

Abstract
Genetic mitochondrial dysfunction is frequently associated with various embryonic developmental defects. However, how mitochondria contribute to early development and cell fate determination is poorly studied, especially in humans. Using human pluripotent stem cells (hPSCs), we established a Dox-induced knockout model with mitochondrial dysfunction and evaluated the effect of mitochondrial dysfunction on human pluripotency maintenance and lineage differentiation. The nucleus-encoded gene TFAM (transcription factor A, mitochondrial), essential for mitochondrial gene transcription and mitochondrial DNA replication, is targeted to construct the mitochondrial dysfunction model. The hPSCs with TFAM depletion exhibit the decrease of mtDNA level and oxidative respiration efficiency, representing a typical mitochondrial dysfunction phenotype. Mitochondrial dysfunction leads to impaired self-renewal in hPSCs due to proliferation arrest. Although the mitochondrial dysfunction does not affect pluripotent gene expression, it results in a severe defect in lineage differentiation. Further study in mesoderm differentiation reveals that mitochondrial dysfunction causes proliferation disability and YAP nuclear translocalization and thus together blocks mesoderm lineage differentiation. These findings provide new insights into understanding the mitochondrial function in human pluripotency maintenance and mesoderm differentiation.
Keywords: TFAM, Mitochondrial dysfunction, hPSCs, Pluripotency maintenance, Mesoderm differentiation
1. Introduction
The mitochondrion is essential as 'energy factories' involved in proliferation, apoptosis, and fate determination in mammalian stem cells. As a semi-autonomous organelle, mitochondria have their genome (mtDNA) encoding part of the functional proteins, including a minority of oxidative phosphorylation (OXPHOS) subunits [1], while the nuclear genome has made a significant contribution to mitochondrial biogenesis and functional maintenance. Mutations in specific genes encoding mitochondrial complex I proteins are associated with many mitochondrial genetic diseases, such as Leber's hereditary optic neuropathy (LHON), Leigh syndrome, which is highly related to specific mitochondrial ND1, ND4, and ND6 and nuclear NDUFAF5 mutations [2]. Therefore, the strategy of replacing mitochondria carrying mtDNA mutations with normal mitochondria in the oocyte to prevent mitochondrial genetic diseases in children has been proposed and attempted [3], despite still being controversial. Meanwhile, the transcription of mtDNA and its function realization is guided by several nuclear coding genes. Mitochondrial transcription factor A (TFAM), which is a nuclear coding mitochondrial gene and regulated by proliferator-activated receptor-γ co-activator 1α (PGC1α) [4,5], makes excellent contributions in the transcription and replication of mtDNA to maintain the stability of mtDNA, and thus is necessary for mitochondrial biogenesis and functional homeostasis, particularly for OXPHOS [6]. Conventional and cardiac-specific (Nkx2.5-Cre-driven) Tfam knockout during middle to late gestational stages [7] exhibit embryonic lethal due to dysfunctional mitochondria, indicating the importance of mitochondria in embryonic development and organogenesis.
In addition, mitochondrial dysfunction is frequently associated with various embryonic developmental defects and failure. Recently, it has been reported that mutations in mtDNA resulted in decreased ovarian primordial and mature follicles in the mouse by impairing the oocyte's NADH/NAD+ redox state [8]. Furthermore, mice lacking Drp1 exhibited abnormal forebrain development and died at E12.5 [9]. Another report revealed that Shmt2 knockout mice showed reduced mitochondrial translation and thus resulted in severe respiration defects contributing to the E13.5 lethal [10]. In addition, Kasahara and colleagues showed that cardiac-specific double knockout of mitochondrial fusion protein Mfn1/Mfn2 caused mouse embryonic lethal between E9.5 and E15.5 due to the severe defects in the myocardial wall [11]. Together, these reports demonstrate that tissue-specific mitochondrial dysfunction usually correlates with severe embryonic developmental defects or intrauterine death. However, the function of mitochondria in maintaining stem cell proliferation and germ layers differentiation during the very early embryonic stage is absent of precise determination.
Due to ethical restrictions and lack of proper experimental materials, studying the early embryonic development of humans has always been a bottleneck in developmental biology. Human pluripotent stem cells (hPSCs), including embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), provide an excellent alternative model for early human development due to the ability of self-renewal and multi-lineage differentiation. Furthermore, the in vitro differentiation of hPSCs reflects a similar response to the critical regulatory factor or pathway to in vivo development, thus helping illustrate the complicated developmental regulatory network in human species. For example, while fibroblast growth factor (FGF) and bone morphogenetic protein (BMP) together induce hPSCs differentiation to cardiovascular mesoderm, BMP10 is critically required for specification and maintenance of human endocardial cells from hPSC-derived cardiovascular mesoderm [12]. In addition, hPSCs from Duchenne patients represent the disease phenotype, including developmental defects [13].
Furthermore, hPSC-derived cardiomyocytes, or micro heart tissues, are considered an ideal drug safety screening model besides mimicking the developmental heart process [14,15]. Of note, hPSCs are also applied to study the role of mitochondria in lineage differentiation. Lees and colleagues reported that dynamic mitochondrial remodeling existed during neural ectoderm differentiation from hPSCs, accompanied by switched glucose metabolic patterns [16]. Importantly, Yamada and colleagues revealed that mitochondrial dysfunction, mediated by knockdown of MFN1 using Mfn1 shRNA lentiviruses, inhibited neural specification from hPSCs [17]. Moreover, Lu and colleagues found that mitochondrial respiration and glutamine oxidation is essential for mesoderm differentiation [18]. Currently, how mitochondria participate in lineage differentiation is largely unknown, probably due to the lack of stable mitochondrial damage models of hPSCs as mitochondrial function is fundamentally required for cell survival.
Very recently and notably, two groups developed a novel strategy that inserting inducible exogenous genes and then knocking out the endogenous genes could completely deplete essential genes such as BACH1 and DICER1in hPSCs [19,20], which make the study of crucial mitochondrial genes more feasible. Here we utilize a similar strategy and generate an hPSC line with Dox (Doxycycline)-induced exogenous TFAM expression and then knock out the endogenous TFAM. Then, we have characterized the pluripotency of the TFAM-depleted hPSCs upon withdrawal of Dox. Moreover, we evaluate the mesoderm differentiation capacity and investigate the underlying mechanism of TFAM-mediated mitochondrial dysfunction in mesoderm differentiation.
