

Summary
Introduction
Our previous experiments showed that the transient sodium current (INa) was abnormally increased in early ischaemia and atorvastatin could inhibit INa. The aim of this study was to observe the time-dependent effects of simulated ischaemia on INa and characterise the direct effects of atorvastatin on ischaemic INa.
Methods
Left ventricular myocytes were isolated from Wistar rats and randomly divided into two groups: a control group (normal to simulated ischaemia) and a statin group (normal to simulated ischaemia with 5 μmol/l atorvastatin). The INa was recorded under normal conditions (as baseline) by wholecell patch clamp and recorded from three to 21 minutes in the next phase of simulated ischaemic conditions.
Results
In the control group, normalised INa (at –40 mV) was increased to the peak (1.15 ± 0.08 mA) at three minutes of ischaemia compared with baseline (0.95 ± 0.04 mA, p < 0.01), it subsequently returned to baseline levels at nine and 11 minutes of ischaemia (0.98 ± 0.12 and 0.92 ± 0.12 mA, respectively), and persistently decreased with prolonged ischaemic time. In the statin group, there were no differences between baseline and the early stages of ischaemia (0.97 ± 0.04 mA at baseline vs 0.92 ± 0.12 mA in ischaemia for three minutes, p > 0.05).
Conclusion
Our results suggest that, in the early stages of ischaemia, changes in INa in ventricular myocytes are timedependent, showing an initial increase followed by a decrease, while atorvastatin inhibited the transient increase in INa and made the change more gradual.
Keywords: ventricular myocytes, sodium, ventricular arrhythmia, membrane potential, statin
Clinically, acute ischaemia is one of the common causes of malignant ventricular arrhythmias.1 A retrospective study showed that 7.5% of patients with acute myocardial infarction developed ventricular arrhythmias, most of which (78%) occurred within the first 48 hours of ischaemic symptoms,2 suggesting that electrical activities are very unstable in the early stage of ventricular ischaemia.
Sodium current (INa) is the starting current of the action potential and affects the shape and conduction of the action potential.3 It is one of the most common targets to cause and treat arrhythmias. Animal experiments found that in an aconitine-induced arrhythmia model,4 increased INa could lead to pre-contraction and even ventricular arrhythmias. Therefore INa plays an important role in arrhythmogenesis.
Previous studies have shown that INa would be decreased or Nav1.5, which is the ion channel protein of INa, would be downregulated in the ischaemic condition.5,6 However in our pre-experiment of simulated ischaemia, peak INa was transiently increased in the very early stage of ischaemia (three to five minutes), suggesting unstable early ischaemic electrical activity. As the decreased INa demonstrated in ischaemia or simulated ischaemia usually needs myocyte exposure for more than 10 minutes,5 this indicates that time is a key factor affecting INa in the ischaemic state.
On the other hand, as the basic therapeutic agents of acute coronary syndrome, statins may reduce the incidence of ischaemic ventricular arrhythmias7,8 and can prevent sudden cardiac death,9 as well as other cardiovascular events. However, the mechanisms are controversial. One view is that electrical protection from the statin is secondary to a decrease in low-density lipoprotein cholesterol, whereas another view is that statins act as an upstream protection on the basis of pleiotropic effects.10 In addition, Vaquero et al.11 confirmed that atorvastatin and simvastatin had an inhibitory effect on atrial plateau currents [hKv1.5 and Kv4.3 channels, while ICa,L (L-type calcium current) could also be blocked by simvastatin acid] at the cellular level. Similarly, there is a direct electrical effect on the INa of ventricular myocytes in the early stage of ischaemia only.
We assumed that the effect of ischaemia on INa was timedependent, that INa may be transiently increased during the first 10 minutes of ischaemia, and that atorvastatin could inhibit this phenomenon. Therefore we used a patch-clamp technique to observe the time-dependent effects of simulated ischaemia on INa in ventricular myocytes by setting the observation interval to two minutes. In addition, we also applied atorvastatin on the above basis, in order to observe its direct effect on INa in the early ischaemic condition.
Methods
Thirty Wistar rats (300 ± 50 g, male and female) were purchased from the Chinese Academy of Medical Sciences Institute of Radiation Medicine Experimental Animal Centre. All study protocols and use of rats were approved by the Institutional Animal Care and Use Committee of Tianjin Medical University (Tianjin, China).
