

Abstract
Summary background
Multidrug-resistant (MDR) tuberculosis (TB) poses an important challenge in TB management and control. Rifampicin resistance (RR) is a solid surrogate marker of MDR-TB. We investigated the RR-TB clustering rates, bacterial population dynamics to infer transmission dynamics, and the impact of changes to patient management on these dynamics over 27 years in Rwanda.
Methods
We analysed whole genome sequences of a longitudinal collection of nationwide RR-TB isolates. The collection covered three important periods: before programmatic management of MDR-TB (PMDT; 1991–2005), the early PMDT phase (2006–2013), in which rifampicin drug-susceptibility testing (DST) was offered to retreatment patients only, and the consolidated phase (2014–2018), in which all bacteriologically confirmed TB patients had rifampicin DST done mostly via Xpert MTB/RIF assay. We constructed clusters based on a 5 SNP cut-off and resistance conferring SNPs. We used Bayesian modelling for dating and population size estimations, TransPhylo to estimate the number of secondary cases infected by each patient, and multivariable logistic regression to assess predictors of being infected by the dominant clone.
Results
Of 308 baseline RR-TB isolates considered for transmission analysis, the clustering analysis grouped 259 (84.1%) isolates into 13 clusters. Within these clusters, a single dominant clone was discovered containing 213 isolates (82.2% of clustered and 69.1% of all RR-TB), which we named the “Rwanda Rifampicin-Resistant clone” (R3clone). R3clone isolates belonged to Ugandan sub-lineage 4.6.1.2 and its rifampicin and isoniazid resistance were conferred by the Ser450Leu mutation in rpoB and Ser315Thr in katG genes, respectively. All R3clone isolates had Pro481Thr, a putative compensatory mutation in the rpoC gene that likely restored its fitness. The R3clone was estimated to first arise in 1987 and its population size increased exponentially through the 1990s’, reaching maximum size (∼84%) in early 2000 s’, with a declining trend since 2014. Indeed, the highest proportion of R3clone (129/157; 82·2%, 95%CI: 75·3–87·8%) occurred between 2000 and 13, declining to 64·4% (95%CI: 55·1-73·0%) from 2014 onward. We showed that patients with R3clone detected after an unsuccessful category 2 treatment were more likely to generate secondary cases than patients with R3clone detected after an unsuccessful category 1 treatment regimen.
Conclusions
RR-TB in Rwanda is largely transmitted. Xpert MTB/RIF assay as first diagnostic test avoids unnecessary rounds of rifampicin-based TB treatment, thus preventing ongoing transmission of the dominant R3clone. As PMDT was intensified and all TB patients accessed rifampicin-resistance testing, the nationwide R3clone burden declined. To our knowledge, our findings provide the first evidence supporting the impact of universal DST on the transmission of RR-TB.
Keywords: Tuberculosis in Rwanda, Rifampicin-resistant tuberculosis control, Universal drug-susceptibility testing, Mycobacterium tubercuslosis transmission
1. Introduction
Multidrug-resistant tuberculosis (MDR-TB; i.e. TB resistant to at least rifampicin and isoniazid) is among the top global challenges in TB control and management [1]. Rifampicin resistance (RR) is a solid surrogate marker of MDR-TB [2]. The direct transmission of resistant strains, also known as primary resistance, is postulated to be the main driver of the MDR-TB epidemic [3], [4]. The majority of RR-TB patients remain undiagnosed and untreated, leaving a large number spreading resistant Mycobacterium tuberculosis complex (MTBC) strains [5]. Moreover, despite recent advances in treatment, the global average of RR-TB treatment success has stagnated around 57% [6].
Besides direct transmission, acquisition of RR occurs through selection of mutant strains during anti-TB treatment, usually due to inappropriate prescription, stock-outs, inadequate adherence or penetration of drugs [4], [7]. The overall higher prevalence of resistance among previously treated TB patients often results from missed primary resistance in combination with acquisition of resistance during the previous treatment episode [8].
To better direct setting-specific programmatic control efforts, understanding the key determinants of RR-TB trends is important given the differences in strategic responses. While acquired resistance can be controlled by supporting adherence to appropriate and optimised therapy [9], the control of primary resistance requires suitable interventions, such as early diagnosis combined with prompt effective therapy [3], [10], [11], and an appropriate contact tracing strategy. However, RR-TB transmission studies and strain population dynamics in endemic countries are scarce, including in the African Great Lakes Region [12], [13].
In Rwanda, the first cases of RR-TB were identified in 1989 [14]. A baseline drug-resistance survey conducted in the early 1990′s showed a low rate of RR-TB: only 1·3% in new and 6·5% among previously treated TB patients [15]. Massive disruptions to the health system and a lack of RR-TB treatment during the 1990′s [14] resulted in increased RR-TB cases. A second drug-resistance survey conducted in 2005 showed a statistically significant increase in RR-TB prevalence: 3·9% in newly diagnosed and 9·4% in previously treated TB patients [16]. Consequently, the programmatic management of RR-TB (PMDT) was initiated in 2005 and integrated as a core component in the national TB control program [14]. The PMDT introduced the standardized long multidrug-resistant (MDR-)TB regimen and the countrywide surveillance of drug-resistant TB. Diagnostic improvements led to shortened delays in initiating MDR-TB treatment and significantly decreased its mortality in Rwanda [17]. Moreover, the prevalence of RR-TB among new TB patients decreased to 1·4% in 2015 [18], likely due to shortened diagnostic and therapeutic delays [17], while no change was observed in previously treated TB patients (10·7%) [18]. However, the contribution of primary versus acquired resistance to these fluctuations in RR-TB prevalence is unknown, making it difficult to evaluate how to further reduce the number of RR-TB cases.
Whole genome sequencing (WGS) has been adopted as the molecular gold standard for outbreak and transmission analysis [19], [20], [21] primarily using SNP-based transmission clustering of closely related MTBC strains, allowing for the level of transmission to be measured by assessing the clustering rate. Through a unique longitudinal collection of nationwide RR-TB isolates spanning a quarter century (1991–2018), we used WGS-based molecular epidemiology to estimate the actual contribution of primary resistance in the RR-TB epidemic in Rwanda.
