

Abstract
The assessment of DNA damage can be a significant diagnostic for precision medicine. DNA double strand break (DSBs) pathways in cancer are the primary targets in a majority of anticancer therapies, yet the molecular vulnerabilities that underlie each tumor can vary widely making the application of precision medicine challenging. Identifying and understanding these interindividual vulnerabilities enables the design of targeted DSB inhibitors along with evolving precision medicine approaches to selectively kill cancer cells with minimal side effects. A major challenge however, is defining exactly how to target unique differences in DSB repair pathway mechanisms. This review comprises a brief overview of the DSB repair mechanisms in cancer and includes results obtained with revolutionary advances such as CRISPR/Cas9 and machine learning/artificial intelligence, which are rapidly advancing not only our understanding of determinants of DSB repair choice, but also how it can be used to advance precision medicine. Scientific innovation in the methods used to diagnose and treat cancer is converging with advances in basic science and translational research. This revolution will continue to be a critical driver of precision medicine that will enable precise targeting of unique individual mechanisms. This review aims to lay the foundation for achieving this goal.
Keywords: DSB repair, nonhomologous end joining, homologous recombination, risk prediction, precision medicine, machine learning, artificial intelligence
Introduction
Of the various types of DNA damage insults, DNA double-strand breaks (DSBs) are considered the most significant type of damage due to their toxicity to the cell. An individual can typically encounter 10-50 DSBs/cell/day and in order to maintain genomic integrity the DSB repair system must be highly regulated [1]. DSBs can arise from both exogenous and endogenous sources and multiple related pathways exist to repair them [2]. Environmental agents and normal cell metabolism subject the genome to constant damaging attacks that can lead to instability. An estimated 10,000 endogenous insults occur daily to the >3 billion base pair genome in each cell of the human body [3]. Although this represents a small percentage of the genome, the inability to regulate this level of insults can have drastic outcomes. In particular, endogenous sources that attack the genome trigger both single and double-strand breaks, such as free-radical species. Free radical species are generated during mitochondrial oxidative metabolism, as well as in response to cytokines, due to chronic inflammation or bacterial infections, and xenobiotics. At high levels, free radicals damage the cellular environment by oxidization of lipids, proteins and DNA. Damage to DNA is the most significant consequence of the oxidative stress caused by free radicals since modified DNA structures lead to mutations and genomic instability. Replication stress is another source of DNA damage where errors in DNA replication occur due to slow or faulty progression of replication forks [4].
Exogenous agents can also be a source of DNA damage particularly through diet, ultraviolet rays from sunlight, smoking and air pollution. Chemotherapeutic drugs and ionizing radiation (IR) or radiomimetic chemicals that mimic the action of IR are additional sources of DSBs [5]. Logically, a healthy cell is one that has a DNA repair capacity that exceeds the rate of DNA damage. As such, robust and efficient response mechanisms to repair damaged DNA is crucial for cells to prevent the loss of genetic material. Genome integrity is therefore protected by multiple coordinated DNA repair systems that recognize DNA lesions and rapidly activate the best repair pathway to minimize damage. Overall, the loss or reduction of DNA repair capacity is driven by mutations that are either hereditary (germline) or sporadic (somatic).
Germline Mutations
Germline mutations, particularly in DNA repair genes, predispose an individual to cancer and account for an estimated 5-10% of all cancers [6]. Genome sequencing technology has become so common and affordable that the general public now has access to their genetic identity. The emergence of several genetic testing companies, such as AncestryDNA, 23andMe, African Ancestry, has enabled individuals to know if they carry variations in genes associated with human diseases. Although this vast amount of genetic information can be useful, it is largely difficult to interpret. In most cases, this knowledge is expected to be used to adjust modifiable lifestyle factors (e.g., diet, exercise, smoking cessation) that can potentially decrease the risk of developing cancer. Somatic mutations however are more common and challenging to identify without multiple sequencing points.
Somatic Mutations
Somatic mutations occur from genomic damage that accrues within a cell over an individual’s lifetime. In this era of genomic medicine, it is envisioned that examining an individual’s genotype will enable prediction of a phenotype. In this context, the strategy is to use clinical trials to investigate molecular features of tumors that may predict response to a drug with a given mechanism of action. Genomic sequencing is the first step in identifying germline or somatic mutations that can be used to predict the risk associated with developing human diseases, such as cancer. However, the limitation is that it does not always specify the precise pathway that becomes altered leading to tumorigenesis. Delineating these complex mechanisms will require integration of complementary studies and advance our knowledge and ability to treat cancer on an individual basis.
Overall, the DNA damage response network (DDR) is intricately integrated in the regulation of cell cycle progression. A major question in the field is what factors determine the DSB repair pathway an individual cell uses to repair these toxic lesions. This is important to explore because cells that cannot repair DSBs typically have two primary outcomes. In one scenario, failure to repair DSBs can result in increased instability and cell death through apoptosis, an essential mechanism for removing pre-cancerous cells [7]. Alternatively, mis-repair of DSBs can lead to mutations (e.g. base pair insertions/deletions), loss of heterozygosity or chromosome rearrangements that result in cancer [8]. The genetic variation resulting from genomic mutations affects the ability of cells to efficiently repair DSBs, therefore knowing DSB mutation status has become critical in cancer care. The premise of precision oncology is to develop targeted treatments directed toward the molecular characteristics of an individual’s tumor. The most effective anticancer strategies are designed to direct cancer cells to self-destruct. Prolonged anticancer therapies often lead to altered DSB repair capacity in cancer that promote drug resistance and the accumulation of mutations. Bypassing cellular self-destruct mechanisms is the hallmark of cancer and the major cause of radio- and chemoresistance and even cancer recurrence. Therefore, the better our understanding of DSB repair pathways, the better we are able to design more effective therapeutics that circumvent cancer cell resistance mechanisms. In this chapter, we focus on integrative approaches in precision medicine needed to further our understanding of DSB repair pathways.
DNA Repair Pathways as Possible Therapeutic Targets
There are two main pathways for the repair of DSBs: ‘classical’ nonhomologous end joining (cNHEJ) and homologous recombination (HR) (Figure 1). NHEJ is the dominant pathway in mammals and multiple lines of evidence demonstrate that it functions throughout the cell cycle [9] and it is believed that the first attempt to repair DSB is through this pathway [10, 11]. If this pathway is unsuccessful however, the DSB becomes resected to produce single-stranded DNA overhangs allowing for activation of other methods of repair [12–14]. HR repair is an evolutionarily fundamental process essential for genome stability and enables the exchange of genetic information between identical or closely related DNA [15]. However, HR availability is restricted to the S and G2 phases when the homologous sequence of the undamaged sister chromatid is accessible. There is a point within the G2 phase of the cell cycle, when both NHEJ and HR can be activated. Although numerous factors influence the decision to repair DSB by NHEJ or HR, evidence suggests these pathways can synergize, as well as compete to maintain genomic integrity, yet the precise mechanisms remain poorly understood [16]. Subsets of these primary pathways have recently been discovered that add to the complex interplay of the system.
Figure 1. DSB repair pathway choice.
