

Abstract

The pyrimidine core has been utilized extensively to construct kinase inhibitors, including eight FDA-approved drugs. Because the pyrimidine hinge-binding motif is accommodated by many human kinases, kinome-wide selectivity of resultant molecules can be poor. This liability was seen as an advantage since it is well tolerated by many understudied kinases. We hypothesized that nonexemplified aminopyrimidines bearing side chains from well-annotated pyrimidine-based inhibitors with off-target activity on understudied kinases would provide us with useful inhibitors of these lesser studied kinases. Our strategy paired mixing and matching the side chains from the 2- and 4-positions of the parent compounds with modifications at the 5-position of the pyrimidine core, which is situated near the gatekeeper residue of the binding pocket. Utilizing this approach, we imparted improved kinome-wide selectivity to most members of the resultant library. Importantly, we also identified potent biochemical and cell-active lead compounds for understudied kinases like DRAK1, BMP2K, and MARK3/4.
Introduction
Pyrimidines represent an important building block in the medicinal chemistry arsenal. Compounds bearing a pyrimidine core have proven to be bioactive and exhibit diverse pharmacology, including anticonvulsant, analgesic, sedative, antidepressive, antipyretic, anti-inflammatory, antiviral, anti-HIV, antimicrobial, and antitumor activities.1 Pyrimidines are very useful as kinase scaffolds that employ a nitrogen to make key hydrogen bonds with the conserved hinge region found in nearly all human kinases. Aminopyrimidines substituted with an NH in the 2-position can make an additional hydrogen bond with the kinase hinge. To date, eight FDA-approved kinase inhibitors employ a pyrimidine as the key kinase hinge-binding motif.2
The work we describe here stems from the synergistic convergence of two separate interests that were satisfied through diversification of the pyrimidine scaffold. The first of these interests centers around the generation and use of kinase inhibitors as tools to build deeper understanding of signaling in neurodegenerative disease. TBK1, a kinase with links to amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), Huntington’s disease, and Alzheimer’s disease (AD), is potently inhibited by several pyrimidine-bearing compounds.3−12 A second interest of ours is identification of high-quality tool molecules for understudied kinase targets. Our pursuit of this interest relies on parallel chemical tool and kinase assay development, efforts that are supported in part by the NIH Illuminating the Druggable Genome (IDG) program. The IDG program aims to catalyze the characterization of all proteins through stimulating research around those that are most poorly studied.13 One arm of the IDG program supports illumination of the dark kinome, which includes development of high-quality chemical tools for these understudied kinases. As we examined the data available for literature pyrimidine-based TBK1 inhibitors, we noticed a range of understudied kinases that were also inhibited by these compounds.
Several of the understudied kinases inhibited by pyrimidines also have been genetically implicated in driving neurodegenerative diseases. The MARK family of kinases, for example, phosphorylates tau protein in its repeat domain and thereby regulates its affinity for microtubules, affecting the aggregation of tau into neurofibrillary tangles. Observations of AD brains show a strong correlation between cognitive dysfunction and cortical neurofibrillary tangle density.14−16 Mutations in tau have also been shown to cause a form of FTD.16 Furthermore, with a characterized role in dendrite branching and spine development, understudied kinase AAK1 is suggested to play a role in several neurodegenerative disorders, including AD and ALS.14,17,18
Figure 1 shows the structures and kinome-wide profiling data generated at DiscoverX (scanMAX or KINOMEscan) for three TBK1-targeting aminopyrimidines.19 The data for TBK1 inhibitors MRT67307 and BX-912 (designed for PDK1 but potent inhibitor of TBK1) are already in the literature (LINCS database).20−24 These two compounds were screened at 10 μM (Figure 1). GSK8612 was recently disclosed by GlaxoSmithKline as a potent and selective TBK1 inhibitor.25 We opted to survey the kinome-wide selectivity of GSK8612 at DiscoverX at 1 μM (Figure 1). While the screening concentrations are different, it is apparent that these scaffolds differentially and potently inhibit many kinases across the kinome and that selectivity can be augmented through structural changes.
Figure 1.

Structures and broad selectivity profiling of aminopyrimidines that served as the basis for library design. All WT kinases inhibited > 90% at 10 μM by MRT67307 and BX-912 and all WT kinases inhibited > 65% at 1 μM by GSK8612 are shown.
Several kinases potently inhibited by aminopyrimidines like BX-912, MRT67307, and/or GSK8612 are members of the IDG nominated list of dark kinases. Thus, this scaffold was considered an excellent starting point from which to design high-quality chemical tools. Development of these tools will enable elucidation of the function of those kinases that have suffered from a dearth of characterization, including those on the IDG list. High-quality chemical tools will also enable further characterization of kinase-mediated signaling in neurodegenerative disease and facilitate the validation of therapeutic hypotheses.
Results and Discussion
Compound Design and Synthesis
Our library design involved mixing and matching the side chains and cores from published aminopyrimidine inhibitors MRT67307, BX-912, and GSK8612 to furnish 21 total analogs. Specifically, we paired mixing and matching the side chains from the 2- and 4-positions of the parent compounds with modifications at the 5-position of the pyrimidine core as shown in the box in Scheme 1. Seven final compounds were prepared with X = H, seven with X = Br, and seven with X = cyclopropyl (Figure 2). Analogs of BX-912 and MRT67307 (and the parent compounds themselves) that varied only at the 5-position (box in Scheme 1) were not prepared. A great deal of effort has been dedicated to making close structural analogs of these two compounds, and we did not want our work to be redundant. Since it was not commercially available at the time and the most selective of the parent scaffolds, GSK8612 and variants with X = H and cyclopropyl were all synthesized. The method used to prepare these analogs is outlined in Scheme 1. Briefly, taking advantage of the inherent reactivity of 2,4-dichloropyrimidines, iterative nucleophilic aromatic displacements were executed, and specific brominated compounds (GSK8612, 6, 9, 10, and 13) were subsequently dehalogenated to furnish all final compounds.
Scheme 1. Library Design and Preparation Strategy.

Step A: Pyrimidine 1, amine 1, DIPEA, ethanol, −10 °C to 50 °C. Step B: Aminopyrimidine 2, amine 2, dioxane × HCl, butanol, 80 °C. Step C: Aminopyrimidine X, 5% Pd/C, H2, TEA, methanol, r.t.
Figure 2.

Library of aminopyrimidine analogs prepared.
We had several expected outcomes from our strategy of mixing and matching the side chains and cores from well exemplified pyrimidine-based inhibitors. First, we intended to develop more narrowly selective compounds by incorporating these distinct side chains and 5-position modifications into new compounds. Next, we wanted to generate preliminary structure–activity relationships (SAR) for several understudied kinases. In doing so, we aimed to leverage these scaffolds with known inhibitory activity of dark kinases to identify more optimal chemical starting points for development of high-quality chemical tools. Finally, we wanted to develop focused SAR around the 5-position (box in Scheme 1), which has not been systematically investigated in the literature yet and is proposed to play a key role in dictating both potency and selectivity due to its proximity to the kinase gatekeeper residue (Figure 3).
Figure 3.
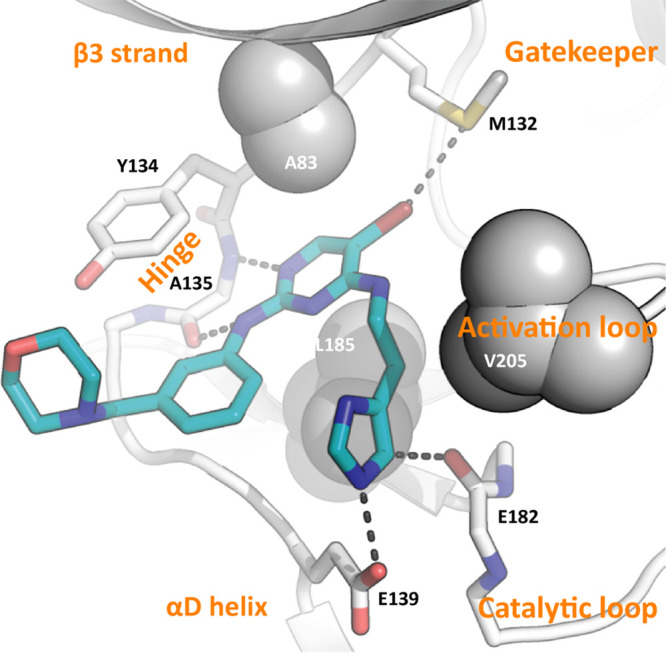
Structural studies support optimization for understudied MARK kinases. X-ray crystallography structure of human MARK3 in complex with aminopyrimidine 9. Compound 9 (PDB code: 7P1L) is shown in teal stick representation. The kinase structural elements mediating the binding are labeled in orange. Parts of the G-rich loop, including I62, were made transparent to provide a better view of the interactions. Key interactions are indicated as dashed lines.
Targeted Kinase Inhibition Profiling
We selected a small panel of representative kinases against which to profile our library of 21 aminopyrimidine analogs. These kinases include some of the original targets for which pyrimidine-based inhibitors were prepared (JAK2, IKKε, TBK1, and ULK1), a more well-studied kinase that is potently inhibited by many analogs within this structural class (AURKB), and several understudied kinases, many of which are both on the IDG list (AAK1, BMP2K, DRAK1–2, MARK1–4, MLK1, MLK3, and NUAK1) and of interest in the neuroscience space.14,15,17,26−29 We profiled our aminopyrimidine series and the parent aminopyrimidines that influenced our design against this kinase panel at a single concentration (1 μM) in radiometric enzymatic assays at Eurofins at the Km = ATP for each kinase. Table S1 shows the results of this study, where % control is reported for each compound for each kinase and lower values indicate greater inhibition. The column labeled “no. kinases ≤ 10 PoC in enzyme panel” in Table 1 captures the number of kinases in this 16-member panel inhibited ≥ 90% by each compound.
Table 1. Kinase Panel Profiling of Aminopyrimidine Library.
| compound | no. kinases ≤ 10 PoC in enzyme panel | S10 (1 μM)b | no. kinases with PoC < 10c |
|---|---|---|---|
| 3 | 0 | 0.005 | 2 |
| 4 | 1 | 0.027 | 11 |
| 5 | 1 | 0.005 | 2 |
| 6 | 4 | 0.047 | 19 |
| 7 | 7 | 0.154 | 62 |
| 8 | 0 | 0.002 | 1 |
| 9 | 13 | 0.233 | 94 |
| 10 | 0 | 0.069 | 28 |
| 11 | 1 | 0.007 | 3 |
| 12 | 0 | 0.002 | 1 |
| 13 | 4 | 0.03 | 12 |
| 14 | 6 | 0.067 | 27 |
| 15 | 0 | 0 | 0 |
| 16 | 0 | 0.002 | 1 |
| 17 | 0 | 0.007 | 3 |
| 18 | 16 | 0.223 | 90 |
| 19 | 0 | 0.007 | 3 |
| 20 | 1 | 0.022 | 9 |
| 21 | 1 | 0.002 | 1 |
| 22 | 9 | 0.074 | 30 |
| MRT67307 | 12 | N.T.a | N.T.a |
| MRT68921 | 13 | N.T.a | N.T.a |
| BX-795 | 16 | N.T.a | N.T.a |
| BX-912 | 14 | N.T.a | N.T.a |
| GSK8612 | 1 | 0.02 | 8 |
N.T.: not tested.
S10 (1 μM): Percentage of screened kinases with PoC values < 10 at 1 μM.
Number of kinases with PoC values < 10 at 1 μM.
We found that, with the exception of two analogs (compounds 9 and 18), all compounds in our library inhibited fewer kinases in this custom kinase panel than published compounds BX-912, BX-795 (analog of BX-912), MRT67307, and MRT68921 (analog of MRT67307).20−23 In most cases, we demonstrated a significant decrease in the number of kinases potently inhibited by our aminopyrimidine analogs. This becomes more meaningful when we consider that the kinases selected in our custom panel represent some of the most common off-target kinases inhibited by the parent compounds. Gratifyingly, unlike the published compounds from the BX and MRT series that elicited potent inhibition of nearly all kinases in the panel, our compounds have cleaned-up profiles, and we were able to dial out inhibition of certain kinases through structural modification. We were pleased to observe potent inhibition of several understudied kinases by analogs that inhibited fewer kinases in this panel, including BMP2K by 6; AAK1, DRAK1, DRAK2, and MARK1 by 7; AAK1 and BMP2K by 13; and MARK1, MARK3, MARK4, MLK1, and NUAK1 by 14.
