

Abstract
The molecular mechanisms leading to resistance to PD-1 blockade are largely unknown. Here, we characterize tumor biopsies from a melanoma patient who displayed heterogeneous responses to anti-PD-1 therapy. We observe that a resistant tumor exhibited a loss-of-function mutation in the tumor suppressor gene FBXW7, while a sensitive tumor from the same patient did not. Consistent with a functional role in immunotherapy response, inactivation of Fbxw7 in murine tumor cell lines caused resistance to anti-PD-1 in immunocompetent animals. Loss of Fbxw7 was associated with altered immune microenvironment, decreased tumor-intrinsic expression of the dsRNA sensors Mda5 and Rig-I, diminished induction of type I interferon and MHC-I expression. In contrast, restoration of dsRNA sensing in Fbxw7-deficient cells was sufficient sensitize them to anti-PD-1. Our results thus establish a new role for the commonly inactivated tumor suppressor FBXW7 in viral sensing and sensitivity to immunotherapy.
INTRODUCTION
Immunotherapies, such as CTLA-4, PD-1 inhibitors, or their combination, have revolutionized the treatment of cancer patients (1). However, a key challenge to optimize the opportunity provided by these therapies is the dramatically varied responses among different patients, or even among different tumors in the same patient (2). Prior studies have identified decreased CD8+ T cell infiltration(3), defects in interferon signaling (4,5) or antigen presentation (6), as well as alteration of viral sensing pathways (7–9) as mechanisms leading to therapeutic resistance. These phenotypes can be altered due to oncogenic events in tumor cells, including activation of β-catenin (10,11), loss-of-function mutations in JAK1/2 (5), or in the tumor suppressor LKB1/STK11 (12). Nevertheless, these mechanisms collectively do not account for the majority of cases of immunotherapy resistance. Thus, the identification of additional molecular mechanisms of resistance has the potential to identify patients who are more likely to benefit from these treatments. Elucidation of resistance pathways could also enable rational therapeutic approaches that restore tumor immunity in genomically-selected patient populations.
F-box and WD repeat domain containing 7 (FBXW7) is a commonly mutated tumor suppressor in diverse tumor types. Missense mutations in FBXW7 are observed in about 6% of cancers, including endometrial, colon, cervical, stomach, skin, urothelial, lung, ovarian, testis, breast, pancreatic, renal, liver, prostate, brain and thyroid cancers (13–15). Approximately 30% of human cancers also have deletions of chromosome 4q32, which includes the FBXW7 locus (15). Inactivating mutations or the genomic loss of FBXW7 disrupts the activity of an evolutionary conserved SKP1, CUL1, F-box protein (SCF) ubiquitin ligase complex(13,16–18), leading to increases in cell proliferation and division proteins such as c-Myc, Cyclin E1, and c-Jun.
While a role of FBXW7 in tumor immunity has not yet been shown, a recent report has described a function of FBXW7 in antiviral immunity through regulating the stability of Retinoic acid-inducible gene I (RIG-I, encoded by DDX58) (19). RIG-I and Melanoma differentiation-associated protein 5 (MDA5, encoded by IFIH1) are two major viral nucleic acid sensors that defend against viral infection and other pathogens (20). Upon detection of double-stranded RNA (dsRNA) in tumor cells, RIG-I and MDA5 associate with Mitochondrial antiviral-signaling protein (MAVS), leading to the recruitment and autophosphorylation of TANK-binding kinase 1 (TBK1). TBK1 phosphorylates the transcription factor Interferon regulatory factor 3 (IRF3), which triggers the expression of type I interferons and proinflammatory cytokines, such as CXCL10. This pathway therefore activates innate immune responses in the tumor microenvironment (21).
In this study, we found that a loss-of-function mutation in FBXW7 was associated with resistance to PD-1 blockade in a melanoma patient. Using an immunocompetent, anti-PD-1 sensitive melanoma mouse model, we found that Fbxw7 deletion or its mutation in tumor cells is sufficient to confer resistance to PD-1 blockade. Tumor-intrinsic Fbxw7 deficiency altered the tumor immune microenvironment by decreasing immune cell infiltration and diminished the activation of viral sensing and interferon signaling pathways in vivo. Fbxw7 was essential for the expression of Rig-I and Mda5, which are both required for Fbxw7-mediated dsRNA sensing. Finally, we have shown that restoration of dsRNA sensing in Fbxw7-deficient cells increased MHC-I expression and sensitized Fbxw7-deficient tumors to anti-PD-1. These findings provide insights into the function of the FBXW7 tumor suppressor gene in tumor immunity and suggest a therapeutic strategy to overcome resistance to PD-1 blockade in a genotype-selected group of patients.
RESULTS
FBXW7 loss-of-function is associated with resistance to pembrolizumab
To uncover oncogenic mutations that confer resistance to PD-1 blockade, we identified metastatic melanoma patients who exhibited resistance of a single tumor site despite responses in other disease sites. One such patient, a 74-year old man with diffusely metastatic melanoma (Figure 1a) exhibited a complete response to pembrolizumab in all lesions within 11 months of treatment, except for a right adrenal mass which did not respond. To identify tumor cell-intrinsic genomic changes associated with resistance to PD-1 blockade, we performed whole-exome sequencing and analysis on a pre-treatment lesion (a cervical lymph node), the right adrenal resistant lesion, and a germline sample (peripheral blood mononuclear cells). We then used ABSOLUTE (22,23) to determine allele fraction of called mutations and allelic copy number information in the pre-treatment and resistant samples. Overall, 1583 somatic variants were shared between both tumors (Figure 1b, Supplementary Table 1). Twenty-eight mutations were unique to the resistant adrenal tumor, whereas 26 mutations were unique to the pre-treatment tumor, suggesting that the resistant lesion evolved from a precursor clone. As expected for melanoma, both the resistant and pre-treatment tumors had a mutational signature consistent with UV exposure (Supplementary Figure 1a). Both the pre-treatment and resistant lesion had similar numbers of mutations, non-synonymous mutations, and predicted neoantigens (Supplementary Figure 1b). The copy number profile between the pre-treatment and resistant lesions was also similar (Supplementary Figure 1c).
Figure 1. An inactivating mutation in FBXW7 is associated with resistance to PD-1 blockade.
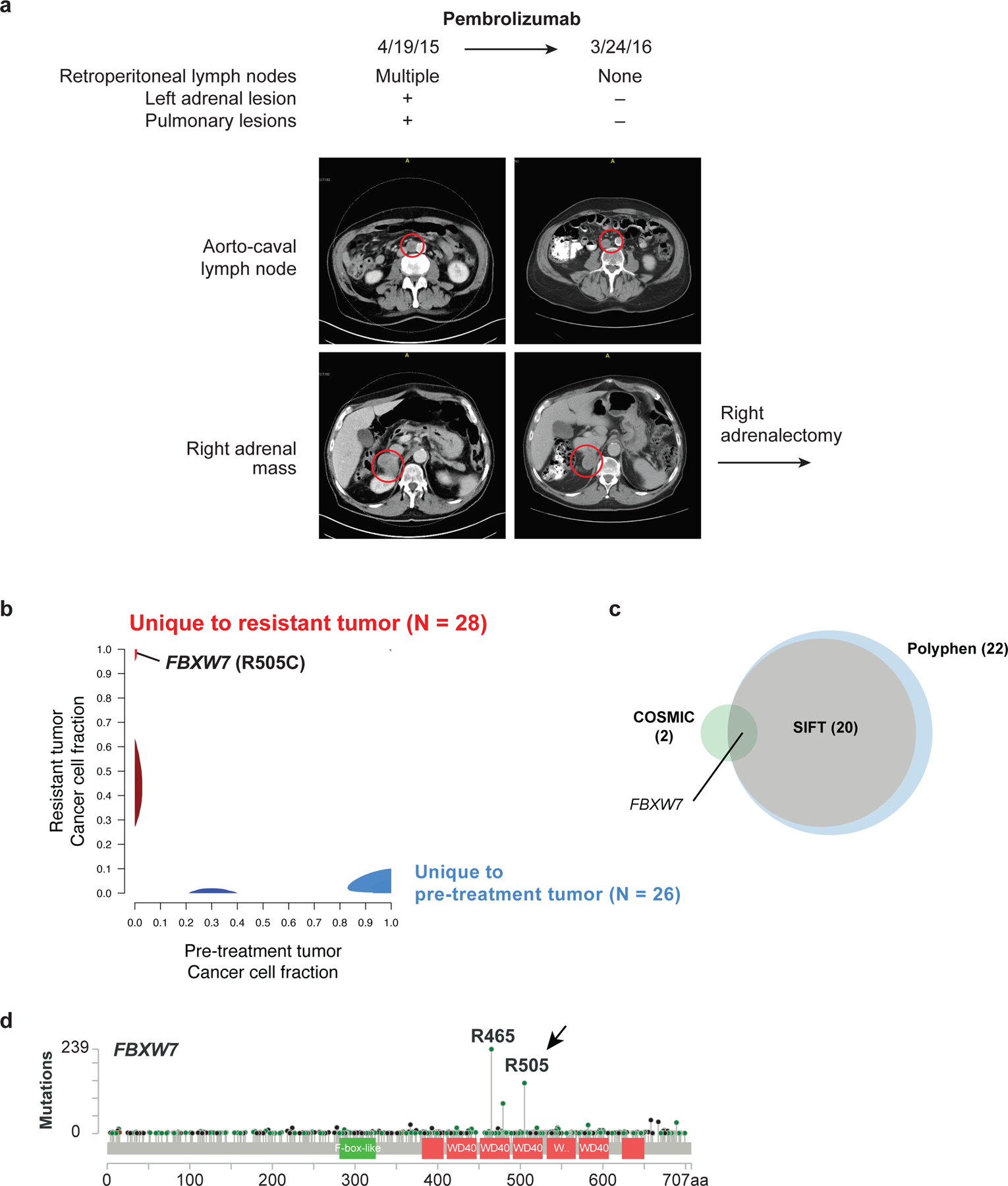
(a) CT scan from a patient with metastatic melanoma with heterogenous response to the PD-1 inhibitor pembrolizumab. The patient presented with diffuse metastatic disease, which responded to treatment with the exception of a right adrenal mass. The patient’s cervical lymph node tumor was biopsied prior to treatment and right adrenal gland was biopsied upon the development of adrenal resistance. (b) Phylogic analysis of the somatic mutations identified by whole exome sequencing of the pre-treatment and resistant (right adrenal) biopsies. (c) Intersection of clonal, deleterious somatic changes identified by SIFT or PolyPhen analysis with clonal somatic mutations listed in the COSMIC database. (d) Frequency of FBXW7 mutations in cancer. R505 and R465 are known oncogenic, loss-of-function mutations in FBXW7. Green: missense mutations; black: nonsense mutations. cBioPortal, 1483 samples. Additional data in Supplementary Figure 1, Supplementary Table 1 and Supplementary Table 2.