2. Results
2.1. Establishment of Dox-induced Exo-TFAM expression system in Endo-TFAM knockout hPSC
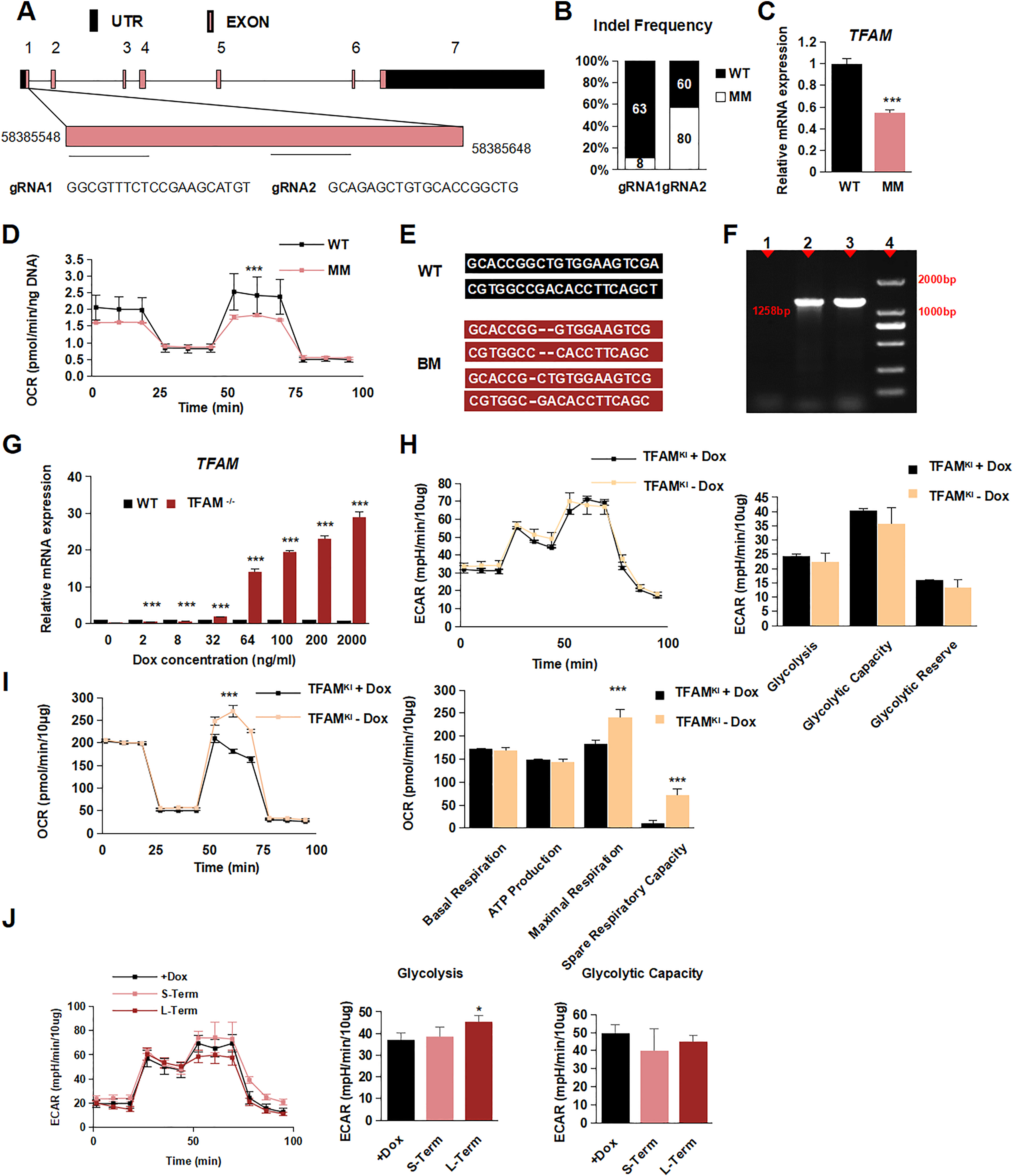
First, we attempted to directly target TFAM exon 1 (encoding 25 amino acids) in hPSCs using the CRISPR/Cas9 system [21]. Two different guide RNAs were designed and cloned to generate TFAM-knockout hPSC with a frameshift mutation (Fig. S1A). After picking up 211 clones (Fig. S1B), we obtained 88 heterozygous TFAM (TFAM+/-) clones but failed to generate any knockout TFAM (TFAM-/-) clone. Compared with wild-type hPSC, TFAM+/- cells remained at about 50% RNA expression level of TFAM (Fig. S1C). In addition, they exhibited a significant decrease in cellular oxygen consumption rate (OCR) (Fig. S1D), demonstrating the effective targeting. These results suggested that TFAM was essential for hPSC long-term maintenance.
To generate an inducible TFAM-knockout hPSC line [22], we first inserted a Dox-inducible Exo-TFAM (exogenous) gene fragment with 3 × FLAG into the AAVS1 locus in a wild-type hPSC line. Then, we used a similar strategy as mentioned above to knockout the Endo-TFAM gene (endogenous) while maintaining Exo-TFAM expression under Dox administration (Fig. 1A). Eventually, 6 Endo-TFAM-/- clones were obtained after picking 48 clones with Dox-induced Exo-TFAM expression. Next, we chose an Endo-TFAM-/- hPSC clone with frameshift mutation named iTFAM-/- in the following study (Fig. S1E and F). In addition, to mimic the equal endogenous TFAM expression level in hPSCs, we titrated the Dox concentrations. We found that 27 ng/mL Dox could induce a similar RNA expression level of Exo-TFAM to Endo-TFAM in hPSCs (Fig. S1G). Upon Dox withdrawal, TFAM mRNA and protein levels were gradually lost over time, along with the decrease of mtDNA levels. The TFAM protein completely disappeared after three days post Dox withdrawal, and the mtDNA copy numbers dropped to ∼5% of the average level after five days (Fig. 1B–D). Taken together, we successfully generated a Dox-induced TFAM-knockout hPSC line.
Fig. 1.

Establishment of TFAM-knockout hPSCs.
(A) Schematic diagram the procedure of generating Dox-induced TFAM-knockout hPSC line.
(B) Quantitative RT-PCR analysis of the expression level of TFAM and mtDNA in TFAM-/- hPSC after Dox withdrawal. Data were shown as Mean ± SEM (n = 3, ***p < 0.001 compared with the +Dox group).
(C) Western blot of the expression of Endo-TFAM and Exo-TFAM (3 × flag tagged) protein in WT, TFAMKI (Exo-TFAM knock-in), and TFAM-/- hPSC with or without Dox. β-actin served as the loading control.
(D) Western blot analysis of Exo-TFAM protein level in TFAM-/- hPSC after Dox removed. β-actin served as the loading control.
(E) Representative transmission electron photomicrographs showing the mitochondrial ultrastructure of TFAM-/- hPSCs treated with or without Dox. Quantification of intact, impaired, and destroyed mitochondria was calculated respectively. Data were shown as Mean ± SEM (n = 15).
(F–G) Representative immunofluorescence images (F) and the quantification (G) for CellRox (green)
/ Mito-tracker (red), DAPI (blue), and JC-A (red)/M (green) in Dox treated or untreated. Scale bars, 20 μm. Data were shown as Mean ± SEM (n = 7, ***p < 0.001 compared with the +Dox group).
(H) Seahorse assay of oxygen consumption rate (OCR) in TFAM-/- hPSCs treated with or without Dox and quantification of mitochondrial functional parameters. Data were shown as Mean ± SEM (n = 3, *p < 0.05, ***p < 0.001 compared with the +Dox group).
(I) The total ATP value in group +Dox, Short-Term, and Long-Term. Data were shown as Mean ± SEM (n = 3, ***p < 0.001 compared with the +Dox group). (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
2.2. Deletion of TFAM disrupts the mitochondrial structure and impairs mitochondrial function
To determine the overall mitochondrial damage caused by TFAM loss, we assessed the ultrastructure morphology of mitochondria under the electron microscope. The intact mitochondrial structure showed a dramatic decrease upon long-term TFAM loss (10 days), but the change was not apparent in the short-term loss (5 days) condition (Fig. 1E). To confirm the mitochondrial dysfunction in TFAM knockout hPSCs, we stained with CellRox and JC-1 to evaluate the ROS and mitochondrial membrane potential level. We found ROS gradually decreased by the time of TFAM loss, suggesting TFAM loss indeed affected mitochondrial oxidation to a certain extent (Fig. 1F and G). In addition, long-term TFAM loss showed a dramatically lower level of oxygen consumption together with a slightly increased glycolysis level. Still, those phenotypes were not observed in the cells with short-term TFAM loss (Fig. 1H and Fig. S1J). The effect of Dox treatment on mitochondrial energy function in hPSCs was excluded (Fig. S1H and I). More importantly, we found the total ATP value in long-term TFAM loss decreased (Fig. 1I), consistent with the change of metabolic pattern. Collectively, the continuous TFAM depletion led to progressive disruption of mitochondrial structure and functional dysregulation, demonstrating the critical role of TFAM in mitochondrial homeostasis and further confirming the successful establishment of mitochondrial dysfunction cell model by inducibly targeting TFAM in hPSCs.
