

Abstract
Psoriasis and atopic dermatitis are chronic inflammatory skin diseases characterized by keratinocyte (KC) hyperproliferation and epidermal acanthosis (hyperplasia). The milieu of disease-associated cytokines and soluble factors is considered a mitogenic factor; however, pinpointing the exact mitogens in this complex microenvironment is challenging. We employed organotypic human epidermal equivalents, faithfully mimicking native epidermal proliferation and stratification, to evaluate the proliferative effects of a broad panel of (literature-based) potential mitogens. The KC GF molecule, the T-helper 2 cytokines IL-4 and IL-13, and the psoriasis-associated cytokine IL-17A caused acanthosis by hyperplasia through a doubling in the number of proliferating KCs. In contrast, IFN-γ lowered proliferation, whereas IL-6, IL-20, IL-22, and oncostatin M induced acanthosis not by hyperproliferation but by hypertrophy. The T-helper 2‒cytokine‒mediated hyperproliferation was Jak/signal transducer and activator of transcription 3 dependent, whereas IL-17A and KC GF induced MAPK/extracellular signal‒regulated kinase kinase/extracellular signal‒regulated kinase‒dependent proliferation. This discovery that key regulators in atopic dermatitis and psoriasis are direct KC mitogens not only adds evidence to their crucial role in the pathophysiological processes but also highlights an additional therapeutic pillar for the mode of action of targeting biologicals (e.g., dupilumab) or small-molecule drugs (e.g., tofacitinib) by the normalization of KC turnover within the epidermal compartment.
Abbreviations: 3D, three-dimensional; AD, atopic dermatitis; EdU, 5-ethynyl-2′-deoxyuridine; ERK, extracellular signal‒regulated kinase; HEE, human epidermal equivalent; KC, keratinocyte; KGF, keratinocyte GF; MEK, MAPK/ extracellular signal‒regulated kinase kinase; STAT, signal transducer and activator of transcription; Th, T helper
Introduction
Keratinocytes (KCs) are the major cell type of the epidermis and maintain its structural integrity, contribute to the barrier function of the skin, and regulate immunological cues as part of the innate immune system. KC proliferation is vital for physiological epidermal renewal and for epidermal regeneration after wounding. In this process, interfollicular epidermal stem cells give rise to transit-amplifying cells in the basal layer of the epidermis. After proliferation, daughter cells exit the cell cycle, start to differentiate, and move upward through the stratum spinosum and stratum granulosum to finally end up as terminally differentiated corneocytes before shedding from the skin surface (Lechler and Fuchs, 2005; Weinstein et al., 1984).
In healthy skin, complete epidermal renewal takes approximately 30 days. Accelerated KC turnover or hyperproliferation is required during skin injury. However, under certain circumstances, this tightly regulated process is disturbed, and basal KCs remain hyperproliferative. Besides being related to malignancies (Frost et al., 1966; Wright, 1985), KC hyperproliferation is a classic feature of several benign skin disorders, including the chronic inflammatory skin diseases psoriasis (Leigh et al., 1985; Vanscott and Ekel, 1963; Weinstein et al., 1985) and atopic dermatitis (AD) (Sapuntsova et al., 2002). Although the underlying pathogenesis and pathophysiological processes are distinct, both diseases are typically characterized by epidermal thickening (acanthosis) owing to hyperplasia (increased cell numbers). The resulting excessive skin scaling is due to both disturbed regulation of epidermal proliferation and the terminal differentiation and desquamation process. These disturbances are generally considered to be caused by the cutaneous milieu of potentially mitogenic factors secreted by the inflammatory tissue microenvironment (Guttman-Yassky and Krueger, 2017).
From the early 80s onward, in vitro studies have addressed KC proliferation aiming to identify mitogenic stimuli such as GFs and cytokines that could be relevant for inflammatory diseases or epidermal repair processes. The results of these studies are conflicting and difficult to interpret because of the various culture systems (monolayers vs. organotypic models, foreskin vs. adult KCs) and different readout parameters (3H-thymidine incorporation, surrogate markers such as keratin 16). This lack of a systematic approach hampered the identification of bona fide KC mitogens. Many of these studies were furthermore not specifically designed to investigate KC (hyper)proliferation but focused on the involvement of the investigated molecules in AD or psoriasis. Clearly, identification of relevant, disease-specific mitogens is ever needed to understand the pathophysiology and to facilitate target drug discovery and development.
We therefore conducted a literature search and screened the identified candidate molecules for their potential as acting KC mitogens using a standardized organotypic epidermal culture system, a concentration series of candidate molecules, and primary epidermal KCs from multiple donors. This comprehensive study uncovered mitogens and calls on the validity of previously reported mitogens. In addition, we provide evidence for the relevance of the signaling pathways associated with the identified mitogens for the induction of KC hyperproliferation.
Results
Selection of candidate molecules involved in KC proliferation
We performed a comprehensive literature search, summarized in Table 1, for selecting candidate mitogens associated with KC hyperproliferation particularly related to psoriasis and AD or wound healing. Studies were screened for information on the effects of soluble factors on KC (hyper)proliferation and epidermal alterations, resulting in a total of 34 potential candidate mitogens that we selected for further functional analysis. This list includes the classical disease-associated cytokines (derived from T helper [Th] 1, Th2, Th17 Th22 cells) but also GFs and serum-derived molecules.
Table 1.
Candidate Mitogens Associated with Keratinocyte Proliferation in Psoriasis and Atopic Dermatitis
| Candidate Mitogen | Species | Model | Link Toward Mitogenicity | Ref. |
|---|---|---|---|---|
| CCL17 | Human | In vivo | Atopic dermatitis | (Hijnen et al., 2004) |
| Human Mouse |
2D, In vivo |
Association with keratinocyte proliferation | (Nakahigashi et al., 2011) | |
| Human | In vivo | Atopic dermatitis | (Fujisawa et al., 2002) | |
| CGRP | Human | In vivo | Psoriasis | (Jiang et al., 1998) |
| Human | 2D | Association with keratinocyte proliferation | (Yu et al., 2009) | |
| Mouse | In vivo | Association with keratinocyte proliferation | (Seike et al., 2002) | |
| CRNN | Human Mouse | 2D, In vivo |
Psoriasis | (Li et al., 2019) |
| EGF | Human | Ex vivo | Association with keratinocyte proliferation and migration | (Bhora et al., 1995) |
| Human | 3D (HSE) | Association with keratinocyte proliferation and migration | (Gibbs et al., 2000) | |
| Et-1 | Human | 2D | Association with keratinocyte proliferation | (Venuti et al., 1997) |
| FCS | Human | 2D | Enhances keratinocyte migration | (Henry et al., 2003) |
| Mouse | 2D | Serum contains components enhancing and inhibiting keratinocyte proliferation | (Bertolero et al., 1986) | |
| GM-CSF | Human | In vivo | Association with keratinocyte proliferation | (Braunstein et al., 1994) |
| Human | 2D, In vivo |
Atopic dermatitis | (Pastore et al., 1997) | |
| Mouse | In vivo | Association with keratinocyte proliferation | (Mann et al., 2001) | |
| IFN-γ | Human | In vivo | Psoriasis | (Uyemura et al., 1993) |
| Human | 2D, Ex vivo |
Decreases keratinocyte proliferation | (Hattori et al., 2002) | |
| IGF-1 | Human | 2D, In vivo |
Elevated IGF-1R expression associated with enhanced keratinocyte proliferation | (Krane et al., 1991) |
| Human | 2D, In vivo |
IGF-1R expression in proliferative keratinocytes | (Tavakkol et al., 1992) | |
| Human Mouse |
2D, In vivo |
IGF-1R inhibition reverses keratinocyte hyperproliferation | (Wraight et al., 2000) | |
| IL-1α | Mouse | 2D, In vivo |
Association with keratinocyte proliferation | (Hobbs et al., 2004) |
| Human | 3D (HSE) | Association with keratinocyte proliferation in co-culture with fibroblasts | (Maas-Szabowski et al., 2000) | |
| Human | 3D (HSE) | Association with keratinocyte proliferation | (Taniguchi et al., 2014) | |
| IL-1β | Human Mouse | In vivo | Psoriasis | (Cai et al., 2019) |
| IL-4 | Human | In vivo | Atopic dermatitis | (Hamid et al., 1994) |
| Human | 2D | Association with keratinocyte proliferation | (Yang et al., 1996) | |
| IL-5 | Human | In vivo | Atopic dermatitis | (Kondo et al., 2001) |
| IL-6 | Human | 2D, In vivo |
Psoriasis | (Grossman et al., 1989) |
| Rat | In vivo | Association with keratinocyte proliferation | (Sawamura et al., 1998) | |
| IL-8 | Human | In vivo | Psoriasis | (Steude et al., 2002) |
| Human | 3D (HSE) | Association with keratinocyte proliferation | (Steude et al., 2002) | |
| IL-12 | Human | In vivo | Atopic dermatitis | (Hamid et al., 1996) |
| Human | In vivo | Psoriasis | (Yawalkar et al., 1998) | |
| IL-13 | Mouse | In vivo | Atopic dermatitis | (Herrick et al., 2003) |
| Human | 2D | Association with keratinocyte proliferation | (Matsumura et al., 2015) | |
| IL-15 | Human | In vivo | Psoriasis | (Rückert et al., 2000) |
| Human | 2D | Enhances keratinocyte proliferation | (Yano et al., 2003) | |
| Human | 2D, In vivo |
Association with keratinocyte proliferation and migration | (Jones et al., 2016) | |
| IL-17A | Human Mouse |
2D, In vivo |
Psoriasis | (Charruyer et al., 2017) |
| Mouse | 2D, In vivo |
Association with keratinocyte proliferation | (Wu et al., 2015) | |
| Human | 2D, In vivo |
Association with keratinocyte proliferation, Psoriasis |
(Ma et al., 2016) | |
| IL-19 | Human Mouse | 2D, In vivo |
Psoriasis | (Witte et al., 2014) |
| Human Mouse | 2D, In vivo |
Wound healing | (Sun et al., 2013) | |
| Human | In vivo | Atopic dermatitis, Cutaneous T-cell lymphoma |
(Oka et al., 2017) | |
| Human Mouse | 2D, In vivo |
Atopic dermatitis | (Bao et al., 2014) | |
| Human | In vivo | Psoriasis | (Li et al., 2005) | |
| IL-20 | Human | In vivo | Psoriasis | (Wei et al., 2005) |
| IL-21 | Human, Mouse | In vivo | Psoriasis | (Caruso et al., 2009) |
| Human | In vivo | Atopic dermatitis | (Mizutani et al., 2017) | |
| IL-22 | Human | 2D, 3D (HEE) |
Enhances epidermal hypertrophy and keratinocyte migration | (Boniface et al., 2005) |
| Human | 2D | Association with keratinocyte proliferation | (Ekman et al., 2019) | |
| Mouse | In vivo | Psoriasis | (Zheng et al., 2007) | |
| IL-23 | Human Mouse |
2D, In vivo |
Induces epidermal hyperplasia | (Chan et al., 2006) |
| Human Mouse |
2D, In vivo |
Psoriasis | (Works et al., 2014) | |
| Human | In vivo | Psoriasis | (Nair et al., 2009) | |
| IL-25 | Human Mouse |
2D, In vivo |
Psoriasis | (Xu et al., 2018) |
| IL-27 | Human | 2D, In vivo |
Psoriasis | (Shibata et al., 2010) |
| Human Human Mouse |
2D Ex vivo In vivo |
Wound healing | (Yang et al., 2017) | |
| IL-31 | Mouse | In vivo | Association with keratinocyte proliferation and epidermal acanthosis | (Singh et al., 2016) |
| Mouse | In vivo | Atopic dermatitis | (Dillon et al., 2004) | |
| Human | 2D, In vivo |
Psoriasis | (Finch et al., 1997) | |
| Human | 3D (HSE) | Association with keratinocyte proliferation in coculture with fibroblasts | (Maas-Szabowski et al., 2000) | |
| KGF | Human | 3D (HSE) | Association with keratinocyte proliferation | (Andreadis et al., 2001) |
| Human | 2D, In vivo |
Psoriasis | (Finch et al., 1997) | |
| Human | 2D | Association with keratinocyte proliferation | (Marchese et al., 1990) | |
| Leptin | Human Mouse |
2D In vivo |
Association with keratinocyte proliferation and involvement in wound healing | (Stallmeyer et al., 2001) |
| OSM | Human | 2D, 3D (HEE), In vivo |
Atopic dermatitis, Psoriasis |
(Boniface et al., 2007) |
| Human | 2D | Association with keratinocyte proliferation | (Wang et al., 2020) | |
| PGE2 | Mouse | In vivo | Receptor expression associated with enhanced keratinocyte proliferation and tumor formation | (Chun et al., 2010) |
| TNF-α | Human | 2D | Decreases keratinocyte proliferation | (Detmar and Orfanos, 1990) |
| Human | 2D, In vivo |
Psoriasis | (Johansen et al., 2006) | |
| TSLP | Human | 2D, In vivo |
Atopic dermatitis | (Soumelis et al., 2002) |
| VIP | Human | In vivo | Atopic dermatitis, psoriasis | (Anand et al., 1991) |
| Human | 2D | Receptor expression associated with enhanced keratinocyte proliferation | (Kakurai et al., 2001) | |
| Human | 2D | Association with keratinocyte proliferation | (Sung et al., 1999) |
Abbreviations: 2D, two-dimensional; 3D, three-dimensional; CRNN, cornulin; FCS, fetal calf serum; HEE, human epidermal equivalent; HSE, human skin equivalent; KGF, keratinocyte GF; OSM, oncostatin; Ref, reference; TSLP, thymic stromal lymphopoietin.
