

Abstract
Purpose:
Adducins interconnect spectrin and actin filaments to form polygonal scaffolds beneath the cell membranes, and form ring-like structures in neuronal axons. Adducins regulate mouse neural development, but their function in the human brain is unknown.
Methods:
We used exome sequencing to uncover ADD1 variants associated with intellectual disability (ID) and brain malformations. We studied ADD1 splice isoforms in mouse and human neocortex development with RNA Sequencing, super resolution imaging and immunoblotting. We investigated four variant ADD1 proteins and heterozygous ADD1 cells for protein expression and ADD1-ADD2 dimerization. We studied Add1 functions in vivo using Add1 knockout mice.
Results:
We uncovered loss-of-function (LoF) ADD1 variants in four unrelated individuals affected by ID and/or structural brain defects. Three additional de novo copy number variations covering the ADD1 locus were associated with ID and brain malformations. ADD1 is highly expressed in the neocortex and the corpus callosum, while ADD1 splice isoforms are dynamically expressed between cortical progenitors and postmitotic neurons. Human variants impair ADD1 protein expression and/or dimerization with ADD2. Add1 knockout mice recapitulate corpus callosum dysgenesis and ventriculomegaly phenotypes.
Conclusion:
Our human and mouse genetics results indicate that pathogenic ADD1 variants cause corpus callosum dysgenesis, ventriculomegaly, and/or intellectual disability.
Keywords: adducin, alternative splicing, membrane-associated periodic ring-like structure (MPS), axon degeneration
INTRODUCTION
The corpus callosum connects corresponding cortical areas of left-right cerebral hemispheres in mammals, and agenesis/dysgenesis of the corpus callosum is associated with intellectual disability and speech delay1,2. The generation of the corpus callosum occurs early in development and is critical for the functional synchronization of the two hemispheres3. Corpus callosum malformation is the most frequently observed structural brain defect4,5, often caused by abnormal neurogenesis, migration, and axon guidance2.
Three adducin genes ADD1/ADD2/ADD3 encode cytoskeleton proteins that are critical for osmotic rigidity and cell shape6,7. Adducins are best known for their association with the junctional complex in erythrocytes, interconnecting spectrin and actin filaments to form polygonal scaffolds beneath the cell membrane8,9. In neurons, adducins have been reported to form a membrane-associated periodic ring-like structure (MPS) with actin and β-spectrin10, and our previous work showed that deletion of Add1 in mice led to increased diameter of the MPS rings and axonal degeneration 11.
ADD1, ADD2, and ADD3 form heterodimers (ADD1/ADD2, ADD1/ADD3), which further form heterotetramers12. ADD1 and ADD3 are expressed in most tissues, while ADD2 is highly expressed in the brain and erythrocytes7. The three adducins have similar protein structure consisting of three main domains: the head, neck, and tail domains. The C-terminal tail domain has a well-conserved myristoylated alanine-rich C kinase substrate (MARCKS)-related domain that targets adducins to lateral membranes and stimulates β-spectrin-actin association13,14.
In Add1 null mice, Add2 and Add3 proteins were also undetectable11,15, indicating a predominant role of Add1 in stabilizing Add2 and Add3. Add1 null mice showed growth retardation, anemia and approximately 50% developed lethal communicating hydrocephalus accompanied by dilation of the ventricles15. Moreover, ADD1 regulates synaptic plasticity through glutamate receptors and is associated with memory performance in humans16. In contrast, Add2 knockout mice did not show structural brain malformation and had increased Add1/Add3 protein levels in the membrane fraction, suggesting dosage compensation17,18. Add2 regulates activity-dependent connectivity formation and Add2 knockout was reported to impair spine turnover in hippocampal neurons19,20.
Here we report four ADD1 variants associated with neurological symptoms, including one recessive missense variant and three de novo variants. The recessive variant is associated with absence of corpus callosum and enlarged lateral ventricles, and the de novo variants are associated with variable degrees of neurological disorders ranging from complete or partial agenesis of corpus callosum, to mild intellectual disability and attention deficit. We show that alternative splicing generates different isoforms of ADD1 between neural progenitors and cortical neurons, and ADD1 variants impair normal protein function. In addition to the previously reported lethal hydrocephaly phenotypes, survived Add1 knockout mice showed reduced thickness of corpus callosum in adulthood. Our human and mouse genetic results indicate that ADD1 loss-of-function is associated with corpus callosum malformation, ventriculomegaly, and/or intellectual disability.
MATERIALS AND METHODS
Human subjects
This study was conducted with the approval of institutional review boards and according to the ethical standards of the participating institutions: Boston Children’s Hospital; Children’s Hospital of Philadelphia; University of Alberta at Edmonton; and Helen DeVos Children’s Hospital. Informed consent was obtained from all subjects involved in this study or from parents of those who were younger than 18 years old. Control postmortem human tissues were obtained from NIH NeuroBank/NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore.
Molecular cloning
For ectopic expression of genes in the neuronal system, the pCAGIG (Addgene, 11159) vector and its previously described derivatives21 were used in this study. ADD1 and ADD2 genes were amplified using PCR and inserted into the AscI and NotI-digested pCAG-HA-Flag-IRES-GFP and pCAG-V5-Flag-IRES-GFP vectors, respectively. Ptbp1 shRNA knockdown constructs were as reported previously21.
Immunoprecipitation
Immunoprecipitation was carried out as instructed by the manufacturer (Thermo Scientific, 88837). Wild-type and mutated ADD1 coding sequences were subcloned into the pCAG-HA-Flag-IRES-GFP vector, and ADD2 was subcloned into the pCAG-V5-Flag-IRES-GFP vector. Then, different versions of mutated ADD1 and ADD2 were co-transfected into HEK293FT cells. 36-48 hours after transfection, cells were lysed with IP buffer (Thermo Scientific, 87787) and 500ug lysate was used for immunoprecipitation with anti-HA magnetic beads for 30min at room temperature. The immunoprecipitated protein was denatured and subjected to western blot.
RNA and protein analysis
DNase-treated total RNA samples were reverse transcribed using SuperScript IV with random hexamers following manufacturer’s instructions (Invitrogen 18-090-050), diluted, PCR amplified (primer sequences in Table S2), and resolved on agarose gels. Protein lysates were resolved on SDS-PAGE gels and western blots were carried out using the LI-COR Odyssey system. For immunofluorescence staining, embryonic mouse brains were dissected, fixed in 4% paraformaldehyde overnight at 4°C, and cryopreserved in 30% sucrose. Coronal sections were stained with primary antibodies at 4°C overnight followed by secondary antibodies for 1hr at room temperature. Primary antibodies used in this study are listed in Table S3.
Cell culture and generation of knockout cells
HEK293FT cells were cultured in DMEM supplemented with 10% FBS. Cells were transfected with sgRNAs and seeded into 10cm culture dishes (1000 cells/dish). Single colonies were picked 7-10 days later and cultured in 96-well plates for genotyping using Sanger sequencing. The ones with no variant were used as wild type controls whereas the ones containing variants were used as mutated cell lines.