2.3. TFAM knockout impedes self-renewal in hPSCs
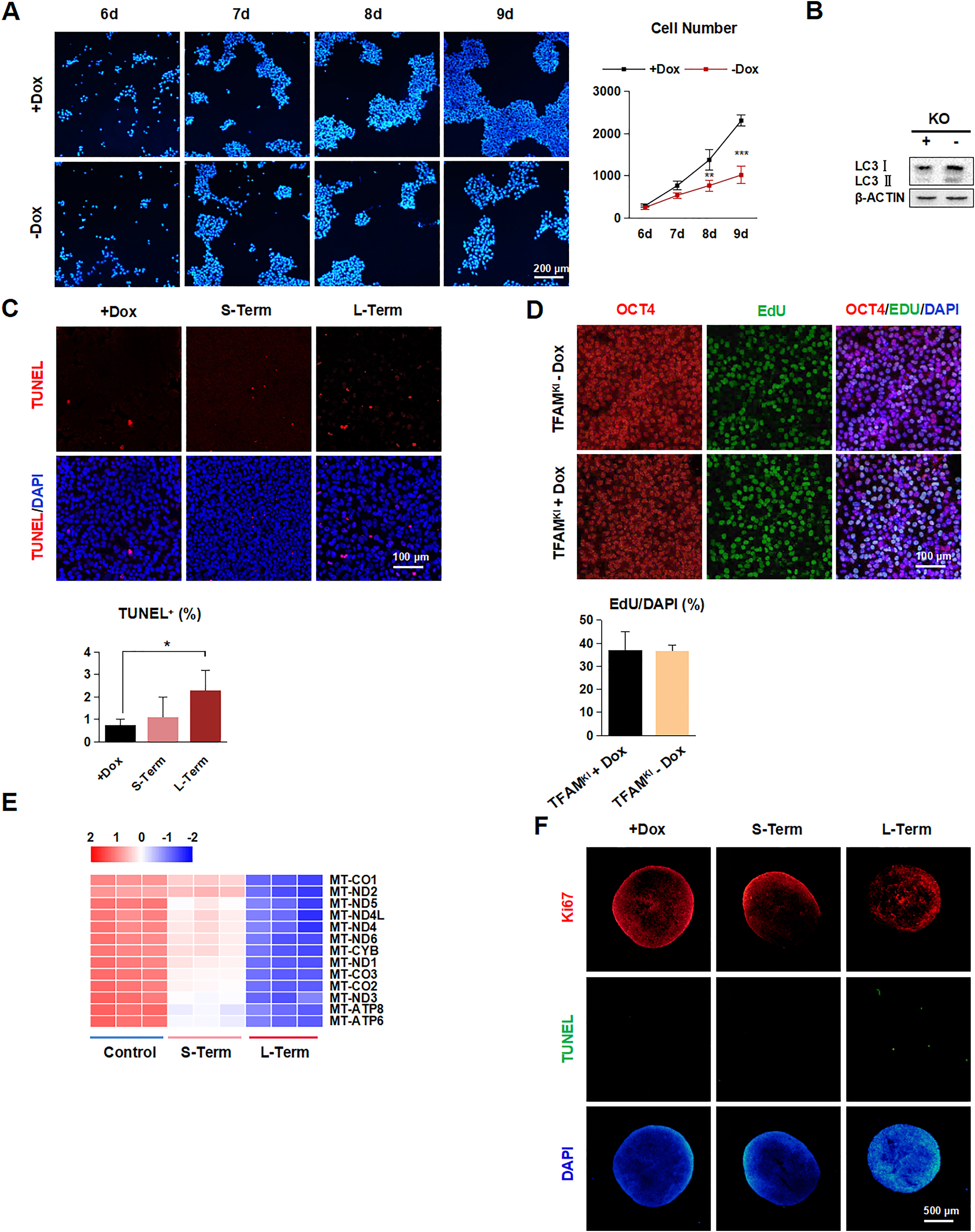
Next, we evaluated whether the pluripotency was affected by TFAM knockout. Results showed that the colony size and growth curve significantly decreased after deleting TFAM in hPSCs (Fig. S2A). Colony formation assay also showed reduced self-renewal ability in both TFAM short-term and long-term depletion in monolayer hPSCs culture conditions (Fig. 2A and B). However, we checked the expression levels of pluripotent markers such as OCT4 and SSEA4, and the results showed no significant change in either RNA or protein level (Fig. 2C and D). To further understand the cause of impaired self-renewal, we then evaluated the cell death and proliferation status. We observed a slightly but significantly increased ratio of TUNEL+ and autophagy marker (LC3Ⅰ/LC3Ⅱ) staining after TFAM deletion (Fig. S2B–C). However, we found up to 40% remarkable decrease in proliferation upon long-term TFAM loss (Fig. 2E and F) despite no significant increase by overexpressing TFAM (Fig. S2D). The results suggested that the proliferation defect, rather than apoptosis or autophagy, predominantly contributes to the impaired self-renewal of hPSCs caused by TFAM knockout.
Fig. 2.

TFAM is critical for hPSC self-renewal
(A-B) Representative staining images (A) and quantification (B) of AP+ (purple) in TFAM-/- hPSCs treated with or without Dox. Scale bars, 2 mm. Data were shown as Mean ± SEM (n = 3, ***p < 0.001 compared with the +Dox group).
(C) Quantitative RT-PCR analysis of the expression level of OCT4 in TFAM-/- hPSCs treated with or without Dox. Data were shown as Mean ± SEM (n = 3).
(D) Representative immunostaining images for OCT4+ (green), SSEA4+ (red) and DAPI (blue) in TFAM-/- hPSCs treated with or without Dox. Scale bars, 100 μm.
(E–F) Representative immunostaining images (E) and quantification (F) of EdU+ (green) and DAPI (blue) in TFAM-/- hPSCs treated with or without Dox. Scale bars, 100 μm. Data were shown as Mean ± SEM (n = 8, *p < 0.05, ***p < 0.001 compared with the +Dox group).
(G) PCA scatter plot of gene expression in group + Dox, Short-Term, and Long-Term. (n = 3).
(H) Venn diagram representation of the common pool of differentially regulated genes in three groups.
(I–J) GO and KEGG analysis in Part Ⅰ (I) and Part Ⅱ (J).
(K) The heatmap displays the Logarithm of fold change (Log2FC) in the G1/S transition of the mitotic cell cycle and DNA replication.
(L) Flow cytometry assessment of cell cycle in group +Dox, Short-Term, and Long-Term. (n = 3). (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
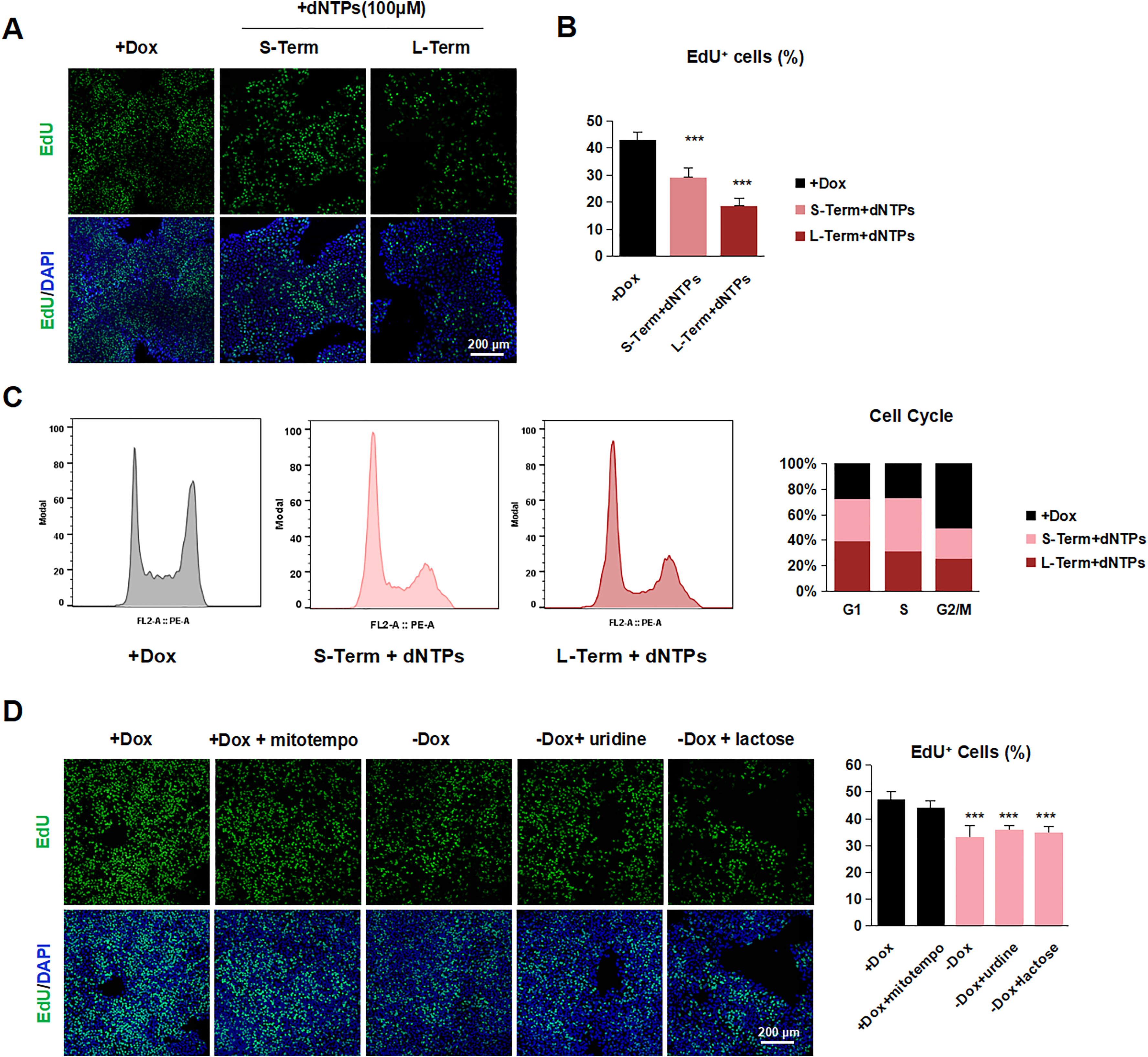
To dissect the underlying molecular events upon TFAM deletion, we performed the RNA-seq using short-term and long-term TFAM-off hPSC samples. Principal component analysis showed these samples clustered clearly between control, TFAM short-term off, and TFAM long-term off hPSCs (Fig. 2G). As expected, mitochondrion-encoded genes exhibited the most significant down-regulation features upon TFAM knockout (Fig. S2E). Of note, genes in DNA replication and G1/S transition in the mitotic cell cycle showed progressively significant expression changes upon TFAM deletion (Fig. 2H–K). Indeed, the cell cycle analysis indicated the G1/S phase arrested, and G2/M phase decreased in TFAM knockout hPSCs (Fig. 2L), despite no difference for the G0 phase, which was also evidenced by the observed undistinguished Ki67-positive cell percentage (Fig. S2F). To further explore the cause of cell cycle arrest, we suspected dNTPs or lack of energy metabolites as the cause. However, when dNTPs, uridine, or lactose were added to TFAM-knockout cells, there was no obvious complement to proliferation (Fig. S3A–D). In summary, TFAM knockout results in the failure of mitochondria-encoded gene expression and ultimately leads to cell cycle arrest in hPSCs, although it does not directly affect the expression of pluripotent genes.
