on gating (see Figure 2)
- # Live Leukocytes: Number of live leukocytes based on gating (see Figure 2)
- Acquired Volume of Beads (μL): Calculated volume based on dividing the # Beads by the Bead Concentration/μL
- Total # Live Leukocytes: Calculated by multiplying the sample volume by the # Live Leukocytes and dividing by acquired volume of beads. (As beads and cells were added at a 1:1 ratio (step 2), the volume of acquired beads and acquired cells should be the same).
- Desired # cells/mL: Desired concentration of cells per mL for the experiment
- Volume to Add (mL): Calculated by dividing the Total # Live Leukocytes/mL by desired # of cells/mL and subtracting the sample volume (Table 1)
Figure 2. Cell counting gating strategy.

Figure showing placement of the bead gate and the leukocyte gate following cell counting on a flow cytometer. The leukocytes are further gated to limit cells to the live leukocytes.
Table 1.
Single-cell suspension from Spleen and Lung
| Sample ID | Sample volume (μL) | Bead concentration/μL | # Beads | # Live Leukocytes | Acquired Volume of Beads | Total # Live Leukocytes | Desired # Cells/mL | Volume to Add (mL) |
|---|---|---|---|---|---|---|---|---|
| 800 | 1040 | 590 | 28,933 | 0.57 | 4.08E7 | 2E7 | ||
| 400 | 1040 | 2,519 | 21,942 | 2.42 | 3.62E6 | 5E6 |
BASIC PROTOCOL 2: Surface and Intracellular Flow Cytometry Staining
In this protocol, an initial 6-hour incubation with a Protein Transport Inhibitor prevents intracellular proteins from being secreted, causing the accumulation of such proteins in cells. This allows for the intracellular proteins to be stained. Following this incubation, anti-mouse CD16/32 is added to block FC-receptors on leukocytes, which prevents non-specific antibody binding. Dead cells are then stained using the viability dye, Zombie-NIR, and surface antibodies are added. To stain for intracellular markers, fixation/permeabilization buffer is added to allow antibodies to pass through the plasma membrane. If this protocol is conducted properly, cells stained with the fluorophore-marker pairs can be visualized on a flow cytometer.
If the panel only contains surface markers, the 6-hour incubation with a Protein Transport Inhibitor and steps with Permeabilization/Fixation can be skipped. If so, cells are simply blocked with anti-CD16/32, stained with Zombie-NIR, and then stained with surface antibodies. Thereafter, cells are washed with FACS Staining Buffer and fixed by incubating with 4% PFA for 20 minutes.
Materials:
Reagents, Solutions
1000X Protein Transport Inhibitor [BD GolgiStop] (BD Biosciences, cat# 554724)
Complete Media (see recipe)
Phosphate Buffered Saline (VWR, cat# 45000–446)
Zombie-NIR Fixable Viability Kit (VWR, cat# 10761–492)
FACS Staining Buffer (see recipe)
Anti-mouse CD16/32 (Biolegend, cat# 101330)
T cell surface antibody cocktail (see recipe)
FMOs (see recipe)
Perm/Fix buffer (see recipe)
1X Permeabilization buffer (see recipe)
T cell intracellular antibody cocktail (see recipe)
Hardware and Instruments
Biosafety cabinet (e.g., Labconco Type A2)
10 μL pipettor (Sigma Aldrich, cat# EP3124000024)
10 μL pipet tips (Thermo Fisher Scientific, cat# 2707454)
Motorized Serological Pipette Filler (SCILOGEX, cat# 740200029999)
5 mL serological pipets (Thermo Fisher Scientific, cat# 170355)
37°C CO2 incubator (e.g., VWR water-jacketed CO2 incubator)
Tabletop Centrifuge (e.g., Beckman Coulter Allegra 6)
Paper towels (Supply Works, cat# SCAHB9201)
200 μL pipettor (Sigma Aldrich, cat# EP3124000083)
200 μL pipet tips (VWR, cat# 53508–810)
5 mL Polypropylene round bottom flow tubes (VWR, cat# 60819–794) [note that polystyrene tubes may need to be used depending on the cytometer]
Protocol steps—Step annotations:
Prepare a 1X solution of Protein Transport Inhibitor (GolgiStop) in Complete Media.
Add 100 μl of above prepared media with the Protein Transport Inhibitor to each well on the 96-well plate containing pelleted cells.
Incubate plate in a CO2 incubator set at 37°C for 6 hours.
-
Remove plate from incubator and centrifuge at 380 G-force for 10 minutes at 4°C.
Note: All centrifugations will be at 380 G-force for 10 min at 4°C.
Remove supernatant by gently tapping the liquid out of the wells onto a paper towel.
Wash cells by adding 100 μL of PBS to each well and centrifuging at 380 G-force for 10 minutes at 4°C.
Remove supernatant by gently tapping all of the liquid out of the wells onto a paper towel, being careful not to disrupt the pellet.
-
Add 100 μl of Zombie-NIR live/dead stain (1:2000) dilution in PBS to each well except for the “Unstained Sample well” and incubate for 15 minutes at room temperature in the dark.
Note: Zombie-NIR must be diluted in PBS rather than FACS Staining Buffer because the FACS Staining Buffer contains Fetal Bovine Serum that quenches the Zombie signal. Note that all further steps should take place in the dark so that the fluorescently labeled antibodies are not photobleached.
Centrifuge cells at 380 G-force for 10 minutes at 4°C and remove the supernatant.
Wash cells by adding 100 μL of FACS Staining Buffer and centrifuge at 380 G-force for 10 minutes at 4°C.
Remove supernatant.