Exome sequencing and data analysis
For Case I, genomic DNA was extracted from peripheral blood and subjected to array capture with the SureSelect Human Exon Kit (Agilent). 76-bp paired-end sequencing was performed on an Illumina HiSeq 2000 at the Broad Institute, yielding ~10 Gb of sequences per sample covering 86% of the target sequence at least 20 times. Sequencing reads were trimmed and aligned to the reference human genome (hg19) with the Burrows-Wheeler Aligner (v.0.5.7), followed by variant calling with the Genome Analysis Toolkit and variant annotation with ANNOVAR. Annotated variants were entered into a MySQL database and filtered with custom queries. Exome of other cases and their parents were performed following comparable procedures and filtered for de novo variants.
Analysis of alternatively spliced exons
Analysis of alternative splicing was performed as reported previously21. RNA-Seq datasets of laser microdissected cortical tissues22 were reanalyzed, and Sashimi plots were generated in IGV23. To validate differentially-spliced adducin exons during human brain development, we microdissected GW15, GW17, and GW18 fetal human cortical tissues and performed RT-PCR. We also harvested dorsal brain tissues from E12.5, E14.5, E16.5, E18.5, P12, and P40 CD1 mice, and extracted RNA with Trizol (Sigma) for RT-PCR.
Hippocampal neuron culture
Mouse and rat hippocampal neuron cultures were performed as previously described 24. Briefly, E18.5 hippocampi were digested in 0.06% trypsin from porcine pancreas solution for 15min (Sigma-Aldrich, T4799) and triturated. 12,500 cells/coverslip were plated onto 50 μg/mL poly-L-lysine hydrobromide (Sigma-Aldrich, P2636-100MG) pre-coated 1.5H glass 13mm rounded coverslips (Marienfeld) in 24-well plates (Nunc). Neurons were then cultured in Neurobasal medium (Thermo Scientific, 21103-049) supplemented with 2% B-27 (Thermo Scientific, 0080085SA), 1% penicillin/streptomycin (Thermo Scientific, 15140-122), and 2mM L-glutamine (Thermo Scientific, 25030024).
Immunostaining
Primary hippocampal neurons were fixed after 10 days in vitro (DIV10) with 4% paraformaldehyde (PFA, in PBS at pH 7.4) for 20min at room temperature. Fixed cells were permeabilized with 0.1% (v/v) Triton X-100 (in PBS) for 5min, quenched with 0.2M ammonium chloride (NH4Cl, Merck, 1.01145.0500), and incubated with blocking buffer (5% FBS in PBS) for 1hr. Primary antibodies diluted in blocking buffer were incubated overnight at 4°C (Table S3). After three 5min washes in PBS, secondary antibodies were incubated for 1hr at room temperature. Images were acquired using a TCS Leica SP8 confocal microscope.
STED imaging
STED imaging was performed on an Abberior Instruments ‘Expert Line’ gated-STED coupled to a Nikon Ti microscope. DIV10 hippocampal neurons were imaged at a fixed distance of 80-100 μm from the cell body, with an oil-immersion 60x 1.4NA Plan-Apo objective (Nikon, Lambda Series) using confocal and STED modes. The system featured 40 MHz modulated excitation (405, 488, 560, and 640nm) and depletion (775nm) lasers. The microscope’s detectors were avalanche photodiode detectors. The 2D vortex STED images with lateral resolution enhancement were recorded with 20nm pixel size in xy, and the pinhole was set to 0.8 Airy units. To analyze ring periodicity, the maximum intensity of peaks was determined and the interpeak distance was measured.
Animals
Add1 KO mice and WT littermates (129S1/ SvlmJ;C57BL/6J) were obtained from heterozygous breeding pairs and genotyped as described15. The protocols described were approved by of the University of Chicago IACUC and/or the IBMC Ethical Committee and by the Portuguese Veterinarian Board. Brains from P0, P14, and adult (9-week old) animals were collected and fixed with 4% paraformaldehyde for 24hrs at 4°C. After dehydration and clearing in toluene, brains were embedded in paraffin and whole brain coronal sections with 6 μm were cut in the microtome Microm HM335E (GMI-Trusted Laboratory Solutions). Cuts from two different regions of the corpus callosum (planes 27-30 and 41-50, according to the Allen Mouse Brain Atlas; https://mouse.brain-map.org/static/atlas) were selected and processed for hematoxylin and eosin staining. Coronal brain images were acquired in the NanoZoomer 2.0-HT slide scanner (Hamamatsu Photonics) with a 20x magnification. Corpus callosum thickness was measured using Qupath (University of Edinburgh) ImageJ software.
Statistics
All statistical tests were performed with GraphPad Prism 6. Specifically, for multiple comparisons, the 1-way ANOVA statistical test was performed followed by Tukey’s post hoc test. A P-value less than 0.05 was considered significant. Statistical tests and sample sizes are indicated in figure legends and significance was defined as *P < 0.05 and **P < 0.01.
RESULTS:
Variants in ADD1 are associated with corpus callosum malformation and neurological symptoms
Case I-1 is a female from a consanguineous family, and she was diagnosed with intellectual disability/mental retardation (Fig. S1, Table S1). At the age of two, Case I-1 showed agenesis of corpus callosum, abnormal sulcation of medial cerebral hemispheres, and grossly enlarged lateral and third ventricles on brain magnetic resonance imaging (MRI, Fig. 1A, Table S1). She also had hypoplasia of white matter and the cerebellar vermis. Exome sequencing of Case I-1 and her parents identified recessive variants in ADD1 and RTKN2 that were predicted to be damaging by PolyPhen2 and PROVEAN (Fig. S1B–S1D). RNA-Seq data from GTEx and other published datasets showed that ADD1 but not RTKN2 was expressed in adult or developing brain tissues (Fig. S1E–S1G). The rare recessive ADD1 variant (Chr4:2877811A>T, hg19, p.Arg57Trp, gnomAD allele frequency 0/313396, Fig. 1B) was considered the best candidate for symptoms in Case I-1.
Figure 1. Variants in ADD1 are associated with agenesis of corpus callosum and ventriculomegaly.