2.4. TFAM knockout blocks multi-lineage differentiation potential of hPSCs
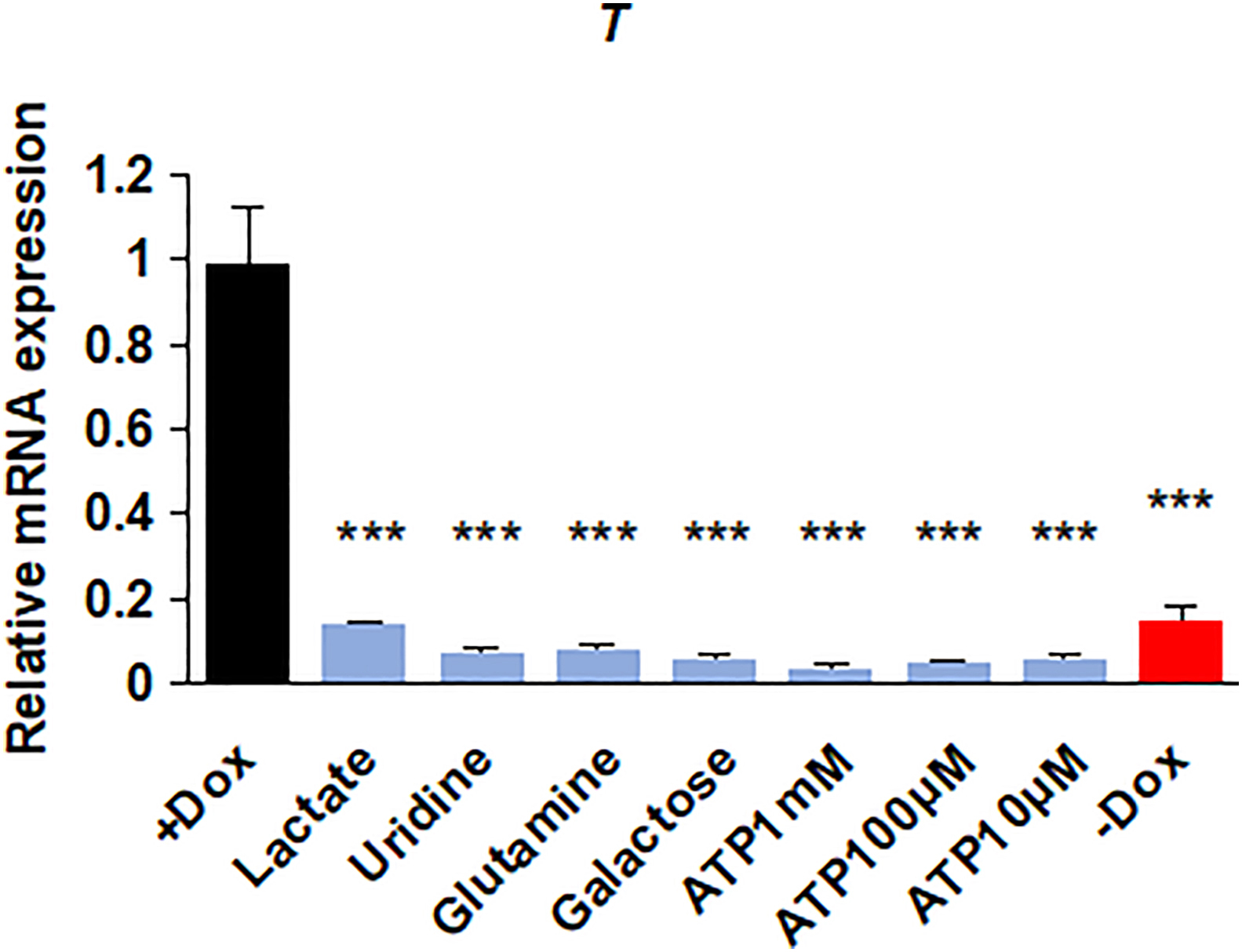
We were then wondering whether TFAM knockout affected the differentiation potential of hPSCs. A significantly smaller embryoid bodies (EBs) from TFAM knockout cells was observed compared to wild-type groups (Fig. 3A). Furthermore, TFAM knockout generally resulted in lower expression of all three germ layers and higher remaining pluripotent markers (Fig. 3B), indicating the failure of lineage differentiation. According to well-established protocols, we further performed directed differentiation toward definitive endoderm [23,24] and observed that TFAM knockout hPSCs exhibited much lower differentiation efficiency measured by SOX17-positive percentage (Fig. 3C). Furthermore, ectoderm differentiation from hPSCs [25,26] in TFAM knockout group expressed much lower SOX1 (ectoderm) (Fig. 3D). Similarly, TFAM knockout hPSCs showed decreased T-positive staining upon differentiation into mesoderm [27] (Fig. 3E). Also, mesoderm differentiation from hPSCs in the TFAM knockout group showed reduced TMRM and ROS, suggesting that the differentiation system was a mitochondria dysfunction model (Fig. 3F and G). The above results are consistent with the EB assay (Fig. 3A). Meanwhile, we noticed that adding metabolites including uridine, galactose, lactate, glutamine, and ATP (10 μM, 100 μM, 1 mM) did not reverse differentiation (Fig. S4A). In general, TFAM knockout hinders the multi-lineage commitment ability of hPSCs.
Fig. 3.

The deletion of TFAM blocks multi-lineage differentiation potential of hPSCs
(A) Schematic diagram of EB differentiation. Representative image and quantification of EBs from hPSCs treated with or without Dox. Data were shown as Mean ± SEM (n = 3, ***p < 0.001 compared with the +Dox group).
(B) Quantitative RT-PCR analysis of the expression level of SOX1, PAX6, FOXA2, GATA4, SOX17, OCT4, and NANOG in EB differentiation treated with or without Dox. Data were shown as Mean ± SEM (n = 3, *p < 0.05, ***p < 0.001 compared with the +Dox group).
(C) Schematic diagram of entoderm differentiation. Representative immunostaining images and quantification of SOX17+ (red) and DAPI (blue) in PSC to entoderm stage treated with or without Dox. Scale bars, 200 μm. Data were shown as Mean ± SEM (n = 3, ***p < 0.001 compared with the +Dox group).
(D) Schematic diagram of ectoderm differentiation. Representative immunostaining images and quantification of SOX1+ (red) and DAPI (blue) in PSC to ectoderm stage treated with or without Dox. Scale bars, 200 μm. Data were shown as Mean ± SEM (n = 3, ***p < 0.001 compared with the +Dox group).
(E) Schematic diagram of mesoderm differentiation. Representative immunostaining images and quantification of T+ (red) and DAPI (blue) in PM (PSC to Mesoderm) stage treated with or without Dox. Scale bars, 200 μm. Data were shown as Mean ± SEM (n = 3, ***p < 0.001 compared with the +Dox group).
(F) Representative immunostaining images and quantification of TMRM (red) and DAPI (blue) in PM stage treated with or without Dox. Scale bars, 20 μm. Data were shown as Mean ± SEM (n = 3, *p < 0.05 compared with the +Dox group).