Wash cells again by adding 100 μL of FACS Staining Buffer and centrifuge at 380 G-force for 10 minutes at 4°C.
Remove supernatant.
-
Incubate cells with FACS Staining Buffer containing 2.5 μg/mL of anti-mouse CD16/32 for 20 minutes at 4°C.
Note: This antibody blocks non-specific FC receptor binding.
Centrifuge cells at 380 G-force for 10 minutes at 4°C and remove the supernatant.
-
Add the T-cell surface marker antibody panel to all the sample wells. Also add the appropriate surface marker FMOs to the wells.
Note: Do not add intracellular antibodies at this point.
Incubate cells at 4°C for 30 minutes in the dark.
Centrifuge cells at 380 G-force for 10 minutes at 4°C and remove the supernatant.
Wash cells by adding 150 μL of FACS Staining Buffer and centrifuge at 380 G-force for 10 minutes at 4°C.
Remove supernatant.
Add 150 μl of 1X Perm/Fix buffer and incubate for 1 hour at room temperature.
Centrifuge the cells at 380 G-force for 10 minutes at 4°C and wash the cells with 150 μl of 1X Permeabilization Buffer.
Remove supernatant.
Centrifuge cells at 380 G-force for 10 minutes at 4°C and wash cells with 150 μl of Permeabilization Buffer again.
Remove supernatant.
Add 100 μl of the T-cell intracellular antibody cocktail and intracellular FMO antibodies cocktail to respective wells.
Incubate plate overnight at 4°C in dark.
The Next Day
Centrifuge cells at 380 G-force for 10 minutes at 4°C.
Wash cells by adding 150 μl of Permeabilization Buffer and centrifuge at 380 G-force for 10 minutes at 4°C.
Remove supernatant.
Suspend cells in 100 μL of Permeabilization Buffer.
Transfer cells to flow cytometry tubes that contain an additional 200 μL of Permeabilization Buffer.
Read samples on a flow cytometer.
SUPPORT PROTOCOL 3: Single-Color Bead Controls
Single-color bead controls, also known as reference controls, are used to visualize the spectral signature for each fluorophore on the flow cytometer. Because spectral unmixing is dependent on clear separation of positive and negative populations and an exact spectra match, beads are often used. Each marker in the flow cytometry panel must have a single-color control made. If this protocol is conducted properly, there should be a positive and negative population with clear separation on the flow cytometer. Further, the spectral signature for each single-color control should be unique. This will allow the flow cytometer to perform spectral unmixing on the samples.
Materials:
Reagents, Solutions, and test animals
Individual stains (see supplemental for list of antibodies)
UltraComp eBeads (Fisher, cat# 01-2222-42)
96-well plates with V-bottom (Sigma Aldrich, cat# M9686)
FACS Staining Buffer (see recipe)
Hardware and Instruments
10 μL pipettor (Sigma Aldrich, cat# EP3124000024)
10 μL pipet tips (Thermo Fisher Scientific, cat# 2707454)
200 μL pipettor (Sigma Aldrich, cat# EP3124000083)
200 μL pipet tips (VWR, cat# 53508–810)
Tabletop Centrifuge (e.g., Beckman Coulter Allegra 6)
5 mL Polypropylene round bottom flow tubes (VWR, cat# 60819–794)
Cytek Aurora Flow Cytometer
Protocol steps—Step annotations:
Prepare individual stains by adding the appropriate antibody dilution to 100 μL of FACS Staining Buffer.
Add 1 drop of UltraComp beads for each single-color control to wells on a 96-well plate.
Centrifuge beads at 380 G-force for 10 minutes at 4°C.
Remove supernatant by gently tapping the liquid out of the wells onto a paper towel.
Add 100 μL of the individual stains to each well.
Incubate for 10 minutes at room temperature.
Centrifuge beads at 380 G-force for 10 minutes at 4°C.
Remove supernatant.
Wash beads by adding 100 μL of FACS Staining Buffer and centrifuge again at 380 G-force for 10 minutes at 4°C.
Remove supernatant by gently tapping the liquid out of the wells onto a paper towel.
Wash beads again by adding 100 μL of FACS Staining Buffer and centrifuge again at 380 G-force for 10 minutes at 4°C.
Remove supernatant.
Add 100 μL FACS Staining buffer to each well and transfer the samples to flow tubes that contain 200 μL FACS Staining buffer.
Read samples on a flow cytometer.
REAGENTS AND SOLUTIONS:
2x DNAse/Liberase
Materials
Liberase (5.2 Wunch units/mg) (Sigma, cat# 5401127001)
DMEM 1x with 4.5g/L glucose without L-glutamine, sodium pyruvate (Corning, cat# 15–017-CV)
DNase I [type IV Bovine] (3,000 units/mg) (Sigma, cat# D5025–150KU)
- 50 mL Conical Tubes (Thermo Fisher Scientific, cat# 12565270)
- Mix 100 mg of Liberase (520 units) and 50 mg of DNase (150,000 units) in DMEM and adjust the total volume to equal 200 mL.
- Mix the solution well and aliquot into conical tubes based on experimental needs taking into consideration that each lung sample will require 1 mL of this solution.
- Store at −80°C.
Complete Media
Materials
DMEM 1x with 4.5g/L glucose without L-glutamine, sodium pyruvate (Corning, cat# 15–017-CV)
MEM Nonessential amino acids 100X (Corning, cat# 25–025-CI)
Penicillin streptomycin (Thermo Fisher Scientific, cat# 15140–122)
L-glutamine (Thermo Fisher Scientific, cat# 25030081)
- Heat-inactivated Fetal Bovine Serum (Thermo Fisher Scientific, cat# MT35011CV)
- Add 4.5 mL MEM amino acids, 4.5 mL Penicillin streptomycin, 4.5 mL L-glutamine, and 45 mL Fetal Bovine Serum to the bottle of 500 mL 1x DMEM in a sterile hood.