Ca2+-free Tyrode solution contained (mM): NaCl 137, KCl 5.4, MgCl2 1, NaH2PO4 0.33, HEPES 10, and glucose 10 (pH 7.4 with NaOH). KB solution contained (mM): L-glutamic acid 50, KCl 40, MgCl2 3, KH2PO4 20, taurine 20, KOH 70, EGTA 0.5, HEPES 10, and glucose 10 (pH 7.4 with KOH). The pipette solution contained (mM): CsCl 140, NaCl 10, EGTA 5, HEPES 5, Na2ATP5 (pH 7.3 with CsOH). The normal extracellular solution contained (mM): choline-Cl 120, NaCl 25, CsOH 4, CaCl2 0.1, CoCl2 2, MgCl2 1, HEPES 10, and glucose 10 (pH 7.4 with CsOH). The simulated ischaemic extracellular solution contained (mM): choline-Cl 120, NaCl 25, CsOH 4, CaCl2 0.1, CoCl2 2, MgCl2 1, HEPES 10, and natrium lacticum 20 (adjusted to pH 6.8 and filled with nitrogen for more than five minutes before using). Atorvastatin calcium (USP Corporation, Lot 344423-98-9) was dissolved in the ischaemic extracellular solution to prepare the drug solution containing 5 μM atorvastatin (usually 3.02 mg of atorvastatin calcium was dissolved in 500 ml of extracellular solution).
For isolation of the myocytes, single ventricular myocytes were dissociated from hearts of Wistar rats using type II collagenase (Gibco). Rats were weighted, heparinised (5 000 UI/ kg), anaesthetised with chloral hydrate (40 mg/kg), the chest was opened and the hearts were removed, and then the rats were euthanised. The heart was immersed in Ca2+-free Tyrode solution (4°C) and immediately clipped.
The heart was cannulated through the aorta and mounted on a Langendorff perfusion apparatus (100% O2, 37°C, perfusion pressure 70 cm H2O). It was retrogradely perfused with Ca2+-free Tyrode solution until the blood was washed out, followed by perfusion with the same Ca2+-free Tyrode solution supplemented with 0.6 mg/ml collagenase II and 0.5 mg/ml albumin bovine serum (68 kD, Roche). As the drip rate reached 20 ml/min and the colour of the heart changed to orange and transparent, the perfusion was complete.
The heart was then removed into KB solution (37°C). The free left ventricular wall was cut into approximately 8 × 2-mm sections with a fine scissors and the endocardium and epicardium were removed in the KB solution. The mid-myocardial section was cut up and agitated with a dropper in order to obtain isolated cells. The cell suspension was then filtered with a strainer (200 mesh). Before recording, the myocytes were placed in filtered KB solution for more than two hours.
INa was recorded at room temperature (25°C) using the whole-cell configuration of the patch-clamp with Axopatch 700B amplifiers and pClamp 10.1 software (Axon Instruments, USA). Pipettes were pulled from borosilicate capillary tubes using a programmable horizontal micro-electrode puller (P-97, Sutter Instruments, USA) and heat polished with a microforge (MF-830, Narishige). Micropipette resistance was kept at 2–5 MΩ when filled with pipette solution and immersed in the extracellular solution.
The cells were placed in normal extracellular solution for rupture of the membrane, compensation for membrane capacitance and series resistor (75%), and the currents were recorded for baseline. Then the cell bath was perfused with the simulated ischaemic solution (control group) or drug solution (statin group) for three minutes (3 ml/min). At this time, the extracellular solution was replaced completely and we considered the time after one minute of perfusion as the zero point for the start of ischaemia. The cells were then left standing for one minute to avoid interference from mechanical vibration. Thereafter INa was recorded every two minutes from three minutes after the start of ischaemia to 21 minutes, in both the statin and the control groups.
The holding potential was maintained at –90 mV and the protocol for recording INa was composed of 50-ms pulses that were imposed in 5-mV increments between –80 and +50 mV, and pulse frequency was 2.5 Hz, which was matched with the rat’s natural heart rate. In order to trace the inactivation curves, a double-pulse protocol was set up: the first 50-ms conditioning pulses were imposed in 5-mV increments between –80 and +50 mV, each of which was followed by a test pulse to +10 mV. Finally, to describe the recovery curves after inactivation, another double-pulse protocol was used: the first conditional pulses were imposed at –40 mV for 50 ms, each of which was followed by a fixed 80-ms test pulse from –90 to –40 mV, and the interval between the two pulses was increased in 2-ms increments from 2 to 76 ms.