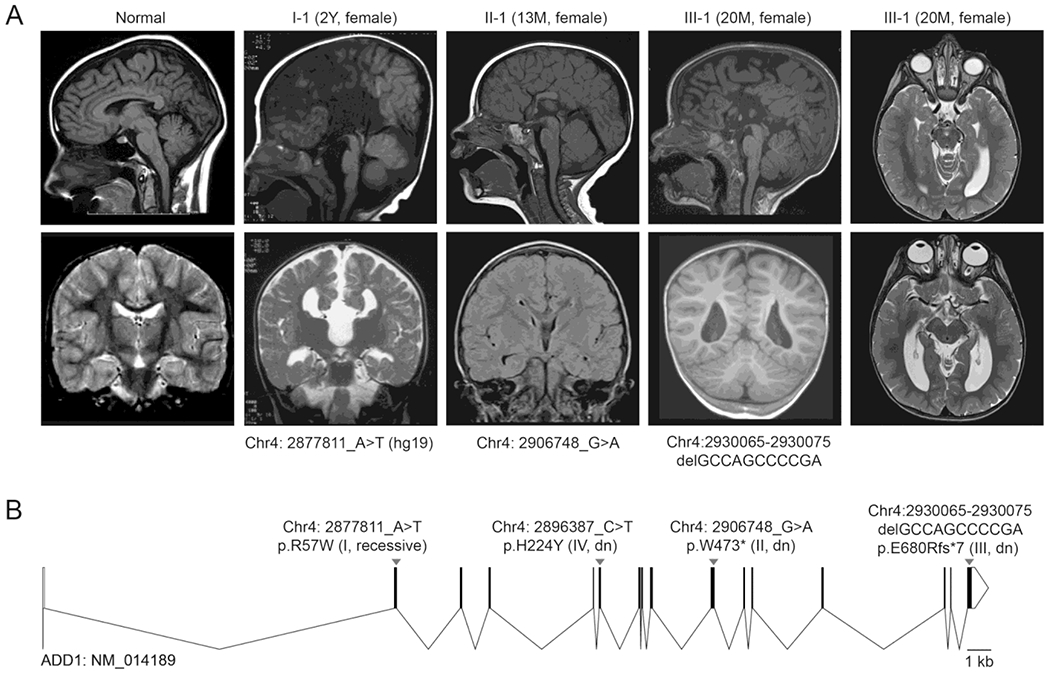
A) Brain MRIs showing that ADD1 variants are associated with agenesis of corpus callosum. Case I-1 carrying the homozygous ADD1 variant Chr4:2877811 A>T (hg19, p.Arg57Trp ) exhibits agenesis of corpus callosum (ACC), a grossly enlarged 3rd ventricle, and hypoplasia of white matter and cerebellar vermis. Case II-1 carrying the de novo truncation variant Chr4:2906748_G>A (p. Trp473*, NM_014189) exhibits partial agenesis of the corpus callosum, with only a segment of the anterior body present; there is also abnormal lobulation and disorganized subluxation and folia of the inferior and lateral aspects of the left cerebellar hemisphere. Case III-1 carrying the de novo truncation variant Chr4:2930065-2930075 delGCCAGCCCCGA, NM_001119, p.Glu680Argfs*7) exhibits complete agenesis of the corpus callosum and enlarged ventricles.
B) ADD1 exon-intron structure and the positions of variants reported in this study.
See also Figure S1.
Case II-1 is a female diagnosed with global developmental delay and intellectual disability. Case II-1 showed partial agenesis of the corpus callosum at the age of 13 months, consisting of only a segment of the anterior body measuring approximately 2cm (Fig. 1A, Table S1). There was abnormal lobulation, and disorganized subluxation and folia of the inferior and lateral aspects of the left cerebellar hemisphere. The patient showed proportional stature, distinctive facial features, generalized hypotonia, and developed seizures controlled with a ketogenic diet. She had a diagnosis of cerebral palsy and autism, and continued to make progress on her speech. Exome sequencing of Case II-1 and her parents identified a single rare de novo truncation variant in ADD1 (Chr4:2906748_G>A, ADD1: NM_014189.3, c.1418G>A, p.Trp473*, not found in gnomAD, Fig. 1A–1B).
Case III-1 is a female born at 40 weeks with normal weight, length and head circumference (35 cm, 80th percentile, Table S1). She was noted in utero to have complete agenesis of the corpus callosum and follow-up brain MRI at day 1 confirmed this. She was hypotonic with right hemiplegia and had failure to thrive as an infant which required a g-tube for 12 months. She had mild motor delays. She sat independently at 10 months and walked at 14 months. She had a seizure of unclear origin at 2 years 11 months, which was suspected to be related to hypernatremia. Subsequent EEG was normal. She also has had multiple staring spells that do not have EEG-correlate. A brain MRI performed at 20 months noted complete agenesis of the corpus callosum with absence of the cingulate gyrus and septum pellucidum (Fig. 1A). There was associated parallel configuration of the lateral ventricles with colpocephaly. The bilateral optic nerve sheath complexes were tortuous and the optic nerves appeared slightly thin. She had a normal muscle biopsy at 5 years old with muscle coenzyme Q10 at 21.1 ug/g (normal 24-33 ug/g). Muscle ETC noted compared to controls complex I deficiency at 26% and complex IV deficiency at 24%. She was started on ubiquinol with some improvement in her fatigue. Her other symptoms include patent foramen ovale, headaches, leg pains, night sweats, leukocytosis, and a qualitative platelet defect. At 8 years old she continues to have gastrointestinal issues with constipation, fatigue, leg and joint pain, and staring spells. She has behavioral outbursts, sensory issues and mild attention issues, and continues to be on the low end of the growth curve for length (121.3 cm, 8th percentile) and weight (21 kg, 6th percentile). Reanalysis of clinical negative exome sequencing of Case III-1 and her parents identified a rare de novo truncation variant in ADD1 (Chr4:2930065-2930075 delGCCAGCCCCGA, NM_001119, c.2029_2039del, p.Glu680Argfs*7, gnomAD allele frequency 0/251484, Fig. 1C), and a missense variant in CLASP1 (Chr2:122218756_C>T, NM_015282, c.G953A, p.R318Q) of unknown significance.
Case IV-1 is a male and presented with seizures beginning at the age of 1 year, along with speech delay, mild intellectual disability, attention-deficit/hyperactivity disorder (ADHD). Brain imaging at the age of 3.5 years did not display noticeable structural abnormalities. Exome sequencing of Case IV-1 and his parents identified a de novo missense variant in ADD1 (Chr4:2896387_C>T, ADD1: NM_001119, c.670C>T, p.His224Tyr, gnomAD allele frequency 0/31320, Fig. 1C). This variant was predicted to be deleterious by PROVEAN (Fig. S1D). The only other variants identified in Case IV-1 were compound heterozygous missense variants in SPTBN2 (NM_003128 c.4022G>A p.Arg1341Gln, and c.1004A>G p.Asn335Ser), the Spectrin Beta Non-Erythrocytic 2/βIII-spectrin gene associated with spinocerebellar ataxia, though Case IV-1 does not have ataxia.
Among the over-120,000 whole exomes on gnomAD, zero homozygous ADD1 LoF variants have been reported, and total observed LoF alleles are 14% (90% confidence interval 7%-30%) of expected (pLI = 0.99)25, indicating that ADD1 is intolerant to LoF variants and that de novo damaging variants can be deleterious. We examined 54 deletion variants affecting the ADD1 locus reported on ClinVar, and the majority of these deletions were associated with developmental delay. Interestingly, one deletion (hg19 chr4:71552-29006745) was associated with ventriculomegaly, agenesis of corpus callosum, Dandy-Walker malformation, and intrauterine growth retardation. Two additional deletions (hg38 chr4:36424-3881330, hg38 chr4:68453-6055026) were associated with microcephaly, delayed speech, muscular hypotonia, and motor delay. ADD1 is located in Chr4p16.3, and the region is associated with Wolf-Hirschhorn syndrome. The variants reported here support that recessive and de novo damaging ADD1 variants are associated with brain malformations and neurological symptoms such as intellectual disability and attention deficit.