- Store at 4°C.
FACS Staining Buffer
Materials
Heat-inactivated Fetal Bovine Serum (Thermo Fisher Scientific, cat# MT35011CV)
Phosphate Buffered Saline (VWR, cat. No. 45000–446)
- Sodium Azide (Fisher Chemical, cat# S2271–25)
- Add 10 mL of Fetal Bovine Serum to a 500mL bottle of PBS.
- Weight 0.25 g of Sodium Azide and add to the PBS/FBS solution.
- Store at 4°C.
FMOs
Materials
- Same materials as those listed in T cell surface antibody cocktail and T cell intracellular antibody cocktail
- Create one FMO for every marker in the flow cytometry panel.
-
Add a 1:10 dilution of Brilliant Stain buffer and the appropriate dilutions of all the markers except for one to FACS Staining Buffer to make up a 100 μL volume.Note: The surface FMOs will be prepared in FACS Staining Buffer and the intracellular FMOs will be prepared in Permeabilization Buffer.
1X Perm/Fix Buffer
Materials
Fixation/Permeabilization Concentrate (Thermo Fisher Scientific, cat# 501129082)
- Fixation/Permeabilization Diluent (Thermo Fisher Scientific, cat# 501129081)
- Add 1-part Fixation/Permeabilization Concentrate to 3-parts Fixation/Permeabilization Diluent. Example: 12.5 mL concentrate + 37.5 mL diluent
- Store at 4°C
1X Permeabilization Buffer
Materials
Permeabilization Buffer 10X (Invitrogen, cat# 00-8333-56)
- Sterile Deionized Water
- Add 1-part Permeabilization Concentrate to 9-parts Sterile Deionized Water. Example: 5 mL Perm buffer + 45 mL water
- Store at 4°C.
T cell surface antibody cocktail
Materials
(See Table 2 for antibody specifics)
Table 2.
T cell surface antibody cocktail
| Fluorophore | Marker | Clone | Effective Concentration |
|---|---|---|---|
| BB515 | Ly-6A/E (Sca-1) | D7 | 0.2 μg/mL |
| Alexa Fluor 532 | CD3 | 17A2 | 4 μg/mL |
| PE Dazzle 594 | CD62L | MEL-14 | 0.4 μg/mL |
| PE Cy5 | CD122 | TM-β1 | 2 μg/mL |
| PerCP Cy5.5 | CD28 | 37.51 | 4 μg/mL |
| PerCP e710 | PD-1 | RMP1–30 | 2 μg/mL |
| APC R700 | CD103 | M290 | 1 μg/mL |
| APC Fire750 | CD44 | IM7 | 0.2 μg/mL |
| BV480 | CD4 | RM4–5 | 2 μg/mL |
| BV570 | CD8 | 53–6.7 | 1 μg/mL |
| BV605 | CD152 (CTLA-4) | UC10–4B9 | 4 μg/mL |
| BV650 | CD27 | LG.3A10 | 2 μg/mL |
| BV711 | CD153 | RM153 | 2 μg/mL |
| BV786 | KLRG-1 | 2F1 | 2 μg/mL |
| - | CD16/32 | 93 | 2.5 μg/mL |
FACS Staining Buffer (see recipe)
Brilliant Stain Buffer (BD Biosciences, cat# 566349)
Rat Anti Ly-6A/E, BB515 (BD Biosciences Cat# 565397, RRID: AB_2739218)
Anti-mouse CD3, Alexa Fluor 532 (Thermo Fisher Scientific Cat# 58-0032-82, RRID: AB_11217479)
Anti-mouse CD62L, PE/Dazzle 594 (BioLegend Cat# 104448, RRID:AB_2566163)
Anti-mouse CD122, PE/Cy5 (BioLegend Cat# 123220, RRID: AB_2715962)
Anti-mouse CD28, PerCP/Cyanine 5.5 (BioLegend Cat# 102114, RRID: AB_2073850)
Anti-mouse PD-1, PerCP-eFluor 710 (Thermo Fisher Scientific Cat# 46-9981-82, RRID: AB_11151142)
Anti-mouse CD103, APC-R700 (BD Biosciences Cat# 565529, RRID: AB_2739282)
Anti-mouse CD44, APC/Fire 750 (BioLegend Cat# 103062, RRID: AB_2616727)
Anti-mouse CD4, Brilliant Violet 480 (BD Biosciences Cat# 565634, RRID: AB_2739312)
Anti-mouse CD8, Brilliant Violet 570 (BioLegend Cat# 100739, RRID: AB_10897645)
Anti-mouse CD152, Brilliant Violet 605 (BioLegend Cat# 106323, RRID: AB_2566467)
Anti-mouse CD27, Brilliant Violet 650 (BioLegend Cat# 124233, RRID: AB_2687192)
Anti-mouse CD153, Brilliant Violet 711 (BD Biosciences Cat# 740751, RRID: AB_2740419)
Anti-mouse KLRG-1, Brilliant Violet 786 (BD Biosciences Cat# 565477, RRID: AB_2739256)
- Anti-mouse CD16/32 (Biolegend, cat# 101330)
-
Prepare surface antibody cocktail in FACS Staining Buffer with a 1:10 dilution of Brilliant Violet Buffer.Note: If two or more brilliant violet dyes are used together, it is important to add Brilliant Violet Buffer and FACS Staining Buffer together before adding antibodies to avoid aggregation of the antibodies labeled with brilliant violet dyes.