When we examined the data generated via screening in the Eurofins kinase panel versus compound structures for our synthetic library, we noticed some predominating trends. When varying the 5-position (X in Figure 2), more kinases were tolerant of the bromo substituent, and thus those analogs bearing X = Br inhibited the most kinases in this panel. When X = Br, an average of 6.4 kinases (range of 0–16 kinases) were inhibited ≥ 90%. The average number of kinases inhibited was calculated by adding the number of kinases inhibited ≤ 10 percent of control (PoC) in the enzyme panel (Table 1, column 2) and dividing by the total number of compounds. For X = Br, this works out to be a sum of 45, divided by 7 total compounds for an average of 6.4 kinases. We will use this equation to calculate and report average kinase inhibition throughout this section. Switching to X = H resulted in compounds that inhibited the fewest kinases in the series, with an average of only 0.1 kinases (range of 0–1 kinase), demonstrating ≥ 90% inhibition in the Eurofins panel at 1 μM. Finally, cyclopropyl at position X was somewhere between H and Br in terms of number of kinases inhibited, with an average of 2.7 kinases (range of 0–9 kinases) inhibited ≥ 90%. When considering the amine side chains in the 2- and 4-positions, some general inhibition trends were also observed. Incorporation of the sulfonamide-bearing side chain in the pyrimidine 4-position (R1 position, Scheme 1) resulted in compounds that inhibited the fewest number of kinases: an average of 1.3 kinases (range of 0–4 kinases) ≥ 90% at 1 μM. In the pyrimidine 2-position, the substituted pyrazole is the R2 substituent that resulted in aminopyrimidines with the narrowest inhibition profiles: an average of 1.1 kinases (range of 0–7 kinases) ≥ 90%. The ortho-methyl group on the pyrazole ring likely contributes to this enhanced inhibition profile when compared to other side chains at the R2 position. While some of the analogs in our library, including those with a 5-position H and/or 4-position sulfonamide, did not inhibit any kinases in our smaller panel, it is difficult to extrapolate whether this translates to loss of all kinase inhibitory activity or a narrower selectivity profile unless broader kinome-wide screening is executed.
Library-Wide Cellular Target Engagement Studies
The data generated by screening our aminopyrimidine analogs in the panel of kinases at Eurofins motivated follow-up cell-based studies. We used cellular target engagement assays to determine whether potent enzyme inhibition corresponded with potent binding in cells. The NanoBRET assay offers a method through which cellular penetrance and binding of a compound to its kinase target in cells can be simultaneously assessed.30 Given the consistently potent inhibition of TBK1 by many compounds in our library (Table S1) and our interest in this kinase, we profiled the entire library using the TBK1 NanoBRET assay in dose–response format (Table 2). We found that potent enzymatic inhibition of TBK1 did not always translate to potent engagement of TBK1 in cells. We note that orthogonal assay formats, such an enzymatic and cellular target engagement assay, can sometimes yield different results and, for this reason, that its best to assess kinase inhibition via multiple methods. All compounds that demonstrated at least 50% inhibition at 10 μM when tested using the TBK1 NanoBRET assay in the first dose–response experiment were followed up with two additional replicates. Two members of our library, 7 and 18, and GSK8612 demonstrated submicromolar IC50 values in the TBK1 NanoBRET assay. These three compounds demonstrated PoC values ≤ 6 in the TBK1 enzymatic assay (Table S1), making them among the most potent inhibitors tested in this assay of those that we synthesized. The validation of GSK8612 as a potent, cell-active compound targeting TBK1 aligns well with the recent publication that described its development and characterization, including validation of its activity in multiple cell-based assays.25 Further, submicromolar activity in a NanoBRET assay has translated to phenotypic results for multiple chemical probes and is one part of the chemical probe criteria defined for SGC-nominated chemical probes.17,31,32
Table 2. NanoBRET Profiling of Entire Aminopyrimidine Library.
| IC50 values (nM) |
||||
|---|---|---|---|---|
| compound | DRAK1b | MARK3c | MARK4c | TBK1b |
| 3 | >10000 | >10000 | >10000 | >10000 |
| 4 | 874 ± 103 | >10000 | >10000 | 2680 ± 230 |
| 5 | >10000 | >10000 | >10000 | 7456 |
| 6 | >10000 | >10000 | >10000 | >10000 |
| 7 | 126 ± 27.6 | >10000 | 3537 | 321 ± 2.49 |
| 8 | >10000 | >10000 | >10000 | >10000 |
| 9 | 638 ± 91.1 | 872 | 214 | >10000 |
| 10 | >10000 | >10000 | 8445 | >10000 |
| 11 | >10000 | >10000 | >10000 | 2050 ± 196 |
| 12 | >10000 | >10000 | >10000 | >10000 |
| 13 | >10000 | >10000 | >10000 | 6170 ± 2190 |
| 14 | 3622 | 4015 | 808 | >10000 |
| 15 | >10000 | >10000 | >10000 | >10000 |
| 16 | >10000 | >10000 | >10000 | >10000 |
| 17 | 2311 | >10000 | >10000 | >10000 |
| 18 | 4.47 ± 0.406 | 137 | 72 | 338 ± 22.5 |
| 19 | >10000 | >10000 | >10000 | >10000 |
| 20 | >10000 | >10000 | >10000 | >10000 |
| 21 | >10000 | >10000 | >10000 | >10000 |
| 22 | 58.2 ± 3.21 | 2001 | 509 | 2580 ± 215 |
| MRT67307 | 43.2 ± 2.69 | 328 ± 38.5 | 319 | N.T.a |
| MRT68921 | N.T.a | 95 | N.T.a | N.T.a |
| BX-795 | N.T.a | 515 | N.T.a | N.T.a |
| BX-912 | N.T.a | 3333 | 338 ± 4.5 | 2327 |
| GSK8612 | >10000 | >10000 | >10000 | 339 ± 39.5 |
N.T.: not tested.
Compounds tested in singlicate (n = 1) in dose–response where error not shown, and compounds with SEM tested in triplicate (n = 3) in dose–response.
Compounds tested in singlicate (n = 1) in dose–response where error not shown, and compounds with SEM tested in duplicate (n = 2) in dose–response.
Shifting our attention to understudied kinases, we also observed that some of our compounds potently inhibited DRAK1, MARK3, and MARK4 (Table S1). As part of the IDG program, we have interest in developing high-quality chemical tools to help elucidate the function of these poorly characterized kinases. For our aminopyrimidine series, dose–response NanoBRET analysis yielded five analogs (4, 7, 9, 18, and 22) and MRT67307 with submicromolar activity in the DRAK1 NanoBRET assay. We repeated the NanoBRET assay in dose–response for these six compounds and found all to maintain activity over three replicates. Gratifyingly, 7, 9 18, 22, and MRT67307 were the most potent compounds in the DRAK1 enzymatic assay (Table S1), and all demonstrated PoC ≤ 9. Finally, the entire series was tested in the MARK3 and MARK4 NanoBRET assays. We found four compounds (9, 14, 18, and 22) with submicromolar IC50 values in the MARK4 NanoBRET assay, two of which (9 and 18) also demonstrated submicromolar IC50 values in the MARK3 NanoBRET assay. Several of the parent compounds also had submicromolar IC50 values in the MARK3/4 NanoBRET assays. For MARK3, the four most potent compounds in the MARK3 enzymatic assay, all with PoC ≤ −1 (Table S1), were the most efficacious in the MARK3 NanoBRET assay (Table 2). For MARK4, compounds 9, 18, MRT67307, and BX-912 were among the most potent compounds in the MARK4 enzymatic assay (Table S1, PoC ≤ 5), and all demonstrated MARK4 NanoBRET IC50 values < 400 nM. In general, we noted very good correlation between the orthogonal enzymatic (Table S1) and cellular target engagement (Table 2) assay formats for these understudied kinases. Based on our 16-kinase enzyme inhibition panel, we felt confident that some of these understudied kinase chemical leads inhibited fewer kinases than their parent compounds, and we chose to assess their kinome-wide selectivities to determine whether they require further optimization in our pursuit of high-quality chemical tools.
Assessment of Kinome-Wide Selectivity
The promising inhibition profiles of our aminopyrimidine series in the custom enzymatic assay panel motivated a broader survey to ascertain the kinome-wide selectivity of our library. All 21 novel analogs were screened at 1 μM via the DiscoverX scanMAX platform, which includes 403 wild-type (WT) human kinases. A selectivity score (S10) for each compound is included in Table 1, representative of the percentage of kinases that exhibit binding with a percent of control (PoC) < 10 at 1 μM. A final column is included in Table 1 that converts this S10 (1 μM) value into the number of kinases bound with a PoC < 10 in the DiscoverX panel. Figure S1 details the specific WT kinases that bound with a PoC < 10 at 1 μM in the DiscoverX scanMAX panel for each aminopyrimidine analog.
This more comprehensive analysis of selectivity surfaced many findings. We learned that trends within our smaller curated enzymatic kinase panel (16 kinases) were generally maintained in this larger profiling effort. Compounds that inhibited the fewest kinases in the Eurofins enzymatic panel were largely those that demonstrated the most favorable selectivity profile in the DiscoverX scanMAX screening. We identified several pyrimidine-based kinase inhibitors with useful selectivity. To provide some context on a useful selectivity threshold for tool compounds, we include kinase inhibitors with an S10 (1 μM) < 0.04 in our kinase chemogenomic set (KCGS) since they are the compounds that when screened can be more easily used to correlate phenotype with kinase target.33,34 Based on their selectivity scores, 14 of 21, or 67%, of our novel inhibitors were found to be KCGS eligible. We confirmed that kinases potently inhibited in the smaller, biased enzymatic panel were also inhibited in the DiscoverX profiling and identified additional kinases that were differentially inhibited by certain analogs, providing fodder for future projects.
When comparing the results from our smaller enzymatic screening with kinome-wide screening, it is apparent that results do not correlate perfectly. It is not unexpected that these orthogonal assay types, enzymatic and binding, would yield slightly different results. General structural trends observed when analyzing the smaller kinase panel, however, were generally conserved in broader profiling. The most selective compounds, inhibiting an average of 2.6 of 403 WT kinases, bear a 5-position H (8, 12, 15, 16, 17, 19, 20). As described for the smaller enzyme panel, average kinase inhibition here is being calculated by summing the number of kinases with PoC < 10 in the kinome-wide profiling for each compound (Table 1, column 4) and then dividing by the total number of compounds. For 5-position H, this works out to be 18 divided by 7, which is equal to an average of 2.6 kinases inhibited. The 5-position cyclopropyl aminopyrimidines (3, 4, 5, 11, 14, 21, and 22) inhibited an average of 10.9 of 403 WT kinases, making them the next most selective when considering just the 5-position substituent. Finally, the remaining compounds (6, 7, 9, 10, 13, 18, and GSK8612) with a 5-position bromo are among the least selective compounds when screened broadly, inhibiting an average of 44.7 of 403 WT kinases. Moving to the 4-position amine side chain, the aminopyrimidine analogs bearing a sulfonamide side chain (5, 6, 11, 12, 13, 16, 19, 21, and GSK8612) were among the most selective compounds profiled and only inhibited an average of 5.6 of 403 WT kinases. Similarly, the 2-position pyrazole side chain imparted some selectivity to analogs 3, 4, 5, 7, 8, 10, 12, 15, and GSK8612, resulting in inhibition of an average of 12.8 of 403 WT kinases when broadly screened.
Taken together, our NanoBRET profiling (Table 2) and kinome-wide screening (Table 1) enabled us to confirm that GSK8612 is the most potent in cells, selective (S10 (1 μM) = 0.02), and the most useful TBK1 inhibitor from all that we tested. We did not improve upon the activity of this published compound through these synthetic efforts. Next, of the DRAK1 active compounds in the NanoBRET assay, only 4 was selective enough to be considered a valuable tool molecule. Given its S10 (1 μM) = 0.027 and submicromolar IC50 value in the DRAK1 NanoBRET assay, compound 4 was nominated as a dark kinase tool that can be used to help illuminate the function of DRAK1. Information for compound 4 has been posted on the Dark Kinase Knowledgebase.35 Finally, selectivity profiling shows that all compounds with submicromolar IC50 values in the MARK3/4 NanoBRET assays require further optimization to reduce the number of off-target kinases that are potently inhibited in addition to MARK3/4 (S10 (1 μM) = 0.067–0.233). Efforts are ongoing to improve the selectivity of analogs we identified as cell-active in the MARK3/4 NanoBRET assays. The weak cellular potency of 4 for DRAK1 and 14/22 for MARK4 make it difficult to judge the selectivity window offered by these compounds. They should be considered chemical starting points in need of further optimization to improve upon this potency while maintaining or improving kinome-wide selectivity.
Orthogonal Validation of scanMAX Results
The scanMAX assay identifies potential targets for our compounds across the kinome. We chose to follow up and validate the scanMAX kinase binding results by further testing of selected actives in enzyme inhibition assays. Our choice of kinases for follow-up varied depending on the selectivity of the compound in question, as measured by S10 (1 μM). Highly selective compounds that are also potent on their kinase target have the potential to be chemical probe candidates, and, as such, we chose to execute thorough enzyme profiling to validate or invalidate potential off-targets as well as further quantify potency. Thus, for the five compounds in our library with an S10 (1 μM) < 0.002 (15, 8, 12, 16, and 21), we followed-up on all kinases with PoC < 35% at 1 μM in the scanMAX platform and/or PoC < 50% in our initial custom enzymatic profiling panel at Eurofins (Table S1). One exception to this was exclusion of AURKA follow-up for 21. For the six compounds in our library with an S10 (1 μM) = 0.005–0.02 (3, 5, 11, 17, 19, and GSK8612), we carried out enzymatic assays on all kinases with PoC < 20% at 1 μM in the scanMAX platform and/or PoC < 50% in our initial enzymatic profiling at Eurofins (Table S1). Given our interest in identifying chemical leads, a few additional understudied kinases with PoC < 35% at 1 μM in the scanMAX platform for these six compounds were also selected for follow-up. Lastly, for a final four compounds (20, 4, 13, and 6) with S10 (1 μM) = 0.022–0.047, we selected only certain kinases with PoC < 20% at 1 μM in the scanMAX platform and/or PoC < 50% in our initial profiling at Eurofins (Table S1) for targeted follow-up, with a bias toward kinases that were frequently inhibited by other analogs in the series as well as understudied kinases of interest. All follow-up enzymatic assays were executed at the Km = ATP for each respective kinase. Results from these studies combined with the single-concentration enzymatic results from Table S1 are displayed in Table 3. Compounds in Table 3 are listed in order of their kinome-wide selectivity scores from most (15) to least (6) selective.