Analysis of KC proliferation in organotypic human epidermis
For evaluating the direct mitogenic potential of relevant factors as listed in Table 1, we employed the widely used and highly standardized organotypic human epidermal equivalent (HEE) model (Niehues et al., 2018), generated from primary adult human KCs in the absence of a dermal substrate to omit any confounding interactions with other cell types. First, we validated that the formation of HEEs using this methodology is based on the proliferation of KCs in the basal layer that generate a complete epidermis over time and not by the stacking of an excess of KCs as known for other models (Mildner et al., 2006; Sen et al., 2010; Truong et al., 2006). Directly after a 24-hour exposure of the HEEs to the thymidine analog 5-ethynyl-2′-deoxyuridine (EdU) on day 5 of the air‒liquid interface culture, solely basal KCs appeared EdU positive. After a wash-out period of 2 days, EdU-positive cells were found in basal cells, indicative of label-retaining transit-amplifying cells, but also in differentiated KCs of the spinous layer, which correspond to the daughter cells that entered the terminal differentiation program (Figure 1a). This process of KC proliferation and subsequent differentiation in our HEE model system resembles epidermal renewal and KC maturation in vivo.
Figure 1.

Keratinocyte proliferation in vitro. (a) EdU was incorporated in proliferating keratinocytes for 24 hours from day 5 to day 6 of air‒liquid interface culture. HEEs were harvested on day 6 (directly after EdU incorporation or on day 8 [2 days after EdU incorporation]) (n = 2). (b) Ki-67 staining of HEEs on days 5 and 8 (n = 8). (c) HEEs harvested on days 12, 16, and 20 of the air‒liquid interface stained with H&E and Ki-67 to evaluate the epidermal thickness and proliferation (n = 1). Bar = 100 μm. EdU, 5-ethynyl-2′-deoxyuridine; HEE, human epidermal equivalent.
KC proliferation in organotypic models versus in native human skin
Before detection of hyperproliferative changes on stimulation with the molecules of interest, we first sought to show the proliferative capacity of the HEE model and compare this with that of in vivo native skin. In general, the proliferation rate of KCs in HEEs was, on the basis of the surrogate proliferation marker Ki-67, higher than that in the native skin (Figure 1b). This elevated proliferation rate is commonly seen in organotypic skin and is in this study related to the 8-day period of the air‒liquid interface before harvesting. On prolonged air‒liquid interface culture periods (up to 20 days), proliferation rates go down, resulting in a thinning of the living epidermis until ultimately all transit-amplifying cells exit the cell cycle marking the end of the organotypic life span (Figure 1c).
To put the mitogenic potential of the tested molecules into perspective regarding hyperproliferation in AD or psoriasis pathophysiology, we compared the epidermal features of normal skin with those of both hyperproliferative skin conditions. The histology of skin biopsies showed the classical morphological hallmarks of AD (e.g., acanthosis, spongiosis) and psoriasis (e.g., acanthosis, rete ridges, parakeratosis) (Figure 2). In normal skin, only a small portion of basal KCs is Ki-67+. In contrast, the acanthotic epidermis of AD and psoriasis skin shows most of all basal KCs as Ki-67+, whereas also in suprabasal cell layers, Ki-67+ cells are found. Semiautomated software‒guided quantification analysis estimates a 5.5-fold higher number of Ki-67+ cells in AD and over a nine-fold higher number of Ki-67+ cells in psoriasis in vivo.
Figure 2.

Keratinocyte proliferation in vivo. H&E and Ki-67 staining of normal skin and AD and psoriasis lesional skin. Beyond increased proliferation, AD and psoriasis show typical features such as acanthosis (both indicated by black lines), spongiosis (AD: indicated by closed arrow), and parakeratosis (psoriasis: indicated by open arrow). Boxes show an enlarged section of Ki-67‒positive cells in the basal layer. Bar = 100 μm. The graph shows the quantification of Ki-67 (P = 0.005) staining of normal skin (n = 4), AD (n = 2), and psoriasis (n = 3) as the number of Ki-67‒positive cells/mm stratum granulosum to correct for basal layer elongation in AD and psoriasis. ∗P < 0.05. Data are presented as mean ± SEM. AD, atopic dermatitis.
Soluble factors affecting proliferation and epidermal morphology in organotypic epidermis
After the validation procedures, mitogenic molecules were individually tested in the HEE cultures. For this first screening, every molecule was tested in a dose series, depicted in Table 2, using HEEs from two different KC donors. General epidermal morphology was assessed, and HEEs were screened for AD and psoriasis phenotypic features. KC proliferation was assessed by Ki-67 protein expression analysis. Results, as summarized in Table 3, indicate that most of the investigated candidates did not alter epidermal morphology nor changed KC proliferation rates. Two candidates (fetal calf serum, TNF-α) detrimentally affected epidermal morphology at higher concentrations. IFN-γ induced hypoplasia and lowered proliferation rates, whereas IL-6, IL-20, IL-22, and oncostatin M induced acanthosis but without any signs of hyperproliferative KCs, illustrating their hypertrophic (cell enlargement) effects on KCs. Epidermal spongiosis (intercellular edema), a typical AD hallmark, was detected for IL-1α and IL-1β. Of the 34 putative mitogens, we found the following four effector molecules to induce hyperproliferation accompanied by acanthosis in our HEE model: KC GF (KGF), IL-4, IL-13, and IL-17A (Table 3 and Figure 3a).
Table 2.
Effector Molecules and Pathway Inhibitors Tested in HEE Model
| Effector/Inhibitor | Manufacturer (Catalog Number) | Concentrations Tested |
|---|---|---|
| CCL17/TARC | PeproTech Recombinant human TARC (CCL17), 300-30 |
10, 30, and 100 ng/ml |
| CGRP | Sigma-Aldrich Calcitonin gene-related peptide, C0167 |
1, 10, and 100 ng/ml |
| CRNN | ProSpec Recombinant human cornulin, Pro-797 |
10, 30, and 100 ng/ml |
| EGF | Sigma-Aldrich EGF from murine submaxillary gland E4127 |
0.5, 1, and 5 ng/ml |
| Et-1 | Sigma-Aldrich Porcine human endothelin 1, E7764 |
10, 30, and 100 ng/ml |
| FBS | Hyclone Celbio FBS, SH30071.03 |
5, 10, and 20% |
| GM-CSF | PeproTech Recombinant human granulocyte-macrophage colony-stimulating Factor, 300-03A |
10, 30, and 100 ng/ml |
| IFN-γ | Hycult Biotech Recombinant Human IFN-gamma, HC2030-01 |
20, 100, and 250 Units/ml |
| IGF-1 | PeproTech Recombinant human insulin-like growth factor-I,100-11 |
10, 30, and 100 ng/ml |
| IL-1α | Stemcell Technologies Recombinant human IL-1α, 78076 |
10, 30, and 100 ng/ml |
| IL-1β | PeproTech Recombinant human IL-1β, 200-01B |
10, 30, and 100 ng/ml |
| IL-4 | PeproTech Recombinant human IL-4, 200-04 |
10, 30, and 100 ng/ml |
| IL-5 | PeproTech Recombinant human IL-5, 200-05 |
10, 30, and 100 ng/ml |
| IL-6 | PeproTech Recombinant human IL-6, 200-06 |
10, 30, and 100 ng/ml |
| IL-8 | PeproTech Recombinant human IL-8 (CXCL8), 200-08 |
10, 30, and 100 ng/ml |
| IL-12 | PeproTech Recombinant human IL-12 p70, 200-12 |
10, 30, and 100 ng/ml |
| IL-13 | PeproTech Recombinant human IL-13, 200-13 |
10, 30, and 100 ng/ml |
| IL-15 | PeproTech Recombinant human IL-15, 200-15A |
1, 10, and 100 ng/ml |
| IL-17A | PeproTech Recombinant human IL-17A, 200-17 |
10, 30, and 100 ng/ml |
| IL-19 | PeproTech Recombinant human IL-19, 200-19 |
10, 30, and 100 ng/ml |
| IL-20 | PeproTech Recombinant human IL-20, 200-20 |
10, 30, 100 ng/ml |
| IL-21 | PeproTech Recombinant human IL-21, 200-21 |
10, 30, and 100 ng/ml |
| IL-22 | PeproTech Recombinant human IL-22, 200-22 |
10, 30, and 100 ng/ml |
| IL-23 | PeproTech Recombinant human IL-23, 200-23 |
10, 30, and 100 ng/ml |
| IL-25 | PeproTech Recombinant human IL-17E, 200-24 |
10, 30, and 100 ng/ml |
| IL-27 | PeproTech Recombinant human IL-27, 200-38 |
10, 30, and 100 ng/ml |
| IL-31 | PeproTech Recombinant human IL-31, 200-31 |
10, 30, and 100 ng/ml |
| KGF | Sigma Aldrich Recombinant human KGF, K1757 |
10, 30, and 100 ng/ml |
| Leptin | PeproTech Recombinant human Leptin, 300-27A |
10, 30, and 100 ng/ml |
| OSM | PeproTech Recombinant human Oncostatin M, 300-10T |
10, 30, and 100 ng/ml |
| PGE2 | Sigma Aldrich Synthetic PGE2, P0409 |
10, 30, and 100 μM |
| TNFα | PeproTech Recombinant human TNF-α, 300-01A |
10, 30, and 100 ng/ml |
| TSLP | PeproTech Recombinant human TSLP, 300-62 |
3, 10, and 30 ng/ml |
| VIP | Sigma Aldrich Porcine human vasoactive intestinal peptide, V3628 |
0.01, 0.1, and 1 μM |
| Dupilumab | Dupixent, Sanofi Genzyme | 10 nM |
| Tofacitinib | Sigma-Aldrich PZ0017 |
450 nM |
| JakI inhibitor | Calbiochem JAK inhibitor I, 420099 |
100 nM |
| MEK/ERK inhibitor | Sigma Aldrich U0126 monoethanolate, U120 |
100 nM |
| p38 MAP Kinase inhibitor IV | Sigma-Aldrich SML0543 |
100 nM |
Abbreviations: CRNN, cornulin; FBS, fetal bovine serum; HEE, human epidermal equivalent; KGF, keratinocyte GF; MEK/ERK, MAPK/extracellular signal‒regulated kinase kinase/extracellular signal‒regulated kinase kinase; OSM, oncostatin; TSLP, thymic stromal lymphopoietin.