Statistical analysis
In order to eliminate the effect of cell size on INa, the INa from different myocytes should be standardised. As atorvastatin may also affect the membrane capacitance, which may become a confounding factor in the current density, we used the relative current value as the normalised INa in order to evaluate the effects of atorvastatin on the peak value of the INa.
The Boltzmann equation was used to fit the activation and inactivation curves, and the recovery curve after inactivation was fitted with an exponential equation. We observed the normalised INa, membrane potential at 50% maximal activation (V1/2,a), offsetting of the activation curve (Ka), membrane potential at 50% maximal inactivation (V1/2,i), offsetting of the inactivation curve (Ki) and recovery constant (τ). The data were analysed by means of variance analysis of repeated measurement data, and the gating characteristics were analysed with the allogeneic paired t-test; p < 0.05 indicated that the difference was statistically significant.
Results
Effect of ischaemia on INa in the early stage after perfusion: Previous experiments showed that ischaemia suppressed the amplitude of INa, but we observed the normalised INa was transiently increased in the very early stage of ischaemia in the pre-experiment. In order to verify the increased current was not associated with the mechanical effect of perfusion, we compared the effect of ischaemic and normal extracellular solutions on INa in the same way. We found compared with normal extracellular solution, normalised INa was transiently increased after perfusion with ischaemic extracellular solution, while simulated ischaemia was for three minutes (0.92 ± 0.04 vs 1.42 ± 0.34 mA, p < 0.01; Fig. 1).
Fig. 1.

Effects of ischaemia on INa in the very early stage after perfusion. (A) Current–voltage (I–V) curves between baseline and ischaemia. When ventricular myocytes were perfused with ischaemic solution, the peak value of INa was voltage-dependently increased in the stage of ischaemia after three minutes. Compared with baseline, †p < 0.05, ‡p < 0.01. (B) Normalised INa after perfusion with normal and ischaemic extracellular solution. In the first 10 minutes after perfusion, normalised INa was transiently increased when perfused with ischaemic solution, whereas there was little change when perfused with normal solution, which excluded the effects of mechanical action on INa. Compared with normal solution, †p < 0.05, ‡p < 0.01.
Effect of atorvastatin on INa in the early stage of ischaemia: When entering the simulated ischaemic state, the whole-cell currents of control and statin groups both changed over time (Fig. 2). Because of the voltage-dependent characteristics, the maximum currents appeared at –40-mV test potential (Fig. 3), which was used to analyse the time-dependent effects of ischaemia and atorvastatin on INa.
Fig. 2.

Trend of whole-cell currents of INa over time. (A) Whole-cell currents in the control group. (B) Whole-cell currents in the statin group. *zero point of simulated ischaemia.
Fig. 3.

Current–voltage (I–V) curves between baseline and ischaemia for three minutes. (A) I–V curve of control group, which was down-shifted in the very early stage of ischaemia, and represented the increase of INa at the test potential from –55 to 50 mV. (B) I–V curve of statin group, which was little changed in the early ischaemic condition, compared to the baseline.
In the control group, the normalised INa increased above baseline in the first three to seven minutes of simulated ischaemia, and peaked at three minutes (Figs 3A, 4). Compared with the three-minute point, the normalised INa decreased at seven minutes (p = 0.0321). At the nine- and 11-minute points, the normalised INa returned to baseline (p = 0.3209 and 0.5505, respectively). With the recording time extended, the normalised INa was lower than baseline (p < 0.05) and gradually decreased from 13 to 21 minutes (13 vs 15 minutes, p = 0.0270; 15 vs 17 minutes, p = 0.0146; 19 vs 21 minutes, p = 0.0014, respectively; Fig. 4).
Fig. 4.