Table 3. Combined Enzymatic Data.
| compound | potently activea | moderately activeb | weakly activec |
|---|---|---|---|
| 15 | MYLK2 = 479 nM | DRAK2: 91% | |
| 8 | MKNK2 = 2866 nM, DRAK2: 60%, YANK2 > 10000 nM | ||
| 12 | CSF1R = 233 nM | ||
| 16 | TYK2 = 33 nM | CSF1R = 305 nM | JAK2: 62% |
| 21 | TBK1 = 128 nM, TRKA = 477 nM | BMPR2 = 2918 nM, AURKB: 61%, JAK2: 107% | |
| 3 | LRRK2 = 89 nM | DRAK2: 59%, NIM1 = 4398 nM, MYLK2 = 1119 nM | |
| 5 | TBK1 = 149 nM | IKKε = 1199 nM | |
| 11 | TBK1 = 187 nM, TRKA = 153 nM | JAK2: 87%, IKKε = 1314 nM, AURKB: 51% | |
| 17 | DRAK1 = 325 nM, DRAK2 = 161 nM, AAK1 = 390 nM, SIK2 = 481 nM | MKNK2 = 769 nM, TYK2 = 720 nM, JAK2 > 10000 nM, BMP2K: 56% | |
| 19 | CSF1R = 91 nM | TYK2 = 276 nM | JAK2: 54%, ERBB2 > 10000 nM, TBK1 = 2737 nM |
| GSK8612 | TBK1 = 37 nM | LRRK2 = 159 nM, MAP2K5: 11%, CSF1R = 264 nM | IKKε = 552 nM, DAPK3 > 10000 nM, NUAK2 = 1151 nM, ULK3 = 946 nM, MKNK2 = 1369 nM |
| 20 | AURKB: 5% | NUAK1 = 176 nM, SIK2 = 185 nM | BMP2K: 58%, DRAK1: 78%, DRAK2: 65%, JAK2: 81%, ACVR1 = 537 nM, BMPR1B = 9028 nM |
| 4 | LRRK2 = 19 nM, DRAK2 = 62 nM | IKKε = 216 nM, DRAK1 = 202 nM, ULK3 = 343 nM, MKNK2 = 380 nM, TBK1 = 192 nM, MAP2K5: 12% | ULK2 = 1742 nM, ULK1: 58% |
| 13 | AURKB: 6%, BMP2K = 40 nM, AAK1 = 74 nM, TYK2 = 48 nM, STK16 = 82 nM | TBK1 = 192 nM, JAK2: 12%, BMPR2 = 488 nM, TRKA = 283 nM, NUAK1 = 317 nM | NUAK2 = 900 nM, CSNK2A2 = 2269 nM, PIP5K1A > 10000 nM, MKNK2 = 1009 nM |
| 6 | TBK1 = 66 nM, JAK2: 3%, BMP2K = 38 nM, STK16 = 88 nM, TYK2 = 67 nM, AURKB: 7%, AAK1 = 80 nM | CSF1R = 192 nM, TRKA = 158 nM, NUAK1 = 171 nM | PRP4 > 10000 nM, PIP5K1A > 10000 nM, CSNK2A2 = 2304 nM, CSNK2A1 > 10000 nM, ULK3 = 1371 nM |
Potently active: IC50 value < 100 nM or < 10% control at 1 μM (Table S1). All IC50 values determined in duplicate.
Moderately active: IC50 value 200–500 nM or 10–49% control at 1 μM (Table S1). All IC50 values determined in duplicate.
Weakly active: IC50 value > 500 nM or > 49% control at 1 μM (Table S1). All IC50 values determined in duplicate.
In examining the data generated and collected in Table 3, some interesting trends emerge. The most selective compounds (S10 (1 μM) < 0.02: 15, 8, 12, 16, and 21) that were comprehensively profiled via enzymatic assays potently inhibited 0–1 kinase (potently active, Table 3). We classify the kinases potently inhibited by these compounds as more well-studied. Except for 17 and GSK8612, the same group of most selective compounds (S10 (1 μM) < 0.02) inhibited 0–2 kinases with moderate potency (Table 3). Some less selective compounds (13 and 6) for which we did selective enzymatic follow-up were found to be potent inhibitors of 5–7 kinases, including members of the understudied NAK family (AAK1, BMP2K, and STK16).17 Several of the aminopyrimidines tested demonstrated IC50 values of 200–500 nM or inhibition equal to 10–49% control at 1 μM for understudied kinases. Given their kinome-wide selectivity scores and modest potency, these compounds represent potential chemical leads for the development of chemical tools to study these poorly characterized kinases. Many kinases were assigned the weakly active designation based on weak potency in the respective enzymatic assays. In some cases, such as with compounds 5 and 13, two kinases that share high structural homology were inhibited with differential potencies. For compound 5, there is an 8-fold difference in potency for TBK1 and IKKε, and for compound 13, there is a nearly 3-fold difference between NUAK1 and NUAK2.
Importantly, we see that the compounds with submicromolar IC50 values in the respective NanoBRET assays corresponded with enzymatic inhibition IC50 values < 250 nM for GSK8612 (TBK1 = 37 nM) and compound 4 (DRAK1 = 202 nM). TBK1, LRRK2, DRAK1/2, CSF1R, TYK2, and TRKA were identified as frequently inhibited kinases by our aminopyrimidine series. This list is comprised of some kinases that pyrimidines are known to potently inhibit (TBK1 and TYK2)36,37 as well as kinases that represent new targets for optimization. Analysis of the compounds that inhibit these kinases could inform next steps in new chemistry to develop specific SAR for these kinases. Before embarking on next synthetic steps, the narrow selectivity profiles of exemplars within our series coupled with potent enzymatic data motivated interrogation of the cell-based activity of some of these compounds in the respective NanoBRET assays.
Selective NanoBRET Assay Follow-Up
For compounds in Table 3 that inhibited kinases with potency < 500 nM, we elected to determine their cellular target engagement via the NanoBRET assay. No more than two kinases were evaluated per compound. Several single digit micromolar inhibitors of specific kinases were identified among these selective aminopyrimidine compounds. Table 4 shows that these compounds were most cell-active in the BMP2K NanoBRET assay. We were excited to identify that weaker inhibition of MYLK2 in the enzymatic assay (IC50 value = 479 nM) translated to single digit micromolar activity in the MYLK2 NanoBRET assay. Compound 15 potently inhibits the fewest kinases of the compounds that we synthesized (Tables 1 and 3), but demonstrates modest MYLK2 inhibitory potency. Given the paucity of literature around MYLK2 and the distinct lack of compound optimization efforts directed at MYLK2, 15 represents one of the most promising chemical starting points for MYLK2 chemical probe development. The single digit micromolar activity in cells will need to be improved upon via medicinal chemistry optimization to yield analogs with a better selectivity window versus off-target kinases. We also observed that compounds that exhibited potent activity in the LRRK2 enzyme assay (3 and GSK8612, IC50 values < 160 nM) and in the TYK2 enzyme assay (16 and 19, IC50 values < 280 nM) did not have any activity in the respective NanoBRET assays when tested at concentrations up to 10 μM. The disparity between our enzymatic potencies and NanoBRET IC50 values prompted us to further investigate compound properties that might impact cell permeability.
Table 4. Selected NanoBRET Follow-Up.
| IC50 values (nM)a |
||||||
|---|---|---|---|---|---|---|
| compound | BMP2K | CSF1R | LRRK2 | MYLK2 | NUAK1 | TYK2 |
| 3 | >10000 | |||||
| 6 | 1020 | 7010 | ||||
| 12 | >10000 | |||||
| 13 | 3000 | >10000 | ||||
| 15 | 3670 | |||||
| 16 | >10000 | >10000 | ||||
| 19 | 5060 | >10000 | ||||
| GSK8612 | >10000 | >10000 | ||||
Compounds tested in singlicate (n = 1) in dose–response.
Assessment of Compound Properties
With exceptions, we observed > 30–300-fold losses in potency when considering enzymatic versus NanoBRET activities. This was true for NanoBRET values reported both in Tables 2 and 4. A < 10-fold loss in potency was only observed in four cases: GSK8612 for TBK1, 15 for MYLK2, and 4 and 17 for DRAK1. The overall trend of biochemical activity not translating to cellular potency made us curious about the physicochemical properties of our aminopyrimidines. To address this, we evaluated the kinetic solubility and permeability (PAMPA) of our library of compounds and included the parent compounds as well (Table 5).
Table 5. Kinetic Solubility and PAMPA Assay Results.
| compound | kinetic solubility (μM) | Pe (cm/s) |
|---|---|---|
| 3 | 171.0 | 3.89 × 10–7 |
| 4 | 190.1 | 5.08 × 10–6 |
| 5 | 33.2 | <LOQa |
| 6 | 4.7 | <LOQa |
| 7 | 40.3 | 3.73 × 10–6 |
| 8 | 174.6 | 2.39 × 10–6 |
| 9 | 159.0 | 1.30 × 10–6 |
| 10 | 195.2 | 1.51 × 10–6 |
| 11 | 118.7 | 7.09 × 10–7 |
| 12 | 167.7 | <LOQa |
| 13 | 20.0 | <LOQa |
| 14 | 173.6 | 5.61 × 10–7 |
| 15 | 176.9 | 6.23 × 10–8 |
| 16 | 147.0 | <LOQa |
| 17 | 169.3 | 8.24 × 10–8 |
| 18 | 135.6 | 8.85 × 10–7 |
| 19 | 176.5 | 4.56 × 10–7 |
| 20 | 199.7 | 1.66 × 10–7 |
| 21 | 85.6 | 2.60 × 10–6 |
| 22 | 164.0 | 5.97 × 10–7 |
| MRT67307 | 164.6 | 2.60 × 10–6 |
| BX-912 | 155.5 | 1.39 × 10–7 |
| GSK8612 | 173.1 | <LOQa |
<LOQ: Below limit of quantitation.
Results in Table 5 demonstrated that our compounds were generally very soluble. With the exception of 5, 6, 7, 11, 13, 16, 18, and 21, the measured solubility was estimated to be > 75% of the dose concentration, and thus the actual solubility may be higher than Table 5 reflects. As nearly all compounds demonstrated solubility > 10 μM, they were not considered poorly soluble, and this was eliminated from consideration as driving their poor cellular potency. The permeability data, however, were a bit more varied for this aminopyrimidine library. Six compounds (5, 6, 12, 13, 16, and GSK8612) were below the limit of quantitation (LOQ) and/or precipitated in the assay media, making it impossible to determine their permeability. Several of these compounds were among the analogs with the lowest kinetic solubility concentrations as well. For reference, Pe (permeability coefficient) values < 1.50 × 10–6 cm/s correlate with the human fraction absorbed < 80% and is a generally accepted cutoff for low permeability. In addition to the compounds already mentioned that were below the LOQ, this cutoff adds the majority of our library as well as some parent compounds to a low permeability category: 3, 9, 11, 14, 15, 17–20, 22, and BX-912.
If we start to consider which structural elements could be compromising the solubility and/or permeability of our compounds, some trends emerge. The sulfonamide side chain is present in most of the least soluble compounds (5, 6, 11, 13, 16, and 21) and all poorly permeable compounds (5, 6, 12, 13, 16, and GSK8612). As compounds with suboptimal solubility (7) or poor permeability (GSK8612) still proved active in our NanoBRET assays, just looking at half of the data is not sufficient. None of the compounds with both problematic solubility and permeability (5, 6, 13, and 16) were active in our NanoBRET assays, leading us to conclude that these two factors together point to compounds that are poorly cell permeable. Since the majority of compounds did not fall into this final category, consideration of physicochemical properties did not explain our cell-based results.
It is worth noting for compounds/kinases where enzymatic data has been published, the control compounds used in the NanoBRET assays can be considered as a benchmark. For CSF1R, dasatinib has a published enzymatic IC50 = 0.57 nM, and we determined its NanoBRET IC50 to be 18.3 nM. For NUAK1, BX-795 has a published IC50 = 5 nM, and we determined its NanoBRET IC50 to be 187 nM.19 In both cases, single digit or subnanomolar enzymatic IC50 values translated to submicromolar NanoBRET IC50 values, and a ∼34-fold loss in potency was observed when moving to the cell-based assay. While not universal, it appears that for this set of compounds and kinase targets, exceptional enzymatic potency is key to achieving cellular potency to overcome the more than 30-fold drop-off in cellular potency.