(G) Representative immunostaining images and quantification of CellRox (green),Mito-tracker (red) and DAPI (blue) in PM stage treated with or without Dox. Scale bars, 20 μm. Data were shown as Mean ± SEM (n = 3, ***p < 0.001 compared with the +Dox group). (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
2.5. TFAM knockout blocks mesoderm differentiation of hPSCs
To understand the underlying mechanism that mitochondrial dysfunction represses lineage differentiation of hPSCs, we focused on mesoderm lineage differentiation. First, we performed RNA-seq to analyze the effect of TFAM-knockout in the process of hPSCs on mesoderm differentiation (Fig. 4A). Gene set enrichment analysis (GSEA) showed that TFAM knockout significantly affected the expression of genes involving mesoderm development (Fig. 4B), consistent with the previous results (Fig. 3E). To further dissect the molecular events during mesoderm differentiation upon TFAM deletion, we performed gene ontology (GO) analysis (Fig. 4C), in which regulation of cell proliferation was enriched (Fig. 4D). Next, we examined whether the limited cell proliferation observed in hPSCs was involved. To this aim, we utilized EdU to repress cell proliferation during mesoderm differentiation. The result of immunofluorescence (Fig. 4E) showed lower mesoderm markers in the EdU-treated group after two days' treatment. However, this phenotype is not obvious in one day's treatment, evidenced by western blotting results (Fig. 4F) and qRT-PCR (Fig. 4G), consistent with the short-term Dox-withdrawal group. These results also indicated that the absence of TFAM affected the fate of hPSCs mesoderm decisions by decreasing proliferation. Consistent with the previous hypothesis, TFAM knockout shared a similar proliferation gene set with EdU treatment on mesoderm induction. We observed similar results by applying another cell proliferation inhibitor ara-C (Fig. S5A–C). We noticed that TFAM knockout, having a higher cell proliferation rate than the EdU-treated group, interestingly exhibited a lower differentiation efficiency, which suggested that other factors besides proliferation contribute to the blocked lineage differentiation.
Fig. 4.

Decreased cell proliferation contributes to the impeded mesodermal differentiation in TFAM-/- cells
(A) Differentially expressed genes in PM stage treated with or without Dox.
(B) GSEA revealed the enrichment of the mesoderm development between hPSCs stage and PM stage treated with or without Dox.
(C) GO and KEGG analysis in the group treated with or without Dox.
(D) The heatmap displays the Log2FC in cell proliferation genes.
(E) Representative immunostaining images and quantification of T+ (red) and DAPI (blue) in PM stage treated with Dox/EdU or without Dox. Scale bars, 200 μm. Data were shown as Mean ± SEM (n = 3, *p < 0.05, n = 3, ***p < 0.001 compared with the +Dox group).
(F) Representative western blot TFAM in PM stage treated with Dox/EdU or without Dox. β-actin served as the loading control.
(G) Quantitative RT-PCR analysis of the expression level of TFAM, T in PM stage treated with Dox/EdU or without Dox. Data were shown as Mean ± SEM (n = 3, ***p < 0.001 compared with the +Dox group). (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
2.6. YAP mediated microenvironment force response contributes to the mesoderm fate determination in the hPSC differentiation model
Interestingly, the extracellular matrix (ECM) term was enriched in the differentially expressed genes between Dox withdrawal and the control group (Fig. 5A and B). In 2006, Engler and colleagues demonstrated that stem cells have different fate-determining tendencies in additional substrate stiffness [28]. The YAP-TEAD1 axis, regulated by ECM stiffening, could direct hPSCs specification by influencing cytoskeleton arrangement and intracellular tension [29], also appeared in our RNA-seq results. Then we tested the YAP-TEAD1 axis in our system and observed a significantly higher YAP nucleus-localized signal in TFAM-knockout cells (Fig. 4C). Interestingly, we found that a highly thick layer of Matrigel (5x normal concentration) coating could quickly rescue mesodermal differentiation in TFAM knockout cells, assayed in mRNA and protein levels (Fig. 5C and D). The nuclear localization events of YAP decreased in the thick Matrigel layer group (Fig. 5E). Further experiments proved that Collagen in Matrigel played a complementary role (Fig. 5F).
Fig. 5.

TFAM knockout leads to abnormal nucleation of YAP
(A) GSEA revealed the enrichment of the extracellular matrix assembly in PM stage treated with or without Dox.
(B) The heatmap displays the Log2FC in cell-matrix genes.
(C) Representative immunostaining images and quantification of T+ (red) and DAPI (blue) in PM stage treated with or without Dox. Scale bars, 200 μm. Data were shown as Mean ± SEM (n = 8, ***p < 0.001 compared with the +Dox group).
(D) Quantitative RT-PCR analysis of the expression level of TFAM, T, and MESP1 in PM stage treated with or without Dox. Data were shown as Mean ± SEM (n = 3, ***p < 0.001 compared with the +Dox group).
(E) Representative immunostaining images and quantification of YAP+ (green) and DAPI (blue) in PM stage treated with or without Dox. Scale bars, 20 μm. Data were shown as Mean ± SEM (n = 8, *p < 0.05, ***p < 0.001 compared with the +Dox group).
(F) Quantitative RT-PCR analysis of the expression level of T in PM stage treated with or without Dox/collagen. Data were shown as Mean ± SEM (n = 3, ***p < 0.001 compared with the -Dox group).
(G) Quantitative RT-PCR analysis of the expression level of T, TFAM and YAP1 in PM stage treated with or without Dox/YAP1-shRNA. Data were shown as Mean ± SEM (n = 3, ***p < 0.001 compared with the -Dox group).
(H) Representative immunostaining images and quantification of T+ (red) and DAPI (blue) in PM stage treated with or without Dox/shRNA. Scale bars, 200 μm. Data were shown as Mean ± SEM (n = 7, ***p < 0.001 compared with the -Dox group). (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
To explore whether YAP signal participates in mesoderm differentiation, we first applied Lats-IN-1, a chemical targeting LATS1/2 kinase and promoting YAP-TEADs pathway [30], and observed that the mesoderm efficiency was significantly decreased (Fig. S6A). To further confirm whether YAP was the target of TFAM during mesoderm differentiation, we applied shRNA targeting YAP in TFAM knockout cells. Importantly, we found knockdown of YAP could significantly rescue the failed mesoderm differentiation phenotype caused by TFAM knockout (Fig. 5G and H). These results suggested that TFAM regulated the YAP-mediated extracellular force response and then contributed to the mesoderm fate determination.
3. Discussion
Recently, the mitochondrion has been annotated as an essential regulator of cell fate determination beyond the fundamental energy provider, thus attracting more and more attention in stem cell biology. Here, we utilized the hPSC model combined with genome editing technology to elucidate the effect of TFAM-mediated mitochondrial dysfunction in pluripotency maintenance and lineage differentiation. We demonstrated that TFAM was necessary for long-term maintenance of hPSCs and proper lineage differentiation as well. Furthermore, we studied the underlying mechanism of TFAM in mesoderm differentiation. We found TFAM-mediated mitochondrial dysfunction affected mesoderm lineage commitment in hPSCs mainly by reducing cell proliferation and inhibiting the YAP-TEAD axis (Fig. 6).
Fig. 6.

Schematic diagram of TFAM depletion contribution in human PSCs self-renewal and mesoderm lineage differentiation regulation.
Due to ethical constraints, it is hard to study the mitochondrial roles in human early embryonic development. Many previous reports have illustrated the importance of mitochondrial morphology and function during mammalian development. For instance, TFAM specifically in cardiac progenitor cells or depletion of Mfn1/Mfn2 led to embryo lethality before E15.5 [7,11], and the mitochondrial dysfunction due to Drp1 deficiency caused defects in mitophagy and thus resulted in lethal heart failure and neurodegeneration in neonatal mice [31]. However, early embryo lethality led by significant mitochondrial genes deficiency makes it very challenging to study early embryos. Therefore, current reports focused on organ development and maturation in the perinatal period. Although the emergence of hPSCs and the application of the differentiation system provide an excellent platform for exploring early development, mitochondria's influence and mechanism on pluripotency and early lineage differentiation are rarely studied. Combined with the new strategy that is inserting inducible exogenous genes and then knocking out endogenous genes, our present study has established a mitochondrial dysfunction model by constructing an inducible TFAM knockout hPSC line (Fig. 1, Fig. 2). To our knowledge, this is the first report about the inducible knockout of a critical gene-regulating mitochondria function. Our present study thus lays a foundation for the subsequent studies on the roles of mitochondria-related genes during early development and lineage differentiation.