We evaluated the 28 mutations in the resistant tumor for known genomic mechanisms of resistance to immunotherapy. However, we found no somatic mutations in antigen presentation or interferon signaling pathways, which are correlated with anti-PD-1 resistance (4–6). We also used the COSMIC database and several variant discovery engines (24–26) to predict oncogenic and deleterious mutations (Figure 1c). The only known oncogenic mutation that distinguished the pre-treatment and the resistant tumor was an arginine-to-cysteine mutation at amino acid 505 (R505C) in the tumor suppressor gene FBXW7 (Figure 1b–c, Supplementary Table 2). The R505 mutation, the second most common mutation in FBXW7 observed in cancer (Figure 1d), is associated with the increased expression of FBXW7 substrates and leads to dominant-negative phenotypes, suggesting that immunotherapy resistance could be associated with the loss of FBXW7 activity.
Fbxw7 is required for the antitumor activity of PD-1 blockade
To test the possibility that FBXW7 inactivation leads to resistance to PD-1 blockade in melanoma, we developed a murine melanoma model lacking Fbxw7. Our model was based on D4M3A, a Braf-mutant, Pten deleted melanoma murine cell line that is 98% genetically identical to C57BL/6 mice (27). D4M3A cells were modified ex vivo to express Cas9 (hereafter denoted D4C9), facilitating the rapid deletion of genes by CRISPR (28). Cells transduced with a control sgRNA grew similarly in immunocompetent C57BL/6 and immunocompromised nude mice (Supplementary Figure 2a–b, Supplementary Figure 9h–i). Anti-PD-1 treatment of immunocompetent mice with D4C9-sgCtrl tumors was associated with durable tumor control (> 100 days), even after only three drug treatments (Supplementary Figure 2c). Anti-PD-1 treatment had no impact on the survival of nude mice (Supplementary Figure 2d).
To determine whether Fbxw7 is required for response to anti-PD-1 therapy, we generated D4C9 derivatives lacking Fbxw7. Three independent sgRNAs (each targeting all Fbxw7 isoforms), decreased Fbxw7 protein with concomitant increases of the Fbxw7 targets c-Myc and Cyclin E1 (Figure 2a). There was no difference in the growth rate of Fbxw7-deficient and control D4C9 cells in vitro (Figure 2b). However, Fbxw7-deficient tumors were resistant to anti-PD-1 treatment compared to isogenic matched wild-type tumors (Figure 2c, Supplementary Figure 9a–d). Animals with Fbxw7-deficient tumors also had significantly poorer survival after anti-PD-1 treatment compared to mice with wild-type tumors (Figure 2d). To evaluate the specificity of these results, we expressed a sgRNA-resistant Fbxw7α cDNA in D4C9 sgFbxw7 cells (Supplementary Figure 2e). Restoration of Fbxw7a increased the number of complete responders to anti-PD-1 as compared to Fbxw7-deficient tumors (Supplementary Figure 2f, Supplementary Figure 9j–l). While the survival of animals bearing tumors with Fbxw7 deletion was significantly poorer relative to control animals, mice with Fbxw7a-restored tumors survived similarly to controls (Supplementary Figure 2g). To evaluate whether the R505C oncogenic mutation also confers resistance to anti-PD-1, we generated D4C9 cells expressing wild-type Fbxw7 or Fbxw7(R505C). Fbxw7(R505C) expression induced the expected increase in c-Myc expression, consistent with its known dominant-negative effect (29) (Figure 2e). While wild-type Fbxw7 significantly delayed tumor growth, expression of Fbxw7(R505C) conferred resistance to PD-1 blockade (Figure 2f–g, Supplementary Figure 9e–g). Finally, to determine whether these findings applied to another cancer model, we generated an Fbxw7-deficient derivative of MC38, a colon carcinoma cell line syngeneic to C57BL/6 mice that is partially sensitive to anti-PD-1 treatment (30) (Supplementary Figure 2h). The deletion of Fbxw7 in this model also significantly diminished the response to anti-PD-1 treatment (Supplementary Figure 2i–m). Together, these data demonstrate that loss of Fbxw7 activity confers resistance to PD-1 blockade.
Figure 2. Fbxw7 is required for PD-1 blockade anti-tumor activity.
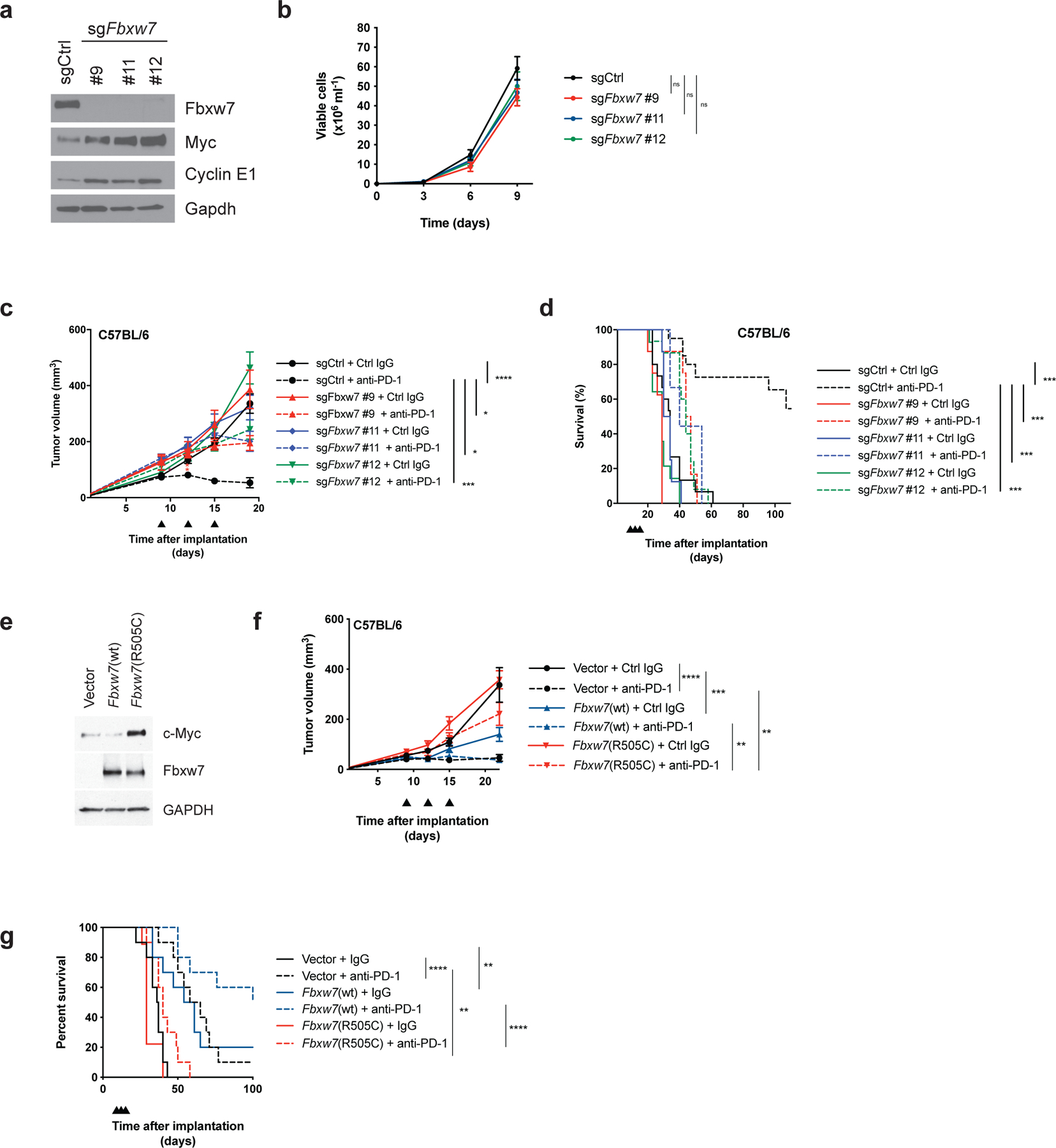
(a) Western blot of D4C9 cells transduced with sgRNAs targeting Fbxw7. (b) Graph showing viable cell number of D4C9 derivatives transduced with sgRNAs targeting Fbxw7 (n = 4 biologically independent samples per group). (c) Growth of D4C9 derivatives transduced with sgRNAs targeting Fbxw7 in C57BL/6 animals. Anti-PD-1 or control immunoglobulin (Ctrl IgG) were administered at days 9, 12, 15 (black arrowheads) after implantation (n ≥16 biologically independent samples per group). Significant differences between groups were calculated by one-way ANOVA with correction with Tukey’s multiple comparison test. ****, P < 0.0001; ***, P < 0.001; *, P<0.05. (d) Survival of C57BL/6 mice implanted with D4C9 derivatives (n ≥ 10 per group). Significant differences between groups were calculated by Log-rank (Mantel-Cox) tests with Bonferroni correction. ***, P < 0.001. (e) Western blot of D4C9 cells transduced with empty vector, wild-type Fbxw7 or Fbxw7(R505C). (f) Growth of D4C9 derivatives transduced with empty vector, wild-type Fbxw7 or Fbxw7(R505C) in C57BL/6 animals. Anti-PD-1 or control immunoglobulin (Ctrl IgG) were administered at days 9, 12, 15 (black arrowheads) after implantation (n = 16 per group). Significant differences between groups were calculated by one-way ANOVA with Sidak’s multiple comparison test. ****, P < 0.0001; ***, P < 0.001; **, P < 0.01. (g) Survival of C57BL/6 mice implanted with D4C9 derivatives (n = 9–10 per group). Significant differences between groups were calculated by Log-rank (Mantel-Cox) tests with Bonferroni correction. **, P < 0.01; ****, P < 0.0001.
Loss of Fbxw7 alters the tumor immune microenvironment
To identify the mechanisms by which Fbxw7 deficiency impairs antitumor immunity, we used the Nanostring nCounter System to measure the expression of immune-related mRNA transcripts in control and Fbxw7-deficient tumors before and after anti-PD-1 treatment (31). We found that Fbxw7 inactivation decreased a specific immune gene signature, including interferon-gamma (Ifnγ) related genes, that has been shown to correlate with responses to pembrolizumab in cancer patients (32) (Figure 3a, Supplementary Table 3). Profiling of Fbxw7 wild-type and deficient models also found that genes expressed in response to viral sensing or type I interferon stimulation were among the most significantly decreased genes in Fbxw7-deficient tumors compared to controls after anti-PD-1 treatment (Figure 3b–c). To validate these findings, we compared the levels of two viral sensing targets in Fbxw7-deficient and control tumors by quantitative PCR (qPCR). Both Cxcl10 and Ifnb1 mRNA levels were significantly decreased in tumors lacking Fbxw7 compared to control tumors (Figure 3d–e).