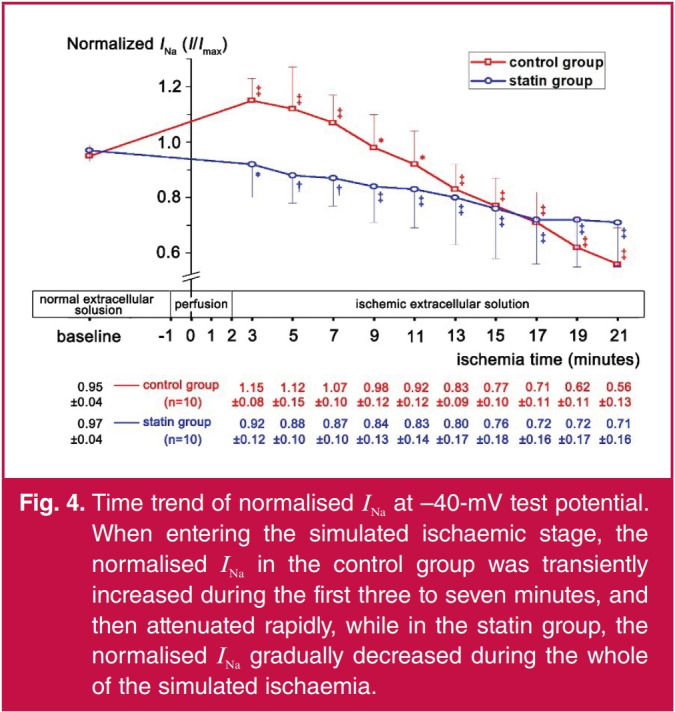
Time trend of normalised INa at –40-mV test potential. When entering the simulated ischaemic stage, the normalised INa in the control group was transiently increased during the first three to seven minutes, and then attenuated rapidly, while in the statin group, the normalised INa gradually decreased during the whole of the simulated ischaemia.
In the statin group, the normalised INa gradually decreased during the whole time of simulated ischaemia. It decreased by 0.09 ± 0.03 mA at five minutes compared with baseline (p = 0.0163), decreased by 0.08 ± 0.03 mA at 13 minutes compared with five minutes (p = 0.0256), and continued decreasing by 0.09 ± 0.02 mA (p = 0.0040) at 21 minutes compared with 13 minutes (Fig. 4).
Comparing normalised INa between the two groups (Fig. 4), there were no differences at baseline and 11 to 19 minutes of ischaemia (p > 0.05). Normalised INa in the statin group was lower than in the control group at three to nine minutes of ischaemia (p < 0.05), while at 21 minutes, INa in the statin group was higher than in the control group (p < 0.05).
Table 1 shows the gating characteristics of the two groups. Compared with baseline, in the three minutes of simulated ischaemia, the curves of activation and inactivation were shifted negatively (Fig. 5A–D), and Ka and τ were decreased in both groups (Table 1, Fig. 5A, B, E, F). At three minutes of simulated ischaemia, Ki in the statin group was lower than in the control group (p < 0.05), and τ in the statin group was higher than in the control group (p < 0.05; Table 1, Fig. 5C–F).
Table 1. Gating characteristics at three minutes of simulated ischaemia (x ± s).
| Activation (n = 8) | Inactivation (n = 8) | Resurrection (n = 9) | |||
| V1/2,a (mV) | Ka (mV) | V1/2,i (mV) | Ki (mV) | τ (ms) | |
| Control group | |||||
| Baseline (A1) | –54.91 ± 4.22 | 1.45 ± 0.48 | –62.84 ± 2.50 | 4.52 ± 0.97 | 34.23 ± 4.40 |
| Ischaemia (B1) | –58.82 ± 3.65 | 0.90 ± 0.31 | –65.19 ± 3.33 | 4.28 ± 1.11 | 25.54 ± 6.41 |
| Value of B1–A1 | –3.90 ± 2.16 | –0.55 ± 0.44 | –2.35 ± 1.71 | –0.23 ± 0.38 | –8.69 ± 4.75 |
| p (A1:B1) | 0.0014 | 0.0090 | 0.0061 | 0.1238 | 0.0006 |
| Statin group | |||||
| Baseline (A2) | –54.70 ± 3.54* | 1.41 ± 0.65* | –63.33 ± 2.24* | 4.92 ± 0.55* | 34.58 ± 8.55* |
| Ischemia (B2) | –59.16 ± 3.53 | 1.03 ± 0.58 | –66.45 ± 1.91 | 4.12 ± 0.56 | 30.22 ± 9.65 |
| Value of B2–A2 | –4.47 ± 1.97 | –0.38 ± 0.35 | –3.12 ± 1.00 | –0.81 ± 0.35† | -4.36 ± 4.82† |
| p (A2:B2) | 0.0004 | 0.0169 | 0.0000 | 0.0004 | 0.0263 |
Compared with the baseline of the control group, *p > 0.2, and compared with the
value of B1–A1, †p < 0.05.