2. Methods
2.1. Study design and population
This longitudinal genome analysis included baseline RR-TB isolates from nationwide patients, collected for diagnostic purposes. Before inception of the PMDT (1991–2005), when drug resistance testing was not yet possible in Rwanda, sputum specimens were sent to the Institute of Tropical Medicine (ITM), Antwerp, Belgium. Afterwards, isolates were sent to ITM for distinct drug-resistance surveys (ie, 1993, 2005, and 2015) [15], [16], [22], and for quality control. During the early phase of the PMDT, 2006–13, the diagnosis of RR-TB mainly relied on phenotypic drug-susceptibility testing (pDST) on Löwenstein-Jensen medium (LJ), and the GenoType MTBDRplus line probe assay (Hain Lifescience, Nehren, Germany) performed in Rwanda, while molecular RR-TB diagnostic assays, mainly Xpert MTB/RIF, were further expanded in the 2014–18 period [17] (Fig. 1).
Fig. 1.

Study population and clustering results.
The sampling fraction and composition for our study depended on available isolates and varied per period (Table 1).
Table 1.
Sampling fraction versus transmission cluster (5 SNPs cut-off, same MDR-resistance SNPs).
| Year | WHO RR-TB notification, corrected* | Included in analysis (%) | unclustered isolates n (%) | Isolates in cluster n (%) | Isolates in the R3clone n (%) |
|---|---|---|---|---|---|
| 1991–2005 | – | 96 | 21 (21·9) | 75 (78·1) | 59 (61·5) |
| 2006–2013 | 479 | 93 (19·4) | 5 (5·4) | 88 (94·6) | 78 (83·9) |
| 2014 | 73 | 12 (16·4) | 4 (33·3) | 8 (66·7) | 6 (50·0) |
| 2015 | 88 | 31 (35·2) | 6 (19·3) | 25 (80·7) | 19 (61·3) |
| 2016 | 69 | 26 (37·7) | 4 (15·4) | 22 (84·6) | 22 (84·6) |
| 2017/2018# | 58 | 50 (86·2%) | 9 (18·0) | 41 (82·0) | 29 (59·2) |
| Total¥ | 767 | 212 (27.6) | 28 (13.2) | 184 (86.8) | 154 (72.6) |
*Based on a recent finding, only 14·3% of RR on Xpert with a very low bacterial load was confirmed as rifampicin-resistant (RR); #including Jan-March (2018/03) with only 9 confirmed RR patients being registered in this period; ¥Exclude 96 isolates collected between 1991 and 2005 as no notification data for that period. MDR = multidrug resistant; SNPs = single nucleotide polymorphisms.
Before 2013, testing for RR-TB was merely undertaken for previously treated patients, thus the majority of isolates during this period were from patients with treatment failure or relapse, sometimes after multiple first-line treatment episodes. Our previous analysis showed that from 2013 onwards, after revising the drug-resistance diagnostic approach particularly the universal DST, most RR-TB was diagnosed among new TB patients, although previously treated patients were still being prioritized for RR-TB testing [17].
2.2. Retrieval of isolates for WGS and data collection
All available isolates registered as RR-TB patients were retrieved from −80 °C freezers and regrown on multiple LJ slants appropriate for genomic DNA extraction. Patients’ clinical and demographic data were collected from the national RR-TB databases and/or patients’ files and were linked to their specific isolates based on patients’ and samples’ unique identifiers assigned during diagnosis.
2.3. Whole genome sequencing, assembly and resistance prediction
Regrown cultures were scraped and heat-killed in 150 µL of 0·5 M Tris-EDTA buffer (pH 8.5) followed by genomic DNA extraction using an in-house optimised protocol [23]. WGS was outsourced to FISABIO (Valencia, Spain) and KU Leuven (Leuven, Belgium) sequencing facilities. Genomic libraries were built using the Nextera XT kit and sequenced on an Illumina MiSeq or NextSeq platform with paired end, 150-bp read lengths (California, USA).
For sequence analysis, non-MTBC reads were filtered out from Fastq datasets using Centrifuge v1.0.3 [24]. Reads mapping to any variant in the MTBC were retained. Samples with<95% reads mapped to the MTBC were excluded from further analysis. Reads were next mapped to the inferred ancestral M. tuberculosis genome [25] with MTBseq [24] using default values (filter for variants was set at 5%). TB-Profiler v2.6 [26] was used to define WGS-based resistance profiles with the tbdb mutation database [26] accessed on 13/07/2020. All isolates without a known RR-conferring mutation, and sequence results showing polyclonal TB infection (i.e. multiple strains in one sample) or heteroresistance (i.e. mixed population of drug-resistant mutants and wildtype, when the mutants population is still < 95%) were excluded from analysis.
2.4. Phylogeny and transmission analysis
A SNP alignment was created using MTBseq after which constant site counts for the remainder of the genome was created using a custom Python script. This data was then input to RAxML-NG v0.9.0 [24] to create a Maximum Likelihood phylogeny with a GTR + G + asc substitution model [26], site-repeat optimisation [26], 10 starting trees and 200 bootstrap replicates.
For transmission analysis, custom Python scripts were used to construct clusters from the SNP alignment based on a 5 SNPs cut-off [20], [27] using a loose cluster definition (ie, isolates in a cluster differed at most 5 SNPs from at least 1 other isolate in the cluster) [27], [28]. MDR-TB transmission clusters were further confirmed by assessing whether the SNPs conferring resistance to rifampicin and isoniazid were the same for all members of a cluster. Moreover, a measurement based on a 12 SNPs cut-off was used to assess the steadiness of the 5 SNPs predefined clusters. All custom python scripts are available at https://github.com/conmeehan/pathophy.
2.5. Estimation of epidemiological parameters using Bayesian phylogenetics
Dating and population size estimations (i.e. an independent measurement of the bacterial population burden which is less affected by sampling fraction differences) were undertaken using BEAST v1.10.4 [29]. The SNP alignment was imported into BEAUti and tips were dated based on the date of isolation. Where only the year of isolation was known, the date was set to January 1st and the uncertainty was set to 1 to allow the Markov chain Monte Carlo (MCMC) process to better incorporate this uncertainty in the final dating estimates. A GTR + G site model was used in conjunction with an uncorrelated relaxed clock [20] with lognormal distribution and a Skyride [20] with time-aware smoothing tree prior. The ucld.mean was set to lognormal with a mean in real space and a starting value of 0·0005 as has been shown to be appropriate for Lineage 4 isolates [27]. The XML document was then manually edited to add the constant site count for each base as an ascertainment bias correction.