ADD1 splicing isoforms are dynamically expressed during cortical development
ADD1 mRNA was broadly expressed in human neural and non-neural tissues (Fig. S1E), while ADD2 mRNA was expressed in the brain (Fig. S2A). We examined the impact of gender on ADD1 expression using the GTEx dataset26 and did not find differential expression of ADD1 between male and female brain tissues (Fig. S1F). RNA-Seq and RT-PCR results showed that ADD1 mRNA was highly expressed in the developing human brain, and ADD1 splicing isoforms were differentially expressed between the ventricular zone (VZ) and cortical plate (CP) (Fig. 2A). Specifically, exon 10 of human ADD1 had an extended 5’ splice site that was preferentially expressed in the CP, which mainly consisted of post-mitotic neurons; exon 15 was selectively included in the CP as well. RNA-Seq data from GTEx confirmed that ADD1 isoform containing extended exon 10 and inserted exon 15 was specifically expressed in brain tissues (Fig. S2B). Hereafter, we refer to ADD1 transcript NM_176801, with extended exon 10 and inclusion of exon 15, as the neuronal isoform; and the ADD1 transcript NM_001119, with shorter exon 10 and exclusion of exon 15, as the neural progenitor cell (NPC) isoform (Fig. 2A–2B).
Figure 2. ADD1 splicing isoforms are differentially expressed in the developing neocortex.

A) Alternative splicing of ADD1 exon10 and exon15 during human and mouse brain development. Top, genome (exon-intron) structure of ADD1 with indicated positions of exons 10 and 15. Left, RNA sequencing results and Sashimi plots showing the alternative 5’ splice site for exon10 and the inclusion/exclusion of exon15 between the cortical plate (CP, mostly neurons) and ventricular zone (VZ, mostly neural progenitor cells or NPCs) of gestational week (GW) 13-16 human fetal brains 40. Top right, RT-PCR results showing that human ADD1 exon10 and exon15 are differentially spliced between microdissected VZ and CP; Bottom right, RT-PCR results showing that mouse Add1 exon10 and exon15 are differentially spliced during dorsal cortex development.
B) ADD1 splice isoforms between neurons (top, NM_176801) and NPCs (bottom, NM_001119). The neuronal isoform has a longer neck domain due to extended exon10, but lacks the MARCKS-related domain due to inclusion of exon15, which introduces an in-frame stop codon. Positions of variants reported in this study are indicated.
C) CLIP-Seq peaks (green) showing that Ptbp1 binds to Add1 intron 14.
D) RT-PCR results showing that Ptbp1 shRNAs promote inclusion of Add1 exon15 in Neuro2a cells.
E) Western blot analysis of Add1 isoforms in E12.5, E14.5, E16.5 and E18.5 mouse dorsal brains, Neuro2a cells, and primary hippocampal neurons (DIV11).
See also Figure S2.
Interestingly, the inclusion of exon 15 introduces an in-frame stop codon, leading to the removal of the C-terminal MARCKS-related domain. Thus, the NPC isoform contains the MARCKS-related domain for localization to lateral membranes, whereas the neuronal isoform lacks this domain and has an extended neck domain (Fig. 2B). Add1 exon 10 and exon 15 were also differentially spliced in the developing mouse neocortex (Fig. 2A and S2C).
The RNA binding protein Ptbp1 is expressed in NPCs and suppresses neuronal exon insertion21,27. The intronic sequence upstream of Add1 exon 15 contains a CU-rich Ptbp1 binding motif and bears a Ptbp1 CLIP-Seq peak in NPCs (Fig. 2C). We infected Neuro2a cells with shRNAs targeting Ptbp1 and found that three different Ptbp1 shRNAs significantly increased the inclusion of Add1 exon 15 (neuronal isoform, Fig. 2D). Interestingly, other genes associated with adducins and β-spectrin, such as Ank2 and Epb4.1l3, were differentially spliced during cortical neurogenesis and coordinately regulated by Ptbp1 (Fig. S2D). These results indicate that Ptbp1 suppresses the Add1 neuronal isoform during cortical neurogenesis.
We examined multiple antibodies to determine their specificity to adducin homologs and isoforms (Fig. S2E–S2F, Fig. 2E and Fig. 3A). Two antibodies specifically recognized the ADD1 N-terminal domain (sc33633, named ADD1(NT) hereafter) or the ADD1 C-terminal domain (HPA035873, named ADD1(CT)) but not those of ADD2 and ADD3; and another antibody (ab51130, pan-ADD) recognized the conserved C-terminal MARCKS-related domain shared by ADD1/ADD2/ADD3 (Fig. S2E–S2F). Western blot with ADD1(NT) antibody confirmed that NPC and neuronal Add1 protein isoforms were dynamically expressed during brain development (Fig. 2E). At postnatal day 7 (P7) in mice, adducins were expressed in the neocortex and highly enriched in callosal axons (Fig. 3B and S3A). At embryonic day 14.5 (E14.5), Add1 and other adducins were expressed broadly in the brain and enriched in the cytosol (Fig. 3C).
Figure 3. Expression of ADD1 splice isoforms in the brain.
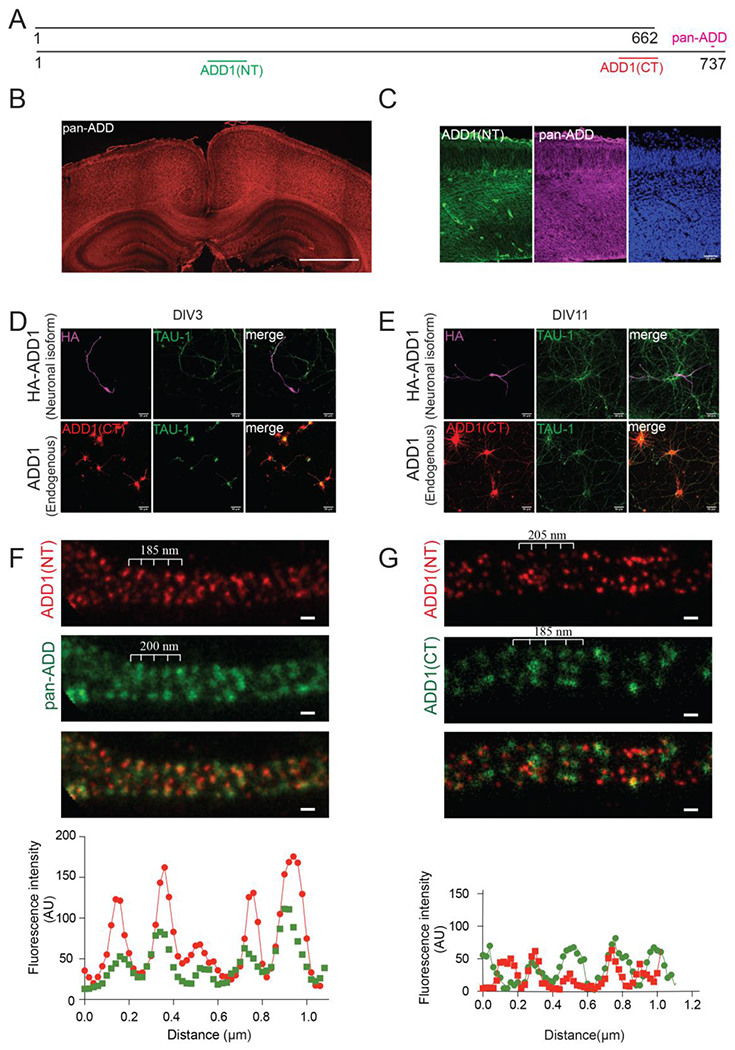
A) Schematic structure of ADD1 isoforms with regions that are recognized by different antibodies. ADD1(NT), sc33633; ADD1(CT), HPA035873; pan-ADD, ab51130.