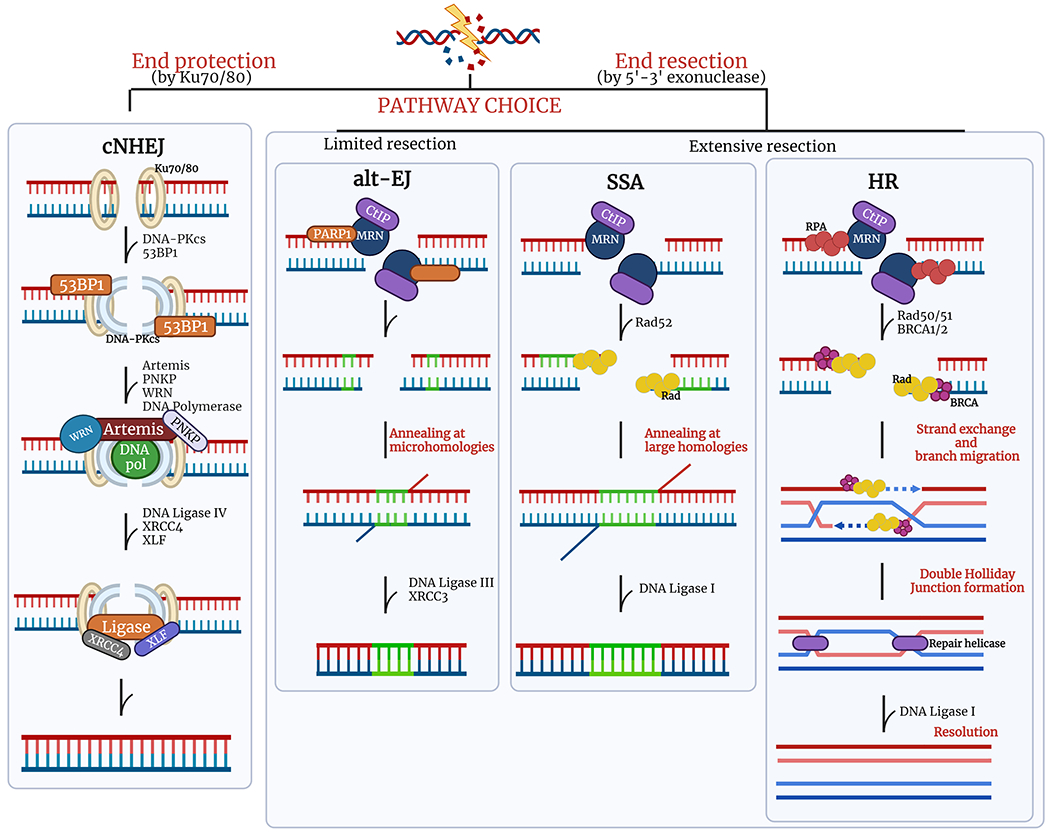
Balancing DSB repair through multiple mechanisms throughout the cell cycle. (A) cNHEJ and alt-EJ can occur at any phase of the cell cycle while HR and SSA are restricted to the S and G2 phase. (B) cNHEJ is initiated with Ku70/80 heterodimer recognizing and protecting the broken DNA ends, then recruits additional proteins to trim the ends and ligate gaps. Without the protection from Ku70/80, broken ends are subjected to resection. When there are limited amounts of resection, mediated by PARP1, the alt-EJ pathway is taken, resulting in annealing at microhomologies which can potentially cause multiple indels. With extensive end resection, HR and SSAare initiated by the MRN complex to make single stranded regions that allow recruitment of DNA repair proteins to mediate strand invasion and annealing at long homologies, respectively. The SSA pathway results in large deletions. Created with Biorender.com.
Classical Non-Homologous End Joining
Classical NHEJ (cNHEJ) accounts for the majority of DSB repair, particularly in the G1 and early part of the S phase of the cell cycle since it can directly ligate broken DNA ends without needing DNA resection [11]. The coordinated activities of macromolecular protein assemblies containing helicase and exonuclease activities, such as the MRN complex composed of MRE11, RAD50 and NBS proteins, can function as a signaling hub that regulates the flow of information leading to distinct cell outcomes [17]. The multifunctionality of the MRN complex illustrates its central role as a DSB sensor, activator of DSB-induced cell cycle signaling and DSB repair effector in both NHEJ and HR pathways [10, 18–20]. The MRN complex processes DNA ends and initiates the signaling cascade for DSB repair. The cNHEJ pathway is activated when the Ku70/80 heterodimer binds to DSB ends [21]. The Ku70/80 heterodimer functions as an asymmetric ring structure that forms a channel with easy access to DNA ends thereby limiting DNA processing [21]. Imaging of live cells illustrate that one Ku70/80 heterodimer binds to each of the exposed DNA ends of the chromosome [22]. With the Ku70/80 serving as a platform, the serine/threonine DNA-protein kinase (DNA-PK) is then recruited to these damage sites to interact with Ku70/80 to synapse the two broken DNA ends. DNA-PK is a viable target in anticancer therapies since inhibitors of its activity increase the efficacy of DSB inducing agents [23, 24]. Once bound to broken DNA ends, DNA-PK becomes activated and its autophosphorylation induces conformational changes leading to the recruitment of additional repair proteins, such as Artemis, polynucleotide kinase/phosphatase (PNKP), Werner syndrome ATP-dependent helicase (WRN) and DNA polymerases. Incorporation of the necessary deoxyribonucleotide triphosphates (dNTPs) and ribonucleotide triphosphates is carried out by Pol X family polymerases [18]. Subsequently, the structural mechanism of DNA Ligase IV has provided molecular details of how this enzyme performs the final DNA nick-sealing step with assistance from the X-ray cross complementing protein 4 (XRCC4) and the XRCC4-like factor (XLF) [25]. When the Ku70/80 heterodimer is removed from DSBs sites alternate pathways can be activated to stimulate DSB repair [21].
Homologous Recombination
HR is important in multiple pathways, such as DNA repair, DNA replication, meiotic chromosome segregation and telomere maintenance [26]. HR is largely functional in the S phase and G2 phase of the cell cycle, as it requires that the homologous sequence in the sister chromatid is in close proximity. This coordinated activity relies on tight regulation. Inaccuracies in the NHEJ pathway can be tolerated by differentiated somatic cells, where a large fraction of the genome is no longer functional, because misalignment of repetitive sequences may complicate HR [27]. These misalignments can lead to deletions and translocations. Therefore, it is advantageous to restrict HR-direct repair to the late S and G2 phases of the cell cycle. Overall, HR can be divided into three key steps: strand exchange, branch migration and resolution [15, 28]. First, DSBs require processing so that the DNA end is resected to degrade the 5’terminal DNA strand in the 5’ to 3’ direction on one strand of the DSB ends to produce terminal 3’-OH single strand DNA (ssDNA) tails or overhang. The first step is catalyzed by the (MRE11, RAD50, NBS1) MRN complex and CtIP [29]. The endonuclease, MRE11, cleaves the 5’-terminated DnA at the break site. This cleavage event is fueled by the ATPase activity of the RAD50 protein with CtIP and NBS1 as cofactors [17, 18, 29]. Downstream of the endonuclease activity, MRE11 has an additional function where it uses its 3’ – 5’ exonuclease activity to work backwards toward the DNA end to generate the 3’ ssDNA tail [29]. This critical step can regulate pathway choice but is also essential to allow homologous DNA sequences to invade a homologous DNA duplex to form a DNA crossover or Holliday junction, which is mediated by RAD51 [17]. (Figure 1) This enables DNA synthesis to initiate thus restoring the missing genetic information at the site of the break. Subsequent to strand invasion, branch migration occurs to extend the region away from the Holliday junction. Cleaving the original pair of crossing strands or by cleaving the non-crossing strands connects the two helices in the resolution step of HR. The separated strands that result from the original non-crossings yield two recombinant chromosomes from the reciprocal exchange of double-stranded DNA.
Alternative End Joining
Alt-EJ requires DNA to be resected to expose microhomologies that are recognized during DSB repair. Alt-EJ is mechanistically similar to both HR and cNHEJ but also mechanistically distinct [30]. Referred to as a back-up pathway when cNHEJ is inactivated, alt-NHEJ can be suppressed by Ku70/80 and functions in the absence of DNA-PK [30]. Compelling evidence illustrates functions of alt-EJ in cNHEJ proficient cells [31]. After DSB end processing and resection, microhomology regions are formed that can range from 1 bp to 25 bp [32]. Poly ADP-ribose polymerase 1 (PARP-1) is implicated as a possible platform for DSB synapsis and recruitment of additional repair proteins. Nevertheless, while this pathway appears to be less efficient, some DSB are repaired by alt-EJ.
Singe-Strand Annealing
Single stand annealing (SSA) is a DNA repair pathway similar to alt-EJ that can be activated depending on the nature of the sequence surrounding the break [33]. SSA is a conserved mechanism found in several organisms, however it causes large deletions and possibly translocations. The initial evidence of SSA was described using DNA plasmid substrates that were used to detect homologous recombination [34]. When transfected into mouse cells, it was observed that a DSB between homologous repeats yielded DSB end resection. The 3’ ssDNA that became exposed suggested there are flanking homologous sequences that are annealed to form an intermediate synapse. Further processing of the intermediate resulted in ligation and filling of DNA gaps by polymerase, thereby proposing a model for SSA.
DSB Repair Pathway Choice
Cell cycle phase, DNA end resection and DSB end structures determine which DSB repair pathway is executed [9, 11, 35]. Cell cycle dependency relies on three phosphoinositide 3 kinase-related protein kinases (PI3KKs); ataxia-telangiectasia mutated (ATM), ATM and RAD3-related (ATR) and DNA-dependent protein kinase catalytic subunit (DNA-PKcs). These PI3KKs function as sensors and set the stage for the recruitment of key DNA repair proteins to sites of DSB breaks [8, 36–38].