Fig. 5.

Gating characteristic curves between baseline and ischaemia for three minutes. (A) Activation curve in the control group. (B) Activation curve in the statin group. (C) Inactivation curve in the control group. (D) Inactivation curve in the statin group. (E) Recovery curve in the control group. (F) Recovery curve in the statin group.
Discussion
Sodium current plays an important role in ischaemic ventricular arrhythmias, which may affect cardiac conductivity and irritation.3 Previous studies have shown that sodium current may decrease in the ischaemic state,5 but in our study, the current transiently increased in the early stage of ischaemia. Ventricular arrhythmias mainly appear in acute myocardial ischaemia in two time periods after birth (0–0.5 and 1.5–9 hours).12
In view of the relationship between ischaemic time and the degree of injury, we hypothesised that the change of INa in simulated ischaemia may be time-dependent. To observe the instantaneous change in INa, the measurement time interval was shortened to two minutes. The results showed that INa was transiently increased and peaked at three minutes after simulated ischaemia. At this time, the V1/2,a and V1/2,i were both decreased, which represented the activation and inactivation thresholds, respectively, and meant that both the activation and inactivation processes would be much easier at the early stage of ischaemia. In addition, decreased Ka and τ indicated that the processes of channel activation and recovery had been changed much faster (Fig. 5).
In summary, these changed gating characteristics indicated that channel transition between open and closed states became more frequent, and the open probability of sodium channels per unit time had been increased. Since Im = i P0 N (where Im is the whole-cell current, i is the single-channel current, P0 is the open probability, and N is the number of channels),13 the whole-cell INa had been consequently increased at three minutes of simulated ischaemia. However, this experiment also showed that INa was gradually attenuated over time after 10 minutes of simulated ischaemia, which was consistent with previous reports.14 A possible reason may be the secondary damage to cells by secondary calcium overload related to increased intracellular sodium concentration.15
The aconitine model has shown16,17 that the abnormally increased INa may result in the increase of 0 phase amplitude of the action potential. As the increased action potentials pass into the adjacent tissue in the relative refractory period, it will cause threshold stimulation, which may lead to premature contraction. In our study, it was observed that INa had first been increased and then decreased in the simulated ischaemic state. Therefore, in the early stage of ischaemia, cardiomyocytes were in a heterogeneous ischaemic state, and the dispersion of INa in ischaemic tissue would be increased with the prolongation of ischaemia, which may be one of the bases for the formation of local abnormal current.
As a basic drug of acute coronary syndrome, statins have been shown to reduce the morbidity of ventricular arrhythmias and the mortality rate.18,19 Therefore we observed the effect of atorvastatin on INa, which was in the early stage of ischaemia, and found that the increased current was inhibited. As we know, before producing pleiotropic effects, statins should inhibit HMG-CoA reductase and then block the important mevalonate pathway.20,21 However, Gerber et al. showed that atorvastatin decreased the HMG-CoA reductase activity in L cells only after incubation with the drug for 18 hours.22 In addition, Vaquero et al. demonstrated the membrane capacitance was not changed by atorvastatin.11 Therefore, non-specific perturbation of the membrane seems a very unlikely mechanism for atorvastatin to be responsible for, otherwise the capacitance would be changed as the dielectric constant had been modified.
As a fat-soluble statin, atorvastatin calcium is slightly soluble in pH 7.4 phosphate buffer, which means that the theoretical maximum range of atorvastatin is 82.68 to 826.8 μmol/l. We used a concentration of 5 μmol/l, which was equivalent to the clinical dose of 20–80 mg/d.23 This could avoid the use of a fat-soluble solvent, which may also influence the membrane currents.
Conclusions
In this study we observed the time-dependent effect of atorvastatin on INa in a simulated ischaemic condition and found that the phenomenon of transiently increased INa disappeared. The gated characteristics showed that atorvastatin reduced Ki and weakened the decline of τ value caused by ischaemia. Therefore the channel inactivation was faster and the recovery was slower, which caused the number of open channels per unit time to decrease, finally resulting in a decrease in whole-cell current.