TFAM is a mitochondrial transcription factor regulating the normal function of mitochondria. A previous study has demonstrated that in mice with TFAM depletion, the copy number of mitochondrial DNA and the level of oxidative phosphorylation were decreased along with the damaged mitochondrial cristate [32], which are similar to our study. In addition, our results suggested a critical role of TFAM in self-renewal and lineage differentiation of hPSCs. TFAM knockout caused impaired self-renewal mainly due to the blockage of the cell cycle in G1/S transition. However, apoptosis in the TFAM-knockout hPSCs is not as severe as the observation in the rodent model [33,34] and human cancer cells [[35], [36], [37], [38]].
Then, we surveyed the function of TFAM in hPSCs mesoderm differentiation. TFAM-deficient hPSCs could not differentiate into mesoderm. Proliferation is also essential for cell differentiation. Many reports have demonstrated that signaling pathways associated with developmental genes are highly enriched in the G1 phase [[39], [40], [41]]. Furthermore, we found proliferation declined in the differentiation process in TFAM knockout cells, which is consistent with the cell stage. Indeed, chemicals inhibiting cell proliferation affected mesoderm differentiation of hPSCs (Fig. 4 and S5). Most interestingly, we detected the cell migration and cytoskeleton dysregulation in RNA-seq analysis of TFAM knockout cells. Previous studies showed that some cellular processes, including migration and differentiation, are related to cytoskeleton dynamics regulated by the YAP-TEAD signaling pathway. Also, a recent study characterized that the regulation of the YAP-TEAD axis in response to substrate rigidity directs hPSC mesoderm specification by controlling cytoskeleton dynamics [29]. Consistent with the above results, we show that TFAM promotes mesoderm formation via maintaining the YAP-TEAD pathway. These results expand our understanding of the role of TFAM and mitochondria in mesoderm commitment. However, since we performed the knockout for the entire mitochondria, the subsequent events are unclear yet about which part of the mitochondria function, such as ROS production, regulation of cell metabolism, Ca2+ dynamics, and cell apoptosis are affected [42].
In summary, our present study provided a platform to study mitochondrial function in hPSCs and revealed the role and mechanism of TFAM-mediated mitochondrial dysfunction in pluripotency maintenance and lineage differentiation (Fig. 6). Limited by our research interest, we focused on mesoderm differentiation and found cell proliferation and YAP signal played a functional role in TFAM-mediated regulation. In addition, we observed the defect in ectoderm and endoderm differentiation, but whether the exact mechanism contributed to the other two lineage differentiation is awaiting further dissections.
4. Materials and methods
4.1. Cell lines and cultures
Two hPSC lines, WTC [43] (Gladstone) and PGP1 [44] (Thomas Gingeras), were cultured in mTeSR1 medium (Stemcell Technologies) with 1% penicillin-streptomycin (Gibco, 15140122). In addition, Matrigel (Corning, 354277) was diluted by KncokOut™ DMEM medium (Gibco, 10829018) in 1:100 and coated on the plate before use. The medium was changed every 24 h. At 80%–90% confluence, cells were dissociated with versene (Stemcell, 15040-066) and passaged with the p160Rho-associated coiled-coil kinase (ROCK) inhibitor Y-27632 (Tocris, 1254, 10 μM).
4.2. Generation of stable TFAMKI and TFAM-/- hPSC lines
TFAMKI cells were generated by electroporation of AAVS1-guide (2 μg) and template vector (4 μg) with an Amaxa nucleofector 2b device (program B-016) and Amaxa human stem cell Nucleofector kit (Lonza). Cells were selected by G418 (SANTA CRUZ, sc-29065A, 200 μg/mL) for five days, and the remaining cells were split into 6-well plates for colony picking in mTeSR1 medium supplemented with Y-27632 (10 μM). Expanded clones were selected by G418 again. Genotyping PCR was performed to detect whether Exo-TFAM has been knocked in.
TFAM-/- cells were generated by electroporation of TFAM gRNA2 (4 μg) as above. Cells were selected by puromycin (0.5 μg/mL) for one day, and the survived cells were cultured with Dox (100 ng/mL, Sigma) until the cells were split into 6-well plates in mTeSR1 medium supplemented with Y-27632 (10 μM) and Dox (Sigma, D9891, 100 ng/mL). Single colonies were then picked up and genotyped by PCR, followed by Sanger sequencing.
The guide sequence information was as follows:
TFAM-g1 (5′-CACCCGTTTCTCCGAAGCATGT-3′); TFAM-g2 (5′-CACCGCAGAGCTGTGCACCGGCTG-3′); AAVS1-g (5′-CACCGGGGCCACTAGGGACAGGAT-3′).
4.3. Generation of stable YAP knockdown hPSC lines
YAP1 knockdown hPSC lines were generated by lentiviral vector carrying small hairpin RNA-1 (5′-GACATCTTCTGGTCAGAGA-3′) or small hairpin RNA-2 (5′-GGTGATACTATCAACCAAA-3′) targeting the YAP1. A negative control with an unrelated human gene sequence (5′-TTCTCCGAACGTGTCACGT-3′) was transfected. Stabled cell lines were selected by puromycin (0.5 μg/mL).
4.4. Cell cycle analysis
Harvested cells were washed twice with DPBS (Gibco, C14190500BT) in a 15 mL centrifuge tube and then the appropriate pre-cooled ethanol (70%) was added. Meanwhile, the centrifuge tubes were placed on the vortex to prevent cells from clumping. After being fixed at 4 °C for 30 min, the cells were washed twice with DPBS. 200 μL DPBS solution containing RNase was added for 30 min, followed by 10 μL PI (final concentration 50 μg/mL) for another 30 min. Tubes were put into flow cytometry (FCM) with the cell reading number set to 50000. The resulting data were analyzed with Flowjo.
4.5. Real-time quantitative PCR analysis
Total RNA from hPSCs and the derivates was extracted using HiPure Total RNA Mini Kit (Magen, R4111-03). RNA (1 μg) was reversely transcribed into cDNA using Hifair™ Ⅱ 1st strand cDNA short Kit (Yeasen, 11123ES60). Every PCR reaction was performed using Hieff ™ qPCR SYBR Green Master Mix (Yeasen, 11201ES08), with primers and cDNA (5 ng). The reactions were run in triplicates in a fluorescence quantitative detection system (BIORAD). Cycle threshold values were normalized to GAPDH mRNA expression. Primers sequences were listed as follows:
GAPDH, 5′-AATGAAGGGGTCATTGATGG-3′ and 5′-AAGGTGAAGGTCGGAGTCAA-3'; TFAM, 5′-GCGCTCCCCCTTCAGTTTTG-3′ and 5′-GTTTTTGCATCTGGGTTCTGAGC-3'; NANOG, 5′-GTCTTCTGCTGAGATGCCTCACA-3′ and 5′-CTTCTGCGTCACACCATTGCTAT-3'; SOX1, 5′-GAGTGGAAGGTCATGTCCGAGG-3′ and 5′-CCTTCTTGAGCAGCGTCTTGGT-3'; PAX6, 5′-TCCGTTGGAACTGATGGAGT-3′ and 5′-GTTGGTATCCGGGGACTTC-3'; FOXA2, 5′-GGAACACCACTACGCCTTCAAC-3′ and 5′-AGTGCATCACCTGTTCGTAGGC-3'; GATA4, 5′-GCGGTGCTTCCAGCAACTCCA-3′ and 5′-ACATCGCACTGACTGAGAACG-3'; SOX17, 5′-GCATGACTCCGGTGTGAATCT-3′ and 5′-TCACACGTCAGGATAGTTGCAGT-3'; OCT4, 5′-CAAAGCAGAAACCCTCGTGC-3′ and 5′-TCTCACTCGGTTCTCGATACTG-3'; T, 5′-GATGATCGTGACCAAGAACGG-3′ and 5′-CCACGAAGTCCAGCAGGAA-3'; mtDNA, 5′-CGAAAGGACAAGAGAAATAAGG-3′ and 5′-CTGTAAAGTTTTAAGTTTTATGCG-3'. YAP1, 5′-TGTCCCAGATGAACGTCACAGC-3′ and 5′-TGGTGGCTGTTTCACTGGAGCA-3'.
4.6. Immunofluorescence staining
Harvested cells were cultured in confocal dishes. After growing to the conditions required for the experiment, the medium was removed, and cells were fixed with 4% paraformaldehyde (PFA, Dingguo, AR-0211) and incubated at room temperature for 30 min. Washed three times with DPBS and then blocked the cells with blocking buffer (DPBS containing 5% donkey serum and 0.2% Triton X-100) at 4 °C overnight. Cells were incubated with 200 μL/well of blocking buffer containing primary antibody at 4 °C overnight. After washing twice with DPBS, cells were incubated at room temperature for 2 h (in the shade) with 200 μL/well of blocking buffer containing secondary antibody. Washed twice or three times with DPBS, the nucleus was then stained with DAPI (Thermo Fisher, D1306, 1:1000) at room temperature of about 10 min. After washing twice or three times with DPBS, Immunofluorescence staining was observed by Zeiss confocal microscope (LSM 980), and image data were analyzed using ImageJ software.