Chromatin structure surrounding DSB sites may limit the access and repair of DSBs. Multiple mechanisms coordinate and communicate cell cycle status to DSB repair sites and involves a large number of genes. In response to DSBs, ATM, ATR and DNA-PKc phosphorylate Ser139 of the histone variant H2AX (γ-H2AX) [37]. The discovery of H2AX was a milestone that helped to revolutionize the study of genome instability [39, 40].
Histone 2A variant has been demonstrated to be a sensitive marker for DNA damage and repair [41]. Bonner and colleagues demonstrated for the first time that γ-H2AX occurs rapidly at DSB break sites and that the dynamics of DSB repair can be studied through microscopic detection of γ-H2AX molecules by immunofluorescence. Today this technique is widely used not only in basic research settings, but also has important clinical applications [42–45]. Checkpoint mediators relay signals from DSB sites and once bound to γ-H2AX, mediator of DNA damage checkpoint protein (MDC1) becomes a multifunction protein that aids HR between sister chromatids and ATM signal amplification through association with MRN [17].
Although knowledge of the complex mechanisms of DSB repair is extensive, there are still gaps in our understanding of repair pathway choice. Altered levels of DNA repair proteins are common in aggressive forms of cancer and enable the proliferation of cancer cells due to imbalances between NHEJ and HR [46–48]. For example, the Breast Cancer susceptibility genes, BRCA1/2, were discovered as critical tumor suppressors against breast and ovarian tumorigenesis among women, although they have different roles in DSB repair [49–51]. Since their discovery, BRCA genes have been studied extensively for their dynamic roles in genome maintenance and are implicated in the development of prostate and pancreatic, as well. A subset of BRCA1-associated breast tumors lost expression of the DNA repair protein p53 Binding protein 1 (53BP1) [52]. Proteins that function as mediators of DSB repair, such as BRCA1 and 53BP1, are recruited to DSB break sites to assemble into key DNA repair complexes [53]. The tumor suppressor function of BRCA1 is critical for HR and in healthy cells it is general considered that there is a balance between HR and NHEJ pathways [15]. Deficiencies in key DNA repair genes however, tip the balance. BRCA1 recruited to DSB promotes HR, while recruitment of 53BP1 promotes NHEJ [53–55]. Deficiencies in BRCA1 causes cells to initiate NHEJ through the actions of 53BP1 and leads to the formation of tumors [56–58]. Conversely, in 53BP1-deficient cells, DSBs are repaired by HR although tumors were still observed likely through defects in other pathways involving 53BP1, such as DNA damage checkpoint [52, 59]. Interestingly, Bunting et al. (2010) report that generating a double knockout of the antagonistic DNA damage mediator proteins, BRCA1 and 53BP1, restores the HR pathway and the balance between the two DSB repair pathways with low or no tumor incidence [57]. These results suggest strategies that simultaneously target antagonizing proteins that favor distinct DSB pathways may be promising targets.
Manipulation of DNA Damage Repair Using Inhibitors
The complexities of DSB repair choice have been shaped by discoveries that have been translated into clinical practices. For instance, synthetic lethality is a strategy that targets the simultaneous mutation of two genes to induce lethality of a cell, while mutation of one gene is viable [60]. The most common example of synthetic lethality that has translated into clinical medicine are inhibitors of the poly (ADP-ribose) polymerase (PARP) enzyme (PARPi) [61, 62]. PARPi have shown great promise in treating BRCA-deficient tumors [60, 61, 63, 64]. Currently, there are four FDA approved PARPi for HR-deficient cancers (breast, ovarian, prostate, pancreatic). The effectiveness of PARPi relies on a non-functional HR-mediated repair system in BRCA-deficient tumors [64]. More specifically, PARP enzymes attach polymers of ADP-ribose (PAR) that serve as molecular tags, to itself and other proteins. PARPs have established roles in DNA repair pathways [61]. PARPi bind within the catalytic active site of the enzyme preventing it from attaching PAR onto itself and protein substrates. The inability of PARP to function in DNA repair thereby induces formation of single- and double-strand breaks within the cell. Cells that are not cancerous, which have HR-directed repair intact are able to repair DNA and survive. BRCA-deficient tumors are sensitive to selective killing due to the inability of cells to repair DNA through HR. The cytotoxicity of PARPi in cells with impaired HR highlight their promise as neoadjuvant agents used to help sensitize cancer cells for other treatment, such as ionizing radiation or conventional chemotherapy.
Olaparib (Lynparza) is a PARPi with FDA approval to treat several types of cancer that harbor BRCA deficiencies. In breast cancer that is HER2 negative and metastasized Olaparib is given with chemotherapy before or after surgery. In advanced ovarian cancer, Olaparib is given to patients already treated with three other types of chemotherapy. It can be used as a first-line therapy in patients who have complete or a partial response to platinum-based chemotherapy for the treatment of ovarian epithelial, fallopian tube or primary peritoneal cancers, as well as those with recurrent disease. In pancreatic cancer, Olarparib is used as maintenance therapy for metastasis after platinum-based chemotherapy. In castration resistant prostate cancer, it is used in patients whose disease has worsened after first-line treatments. Treatment with Olaparib has resulted in longer survival of patients with various forms of cancer and research is ongoing to study its effectiveness in other types of cancer. Despite its promise in treating cancer, prolonged treatment results in activation of resistance mechanisms [65].
PARPi resistance is ubiquitous in the clinic and develops through multiple mechanisms [66]. Prolonged treatment with PARPi leads to restoring HR repair and in some cases stabilization of replication forks [67]. More than 40% of BRCA mutated ovarian cancers failed to benefit from PARPi. Chemoresistance is a frequent outcome in cancer and can be classified in two groups [65]. More than 50% of cancer patients exhibit intrinsic resistance, that is, they do not respond to current treatments. There is an urgent need to identify novel agents that improve response and overcome resistance mechanisms. It is becoming evident that personalized, targeted interventions against a deregulated DSB pathway can be instrumental in overcoming chemoresistance and many examples have been reported [68, 69]. Anticancer strategies have exploited the ability to induce DSB in cancer treatment for decades [70]. Treatment with ionizing radiation and inhibitors of DNA repair proteins, like topoisomerase inhibitors are a common part of anticancer therapies [71]. Topoisomerase inhibitors inactive the enzyme responsible for cleaving single- or double-stranded DNA to help unwind and untangle supercoiled DNA. Several topoisomerase inhibitors have shown promise in the treatment of certain types of cancers (lung, cervical, ovarian and leukemia) [72]. In this era of scientific innovation, strategies for cancer treatment should enable predictions on the potential of chemoresistance. To understand this, correlations that characterize the genetic backgrounds can help to describe the molecular vulnerabilities of interindividual tumors.
Polygenic risk scores (PRS) have helped estimate an individual’s lifetime risk for developing disease. However, PRS maybe more informative at various points in disease progression [73]. While cancer is a genetic disease, risk scores can be grouped into two classes. The first class relates to a single gene associated with disease, and the second class is influenced by multiple genes and environmental factors, such as diet, sleep or smoking). Risk prediction scores have been generated from a variety of cancers [73, 74]. These risk scores enable personalized potential measures and can be used to described behavior of most chronic diseases. Several groups have looked at genetic data to calculate a HR-deficiency score [75]. Taken together, these analyses enable the ability to assign a representative system-level score to reflect efficiency and chromosomal stability and in some cases cancer patient survival. Understanding the factors that lead to chemoresistance mechanisms should be incorporated into risk prediction scores. For example, risk prediction scores need to include not only genes that influence cancer risk, but also genes that include chemoresistance risk. Combining risk scores, gene expression data and multi-omics approaches from cancer patients will continue to further the understanding of the molecular details of DSB repair and also inform future biomarker discoveries.
Heterogeneity of tumors present a challenge in treatment options. Rare cancers typically do not have standard treatment options if surgery is unsuccessful. One of the largest clinical trials, the National Cancer Institute’s Molecular Analysis for Therapy Choice (NCI-MATCH) is a phase 2 clinical trial guiding the development of targeted treatments to patients with distinct gene variants, regardless of cancer type [76]. Patients with rare cancers or solid tumors, lymphoma, or myeloma are not generally eligible for participating in conventional clinical trials. Identifying the genetic abnormalities that drive these types of cancers will have important implications for treatment strategies. The results obtained with NCI-MATCH has expanded to more detailed precision medicine trials, such as ComboMATCH, focused on testing drug combinations, MyeloMATCH, focused on testing treatment options for patients with acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS), which are not studied in NCI-MATCH. Lastly, ImmunoMatch (iMATCH) studies how the tumor’s immune status affects the response to target treatments with immunotherapy. The data collected in these studies will provide the critical framework to build more effective treatment options for cancer patients.