Figure 3. Loss of Fbxw7 is associated with altered immune microenvironment and decreased interferon signaling.
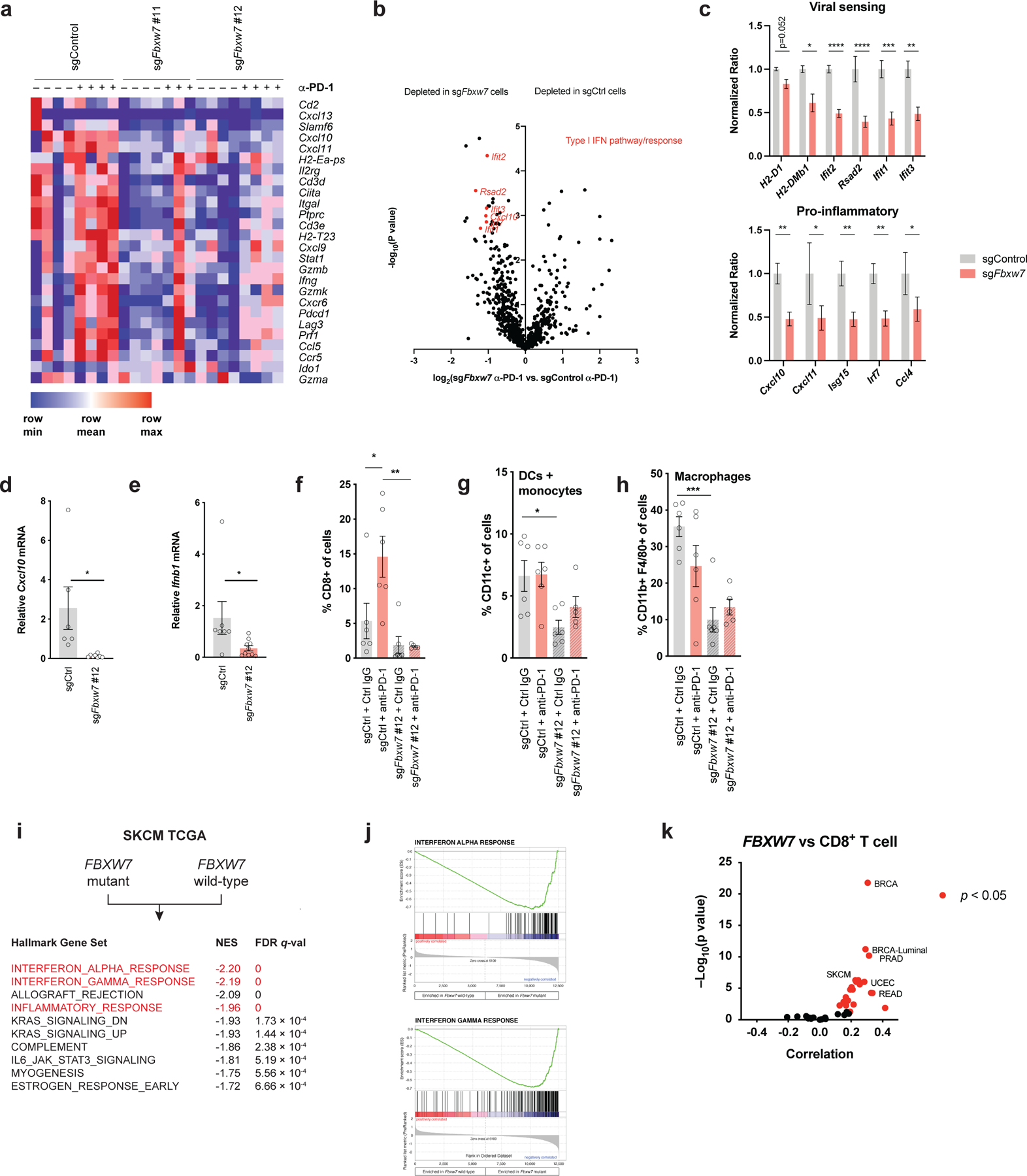
(a) Heat map showing the expression of an immune-related gene signature associated with response to pembrolizumab in Fbxw7-deficient and control D4C9 tumors pre- or post-treatment with anti-PD-1. (b) Volcano plot showing relative expression of Nanostring immune transcripts in Fbxw7-deficient versus control D4C9 tumors after treatment with anti-PD-1. Highlighted in red are genes expressed in response to interferons or viral sensing. The p-value was generated as described in nCounter Analysis System User Manual. (c) Specific viral sensing and pro-inflammatory transcript ratios in Fbxw7-deficient (n=10) and control (n=3) D4C9 tumors after treatment with anti-PD-1. Unpaired t-test was used to determine statistical significance. *, P <0.05; **, P <0.01; ***, P <0.001; ****, P <0.0001. (d-e) Relative (d) Cxcl10 and (e) Ifnb1 mRNA expression in control and Fbxw7-deficient D4C9 tumors. Each dot represents a biological replicate. Unpaired t test was used to determine statistical significance. *, P <0.05. (f-h) Flow cytometry analysis of (f) CD8+ T cell, (g) dendritic cell and monocytes, and (h) macrophage infiltration in Fbxw7-deficient and control D4C9 tumors after treatment with control isotype or anti-PD-1. Each dot represents a biological replicate. One-way ANOVA with Sidak’s multiple comparisons test was used to determine statistical significance. *, P <0.05; **, P <0.01; ***, P <0.001. (i) Gene set enrichment analysis comparing gene expression of FBXW7-mutant and wild-type tumors. Highlighted in red are the decreases of the interferon response and inflammatory signatures in FBXW7-mutated cancer. NES: Normalized Enrichment Score. (j) Enrichment score plots for the indicated gene sets. Vertical bars refer to individual genes in a gene set and their position reflects the contribution of each gene to the ES. (k) Spearman correlation of FBXW7 mRNA levels with CD8A mRNA levels across TCGA cancers. Each dot represents a cancer type in TCGA, red dots indicate significant correlation with P < 0.05. SKCM, skin cutaneous melanoma; BRCA, breast carcinoma; PRAD, prostate adenocarcinoma; UCEC, uterine corpus endometrial carcinoma; READ, rectum adenocarcinoma. Additional data in Supplementary Table 5.
As viral sensing signaling pathways impact immune cell infiltration in tumors (7,8), we evaluated the effects of Fbxw7 inactivation on the immune microenvironment. We measured the intratumoral abundance of immune cell populations in Fbxw7-deficient and control tumors from C57BL/6 mice after anti-PD-1 treatment. We found that loss of Fbxw7 significantly decreased global immune cell infiltration in tumors, altering both lymphocyte and myeloid cell infiltration (Supplementary Figure 3a–c, gating strategy shown in Supplementary Figure 10a). Fbxw7 inactivation led to diminished PD-1 blockade-induced CD8+ T cell infiltration (Figure 3f, Supplementary Figure 3d–f), consistent with the established association of intratumoral CD8+ T cell infiltration with the response to PD-1 blockade (3). We also found that the loss of Fbxw7 decreased the infiltration of dendritic cells and macrophages (Figure 3g, h, Supplementary Figure 3g). Macrophages trended towards a more immunosuppressive M2 phenotype following Fbxw7 inactivation (Supplementary Figure 3h). Together, our results show that the resistance to PD-1 blockade caused by Fbxw7 inactivation is associated with an altered tumor immune microenvironment, including decreased viral sensing and anti-tumor immune cell infiltration.
We next used The Cancer Genome Atlas (TCGA) data sets to determine the relevance of our findings to human cancers. We used gene set enrichment analysis to identify significantly dysregulated gene sets in mutant FBXW7 compared to wild-type FBXW7 melanomas. Loss-of-function mutations in FBXW7 correlated with increased Myc signaling (Supplementary Figure 3i–j) and diminished type I and type II interferon signaling (Figure 3i–j, Supplementary Table 4). We also found that expression of FBXW7 strongly correlated with CD8+ T cell infiltration in many human cancer types, including melanoma (Figure 3k, Supplementary Table 5).
Inactivation of Fbxw7 impairs dsRNA sensing and interferon signaling in tumor cells
To examine the requirement of Fbxw7 for the activation of viral sensing pathways in tumor cells, we transfected cells with low molecular weight (LMW) poly(I:C), a synthetic dsRNA analog (7). Under these conditions, we observed no significant effect of LMW poly(I:C) on cell viability (Supplementary Figure 4a). LMW poly(I:C) activated the Tbk1/Irf3 signaling pathway (Figure 4a) and induced Cxcl10 and Ifnb1 mRNA expression in control cells (Figure 4b–c), but these phenotypes were strongly diminished by the genetic deletion of Fbxw7 (Figure 4a–c). Conversely, exogenous wild-type Fbxw7 further increased LMW poly(I:C)-induced Tbk1 activation, while Fbxw7-R505C suppressed it (Figure 4d). Similarly, Cxcl10 and Ifnb1 mRNA expression were increased following wild-type Fbxw7 overexpression (Figure 4e–f). We found that replication stress, which can increase dsRNA or dsDNA levels, was not observed upon wild-type Fbxw7 overexpression (Supplementary Figure 4b). LMW poly(I:C) also induced MHC-I and Pd-L1 cell surface expression in control D4C9 cells, while Fbxw7 deletion significantly diminished these phenotypes (Figure 4g–h, gating strategy shown in Supplementary Figure 10b). Together, our results establish Fbxw7 as a novel positive regulator of dsRNA sensing, type I interferon production and MHC-I expression in melanoma. To evaluate if Fbxw7 regulates other nucleic acid sensing pathways, we treated control and Fbxw7-deficient cells with the Sting agonist ADU-S100 (33). We found that the deletion of Fbxw7 did not affect ADU-S100-mediated Tbk1/Irf3 activation (Supplementary Figure 4c), or Cxcl10 and Ifnb1 mRNA expression (Supplementary Figure 4d–e). The effects of this agonist were specific, as deletion of Sting (encoded by Tmem173) blocked the induction of Cxcl10 and Ifnb1 mRNA (Supplementary Figure 4f–g).
Figure 4. Fbxw7 is necessary for tumor-intrinsic type I interferon signaling.