BEAST was run in four separate times with an MCMC chain length of 100 M sampling every 10,000 steps. Trees were combined between runs using LogCombiner with a 50% burn-in per run. TreeAnnotator was used to create a maximum clade credibility tree with mean node heights. The Skyride plot was created using the Tracer v1.7.1 [20] demographic reconstruction analysis with default options with the age set to 2018.
2.6. Estimation of secondary cases
To estimate the secondary cases generated from a single isolate, we used a subset of 93 strains from the dominant clone which had detailed data on previous TB treatment history. We used a Bayesian approach to model the influence of previous treatment history on transmission dynamics, specifically the number of secondary cases infected by a primary case. The TreeAnnotator maximum clade credibility tree was input to the R package TransPhylo v1.4.4 [30] to estimate secondary case contact rates for each isolate. All zero length branches were set to 1*10-11 and the last sampled date set to 2018 based on the sampling dates of the isolates. The following parameters were then used for the TransPhylo run, based on those used by Didelot et al. on their MTBC outbreak dataset [30]: w.shape = 1.3; w.scale = 1/0.3; ws/shape = 1.1; ws.scale = 1/0.4; mcmcIterations = 20 M; thinning = 2000; Convergence was confirmed using coda v0.19.4 [31]. After a burn-in of 50%, an approximate per-individual average number of transmissions (offspring) was calculated from the remaining 50% of MCMC samples. Isolates were then grouped by patient’s previous TB treatment history such as new TB (New), failure or relapse to anti-TB category 1 (Cat1), and failure or relapse to anti-TB category 2 (Cat2, i.e. first-line re-treatment regimen for previously treated TB patients). To test for significance in secondary case numbers between previous history on TB treatment groups (New, Cat 1, Cat 2), the corresponding per-individual average number of transmissions were compared using a three-way ANOVA with a Tukey’s Honest Significant Differences post-hoc test to determine which groups differed in secondary case contact rates. The Cohen’s d effect size was then calculated pairwise between the groups (New, Cat1 and Cat2) using the effsize package [32]. Boxplots of the secondary case counts were created using ggplot2 [33].
2.7. Metadata statistical analysis
The Pearson’s chi-square and Fisher's exact tests were used to test for associations between the predominant clone (R3clone, see Results section) and potential predictors. Multivariable logistic regression was used to assess predictors of being infected by the R3clone. STATA version 14·2 (College Station, TX: STATA Corp) was used for metadata analysis.
2.8. Ethics
The study protocol was approved by the Rwanda National Ethical committee, Kigali, Rwanda (IRB 00001497 of IORG0001100; Ref No·0069/RNEC/2017), the Institutional Review Board of the Institute of Tropical Medicine, Antwerp, Belgium (IRB/AB/AC/062; Ref No.1208/17; 19/03/2018), and the Ethics Committee of the Antwerp University Hospital, Universitair Ziekenhuis Antwerpen Ethische Commissie, Antwerp, Belgium (REG No.B300201836458; 14/05/2018).
3. Results
3.1. MTBC isolates and patient characteristics
Overall, 437 individual patients’ MTBC isolates registered as RR-TB were retrieved and sequenced. Of the 437 isolates, 56 were excluded from analysis, either due to poor sequence quality (n = 24) or sequence results demonstrating mixed populations (polyclonal TB infection or heteroresistance) (n = 32) (Fig. 1). In addition, 73 were excluded as no known RR-conferring mutation was identified by WGS. Most of these isolates had a wildtype rpoB gene sequence, and were initially diagnosed via Xpert MTB/RIF assay, thus likely explained by false RR associated with low bacterial load samples tested on Xpert MTB/RIF [34]. Of 308 isolates considered for the transmission analysis, 96 (31·2%) were isolated between 1991 and 2005 (before starting the PMDT), 93 (30·2%) were isolated between 2006 and 2013 (in the early PMDT phase, prior to expanding utilization of rapid molecular RR-TB diagnostic assays), and 119 (38·6%) were isolated between 2014 and 2018 (after expanding MDR/RR-TB molecular diagnostic assays) [17] (Fig. 1).
The sex was documented for 251 of 308 patients. Most were male (151, 60·2%). The median age was 34 years (interquartile range (IQR): 27–44 years). HIV-coinfection status was documented for 164 (53·3%), of whom 77 (47·0%) were HIV-coinfected. Of 181 with data on TB treatment history, 101 (55·8%) were previously treated TB patients, while 80 (44·2%) were new TB patients. All new patients were diagnosed since 2013, after implementing universal DST, the majority (59; 73.8%) using the Xpert MTB/RIF assay.
SNP-based lineage assignment [35] classified strains as Ugandan sub-lineage 4.6.1.2 (240, 79·5%), Ugandan sub-lineage 4.6.1.1 (18, 6·0%), and lineages 4.3 (22, 7·2%), 4.7 (20, 6·5%), 4.4 (1, 0·3%), and 4.8 (1, 0·3%), while four (1·3%) were lineage 3.1.1 (Delhi-CAS), one (0·3%) was lineage 2.2.1 (Beijing strain), and for the first time a single L8 strain was documented from this dataset [36] (S1 Table).
Of the 308 RR isolates, 292 (94·8%) had concomitant resistance to isoniazid, thus were MDR-TB. The majority (274, 89·0%) of RR was due to the Ser450Leu rpoB mutation. Three isolates (all diagnosed before 2005) harboured the rpoB gene Val170Phe mutation, a RR-conferring mutation outside the rifampicin-resistance determining region (RRDR).
The majority (259, 88·7%) of resistance to isoniazid was due to the Ser315Thr katG mutation, while only two isolates had Ser94Ala inhA, a rare isoniazid resistance conferring mutation (Supplementary Table 1). No double mutants (katG and inhA), conferring the highest levels of isoniazid resistance, were seen. Two isolates had fluoroquinolone resistance-conferring mutations in the gyrA gene (Asp94Gly and Asp94Ala), but neither showed genotypic or phenotypic resistance to second-line injectables. There was no mutation known to be associated with novel or repurposed second-line MDR-TB drugs, such as bedaquiline, linezolid, delamanid and clofazimine [37].
3.2. Transmission clusters
Using a 5 SNP cut-off and loose cluster definition, also considering rifampicin and isoniazid resistance-conferring SNPs, grouped 259 (84·1%) isolates into 13 clusters (Table 1; Fig. 2).
Fig. 2.