DSB Repair to Predict Disease and Response to Therapy
The field of precision medicine is constantly evolving, but its foundation aims to use predictive measures to evaluate genetic information to understand phenotype (i.e., how a patient responds to a given drug). Even as we celebrate 20 years since publishing of the first draft sequences of the human genome [77], we realize that our understanding of human genetic variation is not fully understood. Despite having such extensive genomic data, we are challenged with how to best interpret it. A bit costly at the onset, sequencing technology used in the early part of the 1990’s set the stage for complete genome sequencing of all domains of life. Haemophilus influenzae was the first bacterial genome to be sequenced with Saccharomyces cerevisiae being the first eukaryote sequenced within the same year [78, 79]. Shortly thereafter, two revolutionary reports set the stage for not only exploring human health, but also the potential for understanding disease [77, 80]. Completed within its 15-year prediction, the Human Genome Project (HGP) ushered in a new era of biomedicine and the notion that phenotypes may be predicted based on genotype. This significant milestone set the stage for not only the sequencing of genomes of humans from around the world, as well as anatomical subsite sequencing. It also required the revamping of essential policies for conducting this type of research that incorporates the digitizing of genetic information. Since the completion of the HGP in 2003, the sequencing and mapping of thousands of genomes has led to the generation of large biological data sets, including profiles of patient tumors. The rapid growth and development of novel biomolecular techniques and data-dense technologies ushered in what is known as the -omics era, where multiple disciplines are integrated to gain further insight into complex phenotypes and biological phenomena. These heavily technology-driven approaches provide a deeper look into and beyond the central dogma of biology. More importantly, these studies have led to the discovery of relevant tumor specific DSB repair biomarkers having the potential to be exploited as risk predictors.
The success of genome sequencing has been met with some challenges, particularly in the clinical setting. The ability to sequence tumor profiles has given insight into the complexity of cancer and has revealed an increased number of variants of unknown significance (VUS). A VUS is a variation in the sequence of a gene whose impact on cancer risk is not yet known. Classification of VUS fall into five categories: pathogenic, likely pathogenic, uncertain, likely benign and benign. Several studies discuss the importance of ways to interpret VUS in hope that tailored treatments can be made [81–84]. Extensive databases also exist to help correlate functional loss of activity with known and novel variants [85–87]. For example, cBioportal has been used to visualize tumor sequencing profile data obtained from The Cancer Genome Atlas (TCGA). To date, there have been extensive sequencing studies performed and made available in this repository [88]. Similarly, ClinVar is a public archive that reports relationships among genetic variations and phenotypes, linked with supporting information [89]. These repositories play a significant impact in sharing the most up-to-date clinical data between patients, clinicians and scientists.
BRCA1 and BRCA2 were among the first cancer genes to be sequenced [90, 91]. Although it is common practice to link mutations in a given gene to a certain disease phenotype, not all mutations of the same gene cause the same defect or even the same disease. BRCA gene mutations are known for their role in breast cancer development. Vast and continued sequencing has identified thousands of BRCA mutations, which exist across numerous databases. Notably, cBioPortal allows high-quality access to molecular profiles and clinical attributes from several large-scale cancer genomics projects. This access enables clinicians and researchers to visualize, explore and analyze genomic datasets. In more than 184 studies, novel and existing variants (missense, truncating, insertions/deletions, fusions) of the BRCA genes can be visualized including their level of frequency (Figure 2). Not all variants however, have the same clinical significance and in some cases many variants will need to be evaluated. Data across several clinical databases estimates more than 20,000 unique BRCA1 and BRCA2 variants. The BRCA Exchange dataset has classified of a large percentage of these variants, which have been curated by a panel of experts and 3,700 are known to cause disease [86]. Significant effort is underway to constantly assess both novel and known VUS of BRCA genes to interpret their functions and chart effective care management plans across distinct populations [92–94]. The prevalence of germline BRCA mutations have been shown to vary by racial/ethnic groups and in fact, Ashkenazi Jewish women were the first group reported to have a greater frequency of germline BRCA1/2 mutations (8-10%) with a strong association with triple-negative breast cancer (TNBC) [95–97]. TNBC are aggressive tumors that do not express the genes for estrogen receptor (ER-), progesterone receptor (PR-), excess human epidermal growth factor receptor 2 (HER2-), and do not respond to hormone-based therapies thereby limiting effective treatment options. Using public databases, several groups have used computational approaches to evaluate the functions of VUS from BRCA1 and BRCA2. Tram and Ozcelik proposed that VUS located across the entire protein sequence can affect phosphorylation of BRCA proteins and this mechanism may reveal novel targets in treating breast cancers [98]. In assessing the VUS from the BRCA2 DNA-binding domain, 186 missense variants were observed but functional data was missing for 154 of them. The authors were able to generate functional data using HR repair assays and were able to reclassify 39 of the VUSs as likely pathogenic/pathogenic or 93/132 as likely benign/benign [82]. These studies can aid in translating discoveries directly into the clinic. Being able to better correlate mechanistic findings with distinct genetic mutations will allow for development of improved therapeutics and individualized, targeted treatment.
Figure 2. BRCA mutations identified across cancer studies.
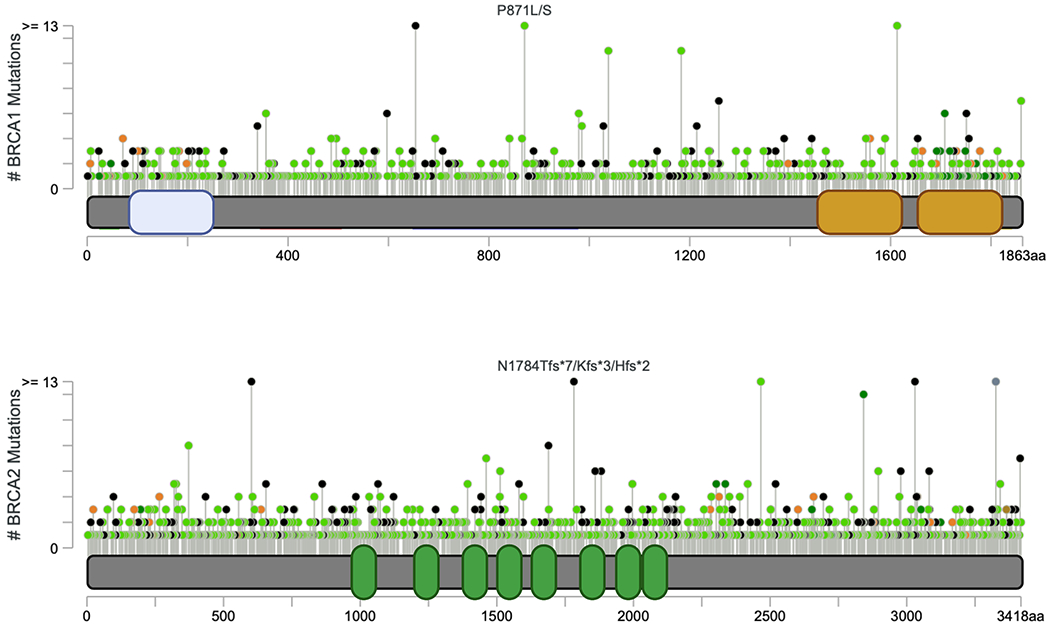
Cancer genomic portal provides a vast amount of information that enables visualization and analysis of molecular profiles and clinical features. Data from 184 studies representing more than 485,604 patient samples can be quickly assessed to determine BRCA mutations identified across all cancers. The somatic mutation frequencies for BRCA1 and BRCA2 are 1.8% and 3.0% respectively. These mutations are grouped between missense (green circles), truncating (black circles), inframe (deletion/insertion) (brown), fusion (purple) or other (pink) and span the entire protein sequence, including known protein domains (x-axis) while also indicating the frequency of each mutation. Image created in cBioPortal (Cerami et al., Cancer Discov. 2012 ; Gao et al., Sci. Signal. 2013) and modified with Biorender.com.