Table 3.
Screening of all Potential Mitogenic Factors in Dosage Range in HEE Model
| Effector | Morphology (H&E) | Proliferation (Ki-67) | Concentrations Tested | Morphological Changes |
|---|---|---|---|---|
| CCL17/TARC | 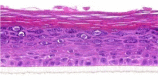 |
 |
10, 30, and 100 ng/ml | ND |
| CGRP | 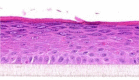 |
 |
1, 10, and 100 ng/ml | ND |
| CRNN |  |
 |
10, 30, and 100 ng/ml | ND |
| EGF | 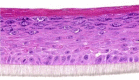 |
 |
0.5, 1, and 5 ng/ml | ND |
| Et-1 |  |
 |
10, 30, and 100 ng/ml | ND |
| FCS | 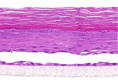 |
 |
5, 10, and 20% | Toxicity |
| GM-CSF | 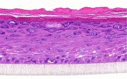 |
 |
10, 30, and 100 ng/ml | ND |
| IFN-γ | 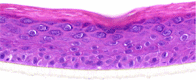 |
 |
20, 100, and 250 Units/ml | Hypoplasia |
| IGF-1 |  |
 |
10, 30, and 100 ng/ml | ND |
| IL-1α |  |
 |
10, 30, and 100 ng/ml | Spongiosis |
| IL-1β | 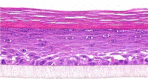 |
 |
10, 30, and 100 ng/ml | Spongiosis |
| IL-4 |  |
 |
10, 30, and 100 ng/ml | Hyperplasia |
| IL-5 |  |
 |
10, 30, and 100 ng/ml | ND |
| IL-6 | 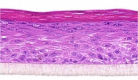 |
 |
10, 30, and 100 ng/ml | Hypertrophy |
| IL-8 |  |
 |
10, 30, and 100 ng/ml | ND |
| IL-12 |  |
 |
10, 30, and 100 ng/ml | ND |
| IL-13 |  |
 |
10, 30, and 100 ng/ml | Hyperplasia |
| IL-15 |  |
 |
1, 10, and 100 ng/ml | ND |
| IL-17A | 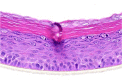 |
 |
10, 30, and 100 ng/ml | Hyperplasia |
| IL-19 |  |
 |
10, 30, and 100 ng/ml | ND |
| IL-20 | 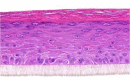 |
 |
10, 30, and 100 ng/ml | Hypertrophy |
| IL-21 | 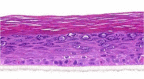 |
 |
10, 30, and 100 ng/ml | ND |
| IL-22 | 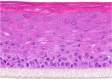 |
 |
10, 30, and 100 ng/ml | Hypertrophy |
| IL-23 | 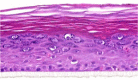 |
 |
10, 30, and 100 ng/ml | ND |
| IL-25 |  |
 |
10, 30, and 100 ng/ml | ND |
| IL-27 |  |
 |
10, 30, and 100 ng/ml | ND |
| IL-31 |  |
 |
10, 30, and 100 ng/ml | ND |
| KGF | 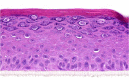 |
 |
10, 30, and 100 ng/ml | Hyperplasia |
| Leptin |  |
 |
10, 30, and 100 ng/ml | ND |
| OSM |  |
 |
10, 30, and 100 ng/ml | Hypertrophy |
| PGE2 |  |
 |
10, 30, and 100 μM | ND |
| TNFα |  |
 |
10, 30, and 100 ng/ml | Toxicity (> 10 ng/mL) |
| TSLP |  |
 |
3, 10, and 30 ng/ml | ND |
| VIP | 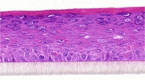 |
 |
0.01, 0.1, and 1 μM | ND |
Abbreviations: CRNN, cornulin; FCS, fetal calf serum; HEE, human epidermal equivalent; KGF, keratinocyte GF; ND, not detected; OSM, oncostatin; TSLP, thymic stromal lymphopoietin.
The photographs show the representative images of two keratinocyte donors tested. Testing of all compounds was performed in several experiments, including one or more test compounds in a dosage range indicated in the table, and all experiments included negative controls (a total of nine keratinocyte donors used). The bold and underlined concentration corresponds to the depicted photograph, and either shows the highest concentration tested without any morphological/mitogenic effect or the lowest effective concentration. Photographs show ×40 magnification.
Figure 3.

Quantification of keratinocyte proliferation on compound stimulation. (a) Ki-67 staining. Graph shows the fold difference (per mm length of the epidermis) on KGF (100 ng/ml, n = 6), IL-4 (10 ng/ml, n = 6), IL-13 (10 ng/ml, n = 3), IL-17A (50 ng/ml, n = 4), IL-22 (30 ng/ml, n = 6), OSM (30 ng/ml, n = 2), and IFN-γ (250 Units/ml, n = 8) stimulation compared with that of the control (set to 1). Data are presented as mean ± SEM. (b) Ki-67 staining of HEEs pretreated for 24 hours with inhibiting agents (100 nM U0126, 450 nM TOF, 100 nM JakI inhibitor, 100 nM p38, 10 nM DUP) and thereafter costimulated with KGF for 72 hours (n = 1). Bar = 100 μm. DUP, dupilumab; HEE, human epidermal equivalent; KGF, keratinocyte GF; OSM, oncostatin M; TOF, tofacitinib.
KGF induces proliferation through MAPK/extracellular signal‒regulated kinase kinase/extracellular signal‒regulated kinase in HEEs
Stimulation of HEEs with KGF induced acanthosis through hyperproliferation as witnessed by increased epidermal thickness and a higher number of Ki-67‒ and EdU-positive KCs from a dosage of 10 ng/ml and higher (Figure 4a‒c). No other apparent morphological changes were observed. The results obtained with the organotypic HEE model used in this study correspond to previous studies showing that KGF is a direct regulator of KC proliferation through MAPK/extracellular signal‒regulated kinase (ERK) kinase (MEK)/ERK signaling. Indeed, pretreatment of HEEs with a selective blocker of MEK signaling before KGF exposure significantly reduced KGF-induced KC proliferation rates (Figure 4d and e). Other blocking agents targeting non-MEK/ERK signaling cascades did not interfere with KGF-dependent hyperproliferation (Figure 3b). Even at higher concentrations (up to 1 μM) the p38 MAPK inhibitor did not inhibit KGF-induced hyperproliferation (data not shown).
Figure 4.

KGF causes keratinocyte hyperproliferation in HEE. HEEs (n = 3) were stimulated with a dosage range of KGF for 72 hours. EdU was incorporated 24 hours before harvesting on day 8 of the air‒liquid interface. (a) H&E, Ki-67, and EdU staining on KGF stimulation and (b) quantification of Ki-67‒ and EdU-positive nuclei/mm length of the HEE. (c) Epidermal thickness (P = 0.008). (d) H&E, Ki-67, and EdU staining and (e) quantification of HEEs pretreated with 100 nM MEK/ERK inhibitor (U0126) for 24 hours and then costimulated with 100 ng/ml KGF for 72 hours (Ki67, P = 0.004; EdU, P = 0.011) (n = 3). ∗P < 0.05. Data are presented as mean ± SEM. Bar = 100 μm. EdU, 5-ethynyl-2′-deoxyuridine; HEE, human epidermal equivalent; KGF, keratinocyte GF; MEK/ERK, MAPK/extracellular signal‒regulated kinase kinase/extracellular signal‒regulated kinase kinase.
Mitogenic effects and signaling pathways of psoriasis- and AD-associated cytokines
In addition to KGF, we found specific ILs classically known to be elevated in psoriasis and/or AD skin to induce KC proliferation in the organotypic HEEs. IL-17A resulted in a higher number of Ki-67+ cells in the basal layer and the presence of Ki-67+ cells in suprabasal layers of the stratum spinosum (Table 3). Results of the initial screening were verified in a dosage range study with two KC donors, and quantification indicated that IL-17A leads to a doubling of total proliferative KC at a concentration of 1 ng/ml and higher (Figure 5).
Figure 5.

IL-17A accelerates keratinocyte proliferation in vitro. (a) H&E, Ki-67, and EdU (24-hour incorporation) staining of HEEs stimulated with a dosage range of IL-17A for 72 hours and (b) quantification presented as the number of Ki-67‒ and EdU-positive nuclei/mm length of the epidermal equivalent (n = 2). Data are presented as mean ± SEM. Bar = 100 μm. EdU, 5-ethynyl-2′-deoxyuridine; HEE, human epidermal equivalent; SEC, secukinumab; TOF, tofacitinib.
To reverse the IL-17A‒induced proliferation and investigate the downstream signaling pathways, HEEs were pretreated with secukinumab (anti‒IL-17A antibody) and diverse inhibiting agents before IL-17A stimulation. Secukinumab and blocking of the MEK/ERK signaling route reduced the number of proliferating KCs, whereas the inhibitors of Jak (tofacitinib) and MAPK signaling (p38) were unable to dampen the IL-17‒induced hyperproliferation (Figure 6a and b). To further dissect the involved signaling events, the phosphorylation (and thus activation) of signal transducer and activator of transcription (STAT) 3 by IL-17A was analyzed. Only, secukinumab and tofacitinib inhibited the phosphorylation of STAT3 (Figure 6c). Because tofacitinib pretreatment did not reduce the numbers of proliferative KCs, IL-17A‒mediated hyperproliferation is not likely a result of the activated Jak/STAT3 signaling pathway.
Figure 6.

Inhibition of IL-17A‒mediated hyperproliferation. (a) H&E, Ki-67, and EdU (24-hour incorporation) staining, (b) quantification (EdU, P = 0.009) and (c) western blot analysis (pSTAT3, P = 0.003) of HEEs pretreated with diverse inhibiting agents (10 nM SEC, 450 nM TOF, 100 nM p38, 100 nM U0126) before costimulation with IL-17A (50 ng/ml) for 72 hours (n = 2). The intensity of the pSTAT3 signal is corrected for β-actin expression, after which the IL-17A‒stimulated HEEs without inhibitor are set at 1 for the relative quantity. Data are presented as mean ± SEM. Bar = 100 μm. EdU, 5-ethynyl-2′-deoxyuridine; HEE, human epidermal equivalent; pSTAT3, phosphorylated signal transducer and activator of transcription 3; SEC, secukinumab; TOF, tofacitinib.