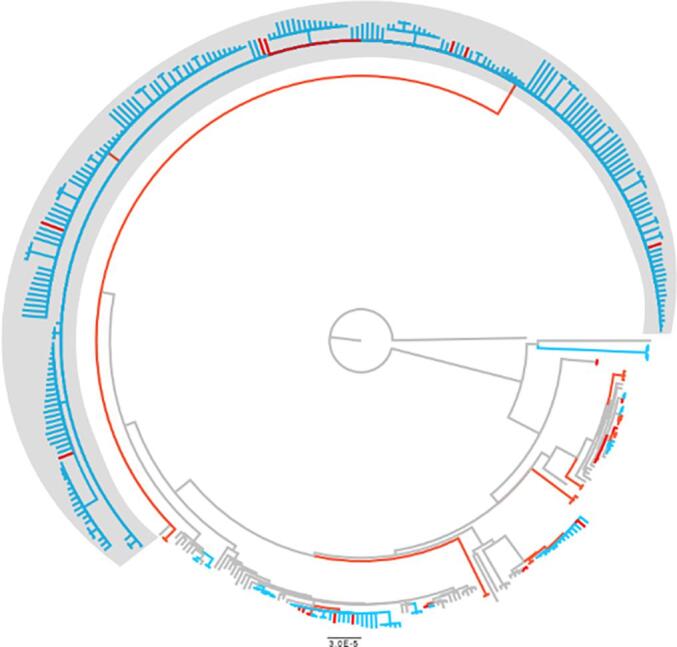
Phylogenetic tree of RR-TB strains in Rwanda. The red leaves show clusters by 12SNP and blue by 5 SNP cut-off. The R3clone is shown in grey box. All branches with bootstrap support below 70% were collapsed into polytomies.
Among 49 unclustered isolates (i.e., unclustered), the majority (37; 75·5%) were from previously treated TB patients, while 12 (24.5%) were from new TB patients. Of the 259 isolates in clusters, 213 (82·2%) belonged to a single dominant clone within Ugandan sub-lineage 4.6.1.2 that we named the “Rwanda rifampicin-resistant clone (R3clone)” (Fig. 2). The remaining 12 clusters had between 2 and 11 isolates each. By increasing the clustering cut-off to 12 SNPs, the R3clone cluster increased by only 17 isolates (S2 Table). Subsequent analyses are based on the 5 SNP cut-off defined clusters.
In the R3clone, resistance to rifampicin and isoniazid was conferred by Ser450Leu rpoB and Ser315Thr katG respectively. Moreover, all isolates of the R3clone, as early as 1991, harboured a putative compensatory mutation Pro481Thr in the rpoC gene. The R3clone is resistant to ethambutol (EmbB Met 306Val or His1002Arg) and all except five were resistant to pyrazinamide (pncA Met175Ile, His43Pro, Gln141pro or Gln10Arg). Thus, most R3clone isolates were resistant to all first-line TB drugs. The mutation at position 80 in gyrA (Thr80Ala) was present in all R3clone isolates.
3.3. R3clone population dynamics
Of the 96 RR-TB isolates from the pre-PMDT period, 59 (61·5%, 95%CI: 51.0–71.2) belonged to the R3clone, increasing to 78 of 93 isolates (83·9%, 95%CI: 74.8–90.7) during the early PMDT phase, followed by a decline to 76/119 (63·9%, 95%CI: 54.6–72.5) in the consolidated PMDT period.
In a multivariable analysis, the odds of being affected by R3clone was significantly higher in the early PMDT phase (adjusted odds ratio [aOR] 2.5, 95%CI 1.1–5.6). Female patients had a significantly higher odds of being affected by the R3clone (aOR 2·6, 95%CI 1·2-5·9), while the odds of having R3clone TB was not significantly different between age groups, Kigali city versus other provinces in Rwanda or HIV-positive versus HIV-negative(Table 2).
Table 2.
Factors associated with R3clone (5 SNP cut-off, same MDR-resistance SNP).
| Total | R3 clone | Univariate analyses | Multivariable analyses | ||
|---|---|---|---|---|---|
| n | n (%) | OR (95% CI) | aOR (95% CI) | ||
| Total | n | 308 | 213 (69·2) | ||
| Sex n = 251 | Male | 151 | 90 (59·6) | reference | reference |
| Female | 100 | 78 (78·0) | 2·4 (1·4-4·2) | 2·6(1.2-5·9) | |
| Unknown | 57 | 57 (18·5) | |||
| Age[years] | <30 | 62 | 46 (74·2) | 2·3 (0·8-6·8) | 1·6 (0·5-5·4) |
| [30], [31], [32], [33], [34], [35], [36], [37], [38], [39], [40], [41], [42], [43], [44] | 75 | 54 (72·0) | 2·1 (0·7-5·9) | 1·6 (0·5-5·0) | |
| [45–54] | 25 | 18 (72·0) | 2·1 (0·6-7·4) | 1·5 (0·4-6·0) | |
| >54 | 19 | 10 (52·6) | reference | reference | |
| Unknown | 127 | 85 (66·9) | – | ||
| TB treatment history* | New | 80 | 46 (57·5) | reference | |
| Previously treated | 101 | 74 (73·3) | 1·5 (0·8-3·0) | ||
| Unknown | 127 | 93 (73·2) | |||
| HIV | Negative | 86 | 65 (76·5) | reference | |
| Positive | 78 | 50 (64·9) | 0·6(0·3-1·2) | ||
| Unknown | 144 | 98 (68·1) | |||
| Resident in Kigali city | Yes | 83 | 66 (79·5) | 2·1 (1·1-4·2) | 1·8 (0·9-3·6) |
| No | 93 | 60 (64·5) | reference | reference | |
| Unknown | 132 | 87 (65·9) | |||
| Period of RR diagnosis | Period1 (1991–2005) | 96 | 59 (61·5) | reference | reference |
| Period 2 (2006–2013) | 93 | 78 (83·9) | 3·3 (1·6-6·5) | 2·5 (1·1-5·6) | |
| Period 3 (2014–2018) | 119 | 76 (63·9) | 1·1 (0·6-1·9) | 1 |
RR = rifampicin-resistant; MDR = multidrug resistant; SNP = single nucleotide polymorphism, *TB treatment history had collinearity with the period of rifampicin-resistant tuberculosis diagnosis, as before 2013, testing for RR-TB was merely undertaken for previously treated patients.
The Bayesian phylogeny of the R3clone estimated that the strain first arose in 1987 (95% HDP: 1981–1991) (Fig. 3), with a steady exponential increase in population size through the 1990 s’, turning into a constant population size in the early 2000 s’, followed by a declining trend starting in 2014 (Fig. 3).