Structural Studies and Lead Optimization Plan for MARK Subfamily
The MARK3 cocrystal structure with 9 corroborates the high affinity that this compound demonstrates in the MARK3 NanoBRET assay (Table 2). Figure 3 shows that the activation segment of MARK3 folds back and packs against compound 9 via residue V205, while maintaining a DFG-in conformation. Several key hydrogen bonds are made between 9 and the binding pocket. In addition, several hydrogen bonds are present between the binding pocket and the 2- and 4-position side chains of 9. The imidazole nitrogen(s) can interact with the main chain carbonyl of I62 in the G-rich loop as well as E182 in the catalytic loop and E139 in the αD helix. It has also been suggested that, although weak, C–H–O hydrogen bonds can exist.38 While inhibitor conformations have been synchronized, the free rotation of the 4-position side chain allows the imidazole to rotate, which perturbs some of these interactions. These hydrogen bonds, therefore, do not simultaneously coexist, but rather different hydrogen-bond patterns emerge due to free rotation of the imidazole ring. With respect to the 2-position side chain, K60 (not shown) is at a distance of 3.4 Å and Y134 is at a distance of 3.2 Å from the morpholine ring. Since the morpholine ring is highly flexible, these distances represent the proximity of the mentioned residues to the ring rather than to a particular atom within it. Unsupported geometries as well as the flexibilities of both residues and the morpholine ring, however, suggest that K60 and Y134 do not form strong hydrogen bonds with the morpholine ring. A weak and likely transient interaction between Y134 and the morpholine ring on 9 is shown in the Figure 3 pose. Essential interactions are made between the 3-position nitrogen of the pyrimidine core and 2-position NH with A135 in the MARK3 hinge. Importantly, the 5-position bromo is oriented such that it makes an interaction with the sulfur within gatekeeper residue M132.
It is interesting to consider this cocrystal structure versus those solved for MRT67307 with TBK1 (PDB codes: 4IM0 and 4IWQ) and ULK2 (PDB code: 6QAU), and BX-320, a close structural analog of BX-912, with PDK1 (PDB code: 1Z5M).23,39,40 The gatekeeper in TBK1 and ULK2, like in MARK3, is a methionine (M86 in TBK1 and M85 in ULK2), while in PDK1 the gatekeeper is a leucine (L86).23,39,40 The proximity of the cyclopropyl group in MRT67307 or bromine in BX-320 to the gatekeeper residue varies, supporting this position influences binding preference and excludes molecules that bear a group that is too large at the 5-position of the pyrimidine ring. Furthermore, the discussion around M85 in ULK2 noted some flexibility of this residue, suggesting a certain degree of plasticity of the back pocket when accommodating bulky hydrophobic residues such as the cyclopropyl group in MRT67307.40 Hinge-binding interactions are maintained in all cases between the pyrimidine nitrogen at position 1 and the amino side chain NH at position 2, supporting that this essential part of the molecule cannot be modified without substantial losses in binding affinity. When MRT67307 binds to ULK2, the pendant morpholine on the 2-position side chain makes hydrogen-bonding interactions via a water molecule with D95 of ULK2.40 The morpholine is not shown to hydrogen bond in TBK1 structures with MRT67307, which could partially explain the loss of affinity of 9 for TBK1 (Table 2) and potent binding of 9 to ULK2 (Figure S1).39 Although the interactions between the morpholine ring and K60/Y134 of MARK3 are weak, these residues may be key in the development of more specific MARK3 binders. If the flexibilities of this 2-position side chain and/or morpholine ring are reduced, it may allow for formation of stronger hydrogen bonds and increase MARK3 binding affinity.
Also, in comparing the structure of 9 bound to MARK3 versus both the ULK2 and TBK1 structures with MRT67307, we observe that the binding mode of 9 induced a “folded” conformation of the activation segment that partially protrudes into the ATP binding pocket. Comparison of the various cocrystal structures suggests that this could be due to a more compact structure of 9, in which the 2-position imidazole stacks onto the 4-position phenyl ring. MRT67307, in comparison, adopts a more elongated binding conformation that likely prevents this “folded” conformation. Six compounds from our library have the 4-position imidazole side chain: 3, 9, 10, 14, 15, and 20. Of these, only 9 and 14 demonstrate submicromolar affinity for MARK3 and/or MARK4 in the corresponding NanoBRET assays. In addition to the 2-position imidazole, both 9 and 14 share the 4-position phenyl ring in their structures. It is possible that the 2-position imidazole cannot efficiently stack with the 4-position pyrazole shared by 3, 10, and 15 to allow for binding in the MARK3 pocket created when the activation segment folds. This unique compact binding mode that seems to be tied to inhibitor side chain substituents should be considered in future design efforts.
If we examine and compare the residues that line or surround the active sites of these kinases (TBK1, ULK2, and PDK1) versus some of our understudied kinases of interest (DRAK1, MARK3/4, and BMP2K; Figure 4), we observe that the gatekeeper is either a methionine or leucine in all cases (gatekeeper, Figure S2). The key hinge-binding residue in all these kinases is maintained as either a cysteine or alanine (hinge donor and outer hydrogen-bond acceptor, Figure S2). Finally, the morpholine-anchoring residue is coming from the αD helix in ULK2 rather than the conserved hinge tyrosine (hinge, Figure S2) utilized to anchor the morpholine in our MARK3 structure, supporting that this residue may be key to the design of MARK-family specific molecules.
Figure 4.

Promising chemical leads for understudied kinases.
Using the MARK3 cocrystal structure as our guide, we will build on the subfamily selectivity observed for compounds 14 and 22 versus 9 and 18. Given that the 5-position of these compounds seems to dictate both subfamily and kinome-wide selectivity, we hypothesize that further exploration of this position is warranted. Y134 within the hinge of MARK family members is not maintained by all kinases, and thus adding interactions with this residue may impart additional selectivity through precluding binding with other kinases where it is another residue (phenylalanine in TBK1, for example). We aim to use these learnings to develop chemical tools that demonstrate specificity within the MARK family in addition to improved kinome-wide selectivity.
Conclusions
In summary, we provide details related to the synthesis and extensive biological evaluation of a library of aminopyrimidines. We have shown that selectivity can be built into the pyrimidine scaffold through design. Several cell-active compounds were discovered that exhibit submicromolar NanoBRET cellular target engagement IC50 values against kinases including TBK1, DRAK1, MARK3, and MARK4. Three of these kinases (DRAK1, MARK3, and MARK4) are understudied, IDG kinases that need high-quality chemical tools to be able to characterize their function. Examples of compounds with sub- or nearly submicromolar activities for understudied kinases and modest kinome-wide selectivity are included in Figure 4. These aminopyrimidines represent promising chemical starting points in our campaign to identify chemical probes to enable elucidation of the biological function(s) of lesser studied kinases. The cellular potency of these compounds will need to be improved via chemical optimization to furnish compounds more suitable for interrogating the biology of these understudied kinases. The aminopyrimidine represents an alternative chemotype to our recently disclosed AAK1/BMP2K chemical probe and, as such, inhibits a different panel of off-target kinases.17,18 Development of this chemotype is expected to yield a chemical probe with a different kinome-wide inhibition profile and thus slightly different biological activities. Results here reiterate the need to employ orthogonal biochemical and cell-based assays to understand the actual selectivity and potency of kinase inhibitors more fully. The methods described herein offer a path for others to identify and develop high-quality inhibitors for understudied kinases to facilitate illumination of the entire druggable kinome.
Experimental Section
Chemistry. General Information
Reagents were purchased from commercial suppliers and used without further characterization or purification. Temperatures are given in degrees Celsius (°C), and unless otherwise stated, operations were carried out at room or ambient temperature (r.t.), typically around 25 °C. Evaporation of solvent was carried out using a rotary evaporator under reduced pressure with a bath temperature not exceeding 60 °C. Thin-layer chromatography (TLC) was used to follow the course of reactions. Intermediates and products exhibited satisfactory 1H NMR and/or microanalytical data. The following conventional abbreviations are also used: equivalents (equiv), mmol (millimoles), mg (milligrams), and h (hours). Reactions were carried out under a blanket of nitrogen unless otherwise stated. Compounds were visualized using a UV (ultraviolet) lamp (254 nM). 1H and 13C NMR spectra were collected in DMSO-d6, acetonitrile-d4, chloroform-d, or methanol-d4 and recorded on Varian Inova 400 Megahertz (MHz), Bruker DRX 500 MHz, Bruker Avance III 400 MHz, Varian VNMRS 500 MHz, or Agilent ProPulse 600 MHz spectrometers, noting the magnet strength in all cases. Peak positions are given in parts per million (ppm) and calibrated based upon the shift of the indicated solvent. Coupling constants (J values) are expressed in hertz (Hz), and multiplicities are reported as follows: singlet (s), doublet (d), doublet(s) of doublets (dd/ddd) or triplets (dt) or quartets (dq), triplet (t), triplet(s) of doublets (td/ttd), quartet (q), pentet (p), pentet of doublets (pd), heptet (h), and multiplet (m). An asterisk (*) was added after the line listing of spectral data for compounds where a single peak could not be unambiguously assigned in the 13C spectrum. Double asterisks (**) were added after the line listing of spectral data for compounds where a single peak as part of a multiplet could not be unambiguously assigned in the 13C spectrum. Purity was assessed via LC–MS using an Agilent mass spectrometer (column: Agilent Poroshell 120 SB-C18, 4.6 mm × 30 mm, 2.7 μm with UHPLC Guard Infinity Lab Poroshell 120 SB-C18, 4.6 mm × 5 mm, 2.7 μm). Syntheses of key intermediates and several final products were performed by ChemSpace LLC. Identity and purity of all final products were confirmed upon arrival.
General Procedure for the Synthesis of Compound B: Procedure A
To a solution of compound 1 (1 equiv) in ethanol (10–20 mL) were added dropwise DIPEA (2 mL) and amine 1 (1 equiv) at −10 °C. The resulting mixture was heated to r.t. then stirred at 50 °C for 16 h. Solvent was next evaporated from the reaction mixture and to the resulting material was added water (30 mL). The obtained precipitate was filtered, washed with water, isopropanol, and hexane, and dried under vacuum at 50 °C to give compounds 2a–2g (amount, yield, purity), which were used in the next step without further purification.
N-(3-((2-Chloropyrimidin-4-yl)amino)propyl)cyclobutanecarboxamide (2a)
330 mg, 50% yield, 95% purity by LC–MS.
N-(3-((5-Bromo-2-chloropyrimidin-4-yl)amino)propyl)cyclobutanecarboxamide (2b)
2.20 g, 50% yield, 95% purity by LC–MS.
N-(3-((2-Chloro-5-cyclopropylpyrimidin-4-yl)amino)propyl) cyclobutanecarboxamide (2c)
570 mg, 67% yield, 95% purity by LC–MS.
4-(((5-Bromo-2-chloropyrimidin-4-yl)amino)methyl)benzenesulfonamide (2d)
6.40 g, 55% yield, 95% purity by LC–MS.
4-(((2-Chloro-5-cyclopropylpyrimidin-4-yl)amino)methyl)benzenesulfonamide (2e)
850 mg, 50% yield, 95% purity by LC–MS.
N-(2-(1H-Imidazol-4-yl)ethyl)-5-bromo-2-chloropyrimidin-4-amine (2f)
2.50 g, 25% yield, 95% purity by LC–MS.
N-(2-(1H-Imidazol-4-yl)ethyl)-2-chloro-5-cyclopropylpyrimidin-4-amine (2g)
420 mg, 50% yield, 95% purity by LC–MS.
General Procedure for the Synthesis of Compounds 3–11, 13, 14, 17, 18, 21, 22, and GSK8612: Procedure B
To a solution of compound 2 (1 equiv) and amine 2 (1 equiv) in butanol (2–5 mL) was added dioxane × HCl (10%w/w) (0.5–2 mL), and the resulting mixture was stirred at 80 °C for 16 h. The reaction mixture was next neutralized with aqueous ammonia and concentrated under vacuum. The resulting material was purified by preparative HPLC (2–7 min, 35–70% methanol (0.1% ammonium hydroxide), 30 mL/min; column: YMC-ACTUS TRIART C18, 20 mm × 100 mm, 5 μm) to give final compounds 3–11, 13, 14, 17, 18, 21, 22, and GSK8612 (amount, yield).
N4-(2-(1H-Imidazol-4-yl)ethyl)-5-cyclopropyl-N2-(3-methyl-1-(2,2,2-trifluoroethyl)-1H-pyrazol-4-yl)pyrimidine-2,4-diamine (3)
71.0 mg, 36% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.86 (s, 1H), 8.11 (s, 2H), 7.56 (s, 2H), 6.84 (d, J = 16.5 Hz, 2H), 5.02–4.91 (m, 2H), 3.63 (q, J = 6.8 Hz, 2H), 2.81 (t, J = 7.4 Hz, 2H), 2.16 (s, 3H), 1.41 (td, J = 10.7, 8.6, 5.9 Hz, 1H), 0.78 (d, J = 8.0 Hz, 2H), 0.40 (t, J = 5.1 Hz, 2H). 13C NMR (151 MHz, acetonitrile-d3) δ 160.51, 156.33, 150.93, 138.16, 134.55*, 132.59, 121.60** (q, J = 280.0 Hz), 120.39, 120.11, 113.02*, 107.40, 49.73 (q, J = 33.9 Hz), 38.56, 24.68, 8.17, 5.05, 1.84. HPLC purity: 95.0%. HRMS (ESI) ([M + H]+) Calcd for C18H22F3N8: 407.1920, found: 407.1918.