Much of the discussion around precision medicine has focused on genomics, yet while genes are a key factor in determining cancer risk, not all mutations are equal, particularly on the protein level. Precision medicine must include the -omics rather than genomics alone. Importantly, proteomic and interactomic approaches play a significant part in achieving precision medicine, especially since the majority of drugs target proteins, rather than genes. Several lines of investigation seek to address why some populations develop cancer and others do not. In a rare case, for example, female identical twins with an inherited BRCA1 mutation (deletion of exons 9-11 out of 24 total) had considerably different outcomes. While the twins seemed to share a similar risk for breast cancer, only one twin developed breast cancer at age 29. Interestingly, the second twin never developed breast cancer, however developed epilepsy and focal subcortical heterotopia at an early age [99, 100]. This example highlights the importance of taking the genetic background of a patient into consideration for interventions, as well as using an integrative approach to determine how genetic variants impact protein function in multiple cellular pathways.
Efforts to accurately classify most novel variants have been a continual challenge and associated risks cannot be assigned with certainty using a single method. The advent of gene editing has enabled assessment of every potential variant to be tested in a suitable system. Using saturation genome editing, Findlay and colleagues assayed 96.5% of all possible single nucleotide variants (SNV) of BRCA1 in 13 exons that encode functional protein domains [101]. The functional scores that were assigned were capable of predicting the pathogenicity of these variants and this information is poised to help with clinical interpretation of BRCA1. The biggest limitation is that for a majority of the BRCA1 protein, there is no structural information to enable assessment of all potential SNV. Exons 11-13 comprise the larger majority of BRCA1 (67%) and includes numerous genetic variants, but the study was not able to test this region predicted to be disordered in the protein. This limitation emphasizes the need to integrate complementary approaches to help elucidate cancer mechanisms and ensure precision medicine for all.
While Genome-Wide Association Studies (GWAS) have contributed greatly to our understanding of how genes influence disease risk, the structural changes that can occur at the protein level as a result of genetic mutations often go overlooked. The central dogma of biology explains it best: a given DNA sequence is read and transcribed into RNA which is then translated into proteins. From there, subcellular interactions, pathways, and whole-organism phenotype can be explored and evaluated. For the promise of precision medicine to become a reality, a deeper mechanistic understanding of how sequence variants translate to alterations in protein structure/conformation and how this can lead to downstream functional defects is required. Along with family history and genetic testing, elucidating the complexity that underlies each individual mutation can lead to better targeted therapeutics. As more variants are identified and characterized in cancer related genes and proteins, machine learning can be applied to better predict the functional outcome of such variants. Although only a few studies have begun to apply this tool to DSB repair, there have been some notable discoveries. In a recent study published by Hart et al., (2020) the researchers trained and evaluated hundreds of machine learning algorithms to assess the functional impact of BRCA1 and/or BRCA2 missense variants and predict whether they are damaging or neutral [102].
The Role of Genomic Editing and Machine Learning in Decoding Cancer
Scientific innovation is leading integration of revolutionary fields that have aided in advancing our understanding of DSB repair and cancer. Genome engineering has enabled site specific editing abilities that enable DNA to be inserted, deleted or modified. The 2012 Nobel Prize in Chemistry was awarded to Emmanuelle Charpentier, a French Microbiologist and Jennifer Doudna, an American biochemist for developing CRISPR/Cas9 technology. Clustered regularly interspaced short palindromic repeats (CRISPR) along with the CRISPR associated protein 9 (Cas9) was discovered as a natural immune process in prokaryotic organisms [103]. Ethical issues aside, small genetic changes that drive human cancers can potentially be eradicated using CRISPR/Cas9 technology, although it is a long way off. In the interim, CRISPR/Cas9 has made significant discoveries in understanding biology. Several groups have demonstrated using CRISPR/Cas9 to identify genes that suppress disease progression [104]. In fact, it is our understanding of DSB repair that enabled the revolutionary breakthrough of CRISPR. The CRISPR/Cas9 world has been filled with excitement, as well as some controversy [105]. Applications of CRISPR are now widespread and has had some interesting questions presented. In some studies, it was suggested that there are off-target effects such as large deletions or mutations of several thousand base pairs away from the target site [106–108]. This has cast some doubt on the effectiveness of DSB repair induced by CRISPR/Cas9 technology. These doubts have led to subsequent studies aimed at improving the efficiency of CRISPR/Cas9 applications. Recently, in an interesting study, Sherwood et. al., set out to predict genotypic products following Cas9 induced DSBs. The authors found that the ability to predict Cas9-mediated products enables increased precision in genome editing research [109]. Therefore, CRISPR itself is evolving, into a diverse tool for studying various DNA repair pathway features. Two groups of researchers utilized CRISPR technology to develop of loss-of-function screens to aid in the identification of key players in the DNA repair process that contribute to PARP inhibitor resistance in BRCA1 deficient cells [110, 111]. In a more recent, elaborately immense study, Roidos et al. (2020) developed a high-throughput assay to assess DSB repair choice, which can transcend understanding DNA repair and testing the efficiency of cancer drugs [112]. This study highlighted that gene editing outcomes can alter DSB repair to favor a specific mechanism. More specifically, this study developed a scalable CRISPR/Cas9-based fluorescent reporter assay, Color Assay Tracing Repair (CAT-R) to screen factors, such as small molecules that target the DNA damage response to visualize DSB repair outcomes. This study measured the rates of end-protection compared to end resection-based repair to assess DSB repair outcomes. Drug compounds currently in clinical trials were also used to target key DSB repair enzymes (ATM, DNA-PK, ATR and PARP1). Importantly, the study discovered a significantly altered gene cluster affected by PARP inhibition that regulates a key step in end resection. This opens the field to further understand the mechanism by which PARP1 regulates end resection.
The advent of machine learning and artificial intelligence offers the ability to recognize patterns within the vast amount of biological data collected over the years. Machine learning can be used in different contexts to enhance our understanding of DSB repair in cancer. For example, DSB breaks formed by Ionizing Radiation-Induced Foci (IRIF) can be quantitated using confocal microscopy, which has been the gold standard for measuring γ -H2AX levels. Vicar et al. (2020) developed a trainable deep learning-based method for counting and analysis of IRIF, called DeepFoci [113]. DeepFoci was as accurate as standard quantitative measure of nuclear foci across different cell lines and can differentiate various classes of cells or pre-malignant subclones. This application of machine learning yields a better understanding of radiation-induced DNA damage and DSB repair. Integrating machine learning of γ -H2AX scoring of patient samples will significantly improve patient-tailored decisions by developing learning algorithms that allow prediction of the appropriate dose of radiation, determine how tumors may respond to all chemotherapeutics, and even help with the identification of pre-cancerous lesions.
This multimodal approach can then be extended to incorporate other factors that may influence disease. Indeed, there are efforts aimed at providing a high-throughput and automated approach to understand DSB repair. Rapid Automated Biodosimetry Tool (RABiT) has been developed to perform radiation dosimetry measurements on blood samples and is being extended to provide direct measurements of the kinetics of DNA repair proteins [114, 115]. The RABiT technology will be a critical way to allow integration of epidemiological factors (e.g., gender, age, race and diet) which may provide the basis for observed variations in inter-individual endogenous γ -H2AX levels leading to a greater understanding of health and disease.
Genetic mapping and sequencing (genomics) allowed for the identification of disease-associated genes. The molecular complexities that underlie each genetic variant highlight the difficulty in drawing direct correlation to individual disease phenotypes. Thus, in order for precision medicine to be achieved, an integrative, multi-omic approach must be taken. The elucidation of molecular mechanism is not often incorporated into the bulk approach of precision medicine. Using computational approaches, we can begin to model the dynamics and interactions downstream of these genetic discoveries. Theoretical models of protein structure and function can then be validated using experimental approaches to create a clearer picture of the molecular defects caused by each genetic variant. Integration of these findings with changes at the RNA level (transcriptomics) and protein level (proteomics) allow for a deeper understanding of how individual genotypes correlate to distinct phenotypes. Each of these “omics” leads to the generation of large amounts of data, which can be nearly impossible for humans to reproducibly analyze manually. Algorithms of machine learning can be created to analyze these data and identify patterns that can be used to analyze future data sets. This can be particularly useful in precision medicine by making predictive models of genetic and/or protein networks (Figure 3).
Figure 3. A path to precision medicine.