- Add the antibodies at the dilution listed above.
-
Store at 4°C in the dark.Note: The antibody cocktail should be protected from light, for instance by wrapping in tin foil.
-
T cell intracellular antibody cocktail
Materials
(See Table 3 for antibody specifics)
Table 3.
T cell intracellular antibody cocktail information
| Fluorophore | Marker | Clone | Effective Concentration |
|---|---|---|---|
| PE | IL-17A | TC11–18H10.1 | 2 μg/mL |
| PE Cy7 | IFN-γ | XMG1.2 | 2 μg/mL |
| BV421 | IL-10 | JES5–16E3 | 1 μg/mL |
| Pacific Blue/e450 | TNF-α | MP6-XT22 | 5 μg/mL |
| - | CD16/32 | 93 | 2.5 μg/mL |
Permeabilization Buffer (see recipe)
Anti-mouse IL-17A, PE (BioLegend Cat# 506904, RRID: AB_315464)
Anti-mouse IFN-γ, PE Cyanine 7 (Thermo Fisher Scientific Cat# 25-7311-82, RRID: AB_469680)
Anti-mouse IL-10, Brilliant Violet 421 (BioLegend Cat# 505021, RRID: AB_10900417)
Anti-mouse TNF-α, Pacific Blue (BioLegend Cat# 506318, RRID: AB_893639)
- Anti-mouse CD16/32 (Biolegend, cat# 101330)
-
Prepare intracellular antibody cocktail in permeabilization buffer.Note: Brilliant Violet Buffer does not need to be added to this cocktail because there is only 1 Brilliant Violet stain present.
- Add the antibodies at the dilution listed above.
-
Store at 4°C in the dark.Note: Protect antibody cocktail from the light.
-
COMMENTARY
BACKGROUND INFORMATION:
Flow cytometry is a technique used to analyze the physical, functional and/or biological properties of cells including antigens, cytokines, size, and granularity. Cells are stained with fluorophore-conjugated antibodies. The cells are then sent single file through the flow cytometer, where a laser light source excites the fluorescently tagged antibodies, which can then emit light that is measured (Becton, 2012).
Traditionally flow cytometry has only been able to examine a handful of parameters at a time. However, with the advent of spectral flow cytometry, there has been a large increase in the number of possible parameters that can be measured. Traditional analysis methods for flow cytometry include manual selection (gating) of cells on two-dimensional plots, often using expensive software such as FlowJo or FCS Express (Verschoor, Lelic, Bramson, & Bowdish, 2015). While these programs are user-friendly, they lack the ability to analyze high-dimensional data quickly and efficiently. Researchers and computational biologists have been working to develop analysis tools that utilize feature engineering, clustering, and dimensionality reduction algorithms to address this complex data. However, to ensure accurate results with these tools, data needs to be acquired following very strict flow cytometry staining procedures, leading to clean and accurate data before analysis. Further, it is crucial that the correct controls are collected, and the flow cytometer is calibrated to a set standard.
With all types of flow cytometry, single-color controls are required for data acquisition. In conventional flow cytometry, these controls are used to build the compensation matrix. In spectral flow cytometry, these controls are used to perform spectral unmixing, which separates the expression of similar fluorophore emissions based on the entire emission spectra of each individual fluorophore. This allows for multiple fluorophore-marker pairs to be used that would be indistinguishable with the same number of lasers on a traditional flow cytometer (Biosciences, 2019).
Fluorescent Minus One controls, or FMOs, are an example of a “best practice” control used in both conventional flow cytometry and spectral flow cytometry (Roederer, 2002). FMOs contain all the markers in a flow cytometry panel except for one. For example, if a panel contains 4 markers (e.g., CD45, CD3, CD4, CD8), then a CD45 FMO would contain CD3, CD4, and CD8, but not CD45. These controls allow for the user to account for any spillover from other fluorophores into the specific marker channel that is of interest (Roederer, 2002). These FMOs allow users to place informed “gates” around populations that are either negative or positive for a marker.
CRITICAL PARAMETERS:
There are multiple factors that influence the success of these protocols.
Protocol 1: Preparation of single-cell suspension for flow cytometry
While using razors to perform crosshatch cuts in the lungs, careful movements should be used to ensure lungs are not cut into pieces. There should only be imprints of the razors into the lungs rather than full cuts through the lungs. If the lungs are digested into small pieces following incubation with DNase/liberase, it is difficult to macerate them through the 70 μm filter.
Special attention should also be paid to the controls used in these experiments. These controls, which include FMOs and single-color beads, are critical in both spectral unmixing on the cytometer and determination of positive and negative populations during data analysis. There should be 1 FMO and 1 single-color control for every fluorophore-marker pair in an experiment.
Protocol 2: Surface and intracellular flow cytometry staining
Panel design is a highly important step prior to flow cytometry staining. Note that the same antibody clone and fluorochrome should be used for subsequent sampling to standardize the fluorescent signal. Ferrer-Font et al. and Mahnke & Roederer et al. describe in detail how to design and optimize a flow cytometry panel (Ferrer-Font et al., 2020; Mahnke & Roederer, 2007).
Performing a pilot optimization study is highly recommended to ensure that the fluorophore-marker pairs do not overlap in emission wavelength on a conventional cytometer or spectral signatures on a spectral cytometer. If the signatures are too similar, there will be a problem with spectral unmixing or signal overlap. In this case, change one of the problematic fluorophores to another that is not in use. Otherwise, the panel’s complexity must be reduced.