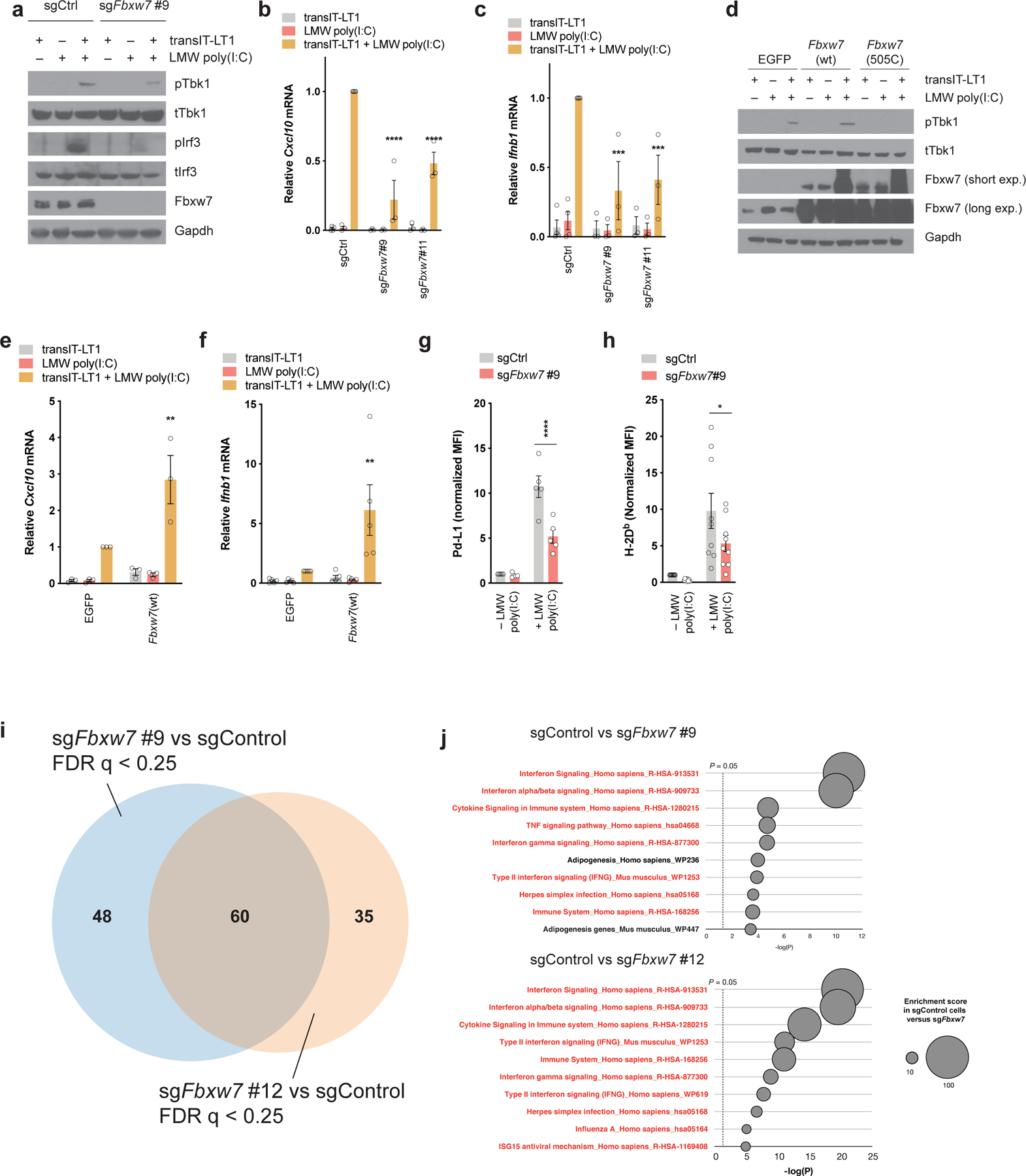
(a) Western blot of Tbk1/Irf3 signaling pathway in control and Fbxw7-deficient D4C9 cells following transfection with LMW poly(I:C) (1μg/ml) for 24h. (b-c) Relative (b) Cxcl10 and (c) Ifnb1 mRNA expression in control and Fbxw7-deficient D4C9 cells following transfection with LMW poly(I:C) (1μg/ml) for 24h. Each dot represents a biological replicate. Two-way ANOVA with Sidak’s multiple comparisons test was used to determine statistical significance, in comparison to D4C9-sgCtrl + transIT-LT1 + LMW poly (I:C). ***, P <0.001; ****, P <0.0001. (d) Western blot of Tbk1 activation in D4C9 cells overexpressing Egfp, wild-type Fbxw7 or Fbxw7(R505C) following transfection with LMW poly(I:C) (1μg/ml) for 24h. (e-f) Relative (e) Cxcl10 and (f) Ifnb1 mRNA expression in D4C9 cells overexpressing Egfp or wild-type Fbxw7, following transfection with LMW poly(I:C) (1μg/ml) for 24h. Each dot represents a biological replicate. Two-way ANOVA with Sidak’s multiple comparisons test was used to determine statistical significance, in comparison to D4C9-EGFP + transIT-LT1 + LMW poly (I:C). **, P <0.01. (g-h) Expression of (g) Pd-L1 and (h) MHC-I (H-2Db) in control and Fbxw7-deficient D4C9 cells following transfection with LMW poly(I:C) (1μg/ml) for 24h. MFI (median fluorescent intensity) was measured relative to sgCtrl. Each dot represents a biological replicate. Two-way ANOVA with Sidak’s multiple comparisons test was used to determine statistical significance. *, P <0.05; ****, P <0.0001. (i-j) RNA-seq analysis of Fbxw7-deficient and control D4C9 cells treated with Ifnγ (10ng/ml) for 24h. (i) Venn diagram showing significantly altered genes sets (false discovery rate q < 0.25) in sgFbxw7#9 and sgFbxw7#12 cells compared to control cells in presence of Ifnγ. (j) Most significantly downregulated gene sets in sgFbxw7#9 and sgFbxw7#12 cells compared to control cells in presence of Ifnγ. Highlighted in red are the gene sets related to viral sensing and interferon signaling.
Recent studies have demonstrated that Ifnγ signaling, which has also been linked to response to immunotherapy, leads to the activation of viral sensing pathways (7,8). We observed that Ifnγ stimulation also increased the abundance of endogenous dsRNA in D4C9 cells (Supplementary Figure 5a–b). Therefore, we evaluated the effect of Fbxw7 deletion on interferon regulated pathways. Ifnγ was sufficient to activate the Tbk1/Irf3 signaling pathway and to induce Cxcl10 and Ifnb1 mRNA expression in control D4C9 cells (Supplementary Figure 5c–e). The Ifnγ-mediated Cxcl10 expression was dependent on Rig-I and Sting (Supplementary Figure 5f). Conversely, Fbxw7 deletion decreased Ifnγ-induced Tbk1/Irf3 signaling and type I interferon production (Supplementary Figure 5c–e). Inactivation of Fbxw7 also suppressed Ifnγ-induced Jak1 and Stat1 phosphorylation (Supplementary Figure 5g), without affecting Jak2 levels or Ifngr1/2 cell surface expression (Supplementary Figure 5g–i). Finally, Fbxw7 deletion was associated with a decrease in Ifnγ-induced expression of MHC-I and Pd-L1 (Supplementary Figure 5g, j–k) and type I interferon-induced MHC-I expression (Supplementary Figure 5l).
To comprehensively evaluate the requirement of Fbxw7 in interferon signaling, we evaluated global gene expression after Ifnγ stimulation of Fbxw7-deficient and control cells. Gene set enrichment analysis (34) was used to identify pathways enriched or depleted in Fbxw7-deficient cells. We found that two independent sgRNAs targeting Fbxw7 affected gene expression similarly (Figure 4i). In both cases, we found that virus sensing, as well as type I and II interferon signaling pathways, were among the most significantly downregulated gene sets in Fbxw7-deficient cells compared to controls (Figure 4j). Collectively, these results establish Fbxw7 as a regulator of the dsRNA sensing and interferon signaling pathways.
Fbxw7 promotes Rig-I- and Mda5-mediated dsRNA sensing
The inactivation of Fbxw7 leads to dysregulated c-Myc, which has been associated with altered tumor immunity. To test the possibility that dysregulated c-Myc underlie the altered viral sensing observed, we suppressed c-Myc in control and Fbxw7-deficient tumors. Consistent with previous findings (35), we found that c-Myc knockdown decreased the expression of Pd-L1, as well as MHC-I. However, c-Myc deletion did not restore Ifnγ-induced Pd-L1 or MHC-I in Fbxw7-deficient tumors (Supplementary Figure 6a), indicating that c-Myc is not required for Fbxw7-mediated interferon signaling.
To determine how Fbxw7 regulates dsRNA sensing, we generated D4C9 derivatives lacking Tbk1 or Mavs using CRISPR. As a control, we generated D4C9 derivatives lacking Tmem173 (Figure 5a). Whereas expression of wild-type Fbxw7 was sufficient to enhance Ifnb1 production following transfection of dsRNA (LMW poly(I:C)) or dsDNA (poly(dA-dT)) analogs, we found that deletion of Tbk1 or Mavs significantly diminished wild-type Fbxw7-mediated Ifnb1 mRNA expression (Figure 5b–c). Similar results were obtained upon transfection with dsRNA or dsDNA, consistent with the fact that dsDNA can be converted to dsRNA by RNA polymerase III (36). In contrast, the loss of Tmem173 did not decrease wild-type Fbxw7-mediated dsRNA or dsDNA sensing (Figure 5b–c). These data confirm that Fbxw7 does not regulate the Sting-mediated dsDNA sensing pathway, and demonstrate that Tbk1 and Mavs are necessary for Fbxw7-dependent type I interferon production. Next, we functionally evaluated the requirement of Rig-I and Mda5, two upstream activators of Mavs, for Fbxw7-mediated dsRNA sensing. We generated D4C9 derivatives lacking Ddx58 or Ifih1 (Figure 5d). Cells were then transfected either with LMW poly(I:C), preferentially recognized by Rig-I, or with high molecular weight (HMW) poly(I:C), preferentially recognized by Mda5 (37). We observed that loss of Mda5 or Rig-I strongly diminished dsRNA-induced, wild-type Fbxw7-mediated Ifnb1 mRNA expression (Figure 5e–f).
Figure 5. Requirement of viral sensing proteins for Fbxw7-dependent regulation of type I interferon signaling.