N-(3-((5-Cyclopropyl-2-((3-methyl-1-(2,2,2-trifluoroethyl)-1H-pyrazol-4-yl)amino)pyrimidin-4-yl)amino)propyl)cyclobutanecarboxamide (4)
57.0 mg, 11% yield. 1H NMR (400 MHz, chloroform-d) δ 8.01 (s, 1H), 7.70 (s, 1H), 6.21 (s, 1H), 5.94 (s, 1H), 5.59 (s, 1H), 4.62 (q, J = 8.5 Hz, 2H), 3.53 (q, J = 5.9 Hz, 2H), 3.36 (q, J = 6.3 Hz, 2H), 2.96 (p, J = 8.2 Hz, 1H), 2.31–2.19 (m, 5H), 2.18–2.10 (m, 2H), 2.03–1.82 (m, 2H), 1.77 (p, J = 6.7 Hz, 2H), 1.51–1.40 (m, 1H), 1.25 (s, 1H), 0.91 (dq, J = 5.7, 3.9 Hz, 2H), 0.53–0.45 (m, 2H). 13C NMR (101 MHz, chloroform-d) δ 175.72, 162.75, 157.86, 152.71, 140.54, 123.13 (q, J = 280 Hz), 122.38, 121.92, 110.04, 52.97 (q, J = 34.7 Hz), 39.91, 37.24, 36.04, 29.84, 25.43, 18.09, 10.99, 7.41, 4.56. HPLC purity: 100%. HRMS (ESI) ([M + H]+) Calcd for C21H29F3N7O: 452.2386, found: 452.2385.
4-(((5-Cyclopropyl-2-((3-methyl-1-(2,2,2-trifluoroethyl)-1H-pyrazol-4-yl)amino)pyrimidin-4-yl)amino)methyl)benzenesulfonamide (5)
74.0 mg, 15% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.01 (s, 1H), 7.79–7.70 (m, 3H), 7.59 (s, 1H), 7.46 (d, J = 8.0 Hz, 2H), 7.38–7.33 (m, 1H), 7.26 (s, 2H), 4.87 (q, J = 9.2 Hz, 2H), 4.68 (d, J = 6.1 Hz, 2H), 2.08 (s, 3H), 1.52 (s, 1H), 0.83 (d, J = 8.0 Hz, 2H), 0.48 (d, J = 5.0 Hz, 2H). 13C NMR (151 MHz, acetonitrile-d3) δ 160.37, 156.47, 151.51, 143.05, 139.49, 138.98, 125.27, 123.81, 123.0 (q, J = 279 Hz), 120.72, 119.89, 107.57, 49.90 (q, J = 34.0 Hz), 41.03, 8.12, 5.08, 1.98. HPLC purity: 100%. HRMS (ESI) ([M + H]+) Calcd for C20H23F3N7O2S: 482.1588, found: 482.1582.
4-(((5-Bromo-2-((3-(morpholinomethyl)phenyl)amino)pyrimidin-4-yl)amino)methyl)benzenesulfonamide (6)
128 mg, 24% yield. 1H NMR (400 MHz, DMSO-d6)δ 9.19 (s, 1H), 8.06 (s, 1H), 7.73 (dd, J = 15.3, 7.0 Hz, 4H), 7.60 (s, 1H), 7.50 (d, J = 8.0 Hz, 2H), 7.43 (d, J = 8.2 Hz, 1H), 7.26 (s, 2H), 7.10 (t, J = 7.8 Hz, 1H), 6.81 (d, J = 7.4 Hz, 1H), 4.70 (d, J = 6.1 Hz, 2H), 3.52 (d, J = 4.8 Hz, 4H), 3.17 (d, J = 5.2 Hz, 2H), 2.28 (s, 4H). 13C NMR (126 MHz, acetonitrile-d3) δ 158.62, 156.44, 145.33*, 144.50, 141.96*, 140.19, 133.48, 128.40, 127.58, 126.11, 122.70, 121.44, 119.72, 93.03*, 66.46, 62.67, 60.58, 53.36, 43.67, 24.26*. HPLC purity: 98.4%. HRMS (ESI) ([M + H]+) Calcd for C22H26BrN6O3S: 533.0970, found: 533.0955.
N-(3-((5-Bromo-2-((3-methyl-1-(2,2,2-trifluoroethyl)-1H-pyrazol-4-yl)amino)pyrimidin-4-yl)amino)propyl)cyclobutanecarboxamide (7)
83.0 mg, 17% yield. 1H NMR (400 MHz, chloroform-d) δ 7.95 (s, 2H), 6.34 (s, 1H), 6.04–5.99 (m, 1H), 5.58–5.53 (m, 1H), 4.64 (q, J = 8.5 Hz, 2H), 3.49 (q, J = 6.2 Hz, 2H), 3.35 (q, J = 6.4 Hz, 2H), 2.98 (p, J = 8.5 Hz, 1H), 2.33–2.20 (m, 5H), 2.19–2.08 (m, 2H), 2.04–1.80 (m, 2H), 1.76 (p, J = 6.3 Hz, 2H). 13C NMR (151 MHz, acetonitrile-d3) δ 173.06, 156.65, 156.48, 153.76, 138.90, 121.50** (q, J = 279.8), 120.99, 119.51, 119.43, 49.88 (q, J = 34.2 Hz), 37.32, 35.47, 33.62, 26.93, 22.78, 15.55, 8.19.HPLC purity: 97.8%. HRMS (ESI) ([M + H]+) Calcd for C18H24BrF3N7O: 490.1178, found: 490.1176.
N-(3-((2-((3-Methyl-1-(2,2,2-trifluoroethyl)-1H-pyrazol-4-yl)amino)pyrimidin-4-yl)amino)-propyl)cyclobutanecarboxamide (8)
75.0 mg, 15% yield. 1H NMR (400 MHz, chloroform-d) δ 7.98 (s, 1H), 7.83 (d, J = 5.9 Hz, 1H), 6.33 (s, 1H), 5.82 (d, J = 5.6 Hz, 1H), 5.56 (s, 2H), 4.59 (q, J = 8.5 Hz, 2H), 3.41–3.25 (m, 4H), 2.95 (p, J = 8.6 Hz, 1H), 2.31–2.18 (m, 5H), 2.13 (q, J = 9.1, 8.3 Hz, 2H), 2.01–1.79 (m, 3H), 1.72 (p, J = 6.3 Hz, 2H). 13C NMR (151 MHz, acetonitrile-d3) δ 172.67, 161.10, 157.78, 153.04*, 138.64*, 125.45*, 121.63** (q, J = 280), 120.78, 119.87, 118.98, 49.87 (q, J = 34.0 Hz), 37.31, 33.92, 26.98, 22.74, 15.56, 8.19. HPLC purity: 100%. HRMS (ESI) ([M + H]+) Calcd for C18H25F3N7O: 412.2073, found: 412.2069.
N4-(2-(1H-Imidazol-4-yl)ethyl)-5-bromo-N2-(3-(morpholinomethyl)phenyl)pyrimidine-2,4-diamine (9)
190 mg, 11% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.83 (s, 1H), 9.21 (s, 1H), 8.01 (s, 1H), 7.75 (s, 1H), 7.63–7.54 (m, 2H), 7.14 (t, J = 7.8 Hz, 2H), 6.83 (d, J = 7.5 Hz, 2H), 3.68 (q, J = 6.9 Hz, 2H), 3.54 (t, J = 4.6 Hz, 4H), 3.31 (s, 2H), 2.87–2.78 (m, 2H), 2.29 (s, 4H). 13C NMR (101 MHz, methanol-d4) δ 163.42*, 158.65, 158.56, 155.08, 140.37, 137.30, 134.65, 128.08, 122.67, 120.21, 118.15, 116.24*, 92.60, 66.28, 63.13, 53.22, 40.58, 26.34. HPLC purity: 95.9%. HRMS (ESI) ([M + H]+) Calcd for C20H25BrN7O:458.1304, found: 458.1300.
N4-(2-(1H-Imidazol-5-yl)ethyl)-5-bromo-N2-(3-methyl-1-(2,2,2-trifluoroethyl)-1H-pyrazol-4-yl)pyrimidine-2,4-diamine (10)
325 mg, 17% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.84 (s, 1H), 8.55 (s, 1H), 8.09 (s, 1H), 7.95 (s, 1H), 7.55 (s, 1H), 7.08 (s, 1H), 6.87 (s, 1H), 4.98 (s, 2H), 3.60 (q, J = 6.9, 6.5 Hz, 2H), 2.78 (t, J = 7.1 Hz, 2H), 2.16 (s, 3H). 13C NMR (151 MHz, acetonitrile-d3) δ 156.58, 156.46, 153.77, 144.88*, 143.51*, 138.62, 132.63, 121.54 (q, J = 279.9 Hz), 120.99, 119.43, 117.93, 49.74 (q, J = 34.1 Hz), 38.91, 24.47, 8.21. HPLC purity: 96.6%. HRMS (ESI) ([M + H]+) Calcd for C15H17BrF3N8:445.0712, found: 445.0709.
4-(((5-Cyclopropyl-2-((3-(morpholinomethyl)phenyl)amino)pyrimidin-4-yl)amino)methyl)-benzenesulfonamide (11)
18.0 mg, 4% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.83 (s, 1H), 7.75 (d, J = 8.1 Hz, 2H), 7.66 (s, 2H), 7.52 (d, J = 8.0 Hz, 2H), 7.50–7.42 (m, 3H), 7.25 (s, 2H), 7.06 (t, J = 7.9 Hz, 1H), 6.75 (d, J = 7.7 Hz, 1H), 4.75 (d, J = 5.9 Hz, 2H), 3.57–3.50 (m, 5H), 3.17 (d, J = 5.5 Hz, 2H), 2.32–2.25 (m, 4H), 1.61–1.50 (m, 1H), 0.88–0.83 (m, 2H), 0.52 (d, J = 4.7 Hz, 2H). 13C NMR (101 MHz, methanol-d4) δ 162.60, 158.31, 152.19, 144.97, 142.02, 140.52, 137.00, 128.03, 127.12, 125.89, 122.35, 120.11, 118.04, 110.30, 66.24, 63.12, 53.13, 43.32, 39.00, 7.14, 3.92. HPLC purity: 100%. HRMS (ESI) ([M + H]+) Calcd for C25H31N6O3S:495.2178, found: 495.2162.
N-(3-((5-Bromo-4-((4-sulfamoylbenzyl)amino)pyrimidin-2-yl)amino)phenyl)pyrrolidine-1-carboxamide (13)
383 mg, 5% yield. 1H NMR (500 MHz, methanol-d4) δ 7.95 (d, J = 2.2 Hz, 1H), 7.80 (dd, J = 8.4, 2.2 Hz, 2H), 7.71 (d, J = 2.4 Hz, 1H), 7.49–7.43 (m, 2H), 7.08 (dt, J = 4.6, 1.8 Hz, 2H), 6.95 (td, J = 4.3, 1.8 Hz, 1H), 4.77 (d, J = 2.0 Hz, 2H), 3.42 (dt, J = 6.8, 3.5 Hz, 4H), 1.94 (h, J = 2.5 Hz, 4H). 13C NMR (101 MHz, methanol-d4) δ 158.61, 156.19, 155.36, 153.60, 144.23, 141.95, 140.23, 139.55, 128.02, 127.27, 125.75, 114.94, 114.62, 112.77, 92.26, 45.57, 43.56, 25.01. HPLC purity: 92.6%. HRMS (ESI) ([M + H]+) Calcd forC22H25BrN7O3S:546.0923, found: 546.0914.
N4-(2-(1H-Imidazol-4-yl)ethyl)-5-cyclopropyl-N2-(3-(morpholinomethyl)phenyl)pyrimidine-2,4-diamine (14)
73.0 mg, 24% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.82 (s, 1H), 8.85 (s, 1H), 7.81 (s, 1H), 7.64–7.54 (m, 3H), 7.11 (t, J = 7.8 Hz, 1H), 6.90–6.81 (m, 2H), 6.77 (d, J = 7.5 Hz, 1H), 3.69 (q, J = 6.7 Hz, 2H), 3.54 (t, J = 4.6 Hz, 4H), 3.30–3.20 (m, 1H), 3.17 (d, J = 3.9 Hz, 1H), 2.85 (t, J = 7.2 Hz, 2H), 2.29 (d, J = 4.8 Hz, 4H), 1.44 (q, J = 4.8, 2.9 Hz, 1H), 0.79 (dt, J = 8.3, 3.1 Hz, 2H), 0.51–0.38 (m, 2H). 13C NMR (151 MHz, acetonitrile-d3) δ 160.24, 156.35, 150.71, 139.02, 136.45, 132.53, 126.05, 119.42, 116.81, 115.42, 114.90, 108.19, 64.35, 60.97, 51.28, 38.46, 24.40, 5.12, 1.85. HPLC purity: 100%. HRMS (ESI) ([M + H]+) Calcd for C23H30N7O:420.2512, found: 420.2506.
N-(3-((4-((3-(Cyclobutanecarboxamido)propyl)amino)pyrimidin-2-yl)amino)phenyl)pyrrolidine-1-carboxamide (17)
19.0 mg, 10% yield. 1H NMR (400 MHz, chloroform-d) δ 7.94 (s, 1H), 7.82 (s, 1H), 7.24 (s, 1H), 7.22–7.03 (m, 3H), 6.95 (d, J = 8.2 Hz, 1H), 6.13 (s, 2H), 5.80 (d, J = 6.5 Hz, 1H), 5.56 (s, 1H), 3.41 (m, 6H), 3.26 (m, 2H), 2.95–2.78 (m, 1H), 2.19 (d, J = 9.8 Hz, 2H), 2.07–1.89 (m, 9H), 1.68 (d, J = 6.7 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 174.29, 162.95, 160.25, 154.80, 154.49, 141.70, 140.83, 128.16, 113.36, 113.15, 111.41, 98.08, 55.31, 46.08, 39.17, 36.78, 29.55, 25.47, 25.11, 18.22. HPLC purity: 100%. HRMS (ESI) ([M + H]+) Calcd for C23H32N7O2: 438.2617, found: 438.2617.