B) Immunostaining of P7 mouse brain with pan-ADD antibody showing that adducins are expressed in the cortex and enriched in the corpus callosum. Scale bar 1000μm.
C) Immunostaining of E14.5 mouse cortex with pan-ADD and Add1-specific antibody (ADD1(NT)) showing that Add1 is expressed in the developing mouse brain.
D) Immunostaining of transfected and endogenous Add1 showing localization in axons of DIV3 primary mouse hippocampal neurons.
E) Immunostaining of Add1 showing localization in axons of rat DIV11 primary hippocampal neurons.
F) STED imaging of Add1 isoforms in primary cultured rat hippocampal neurons showing periodic signals of Add1(ADD1(NT)) and pan-adducins (pan-ADD) in the axon. Scale bar 0.2 μm.
G) STED imaging of Add1 isoforms in primary cultured rat hippocampal neurons showing periodic signals of Add1 (ADD1(NT), ADD1(CT)) in the axon. Scale bar 0.2 μm.
See also Figure S3.
The lack of the C-terminal MARCKS-related domain in the tail of neuronal Add1 suggests that previously-detected MPS signals with the pan-ADD antibody are likely Add2 or Add3 (Fig. 2E and Fig. S2E–S2F)10. Thus, it remained unclear whether the Add1 neuronal isoform is associated with the MPS. We examined endogenous and transfected Add1 neuronal isoform in primary hippocampal neurons, and found that the Add1 neuronal isoform was localized in the axons (Fig. 3D–3E). Next, we performed STED imaging by staining adducins with the pan-ADD antibody and ADD1-specific antibodies in primary cultured rat hippocampal neurons. Both the pan-ADD and ADD1-specific antibodies highlighted periodic structures, though the ADD1-specific antibodies showed a less distinct signal (Fig. S3B and Fig. 3F–3G). In summary, the Add1 neuronal isoform is expressed in axons and associates with the MPS.
ADD1 variants disrupt protein expression and adducin dimerization
To determine the effect of ADD1 variants on protein expression, we expressed ADD1 NPC and neuronal isoforms carrying the recessive variant (Chr4:2877811A>T, p.Arg57Trp) in Neuro2a cells. Although the protein level was not significantly affected, truncated protein products were observed when the p.Arg57Trp mutant was expressed in either the NPC or neuronal isoform (Fig 4A), suggesting that the p.Arg57Trp variant may lead to aberrant splicing or protein translation/cleavage. Add1 mRNA level was significantly decreased in Add1 heterozygous knockout mice compared to wild type15, which strongly suggests the possibility of a dosage effect. To examine whether de novo truncation ADD1 alleles affect protein expression, we generated ADD1 heterozygous HEK293FT cells (ADD1+/−, Fig. S4A) and found that ADD1+/− led to decreased ADD1 protein levels (Fig. 4B). Our results indicate that the p.Arg57Trp missense and truncating ADD1 variants affect ADD1 protein expression.
Figure 4. ADD1 variants disrupt protein functions.
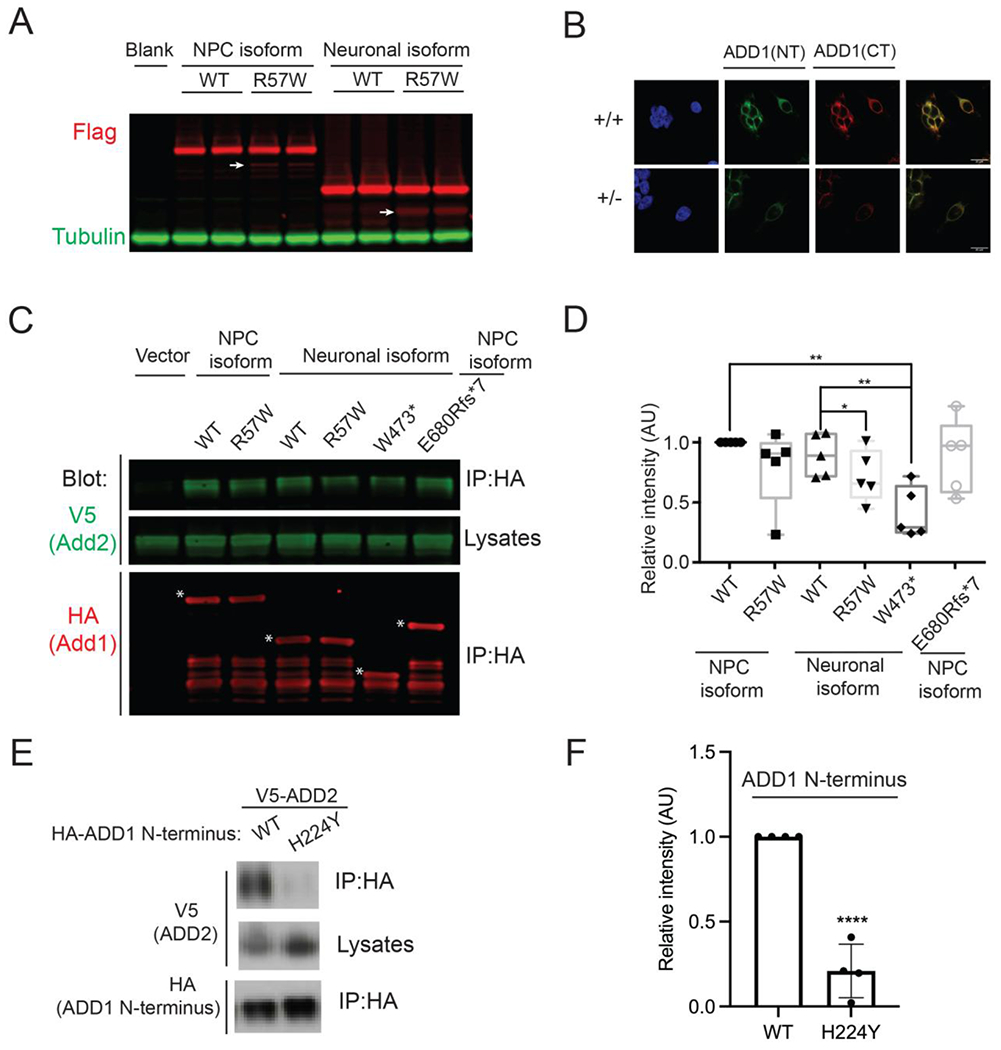
A) Expression of the ADD1 wildtype and mutant forms in Neuro2a cells showing that the Chr4:2877811 A>T (hg19, p.R57W) variant leads to a noticeable amount of truncated proteins (white arrows).
B) Immunostaining results using antibodies against ADD1 N terminal (ADD1(NT), green) and C terminal (ADD1(CT), red)) antibodies showing reduced protein level in ADD1 heterozygous (+/−) HEK293FT cells. Scale bar: 20μm.