For IL-4 and IL-13 treatment, KC hyperproliferation was also observed (Table 3), and again, an extended dose range study was performed to address the potency of both cytokines. For IL-4 and IL-13, the lowest effective concentration leading to increased Ki-67+ cells was 3 ng/ml (Figure 7a and b). Similar to that of IL-17, Ki-67+ cells were also frequently detected in suprabasal layers. These Th2 cytokines increased the number of proliferating KCs by two-fold in the range of 3 to 30 ng/ml (Figure 7c and d). Next, we evaluated whether hyperproliferation also occurred in a more matured HEE culture after 12, 16, or even 20 days of air‒liquid interface culture. In this experiment, the basal proliferation rate is decreasing in time, whereas terminal differentiation and cornification, as witnessed by a thickening of the stratum corneum, are steadily increasing (Figure 8). The Th2 cytokine‒mediated hyperproliferation was observed in all conditions and thus occurred irrespective of the basal proliferation rate or differentiation status of the organotypic model.
Figure 7.

Th2 cytokines IL-4 and IL-13 induce hyperproliferation in HEE. A dosage range of IL-4 and IL-13 added during the last 72 hours of air-exposed culture shows their effect on keratinocyte proliferation. (a, b) H&E, Ki-67, and EdU (24-hour incorporation) staining and (c, d) quantification presented as Ki-67 (IL-4, P = 0.038; IL-13, P = 0.05) and EdU (IL-4, P = 0.02; IL-13, P = 0.011)-positive nuclei/mm length of the epidermal equivalent (n = 3). Data are presented as mean ± SEM. Bar = 100 μm. EdU, 5-ethynyl-2′-deoxyuridine; HEE, human epidermal equivalent; Th2, T helper 2.
Figure 8.

Prolonged HEEs cultures stimulated with IL-4. HEEs were stimulated with 10 ng/ml IL-4 from day 1 of the air‒liquid interface and harvested on days 12, 16, or 20 of culture. H&E and Ki-67 staining show morphological changes and keratinocyte proliferation during long-term epidermal differentiation (n = 1). Bar = 100 μm. HEE, human epidermal equivalent.
To verify that the Th2 cytokine‒mediated hyperproliferation in KCs was directly related to the IL-4 receptor‒activated Jak/STAT signaling cascade (Rawlings et al., 2004), cytokine stimulation was combined with pretreatment of dupilumab (monoclonal anti‒IL-4R) and tofacitinib (Jak inhibitor). Both dupilumab and tofacitinib prevented IL-4‒ and IL-13‒mediated hyperproliferation through the inhibition of STAT3/6 phosphorylation in HEEs. Tofacitinib reduced STAT3 phosphorylation even below endogenous levels, but this did not correspond to lowered numbers of proliferating KCs (Figure 9 and Figure 10a and b). Furthermore, inhibition of MEK/ERK or MAPK signaling did not reverse IL-4‒induced hyperproliferation (Figure 10c and d). From these results, we can conclude that the Th2 family cytokines IL-4 and IL-13 are direct KC mitogens and act through the activation of Jak/STAT6 signaling but not STAT3 considering the ineffectiveness of phosphorylated STAT3 inhibition on KC proliferation after IL-17 exposure.
Figure 9.

Effect of IL-4/IL-13 signaling inhibition. HEEs were pretreated with DUP (10 nM) and TOF (450 nM) 24 hours before (a) IL-4 or (b) IL-13 stimulation and then costimulated for another 72 hours. H&E, Ki-67, and EdU staining show an effect on epidermal morphology and keratinocyte proliferation. Quantification presented as Ki-67‒ (IL-13, P = 0.011) and EdU- (IL-4, P = 0.022) positive nuclei/mm length of the epidermal equivalent for (c) IL-4 and (d) IL-13 (n = 3). ∗P < 0.05. Data are presented as mean ± SEM. Bar = 100 μm. DUP, dupilumab; EdU, 5-ethynyl-2′-deoxyuridine; HEE, human epidermal equivalent; TOF, tofacitinib.
Figure 10.

Inhibition of IL-4‒/IL-13‒induced proliferation. Western blot analysis and quantification of HEEs shown in Figure 9 for (a) IL-4 and (b) IL-13), stained for pSTAT3 (IL-4, P = 0.015; IL-13, P = 0.002) and pSTAT6 (IL-4, P = 0.004; IL-13, P = 0.008) (corrected for β-actin expression, IL-4‒/IL-13‒stimulated HEEs set at 1) (n = 3). ∗P < 0.05. Data are presented as mean ± SEM. (c) Ki-67 staining and (d) quantification (P = 0.037) of HEEs treated with inhibiting agents (450 nM TOF, 100 nM JakI inhibitor, 100 nM p38, 100 nM U0126) 24 hours before costimulation with 10 ng/ml IL-4 for 72 hours (n = 3). Data are presented as mean ± SEM. Bar = 100 μm. DUP, dupilumab; EdU, 5-ethynyl-2′-deoxyuridine; HEE, human epidermal equivalent; inh, inhibitor; pSTAT3/6, phosphorylated signal transducer and activator of transcription 3/6; TOF, tofacitinib.
Discussion
In this study, we aimed to find the direct drivers of KC hyperproliferation in psoriasis and AD. Of the 34 tested candidates, KGF, IL-4, IL-13, and IL-17A met the criteria for acting as KC mitogens and could thus serve as potential biomarkers for epidermal hyperproliferation.
In this study, we have chosen a three-dimensional (3D) organotypic HEE model purely composed of KCs to selectively identify direct KC mitogens. The benefits of such models over conventional monolayer cultures are that the epidermal tissue structure allows for the monitoring of differential KC turnover induced by the selected factors and that it allows for the detection of morphological features related to proliferation. Furthermore, in conventional monolayer culture models, KCs already reside in an almost maximum proliferative state, which minimizes the window of opportunity for detection of increased KC proliferation (Rasmussen et al., 2013). One limitation of the HEE model is that the molecules tested negative in our screen could still be indirect mitogens through the activation of other cell types (e.g., immune cells) in the skin that in turn secrete mitogenic factors for KCs. In addition, mitogens that strongly act in concert with other factors are not identified in this study. Furthermore, possible autocrine regulation by molecules produced on cytokine exposure may contribute to the observed effects. Therefore, our study highlights the potency of KGF, IL-4, IL-13, and IL-17A in the pathophysiological processes of AD and psoriasis but does not rule out the involvement of other molecules tested in processes that ultimately lead to KC hyperproliferation.
We found KGF to elevate proliferation in an epidermal (KC-only) model. Previously, KGF was also found to directly stimulate KC proliferation in monolayer cultures and in 3D full-thickness models (Andreadis et al., 2001; Erdag et al., 2004; Finch et al., 1997; Marchese et al., 1990). In vivo, after paracrine secretion of KGF by fibroblasts, it binds with high affinity to the KGF receptor, which is solely expressed on epithelial cells (Miki et al., 1992). Studies revealed the involvement of KGF during wound healing (Marchese et al., 1995; Staiano-Coico et al., 1993; Werner et al., 1994, 1992) and in psoriasis (Finch et al., 1997; Kovacs et al., 2005). Furthermore, both IL-19 and IL-20, whose expression is increased in psoriatic epidermis, induce KGF transcripts in CD8+ T cells (Li et al., 2005; Wei et al., 2005). KGF may thus be an important contributor to KC hyperproliferation in psoriatic epidermis.
Psoriasis is considered a T-cell‒mediated disease, and many of these Th cells and cytokines derived thereof can nowadays be targeted by mAbs, the so-called biologicals. The most recent developments are within the Th17 immune cell axis, and the involvement of IL-17 in psoriasis pathophysiology is widely accepted. The immune cell infiltrates (e.g., Th17 cells, neutrophils) in psoriasis release IL-17, mainly IL-17A, as key effector cytokine and further promote skin inflammatory processes and epidermal alterations typical for psoriasis (Brembilla et al., 2018). Specific evidence toward the role of IL-17A in epidermal hyperproliferation is yet scarce (Lai et al., 2012; Langowski et al., 2006; Wu et al., 2015). In this study, we found IL-17A to double the rate of KC proliferation. In psoriasis, KC hyperproliferation may thus be a consequence of local IL-17A presence near the epidermis. Interestingly, recent studies also call for a pathogenic role of Th17 cells in certain AD endotypes (Suárez-Fariñas et al., 2013), with IL-17 expression elevated in lesional skin (Gittler et al., 2012) and subsets of CD4+ Th2 cells may produce IL-17 (Wang et al., 2010). These and our data could suggest a role for IL-17 in AD-related KC hyperproliferation, although selective therapeutic targeting using secukinumab in patients with AD was ineffective (Ungar et al., 2021). In contrast, targeting of the Th2 immune axis by the IL-4R‒targeting antibody, dupilumab, is considered effective in AD treatment and widely accepted in daily clinical practice (Beck et al., 2014; Hamilton et al., 2014).
Two members of the Th2 cytokine family, IL-4 and IL-13, have been described not only to drive the inflammatory process that contributes to AD but also to cause reduction of expression of epidermal proteins that are important for epidermal differentiation and barrier formation (Kim et al., 2008; Omori-Miyake et al., 2014). Recently, it was shown that mainly IL-13 signaling pathways dominate in AD and not IL-4 (Tsoi et al., 2019). In this study, we now show that both cytokines are direct KC mitogens in organotypic epidermal models. To our knowledge, it was previously unreported for IL-13, whereas for IL-4, increased KC proliferation in vitro has been described but only in monolayer cultures (Yang et al., 1996). Our results indicate a prime role for the IL-4R‒Jak/STAT6 signaling cascade in KC hyperproliferation for both IL-4 and IL-13 because the pretreatment with dupilumab or tofacitinib reduced STAT6 phosphorylation and inverted the Th2 cytokine‒induced hyperproliferation. Normalization of epidermal turnover and thereby reduction of acanthosis and scaling may thus be considered a direct therapeutic effect of both.
For IL-17A, previous studies investigating signaling events showed a key role for Act1 and MEK/ERK5 involvement in the proliferation of KCs after imiquimod-induced psoriasis-like inflammation in mice (Ha et al., 2014). In our studies, we also aimed to put the observed in vitro hyperproliferation into perspective concerning the actual hyperproliferation that is seen in lesional skin. Similar to recently described (Damen et al., 2021), proliferative KCs were found both in the basal and suprabasal layers of the HEE. The maximum increase in the number of proliferating cells in the organotypic model is two-fold, which is lower than we found in vivo (psoriasis: nine-fold, AD: seven-fold). One explanation could be of biological origin, namely that the complexity and costimulation of other factors in vivo further drive proliferation rates compared with only by a single exposure to IL-17A, IL-4, or IL-13. In preliminary follow-up studies, we combined cytokine stimulations, yet proliferation rates never exceeded two folds. The in vitro model system may thus be a rate-limiting factor given the time point of cytokine stimulation (between days 5 and 8 of air‒liquid interface culture): in this condition, KC turnover is higher than in native healthy skin. This may reduce the detection window, although even after exhaustive culture of 20 days when proliferation rates in the HEE are lower, we again observed a doubled number in proliferation KCs. We postulate that the flat and rigid polycarbonate membrane does not allow for the formation of rete ridges and extension of the basal layer surface such as in native skin to accommodate a larger number of proliferating cells, which hampers the fold increase in proliferating cells and limits acanthosis by proliferation in vitro.