Low or no signal in certain markers can be due to low marker expression on cells. In this case, the cells can be stimulated with PMA-ionomycin. Another check for the low signal would be to perform antibodies dilution series (example dilutions: 1:50, 1:100, 1:200, 1:400, 1:800), to rule out insufficient antibody concentration. If there is still low expression, the fluorophore could be too dim for the marker. It may be best to try a brighter fluorophore-marker pair (Mahnke & Roederer, 2007).
Particularly when using fixation/permeabilization steps, there can be an increase in cell autofluorescence. This autofluorescence can mask signal of other markers, particularly dim ones, if the proper controls are not used. The unstained sample should be used to assess autofluorescence, meaning that all steps (except for the addition of fluorescent antibodies) should be performed on these cells, including the fixation/permeabilization, anti-mouse CD16/32, and washes.
Tandem dyes, such as PE/Cy5 or APC/Fire750, are fluorescent molecules comprised of two covalently bound fluorophores, in which the energy emitted by one fluorophore excites the second one. While these dyes have largely increased the number of fluorophores available for use in flow cytometry, special attention should be paid to their properties. It is not uncommon for tandem dyes to degrade or decouple. This can occur when the fluorophores are exposed to light, long fixation and permeabilization steps, or if the tandem dyes are not properly stored at 4°C (Biologicals).
During the staining procedure, extra precaution should be used in the washing steps. If a washing step is omitted, there may not be good separation between populations. Particularly in the dimension of Zombie-NIR, there cannot be any protein present in the diluent. Proteins, such as those from fetal bovine serum (FBS), Bovine Serum Albumin (BSA), or those released during red blood cell lysis can bind to Zombie-NIR, therefore lowering its effective concentration. Hence, washing cells with PBS prior to adding Zombie-NIR is a crucial step.
TROUBLESHOOTING:
STATISTICAL ANALYSIS:
The flow cytometry data acquired following this protocol can be used with almost any type of statistical analysis. The single-color controls are used either for spectral unmixing in the case of spectral flow cytometry, or the development of a compensation matrix in conventional flow cytometry. The FMOs can be utilized either in manual gating strategies in FlowJo or FCSExpress, or in the novel analysis pipeline, cyto-feature engineering (Fox et al., Under Revision). The cyto-feature engineering is an end-to-end analysis pipeline that can further form correlations between flow cytometry data and additional readouts. The data can also be analyzed with other tools such as OpenCyto (Finak et al., 2014), which implements automated gating, FlowSOM (Van Gassen et al., 2015), which creates self-organizing maps, or t-SNE (Eshghi et al., 2019) which performs dimensionality reduction. To compare differences in marker expression or cell populations (defined by combinations of markers) between experimental groups, statistical tests such as Analysis of Variance (ANOVA) and Tukey Honest Significant Difference (HSD) can be used.
UNDERSTANDING RESULTS:
After gating each sample and FMO on single cells, leukocytes, and live cells, the gates on the FMOs can be placed. An example of a good FMO (Figure 3) is shown with CD4. Note that the cells are concentrated in a cluster and are not very spread out. When this FMO gate is placed on a sample, there is also clear separation between the two populations (Figure 3).
Figure 3. Good FMO sample and gating strategy.

An example of a good FMO is shown (left), in which the cells are tightly clustered and there is no expression of CD4 (as expected). The gate is placed on the negative CD4 population in the CD4 FMO and copied onto subsequent samples. There is further good separation between the positive and negative populations in the sample (right).
An example of spillover is shown in Figure 4. This problem can be diagnosed by plotting the marker of question against the other markers. A hallmark sign of spillover is signal expression along the line Y=X. In this case, a different fluorophore must be substituted.
Figure 4. Bad FMO sample due to spillover.
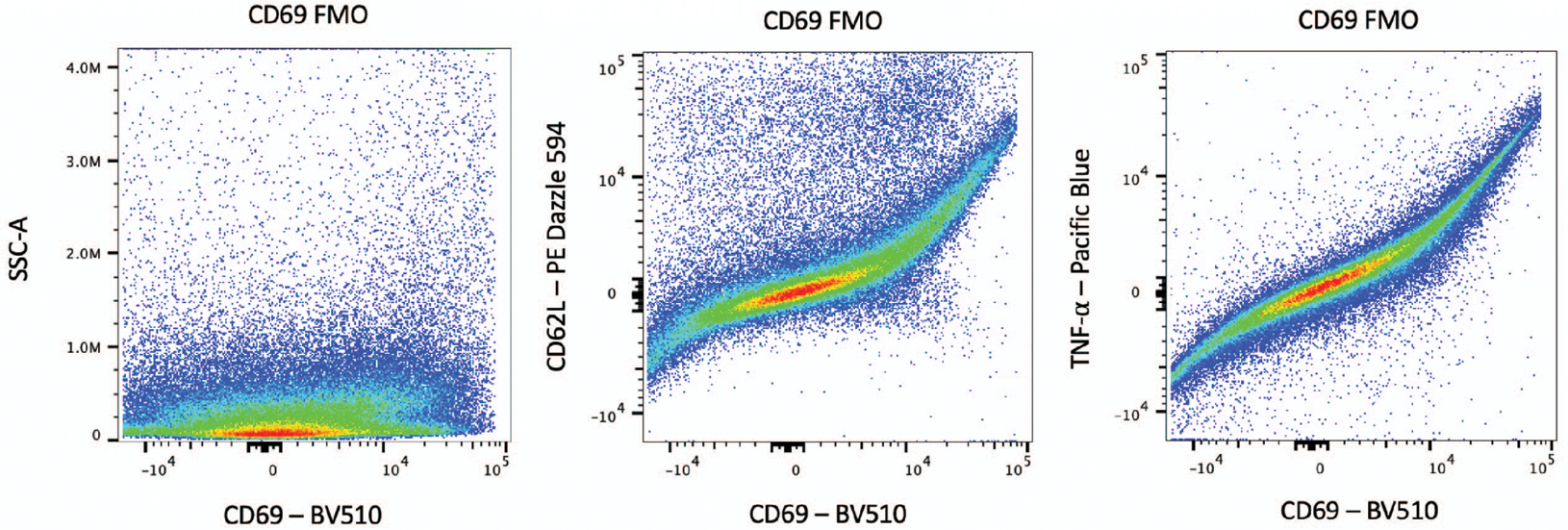
An example of a bad FMO, in which the population is spread across the x-axis (left). The source of the problem can be diagnosed by visualizing the marker versus other panel markers (middle and right). The curved shape of the CD69 in the BV510 channel vs. PE/Dazzle 594 channel and the Pacific Blue channel shows that there is spillover from the other channels.