Primary antibodies used in this study were as follows: anti-OCT4 (Santa Cruz, SC8628, 1:200); anti-SSEA4 (Millipore, MAB4304, 1:200); anti-SOX1 (Cell Signaling, 4194s, 1:200); anti-SOX17 (CUSABIO, CSB-PA887989LA01HU, 1:200); anti-T (CUSABI, CSB-PA16539A0Rb, 1:200); anti-Ki67 (ZSGB-BIO, 17A11203, 1:200). Donkey derived secondary antibodies were as follows: Anti-Rabbit IgG (H + L), Alexa Fluor 488 (Thermo Fisher, A-21206, 1:1000); Anti-Mouse IgG (H + L), Alexa Fluor 488 (Thermo Fisher, A-21202, 1:1000); Anti-Goat IgG (H + L), Alexa Fluor 488 (Thermo Fisher, A-11055, 1:1000); Anti-Rabbit IgG (H + L), Alexa Fluor 594 (Thermo Fisher, A-21207, 1:1000). Anti-Mouse IgG (H + L), Alexa Fluor 594 (Thermo Fisher, A-21203, 1:1000); Dyes were as follows: JC-1 (Thermo Fisher, T3168, 1:1000); TMRM (Thermo Fisher, I34361, 1:1000); Cell-ROX (Thermo Fisher, C10422, 1:1000); Mito-tracker (Thermo Fisher, M7512, 1:1000); Mito-tracker (Thermo Fisher, M7512, 1:2000); Hoechst (Thermo Fisher, H21492, 1:2000); PI (Beyotime, ST512, 1:300). KFluor488-EdU cell proliferation detection kit (KeyGEN BioTECH, KGA331-500) and TUNEL Apoptosis Detection Kit (Alexa Fluor 488) (Yeasen, 40307ES50) were also used in this study.
4.7. Western blotting
Harvested cells were lysed with RIPA lysis buffer (Beyotime, P0013K) and then the protease inhibitor PMSF (Sinopharm, xw020003) was added. After breaking the cells with Ultrasonic Cell Disruptor, proteins were quantified using Takara BCA Protein Assay Kit (Takara, T9300A). Protein samples (10 μg) were separated with SDS-PAGE (12%) and transferred to PDVF membrane (BIORAD). After blocking with TBST solution containing skimmed milk (Yili, 232100, 5%) or BSA (BIOFRO, BIOFROXX200010, 5%) for 1–2 h, the primary antibody was incubated at 4 °C overnight and then washed three times for 10 min each time with TBST. The secondary antibody was incubated at room temperature for 90 min. After washing the secondary antibody with TBST, membranes were incubated for 2 min by Pierce ™ ECL Western Blotting Substrate (Thermo). Membranes were imaged using the ChemiDoc ™ (BIORAD).
Primary antibodies were as follows: anti-β-actin (Proteintech, 23660-1AP, 1:10000); anti-mtTFA (Abcam, 176558, 1:7500); anti-T (CST#81694, 1:200); anti-LC3 (SANTA CRUZ, sc-398822, 1:1000). Secondary antibodies were as follows: HRP-conjugated Affinipure Goat Anti-Rabbit IgG (H + L) (Proteintech, SA00001-2, 1:2000); HRP-conjugated Affinipure Goat Anti-Mouse IgG (H + L) (Proteintech, SA00001-1, 1:2000).
4.8. Seahorse
After a preliminary experiment determining the proper cell density, the cells were seeded on Cell Culture Microplates (Agilent) and Calibrate Sensor Cartridges were treated with Calibrant for 10 h, followed by twice washing using OCR buffer solution or ECAR buffer solution, respectively. Next, the culture plate was placed under the Calibrate Sensor Cartridges and OCR detection reagent (2 μM oligomycin, 1 μM FCCP, 0.5 μM rotenone) or ECR detection reagent (10 mM glucose, 1 μM oligomycin, 50 mM 2-DG) was added in Calibrate Sensor Cartridges. The values were measured in Seahorse XFe24 (Agilent). At last, the cells were lysed with RIPA for BCA quantification to normalize the total cell numbers.
4.9. Cell differentiation
hPSCs were reseeded onto Matrigel-coated dishes at 3 × 104 cells/cm2 and cultured in mTeSR1 medium for 3 days until colonies density reached 80% confluence.
For mesoderm differentiation, hPSCs were incubated with differentiation basal medium, comprised RPMI 1640 medium (Gibco, C11875500BT) and B-27 supplement minus insulin (Gibco, A1895601). On day 0, CHIR99021 (Tocris, 4423, 7.5 μM) was added to the differentiation basal medium. On day 2, CHIR99021 was removed from the medium.
For definitive endoderm differentiation, hPSCs were incubated with a differentiation basal medium, comprised RPMI 1640 medium (Gibco, C11875500BT) and B-27 supplement minus vitamin A (Basal Media, S440J7). On day 0, CHIR99021 (3 μM) and Activin A (Stemcell, 78001, 100 μg/mL) were added to the differentiation basal medium. On day 1, the medium was replaced with a differentiation basal medium supplement with Activin A (100 μg/mL) every day until day 4.
For neural ectoderm differentiation, hPSCs were reseeded onto Matrigel-coated dishes at 4 × 104 cells/cm2 and cultured in mTeSR1 medium for 3 days until colonies density reached 95% confluence. hPSCs were incubated with ectoderm differentiation medium which comprised DMEM/F12 (Gibco, C11995500BT), 0.5x N2 (BasalMedia, S430J4), 0.5x B27 (BasalMedia, S441J7), 1% Gluta-max, 1% NEAA, 0.1% β-mercaptoethanol) supplemented with 10 μM SB431542 (Selleck, S1067) and 0.1 μM LDN (Selleck, 7507). On day 5, the TGF-β inhibitor SB431542 was withdrawn from the ectoderm differentiation medium until day 7. The medium was changed every day until day 7.
4.10. RNA sequencing and data analysis
Raw RNA-seq data will be publicly available on https://ngdc.cncb.ac.cn/ (HRA001146). Cutoffs of P-value > 0.05 and log2(fold-change) ≥ 1 or ≤ −1 were used for differentially upregulated or downregulated genes. RNA-seq PCA plots were created using PCA tools (https://www.omicstudio.cn/index). Heatmaps were made using Heatmapper (http://www.heatmapper.ca/). All genes reported with log2(fold-change) > |0.3| were included in the analysis for stem cell stage data. KEGG pathway and Gene Ontology Biological Processes analysis were performed using the DAVID gene set enrichment tool.
4.11. Statistical analyses
All data are reported as means ± SEM. Results were analyzed using Student's t-test. Differences were considered statistically significant at p < 0.05. At least three independent experiments were performed as replicates for statistical analyses.
Funding
DZ was funded by National Science and Technology Major Project (grant no. 2018YFA0109100), National Natural Science Foundation of China (grant no. 31871496) and Hubei Science Fund for Distinguished Young Scholars (grant no. 2019CFA091).
WJ was funded by National Natural Science Foundation of China (grant no. 31970608) and Hubei Science Fund for Distinguished Young Scholars (grant no. 2021CFA049).
YL was funded by National Natural Science Foundation of China (grant no. 81700360) and the Technology Project of Sichuan Province of China (grant no. 2020YFS0102).
Author contributions
YL and DZ conceptualized the study; YQ, YY, WJiang, YL, and DZ designed the experiments; YQ, YY, RW, SY, YZ, JL and WJin performed experiments; YQ, YY, YZ, JL, WJin, SX, WJiang and YL analyzed data of experiments; YQ, YL, DZ and WJiang wrote or edited the manuscript.
Correspondence and requests for materials should be addressed to DZ.
Data availability
All the sequencing data has been deposited at GSA (https://ngdc.cncb.ac.cn/gsub/, HRA001146. Other data sets used and/or analyzed during the current study are available from reasonable request to corresponding authors.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2022.102248.
Contributor Information
Wei Jiang, Email: jiangw.mri@whu.edu.cn.
Yifei Li, Email: liyfwcsh@scu.edu.cn.