C) Coimmunoprecipitation of V5-ADD2 transfected with indicated versions of HA-ADD1 (white stars) in HEK293FT cells, showing that ADD1 p.R57W and p.W473* reduced ADD1-ADD2 protein interaction.
D) Statistical analysis of signals in C) *P < 0.05, ** P < 0.01, t-test.
E) Coimmunoprecipitation of V5-ADD2 transfected with indicated versions of HA tagged ADD1 N-terminus (1-430 amino acids) in Neuro2a cells showing that p.H224Y reduced ADD1-ADD2 protein interaction.
F) Statistical analysis of signals in E) ****P< 0.0001, t-test.
See also Figure S4.
The N-terminal and C-terminal domains of ADD1 have been reported to mediate ADD1-ADD2 dimerization28. We expressed ADD1 mutant proteins and tested their efficiency in pulling down ADD2 through immunoprecipitation. We found that the missense variants (p.Arg57Trp and p.His224Tyr) in the head domain and the p.Trp473* truncation variant impaired ADD1-ADD2 interaction (Fig. 4C–4F). These results indicate that ADD1 variants caused damaging effects by decreasing protein levels and/or disrupting adducin complex formation.
Add1 knockout mice display ventriculomegaly and corpus callosum malformation
Homozygous Add1 knockout (Add1−/−) mice have been reported to show lethal hydrocephalus at 50% penetrance15, and axonal degeneration occurs in Add1−/− optic nerves and dorsal root ganglion neurons11. Here we examined lateral ventricles and formation of the corpus callosum in Add1−/− mice that did not show lethal hydrocephalus (Fig. 5 and S5). Add1−/− mice displayed ventriculomegaly at neonatal, P14, and adult stages (Fig. 5A and S5A). In 9-week old Add1−/− mice, the thickness of the corpus callosum was significantly decreased in the rostral brain when compared with Add1+/+ littermates (n=3, p<0.01); the corpus callosum in Add1+/− heterozygotes appeared thinner but not statistically significant. In the caudal brain, the thickness of corpus callosum was significantly decreased in Add1−/− samples (Fig. 5A–5C). At P0 and P14, the corpus callosum also showed the trend of thinning in Add1−/− mice (Fig. S5A–S5D). These results indicate that deletion of Add1 is associated with ventriculomegaly and corpus callosum degeneration in mice.
Figure 5. Corpus callosum thickness is decreased in adult mice in the absence of Add1.
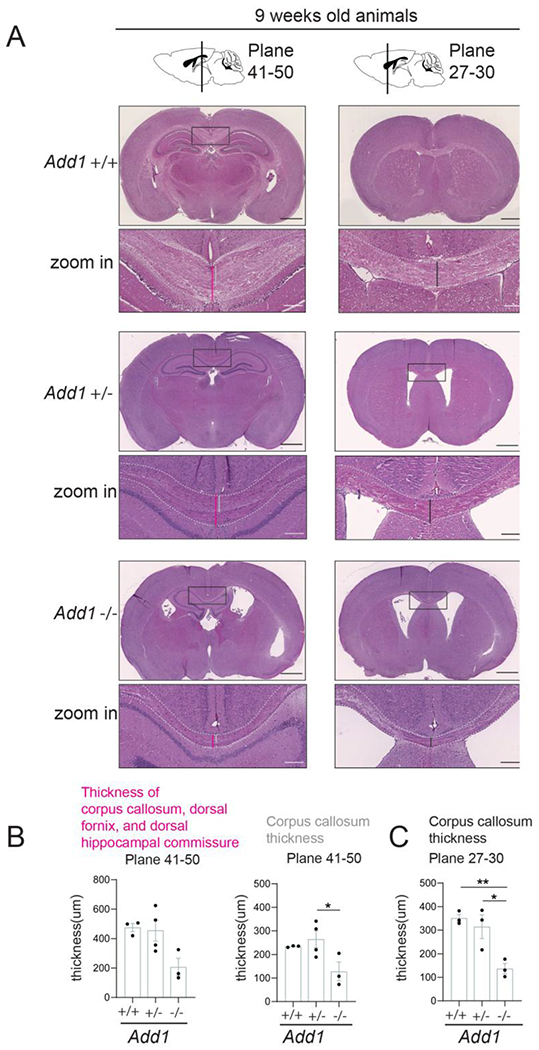
A) Hematoxylin and eosin staining of brain coronal sections from adult wild-type (Add1 +/+), heterozygous (Add1 +/−), and knock-out (Add1 −/−) Adducin1 animals. Scale bar: 500 μm; zoom-in scale bar: 100 μm.
B) Quantification of the thickness of corpus callosum, dorsal fornix, and dorsal hippocampal commissure (indicated by pink lines in A)) and of corpus callosum only (indicated by grey lines in A)) in coronal brain plates 41-50. Data represent mean ± SEM (n = 3-4 animals/condition). *P < 0.05 by 1-way ANOVA with Tukey’s post hoc test.
C) Quantification of corpus callosum thickness (indicated by black lines in A)) in coronal brain plates 27-30. Data represent mean ± SEM (n = 3 animals/condition). *P < 0.05, **P < 0.01 by 1-way ANOVA with Tukey’s post hoc test.
See also Figure S5.
DISCUSSION:
Our results show that ADD1 is differentially spliced during neurogenesis, and loss-of-function variants in ADD1 are associated with corpus callosum malformation, ventriculomegaly, ID and attention deficit. Add1 knockout mice displayed absence/degeneration of corpus callosum and lethal ventriculomegaly (i.e., hydrocephalus), highly similar to the affected individual with the recessive ADD1 variant. Furthermore, we show that missense and de novo variants in ADD1 impair ADD1-ADD2 dimerization and decrease ADD1 levels. These data provide strong support that LoF variants in ADD1 cause malformations of the corpus callosum, ventriculomegaly, and neurological symptoms in humans.
Among the four variants reported here, the recessive missense variants p.Arg57Trp and p.His224Tyr fall in the core (head and neck) domain of ADD1, which mediates oligomerization28. Consistent with this, we found that the p.Arg57Trp and p.His224Tyr variants weakened the association of ADD1 with ADD2 (Fig. 4). The tail domain of ADD1 has also been reported to regulate dimerization12, and the de novo truncation variant p.Trp473* significantly reduced the association between ADD1 and ADD2. It was intriguing that de novo variants in ADD1 caused variable but closely related neurological symptoms compared to the recessive variant. We noticed that Add1 mRNA levels were decreased in Add1+/− mice15 and hypothesized that the heterozygous ADD1 variants had dosage effects. Heterozygous human cells harboring premature stop codons indeed decreased ADD1 protein levels (Fig. 4B), suggesting that de novo truncating variants led to reduced amounts of ADD1 protein. Interestingly, the ADD1 truncation variant affecting only the neuronal isoform (Case II-1, p.Trp473*) was associated with ACC but not ventriculomegaly, while the ADD1 truncation affecting only the NPC isoform (Case III-1, p.Glu680Argfs*7) associated with both ACC and ventriculomegaly (Fig. 1A and Fig. 2B), suggesting that ventriculomegaly relates to the function of ADD1 NPC isoform.