The single-color reference controls should display low signal in the negative population and a positive spectral signature identical to the fluorophore of interest (Figure 5). If there is signal in the negative population, then there may be non-specific binding of the antibody to the negative beads (Figure 6). Further, if the spectral signature does not match the expected signature exactly, there may be contamination from other fluorophores (Figure 7). In both of these cases, the single-color controls should be re-run.
Figure 5. Good single-color control with beads.
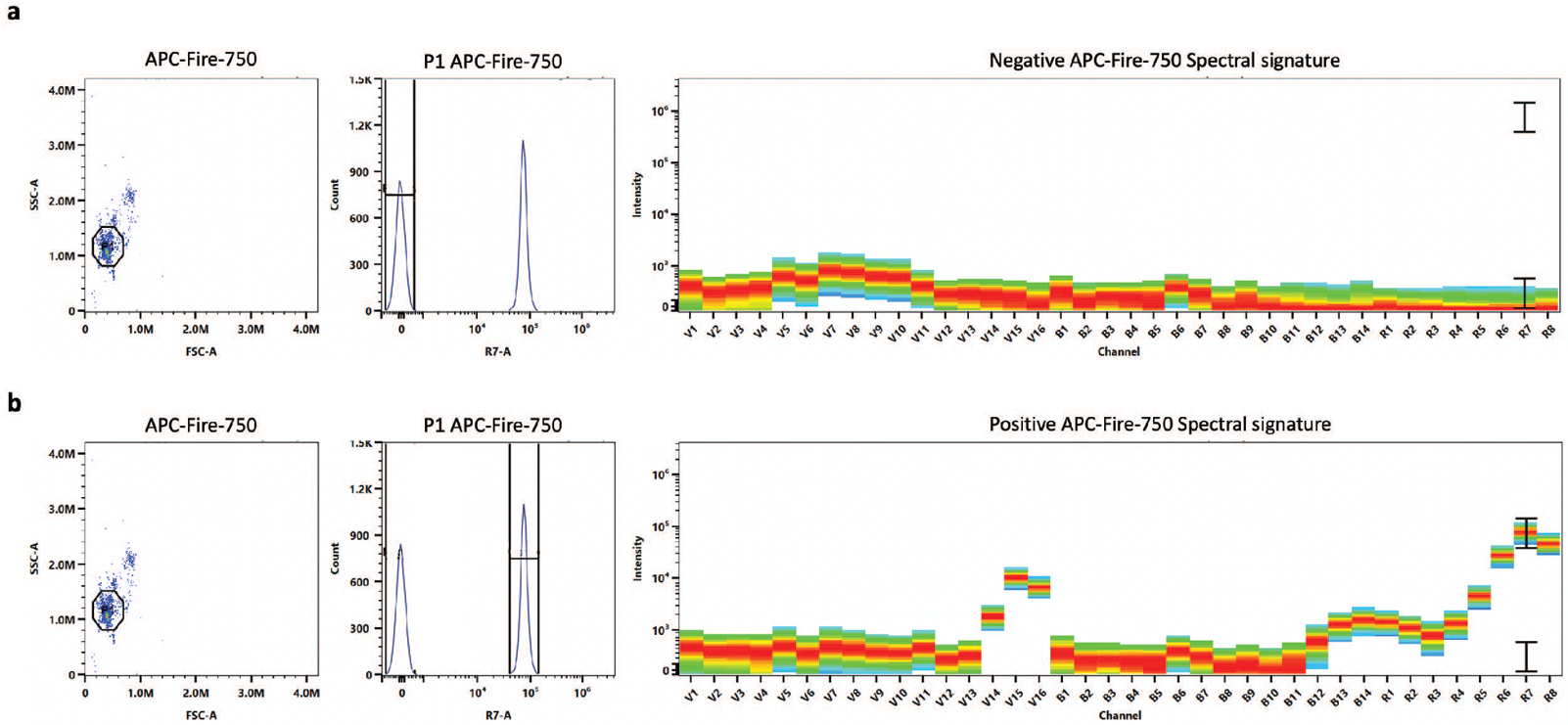
A good APC Fire 750 single-color control with beads is shown. The negative spectral signature should not show any signal (a). The positive spectral signature should match the expected fluorophore spectral signature exactly (b).
Figure 6. Bad single-color control due to fluorescent signal in negative population.