Fig. 3.

Bayesian phylogenetic tree of R3clone and associated Skyride plot. Date of emergence of the clone is indicated at the root node. The mean estimate of population size changes is indicated by the solid black line with the High Posterior Density Interval of this estimate in the shaded blue area. The bush-like topology suggests continuous spread as opposed to consecutive single transmission events.
The estimation of secondary case contact rates grouped by previous TB treatment history, revealed that isolates from unsuccessful treatment on Cat2 treatment were more likely to have linked offspring (i.e. more transmissions) than those isolated from unsuccessful treatment Cat1 or new patients (p-value < 0·0001; Cohen’s d effect size: large; Fig. 4). There was no significant difference observed between isolates from new versus Cat1 patients and Cohen’s d effect size was negligible.
Fig. 4.
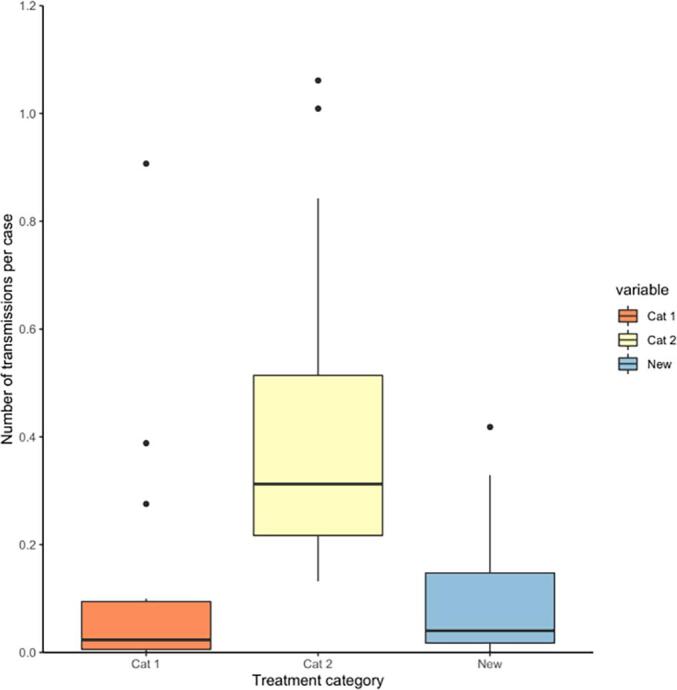
Number of secondary transmissions per case for 93 selected R3clone isolates grouped by tuberculosis (TB) treatment history. Cat 1: patients diagnosed with rifampicin-resistant TB after failure or relapse to category 1 anti-TB treatment; Cat 2: patients diagnosed with rifampicin-resistant TB after failure or relapse to category 2 anti-TB treatment.
4. Discussion
Our data showed that RR-TB disease in Rwanda is largely driven by a single dominant “R3clone”, estimated to have arisen in 1987, only four years after the introduction of rifampicin into the African Great Lakes region [36]. The estimated bacterial population size of the R3clone increased until ‘universal DST’ became implemented. To the best of our knowledge, this is the first molecular evidence to demonstrate a clear association between RR-TB transmission and delays in starting appropriate treatment. We showed that patients in Cat 2 with the R3clone were more likely to generate secondary cases. Such patients likely with primary RR-TB, received multiple rounds of ineffective first-line anti-TB treatment before being diagnosed correctly, while they continued to spread RR-TB strains. The specific genetic features of the R3clone, such as full resistance to first-line drugs, the combination of the Ser450Leu rpoB gene mutation, which has the smallest associated fitness cost of all RR-conferring rpoB mutants [38], - and the Pro481Thr rpoC putative fitness compensating mutation [39], [40], present since the emergence of the clone, enabled the ongoing propagation of the R3clone over more than a quarter century, especially during the early years when rifampicin DST was only offered to retreatment patients. This finding corroborates the recent global genomic analysis that showed decades old resistance likely associated with ongoing transmission by fit clones in most countries [41].
We showed that the nationwide R3clone bacterial population increased in the 1990 s’ through the 2000 s’, explaining much of the increased prevalence of RR-TB among new TB patients observed in the 2005 drug-resistance survey [15], [16]. This was driven by the weak health system during the genocide period [14]. The programmatic efforts that resulted in a drastically shortened diagnostic and treatment delay [17] interrupted the active propagation of this R3clone pathogen population growth. Although it seems logical that RR-TB transmission could be interrupted by swift diagnosis and appropriate treatment, so far there was no such public health evidence to support this aspect of universal DST. Similar to a recent analysis that showed association of health-care delays with an increased rate of secondary TB among dependents [42], Cat 2 patients had several years likely with primary RR-TB before being diagnosed and switched to appropriate MDR-TB treatment, while expected that many died before being diagnosed. This finding underscores that rapid universal DST may interrupt RR-TB transmission, as observed in Rwanda where new patients became eligible for rapid molecular rifampicin DST since 2013 [14].
Our findings confirm what a previous modelling study posited: transmission accounts for a median of 96% of all incident MDR-TB [3], including 61% in previously-treated patients [3]. It is also consistent with the findings in high MDR-TB endemic settings, such as the former Soviet Union, where the most frequent and fit clones harbour compensatory mutations in the rpoA and/or rpoC genes [43], [44]. Novel approaches that identify the main RR-TB transmission hotspots, followed by well-designed targeted active case finding, can further help to decrease RR-TB in Rwanda and be an example of such programmes for other TB endemic countries.
The predominance of the R3clone population as the driver of the RR-TB epidemic might not be unique for Rwanda, and rather reflect a regional problem. However, limited WGS data from the Great Lakes region is available, and no R3clone was identified among 32 RR-TB genomes from the Ugandan isolates collected during a drug-resistance survey conducted between 2008 and 2011 [13].
Consistent with the routine pDST-based drug-resistance surveillance [45], as well as the 2015 drug-resistance survey [17], only two isolates among successfully sequenced RR-TB isolates had a mutation conferring resistance to fluoroquinolones. Overall, the very low rate of fluoroquinolone resistance could partly explain the high treatment success in Rwanda observed with the two WHO-endorsed long and short MDR-TB regimens [17], [46]. While a combination of Thr80Ala and Ala90Gly mutations in the gyrA gene was shown to induce hyper-susceptibility to fluoroquinolones [47], the R3clone isolates only had theThr80Ala mutation, typical for the lineage 4.6.2, which has been associated with a slightly increased minimal inhibitory concentration compared to wildtype gyrA strains [47].