Atorvastatin inhibited the abnormal increase of INa during the early stage of simulated ischaemia by acting on the processes of inactivation and recovery. As statins can block the activity of a voltage-gated calcium channel,24 atorvastatin could also transiently block the sodium channel when entering the cell during the first three to seven minutes of ischaemia. Interestingly, atorvastatin appeared to prevent a further decrease in INa as the ischaemic time extended to more than 19 minutes, indicating another cardioprotective effect of atorvastatin, in preventing further ischaemic injury (such as ischaemic postconditioning of statins25). Therefore atorvastatin played a role only as a buffer in abating rapid changes in INa over time during early ischaemia, which helped to reduce the electrical heterogeneity of the ischaemic myocardium26,27 and improve the cardiac arrhythmia matrix effect.
References
- 1.Glinge C, Sattler S, Jabbari R, Tfelt-Hansen J. Epidemiology and genetics of ventricular fibrillation during acute myocardial infarction. J Geriatr Cardiol. 2016;13(9):789–797. doi: 10.11909/j.issn.1671-5411.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Henkel DM, Witt BJ, Gersh BJ, Jacobsen SJ, Weston SA, Meverden RA. et al. Ventricular arrhythmias after acute myocardial infarction: a 20-year community study. Am Heart J. 2006;151(4):806–812. doi: 10.1016/j.ahj.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 3.Veerman CC, Wilde AA, Lodder EM. The cardiac sodium channel gene SCN5A and its gene product NaV1.5: Role in physiology and pathophysiology. Gene. 2015;573(2):177–187. doi: 10.1016/j.gene.2015.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao Z, Yin Y, Wu H, Jiang M, Lou J, Bai G. et al. Arctigenin, a potential anti-arrhythmic agent, inhibits aconitine-induced arrhythmia by regulating multi-ion channels. Cell Physiol Biochem. 2013;32(5):1342–1353. doi: 10.1159/000354532. [DOI] [PubMed] [Google Scholar]
- 5.Ding C, Fu XH, He ZS, Chen HX, Xue L, Li JX. Cardioprotective effects of simvastatin on reversing electrical remodeling induced by myocardial ischemia–reperfusion in normocholesterolemic rabbits. Chin Med J (Engl) 2008;121(6):551–556. [PubMed] [Google Scholar]
- 6.Wei X, Zhu A, Zhang Y, Yao S, Mao W. Pre- and delayed treatments with ranolazine ameliorate ventricular arrhythmias and Nav1.5 downregulation in ischemic/reperfused rat hearts. J Cardiovasc Pharmacol. 2016;68(4):269–279. doi: 10.1097/FJC.0000000000000412. [DOI] [PubMed] [Google Scholar]
- 7.Das MK, Zipes DP. Antiarrhythmic and nonantiarrhythmic drugs for sudden cardiac death prevention. J Cardiovasc Pharmacol. 2010;55(5):438–449. [PubMed] [Google Scholar]
- 8.Apiyasawat S, Sritara P, Ngarmukos T, Sriratanasathavorn C, Kasemsuwan P. Association of statin therapy with ventricular arrhythmias among patients with acute coronary syndrome. Heart Asia. 2013;5(1):39–41. doi: 10.1136/heartasia-2012-010225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Al-Khatib SM, Stevenson WG, Ackerman MJ, Bryant WJ, Callans DJ, Curtis AB. et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: A report of the American College of Cardiology/ American Heart Association task force on clinical practice guidelines and the Heart Rhythm Society. Circulation. 2018;138:e272–e391. doi: 10.1161/CIR.0000000000000549. [DOI] [PubMed] [Google Scholar]
- 10.Oesterle A, Laufs U, Liao JK. Pleiotropic effects of statins on the cardiovascular system. Circ Res. 2017;120(1):229–243. doi: 10.1161/CIRCRESAHA.116.308537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vaquero M, Caballero R, Gomez R, Nunez L, Tamargo J, Delpon E. Effects of atorvastatin and simvastatin on atrial plateau currents. J Mol Cell Cardiol. 2007;42(5):931–945. doi: 10.1016/j.yjmcc.2007.03.807. [DOI] [PubMed] [Google Scholar]