Adducins promote the assembly of β-spectrin and actin29,30, and in neurons, adducins form the MPS with actin and β-spectrin tetramers10. Previously, we showed that Add1 knockout led to increased diameters of MPS rings and axonal degeneration11. Our current work in humans and knockout mice suggests that ADD1 is required in the brain for balancing cerebrospinal fluid and maintaining intact axon structure. Interestingly, neonatal βII-spectrin knockout mice displayed completely absent or significantly diminished interhemispheric axonal bundles including the corpus callosum, while juvenile βII-spectrin mutants showed a significant increase in the diameter of myelinated axons and signs of axonal degeneration31. Very recently, dominant variants in βII-spectrin SPTBN1 were associated with a neurodevelopmental syndrome32. Thus, the adducin-β-spectrin complex plays an essential role in the mouse and human brain development.
Ankyrins anchor proteins such as ion channels to the spectrin-actin-based membrane cytoskeleton through direct interaction with spectrin tetramers6. In neurons, the levels of AnkyrinB/ANK2 and βII-spectrin/SPTBN1 critically regulate MPS formation in axon development33. Loss of βII-spectrin led to decreased ANK2 levels in mice, and de novo LoF variants in the ANK2 gene have been repeatedly associated with autism in human genetics studies34. Consistent with the ANK2 LoF phenotype, Case IV-1 carrying a de novo ADD1 missense variant displayed speech delay, mild ID, and ADHD. The association of ADD1 truncation variants with structural brain malformation, and the association of missense ADD1 variant with mild ID and ADHD suggest that ADD1 protein dosage is critical in neurological functions. This is consistent with the pleiotropic hydrocephalus and axonal degeneration phenotypes observed in Add1 knockout mice (Fig. 5)11. These observations suggest that variants in components of the adducin-actin-spectrin-ankyrin cytoskeleton network can cause dosage-dependent and pleiotropic neurological symptoms.
Adducins associate with β-spectrin through their C-terminal conserved MARCKS-related domain12, and a pan-adducin antibody (ab51130) against this conserved domain showed periodic ring-like structures (MPS) in axons10. Given that the pan-adducin antibody recognizes ADD1, ADD2 and ADD3 (Fig. S2F), it was unclear which adducin(s) was associated with the MPS, let alone which splice isoform. Here we show that ADD1 undergoes alternative splicing between NPCs and neurons, and the neuronal isoform lacks the conserved MARCKS-related domain (Fig. 2). Our data suggest that although ADD1 was expressed in neuronal axons, the previously reported signal using the pan-ADD antibody was probably from ADD2 and/or ADD3. Intriguingly, our super-resolution imaging using STED showed that the truncated neuronal ADD1 isoform was still associated with the MPS, implying that the C-terminal MARCKS-related domain is not required for ADD1’s association with the MPS.
Alternative splicing of neuronal genes has been increasingly associated with neurological disorders35. Here we show that Add1 is differentially spliced during cortical neurogenesis: the neuronal isoform lacks the C-terminal MARCKS-related domain and is suppressed by Ptbp1 in NPCs. Interestingly, our current and previous works showed that Ank2, Epb4.1l1, Epb4.1l3, and Tpm2 were also coordinately and differentially spliced during brain development (Fig. S2D)21. Furthermore, Ptbp1 knockout mice displayed a lethal hydrocephalus phenotype36 that is comparable to Add1 knockouts, suggesting that Ptbp1-mediated splicing of Add1 has physiological consequences. Together, these observations suggest that the adducin-actin-spectrin-ankyrin cytoskeletal protein network undergoes coordinated alternative splicing during neurogenesis and neuronal differentiation, promoting the restructuring of the membrane cytoskeleton from a polygonal scaffold to the ring-like MPS in neurons.
Common adducin genetic variants were associated with cognitive deficiency in schizophrenia37, and mixed evidence showed that polymorphisms in ADD1, especially p.Gly460Trp, associated with essential hypertension38 and cardiovascular disease in hypertensive individuals39. Interestingly, cases reported here showed developmental delay (II-1) and symptoms in other tissues such as persistent ketoacidosis, lactic acidosis and a qualitative platelet defect (III-1, Table S1). ADD1 is expressed in the heart, brain, and broadly in other human tissues (Fig. S1E), and deletion of Add1 in mice caused compensated hemolytic anemia15. These observations suggest that ADD1 may have essential functions in other tissues in addition to the brain. Further studies are required to gain a complete understanding of ADD1 variant-related clinical presentations.
Supplementary Material
Acknowledgements
We would like to thank Prof. Soma Das for insightful comments on the manuscript, thank all affected individuals and families in this study, and thank Dr. Nahit Motavalli Mukaddes for referring Cases. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institute of Health and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. This work is supported by grants from NIMH (K01 MH109747) and NIGMS (DP2 GM137423) to X.Z, and by the Manton Center for Orphan Disease Research and grants from NINDS (R01 NS035129 and R01 NS032457) to C.A.W. C.A.W. is a Distinguished Investigator of the Paul G. Allen Family Foundation, an Investigator of the Howard Hughes Medical Institute. M.M.S is supported by Fundo Europeu de Desenvolvimento Regional through the Norte Portugal Regional Operational Programme, Portugal 2020, and by FCT - Fundação para a Ciência e a Tecnologia/Ministério da Ciência, Tecnologia e Ensino Superior (NORTE-01-0145-FEDER-028623; PTDC/MED-NEU/28623/2017). C.F was supported by the Region of Southern Denmark.
Footnotes
Disclosure
R.D.G receives consulting fees from Minovia Therapeutics. The other authors declare no conflict of interest.
Ethics Declaration:
This study was conducted with the approval of institutional review boards and according to the ethical standards of the participating institutions: Boston Children’s Hospital; Children’s Hospital of Philadelphia; University of Alberta at Edmonton; and Helen DeVos Children’s Hospital. Informed consent was obtained from all subjects involved in this study or from parents of those who were younger than 18 years old. Control postmortem human tissues were obtained from NIH NeuroBioBank (https://neurobiobank.nih.gov/about-best-practices/). The protocols described were approved by of the University of Chicago IACUC and/or the IBMC Ethical Committee and by the Portuguese Veterinarian Board.
Data availability
Under institutional privacy policies and institutional review boards, further data and experimental details are available upon request.