Notwithstanding the potential importance of molecules for which we were unable to detect any effects in our studies, in the following lines, we highlight the few molecules that did increase epidermal thickness, although proliferation rates remained normal. In several KC models, IL-22 (related to both psoriasis and AD) has been shown to increase epidermal thickness (Boniface et al., 2005; Zheng et al., 2007). We also found IL-22 to induce acanthosis; however, this was not due to increased proliferation resulting in hyperplasia but rather by hypertrophy or cell swelling. Remarkably, we even find acanthosis to be more strongly correlated to cell hypertrophy than to hyperplasia because maximum epidermal thickness is found after exposure to nonmitogenic cytokines, such as IL-22 and oncostatin M. Mitogens, such as KGF or Th2 cytokines, increase epidermal thickness to a much lesser extent. Apparently, epidermal acanthosis in hyperproliferative skin diseases may not solely be caused by a higher KC turnover but also by cell enlargement. Strikingly, all hypertrophy-inducing cytokines, IL-6, IL-20, IL-22, and oncostatin M, are known activators of STAT3 signaling (Nguyen et al., 2015). Studies in psoriatic mice have already shown that STAT3 inhibition led to the normalization of epidermal thickness (Andrés et al., 2013; Miyoshi et al., 2011). Indeed, proliferation rates of murine KC monolayer cultures are higher after IL-17A exposure (Wu et al., 2015). Together with our data on human reconstituted epidermis, the mitogenic effects of IL-17A in the skin are now firmly established.
In summary, our study points at a small set of cytokines that act as direct KC mitogens within the plethora and complexity of the skin inflammatory milieu and elaborates on the epidermis-specific mechanism of action of targeting therapies benefitting their positioning for specific disease endotypes.
Materials and Methods
3D HEE culture
Primary human KC isolation and generation of 3D HEEs were performed as described previously (Rikken et al., 2020) and according to the principles of the Declaration of Helsinki. Briefly, primary human KCs were seeded with a density of 150,000 cells into a 24-well transwell system (Nunc; Thermo Fischer Scientific, Waltham, MA) and cultured submerged for 2 days in a proliferation medium (CnT-PR; CELLnTEC, Bern, Switzerland). After 1 more day of submerged culture in differentiation promoting medium (CnT-PR-3D; CELLnTEC), the cultures were lifted to the air‒liquid interface and cultured thereafter for a total of 8 days, with refreshing of the medium every other day (Niehues et al., 2017). Cytokines and other tested factors supplemented to the culture medium during days 5‒8 of the culture, with refreshing at day 7, in different concentrations are depicted in Table 2. Concentrations of all tested factors were chosen on the basis of experience as well as literature. For signaling studies, inhibitors were added 24 hours before cytokine stimulation and left for costimulation until day 8. To label KC proliferation in time, 10 μM EdU was added into the medium for 24 hours before harvesting at the indicated time points.
Immunohistochemistry
HEEs were fixed in formalin and processed for routine histology. Paraffin sections (6 μm) were stained with antibodies using an indirect immunoperoxidase or immunofluorescence technique (Vectastain, Vector Laboratories, Burlingame, CA) to visualize KC proliferation. Details on antibodies are presented in Table 4.
Table 4.
Antibodies/Chemicals Used for Immunohistochemistry/Immunofluorescence
| Target | Antibody Clone | Dilution |
|---|---|---|
| Ki-67 | SP6, Abcam ab16667 | 1:200 |
| Chromatin | 4′,6-diamidino-2-fenylindool, Boehringer Mannheim | 1:3,000 |
| EdU | Click-iT EdU Alexa Fluor 488 azide, Thermo Fischer Scientific | 1:1,000 |
Abbreviation: EdU: 5-ethynyl-2′-deoxyuridine.
Quantification of protein expression in immunohistochemical biopsy sections
Image acquisition of immunohistochemical-stained HEE tissue sections was performed by a ZEISS Axio Imager equipped with a ZEISS Axiocam 105 color Digital Camera (Zeiss, Oberkochen, Germany) and a ×10 or ×20 objective. The ZEISS Axiocam 105 color is a compact 5-megapixel camera (2,560 ×1,920 pixels) for high-resolution images with a 1/2.5” sensor. The images were chosen as representative of the whole culture and stored in CZI format. The number of fields ranges from one to three images per slide (depending on the variation of proliferation in the HEE). The images were analyzed with the cell image analysis software CellProfiler (Broad Institute, Cambridge, MA) (McQuin et al., 2018). In CellProfiler, different algorithms are available that can be modified and placed in sequential order to form a pipeline for image analysis. Pipelines for Ki-67 and EdU analysis were created (available on request). Data visualization and statistical analysis were performed using GraphPad Prism (GraphPad software, San Diego, CA). To determine statistical significance between multiple groups (n ≥ 3 groups), the Kruskal‒Wallis test (a ranked-based nonparametric test) was used for all data obtained with CellProfiler. If significant, Dunn’s multiple comparison posthoc test was performed (∗P < 0.05). In each HEE culture experiment, the human KC donors used are biological replicates.
Western blotting
HEEs were lysed, and after a single cycle of freeze thawing, the lysates were centrifuged at maximum speed for 10 minutes at 4 °C. Actin antibody (Sigma-Aldrich, St. Louis, MO) was used to control equal protein loading. Before immunoblotting, proteins were separated by SDS-PAGE and transferred to polyvinylidene fluoride membranes using the NuPAGE system (Life Technologies, Carlsbad, CA). SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific, Waltham, MA) was used for detection by the Bio-Rad Universal Hood Gel Imager (Bio-Rad Laboratories, Hercules, CA). All antibodies used are listed in Table 5. Image analysis and quantification of the protein expressions were performed with the Bio-Rad Image Laboratory Software. To determine statistical significance, the Kruskal‒Wallis test was used followed by Dunn’s multiple comparison posthoc test.
Table 5.
Antibodies/Chemicals Used for Western Blot Analysis
| Target | Antibody Clone | Dilution |
|---|---|---|
| β-Actin | AC-15, 121M 4846, Sigma Aldrich | 1:100,000 |
| pSTAT3 | Tyr705, Cell Signaling | 1:2,000 |
| pSTAT6 | pY641, BD Biosciences | 1:1,000 |
Abbreviation: pSTAT, phosphorylated signal transducer and activator of transcription.
Data availability statement
No datasets were generated or analyzed during this study.
ORCIDS
Hanna Niehues: http://orcid.org/0000-0002-6954-6955
Gijs Rikken: http://orcid.org/0000-0003-0535-5399
Ivonne M.J.J. van Vlijmen-Willems: http://orcid.org/0000-0002-3522-2573
Diana Rodijk-Olthuis: http://orcid.org/0000-0002-7752-6209
Piet E.J. van Erp: http://orcid.org/0000-0002-6955-8817
Patrick L.J.M. Zeeuwen: http://orcid.org/0000-0002-6878-2438
Joost Schalkwijk: http://orcid.org/0000-0002-1308-1319
Ellen H. van den Bogaard: http://orcid.org/0000-0003-4846-0287
Acknowledgments
Conflicts of Interest
This research did not receive any specific grant from funding agencies in the public, commercial, or nonprofit sectors. The remaining authors state no conflicts of interest.
Acknowledgments
This study was supported by Meer Kennis met Minder Dieren funding initiative of ZonMw (Nederlandse organisatie voor gezondheidsonderzoek en zorginnovatie) with the grant number ZonMw-MKMD 40-42600-98-16102.
Author Contributions
Conceptualization: HN, GR, PLJMZ, JS, EHvdB; Data Curation: HN, GR, PEJvE; Formal Analysis: HN, GR, PEJvE; Funding Acquisition: JS, EHvdB; Investigation: HN, GR, IMJJvVW, DRO; Methodology: HN, GR, DRO, IMJJvVW, EHvdB; Project Administration: HN, GR, EHvdB; Software: PEJvE; Supervision: PLJMZ, JS, EHvdB; Validation: HN, GR, IMJJvVW, DRO; Visualization: HN, GR, PEJvE, PLJMZ, EHvdB; Writing - Original Draft Preparation: HN, GR; Writing - Review and Editing: PLJMZ, JS, EHvdB
accepted manuscript published online 22 October 2021; corrected proof published online 2 December 2021
Footnotes
Cite this article as: JID Innovations 2022;2:100066
References
- Anand P., Springall D.R., Blank M.A., Sellu D., Polak J.M., Bloom S.R. Neuropeptides in skin disease: increased VIP in eczema and psoriasis but not axillary hyperhidrosis. Br J Dermatol. 1991;124:547–549. doi: 10.1111/j.1365-2133.1991.tb04948.x. [DOI] [PubMed] [Google Scholar]
- Andreadis S.T., Hamoen K.E., Yarmush M.L., Morgan J.R. Keratinocyte growth factor induces hyperproliferation and delays differentiation in a skin equivalent model system. FASEB J. 2001;15:898–906. doi: 10.1096/fj.00-0324com. [DOI] [PubMed] [Google Scholar]
- Andreadis T.G., Anderson J.F., Munstermann L.E., Wolfe R.J., Florin D.A. Discovery, distribution, and abundance of the newly introduced mosquito Ochlerotatus japonicus (Diptera: Culicidae) in Connecticut, USA. J Med Entomol. 2001;38:774–779. doi: 10.1603/0022-2585-38.6.774. [DOI] [PubMed] [Google Scholar]
- Andrés R.M., Montesinos M.C., Navalón P., Payá M., Terencio M.C. NF-κB and STAT3 inhibition as a therapeutic strategy in psoriasis: in vitro and in vivo effects of BTH. J Invest Dermatol. 2013;133:2362–2371. doi: 10.1038/jid.2013.182. [DOI] [PubMed] [Google Scholar]
- Bao L., Alexander J.B., Shi V.Y., Mohan G.C., Chan L.S. Interleukin-4 up-regulation of epidermal interleukin-19 expression in keratinocytes involves the binding of signal transducer and activator of transcription 6 (Stat6) to the imperfect Stat6 sites. Immunology. 2014;143:601–608. doi: 10.1111/imm.12339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck L.A., Thaçi D., Hamilton J.D., Graham N.M., Bieber T., Rocklin R., et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. 2014;371:130–139. doi: 10.1056/NEJMoa1314768. [DOI] [PubMed] [Google Scholar]