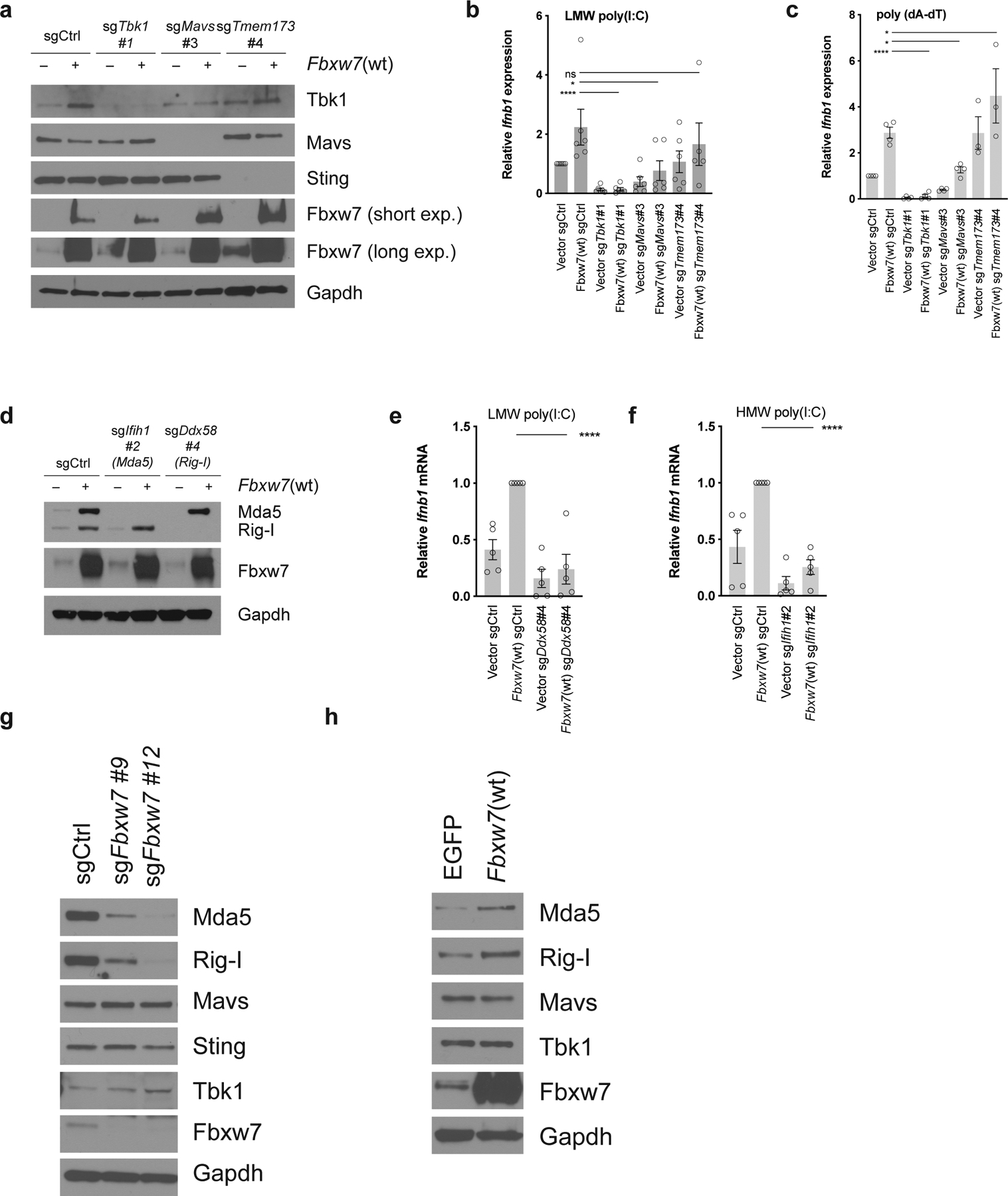
(a) Evaluation of Tbk1, Mavs and Tmem173 knock-out in D4C9 cells overexpressing Egfp or Fbxw7(wt) by western blot. (b-c) Relative Ifnb1 mRNA expression in D4C9 cells overexpressing Egfp or Fbxw7(wt) and transduced with a sgRNA targeting Tbk1, Mavs, Tmem173, or a non-targeting sgRNA, following transfection with (b) LMW poly(I:C) (1μg/ml) or (c) poly(dA-dT) (1μg/ml) for 24h. Each dot represents a biological replicate. One-way ANOVA with Sidak’s multiple comparisons test was used to determine statistical significance. *, P <0.05; ****, P <0.0001. (d) Evaluation of Ifih1 and Ddx58 knock-out in D4C9 cells overexpressing Egfp or Fbxw7(wt) by western blot. (e-f) Relative Ifnb1 mRNA expression in D4C9 cells overexpressing Egfp or Fbxw7(wt) and transduced with a sgRNA targeting Ifih1, Ddx58, or a non-targeting sgRNA, following transfection with (e) LMW poly(I:C) (1μg/ml) or (f) HMW poly(I:C) (1μg/ml) for 24h. Each dot represents a biological replicate. One-way ANOVA with Sidak’s multiple comparisons test was used to determine statistical significance. ****, P <0.0001. (g) Protein expression of components of the viral sensing pathways in control and Fbxw7-deficient D4C9 cells. (h) Protein expression of components of the dsRNA sensing pathway in D4C9 cells overexpressing Egfp or wild-type Fbxw7(wt).
In the context of anti-viral responses, Fbxw7 has been shown to promote Rig-I protein stability (19). Therefore, we evaluated the requirement of Fbxw7 for the expression of viral sensing proteins in tumor cells. The deletion of Fbxw7 led to diminished Rig-I and Mda5 protein expression without affecting Mavs, Tbk1 and Sting levels (Figure 5g). In addition, wild-type Fbxw7 was sufficient to increase Rig-I and Mda5 protein levels (Figure 5h). Together, our findings demonstrate that Fbxw7 is essential for the basal expression of viral sensors that are required for Fbxw7-mediated dsRNA sensing.
To determine whether Fbxw7 regulates Rig-I and Mda5 at the transcriptional or post-transcriptional level, we generated D4C9 cells expressing V5-tagged Ddx58 or Ifih1. We observed that Fbxw7 inactivation in these cells decreased exogenous Rig-I and Mda5 expression (Supplementary Figure 6b), suggesting that Fbxw7 controls their expression at the post-transcriptional level. Song et al (19) showed that Fbxw7 mediates the degradation of Shp2, which promotes the degradation of Rig-I. However, we found that the deletion of Ptpn11 (encoding Shp2) did not restore Rig-I or Mda5 protein expression in Fbxw7-deficient cells (Supplementary Figure 6c), and did not increase the stability of these proteins (Supplementary Figure 6d).
Restoration of dsRNA sensing increases interferon signaling and sensitizes Fbxw7-deficient tumors to anti-PD-1
To test whether altered dsRNA sensing in tumor cells causes resistance to anti-PD-1, we implanted D4C9 derivatives lacking Ddx58, Ifih1, or Mavs in C57BL/6 mice before treating animals with control IgG or anti-PD-1. We observed that loss of Ddx58, Ifih1, or Mavs impaired sensitivity of D4C9 tumors to PD-1 blockade (Figure 6a, Supplementary Figure 7a–b, Supplementary Figure 9m–p). Therefore, we hypothesized that the resistance of Fbxw7-deficient tumors to anti-PD-1 therapy is due to altered dsRNA sensing. To test this, we evaluated whether restoration of the dsRNA sensing pathway was sufficient to sensitize Fbxw7-deficient tumors to PD-1 blockade. As both Mda5 and Rig-I deletion diminished Fbxw7-mediated dsRNA sensing (see Figure 5e–f), we evaluated whether the expression of Mavs could restore type I interferon production in Fbxw7-deficient cells. We generated Fbxw7-deficient and control D4C9 cells expressing either an empty vector, Egfp, or exogenous Mavs (Figure 6b). Expressed Mavs formed aggregates (Supplementary Figure 7c) and was sufficient to induce Cxcl10 and Ifnb1 mRNA expression (Figure 6c–d) and MHC-I and Pd-l1 cell surface expression (Figure 6e–g), consistent with its functional activation. Mavs overexpression did not impact cell viability (Supplementary Figure 7d–e). The impact of Mavs overexpression was milder in Fbxw7-deficient cells compared to controls. This could be due to the required post-translational modification of viral sensors for their activation (38). To evaluate whether restoration of the dsRNA sensing pathway was sufficient to sensitize Fbxw7-deficient tumors to anti-PD-1, we then implanted control and Fbxw7-deficient D4C9 cells overexpressing Mavs or a control vector in C57BL/6 mice. Consistent with our data showing that wild-type Fbxw7 delays tumor growth (see Figure 2f), we found that Mavs overexpression was sufficient to delay the growth of control and Fbxw7-deficient tumors. Importantly, Mavs overexpression sensitized Fbxw7-deficient tumors to PD-1 blockade (Figure 6h, Supplementary Figure 9q–t) and prolonged the survival of anti-PD-1 treated, Fbxw7-deficient tumor-bearing mice (Figure 6i). This was associated with significant changes in the tumor immune microenvironment, such as an increase in CD11c+ cells in both control tumors and Fbxw7-deficient tumors treated with anti-PD-1 (Supplementary Figure 7f–h). Overall, our results suggest that intact dsRNA sensing promotes anti-tumor immunity and response to immunotherapy.
Figure 6. Restoration of viral sensing increases interferon signaling and sensitizes Fbxw7-deficient tumors to anti-PD-1.