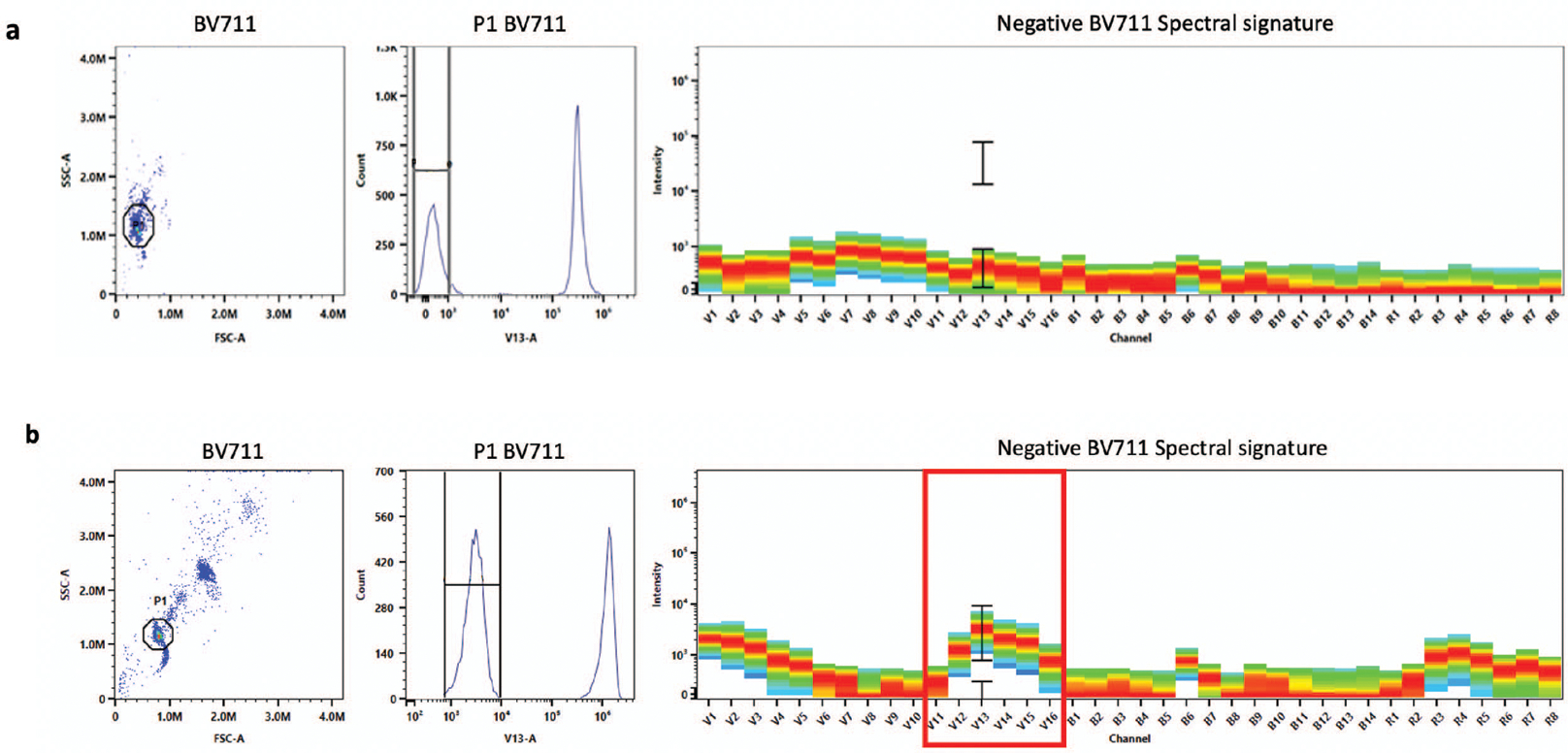
An example where the fluorescent signal in the negative population is low indicating that the single-color control has been run properly (a). There is fluorescent signal (indicated in the red box) in the negative population indicating that there may be non-specific binding of the fluorophore (b). The BV711 single-color control should be prepared again and rerun.
Figure 7. Bad single-color control due to incorrect spectral signature.

The expected APC Fire 750 spectral signature is shown in (a). In the sample, the spectral signatures do not match (b). This is an example of APC signal in the APC Fire-750 single-color control. The control should be prepared again.
Complete analysis of the data acquired using this protocol can be found in the manuscript currently under review at “Cyto-feature engineering: a pipeline for flow cytometry analysis to uncover immune populations and associations with disease.” The code to analyze the data can be found here: https://github.com/aef1004/cyto-feature_engineering.
TIME CONSIDERATIONS:
The time required for these protocols depends on the number and type of samples. In protocol 1, it takes about 1.5 hours to prepare one sample from tissue harvesting to adding cells to a plate for staining. However, if it is the lung tissue being stained, 30 minutes should be added to this time due to the incubation with DNAse/liberase.
To perform intracellular cytokine staining, an initial incubation step of 6 hours must occur. Following this incubation, it takes approximately 4.5–5 hours to stain. The plate must then sit overnight with the intracellular antibodies. The next day washing steps take approximately 30 minutes. Finally, it takes approximately 1–2 minutes to read one sample with 100,000 cells (with an initial concentration of 500,000 cells per tube).
Table 4.