- Bertolero F., Kaighn M.E., Camalier R.F., Saffiotti U. Effects of serum and serum-derived factors on growth and differentiation of mouse keratinocytes. In Vitro Cell Dev Biol. 1986;22:423–428. doi: 10.1007/BF02623533. [DOI] [PubMed] [Google Scholar]
- Bhora F.Y., Dunkin B.J., Batzri S., Aly H.M., Bass B.L., Sidawy A.N., et al. Effect of growth factors on cell proliferation and epithelialization in human skin. J Surg Res. 1995;59:236–244. doi: 10.1006/jsre.1995.1160. [DOI] [PubMed] [Google Scholar]
- Boniface K., Bernard F.X., Garcia M., Gurney A.L., Lecron J.C., Morel F. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J Immunol. 2005;174:3695–3702. doi: 10.4049/jimmunol.174.6.3695. [DOI] [PubMed] [Google Scholar]
- Boniface K., Diveu C., Morel F., Pedretti N., Froger J., Ravon E., et al. Oncostatin M secreted by skin infiltrating T lymphocytes is a potent keratinocyte activator involved in skin inflammation. J Immunol. 2007;178:4615–4622. doi: 10.4049/jimmunol.178.7.4615. [DOI] [PubMed] [Google Scholar]
- Braunstein S., Kaplan G., Gottlieb A.B., Schwartz M., Walsh G., Abalos R.M., et al. GM-CSF activates regenerative epidermal growth and stimulates keratinocyte proliferation in human skin in vivo. J Invest Dermatol. 1994;103:601–604. doi: 10.1111/1523-1747.ep12396936. [DOI] [PubMed] [Google Scholar]
- Brembilla N.C., Senra L., Boehncke W.H. The IL-17 family of cytokines in psoriasis: IL-17A and beyond. Front Immunol. 2018;9:1682. doi: 10.3389/fimmu.2018.01682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y., Xue F., Quan C., Qu M., Liu N., Zhang Y., et al. A critical role of the IL-1β-IL-1R signaling pathway in skin inflammation and psoriasis pathogenesis. J Invest Dermatol. 2019;139:146–156. doi: 10.1016/j.jid.2018.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruso R., Botti E., Sarra M., Esposito M., Stolfi C., Diluvio L., et al. Involvement of interleukin-21 in the epidermal hyperplasia of psoriasis. Nat Med. 2009;15:1013–1015. doi: 10.1038/nm.1995. [DOI] [PubMed] [Google Scholar]
- Chan J.R., Blumenschein W., Murphy E., Diveu C., Wiekowski M., Abbondanzo S., et al. IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2-dependent mechanisms with implications for psoriasis pathogenesis. J Exp Med. 2006;203:2577–2587. doi: 10.1084/jem.20060244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charruyer A., Fong S., Vitcov G.G., Sklar S., Tabernik L., Taneja M., et al. Brief report: interleukin-17A-dependent asymmetric stem cell divisions are increased in human psoriasis: a mechanism underlying benign hyperproliferation. Stem Cells. 2017;35:2001–2007. doi: 10.1002/stem.2656. [DOI] [PubMed] [Google Scholar]
- Chun K.S., Lao H.C., Langenbach R. The prostaglandin E2 receptor, EP2, stimulates keratinocyte proliferation in mouse skin by G protein-dependent and {beta}-arrestin1-dependent signaling pathways. J Biol Chem. 2010;285:39672–39681. doi: 10.1074/jbc.M110.117689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damen M., Wirtz L., Soroka E., Khatif H., Kukat C., Simons B.D., et al. High proliferation and delamination during skin epidermal stratification. Nat Commun. 2021;12:3227. doi: 10.1038/s41467-021-23386-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detmar M., Orfanos C.E. Tumor necrosis factor-alpha inhibits cell proliferation and induces class II antigens and cell adhesion molecules in cultured normal human keratinocytes in vitro. Arch Dermatol Res. 1990;282:238–245. doi: 10.1007/BF00371643. [DOI] [PubMed] [Google Scholar]
- Dillon S.R., Sprecher C., Hammond A., Bilsborough J., Rosenfeld-Franklin M., Presnell S.R., et al. Interleukin 31, a cytokine produced by activated T cells, induces dermatitis in mice [published correction appears in Nat Immunol 2005;6:114] Nat Immunol. 2004;5:752–760. doi: 10.1038/ni1084. [DOI] [PubMed] [Google Scholar]
- Ekman A.K., Bivik Eding C., Rundquist I., Enerbäck C. IL-17 and IL-22 promote keratinocyte stemness in the germinative compartment in psoriasis. J Invest Dermatol. 2019;139:1564–1573.e8. doi: 10.1016/j.jid.2019.01.014. [DOI] [PubMed] [Google Scholar]
- Erdag G., Medalie D.A., Rakhorst H., Krueger G.G., Morgan J.R. FGF-7 expression enhances the performance of bioengineered skin. Mol Ther. 2004;10:76–85. doi: 10.1016/j.ymthe.2004.04.013. [DOI] [PubMed] [Google Scholar]
- Finch P.W., Murphy F., Cardinale I., Krueger J.G. Altered expression of keratinocyte growth factor and its receptor in psoriasis. Am J Pathol. 1997;151:1619–1628. [PMC free article] [PubMed] [Google Scholar]
- Frost P., Weinstein G.D., Van Scott E.J. The ichthyosiform dermatoses. II. Autoradiographic studies of epidermal proliferation. J Invest Dermatol. 1966;47:561–567. doi: 10.1038/jid.1966.185. [DOI] [PubMed] [Google Scholar]
- Fujisawa T., Fujisawa R., Kato Y., Nakayama T., Morita A., Katsumata H., et al. Presence of high contents of thymus and activation-regulated chemokine in platelets and elevated plasma levels of thymus and activation-regulated chemokine and macrophage-derived chemokine in patients with atopic dermatitis. J Allergy Clin Immunol. 2002;110:139–146. doi: 10.1067/mai.2002.126079. [DOI] [PubMed] [Google Scholar]
- Gibbs S., Silva Pinto A.N., Murli S., Huber M., Hohl D., Ponec M. Epidermal growth factor and keratinocyte growth factor differentially regulate epidermal migration, growth, and differentiation. Wound Repair Regen. 2000;8:192–203. doi: 10.1046/j.1524-475x.2000.00192.x. [DOI] [PubMed] [Google Scholar]
- Gittler J.K., Shemer A., Suárez-Fariñas M., Fuentes-Duculan J., Gulewicz K.J., Wang C.Q., et al. Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J Allergy Clin Immunol. 2012;130:1344–1354. doi: 10.1016/j.jaci.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman R.M., Krueger J., Yourish D., Granelli-Piperno A., Murphy D.P., May L.T., et al. Interleukin 6 is expressed in high levels in psoriatic skin and stimulates proliferation of cultured human keratinocytes. Proc Natl Acad Sci USA. 1989;86:6367–6371. doi: 10.1073/pnas.86.16.6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman-Yassky E., Krueger J.G. Atopic dermatitis and psoriasis: two different immune diseases or one spectrum? Curr Opin Immunol. 2017;48:68–73. doi: 10.1016/j.coi.2017.08.008. [DOI] [PubMed] [Google Scholar]
- Ha H.L., Wang H., Pisitkun P., Kim J.C., Tassi I., Tang W., et al. IL-17 drives psoriatic inflammation via distinct, target cell-specific mechanisms. Proc Natl Acad Sci USA. 2014;111:E3422–E3431. doi: 10.1073/pnas.1400513111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamid Q., Boguniewicz M., Leung D.Y. Differential in situ cytokine gene expression in acute versus chronic atopic dermatitis. J Clin Invest. 1994;94:870–876. doi: 10.1172/JCI117408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamid Q., Naseer T., Minshall E.M., Song Y.L., Boguniewicz M., Leung D.Y. In vivo expression of IL-12 and IL-13 in atopic dermatitis. J Allergy Clin Immunol. 1996;98:225–231. doi: 10.1016/s0091-6749(96)70246-4. [DOI] [PubMed] [Google Scholar]
- Hamilton J.D., Suárez-Fariñas M., Dhingra N., Cardinale I., Li X., Kostic A., et al. Dupilumab improves the molecular signature in skin of patients with moderate-to-severe atopic dermatitis. J Allergy Clin Immunol. 2014;134:1293–1300. doi: 10.1016/j.jaci.2014.10.013. [DOI] [PubMed] [Google Scholar]
- Hattori N., Komine M., Yano S., Kaneko T., Hanakawa Y., Hashimoto K., et al. Interferon-gamma, a strong suppressor of cell proliferation, induces upregulation of keratin K6, one of the inflammatory- and proliferation-associated keratins. J Invest Dermatol. 2002;119:403–410. doi: 10.1046/j.1523-1747.2002.01843.x. [DOI] [PubMed] [Google Scholar]
- Henry G., Li W., Garner W., Woodley D.T. Migration of human keratinocytes in plasma and serum and wound re-epithelialisation [published correction appears in Lancet 2003;361:1660] Lancet. 2003;361:574–576. doi: 10.1016/S0140-6736(03)12510-X. [DOI] [PubMed] [Google Scholar]
- Herrick C.A., Xu L., McKenzie A.N., Tigelaar R.E., Bottomly K. IL-13 is necessary, not simply sufficient, for epicutaneously induced Th2 responses to soluble protein antigen. J Immunol. 2003;170:2488–2495. doi: 10.4049/jimmunol.170.5.2488. [DOI] [PubMed] [Google Scholar]
- Hijnen D., De Bruin-Weller M., Oosting B., Lebre C., De Jong E., Bruijnzeel-Koomen C., et al. Serum thymus and activation-regulated chemokine (TARC) and cutaneous T cell- attracting chemokine (CTACK) levels in allergic diseases: TARC and CTACK are disease-specific markers for atopic dermatitis. J Allergy Clin Immunol. 2004;113:334–340. doi: 10.1016/j.jaci.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Hobbs R.M., Silva-Vargas V., Groves R., Watt F.M. Expression of activated MEK1 in differentiating epidermal cells is sufficient to generate hyperproliferative and inflammatory skin lesions. J Invest Dermatol. 2004;123:503–515. doi: 10.1111/j.0022-202X.2004.23225.x. [DOI] [PubMed] [Google Scholar]
- Jiang W.Y., Raychaudhuri S.P., Farber E.M. Double-labeled immunofluorescence study of cutaneous nerves in psoriasis. Int J Dermatol. 1998;37:572–574. doi: 10.1046/j.1365-4362.1998.00533.x. [DOI] [PubMed] [Google Scholar]
- Johansen C., Funding A.T., Otkjaer K., Kragballe K., Jensen U.B., Madsen M., et al. Protein expression of TNF-alpha in psoriatic skin is regulated at a posttranscriptional level by MAPK-activated protein kinase 2. J Immunol. 2006;176:1431–1438. doi: 10.4049/jimmunol.176.3.1431. [DOI] [PubMed] [Google Scholar]
- Jones A.M., Griffiths J.L., Sanders A.J., Owen S., Ruge F., Harding K.G., et al. The clinical significance and impact of interleukin 15 on keratinocyte cell growth and migration. Int J Mol Med. 2016;38:679–686. doi: 10.3892/ijmm.2016.2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakurai M., Fujita N., Murata S., Furukawa Y., Demitsu T., Nakagawa H. Vasoactive intestinal peptide regulates its receptor expression and functions of human keratinocytes via type I vasoactive intestinal peptide receptors. J Invest Dermatol. 2001;116:743–749. doi: 10.1046/j.1523-1747.2001.01306.x. [DOI] [PubMed] [Google Scholar]