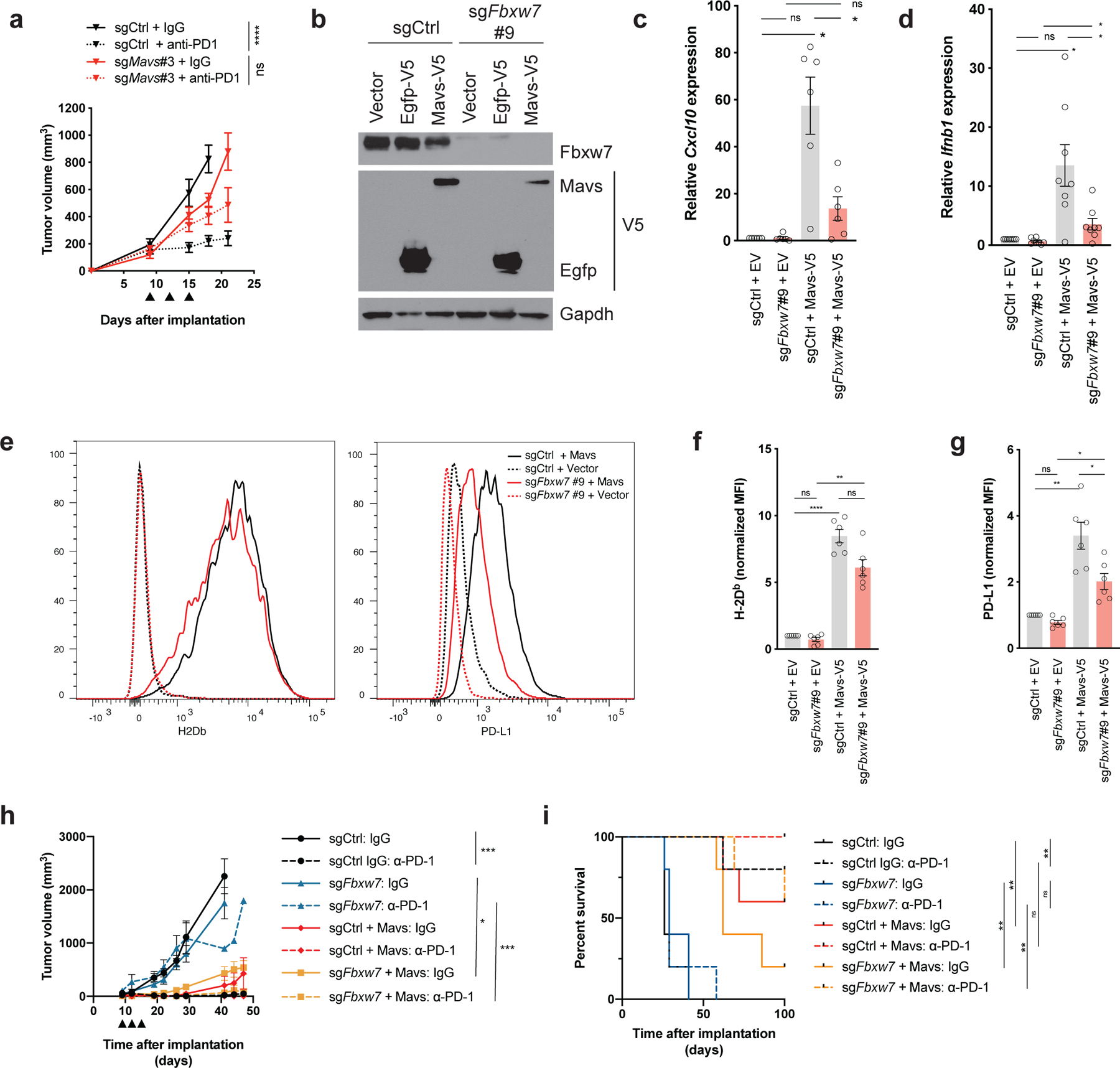
(a) Growth of control or Mavs-deficient D4C9 cells in C57BL/6 mice. Anti-PD-1 or control immunoglobulin were administered at days 9, 12, 15 (black arrowheads) after implantation (n ≥ 8 tumors per group). One-way ANOVA with Sidak’s multiple comparison test was used to determine statistical significance. ****, P<0.0001. (b) Western blot of control and Fbxw7-deficient D4C9 cells transduced with Egfp- or Mavs-expressing virus. (c, d) Relative (c) Cxcl10 or (d) Ifnb1 mRNA expression in control and Fbxw7-deficient D4C9 cells overexpressing Mavs or an empty vector. Each dot represents a biological replicate. RM one-way ANOVA with Sidak’s multiple comparison test was used to determine statistical significance *, P <0.05. (e-g) Expression of (e, f) MHC-I (H-2Db) and (e, g) Pd-L1 in control and Fbxw7-deficient D4C9 cells overexpressing Mavs or an empty vector. (e) One representative experiment is shown. (f, g) MFI (median fluorescent intensity) was measured relative to sgCtrl + EV (Empty Vector). Each dot represents a biological replicate. RM one-way ANOVA with Sidak’s multiple comparison test was used to determine statistical significance. ****, P <0.0001; **, P <0.01; *, P <0.05. (h) Growth of control and Fbxw7-deficient D4C9 cells transduced with Mavs cDNA or an empty vector, in C57BL/6 animals. Anti-PD-1 or control immunoglobulin were administered at days 9, 12, 15 (black arrowheads) after implantation (n ≥ 8 biologically independent samples per group). One-way ANOVA with Sidak’s multiple comparison test was used to determine statistical significance. ***, P<0.001, *, P <0.05. (i) Survival of C57BL/6 mice implanted with D4C9 derivatives (n = 5 per group). Significant differences between groups were calculated by Log-rank (Mantel-Cox) tests with Bonferroni correction. **, P < 0.01.
To further investigate whether restoration of interferon signaling was sufficient to sensitize Fbxw7-deficient tumors to PD-1 blockade, we overexpressed Irf1 in Fbxw7-deficient and control cells (Supplementary Figure 7i). Irf1 expression was sufficient to increase the levels of Pd-L1 and MHC-I expression (Supplementary Figure 7j). Whereas Fbxw7-deficient tumors were resistant to PD-1 blockade, Fbxw7-deficient tumors overexpressing Irf1 were sensitive to this treatment, similar to control tumors (Supplementary Figure 7k–l, Supplementary Figure 9u–w). Taken together, these findings show that Mavs and Irf1 overexpression similarly induce MHC-I and Pd-L1 expression and sensitize Fbxw7-deficient tumors to PD-1 blockade. They also demonstrate that intact dsRNA sensing further impacts anti-tumor immunity by delaying the growth of D4C9 tumors.
DISCUSSION
This study establishes, for the first time, a role for the tumor suppressor gene FBXW7 in anti-tumor immunity. Using a syngeneic, immunocompetent anti-PD-1 sensitive melanoma mouse model, we have demonstrated that Fbxw7 is required for dsRNA sensing and response to PD-1 blockade therapy (Supplementary Figure 8). Importantly, our findings suggest that restoration of the viral sensing signaling pathway by overexpression of Mavs, sensitizes Fbxw7-deficient tumors to anti-PD-1.
By functionally characterizing a single exceptional responder to pembrolizumab, we provided biologic insight into mechanisms of immunotherapy responses, and identified Fbxw7 loss of function as a mechanism of resistance to PD-1 blockade. Although our work nominates Fbxw7 as a putative, clinically relevant biomarker of anti-PD-1 response, larger studies in patient populations are needed to evaluate its predictive value reliably. Similarly, the dysregulation of viral sensing pathways in large patient cohorts could be investigated.
Song et al. (19) have previously observed that Fbxw7 regulates the stability of Rig-I in the context of viral stimulation. They found that Fbxw7 mediates the degradation of Shp2, a negative regulator of Rig-I. Consistent with their findings, we observe that Fbxw7 is required for the expression of Rig-I. However, we found that Fbxw7 loss also affects the levels of Mda5, which was not seen in their model. It is notable that many regulators of Mda5 and Rig-I stability are shared, due to their significant protein similarity. Our findings suggest that Shp2 does not mediate the degradation of Rig-I. The mechanism by which Fbxw7 regulates Rig-I and Mda5 in tumor cells remains to be fully elucidated. The identification of a direct modulator of their expression could enable rational therapeutics that restore dsRNA sensing and immunotherapy response in genomically-defined patient populations.
In agreement with recent studies (8,9,39), our findings demonstrate the requirement of intact dsRNA sensing for MHC-I expression and response to immune checkpoint blockade. Furthermore, our in vivo experiments show that overexpression of Mavs is sufficient to delay the growth of tumors even in the absence of anti-PD-1 therapy. Therefore, it will be of therapeutic interest to comprehensively understand the mechanisms by which the activation of viral sensing pathways promotes tumor immunity.
Our findings have important therapeutic implications. First, they reinforce the value of activating the dsRNA sensing pathways to exert an anti-tumor effect in solid cancers. Therapeutic strategies such as synthetic TLR3, MDA5, or RIG-I agonists (40) are currently being tested in clinical trials for this purpose (41). Furthermore, we provide potential therapeutic strategies for sensitizing tumors with altered viral sensing to PD-1 blockade. However, our results suggest that such therapies may need to be selected for specific patient populations. For example, in Fbxw7-deficient tumors, combining anti-PD-1 with an agonist activating either a dsDNA or a putative dsRNA sensor that is not under the control of Fbxw7, such as Sting (42) may be advantageous. Reactivation of viral sensing signaling could also be achieved by using viruses (43), or epigenetic modulators such as DNMT inhibitors (44).
Other genomic aberrations, beyond Fbxw7 loss of function, have been shown to alter viral sensing pathways and impact the efficacy of immune checkpoint inhibitors. Recent studies demonstrated that inactivation of the tumor suppressor STK11/LKB1 decreases STING expression and dsDNA sensing (45), while the loss of Stk11/Lkb1 in tumor cells promotes resistance to PD-1 blockade (12). Together, these findings suggest that the regulation of viral sensing pathways may be a common mechanism by which tumor suppressors impact tumor immunity. They provide further rationale for combining immune checkpoint blockade therapy with selected dsDNA or dsRNA agonists to overcome resistance to immunotherapy in genomically-defined cancer patient populations.
METHODS
Patient biopsies
We obtained prior, written informed consent from each patient for biopsy collection and analysis. The study was conducted in accordance with recognized ethical guidelines and approved by the Dana-Farber Cancer Institute Institutional Review Board.
Cell lines
D4M3A cells (a gift of David Fisher, Massachusetts General Hospital) were cultured in DMEM supplemented with 5% fetal bovine serum (FBS) and penicillin-streptomycin (PS). MC38 cells (gift of Robin Riley, National Institutes of Health) were grown in RPMI-1640 supplemented with 5% FBS, PS and glutamine. Cells were incubated at 37°C at 5% CO2. Cells were tested monthly and found to be free of mycoplasma using PCR-based screening (Applied Biological Materials). Experiments were performed within 25 cell passages after thawing.
Cell treatment and transfection
Mouse Ifnγ (StemCell), reconstituted in PBS 0.1% BSA was used at a final concentration of 0.1ng/ml or 10mg/ml. ADU-S100 (Chemietek) reconstituted in water was used at final concentration of 10μM. LMW-poly(I:C), HMW-poly(I:C) and poly(dA-dT) (Invivogen) and reconstituted in sterile endotoxin-free water. These compounds were transfected at a final concentration of 1μg/ml using the TransIT-LT1 Transfection Reagent (Mirus).
Generation of D4M3A-Cas9 (D4C9) and MC38-Cas9 cells and derivatives
The lentiviral Cas9-blast (pXPR_101; Broad Institute of Harvard and MIT) vector was co-transfected with packaging plasmids PAX2 and pMD2.G into Lenti-X cells (Clontech) using TransIT-LT1. D4M3A and MC38 cells were infected with Cas9-blast lentivirus, then selected with 10μg/ml of blasticidin (InvivoGen). Single-cell clones were selected for high efficiency editing. sgRNA oligos (sequences in Supplementary Table 6) were annealed and cloned into LentiGuide-hygro (a derivative of LentiGuide-puro). Cas9-expressing clones were infected with lentivirus, followed by drug selection in 150μg/ml hygromycin (InvivoGen). An early passage, pooled hygromycin-resistant population of cells were used for all experiments.