Troubleshooting problems and solutions
| Problem | Potential Cause | Potential Solution |
|---|---|---|
| No cells on flow cytometer | Cells were knocked out of the plate when performing the wash steps Cytometer is clogged |
When decanting supernatant, tap more gently Run 10% bleach or 33% Contrad through the cytometer until the clog is removed |
| Fluorescent signal in the negative population of the single-color bead control | Non-specific binding of antibody to negative beads | Wash the beads with FACS Staining Buffer again to remove non-specific binding on the negative beads |
| Spectral signature of single-color control does not match expected signature for the fluorophore | Contamination of control with another fluorescent antibody Tandem dye degraded |
Re-prepare control Replace tandem dye with new vial |
| High proportion of cells are dead | Cells left in DNAse/liberase for too long Cells left in RBC lysing buffer for too long Centrifuged too long and too hard Cells incubated with Protein Transport Inhibitor for too long or too high of a concentration |
Reduce the amount of time in the DNAse/liberase Reduce the amount of time in the RBC lysing buffer Reduce the centrifuge speed and/or time Reduce the amount of time or concentration of Protein Transport Inhibitor |
| Fluorophore spill-over into other channels | Spectra of two markers is indistinguishable | Substitute a different fluorophore-marker pair |
| Low signal in certain markers | Marker expression might be too low or inexistent Experimental design (i.e., timepoint or vaccine stimulation) does not elicit certain markers Markers chosen were too dim Tandem dyes degraded or decoupled |
Check that the marker is supposed to be expressed in the particular cells or animal model Choose a time point closer to vaccination or stimulate the cells with PMA-ionomycin Choose a brighter fluorophore for the specific marker or optimize dilution used Use a new vial of the tandem dye |
| Not a clear separation in the live/dead sample stain | Residual protein left in the media is binding to and quenching Zombie-NIR Single-color control does not contain both positive and negative populations |
Add additional washing step with PBS prior to staining with Zombie-NIR Use ArC Amine Reactive Compensation Bead Kit (Thermo Fisher, cat# A10346) to run single-color live-dead control |
| Bad separation between negative and positive populations | Antibody concentration is either too high or too low Unbound antibodies were not adequately washed from the samples |
Optimize dilution of antibodies Add additional centrifugation and PBS wash |
SIGNIFICANCE STATEMENT:
With new high-throughput flow cytometry, data analysis has become highly complex. Using open-source software, it is now possible to explore these large datasets, simplifying the seemingly complex data. However, in order to perform these analyses, sample preparation, staining procedure, and use of controls must follow rigorous protocols. In this Current Protocols article, we describe the best practices for preparation and acquisition of spectral flow cytometry samples. Following this protocol will lead to clean results that can be used with the cyto-feature engineering data analysis pipeline described previously (Fox et al., Under Revision).
ACKNOWLEDGEMENTS:
This panel optimization and protocol work was supported by NIH grant 1R01 AI127475-01A1 and National Science Foundation grant (DGE-1450032). We would also like to thank the Colorado State University Flow Core Facility for the use of their flow cytometers.
Footnotes
INTERNET RESOURCES:
This website contains the code to analyze flow cytometry data using the Cyto-feature engineering analysis method.
Contributor Information
G. Brooke Anderson, 350 W. Lake Street, Fort Collins, CO 80523.
Marcela Henao-Tamayo, 401 W. Pitkin St., Fort Collins, CO 80521.
LITERATURE CITED:
- Becton D. a. C. (2012). Introduction to Flow Cytometry: A Learning Guide. In. [Google Scholar]
- Biologicals N. Tandem Dye Conjugated Antibodies In. [Google Scholar]
- Biosciences C. (2019). Spectral Analysis Meets Flow Cytometry. In. [Google Scholar]
- Eshghi ST, Au-Yeung A, Takahashi C, Bolen CR, Nyachienga MN, Lear SP, … O’Gorman WE (2019). Quantitative Comparison of Conventional and t-SNE-guided Gating Analyses. Frontiers in Immunology, 10. doi: 10.3389/fimmu.2019.01194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Font L, Pellefigues C, Mayer JU, Small SJ, Jaimes MC, & Price KM (2020). Panel Design and Optimization for High-Dimensional Immunophenotyping Assays Using Spectral Flow Cytometry. Current protocols in cytometry, 92(1), e70–e70. doi: 10.1002/cpcy.70 [DOI] [PubMed] [Google Scholar]
- Finak G, Frelinger J, Jiang WX, Newell EW, Ramey J, Davis MM, … Gottardo R (2014). OpenCyto: An Open Source Infrastructure for Scalable, Robust, Reproducible, and Automated, End-to-End Flow Cytometry Data Analysis. Plos Computational Biology, 10(8). doi: 10.1371/journal.pcbi.1003806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- FluoroFinder. (2020). FluoroFinder Flow Cytometry Panel Builder. Retrieved from https://fluorofinder.com/
- Fox A, Dutt TS, Karger B, Mauricio RL, Obregon-Henao A, Anderson GB, & Henao-Tamayo M (Under Revision). Cyto-Feature Engineering: A Pipeline for Flow Cytometry Analysis to Uncover Immune Populations and Association with Disease. In (pp. [In Revision]). Scientific Reports. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahnke YD, & Roederer M (2007). Optimizing a multicolor immunophenotyping assay. Clinics in Laboratory Medicine, 27(3), 469-+. doi: 10.1016/j.cII.2007.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roederer M (2002). Compensation in flow cytometry. Current protocols in cytometry, Chapter 1, Unit 1.14-Unit 11.14. doi: 10.1002/0471142956.cy0114s22 [DOI] [PubMed] [Google Scholar]
- Van Gassen S, Callebaut B, Van Helden MJ, Lambrecht BN, Demeester P, Dhaene T, & Saeys Y (2015). FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytometry Part A, 87A(7), 636–645. doi: 10.1002/cyto.a.22625 [DOI] [PubMed] [Google Scholar]
- Verschoor CP, Lelic A, Bramson JL, & Bowdish DME (2015). An introduction to automated flow cytometry gating tools and their implementation. Frontiers in Immunology, 6. doi: 10.3389/fimmu.2015.00380 [DOI] [PMC free article] [PubMed] [Google Scholar]