Our study has important strengths. This analysis included strains isolated over 27 years, including the first RR-TB diagnosed in Rwanda, thus representing the reality of the RR-TB population dynamics in Rwanda. Moreover, this is the first study analysing RR-TB transmission using WGS within the Great Lakes region. The finding of transmission driving the RR-TB epidemic in Rwanda is important to neighbouring countries to conduct similar molecular epidemiological analyses to guide regional RR-TB control efforts.
This analysis also had limitations. Some selection bias may have occurred, as the RR-TB detection rate increased over time as better diagnostic tools became available and accessible. Moreover, the sampling fraction depended on availability of isolates (e.g. in 2016, the proportion of R3clone represented almost 85%, as the analysis included only 26 out of 69 RR-TB notified). <50% of all RR-TB cases notified to the WHO were included for 1991–2013, while 86% of isolates were included for 2014–2018. However, the Bayesian analysis used to estimate the size of the entire circulating bacterial population is robust to low sampling fractions due to the use of a coalescent process. To assess occurrence of secondary cases, we used only 93 (43.7%) R3clone strains which had detailed patient information. A larger dataset would allow for differentiation between new and Cat 1 patients. The samples were collected from different timepoints with improved conditions of life in later years when most new cases were diagnosed. Moreover, the time between sampling and occurrence of potential secondary cases might be shorter for recent cases. Finally, this analysis did not include rifampicin-susceptible TB isolates. While the large contribution of primary resistance can be inferred based on the 5-SNPs clustering and the fact of having R3clone in new TB patients, the exact contribution of acquired resistance cannot be fully determined. Data on socio-economic factors were not collected, while living conditions may have changed in the study period.
In conclusion, transmission mostly driven by R3clone has been the determinant of the RR-TB epidemic in Rwanda. Improved RR-TB patient management, resulting in shortened diagnostic and treatment delays, resulted in a reduction of RR-TB transmission. For further enhanced control, in addition to rapid identification of RR-TB patients followed by effective treatment, well-designed and targeted interventions will be important.
CRediT authorship contribution statement
Jean Claude S. Ngabonziza: Conceptualization, Methodology, Formal analysis, Investigation, Resources, Data curation, Writing – original draft, Writing – review & editing, Visualization, Visualization, Funding acquisition. Leen Rigouts: Conceptualization, Methodology, Investigation, Data curation, Writing – review & editing, Supervision, Project administration, Funding acquisition. Gabriela Torrea: Conceptualization, Investigation, Writing – review & editing. Tom Decroo: Methodology, Investigation, Writing – review & editing. Eliane Kamanzi: Investigation, Resources, Data curation. Pauline Lempens: Investigation. Aniceth Rucogoza: Investigation, Resources, Data curation. Yves M. Habimana: Investigation, Resources, Data curation. Lies Laenen: Investigation, Data curation. Belamo E. Niyigena: Investigation, Resources, Data curation. Cécile Uwizeye: Investigation, Data curation. Bertin Ushizimpumu: Investigation, Resources, Data curation. Wim Mulders: Formal analysis, Investigation, Data curation. Emil Ivan: Investigation, Resources, Data curation, Writing – review & editing. Oren Tzfadia: Investigation. Claude Mambo Muvunyi: Investigation, Data curation. Patrick Migambi: Investigation, Resources, Data curation. Emmanuel Andre: Investigation, Data curation. Jean Baptiste Mazarati: Investigation, Resources. Dissou Affolabi: Investigation, Funding acquisition. Alaine N. Umubyeyi: Investigation, Resources, Data curation. Sabin Nsanzimana: Investigation, Resources, Data curation. Françoise Portaels: Investigation, Data curation. Michel Gasana: Investigation, Resources, Data curation. Bouke C. de Jong: Conceptualization, Methodology, Formal analysis, Investigation, Data curation, Writing – review & editing, Visualization, Supervision, Project administration, Funding acquisition. Conor J. Meehan: Conceptualization, Methodology, Formal analysis, Investigation, Data curation, Writing – original draft, Writing – review & editing, Supervision.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Acknowledgements
This work was supported by EDCTP2 grant DRIA2014-326—DIAMA of the European Union, the Belgian General Directorate for Development Cooperation (PhD fellowship to JCSN), ERC grant [INTERRUPTB; no. 311725], and InnoR3TB study with financial support from ITM’s SOFI programme supported by the Flemish Government, Science & Innovation. The work is also supported by the Academy of Medical Sciences (AMS), the Wellcome Trust, the Government Department of Business, Energy and Industrial Strategy (BEIS), the British Heart Foundation and Diabetes UK and the Global Challenges Research Fund (GCRF) via a Springboard grant [SBF006\1090] awarded to CJM. The views and opinions of authors expressed herein do not necessarily state or reflect those of EDCTP. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Data sharing
Raw read data for all WGS samples can be retrieved from the ENA using the accession code PRJEB43270.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jctube.2022.100299.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Singh R., Dwivedi S.P., Gaharwar U.S., Meena R., Rajamani P., Prasad T. Recent updates on drug resistance in Mycobacterium tuberculosis. J Appl Microbiol. 2020;128(6):1547–1567. doi: 10.1111/jam.14478. [DOI] [PubMed] [Google Scholar]
- 2.Manson A.L., Cohen K.A., Abeel T., Desjardins C.A., Armstrong D.T., Barry C.E., et al. Genomic analysis of globally diverse Mycobacterium tuberculosis strains provides insights into the emergence and spread of multidrug resistance. Nat Genet. 2017;49(3):395–402. doi: 10.1038/ng.3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kendall E.A., Fofana M.O., Dowdy D.D.W., WHO, Dye C., Garnett G., et al. Burden of transmitted multidrug resistance in epidemics of tuberculosis: A transmission modelling analysis. Lancet. Respir Med. 2015;3:963–972. doi: 10.1016/S2213-2600(15)00458-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dheda K., Gumbo T., Maartens G., Dooley K.E., McNerney R., Murray M., et al. The epidemiology, pathogenesis, transmission, diagnosis, and management of multidrug-resistant, extensively drug-resistant, and incurable tuberculosis. Lancet Respir Med. 2017;5(4):291–360. doi: 10.1016/S2213-2600(17)30079-6. [DOI] [PubMed] [Google Scholar]
- 5.Dean A., Cox H., Zignol M. Epidemiology of Drug-Resistant Tuberculosis. Adv Exp Med Biol. 2017;1019:209–220. doi: 10.1007/978-3-319-64371-7_11. [DOI] [PubMed] [Google Scholar]
- 6.WHO | Global tuberculosis report 2020. WHO. 2020 [cited 26 Oct 2020]. Available: http://www.who.int/tb/publications/global_report/en/.