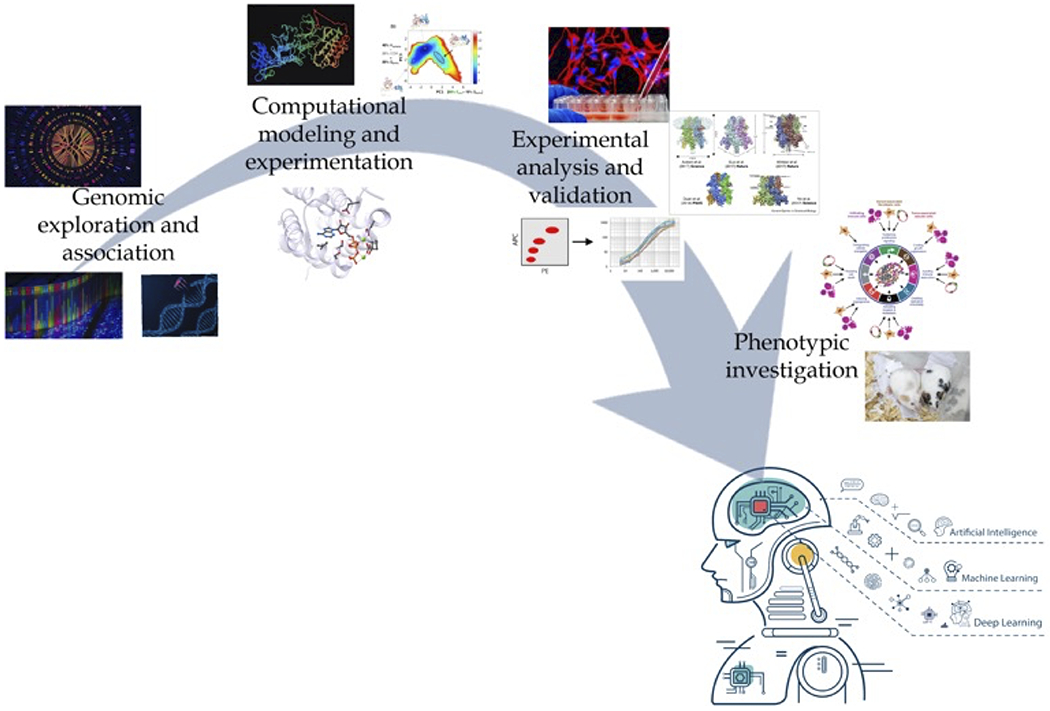
Traditional gene-disease associations, as identified by GWAS data, existed solely based on the presence of genetic mutations. However, with the large amount of data that has and continues to be collected from the multiple -omics (epigenomics, transcriptomics, proteomics, interactomics, and metabolomics) researchers and clinicians can gain a more precise understanding of how individual genetic mutations correlate to distinct disease phenotypes. To actualize on the promise of precision medicine, an integrated approach from gene association to phenotype investigation must be taken. Computational modeling can be used to predict consequences of specific gene mutations at the protein level (i.e. altered conformation, protein-protein interactions etc.). These findings could then be experimentally validated in vitro and/or in vivo to provide a deeper insight into the molecular mechanism(s) that may be altered and how these affect the distinct phenotype observed as a result of each mutation. The big data that can be produced at each step along this path can be integrated and applied to algorithms of machine learning to better predict individualized disease risk, drug sensitivity, and prognosis.
Large databases such as the Cancer Genome Project (CGP) and Cancer Cell Line Encyclopedia (CCLE) contain multitudes of information specific to different cancer and tumor types. These data can be analyzed using available data on cancer treating compounds to identify associations between gene expression and drug sensitivity. However, such associations are not able to take into account the mechanism of action of the drug compounds or the molecular consequence of genetic variants. Combining computational and experimental data from genomic, epigenomic, transcriptomic, and proteomic analyses offer deeper insight into the specific drug response pathways that are alike or divergent across cancer subtypes and within tumor types [116]. These data can then be used to predict clinical response on an individual basis. A recent study conducted by Lee et. al, utilized readily available multi-omics data to generate a machine learning algorithm that identified distinct molecular markers in acute myeloid leukemia and predicted drug sensitivity to known cancer therapeutics [117]. In this study, the authors created a computational method called MERGE (mutation, expression hubs, known regulators, genomic CNV, and methylation) that learns from integrated sets of validated data to make robust gene-drug associations. As the availability of experimentally validated GWAS data and mechanistic studies continues to grow, so does the potential for machine learning that can improve and expand precision medicine. With DSB repair pathways being clinically relevant in many cancers, combinatorial approaches will also be advantages. For example, HR-proficient ovarian cancers that have cyclin E amplifications are resistant to PARPi. Wilson et al. (2016) showed that positive correlations (Spearman) obtained from TCGA and CCLE were identified between Cyclin E E2F1 and E2F1 gene targets related to DNA repair, such as BRCA1 and RAD51. The authors showed that the HDAC inhibitor, Panobinostat, was effective at overcoming tumor resistance induced by DNA damaging drugs. This type of data will be essential to include in machine learning efforts, so that physicians presented with patients with similar genetic backgrounds have more anticancer treatment strategies that increase overall survival [118].
Future Direction
Significant advances have been made over the years that have resulted in an overall 29% decline in the cancer death rate, meaning the survival of an estimated 3 million people [119]. Progress in precision medicine has been steady, however it is not yet part of routine cancer care. Several precision medicine clinical trials are currently being tested. As novel variants in DSB repair pathways are being discovered, what hope is there for patients whose cancer is not aligned with currently approved targeted treatment option? Interindividual genetic variants can alter the interconnectedness of DSB repair, yet these vulnerabilities also represent potential druggable targets. The goal for precision medicine is to establish potent inhibitors for each DSB repair pathway for each genetic scenario. The advent of technological advances and classic experimental approaches will benefit a wide range of cancers, moving closer to the promise of precision medicine for all. Therefore, deciphering DSB repair mechanisms in cancer needs more rigorous and integrative methodology. Groundbreaking discoveries are constantly reshaping our perceptions and understanding of the dynamic and complex DSB repair systems in human health. More so, we are rapidly adapting and translating these discoveries into clinical practice. All genetic changes associated with cancer development have not yet been identified and computational approaches enable reliable predictions to be made. CRISPR/Cas9 screening coupled with machine learning and artificial intelligence has also demonstrated that its utility in speeding up the process to making more accurate predictions is possible. The priority for DSB repair research is the development of integrative models that incorporate data and knowledge from the clinics and basic science applicable to individual patients. The objective is to define mechanistic knowledge of the pathologies of distinct cancers and chart optimization of precise therapies. This approach will require more investment in charting the methodical and systematic strategies needed to unlock precision medicine for all.
References:
- 1.Vilenchik MM and Knudson AG, Endogenous DNA double-strand breaks: production, fidelity of repair, and induction of cancer. Proc Natl Acad Sci U S A, 2003. 100(22): p. 12871–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schär P, Spontaneous DNA damage, genome instability, and cancer--when DNA replication escapes control. Cell, 2001. 104(3): p. 329–32. [DOI] [PubMed] [Google Scholar]
- 3.Jackson SP and Bartek J, The DNA-damage response in human biology and disease. Nature, 2009. 461(7267): p. 1071–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Couch FB, et al. , ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev, 2013. 27(14): p. 1610–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mehta A and Haber JE, Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb Perspect Biol, 2014. 6(9): p. a016428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Institute, N.C. The Genetics of Cancer. Available from: https://www.cancer.gov/about-cancer/causes-prevention/genetics.