Donghui Zhang, Email: dongh.zhang@hubu.edu.cn.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- 1.Gustafsson C.M., et al. Maintenance and expression of mammalian mitochondrial DNA. Annu. Rev. Biochem. 2016;85:133–160. doi: 10.1146/annurev-biochem-060815-014402. [DOI] [PubMed] [Google Scholar]
- 2.Hahn A., Zuryn S. The cellular mitochondrial genome landscape in disease. Trends Cell Biol. 2019;29(3):227–240. doi: 10.1016/j.tcb.2018.11.004. [DOI] [PubMed] [Google Scholar]
- 3.Kang E., et al. Mitochondrial replacement in human oocytes carrying pathogenic mitochondrial DNA mutations. Nature. 2016;540(7632):270–275. doi: 10.1038/nature20592. [DOI] [PubMed] [Google Scholar]
- 4.Fernandez-Marcos P.J., Auwerx J. Regulation of PGC-1alpha, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011;93(4):884S–890S. doi: 10.3945/ajcn.110.001917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Picca A., et al. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat. Rev. Cardiol. 2018;15(9):543–554. doi: 10.1038/s41569-018-0059-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Falkenberg M., et al. DNA replication and transcription in mammalian mitochondria. Annu. Rev. Biochem. 2007;76:679–699. doi: 10.1146/annurev.biochem.76.060305.152028. [DOI] [PubMed] [Google Scholar]
- 7.Zhang D., et al. Mitochondrial cardiomyopathy caused by elevated reactive oxygen species and impaired cardiomyocyte proliferation. Circ. Res. 2018;122(1):74–87. doi: 10.1161/CIRCRESAHA.117.311349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang L., et al. Mitochondrial DNA mutation exacerbates female reproductive aging via impairment of the NADH/NAD(+) redox. Aging Cell. 2020;19(9) doi: 10.1111/acel.13206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ishihara N., et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 2009;11(8):958–966. doi: 10.1038/ncb1907. [DOI] [PubMed] [Google Scholar]
- 10.Tani H., et al. Mice deficient in the Shmt2 gene have mitochondrial respiration defects and are embryonic lethal. Sci. Rep. 2018;8(1):425. doi: 10.1038/s41598-017-18828-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kasahara A., et al. Mitochondrial fusion directs cardiomyocyte differentiation via calcineurin and Notch signaling. Science. 2013;342(6159):734–737. doi: 10.1126/science.1241359. [DOI] [PubMed] [Google Scholar]
- 12.Mikryukov A.A., et al. BMP10 signaling promotes the development of endocardial cells from human pluripotent stem cell-derived cardiovascular progenitors. Cell Stem Cell. 2021;28(1):96–111. doi: 10.1016/j.stem.2020.10.003. e7. [DOI] [PubMed] [Google Scholar]
- 13.Chal J., et al. Differentiation of pluripotent stem cells to muscle fiber to model Duchenne muscular dystrophy. Nat. Biotechnol. 2015;33(9):962–969. doi: 10.1038/nbt.3297. [DOI] [PubMed] [Google Scholar]
- 14.Hoang P., et al. Generation of spatial-patterned early-developing cardiac organoids using human pluripotent stem cells. Nat. Protoc. 2018;13(4):723–737. doi: 10.1038/nprot.2018.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thavandiran N., et al. Design and formulation of functional pluripotent stem cell-derived cardiac microtissues. Proc. Natl. Acad. Sci. U. S. A. 2013;110(49):E4698–E4707. doi: 10.1073/pnas.1311120110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lees J.G., et al. Mitochondrial and glycolytic remodeling during nascent neural differentiation of human pluripotent stem cells. Development. 2018;145(20) doi: 10.1242/dev.168997. [DOI] [PubMed] [Google Scholar]
- 17.Yamada S., et al. Chlorpyrifos inhibits neural induction via Mfn1-mediated mitochondrial dysfunction in human induced pluripotent stem cells. Sci. Rep. 2017;7:40925. doi: 10.1038/srep40925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu V., et al. Mitochondrial metabolism and glutamine are essential for mesoderm differentiation of human pluripotent stem cells. Cell Res. 2019;29(7):596–598. doi: 10.1038/s41422-019-0191-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei X., et al. Bach1 regulates self-renewal and impedes mesendodermal differentiation of human embryonic stem cells. Sci. Adv. 2019;5(3) doi: 10.1126/sciadv.aau7887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teijeiro V., et al. DICER1 is essential for self-renewal of human embryonic stem cells. Stem Cell. Rep. 2018;11(3):616–625. doi: 10.1016/j.stemcr.2018.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cong L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mandegar M.A., et al. CRISPR interference efficiently induces specific and reversible gene silencing in human iPSCs. Cell Stem Cell. 2016;18(4):541–553. doi: 10.1016/j.stem.2016.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Loh K.M., et al. Efficient endoderm induction from human pluripotent stem cells by logically directing signals controlling lineage bifurcations. Cell Stem Cell. 2014;14(2):237–252. doi: 10.1016/j.stem.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang J., et al. GATA6-AS1 regulates GATA6 expression to modulate human endoderm differentiation. Stem Cell. Rep. 2020;15(3):694–705. doi: 10.1016/j.stemcr.2020.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tchieu J., et al. A modular platform for differentiation of human PSCs into all major ectodermal lineages. Cell Stem Cell. 2017;21(3):399–410. doi: 10.1016/j.stem.2017.08.015. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ding S., et al. CHD8 safeguards early neuroectoderm differentiation in human ESCs and protects from apoptosis during neurogenesis. Cell Death Dis. 2021;12(11):981. doi: 10.1038/s41419-021-04292-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lian X., et al. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/beta-catenin signaling under fully defined conditions. Nat. Protoc. 2013;8(1):162–175. doi: 10.1038/nprot.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Engler A.J., et al. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126(4):677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 29.Pagliari S., et al. YAP-TEAD1 control of cytoskeleton dynamics and intracellular tension guides human pluripotent stem cell mesoderm specification. Cell Death Differ. 2021;28(4):1193–1207. doi: 10.1038/s41418-020-00643-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kastan N., et al. Small-molecule inhibition of Lats kinases may promote Yap-dependent proliferation in postmitotic mammalian tissues. Nat. Commun. 2021;12(1):3100. doi: 10.1038/s41467-021-23395-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kageyama Y., et al. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J. 2014;33(23):2798–2813. doi: 10.15252/embj.201488658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Larsson N.G., et al. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 1998;18(3):231–236. doi: 10.1038/ng0398-231. [DOI] [PubMed] [Google Scholar]
- 33.Wallace D.C. Mitochondrial diseases in man and mouse. Science. 1999;283(5407):1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- 34.Vernochet C., et al. Adipose-specific deletion of TFAM increases mitochondrial oxidation and protects mice against obesity and insulin resistance. Cell Metabol. 2012;16(6):765–776. doi: 10.1016/j.cmet.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J., et al. Increased in vivo apoptosis in cells lacking mitochondrial DNA gene expression. Proc. Natl. Acad. Sci. U. S. A. 2001;98(7):4038–4043. doi: 10.1073/pnas.061038798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guo J., et al. Frequent truncating mutation of TFAM induces mitochondrial DNA depletion and apoptotic resistance in microsatellite-unstable colorectal cancer. Cancer Res. 2011;71(8):2978–2987. doi: 10.1158/0008-5472.CAN-10-3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen X., et al. KLF16 suppresses human glioma cell proliferation and tumourigenicity by targeting TFAM. Artif. Cell Nanomed. Biotechnol. 2018;46(sup1):608–615. doi: 10.1080/21691401.2018.1431654. [DOI] [PubMed] [Google Scholar]
- 38.Yang M., et al. TFAM is a novel mediator of immunogenic cancer cell death. OncoImmunology. 2018;7(6) doi: 10.1080/2162402X.2018.1431086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dalton S. Linking the cell cycle to cell fate decisions. Trends Cell Biol. 2015;25(10):592–600. doi: 10.1016/j.tcb.2015.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu L., et al. The cell cycle in stem cell proliferation, pluripotency and differentiation. Nat. Cell Biol. 2019;21(9):1060–1067. doi: 10.1038/s41556-019-0384-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pauklin S., Vallier L. The cell-cycle state of stem cells determines cell fate propensity. Cell. 2014;156(6):1338. doi: 10.1016/j.cell.2014.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao Q., et al. Complex regulation of mitochondrial function during cardiac development. J. Am. Heart Assoc. 2019;8(13) doi: 10.1161/JAHA.119.012731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miyaoka Y., et al. Isolation of single-base genome-edited human iPS cells without antibiotic selection. Nat. Methods. 2014;11(3):291–293. doi: 10.1038/nmeth.2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang G., et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med. 2014;20(6):616–623. doi: 10.1038/nm.3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the sequencing data has been deposited at GSA (https://ngdc.cncb.ac.cn/gsub/, HRA001146. Other data sets used and/or analyzed during the current study are available from reasonable request to corresponding authors.