- 7.Prideaux B., Via L.E., Zimmerman M.D., Eum S., Sarathy J., O'Brien P., et al. The association between sterilizing activity and drug distribution into tuberculosis lesions. Nat Med. 2015;21(10):1223–1227. doi: 10.1038/nm.3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Deun A., Decroo T., Kya Jai Maug A., Hossain M.A., Gumusboga M., Mulders W., et al. The perceived impact of isoniazid resistance on outcome of first-line rifampicinthroughout regimens is largely due to missed rifampicin resistance. PLoS ONE. 2020;15(5):e0233500. doi: 10.1371/journal.pone.0233500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dye C., Williams B.G., Espinal M.A., Raviglione M.C. Erasing the world’s slow stain: Strategies to beat multidrug-resistant tuberculosis. Science. 2002;295(5562):2042–2046. doi: 10.1126/science.1063814. [DOI] [PubMed] [Google Scholar]
- 10.Dharmadhikari A.S., Mphahlele M., Venter K., Stoltz A., Mathebula R., Masotla T., et al. Rapid impact of effective treatment on transmission of multidrug-resistant tuberculosis. Int J Tuberc Lung Dis. 2014;18(9):1019–1025. doi: 10.5588/ijtld.13.0834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fox G.J., Schaaf H.S., Mandalakas A., Chiappini E., Zumla A., Marais B.J. Preventing the spread of multidrug-resistant tuberculosis and protecting contacts of infectious cases. Clin Microbiol Infect. 2017;23(3):147–153. doi: 10.1016/j.cmi.2016.08.024. [DOI] [PubMed] [Google Scholar]
- 12.Chisompola N.K., Streicher E.M., Muchemwa C.M.K., Warren R.M., Sampson S.L. Molecular epidemiology of drug resistant Mycobacterium tuberculosis in Africa: A systematic review. BMC Infect Dis. 2020;20(1) doi: 10.1186/s12879-020-05031-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ssengooba W., Meehan C.J., Lukoye D., Kasule G.W., Musisi K., Joloba M.L., et al. Whole genome sequencing to complement tuberculosis drug resistance surveys in Uganda. Infect Genet Evol. 2016;40:8–16. doi: 10.1016/j.meegid.2016.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klinkenberg E. Epidemiological review and impact analysis of tuberculosis in Rwanda. 2014. Available: http://www.rbc.gov.rw/fileadmin/user_upload/rbc/surveillance_system_tb_epidemiological_impact_assessment_rwanda_2014.pdf.
- 15.Carpels G., Fissette K., Limbana V., Van Deun A., Vandenbulcke W., Portaels F. Drug resistant tuberculosis in sub-Saharan Africa: an estimation of incidence and cost for the year 2000. Tuber Lung Dis. 1995;76(6):480–486. doi: 10.1016/0962-8479(95)90522-7. [DOI] [PubMed] [Google Scholar]
- 16.Umubyeyi A.N., Vandebriel G., Gasana M., Basinga P., Zawadi J.P., Gatabazi J., et al. Results of a national survey on drug resistance among pulmonary tuberculosis patients in Rwanda. Int J Tuberc Lung Dis. 2007;11:189–194. http://www.ncbi.nlm.nih.gov/pubmed/17263290 Available. [PubMed] [Google Scholar]
- 17.Ngabonziza J.-C.- S., Habimana Y.M., Decroo T., Migambi P., Dushime A., Mazarati J.B., et al. Reduction of diagnostic and treatment delays reduces rifampicin-resistant tuberculosis mortality in Rwanda. Int J Tuberc Lung Dis. 2020;24(3):329–339. doi: 10.5588/ijtld.19.0298. [DOI] [PubMed] [Google Scholar]
- 18.Global Tuberculosis Report. 2016 [Google Scholar]
- 19.Meehan C.J., Goig G.A., Kohl T.A., Verboven L., Dippenaar A., Ezewudo M., et al. Whole genome sequencing of Mycobacterium tuberculosis: current standards and open issues. Nat Rev Microbiol. 2019;17(9):533–545. doi: 10.1038/s41579-019-0214-5. [DOI] [PubMed] [Google Scholar]
- 20.Walker T.M., Ip C.LC., Harrell R.H., Evans J.T., Kapatai G., Dedicoat M.J., et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: A retrospective observational study. Lancet Infect Dis. 2013;13(2):137–146. doi: 10.1016/S1473-3099(12)70277-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roetzer A., Diel R., Kohl T.A., Rückert C., Nübel U., Blom J., et al. Whole genome sequencing versus traditional genotyping for investigation of a Mycobacterium tuberculosis outbreak: a longitudinal molecular epidemiological study. PLoS Med. 2013;10(2):e1001387. doi: 10.1371/journal.pmed.1001387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.World Health Organization, WHO. WHO | Global tuberculosis report 2016. WHO. 2019 [cited 26 Dec 2016]. Available: https://www.who.int/tb/publications/global_report/en/.
- 23.Lempens P., Meehan C.J., Vandelannoote K., Fissette K., de Rijk P., Van Deun A., et al. Isoniazid resistance levels of Mycobacterium tuberculosis can largely be predicted by high-confidence resistance-conferring mutations. Sci Rep. 2018;8(1) doi: 10.1038/s41598-018-21378-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim D., Song L.i., Breitwieser F.P., Salzberg S.L. Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res. 2016;26(12):1721–1729. doi: 10.1101/gr.210641.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Comas I. Genome of the inferred most recent common ancestor of the Mycobacterium tuberculosis complex. 2019 [cited 31 Mar 2021]. 10.5281/ZENODO.3497110.