- 12.Opitz CF, Mitchell GF, Pfeffer MA, Pfeffer JM. Arrhythmias and death after coronary artery occlusion in the rat. Continuous telemetric ECG monitoring in conscious, untethered rats. Circulation. 1995;92(2):253–261. doi: 10.1161/01.cir.92.2.253. [DOI] [PubMed] [Google Scholar]
- 13.Karmazinova M, Lacinova L. Measurement of cellular excitability by whole cell patch clamp technique. Physiol Res. 2010;59(Suppl 1):S1–7. doi: 10.33549/physiolres.932000. [DOI] [PubMed] [Google Scholar]
- 14.Carmeliet E. Cardiac ionic currents and acute ischemia: from channels to arrhythmias. Physiol Rev. 1999;79(3):917–1017. doi: 10.1152/physrev.1999.79.3.917. [DOI] [PubMed] [Google Scholar]
- 15.Huang CL. Murine electrophysiological models of cardiac arrhythmogenesis. Physiol Rev. 2017;97(1):283–409. doi: 10.1152/physrev.00007.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan TY. Aconite poisoning. Clin Toxicol (Phila) 2009;47(4):279–285. doi: 10.1080/15563650902904407. [DOI] [PubMed] [Google Scholar]
- 17.Coulson JM, Caparrotta TM, Thompson JP. The management of ventricular dysrhythmia in aconite poisoning. Clin Toxicol (Phila) 2017;55(5):313–321. doi: 10.1080/15563650.2017.1291944. [DOI] [PubMed] [Google Scholar]
- 18.Beri A, Contractor T, Gardiner JC, Ardhanari S, Thakur R. Reduction in the intensity rate of appropriate shocks for ventricular arrhythmias with statin therapy. J Cardiovasc Pharmacol. 2010;56(2):190–194. doi: 10.1097/FJC.0b013e3181e74d4f. [DOI] [PubMed] [Google Scholar]
- 19.Chung CM, Lin MS, Chang CH, Cheng HW, Chang ST, Wang PC. et al. Moderate to high intensity statin in dialysis patients after acute myocardial infarction: A national cohort study in Asia. Atherosclerosis. 2017;267:158–166. doi: 10.1016/j.atherosclerosis.2017.09.018. [DOI] [PubMed] [Google Scholar]
- 20.Margaritis M, Sanna F, Antoniades C. Statins and oxidative stress in the cardiovascular system. Curr Pharm Des 2017 Sep 26. [Epub ahead of print]. doi: 10.2174/1381612823666170926130338. [DOI] [PubMed] [Google Scholar]
- 21.Fang SY, Roan JN, Luo CY, Tsai YC, Lam CF. Pleiotropic vascular protective effects of statins in perioperative medicine. Acta Anaesthesiol Taiwan. 2013;51(3):120–126. doi: 10.1016/j.aat.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 22.Gerber R, Ryan JD, Clark DS. Cell-based screen of HMG-CoA reductase inhibitors and expression regulators using LC-MS. Anal Biochem. 2004;329(1):28–34. doi: 10.1016/j.ab.2004.03.023. [DOI] [PubMed] [Google Scholar]
- 23.Lennernas H. Clinical pharmacokinetics of atorvastatin. Clin Pharmacokinet. 2003;42(13):1141–1160. doi: 10.2165/00003088-200342130-00005. [DOI] [PubMed] [Google Scholar]
- 24.Ali N, Begum R, Faisal MS, Khan A, Nabi M, Shehzadi G. et al. Current statins show calcium channel blocking activity through voltage gated channels. BMC Pharmacol Toxicol. 2016;17(1):43–43. doi: 10.1186/s40360-016-0086-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferdinandy P, Hausenloy DJ, Heusch G, Baxter GF, Schulz R. Interaction of risk factors, comorbidities, and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning. Pharmacol Rev. 2014;66(4):1142–1174. doi: 10.1124/pr.113.008300. [DOI] [PubMed] [Google Scholar]
- 26.Waks JW, Tereshchenko LG. Global electrical heterogeneity: A review of the spatial ventricular gradient. J Electrocardiol. 2016;49(6):824–830. doi: 10.1016/j.jelectrocard.2016.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kessler EL, Boulaksil M, van Rijen HV, Vos MA, van Veen TA. Passive ventricular remodeling in cardiac disease: focus on heterogeneity. Front Physiol. 2014;5:482–482. doi: 10.3389/fphys.2014.00482. [DOI] [PMC free article] [PubMed] [Google Scholar]