- 7.Labi V and Erlacher M, How cell death shapes cancer. Cell Death Dis, 2015. 6: p. e1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jackson SP, DNA damage detection by DNA dependent protein kinase and related enzymes. Cancer Surv, 1996. 28: p. 261–79. [PubMed] [Google Scholar]
- 9.Scully R, et al. , DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol, 2019. 20(11): p. 698–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ceccaldi R, Rondinelli B, and D’Andrea AD, Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shibata A, et al. , Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J, 2011. 30(6): p. 1079–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karanam K, et al. , Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol Cell, 2012. 47(2): p. 320–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mao Z, et al. , Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair (Amst), 2008. 7(10): p. 1765–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rothkamm K, et al. , Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol, 2003. 23(16): p. 5706–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heyer WD, Ehmsen KT, and Liu J, Regulation of homologous recombination in eukaryotes. Annu Rev Genet, 2010. 44: p. 113–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kass EM and Jasin M, Collaboration and competition between DNA double-strand break repair pathways. FEBS Lett, 2010. 584(17): p. 3703–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Syed A and Tainer JA, The MRE11-RAD50-NBS1 Complex Conducts the Orchestration of Damage Signaling and Outcomes to Stress in DNA Replication and Repair. Annu Rev Biochem, 2018. 87: p. 263–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taylor EM, et al. , The Mre11/Rad50/Nbs1 complex functions in resection-based DNA end joining in Xenopus laevis. Nucleic Acids Res, 2010. 38(2): p. 441–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zha S, Boboila C, and Alt FW, Mre11: roles in DNA repair beyond homologous recombination. Nat Struct Mol Biol, 2009. 16(8): p. 798–800. [DOI] [PubMed] [Google Scholar]
- 20.Trujillo KM, et al. , Nuclease activities in a complex of human recombination and DNA repair factors Rad50, Mre11, and p95. J Biol Chem, 1998. 273(34): p. 21447–50. [DOI] [PubMed] [Google Scholar]
- 21.Walker JR, Corpina RA, and Goldberg J, Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature, 2001. 412(6847): p. 607–14. [DOI] [PubMed] [Google Scholar]
- 22.Britton S, Coates J, and Jackson SP, A new method for high-resolution imaging of Ku foci to decipher mechanisms of DNA double-strand break repair. J Cell Biol, 2013. 202(3): p. 579–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ciszewski WM, et al. , DNA-PK inhibition by NU7441 sensitizes breast cancer cells to ionizing radiation and doxorubicin. Breast Cancer Res Treat, 2014. 143(1): p. 47–55. [DOI] [PubMed] [Google Scholar]
- 24.Gavande NS, et al. , Discovery and development of novel DNA-PK inhibitors by targeting the unique Ku-DNA interaction. Nucleic Acids Res, 2020. 48(20): p. 11536–11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaminski AM, et al. , Structures of DNA-bound human ligase IV catalytic core reveal insights into substrate binding and catalysis. Nat Commun, 2018. 9(1): p. 2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sung P and Klein H, Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat Rev Mol Cell Biol, 2006. 7(10): p. 739–50. [DOI] [PubMed] [Google Scholar]
- 27.Kanaar R, Hoeijmakers JH, and van Gent DC, Molecular mechanisms of DNA double strand break repair. Trends Cell Biol, 1998. 8(12): p. 483–9. [DOI] [PubMed] [Google Scholar]
- 28.Li X and Heyer WD, Homologous recombination in DNA repair and DNA damage tolerance. Cell Res, 2008. 18(1): p. 99–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.D’Amours D and Jackson SP, The Mre11 complex: at the crossroads of dna repair and checkpoint signalling. Nat Rev Mol Cell Biol, 2002. 3(5): p. 317–27. [DOI] [PubMed] [Google Scholar]
- 30.Bennardo N, et al. , Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet, 2008. 4(6): p. e1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Corneo B, et al. , Rag mutations reveal robust alternative end joining. Nature, 2007. 449(7161): p. 483–6. [DOI] [PubMed] [Google Scholar]
- 32.Della-Maria J, et al. , Human Mre11/human Rad50/Nbs1 and DNA ligase IIIalpha/XRCC1 protein complexes act together in an alternative nonhomologous end joining pathway. J Biol Chem, 2011. 286(39): p. 33845–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bhargava R, Onyango DO, and Stark JM, Regulation of Single-Strand Annealing and its Role in Genome Maintenance. Trends Genet, 2016. 32(9): p. 566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin FL, Sperle K, and Sternberg N, Model for homologous recombination during transfer of DNA into mouse L cells: role for DNA ends in the recombination process. Mol Cell Biol, 1984. 4(6): p. 1020–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Her J and Bunting SF, How cells ensure correct repair of DNA double-strand breaks. J Biol Chem, 2018. 293(27): p. 10502–10511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stucki M and Jackson SP, gammaH2AX and MDC1: anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair (Amst), 2006. 5(5): p. 534–43. [DOI] [PubMed] [Google Scholar]
- 37.Burma S, et al. , ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem, 2001. 276(45): p. 42462–7. [DOI] [PubMed] [Google Scholar]
- 38.Kinner A, et al. , Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res, 2008. 36(17): p. 5678–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rogakou EP, et al. , DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem, 1998. 273(10): p. 5858–68. [DOI] [PubMed] [Google Scholar]
- 40.Bonner WM, et al. , GammaH2AX and cancer. Nat Rev Cancer, 2008. 8(12): p. 957–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharma A, Singh K, and Almasan A, Histone H2AX phosphorylation: a marker for DNA damage. Methods Mol Biol, 2012. 920: p. 613–26. [DOI] [PubMed] [Google Scholar]
- 42.Redon CE, et al. , γ-H2AX detection in peripheral blood lymphocytes, splenocytes, bone marrow, xenografts, and skin. Methods Mol Biol, 2011. 682: p. 249–70. [DOI] [PubMed] [Google Scholar]
- 43.Redon CE, et al. , γ-H2AX and other histone post-translational modifications in the clinic. Biochim Biophys Acta, 2012. 1819(7): p. 743–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weyemi U, et al. , Twist1 and Slug mediate H2AX-regulated epithelial-mesenchymal transition in breast cells. Cell Cycle, 2016. 15(18): p. 2398–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weyemi U, Redon CE, and Bonner WM, H2AX and EMT: deciphering beyond DNA repair. Cell Cycle, 2016. 15(10): p. 1305–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ribeiro E, et al. , Triple negative breast cancers have a reduced expression of DNA repair genes. PLoS One, 2013. 8(6): p. e66243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tobin LA, et al. , Targeting abnormal DNA repair in therapy-resistant breast cancers. Mol Cancer Res, 2012. 10(1): p. 96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chibon F, Cancer gene expression signatures - the rise and fall? Eur J Cancer, 2013. 49(8): p. 2000–9. [DOI] [PubMed] [Google Scholar]
- 49.Tutt A and Ashworth A, The relationship between the roles of BRCA genes in DNA repair and cancer predisposition. Trends Mol Med, 2002. 8(12): p. 571–6. [DOI] [PubMed] [Google Scholar]
- 50.Friedman LS, et al. , Confirmation of BRCA1 by analysis of germline mutations linked to breast and ovarian cancer in ten families. Nat Genet, 1994. 8(4): p. 399–404. [DOI] [PubMed] [Google Scholar]
- 51.Roy R, Chun J, and Powell SN, BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer, 2011. 12(1): p. 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bouwman P, et al. , 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol, 2010. 17(6): p. 688–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Feng L, et al. , Cell cycle-dependent inhibition of 53BP1 signaling by BRCA1. Cell Discov, 2015. 1: p. 15019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zimmermann M and de Lange T, 53BP1: pro choice in DNA repair. Trends Cell Biol, 2014. 24(2): p. 108–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Panier S and Boulton SJ, Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol, 2014. 15(1): p. 7–18. [DOI] [PubMed] [Google Scholar]
- 56.Bunting SF, et al. , 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell, 2010. 141(2): p. 243–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cao L, et al. , A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol Cell, 2009. 35(4): p. 534–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jaspers JE, et al. , Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov, 2013. 3(1): p. 68–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ward IM, et al. , p53 Binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol Cell Biol, 2003. 23(7): p. 2556–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aly A and Ganesan S, BRCA1, PARP, and 53BP1: conditional synthetic lethality and synthetic viability. J Mol Cell Biol, 2011. 3(1): p. 66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hilton JF, et al. , Poly(ADP-ribose) polymerase inhibitors as cancer therapy. Front Biosci (Landmark Ed), 2013. 18: p. 1392–406. [DOI] [PubMed] [Google Scholar]
- 62.Buchtel KM, et al. , FDA Approval of PARP Inhibitors and the Impact on Genetic Counseling and Genetic Testing Practices. J Genet Couns, 2018. 27(1): p. 131–139. [DOI] [PubMed] [Google Scholar]
- 63.Comen EA and Robson M, Poly(ADP-ribose) polymerase inhibitors in triple-negative breast cancer. Cancer J, 2010. 16(1): p. 48–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee JM, Ledermann JA, and Kohn EC, PARP Inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies. Ann Oncol, 2014. 25(1): p. 32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li H, et al. , PARP inhibitor resistance: the underlying mechanisms and clinical implications. Mol Cancer, 2020. 19(1): p. 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chiappa M, et al. , Overcoming PARPi resistance: Preclinical and clinical evidence in ovarian cancer. Drug Resist Updat, 2021: p. 100744. [DOI] [PubMed] [Google Scholar]