N-(3-((5-Bromo-4-((3-(cyclobutanecarboxamido)propyl)amino)pyrimidin-2-yl)amino)phenyl)pyrrolidine-1-carboxamide (18)
335 mg, 13% yield. 1H NMR (400 MHz, DMSO-d6) δ 9.07 (s, 1H), 7.97 (s, 1H), 7.87 (t, J = 2.1 Hz, 1H), 7.62 (t, J = 5.4 Hz, 1H), 7.25 (d, J = 8.1 Hz, 1H), 7.06 (t, J = 8.0 Hz, 1H), 7.00–6.91 (m, 2H), 3.42 (q, J = 6.6 Hz, 2H), 3.33 (d, J = 6.4 Hz, 4H), 3.07 (q, J = 6.5 Hz, 2H), 2.93 (p, J = 8.4 Hz, 1H), 2.14–2.02 (m, 4H), 1.96 (q, J = 9.8 Hz, 2H), 1.90–1.79 (m, 4H), 1.78–1.60 (m, 3H). 13C NMR (101 MHz, methanol-d4) δ 176.54, 158.67, 158.61, 155.60, 155.03, 140.53, 139.71, 128.13, 114.73, 114.19, 112.40, 92.61, 45.63, 39.47, 37.91, 36.26, 28.86, 25.06, 24.84, 17.58. HPLC purity: 98.1%. HRMS (ESI) ([M + H]+) Calcd for C23H31BrN7O2: 516.1723, found: 516.1707.
N-(3-((5-Cyclopropyl-4-((4-sulfamoylbenzyl)amino)pyrimidin-2-yl)amino)phenyl)pyrrolidine-1-carboxamide (21)
16.0 mg, 8% yield. 1H NMR (400 MHz, methanol-d4) δ 7.80 (d, J = 8.3 Hz, 2H), 7.70 (t, J = 2.1 Hz, 1H), 7.47–7.41 (m, 3H), 7.22 (t, J = 8.0 Hz, 1H), 7.15 (dt, J = 8.0, 1.5 Hz, 1H), 6.97 (dt, J = 8.0, 1.6 Hz, 1H), 4.82 (s, 2H), 3.46–3.41 (m, 4H), 1.98–1.92 (m, 4H), 1.57 (td, J = 7.9, 4.0 Hz, 1H), 1.02–0.97 (m, 2H), 0.61–0.56 (m, 2H). 13C NMR (101 MHz, methanol-d4) δ 163.32, 155.39, 152.73, 142.84, 142.38, 140.50, 139.89, 136.96, 128.85, 127.60, 125.88, 117.51, 117.11, 115.24, 112.53, 45.66, 43.99, 25.01, 23.61, 6.92, 4.27. HPLC purity: 100%. HRMS (ESI) ([M + H]+) Calcd for C25H30N7O3S:508.2131, found: 508.2116.
N-(3-((4-((3-(Cyclobutanecarboxamido)propyl)amino)-5-cyclopropylpyrimidin-2-yl)amino)-phenyl)pyrrolidine-1-carboxamide (22)
16.0 mg, 5% yield. 1H NMR (400 MHz, methanol-d4) δ 7.81 (t, J = 2.1 Hz, 1H), 7.55 (d, J = 1.1 Hz, 1H), 7.25 (ddd, J = 8.1, 2.2, 1.1 Hz, 1H), 7.15 (t, J = 8.1 Hz, 1H), 6.97 (ddd, J = 8.0, 2.1, 1.0 Hz, 1H), 3.58 (t, J = 6.6 Hz, 2H), 3.47–3.42 (m, 5H), 3.24 (t, J = 6.7 Hz, 2H), 2.96 (pd, J = 8.6, 1.0 Hz, 1H), 2.24–2.14 (m, 2H), 2.10–2.01 (m, 2H), 1.98–1.93 (m, 5H), 1.84–1.75 (m, 3H), 1.46 (ttd, J = 8.1, 5.2, 1.1 Hz, 1H), 0.92–0.87 (m, 2H), 0.49–0.44 (m, 2H). 13C NMR (101 MHz, methanol-d4) δ 176.49, 162.67, 158.29, 155.62, 151.52, 140.97, 139.70, 128.11, 114.28, 113.88, 112.08, 110.40, 45.61, 39.47, 39.00, 37.37, 36.13, 29.14, 25.06, 24.83, 17.57, 7.07, 3.81. HPLC purity: 100%. HRMS (ESI) ([M + H]+) Calcd for C26H36N7O2:478.2930, found: 478.2914.
4-(((5-Bromo-2-((3-methyl-1-(2,2,2-trifluoroethyl)-1H-pyrazol-4-yl)amino)pyrimidin-4-yl)-amino)methyl)benzenesulfonamide (GSK8612)
379 mg, 73% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.43 (s, 1H), 7.98 (s, 1H), 7.73 (d, J = 8.0 Hz, 3H), 7.62–7.53 (m, 1H), 7.41 (s, 2H), 7.27 (s, 2H), 4.90 (q, J = 9.1 Hz, 2H), 4.61 (s, 2H), 2.08 (s, 3H). 13C NMR (151 MHz, acetonitrile-d3) δ 156.82, 156.44, 154.25, 142.15, 139.72, 139.67, 125.39, 123.87, 123.25 (q, J = 281 Hz), 121.58, 119.20, 49.92 (q, J = 34.0 Hz), 41.30, 8.17. HPLC purity: 100%. HRMS (ESI) ([M + H]+) Calcd for C17H18BrF3N7O2S: 520.0378, found: 520.0378.
General Procedure for the Synthesis of Compounds 12, 15, 16, 19, and 20: Procedure C
To a solution of 6, 9, 10, 13, or GSK8612 (1 equiv) in methanol (20 mL) were added 5% Pd/C (0.5 equiv) and triethylamine (TEA, 3 equiv). The resulting mixture was stirred under H2 (hydrogen) atmosphere at r.t. for 16 h. Next the reaction mixture was filtered, and the filtrate was concentrated in vacuo. The crude product was purified via preparative HPLC (2–7 min, 30–55% acetonitrile, 30 mL/min; column: SunFire C18, 19 mm × 100 mm, 5 μm) to give 12, 15, 16, 19, and 20 (amount, yield).
4-(((2-((3-Methyl-1-(2,2,2-trifluoroethyl)-1H-pyrazol-4-yl)amino)pyrimidin-4-yl)amino)methyl)benzenesulfonamide (12)
108 mg, 36% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.18 (s, 1H), 7.87 (s, 1H), 7.77 (dd, J = 8.9, 6.9 Hz, 3H), 7.66 (s, 1H), 7.46 (d, J = 8.0 Hz, 2H), 7.30 (s, 2H), 5.92 (s, 1H), 4.90 (q, J = 9.1 Hz, 2H), 4.57 (s, 2H), 2.11 (s, 3H). 13C NMR (151 MHz, acetonitrile-d3) δ 161.02, 157.98, 153.76, 142.49, 139.69, 139.23, 125.44, 125.24, 123.88, 123.79, 121.45 (q, J = 279 Hz), 121.16, 119.67, 94.31*, 49.96 (q, J = 34.4 Hz), 41.30, 8.11. HPLC purity: 96.6%. HRMS (ESI) ([M + H]+) Calcd for C17H19F3N7O2S:442.1273, found: 442.1270.
N4-(2-(1H-Imidazol-5-yl)ethyl)-N2-(3-methyl-1-(2,2,2-trifluoroethyl)-1H-pyrazol-4-yl)pyrimidine-2,4-diamine (15)
86.0 mg, 10% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.81 (s, 1H), 8.15 (d, J = 35.4 Hz, 2H), 7.73 (s, 1H), 7.53 (s, 1H), 7.18 (s, 1H), 6.83 (s, 1H), 5.85 (d, J = 5.8 Hz, 1H), 4.95 (q, J = 9.2 Hz, 2H), 3.51 (s, 2H), 2.75 (t, J = 7.5 Hz, 2H), 2.16 (s, 3H). 13C NMR (151 MHz, acetonitrile-d3) δ 161.05, 157.72, 153.16*, 138.57*, 132.48, 122.48, 121.55 (q, J = 280 Hz), 120.74, 119.80, 116.93*, 114.19, 49.77 (q, J = 32.4 Hz), 38.55, 24.75, 8.19. HPLC purity: 100%. HRMS (ESI) ([M + H]+) Calcd for C15H18F3N8:367.1607, found: 367.1596.
N-(3-((4-((4-Sulfamoylbenzyl)amino)pyrimidin-2-yl)amino)phenyl)pyrrolidine-1-carboxamide (16)
23.0 mg, 8% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.82 (s, 1H), 7.93 (s, 1H), 7.88 (s, 1H), 7.80 (d, J = 5.8 Hz, 1H), 7.74 (m, 3H), 7.48 (d, J = 8.3 Hz, 2H), 7.26 (s, 2H), 7.18 (s, 1H), 6.99 (t, J = 8.0 Hz, 1H), 6.92 (d, J = 8.7 Hz, 1H), 5.97 (s, 1H), 4.63 (s, 2H), 3.31 (d, J = 7.5 Hz, 4H), 1.82–1.78 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 162.86, 160.14, 155.30*, 154.46, 144.67, 142.92, 141.44, 140.84, 128.18, 127.94, 126.07, 113.46, 113.27, 111.51, 97.85*, 46.09, 40.86, 25.46. HPLC purity: 95.0%. HRMS (ESI) ([M + H]+) Calcd for C22H26N7O3S:468.1818, found: 468.1814.
4-(((2-((3-(Morpholinomethyl)phenyl)amino)pyrimidin-4-yl)amino)methyl)benzenesulfonamide (19)
18.8 mg, 38% yield. 1H NMR (400 MHz, methanol-d4) δ 7.84 (d, J = 8.4 Hz, 2H), 7.77 (d, J = 6.0 Hz, 1H), 7.59 (t, J = 2.0 Hz, 1H), 7.52–7.47 (m, 2H), 7.36 (d, J = 8.2 Hz, 1H), 7.14 (t, J = 7.8 Hz, 1H), 6.90 (dt, J = 7.5, 1.4 Hz, 1H), 6.01 (d, J = 6.0 Hz, 1H), 4.70 (s, 2H), 3.61 (t, J = 4.7 Hz, 4H), 3.35 (s, 2H), 2.36 (t, J = 4.5 Hz, 4H). 13C NMR (101 MHz, methanol-d4) δ 163.16, 159.74, 154.25, 144.48, 142.16, 140.34, 137.04, 128.07, 127.30, 125.95, 122.68, 120.45, 118.38, 97.18, 66.23, 63.11, 53.14, 43.26. HPLC purity: 100%. HRMS (ESI) ([M + H]+) Calcd for C22H27N6O3S: 455.1865, found: 455.1851.
N4-(2-(1H-Imidazol-5-yl)ethyl)-N2-(3-(morpholinomethyl)phenyl)pyrimidine-2,4-diamine (20)
21.0 mg, 19% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.81 (s, 1H), 8.92 (s, 1H), 7.80 (d, J = 16.9 Hz, 2H), 7.61 (s, 1H), 7.54 (s, 1H), 7.24 (s, 1H), 7.12 (t, J = 7.8 Hz, 1H), 6.79 (d, J = 10.4 Hz, 2H), 5.92 (d, J = 5.8 Hz, 1H), 3.54 (m, 6H), 3.31 (s, 2H), 2.79 (m, 2H), 2.29 (s, 4H). 13C NMR (101 MHz, Methanol-d4) δ 163.14, 159.75, 153.70, 140.60, 137.24, 134.91, 134.58, 128.07, 122.50, 120.29, 118.18, 116.44, 97.18, 66.28, 63.20, 53.24, 40.31, 26.50. HPLC purity: 100%. HRMS (ESI) ([M + H]+) Calcd for C20H26N7O: 380.2199, found: 380.2186.
Biological Evaluation: Enzymatic Assays
Eurofins kinase enzymatic radiometric assays were carried out at the Km = ATP at a single concentration (1 μM) in duplicate for each kinase in Table S1. Eurofins kinase enzymatic radiometric assays were carried out at the Km = ATP in dose–response (9-pt curve in duplicate) for each kinase with an IC50 value listed in Table 3. Details about the substrate used, protein constructs, controls, and assay protocol for each kinase assay can be found at the Eurofins Web site: https://www.eurofinsdiscoveryservices.com.
Library-Wide NanoBRET Assays
Human embryonic kidney (HEK293) cells (hypotriploid, female, fetal) were purchased from ATCC and grown in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10% (v/v) fetal bovine serum (FBS, Corning). Cells were incubated in 5% CO2 at 37 °C and passaged every 72 h with trypsin. They were not allowed to reach confluency.
Constructs for NanoBRET measurements of DRAK1 (DRAK1-NLuc), MARK3 (NLuc-MARK3), MARK4 (NLuc-MARK4), and TBK1 (NLuc-TBK1) included in Table 2 were kindly provided by Promega. NanoBRET assays were executed as described previously.17 Preferred NLuc orientations are indicated in parentheses after each construct. Assays were carried out in dose–response as described by the manufacturer using 0.5 μM of tracer K-9 for DRAK1 and MARK3 and 0.5 μM of tracer K-5 for MARK4 and TBK1. Respective tracer titration curves that we generated for DRAK1, MARK3, and MARK4 can be found at https://darkkinome.org/data.35 Tracer titration curves for MARK3, MARK4, and TBK1 can also be found on the Promega Web site.