The murine wild-type Fbxw7α cDNA in the pENTR1A vector was purchased from GenScript (Piscataway, NJ). The pDONR221 Egfp was purchased from Addgene (#25899). The empty vector control for viruses was from Eric Campeau & Paul Kaufman (pENTR4 no ccdB, Addgene #17424). The pCMV-SPORT6-Mavs was purchased from PlasmID (Dana-Farber/Harvard Cancer Center). The murine Irf1 cDNA cloned in the pDONR vector was obtained from GeneCopoeia, Inc (Rockville, MD). Site-directed mutagenesis was performed using In-Fusion HD (Clontech). Primer sequences are in Supplementary Table 6. Expression constructs in the pLX304_zeo vector were generated using the Gateway cloning system. All plasmids and their derivatives were verified by sequencing. D4C9 cells were stably infected with lentivirus, then selected with 400 μg/ml of zeocin (InvivoGen).
Cell proliferation and viability
Cells were seeded at day 0 and harvested at days 3, 6 and 9, or transfected with LMW poly(I:C) at day 1 and harvested at days 2 and 3. Trypan Blue was added to measure viability. Cell proliferation and viability were measured using the Vi-Cell XR (Beckman Coulter).
Animal experiments
5 × 105 D4M3A cells (or derivatives) or 1.2 × 105 MC38 cells (or derivatives) were subcutaneously injected into both flanks of 6 weeks-old male C57BL/6 mice (Charles River Laboratories). Nine days after implantation, mice were randomized into two groups and treated with 200μg of either control Rat IgG2α (BioXCell) or anti-PD-1 (clone 29F.1A12 (46)) by intraperitoneal injection at days 9, 12 and 15. Tumor volume was measured twice a week using a digital caliper. Individual growth curves are shown in Supplementary Figure 9. Mice were sacrificed when tumor volume reached 1000mm3 and overall survival was monitored. All experiments were performed in compliance with federal laws and institutional guidelines and were approved by the Animal Care and Use Committee of the Dana-Farber Cancer Institute.
Antibodies
Please refer to Supplementary Table 7 for antibodies used.
Western blot and immunoprecipitation
SDS-PAGE, Western blots and immunoprecipitation were conducted as described (47). SDD–AGE was performed according to published protocols (48) with minor changes. Cells were isolated in Buffer A (10 mM Tris-HCl, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 0.25 M D-mannitol, and protease inhibitor cocktail) and homogenized using a 28G needle syringe, prior to centrifugation at 700g for 10 min at 4 °C. Supernatant was transferred and centrifuged at 10,000g for 30 min at 4 °C. The pellet, containing the crude mitochondria was resuspended in 4x sample buffer and loaded onto a 1.5% agarose gel. After electrophoresis in the running buffer (1x TBE, 0.1% SDS), the proteins were transferred to a nitrocellulose membrane (49).
Flow cytometry
Cells were treated with murine Ifnγ or transfected with LMW poly(I:C) at the indicated concentrations. 24 hours later, cells were stained with antibodies for the proteins of interest or with a control IgG, followed by FACS analysis. BD FACSCanto II or BD LSRFortessa were used for data acquisition and FlowJo was used for data analysis. The median of fluorescence intensity (MFI) was calculated using FlowJo. Gating strategy is provided in Supplementary Figure 10.
mRNA extraction and quantitative RT-PCR
Total RNA was extracted from tumors using the RNeasy Plus Mini kit (Qiagen). Total RNA was extracted from cells using the TRIzol reagent (Ambion). RT-PCR was performed using the iTaq Universal SYBR Green One-Step kit (Bio-Rad), and amplification was measured with a LightCycler 96 (Roche). Expression levels were normalized to 18s. Primer sequences are in Supplementary Table 6.
NanoString assay
Nine days after subcutaneous injection of tumor cells (see above), mice were randomized into two groups and either sacrificed (pre-treatment) or treated with 200 μg of anti-PD-1 at days 9, 12 and 15 prior to tumor harvest at day 16 (post-treatment). Total RNA from tumors was submitted to the CAMD Research Core at Brigham and Women’s Hospital for mRNA profiling using the nCounter Mouse PanCancer Immune Profiling Panel (NanoString Technologies). The analysis was done using the Advanced Analysis Module of nSolver.
Immunofluorescence
D4C9-sgCtrl cells were plated on glass coverslips and treated with Ifnγ (10ng/ml) for 24h. Cells were fixed in 5% (v/v) formaldehyde, 0.25% (w/v) Triton X-100 and incubated overnight at 4 °C with an anti-dsRNA antibody, prior to incubation with an anti-mouse IgG2α conjugated with Alexa Fluor 488, and staining with 6-diamidino-2-phenylindole (DAPI). Slides were imaged with a NIS-Element BR 4.60.00 (Nikon). Intensity of signal was measured using the NIS-Element BR 4.60.00 analysis software. Quantification of cell number was performed by Image J. Signal intensity was normalized by cell number.
Immunohistochemistry
Nine days after implantation, mice were randomized into two groups and either sacrificed (pre-treatment) or treated with 200μg of anti-PD-1 as above. Post-treatment tumors were obtained at day. Harvested tumors were processed as described (47). Analysis was performed by counting the number of CD3, CD8 and F4/80 positive cells per mm2 of tumor.
Tumor digestion and multi-parameter flow cytometry
Mice harboring tumors were treated with 200μg of either control isotype or anti-PD-1 at days 13, 16 and 19 prior to sacrifice at day 20. Tumors were harvested, minced, blended with the gentleMACS Dissociator (Miltenyi Biotec), and digested with the MACS Miltenyi Tumor Dissociation Kit (Miltenyi Biotec) according to the manufacturer’s instructions. Tumor cells were washed with RPMI-1640 medium and lysed with RBC Lysis Solution (Qiagen), prior to resuspension in FACS buffer: PBS (Life Technologies) containing 0.5% BSA and 2 mmol/L EDTA (Sigma-Aldrich). The Zombie Aqua Fixable Viability Kit (Biolegend) was applied to cells in combination with anti-mouse CD16/CD32 blocking antibody (Biolegend) for 20 minutes in the dark on ice, prior to incubation with primary antibodies for 1 hour in the dark on ice. Cells were fixed and permeabilized using the FOXP3/Transcription Factor Staining Buffer Set (eBiosciences), according to the manufacturer’s guidelines, and incubated with antibodies for intracellular antigens 30 minutes in the dark at room temperature. Cells were washed, resuspended in FACS buffer, and analyzed using a BD LSRFortessa flow cytometer. Compensation was performed manually on BD FACSDiva using single color and unstained controls. Signal threshold definition was defined using all-stained and unstained controls. Analysis was performed on FlowJo V10.5.0. Gating strategy is provided in Supplementary Figure 10.
Whole exome analysis
DNA extraction, sequencing, and whole exome analysis were performed as previously described (50–52). Somatic nucleotide polymorphisms (SNPs) were identified by MuTect. Mutational clonality was estimated by ABSOLUTE, which uses allelic fraction of called mutations and allelic copy number information to determine mutational clonality and overall tumor purity and ploidy. Clonal mutations were defined as those with estimated cancer cell fraction (CCF) of 1 or those whose probability of being clonal exceeded the probability of being subclonal. For copy number analysis, copy ratios were calculated for each captured target by dividing the tumor coverage by the median coverage obtained in a set of reference normal samples. Mutational signature deconvolution was conducted using a non-negative matrix factorization technique as previously described(52). Mutational signatures were chosen from those previously described in COSMIC (http://cancer.sanger.ac.uk/cosmic/signatures). For neoantigen prediction, the 4-digit HLA type for each sample was inferred using Polysolver. Putative neoantigens were predicted by defining all novel amino acid 9-mers and 10-mers resulting from each somatic nonsynonymous point mutation and determining whether the predicted binding rank—a proxy for predicted binding affinity to the patient’s germline HLA alleles—was < 2%(53). Strong binders had rank < 0.5%, while weak binders had rank between 0.5% and 2% using NetMHCpan (v3.0).
Association of FBXW7 mutations with survival and gene expression
For analyzing correlation of FBXW7 with cytotoxic T lymphocytes, we calculated Spearman’s correlation and estimated statistical significance using TIMER. To evaluate the correlation of FBXW7 mutations with gene expression, we compared Gene Set enrichment analysis (hallmark gene set) on ranked list of genes correlated with FBXW7 mutant melanomas versus non-mutated melanomas.
RNA sequencing analysis
Control and Fbxw7-deficient cells were stimulated with mouse Ifnγ 10ng/ml or vehicle for 24h. Total RNA was extracted using the RNeasy Plus Mini Kit (Qiagen) following the manufacturer’s protocol. Total RNA was submitted to the Molecular Biology Core Facility at DFCI for sequencing. Raw reads were aligned using Gene counts to produce count tables for each gene. Differential gene analysis was performed on gene raw counts with edgeR package bioConductor. Read count table was filtered so that each gene had a minimum of 1 count across all conditions. Other analyses, including Gene Set Enrichment Analysis were performed using Bioconductor. Raw RNA sequencing will be deposited in the GEO database.
Statistical Analysis
The results are presented as the mean ± s.e.m. Statistical significance was assessed using GraphPad Prism software. P<0.05 was considered statistically significant. ns: statistically non significant.
Supplementary Material
SIGNIFICANCE.
Our findings establish a role of the commonly inactivated tumor suppressor FBXW7 as a genomic driver of response to anti-PD-1 therapy. Fbxw7 loss promotes resistance to anti-PD-1 through the downregulation of viral sensing pathways, suggesting that therapeutic reactivation of these pathways could improve clinical responses to checkpoint inhibitors in genomically-defined cancer patient populations.
ACKOWLEDGMENTS
R.H. acknowledges funding from the Melanoma Research Foundation, the O’Connor-Macgregor Fund for Melanoma Research, and a Stand Up To Cancer (SU2C) Innovative Research Grant (Grant #SU2C-AACR-IRG 16-17). Stand Up to Cancer is a division of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C. E.V.A. acknowledges funding from BroadNext10 and NIH R01CA227388. D.L. is funded by the Damon Runyon Cancer Foundation Physician Scientist Training Grant, the Conquer Cancer Foundation, and the Society for Immunotherapy of Cancer-Bristol-Myers Squibb Postdoctoral Cancer Immunotherapy Translational Fellowship. G.J.F. acknowledges funding from NCI (P50CA101942). W.M. is funded by the Damon Runyon-Rachleff Innovator Award.