- 67.Haynes B, Murai J, and Lee JM, Restored replication fork stabilization, a mechanism of PARP inhibitor resistance, can be overcome by cell cycle checkpoint inhibition. Cancer Treat Rev, 2018. 71: p. 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weber AM and Ryan AJ, ATM and ATR as therapeutic targets in cancer. Pharmacol Ther, 2015. 149: p. 124–38. [DOI] [PubMed] [Google Scholar]
- 69.Bradbury A, et al. , Targeting ATR as Cancer Therapy: A new era for synthetic lethality and synergistic combinations? Pharmacol Ther, 2020. 207: p. 107450. [DOI] [PubMed] [Google Scholar]
- 70.Khanna KK and Jackson SP, DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet, 2001. 27(3): p. 247–54. [DOI] [PubMed] [Google Scholar]
- 71.Elliott B and Jasin M, Double-strand breaks and translocations in cancer. Cell Mol Life Sci, 2002. 59(2): p. 373–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.You F and Gao C, Topoisomerase Inhibitors and Targeted Delivery in Cancer Therapy. Curr Top Med Chem, 2019. 19(9): p. 713–729. [DOI] [PubMed] [Google Scholar]
- 73.Lewis CM and Vassos E, Polygenic risk scores: from research tools to clinical instruments. Genome Med, 2020. 12(1): p. 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hall JM, et al. , Linkage of early-onset familial breast cancer to chromosome 17q21. Science, 1990. 250(4988): p. 1684–9. [DOI] [PubMed] [Google Scholar]
- 75.Takaya H, et al. , Homologous recombination deficiency status-based classification of high-grade serous ovarian carcinoma. Sci Rep, 2020. 10(1): p. 2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Do K, O’Sullivan Coyne G, and Chen AP, An overview of the NCI precision medicine trials-NCI MATCH and MPACT. Chin Clin Oncol, 2015. 4(3): p. 31. [DOI] [PubMed] [Google Scholar]
- 77.Lander ES, et al. , Initial sequencing and analysis of the human genome. Nature, 2001. 409(6822): p. 860–921. [DOI] [PubMed] [Google Scholar]
- 78.Dujon B, et al. , The nucleotide sequence of Saccharomyces cerevisiae chromosome XV. Nature, 1997. 387(6632 Suppl): p. 98–102. [PubMed] [Google Scholar]
- 79.Jacq C, et al. , The nucleotide sequence of Saccharomyces cerevisiae chromosome IV. Nature, 1997. 387(6632 Suppl): p. 75–8. [PubMed] [Google Scholar]
- 80.Venter JC, et al. , The sequence of the human genome. Science, 2001. 291(5507): p. 1304–51. [DOI] [PubMed] [Google Scholar]
- 81.Lyra PCM, et al. , Integration of functional assay data results provides strong evidence for classification of hundreds of BRCA1 variants of uncertain significance. Genet Med, 2021. 23(2): p. 306–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Richardson ME, et al. , Strong functional data for pathogenicity or neutrality classify BRCA2 DNA-binding-domain variants of uncertain significance. Am J Hum Genet, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Guidugli L, et al. , Assessment of the Clinical Relevance of BRCA2 Missense Variants by Functional and Computational Approaches. Am J Hum Genet, 2018. 102(2): p. 233–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Eccles DM, et al. , BRCA1 and BRCA2 genetic testing-pitfalls and recommendations for managing variants of uncertain clinical significance. Ann Oncol, 2015. 26(10): p. 2057–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jhuraney A, et al. , BRCA1 Circos: a visualisation resource for functional analysis of missense variants. J Med Genet, 2015. 52(4): p. 224–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.BRCA Exchange Launches. Cancer Discov, 2019. 9(3): p. 311–312. [DOI] [PubMed] [Google Scholar]
- 87.Cline MS, et al. , BRCA Challenge: BRCA Exchange as a global resource for variants in BRCA1 and BRCA2. PLoS Genet, 2018. 14(12): p. e1007752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gao J, et al. , Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal, 2013. 6(269): p. pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Landrum MJ, et al. , ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res, 2014. 42(Database issue): p. D980–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Collins FS, BRCA1--lots of mutations, lots of dilemmas. N Engl J Med, 1996. 334(3): p. 186–8. [DOI] [PubMed] [Google Scholar]
- 91.Plon SE, et al. , Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat, 2008. 29(11): p. 1282–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li D, et al. , Retrospective reinterpretation and reclassification of BRCA1/2 variants from Chinese population. Breast Cancer, 2020. 27(6): p. 1158–1167. [DOI] [PubMed] [Google Scholar]
- 93.Cragun D, et al. , Racial disparities in BRCA testing and cancer risk management across a population-based sample of young breast cancer survivors. Cancer, 2017. 123(13): p. 2497–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Solano AR, et al. , Spectrum of BRCA1/2 variants in 940 patients from Argentina including novel, deleterious and recurrent germline mutations: impact on healthcare and clinical practice. Oncotarget, 2017. 8(36): p. 60487–60495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hall MJ, et al. , BRCA1 and BRCA2 mutations in women of different ethnicities undergoing testing for hereditary breast-ovarian cancer. Cancer, 2009. 115(10): p. 2222–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.John EM, et al. , Prevalence of pathogenic BRCA1 mutation carriers in 5 US racial/ethnic groups. JAMA, 2007. 298(24): p. 2869–76. [DOI] [PubMed] [Google Scholar]
- 97.Antoniou A, et al. , Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet, 2003. 72(5): p. 1117–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tram E, Savas S, and Ozcelik H, Missense variants of uncertain significance (VUS) altering the phosphorylation patterns of BRCA1 and BRCA2. PLoS One, 2013. 8(5): p. e62468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Eccles D, et al. , BRCA1 mutation and neuronal migration defect: implications for chemoprevention. J Med Genet, 2005. 42(7): p. e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Eccles D, et al. , BRCA1 mutation and neuronal migration defect: implications for chemoprevention. J Med Genet, 2005. 42(5): p. e24. [PMC free article] [PubMed] [Google Scholar]
- 101.Findlay GM, et al. , Accurate classification of BRCA1 variants with saturation genome editing. Nature, 2018. 562(7726): p. 217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hart SN, et al. , Prediction of the functional impact of missense variants in BRCA1 and BRCA2 with BRCA-ML. NPJ Breast Cancer, 2020. 6: p. 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Doudna JA and Charpentier E, Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science, 2014. 346(6213): p. 1258096. [DOI] [PubMed] [Google Scholar]
- 104.Marinaccio C, et al. , LKB1/STK11 is a tumor suppressor in the progression of myeloproliferative neoplasms. Cancer Discov, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Napoletano S, Piersanti V, and Rallo G, CRISPR -Cas9: a groundbreaking new technique which ushers in new prospects and just as many doubts. Clin Ter, 2021. 171(1): p. e52–e54. [DOI] [PubMed] [Google Scholar]
- 106.Wang L, et al. , Large genomic fragment deletion and functional gene cassette knock-in via Cas9 protein mediated genome editing in one-cell rodent embryos. Sci Rep, 2015. 5: p. 17517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kosicki M, Tomberg K, and Bradley A, Erratum: Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol, 2018. 36(9): p. 899. [DOI] [PubMed] [Google Scholar]
- 108.Kosicki M, Tomberg K, and Bradley A, Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol, 2018. 36(8): p. 765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shen MW, et al. , Predictable and precise template-free CRISPR editing of pathogenic variants. Nature, 2018. 563(7733): p. 646–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Barazas M, et al. , The CST Complex Mediates End Protection at Double-Strand Breaks and Promotes PARP Inhibitor Sensitivity in BRCA1-Deficient Cells. Cell Rep, 2018. 23(7): p. 2107–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zimmermann M, et al. , CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature, 2018. 559(7713): p. 285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Roidos P, et al. , A scalable CRISPR/Cas9-based fluorescent reporter assay to study DNA double-strand break repair choice. Nat Commun, 2020. 11(1): p. 4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Vicar T, et al. , DeepFoci: Deep Learning-Based Algorithm for Fast Automatic Analysis of DNA Double Strand Break Ionizing Radiation-Induced Foci. 2020, BioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sharma PM, et al. , High throughput measurement of γH2AX DSB repair kinetics in a healthy human population. PLoS One, 2015. 10(3): p. e0121083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Turner HC, et al. , The RABiT: high-throughput technology for assessing global DSB repair. Radiat Environ Biophys, 2014. 53(2): p. 265–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Heiser LM, et al. , Subtype and pathway specific responses to anticancer compounds in breast cancer. Proc Natl Acad Sci U S A, 2012. 109(8): p. 2724–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lee SI, et al. , A machine learning approach to integrate big data for precision medicine in acute myeloid leukemia. Nat Commun, 2018. 9(1): p. 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wilson AJ, et al. , Panobinostat sensitizes cyclin E high, homologous recombination-proficient ovarian cancer to olaparib. Gynecol Oncol, 2016. 143(1): p. 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Siegel RL, Miller KD, and Jemal A, Cancer statistics, 2020. CA Cancer J Clin, 2020. 70(1): p. 7–30. [DOI] [PubMed] [Google Scholar]