- Kim B.E., Leung D.Y., Boguniewicz M., Howell M.D. Loricrin and involucrin expression is down-regulated by Th2 cytokines through STAT-6. Clin Immunol. 2008;126:332–337. doi: 10.1016/j.clim.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo S., Yazawa H., Jimbow K. Reduction of serum interleukin-5 levels reflect clinical improvement in patients with atopic dermatitis. J Dermatol. 2001;28:237–243. doi: 10.1111/j.1346-8138.2001.tb00124.x. [DOI] [PubMed] [Google Scholar]
- Kovacs D., Falchi M., Cardinali G., Raffa S., Carducci M., Cota C., et al. Immunohistochemical analysis of keratinocyte growth factor and fibroblast growth factor 10 expression in psoriasis. Exp Dermatol. 2005;14:130–137. doi: 10.1111/j.0906-6705.2005.00261.x. [DOI] [PubMed] [Google Scholar]
- Krane J.F., Murphy D.P., Carter D.M., Krueger J.G. Synergistic effects of epidermal growth factor (EGF) and insulin-like growth factor I/somatomedin C (IGF-I) on keratinocyte proliferation may be mediated by IGF-I transmodulation of the EGF receptor. J Invest Dermatol. 1991;96:419–424. doi: 10.1111/1523-1747.ep12469799. [DOI] [PubMed] [Google Scholar]
- Lai Y., Li D., Li C., Muehleisen B., Radek K.A., Park H.J., et al. The antimicrobial protein REG3A regulates keratinocyte proliferation and differentiation after skin injury. Immunity. 2012;37:74–84. doi: 10.1016/j.immuni.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langowski J.L., Zhang X., Wu L., Mattson J.D., Chen T., Smith K., et al. IL-23 promotes tumour incidence and growth. Nature. 2006;442:461–465. doi: 10.1038/nature04808. [DOI] [PubMed] [Google Scholar]
- Lechler T., Fuchs E. Asymmetric cell divisions promote stratification and differentiation of mammalian skin. Nature. 2005;437:275–280. doi: 10.1038/nature03922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh I.M., Pulford K.A., Ramaekers F.C., Lane E.B. Psoriasis: maintenance of an intact monolayer basal cell differentiation compartment in spite of hyperproliferation. Br J Dermatol. 1985;113:53–64. doi: 10.1111/j.1365-2133.1985.tb02044.x. [DOI] [PubMed] [Google Scholar]
- Li C., Xiao L., Jia J., Li F., Wang X., Duan Q., et al. Cornulin is induced in psoriasis lesions and promotes keratinocyte proliferation via phosphoinositide 3-kinase/Akt pathways. J Invest Dermatol. 2019;139:71–80. doi: 10.1016/j.jid.2018.06.184. [DOI] [PubMed] [Google Scholar]
- Li H.H., Lin Y.C., Chen P.J., Hsiao C.H., Lee J.Y., Chen W.C., et al. Interleukin-19 upregulates keratinocyte growth factor and is associated with psoriasis. Br J Dermatol. 2005;153:591–595. doi: 10.1111/j.1365-2133.2005.06665.x. [DOI] [PubMed] [Google Scholar]
- Ma W.Y., Jia K., Zhang Y. IL-17 promotes keratinocyte proliferation via the downregulation of C/EBPα. Exp Ther Med. 2016;11:631–636. doi: 10.3892/etm.2015.2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas-Szabowski N., Stark H.J., Fusenig N.E. Keratinocyte growth regulation in defined organotypic cultures through IL-1-induced keratinocyte growth factor expression in resting fibroblasts. J Invest Dermatol. 2000;114:1075–1084. doi: 10.1046/j.1523-1747.2000.00987.x. [DOI] [PubMed] [Google Scholar]
- Mann A., Breuhahn K., Schirmacher P., Blessing M. Keratinocyte-derived granulocyte-macrophage colony stimulating factor accelerates wound healing: stimulation of keratinocyte proliferation, granulation tissue formation, and vascularization. J Invest Dermatol. 2001;117:1382–1390. doi: 10.1046/j.0022-202x.2001.01600.x. [DOI] [PubMed] [Google Scholar]
- Marchese C., Chedid M., Dirsch O.R., Csaky K.G., Santanelli F., Latini C., et al. Modulation of keratinocyte growth factor and its receptor in reepithelializing human skin. J Exp Med. 1995;182:1369–1376. doi: 10.1084/jem.182.5.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchese C., Rubin J., Ron D., Faggioni A., Torrisi M.R., Messina A., et al. Human keratinocyte growth factor activity on proliferation and differentiation of human keratinocytes: differentiation response distinguishes KGF from EGF family. J Cell Physiol. 1990;144:326–332. doi: 10.1002/jcp.1041440219. [DOI] [PubMed] [Google Scholar]
- Matsumura S., Terao M., Murota H., Katayama I. Th2 cytokines enhance TrkA expression, upregulate proliferation, and downregulate differentiation of keratinocytes. J Dermatol Sci. 2015;78:215–223. doi: 10.1016/j.jdermsci.2015.02.021. [DOI] [PubMed] [Google Scholar]
- McQuin C., Goodman A., Chernyshev V., Kamentsky L., Cimini B.A., Karhohs K.W., et al. CellProfiler 3.0: next-generation image processing for biology. PLoS Biol. 2018;16 doi: 10.1371/journal.pbio.2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki T., Bottaro D.P., Fleming T.P., Smith C.L., Burgess W.H., Chan A.M., et al. Determination of ligand-binding specificity by alternative splicing: two distinct growth factor receptors encoded by a single gene. Proc Natl Acad Sci USA. 1992;89:246–250. doi: 10.1073/pnas.89.1.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mildner M., Ballaun C., Stichenwirth M., Bauer R., Gmeiner R., Buchberger M., et al. Gene silencing in a human organotypic skin model. Biochem Biophys Res Commun. 2006;348:76–82. doi: 10.1016/j.bbrc.2006.07.035. [DOI] [PubMed] [Google Scholar]
- Miyoshi K., Takaishi M., Nakajima K., Ikeda M., Kanda T., Tarutani M., et al. Stat3 as a therapeutic target for the treatment of psoriasis: a clinical feasibility study with STA-21, a Stat3 inhibitor. J Invest Dermatol. 2011;131:108–117. doi: 10.1038/jid.2010.255. [DOI] [PubMed] [Google Scholar]
- Mizutani H., Tamagawa-Mineoka R., Nakamura N., Masuda K., Katoh N. Serum IL-21 levels are elevated in atopic dermatitis patients with acute skin lesions. Allergol Int. 2017;66:440–444. doi: 10.1016/j.alit.2016.10.010. [DOI] [PubMed] [Google Scholar]
- Nair R.P., Duffin K.C., Helms C., Ding J., Stuart P.E., Goldgar D., et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet. 2009;41:199–204. doi: 10.1038/ng.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahigashi K., Kabashima K., Ikoma A., Verkman A.S., Miyachi Y., Hara-Chikuma M. Upregulation of aquaporin-3 is involved in keratinocyte proliferation and epidermal hyperplasia. J Invest Dermatol. 2011;131:865–873. doi: 10.1038/jid.2010.395. [DOI] [PubMed] [Google Scholar]
- Nguyen P.M., Putoczki T.L., Ernst M. STAT3-activating cytokines: a therapeutic opportunity for inflammatory bowel disease? J Interferon Cytokine Res. 2015;35:340–350. doi: 10.1089/jir.2014.0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehues H., Bouwstra J.A., El Ghalbzouri A., Brandner J.M., Zeeuwen P.L.J.M., van den Bogaard E.H. 3D skin models for 3R research: the potential of 3D reconstructed skin models to study skin barrier function. Exp Dermatol. 2018;27:501–511. doi: 10.1111/exd.13531. [DOI] [PubMed] [Google Scholar]
- Niehues H., Schalkwijk J., van Vlijmen-Willems I.M.J.J., Rodijk-Olthuis D., van Rossum M.M., Wladykowski E., et al. Epidermal equivalents of filaggrin null keratinocytes do not show impaired skin barrier function. J Allergy Clin Immunol. 2017;139:1979–1981.e13. doi: 10.1016/j.jaci.2016.09.016. [DOI] [PubMed] [Google Scholar]
- Oka T., Sugaya M., Takahashi N., Nakajima R., Otobe S., Kabasawa M., et al. Increased interleukin-19 expression in cutaneous T-cell lymphoma and atopic dermatitis. Acta Derm Venereol. 2017;97:1172–1177. doi: 10.2340/00015555-2723. [DOI] [PubMed] [Google Scholar]
- Omori-Miyake M., Yamashita M., Tsunemi Y., Kawashima M., Yagi J. In vitro assessment of IL-4- or IL-13-mediated changes in the structural components of keratinocytes in mice and humans [published correction appears in J Invest Dermatol 2018;138:472–3] J Invest Dermatol. 2014;134:1342–1350. doi: 10.1038/jid.2013.503. [DOI] [PubMed] [Google Scholar]
- Pastore S., Fanales-Belasio E., Albanesi C., Chinni L.M., Giannetti A., Girolomoni G. Granulocyte macrophage colony-stimulating factor is overproduced by keratinocytes in atopic dermatitis. Implications for sustained dendritic cell activation in the skin. J Clin Invest. 1997;99:3009–3017. doi: 10.1172/JCI119496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen C., Thomas-Virnig C., Allen-Hoffmann B.L. Classical human epidermal keratinocyte cell culture. Methods Mol Biol. 2013;945:161–175. doi: 10.1007/978-1-62703-125-7_11. [DOI] [PubMed] [Google Scholar]
- Rawlings J.S., Rosler K.M., Harrison D.A. The JAK/STAT signaling pathway. J Cell Sci. 2004;117:1281–1283. doi: 10.1242/jcs.00963. [DOI] [PubMed] [Google Scholar]
- Rikken G., Niehues H., van den Bogaard E.H. Organotypic 3D skin models: human epidermal equivalent cultures from primary keratinocytes and immortalized keratinocyte cell lines. Methods Mol Biol. 2020;2154:45–61. doi: 10.1007/978-1-0716-0648-3_5. [DOI] [PubMed] [Google Scholar]
- Rückert R., Asadullah K., Seifert M., Budagian V.M., Arnold R., Trombotto C., et al. Inhibition of keratinocyte apoptosis by IL-15: a new parameter in the pathogenesis of psoriasis? J Immunol. 2000;165:2240–2250. doi: 10.4049/jimmunol.165.4.2240. [DOI] [PubMed] [Google Scholar]
- Sapuntsova S.G., Mel'nikova N.P., Deigin V.I., Kozulin E.A., Timoshin S.S. Proliferative processes in the epidermis of patients with atopic dermatitis treated with thymodepressin. Bull Exp Biol Med. 2002;133:488–490. doi: 10.1023/a:1019874023845. [DOI] [PubMed] [Google Scholar]
- Sawamura D., Meng X., Ina S., Sato M., Tamai K., Hanada K., et al. Induction of keratinocyte proliferation and lymphocytic infiltration by in vivo introduction of the IL-6 gene into keratinocytes and possibility of keratinocyte gene therapy for inflammatory skin diseases using IL-6 mutant genes. J Immunol. 1998;161:5633–5639. [PubMed] [Google Scholar]
- Seike M., Ikeda M., Morimoto A., Matsumoto M., Kodama H. Increased synthesis of calcitonin gene-related peptide stimulates keratinocyte proliferation in murine UVB-irradiated skin. J Dermatol Sci. 2002;28:135–143. doi: 10.1016/s0923-1811(01)00155-4. [DOI] [PubMed] [Google Scholar]