REFERENCES
- 1.Paul LK et al. Agenesis of the corpus callosum: genetic, developmental and functional aspects of connectivity. Nat Rev Neurosci 8, 287–299, doi: 10.1038/nrn2107 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Edwards TJ, Sherr EH, Barkovich AJ & Richards LJ Clinical, genetic and imaging findings identify new causes for corpus callosum development syndromes. Brain 137, 1579–1613, doi: 10.1093/brain/awt358 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Richards LJ, Plachez C & Ren T Mechanisms regulating the development of the corpus callosum and its agenesis in mouse and human. Clin Genet 66, 276–289, doi: 10.1111/j.1399-0004.2004.00354.x (2004). [DOI] [PubMed] [Google Scholar]
- 4.Jeret JS, Serur D, Wisniewski K & Fisch C Frequency of agenesis of the corpus callosum in the developmentally disabled population as determined by computerized tomography. Pediatr Neurosci 12, 101–103, doi: 10.1159/000120229 (1985). [DOI] [PubMed] [Google Scholar]
- 5.Parrini E, Conti V, Dobyns WB & Guerrini R Genetic Basis of Brain Malformations. Mol Syndromol 7, 220–233, doi: 10.1159/000448639 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bennett V & Baines AJ Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol Rev 81, 1353–1392, doi: 10.1152/physrev.2001.81.3.1353 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Matsuoka Y, Li X & Bennett V Adducin: structure, function and regulation. Cell Mol Life Sci 57, 884–895, doi: 10.1007/PL00000731 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bennett V & Gilligan DM The spectrin-based membrane skeleton and micron-scale organization of the plasma membrane. Annu Rev Cell Biol 9, 27–66, doi: 10.1146/annurev.cb.09.110193.000331 (1993). [DOI] [PubMed] [Google Scholar]
- 9.Pan L, Yan R, Li W & Xu K Super-Resolution Microscopy Reveals the Native Ultrastructure of the Erythrocyte Cytoskeleton. Cell Rep 22, 1151–1158, doi: 10.1016/j.celrep.2017.12.107 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Xu K, Zhong G & Zhuang X Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science 339, 452–456, doi: 10.1126/science.1232251 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leite SC et al. The Actin-Binding Protein alpha-Adducin Is Required for Maintaining Axon Diameter. Cell Rep 15, 490–498, doi: 10.1016/j.celrep.2016.03.047 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hughes CA & Bennett V Adducin: a physical model with implications for function in assembly of spectrin-actin complexes. J Biol Chem 270, 18990–18996, doi: 10.1074/jbc.270.32.18990 (1995). [DOI] [PubMed] [Google Scholar]
- 13.Abdi KM & Bennett V Adducin promotes micrometer-scale organization of beta2-spectrin in lateral membranes of bronchial epithelial cells. Mol Biol Cell 19, 536–545, doi: 10.1091/mbc.E07-08-0818 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li X, Matsuoka Y & Bennett V Adducin preferentially recruits spectrin to the fast growing ends of actin filaments in a complex requiring the MARCKS-related domain and a newly defined oligomerization domain. J Biol Chem 273, 19329–19338, doi: 10.1074/jbc.273.30.19329 (1998). [DOI] [PubMed] [Google Scholar]
- 15.Robledo RF et al. Targeted deletion of alpha-adducin results in absent beta- and gamma-adducin, compensated hemolytic anemia, and lethal hydrocephalus in mice. Blood 112, 4298–4307, doi: 10.1182/blood-2008-05-156000 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vukojevic V et al. A role for alpha-adducin (ADD-1) in nematode and human memory. EMBO J 31, 1453–1466, doi: 10.1038/emboj.2012.14 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gilligan DM et al. Targeted disruption of the beta adducin gene (Add2) causes red blood cell spherocytosis in mice. Proc Natl Acad Sci U S A 96, 10717–10722, doi: 10.1073/pnas.96.19.10717 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yaguchi H et al. Sez6l2 regulates phosphorylation of ADD and neuritogenesis. Biochem Biophys Res Commun 494, 234–241, doi: 10.1016/j.bbrc.2017.10.047 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Bednarek E & Caroni P beta-Adducin is required for stable assembly of new synapses and improved memory upon environmental enrichment. Neuron 69, 1132–1146, doi: 10.1016/j.neuron.2011.02.034 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Ruediger S et al. Learning-related feedforward inhibitory connectivity growth required for memory precision. Nature 473, 514–518, doi: 10.1038/nature09946 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Zhang X et al. Cell-Type-Specific Alternative Splicing Governs Cell Fate in the Developing Cerebral Cortex. Cell 166, 1147–1162 e1115, doi: 10.1016/j.cell.2016.07.025 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fietz SA et al. Transcriptomes of germinal zones of human and mouse fetal neocortex suggest a role of extracellular matrix in progenitor self-renewal. Proc Natl Acad Sci U S A 109, 11836–11841, doi: 10.1073/pnas.1209647109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robinson JT et al. Integrative genomics viewer. Nat Biotechnol 29, 24–26, doi: 10.1038/nbt.1754 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaech S & Banker G Culturing hippocampal neurons. Nature protocols 1, 2406–2415, doi: 10.1038/nprot.2006.356 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Lek M et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291, doi: 10.1038/nature19057 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oliva M et al. The impact of sex on gene expression across human tissues. Science 369, doi: 10.1126/science.aba3066 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vuong CK, Black DL & Zheng S The neurogenetics of alternative splicing. Nat Rev Neurosci 17, 265–281, doi: 10.1038/nrn.2016.27 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joshi R & Bennett V Mapping the domain structure of human erythrocyte adducin. The Journal of biological chemistry 265, 13130–13136 (1990). [PubMed] [Google Scholar]
- 29.Mische SM, Mooseker MS & Morrow JS Erythrocyte adducin: a calmodulin-regulated actin-bundling protein that stimulates spectrin-actin binding. J Cell Biol 105, 2837–2845, doi: 10.1083/jcb.105.6.2837 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gardner K & Bennett V Modulation of spectrin-actin assembly by erythrocyte adducin. Nature 328, 359–362, doi: 10.1038/328359a0 (1987). [DOI] [PubMed] [Google Scholar]
- 31.Lorenzo DN et al. betaII-spectrin promotes mouse brain connectivity through stabilizing axonal plasma membranes and enabling axonal organelle transport. Proc Natl Acad Sci U S A 116, 15686–15695, doi: 10.1073/pnas.1820649116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cousin MA et al. Pathogenic SPTBN1 variants cause an autosomal dominant neurodevelopmental syndrome. Nat Genet 53, 1006–1021, doi: 10.1038/s41588-021-00886-z (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhong G et al. Developmental mechanism of the periodic membrane skeleton in axons. Elife 3, doi: 10.7554/eLife.04581 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iossifov I et al. De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299, doi: 10.1016/j.neuron.2012.04.009 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parikshak NN et al. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 540, 423–427, doi: 10.1038/nature20612 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shibasaki T et al. PTB deficiency causes the loss of adherens junctions in the dorsal telencephalon and leads to lethal hydrocephalus. Cereb Cortex 23, 1824–1835, doi: 10.1093/cercor/bhs161 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Bosia M et al. ADDing a piece to the puzzle of cognition in schizophrenia. Eur J Med Genet 59, 26–31, doi: 10.1016/j.ejmg.2015.12.012 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Cusi D et al. Polymorphisms of alpha-adducin and salt sensitivity in patients with essential hypertension. Lancet 349, 1353–1357, doi: 10.1016/S0140-6736(97)01029-5 (1997). [DOI] [PubMed] [Google Scholar]
- 39.Morrison AC, Bray MS, Folsom AR & Boerwinkle E ADD1 460W allele associated with cardiovascular disease in hypertensive individuals. Hypertension 39, 1053–1057, doi: 10.1161/01.hyp.0000019128.94483.3a (2002). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Under institutional privacy policies and institutional review boards, further data and experimental details are available upon request.