Kinome Screening
The scanMAX assay platform was used to assess the selectivity of each aminopyrimidine analog at 1 μM at Eurofins DiscoverX Corporation. As described previously, this commercial assay platform screens against 403 WT human kinases and provides percent of control values.19 These percent of control values are captured in Table 1.
Specific NanoBRET Assay Follow-Up
NanoBRET assays for the six kinases in Table 4 were carried out in dose–response in singlicate by Carna Biosciences. Assays were carried out according to the manufacturer’s protocol.
Kinetic Solubility and Permeability (PAMPA)
Kinetic solubility analysis was carried out from 10 mM DMSO stocks of compounds in phosphate buffered saline solution (PBS) at pH 7.4 by Analiza, Inc. Following 24 h incubation in a Millipore solubility filter plate, samples were vacuum filtered, and the filtrates collected for analysis. Filtrates were injected into the nitrogen detector for quantification via total chemiluminescent nitrogen determination (CLND). Filtrates were quantified with respect to a calibration curve generated using standards that span the dynamic range of the instrument. Calculated solubility values are corrected for background nitrogen present in the DMSO and the media.
PAMPA analysis was carried out by Analiza, Inc. using a Corning Gentest Precoated PAMPA plate. DMSO stocks of compound diluted in PBS at pH 7.4 were added to the donor compartment of the plate, PBS at pH 7.4 was added to the acceptor compartment, and the plate was left to incubate for 5h. Both the donor and acceptor compartments were collected and analyzed by CLND. Donor and acceptor samples were quantified using the calibration curve generated using standards that span the dynamic range of the instrument. Measured concentrations are corrected for background nitrogen present in the DMSO and the media. Concentration values from the donor and acceptor compartment are used in the calculation of the effective permeability of the compound. Solubility of the compound is determined experimentally rather than assuming full solubility.
Statistics
Standard error of the mean (SEM) was calculated for NanoBRET assays executed more than once. Calculated SEM is included alongside IC50 values in Table 2.
Crystallization and Structure Determination
The coding sequence for the MARK3 residues 48–366 was cloned into the vector pNIC-CT10HF. Accordingly, the expressed construct comprised a TEV-cleavable His6 tag in its C-terminus. Expression in E. coli Rosetta (DE3) was performed as previously described.41 For purification, the pellet was resuspended in lysis buffer (50 mM HEPES/NaOH pH 7.4, 500 mM NaCl, 0.5 mM TCEP, 5% glycerol), and the cells lysed by sonication. After clearance by centrifugation, the lysate was loaded onto a Ni-NTA column. The bound His6-tagged protein was eluted in lysis buffer containing 300 mM imidazole. TEV cleavage was performed while dialyzing the sample overnight at 4 °C. The cleaved tag, TEV, and contaminating proteins were removed by another Ni-NTA step. Finally, MARK3 was subjected to gel filtration using an AKTA Xpress system combined with an S200 column in KGF150 buffer (20 mM HEPES/NaOH pH 7.4, 150 mM NaCl, 0.5 mM TCEP, 5% glycerol).
100 nL of a solution containing the protein–ligand complex (14 mg/mL MARK348–366, 500 μM 9) was transferred to a 3-well crystallization plate (Swissci), mixed with 50 nL of precipitant solution (0.1 M sodium formate pH 7.0, 24% PEG3350), and incubated at 4 °C. Crystals were spotted after 3 days and did not change appearance after 6 days. They were mounted in precipitant solution cryoprotected with additional 25% ethylene glycol. Data were collected at Swiss Light Source (SLS) X06SA and analyzed, scaled, and merged with the SLS automated data processing (adp) pipeline.42 The structure was solved by molecular replacement with Phaser43 using a MARK3 model as a template (PDB code: 2QNJ)44 and refined with Phenix.45 The model and structure factors have been deposited to the PDB with the code 7P1L (crystallographic parameters are included in Table S2).
Acknowledgments
Constructs for NanoBRET measurements of DRAK1, MARK3, MARK4, and TBK1 were kindly provided by Promega. Coral was used to make the kinome tree depicted in our Table of Contents graphic. Coral was developed in the Phanstiel Lab at UNC: http://phanstiel-lab.med.unc.edu/CORAL.46 We used the TREEspot kinase interaction mapping software to prepare the kinome trees in Figure 1 and Supplemental Information: http://treespot.discoverx.com. We thank ChemSpace LLC for synthetic support and Stefan Knapp for guidance on x-ray crystallographic studies. We thank the Department of Chemistry Mass Spectrometry Core Laboratory at the University of North Carolina for their assistance with mass spectrometry analysis. The Structural Genomics Consortium is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Genentech, Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck KGaA Darmstadt Germany, MSD, Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and Wellcome [106169/ZZ14/Z]. Research reported in this publication was supported in part by the NC Biotechnology Center Institutional Support grant 2018-IDG-1030, NIH 1U24DK116204 and U54AG065187, DoD ALSRP award AL190107, and ALS Association grant ID wa1127.
Glossary
Abbreviations Used
- DIPEA
N,N-diisopropylethylamine
- DMSO
dimethyl sulfoxide
- HCl
hydrochloric acid
- HPLC
high-performance liquid chromatography
- IC50
half maximal inhibitory concentration
- LC–MS
liquid chromatography–mass spectrometry
- Km
Michaelis constant
- LINCS
Library of Integrated Network-Based Cellular Signatures
- NanoBRET
bioluminescence resonance energy transfer using nanoluciferase
- Nluc
nanoluciferase
- NMR
nuclear magnetic resonance
- PAMPA
parallel artificial membrane permeability assay
- Pd/C
palladium on carbon
- v/v
volume for volume
- w/w
weight for weight
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c00440.
Eurofins 16-kinase panel enzymatic data, an additional figure that details kinome-wide selectivities of all compounds, sequence alignment of kinases potently inhibited by aminopyrimidines, X-ray crystallographic data, LC chromatograms, and NMR spectra are included (PDF)
Molecular formula strings (CSV)
The authors declare no competing financial interest.
Supplementary Material
References
- Meeta S.; Nadeem S. A review on biological importance of pyrimidines in the new era. Int. J. Pharm. Pharm. Sci. 2016, 8, 8–21. [Google Scholar]
- Roskoski R. Jr Properties of FDA-approved small molecule protein kinase inhibitors: A 2020 update. Pharmacol. Res. 2020, 152, 104609. 10.1016/j.phrs.2019.104609. [DOI] [PubMed] [Google Scholar]
- Richters A.; Basu D.; Engel J.; Ercanoglu M. S.; Balke-Want H.; Tesch R.; Thomas R. K.; Rauh D. Identification and further development of potent TBK1 inhibitors. ACS Chem. Biol. 2015, 10, 289–298. 10.1021/cb500908d. [DOI] [PubMed] [Google Scholar]
- Llona-Minguez S.; Baiget J.; Mackay S. P. Small-molecule inhibitors of IκB kinase (IKK) and IKK-related kinases. Pharm. Pat. Anal. 2013, 2, 481–498. 10.4155/ppa.13.31. [DOI] [PubMed] [Google Scholar]
- Perrior T. R.; Newton G. K.; Stewart M. R.; Aquil R. Pyrimidine compounds as inhibitors of protein kinases IKK epsilon and/or TBK-1, processes for their preparation, and pharmaceutical compositions containing them. International Patent WO2012010826A1, 2012.
- Muvaffak A.; Pan Q.; Yan H.; Fernandez R.; Lim J.; Dolinski B.; Nguyen T. T.; Strack P.; Wu S.; Chung R.; Zhang W.; Hulton C.; Ripley S.; Hirsch H.; Nagashima K.; Wong K.-K.; Jánne P. A.; Seidel-Dugan C.; Zawel L.; Kirschmeier P. T.; Middleton R. E.; Morris E. J.; Wang Y. Evaluating TBK1 as a therapeutic target in cancers with activated IRF3. Mol. Cancer Res. 2014, 12, 1055–1066. 10.1158/1541-7786.MCR-13-0642. [DOI] [PubMed] [Google Scholar]
- Hutti J. E.; Porter M. A.; Cheely A. W.; Cantley L. C.; Wang X.; Kireev D.; Baldwin A. S.; Janzen W. P. Development of a high-throughput assay for identifying inhibitors of TBK1 and IKKε. PLoS One 2012, 7, e41494 10.1371/journal.pone.0041494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crew A. P.; Raina K.; Dong H.; Qian Y.; Wang J.; Vigil D.; Serebrenik Y. V.; Hamman B. D.; Morgan A.; Ferraro C.; Siu K.; Neklesa T. K.; Winkler J. D.; Coleman K. G.; Crews C. M. Identification and characterization of von Hippel-Lindau-recruiting proteolysis targeting chimeras (PROTACs) of TANK-binding kinase 1. J. Med. Chem. 2018, 61, 583–598. 10.1021/acs.jmedchem.7b00635. [DOI] [PubMed] [Google Scholar]
- Oakes J. A.; Davies M. C.; Collins M. O. TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol. Brain 2017, 10, 5. 10.1186/s13041-017-0287-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freischmidt A.; Wieland T.; Richter B.; Ruf W.; Schaeffer V.; Muller K.; Marroquin N.; Nordin F.; Hubers A.; Weydt P.; Pinto S.; Press R.; Millecamps S.; Molko N.; Bernard E.; Desnuelle C.; Soriani M. H.; Dorst J.; Graf E.; Nordstrom U.; Feiler M. S.; Putz S.; Boeckers T. M.; Meyer T.; Winkler A. S.; Winkelman J.; de Carvalho M.; Thal D. R.; Otto M.; Brannstrom T.; Volk A. E.; Kursula P.; Danzer K. M.; Lichtner P.; Dikic I.; Meitinger T.; Ludolph A. C.; Strom T. M.; Andersen P. M.; Weishaupt J. H. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015, 18, 631–636. 10.1038/nn.4000. [DOI] [PubMed] [Google Scholar]
- Hegde R. N.; Chiki A.; Petricca L.; Martufi P.; Arbez N.; Mouchiroud L.; Auwerx J.; Landles C.; Bates G. P.; Singh-Bains M. K.; Dragunow M.; Curtis M. A.; Faull R. L.; Ross C. A.; Caricasole A.; Lashuel H. A. TBK1 phosphorylates mutant Huntingtin and suppresses its aggregation and toxicity in Huntington’s disease models. EMBO J. 2020, 39, e104671 10.15252/embj.2020104671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheijen J.; van der Zee J.; Gijselinck I.; Van den Bossche T.; Dillen L.; Heeman B.; Gómez-Tortosa E.; Lladó A.; Sanchez-Valle R.; Graff C.; Pastor P.; Pastor M. A.; Benussi L.; Ghidoni R.; Binetti G.; Clarimon J.; de Mendonça A.; Gelpi E.; Tsolaki M.; Diehl-Schmid J.; Nacmias B.; Almeida M. R.; Borroni B.; Matej R.; Ruiz A.; Engelborghs S.; Vandenberghe R.; De Deyn P. P.; Cruts M.; Van Broeckhoven C.; Sleegers K.; et al. Common and rare TBK1 variants in early-onset Alzheimer disease in a European cohort. Neurobiol. Aging 2018, 62, 245.e1–245.e7. 10.1016/j.neurobiolaging.2017.10.012. [DOI] [PubMed] [Google Scholar]
- Rodgers G.; Austin C.; Anderson J.; Pawlyk A.; Colvis C.; Margolis R.; Baker J. Glimmers in illuminating the druggable genome. Nat. Rev. Drug Discovery 2018, 17, 301–302. 10.1038/nrd.2017.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krahn A. I.; Wells C.; Drewry D. H.; Beitel L. K.; Durcan T. M.; Axtman A. D. Defining the neural kinome: strategies and opportunities for small molecule drug discovery to target neurodegnerative diseases. ACS Chem. Neurosci. 2020, 11, 1871–1886. 10.1021/acschemneuro.0c00176. [DOI] [PubMed] [Google Scholar]
- Annadurai N.; Agrawal K.; Džubák P.; Hajdúch M.; Das V. Microtubule affinity-regulating kinases are potential druggable targets for Alzheimer’s disease. Cell. Mol. Life Sci. 2017, 74, 4159–4169. 10.1007/s00018-017-2574-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz J. D.; Haidle A.; Childers K. K.; Zabierek A. A.; Jewell J. P.; Hou Y.; Altman M. D.; Szewczak A.; Chen D.; Harsch A.; Hayashi M.; Warren L.; Hutton M.; Nuthall H.; Su H. P.; Munshi S.; Stanton M. G.; Davies I. W.; Munoz B.; Northrup A. Structure guided design of a series of selective pyrrolopyrimidinone MARK inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 114–120. 10.1016/j.bmcl.2016.08.068. [DOI] [PubMed] [Google Scholar]