- 26.Phelan J.E., O’Sullivan D.M., Machado D., Ramos J., Oppong Y.E.A., Campino S., et al. Integrating informatics tools and portable sequencing technology for rapid detection of resistance to anti-tuberculous drugs. Genome Med. 2019;11(1) doi: 10.1186/s13073-019-0650-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meehan C.J., Moris P., Kohl T.A., Pečerska J., Akter S., Merker M., et al. The relationship between transmission time and clustering methods in Mycobacterium tuberculosis epidemiology. EBioMedicine. 2018;37:410–416. doi: 10.1016/j.ebiom.2018.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walker T.M., Merker M., Kohl T.A., Crook D.W., Niemann S., Peto T.E.A. Whole genome sequencing for M/XDR tuberculosis surveillance and for resistance testing. Clin Microbiol Infect. 2017;23(3):161–166. doi: 10.1016/j.cmi.2016.10.014. [DOI] [PubMed] [Google Scholar]
- 29.Suchard M.A., Lemey P., Baele G., Ayres D.L., Drummond A.J., Rambaut A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018;4:Available. doi: 10.1093/ve/vey016. https://academic.oup.com/ve/article/doi/10.1093/ve/vey016/5035211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Didelot X., Fraser C., Gardy J., Colijn C. Genomic infectious disease epidemiology in partially sampled and ongoing outbreaks. Mol Biol Evol. 2017;34:msw075. doi: 10.1093/molbev/msw275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Plummer M, Best N, Cowles K, Vines K. CODA: convergence diagnosis and output analysis for MCMC. undefined. 2006.
- 32.Torchiano M. effsize: Efficient Effect Size Computation. 2020 [cited 3 May 2021]. Available: https://cran.r-project.org/web/packages/ggplot2/citation.html.
- 33.Wickham H. ggplot2: Elegant Graphics for Data Analysis. In: Springer-Verlag New York. ISBN 978-3-319-24277-4 [Internet]. 2016 [cited 3 May 2021]. Available: https://cran.r-project.org/web/packages/ggplot2/citation.html.
- 34.Ngabonziza J.C.S., Decroo T., Migambi P., Habimana Y.M., Van Deun A., Meehan C.J., et al. Prevalence and drivers of false-positive rifampicin-resistant Xpert MTB/RIF results: a prospective observational study in Rwanda. The Lancet Microbe. 2020;1(2):e74–e83. doi: 10.1016/S2666-5247(20)30007-0. [DOI] [PubMed] [Google Scholar]
- 35.Coll F., McNerney R., Guerra-Assunção J.A., Glynn J.R., Perdigão J., Viveiros M., et al. A robust SNP barcode for typing Mycobacterium tuberculosis complex strains. Nat Commun. 2014;5(1) doi: 10.1038/ncomms5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ngabonziza J.C.S., Loiseau C., Marceau M., Jouet A., Menardo F., Tzfadia O., et al. A sister lineage of the Mycobacterium tuberculosis complex discovered in the African Great Lakes region. Nat Commun. 2020;11(1) doi: 10.1038/s41467-020-16626-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.S K, N K, M N, H Z, G T, CU K, et al. Systematic review of mutations associated with resistance to the new and repurposed Mycobacterium tuberculosis drugs bedaquiline, clofazimine, linezolid, delamanid and pretomanid. J Antimicrob Chemother. 2020;75: 2031–2043. 10.1093/JAC/DKAA136. [DOI] [PMC free article] [PubMed]
- 38.Gagneux S., Long C.D., Small P.M., Van T., Schoolnik G.K., Bohannan B.J.M. The competitive cost of antibiotic resistance in Mycobacterium tuberculosis. Science. 2006;312(5782):1944–1946. doi: 10.1126/science.1124410. [DOI] [PubMed] [Google Scholar]
- 39.de Vos M., Muller B., Borrell S., Black P.A., van Helden P.D., Warren R.M., et al. Putative compensatory mutations in the rpoc gene of rifampin-resistant mycobacterium tuberculosis are associated with ongoing transmission. Antimicrob Agents Chemother. 2013;57(2):827–832. doi: 10.1128/AAC.01541-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vargas A., Rios A., Grandjean L., Kirwan D., Gilman R., Sheen P., et al. Determination of potentially novel compensatory mutations in rpoc associated with rifampin resistance and rpob mutations in Mycobacterium tuberculosis Clinical isolates from peru. Int J Mycobacteriology. 2020;9:121–137. doi: 10.4103/ijmy.ijmy_27_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ektefaie Y., Dixit A., Freschi L., Farhat M.R. Globally diverse Mycobacterium tuberculosis resistance acquisition: a retrospective geographical and temporal analysis of whole genome sequences. The Lancet Microbe. 2021;2(3):e96–e104. doi: 10.1016/s2666-5247(20)30195-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.El Halabi J., Palmer N., McDuffie M., Golub J.J., Fox K., Kohane I., et al. Measuring health-care delays among privately insured patients with tuberculosis in the USA: an observational cohort study. Lancet Infect Dis. 2021 doi: 10.1016/s1473-3099(20)30732-5. [DOI] [PubMed] [Google Scholar]
- 43.Merker M., Barbier M., Cox H., Rasigade J.-P., Feuerriegel S., Kohl T.A., et al. Compensatory evolution drives multidrug-resistant tuberculosis in Central Asia. Elife. 2018;7 doi: 10.7554/eLife.38200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Comas I., Borrell S., Roetzer A., Rose G., Malla B., Kato-Maeda M., et al. Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat Genet. 2012;44(1):106–110. doi: 10.1038/ng.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ngabonziza J.C.S., Van Deun A., Migambi P., Niyigena E.B., Dusabe T., Habimana Y.M., et al. Case Report: Dynamics of Acquired Fluoroquinolone Resistance under Standardized Short-Course Treatment of Multidrug-Resistant Tuberculosis. Am J Trop Med Hyg. 2020;103(4):1443–1446. doi: 10.4269/ajtmh.20-0201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Muvunyi C.M., Ngabonziza J.C.S., Uwimana I., Harelimana J.D.D., Mucyo Y., Sebatunzi O.R., et al. Highly successful treatment outcome of multidrug-resistant and genetic diversity of multidrug-resistant Mycobacterium tuberculosis strains in Rwanda. Trop Med Int Heal. 2019 doi: 10.1111/tmi.13245. [DOI] [PubMed] [Google Scholar]
- 47.Aubry A., Veziris N., Cambau E., Truffot-Pernot C., Jarlier V., Fisher L.M. Novel gyrase mutations in quinolone-resistant and -hypersusceptible clinical isolates of Mycobacterium tuberculosis: Functional analysis of mutant enzymes. Antimicrob Agents Chemother. 2006;50(1):104–112. doi: 10.1128/AAC.50.1.104-112.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.