- Sen G.L., Reuter J.A., Webster D.E., Zhu L., Khavari P.A. DNMT1 maintains progenitor function in self-renewing somatic tissue. Nature. 2010;463:563–567. doi: 10.1038/nature08683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata S., Tada Y., Kanda N., Nashiro K., Kamata M., Karakawa M., et al. Possible roles of IL-27 in the pathogenesis of psoriasis. J Invest Dermatol. 2010;130:1034–1039. doi: 10.1038/jid.2009.349. [DOI] [PubMed] [Google Scholar]
- Singh B., Jegga A.G., Shanmukhappa K.S., Edukulla R., Khurana Hershey G.H., Medvedovic M., et al. IL-31-driven skin remodeling involves epidermal cell proliferation and thickening that lead to impaired skin-barrier function [published correction appears in PLoS One 2017;12:e0170446] PLoS One. 2016;11 doi: 10.1371/journal.pone.0161877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soumelis V., Reche P.A., Kanzler H., Yuan W., Edward G., Homey B., et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol. 2002;3:673–680. doi: 10.1038/ni805. [DOI] [PubMed] [Google Scholar]
- Staiano-Coico L., Krueger J.G., Rubin J.S., D'Limi S., Vallat V.P., Valentino L., et al. Human keratinocyte growth factor effects in a porcine model of epidermal wound healing. J Exp Med. 1993;178:865–878. doi: 10.1084/jem.178.3.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallmeyer B., Kämpfer H., Podda M., Kaufmann R., Pfeilschifter J., Frank S. A novel keratinocyte mitogen: regulation of leptin and its functional receptor in skin repair. J Invest Dermatol. 2001;117:98–105. doi: 10.1046/j.0022-202x.2001.01387.x. [DOI] [PubMed] [Google Scholar]
- Steude J., Kulke R., Christophers E. Interleukin-1-stimulated secretion of interleukin-8 and growth-related oncogene-alpha demonstrates greatly enhanced keratinocyte growth in human raft cultured epidermis. J Invest Dermatol. 2002;119:1254–1260. doi: 10.1046/j.1523-1747.2002.19616.x. [DOI] [PubMed] [Google Scholar]
- Suárez-Fariñas M., Dhingra N., Gittler J., Shemer A., Cardinale I., de Guzman Strong C., et al. Intrinsic atopic dermatitis shows similar TH2 and higher TH17 immune activation compared with extrinsic atopic dermatitis. J Allergy Clin Immunol. 2013;132:361–370. doi: 10.1016/j.jaci.2013.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D.P., Yeh C.H., So E., Wang L.Y., Wei T.S., Chang M.S., et al. Interleukin (IL)-19 promoted skin wound healing by increasing fibroblast keratinocyte growth factor expression. Cytokine. 2013;62:360–368. doi: 10.1016/j.cyto.2013.03.017. [DOI] [PubMed] [Google Scholar]
- Sung K.J., Chang S.E., Paik E.M., Lee M.W., Choi J.H. Vasoactive intestinal polypeptide stimulates the proliferation of HaCaT cell via TGF-alpha. Neuropeptides. 1999;33:435–446. doi: 10.1054/npep.1999.0042. [DOI] [PubMed] [Google Scholar]
- Taniguchi K., Arima K., Masuoka M., Ohta S., Shiraishi H., Ontsuka K., et al. Periostin controls keratinocyte proliferation and differentiation by interacting with the paracrine IL-1α/IL-6 loop. J Invest Dermatol. 2014;134:1295–1304. doi: 10.1038/jid.2013.500. [DOI] [PubMed] [Google Scholar]
- Tavakkol A., Elder J.T., Griffiths C.E., Cooper K.D., Talwar H., Fisher G.J., et al. Expression of growth hormone receptor, insulin-like growth factor 1 (IGF-1) and IGF-1 receptor mRNA and proteins in human skin. J Invest Dermatol. 1992;99:343–349. doi: 10.1111/1523-1747.ep12616668. [DOI] [PubMed] [Google Scholar]
- Truong A.B., Kretz M., Ridky T.W., Kimmel R., Khavari P.A. p63 regulates proliferation and differentiation of developmentally mature keratinocytes. Genes Dev. 2006;20:3185–3197. doi: 10.1101/gad.1463206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoi L.C., Rodriguez E., Degenhardt F., Baurecht H., Wehkamp U., Volks N., et al. Atopic dermatitis is an IL-13-dominant disease with greater molecular heterogeneity compared to psoriasis. J Invest Dermatol. 2019;139:1480–1489. doi: 10.1016/j.jid.2018.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungar B., Pavel A.B., Li R., Kimmel G., Nia J., Hashim P., et al. Phase 2 randomized, double-blind study of IL-17 targeting with secukinumab in atopic dermatitis. J Allergy Clin Immunol. 2021;147:394–397. doi: 10.1016/j.jaci.2020.04.055. [DOI] [PubMed] [Google Scholar]
- Uyemura K., Yamamura M., Fivenson D.F., Modlin R.L., Nickoloff B.J. The cytokine network in lesional and lesion-free psoriatic skin is characterized by a T-helper type 1 cell-mediated response. J Invest Dermatol. 1993;101:701–705. doi: 10.1111/1523-1747.ep12371679. [DOI] [PubMed] [Google Scholar]
- Vanscott E.J., Ekel T.M. Kinetics of hyperplasia in psoriasis. Arch Dermatol. 1963;88:373–381. [PubMed] [Google Scholar]
- Venuti A., Marcante M.L., Flamini S., Di Castro V., Bagnato A. The autonomous growth of human papillomavirus type 16-immortalized keratinocytes is related to the endothelin-1 autocrine loop. J Virol. 1997;71:6898–6904. doi: 10.1128/jvi.71.9.6898-6904.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Lei L., Hu J., Li Y. Oncostatin M upregulates livin to promote keratinocyte proliferation and survival via ERK and STAT3 signaling pathways. Exp Physiol. 2020;105:1151–1158. doi: 10.1113/EP088584. [DOI] [PubMed] [Google Scholar]
- Wang Y.H., Voo K.S., Liu B., Chen C.Y., Uygungil B., Spoede W., et al. A novel subset of CD4(+) T(H)2 memory/effector cells that produce inflammatory IL-17 cytokine and promote the exacerbation of chronic allergic asthma. J Exp Med. 2010;207:2479–2491. doi: 10.1084/jem.20101376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C.C., Chen W.Y., Wang Y.C., Chen P.J., Lee J.Y., Wong T.W., et al. Detection of IL-20 and its receptors on psoriatic skin. Clin Immunol. 2005;117:65–72. doi: 10.1016/j.clim.2005.06.012. [DOI] [PubMed] [Google Scholar]
- Weinstein G.D., McCullough J.L., Ross P. Cell proliferation in normal epidermis. J Invest Dermatol. 1984;82:623–628. doi: 10.1111/1523-1747.ep12261462. [DOI] [PubMed] [Google Scholar]
- Weinstein G.D., McCullough J.L., Ross P.A. Cell kinetic basis for pathophysiology of psoriasis. J Invest Dermatol. 1985;85:579–583. doi: 10.1111/1523-1747.ep12283594. [DOI] [PubMed] [Google Scholar]
- Werner S., Peters K.G., Longaker M.T., Fuller-Pace F., Banda M.J., Williams L.T. Large induction of keratinocyte growth factor expression in the dermis during wound healing. Proc Natl Acad Sci USA. 1992;89:6896–6900. doi: 10.1073/pnas.89.15.6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner S., Smola H., Liao X., Longaker M.T., Krieg T., Hofschneider P.H., et al. The function of KGF in morphogenesis of epithelium and reepithelialization of wounds. Science. 1994;266:819–822. doi: 10.1126/science.7973639. [DOI] [PubMed] [Google Scholar]
- Witte E., Kokolakis G., Witte K., Philipp S., Doecke W.D., Babel N., et al. IL-19 is a component of the pathogenetic IL-23/IL-17 cascade in psoriasis. J Invest Dermatol. 2014;134:2757–2767. doi: 10.1038/jid.2014.308. [DOI] [PubMed] [Google Scholar]
- Works M.G., Yin F., Yin C.C., Yiu Y., Shew K., Tran T.T., et al. Inhibition of TYK2 and JAK1 ameliorates imiquimod-induced psoriasis-like dermatitis by inhibiting IL-22 and the IL-23/IL-17 axis. J Immunol. 2014;193:3278–3287. doi: 10.4049/jimmunol.1400205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wraight C.J., White P.J., McKean S.C., Fogarty R.D., Venables D.J., Liepe I.J., et al. Reversal of epidermal hyperproliferation in psoriasis by insulin-like growth factor I receptor antisense oligonucleotides. Nat Biotechnol. 2000;18:521–526. doi: 10.1038/75382. [DOI] [PubMed] [Google Scholar]
- Wright N.A. Changes in epidermal cell proliferation in proliferative skin disease. Curr Top Pathol. 1985;74:141–166. doi: 10.1007/978-3-642-69574-2_4. [DOI] [PubMed] [Google Scholar]
- Wu L., Chen X., Zhao J., Martin B., Zepp J.A., Ko J.S., et al. A novel IL-17 signaling pathway controlling keratinocyte proliferation and tumorigenesis via the TRAF4-ERK5 axis. J Exp Med. 2015;212:1571–1587. doi: 10.1084/jem.20150204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M., Lu H., Lee Y.H., Wu Y., Liu K., Shi Y., et al. An interleukin-25-mediated autoregulatory circuit in keratinocytes plays a pivotal role in psoriatic skin inflammation. Immunity. 2018;48:787–798.e4. doi: 10.1016/j.immuni.2018.03.019. [DOI] [PubMed] [Google Scholar]
- Yang B., Suwanpradid J., Sanchez-Lagunes R., Choi H.W., Hoang P., Wang D., et al. IL-27 facilitates skin wound healing through induction of epidermal proliferation and host defense. J Invest Dermatol. 2017;137:1166–1175. doi: 10.1016/j.jid.2017.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Yoo H.M., Choi I., Pyun K.H., Byun S.M., Ha H. Interleukin 4-induced proliferation in normal human keratinocytes is associated with c-myc gene expression and inhibited by genistein. J Invest Dermatol. 1996;107:367–372. doi: 10.1111/1523-1747.ep12363346. [DOI] [PubMed] [Google Scholar]
- Yano S., Komine M., Fujimoto M., Okochi H., Tamaki K. Interleukin 15 induces the signals of epidermal proliferation through ERK and PI 3-kinase in a human epidermal keratinocyte cell line, HaCaT. Biochem Biophys Res Commun. 2003;301:841–847. doi: 10.1016/s0006-291x(03)00060-3. [DOI] [PubMed] [Google Scholar]
- Yawalkar N., Karlen S., Hunger R., Brand C.U., Braathen L.R. Expression of interleukin-12 is increased in psoriatic skin. J Invest Dermatol. 1998;111:1053–1057. doi: 10.1046/j.1523-1747.1998.00446.x. [DOI] [PubMed] [Google Scholar]
- Yu X.J., Li C.Y., Xu Y.H., Chen L.M., Zhou C.L. Calcitonin gene-related peptide increases proliferation of human HaCaT keratinocytes by activation of MAP kinases. Cell Biol Int. 2009;33:1144–1148. doi: 10.1016/j.cellbi.2009.07.003. [DOI] [PubMed] [Google Scholar]
- Zheng Y., Danilenko D.M., Valdez P., Kasman I., Eastham-Anderson J., Wu J., et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648–651. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No datasets were generated or analyzed during this study.