- Wells C.; Couñago R. M.; Limas J. C.; Almeida T. L.; Cook J. G.; Drewry D. H.; Elkins J. M.; Gileadi O.; Kapadia N. R.; Lorente-Macias A.; Pickett J. E.; Riemen A.; Ruela-de-Sousa R. R.; Willson T. M.; Zhang C.; Zuercher W. J.; Zutshi R.; Axtman A. D. SGC-AAK1–1: A chemical probe targeting AAK1 and BMP2K. ACS Med. Chem. Lett. 2020, 11, 340–345. 10.1021/acsmedchemlett.9b00399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agajanian M. J.; Walker M. P.; Axtman A. D.; Ruela-de-Sousa R. R.; Serafin D. S.; Rabinowitz A. D.; Graham D. M.; Ryan M. B.; Tamir T.; Nakamichi Y.; Gammons M. V.; Bennett J. M.; Counago R. M.; Drewry D. H.; Elkins J. M.; Gileadi C.; Gildadi O.; Godoi P. H.; Kapadia N.; Muller S.; Santiago A. S.; Sorrell F. J.; Wells C. I.; Fedorov O.; Willson T. M.; Zuercher W. J.; Major M. B. WNT activates the AAK1 kinase to promote clathrin-mediated endocytosis of LRP6 and establish a negative feedback loop. Cell Rep. 2019, 26, 79–93.e8. 10.1016/j.celrep.2018.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. I.; Hunt J. P.; Herrgard S.; Ciceri P.; Wodicka L. M.; Pallares G.; Hocker M.; Treiber D. K.; Zarrinkar P. P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- Clark K.; Peggie M.; Plater L.; Sorcek R. J.; Young E. R. R.; Madwed J. B.; Hough J.; McIver E. G.; Cohen P. Novel cross-talk within the IKK family controls innate immunity. Biochem. J. 2011, 434, 93–104. 10.1042/BJ20101701. [DOI] [PubMed] [Google Scholar]
- McIver E. G.; Bryans J.; Birchall K.; Chugh J.; Drake T.; Lewis S. J.; Osborne J.; Smiljanic-Hurley E.; Tsang W.; Kamal A.; Levy A.; Newman M.; Taylor D.; Arthur J. S. C.; Clark K.; Cohen P. Synthesis and structure-activity relationships of a novel series of pyrimidines as potent inhibitors of TBK1/IKKε kinases. Bioorg. Med. Chem. Lett. 2012, 22, 7169–7173. 10.1016/j.bmcl.2012.09.063. [DOI] [PubMed] [Google Scholar]
- Petherick K. J.; Conway O. J.; Mpamhanga C.; Osborne S. A.; Kamal A.; Saxty B.; Ganley I. G. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J. Biol. Chem. 2015, 290, 11376–11383. 10.1074/jbc.C114.627778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman R. I.; Wu J. M.; Polokoff M. A.; Kochanny M. J.; Dinter H.; Zhu D.; Biroc S. L.; Alicke B.; Bryant J.; Yuan S.; Buckman B. O.; Lentz D.; Ferrer M.; Whitlow M.; Adler M.; Finster S.; Chang Z.; Arnaiz D. O. Novel small molecule inhibitors of 3-phosphoinositide-dependent kinase-1. J. Biol. Chem. 2005, 280, 19867–19874. 10.1074/jbc.M501367200. [DOI] [PubMed] [Google Scholar]
- Stathias V.; Turner J.; Koleti A.; Vidovic D.; Cooper D.; Fazel-Najafabadi M.; Pilarczyk M.; Terryn R.; Chung C.; Umeano A.; Clarke D. J. B.; Lachmann A.; Evangelista J. E.; Ma’ayan A.; Medvedovic M.; Schürer S. C. LINCS Data Portal 2.0: next generation access point for perturbation-response signatures. Nucleic Acids Res. 2020, 48, D431–D439. 10.1093/nar/gkz1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson D. W.; Poeckel D.; Zinn N.; Rau C.; Strohmer K.; Wagner A. J.; Graves A. P.; Perrin J.; Bantscheff M.; Duempelfeld B.; Kasparcova V.; Ramanjulu J. M.; Pesiridis G. S.; Muelbaier M.; Bergamini G. Discovery of GSK8612, a highly selective and potent TBK1 inhibitor. ACS Med. Chem. Lett. 2019, 10, 780–785. 10.1021/acsmedchemlett.9b00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faisal M.; Kim J. H.; Yoo K. H.; Roh E. J.; Hong S. S.; Lee S. H. Development and therapeutic potential of NUAKs inhibitors. J. Med. Chem. 2021, 64, 2–25. 10.1021/acs.jmedchem.0c00533. [DOI] [PubMed] [Google Scholar]
- Drewes G.; Ebneth A.; Preuss U.; Mandelkow E.-M.; Mandelkow E. MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell 1997, 89, 297–308. 10.1016/S0092-8674(00)80208-1. [DOI] [PubMed] [Google Scholar]
- Lasagna-Reeves C. A.; de Haro M.; Hao S.; Park J.; Rousseaux M. W.; Al-Ramahi I.; Jafar-Nejad P.; Vilanova-Velez L.; See L.; De Maio A.; Nitschke L.; Wu Z.; Troncoso J. C.; Westbrook T. F.; Tang J.; Botas J.; Zoghbi H. Y. Reduction of Nuak1 decreases tau and reverses phenotypes in a tauopathy mouse model. Neuron 2016, 92, 407–418. 10.1016/j.neuron.2016.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi B.; Conner S. D.; Liu J. Dysfunction of endocytic kinase AAK1 in ALS. Int. J. Mol. Sci. 2014, 15, 22918–22932. 10.3390/ijms151222918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasta J. D.; Corona C. R.; Wilkinson J.; Zimprich C. A.; Hartnett J. R.; Ingold M. R.; Zimmerman K.; Machleidt T.; Kirkland T. A.; Huwiler K. G.; Ohana R. F.; Slater M.; Otto P.; Cong M.; Wells C. I.; Berger B. T.; Hanke T.; Glas C.; Ding K.; Drewry D. H.; Huber K. V. M.; Willson T. M.; Knapp S.; Muller S.; Meisenheimer P. L.; Fan F.; Wood K. V.; Robers M. B. Quantitative, wide-spectrum kinase profiling in live cells for assessing the effect of cellular ATP on target engagement. Cell Chem. Biol. 2018, 25, 206–214. 10.1016/j.chembiol.2017.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asquith C. R. M.; Berger B.-T.; Wan J.; Bennett J. M.; Capuzzi S. J.; Crona D. J.; Drewry D. H.; East M. P.; Elkins J. M.; Fedorov O.; Godoi P. H.; Hunter D. M.; Knapp S.; Müller S.; Torrice C. D.; Wells C. I.; Earp H. S.; Willson T. M.; Zuercher W. J. SGC-GAK-1: A chemical probe for cyclin G associated kinase (GAK). J. Med. Chem. 2019, 62, 2830–2836. 10.1021/acs.jmedchem.8b01213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells C. I.; Drewry D. H.; Pickett J. E.; Tjaden A.; Krämer A.; Müller S.; Gyenis L.; Menyhart D.; Litchfield D. W.; Knapp S.; Axtman A. D. Development of a potent and selective chemical probe for the pleiotropic kinase CK2. Cell Chem. Biol. 2021, 28, 546–558. 10.1016/j.chembiol.2020.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells C. I.; Al-Ali H.; Andrews D. M.; Asquith C. R. M.; Axtman A. D.; Dikic I.; Ebner D.; Ettmayer P.; Fischer C.; Frederiksen M.; Futrell R. E.; Gray N. S.; Hatch S. B.; Knapp S.; Lücking U.; Michaelides M.; Mills C. E.; Müller S.; Owen D.; Picado A.; Saikatendu K. S.; Schröder M.; Stolz A.; Tellechea M.; Turunen B. J.; Vilar S.; Wang J.; Zuercher W. J.; Willson T. M.; Drewry D. H. The Kinase Chemogenomic Set (KCGS): An open science resource for kinase vulnerability identification. Int. J. Mol. Sci. 2021, 22, 566. 10.3390/ijms22020566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drewry D. H.; Wells C. I.; Andrews D. M.; Angell R.; Al-Ali H.; Axtman A. D.; Capuzzi S. J.; Elkins J. M.; Ettmayer P.; Frederiksen M.; Gileadi O.; Gray N.; Hooper A.; Knapp S.; Laufer S.; Luecking U.; Michaelides M.; Muller S.; Muratov E.; Denny R. A.; Saikatendu K. S.; Treiber D. K.; Zuercher W. J.; Willson T. M. Progress towards a public chemogenomic set for protein kinases and a call for contributions. PLoS One 2017, 12, e0181585 10.1371/journal.pone.0181585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berginski M. E; Moret N.; Liu C.; Goldfarb D.; Sorger P. K; Gomez S. M The Dark Kinase Knowledgebase: an online compendium of knowledge and experimental results of understudied kinases. Nucleic Acids Res. 2021, 49, D529–D535. 10.1093/nar/gkaa853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fensome A.; Ambler C. M.; Arnold E.; Banker M. E.; Clark J. D.; Dowty M. E.; Efremov I. V.; Flick A.; Gerstenberger B. S.; Gifford R. S.; Gopalsamy A.; Hegen M.; Jussif J.; Limburg D. C.; Lin T. H.; Pierce B. S.; Sharma R.; Trujillo J. I.; Vajdos F. F.; Vincent F.; Wan Z.-K.; Xing L.; Yang X.; Yang X. Design and optimization of a series of 4-(3-azabicyclo[3.1.0]hexan-3-yl)pyrimidin-2-amines: Dual inhibitors of TYK2 and JAK1. Bioorg. Med. Chem. 2020, 28, 115481. 10.1016/j.bmc.2020.115481. [DOI] [PubMed] [Google Scholar]
- Fensome A.; Ambler C. M.; Arnold E.; Banker M. E.; Brown M. F.; Chrencik J.; Clark J. D.; Dowty M. E.; Efremov I. V.; Flick A.; Gerstenberger B. S.; Gopalsamy A.; Hayward M. M.; Hegen M.; Hollingshead B. D.; Jussif J.; Knafels J. D.; Limburg D. C.; Lin D.; Lin T. H.; Pierce B. S.; Saiah E.; Sharma R.; Symanowicz P. T.; Telliez J.-B.; Trujillo J. I.; Vajdos F. F.; Vincent F.; Wan Z.-K.; Xing L.; Yang X.; Yang X.; Zhang L. Dual inhibition of TYK2 and JAK1 for the treatment of autoimmune diseases: Discovery of ((S)-2,2-difluorocyclopropyl)((1R,5S)-3-(2-((1-methyl-1H-pyrazol-4-yl)amino)pyrimidin-4-yl)-3,8-diazabicyclo[3.2.1]octan-8-yl)methanone (PF-06700841). J. Med. Chem. 2018, 61, 8597–8612. 10.1021/acs.jmedchem.8b00917. [DOI] [PubMed] [Google Scholar]
- Gu Y.; Kar T.; Scheiner S. Fundamental Properties of the CH···O Interaction: Is It a True Hydrogen Bond?. J. Am. Chem. Soc. 1999, 121, 9411–9422. 10.1021/ja991795g. [DOI] [Google Scholar]
- Larabi A.; Devos J. M.; Ng S. L.; Nanao M. H.; Round A.; Maniatis T.; Panne D. Crystal structure and mechanism of activation of TANK-binding kinase 1. Cell Rep. 2013, 3, 734–746. 10.1016/j.celrep.2013.01.034. [DOI] [PubMed] [Google Scholar]
- Chaikuad A.; Koschade S. E.; Stolz A.; Zivkovic K.; Pohl C.; Shaid S.; Ren H.; Lambert L. J.; Cosford N. D. P.; Brandts C. H.; Knapp S. Conservation of structure, function and inhibitor binding in UNC-51-like kinase 1 and 2 (ULK1/2). Biochem. J. 2019, 476, 875–887. 10.1042/BCJ20190038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess-Brown N. A.; Mahajan P.; Strain-Damerell C.; Gileadi O.; Graslund S. Medium-throughput production of recombinant human proteins: protein production in E. coli. Methods Mol. Biol. 2014, 1091, 73–94. 10.1007/978-1-62703-691-7_5. [DOI] [PubMed] [Google Scholar]
- Wojdyla J. A.; Kaminski J. W.; Panepucci E.; Ebner S.; Wang X.; Gabadinho J.; Wang M. DA+ data acquisitino and analysis software at the Swiss Light Source macromolecular crystallography beamlines. J. Synchrotron Radiat. 2018, 25, 293–303. 10.1107/S1600577517014503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Storoni L. C.; Read R. J. Likelihood-enhanced fast translation functions. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2005, 61, 458–464. 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- Murphy J. M.; Korzhnev D. M.; Ceccarelli D. F.; Briant D. J.; Zarrine-Afsar A.; Sicheri F.; Kay L. E.; Pawson T. Conformational instability of the MARK3 UBA domain compromises ubiquitin recognition and promotes interaction with the adjacent kinase domain. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 14336–14341. 10.1073/pnas.0703012104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D.; Afonine P. V.; Baker M. L.; Bunkoczi G.; Chen V. B.; Croll T. I.; Hintze B.; Hung L. W.; Jain S.; McCoy A. J.; Moriarty N. W.; Oeffner R. D.; Poon B. K.; Prisant M. G.; Read R. J.; Richardson J. S.; Richardson D. C.; Sammito M. D.; Sobolev O. V.; Stockwell D. H.; Terwilliger T. C.; Urzhumtsev A. G.; Videau L. L.; Williams C. J.; Adams P. D. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct Biol. 2019, 75, 861–877. 10.1107/S2059798319011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz K. S.; Deoudes E. M.; Berginski M. E.; Jimenez-Ruiz I.; Aksoy B. A.; Hammerbacher J.; Gomez S. M.; Phanstiel D. H. Coral: Clear and customizable visualization of human kinome data. Cell Syst 2018, 7, 347–350. 10.1016/j.cels.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.