The authors would like to acknowledge the DFCI Oncology Data Retrieval System (OncDRS) for the aggregation, management and delivery of the clinical and operational research data used in this project. We also thank Jennifer L. Guerriero and Adam N. R. Cartwright for help with multi-parameter flow cytometry and immunological aspects of this work. The content is solely the responsibility of the authors.
Conflict of interest disclosures:
R.H. has received research grants from Bristol-Myers- Squibb and Novartis, and is a consultant for Tango Therapeutics. G.J.F. has patents and/or pending royalties on the PD-1/PD-L1 pathway from Roche, Merck MSD, Bristol-Myers-Squibb, Merck KGA, Boehringer-Ingelheim, AstraZeneca, Dako, Leica, Mayo Clinic, and Novartis, and has served on advisory boards for Roche, Bristol-Myers-Squibb, Xios, Origimed, Triursus, iTeos, and NextPoint. G.J.F. has equity in Nextpoint, Triursus, and Xios. D.B. has received research grants from Bristol-Myers- Squibb, Lilly and Novartis, and is a consultant for Tango Therapeutics F.S.H. is a consultant for Bristol-Myers Squibb, Merck, EMD Serono, Novartis, Genetech, Bayer, Aduro, Partners Therapeutics, Sanofi, Pfizer, Kairos, Psioxus Therapeutics, Pieris Therapeutics; member of scientific advisory board of Apricity, Torque, Bicara; member of the advisory board of Takeda, Surface, Compass Therapeutics, Pionyr, 7 Hills Pharma, Verastem, Rheos, Amgen.
REFERENCES
- 1.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science 2018;359(6382):1350–5 doi 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017;168(4):707–23 doi 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515(7528):568–71 doi 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-gamma Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016;167(2):397–404 e9 doi 10.1016/j.cell.2016.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med 2016;375(9):819–29 doi 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane JP, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun 2017;8(1):1136 doi 10.1038/s41467-017-01062-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Canadas I, Thummalapalli R, Kim JW, Kitajima S, Jenkins RW, Christensen CL, et al. Tumor innate immunity primed by specific interferon-stimulated endogenous retroviruses. Nat Med 2018;24(8):1143–50 doi 10.1038/s41591-018-0116-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ishizuka JJ, Manguso RT, Cheruiyot CK, Bi K, Panda A, Iracheta-Vellve A, et al. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 2019;565(7737):43–8 doi 10.1038/s41586-018-0768-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell 2018;174(3):549–63 e19 doi 10.1016/j.cell.2018.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015;523(7559):231–5 doi 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 11.Sweis RF, Spranger S, Bao R, Paner GP, Stadler WM, Steinberg G, et al. Molecular Drivers of the Non-T-cell-Inflamed Tumor Microenvironment in Urothelial Bladder Cancer. Cancer Immunol Res 2016;4(7):563–8 doi 10.1158/2326-6066.CIR-15-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov 2018;8(7):822–35 doi 10.1158/2159-8290.CD-18-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer 2008;8(2):83–93 doi 10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- 14.Aydin IT, Melamed RD, Adams SJ, Castillo-Martin M, Demir A, Bryk D, et al. FBXW7 mutations in melanoma and a new therapeutic paradigm. J Natl Cancer Inst 2014;106(6):dju107 doi 10.1093/jnci/dju107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yeh CH, Bellon M, Nicot C. FBXW7: a critical tumor suppressor of human cancers. Mol Cancer 2018;17(1):115 doi 10.1186/s12943-018-0857-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akhoondi S, Sun D, von der Lehr N, Apostolidou S, Klotz K, Maljukova A, et al. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res 2007;67(19):9006–12 doi 10.1158/0008-5472.CAN-07-1320. [DOI] [PubMed] [Google Scholar]
- 17.Davis RJ, Welcker M, Clurman BE. Tumor suppression by the Fbw7 ubiquitin ligase: mechanisms and opportunities. Cancer Cell 2014;26(4):455–64 doi 10.1016/j.ccell.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.King B, Trimarchi T, Reavie L, Xu L, Mullenders J, Ntziachristos P, et al. The ubiquitin ligase FBXW7 modulates leukemia-initiating cell activity by regulating MYC stability. Cell 2013;153(7):1552–66 doi 10.1016/j.cell.2013.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song Y, Lai L, Chong Z, He J, Zhang Y, Xue Y, et al. E3 ligase FBXW7 is critical for RIG-I stabilization during antiviral responses. Nat Commun 2017;8:14654 doi 10.1038/ncomms14654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gurtler C, Bowie AG. Innate immune detection of microbial nucleic acids. Trends Microbiol 2013;21(8):413–20 doi 10.1016/j.tim.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elion DL, Cook RS. Harnessing RIG-I and intrinsic immunity in the tumor microenvironment for therapeutic cancer treatment. Oncotarget 2018;9(48):29007–17 doi 10.18632/oncotarget.25626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 2012;30(5):413–21 doi 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brastianos PK, Carter SL, Santagata S, Cahill DP, Taylor-Weiner A, Jones RT, et al. Genomic Characterization of Brain Metastases Reveals Branched Evolution and Potential Therapeutic Targets. Cancer Discov 2015;5(11):1164–77 doi 10.1158/2159-8290.CD-15-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015;31(16):2745–7 doi 10.1093/bioinformatics/btv195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009;4(7):1073–81 doi 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 26.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7(4):248–9 doi 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jenkins MH, Steinberg SM, Alexander MP, Fisher JL, Ernstoff MS, Turk MJ, et al. Multiple murine BRaf(V600E) melanoma cell lines with sensitivity to PLX4032. Pigment Cell Melanoma Res 2014;27(3):495–501 doi 10.1111/pcmr.12220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cong L CRISPR: Groundbreaking technology for RNA-guided genome engineering. Anal Biochem 2017;532:87–9 doi 10.1016/j.ab.2017.05.005. [DOI] [PubMed] [Google Scholar]
- 29.Bailey ML, Singh T, Mero P, Moffat J, Hieter P. Dependence of Human Colorectal Cells Lacking the FBW7 Tumor Suppressor on the Spindle Assembly Checkpoint. Genetics 2015;201(3):885–95 doi 10.1534/genetics.115.180653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Juneja VR, McGuire KA, Manguso RT, LaFleur MW, Collins N, Haining WN, et al. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med 2017;214(4):895–904 doi 10.1084/jem.20160801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Danaher P, Warren S, Dennis L, D’Amico L, White A, Disis ML, et al. Gene expression markers of Tumor Infiltrating Leukocytes. J Immunother Cancer 2017;5:18 doi 10.1186/s40425-017-0215-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 2017;127(8):2930–40 doi 10.1172/JCI91190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol 2015;15(12):760–70 doi 10.1038/nri3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545–50 doi 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016;352(6282):227–31 doi 10.1126/science.aac9935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chiu YH, Macmillan JB, Chen ZJ. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 2009;138(3):576–91 doi 10.1016/j.cell.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, et al. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med 2008;205(7):1601–10 doi 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang X, Kinch LN, Brautigam CA, Chen X, Du F, Grishin NV, et al. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity 2012;36(6):959–73 doi 10.1016/j.immuni.2012.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ruzicka M, Koenig LM, Formisano S, Boehmer DFR, Vick B, Heuer EM, et al. RIG-I-based immunotherapy enhances survival in preclinical AML models and sensitizes AML cells to checkpoint blockade. Leukemia 2020;34(4):1017–1026 doi 10.1038/s41375-019-0639-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aznar MA, Planelles L, Perez-Olivares M, Molina C, Garasa S, Etxeberria I, et al. Immunotherapeutic effects of intratumoral nanoplexed poly I:C. J Immunother Cancer 2019;7(1):116 doi 10.1186/s40425-019-0568-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vanpouille-Box C, Hoffmann JA, Galluzzi L. Pharmacological modulation of nucleic acid sensors - therapeutic potential and persisting obstacles. Nat Rev Drug Discov 2019;18(11):845–867 doi 10.1038/s41573-019-0043-2. [DOI] [PubMed] [Google Scholar]
- 42.Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med 2015;7(283):283ra52 doi 10.1126/scitranslmed.aaa4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cai H, Wang C, Shukla S, Steinmetz NF. Cowpea Mosaic Virus Immunotherapy Combined with Cyclophosphamide Reduces Breast Cancer Tumor Burden and Inhibits Lung Metastasis. Adv Sci (Weinh) 2019;6(16):1802281 doi 10.1002/advs.201802281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015;162(5):974–86 doi 10.1016/j.cell.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kitajima S, Ivanova E, Guo S, Yoshida R, Campisi M, Sundararaman SK, et al. Suppression of STING Associated with LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discov 2019;9(1):34–45 doi 10.1158/2159-8290.CD-18-0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rodig N, Ryan T, Allen JA, Pang H, Grabie N, Chernova T, et al. Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis. Eur J Immunol 2003;33(11):3117–26 doi 10.1002/eji.200324270. [DOI] [PubMed] [Google Scholar]
- 47.Montero J, Gstalder C, Kim DJ, Sadowicz D, Miles W, Manos M, et al. Destabilization of NOXA mRNA as a common resistance mechanism to targeted therapies. Nat Commun 2019;10(1):5157 doi 10.1038/s41467-019-12477-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu B, Zhang M, Chu H, Zhang H, Wu H, Song G, et al. The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63-linked polyubiquitination. Nat Immunol 2017;18(2):214–24 doi 10.1038/ni.3641. [DOI] [PubMed] [Google Scholar]
- 49.Halfmann R, Lindquist S. Screening for amyloid aggregation by Semi-Denaturing Detergent-Agarose Gel Electrophoresis. J Vis Exp 2008(17):e838 doi 10.3791/838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu D, Schilling B, Liu D, Sucker A, Livingstone E, Jerby-Amon L, et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat Med 2019;25(12):1916–27 doi 10.1038/s41591-019-0654-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.George S, Miao D, Demetri GD, Adeegbe D, Rodig SJ, Shukla S, et al. Loss of PTEN Is Associated with Resistance to Anti-PD-1 Checkpoint Blockade Therapy in Metastatic Uterine Leiomyosarcoma. Immunity 2017;46(2):197–204 doi 10.1016/j.immuni.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim J, Mouw KW, Polak P, Braunstein LZ, Kamburov A, Kwiatkowski DJ, et al. Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat Genet 2016;48(6):600–6 doi 10.1038/ng.3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miao D, Margolis CA, Vokes NI, Liu D, Taylor-Weiner A, Wankowicz SM, et al. Genomic correlates of response to immune checkpoint blockade in microsatellite-stable solid tumors. Nat Genet 2018;50(9):1271–81 doi 10.1038/s41588-018-0200-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.