

Abstract
The year 2021 marks the centennial of Banting and Best’s landmark description of the discovery of insulin. This discovery and insulin’s rapid clinical deployment effectively transformed type 1 diabetes from a fatal diagnosis into a medically manageable chronic condition. In this Review, we describe key accomplishments leading to and building on this momentous occasion in medical history, including advancements in our understanding of the role of insulin in diabetes pathophysiology, the molecular characterization of insulin and the clinical use of insulin. Achievements are also viewed through the lens of patients impacted by insulin therapy and the evolution of insulin pharmacokinetics and delivery over the past 100 years. Finally, we reflect on the future of insulin therapy and diabetes treatment, as well as challenges to be addressed moving forward, so that the full potential of this transformative discovery may be realized.
In 2021, the world celebrates the 100th anniversary of the discovery of insulin, a treatment that transformed type 1 diabetes from a once-fatal diagnosis into a chronic, medically manageable condition. Beyond its immediate therapeutic impact, insulin has served as the centerpiece for incredible advances in the fields of crystallography, molecular biology, prohormone processing, autoimmunity, physiology, and precision health and genetics, while forming the basis for four Nobel Prizes. In honor of this centennial, we commemorate the unlikely scientific journey that led to insulin’s discovery, chronicle the subsequent molecular characterization of the insulin molecule, which has permitted new insulin-based therapeutics, and describe the parallel clinical discoveries that have forged our contemporary understanding of diabetes classification and etiology.
The discovery of insulin
The events surrounding the discovery of insulin are well chronicled. Michael Bliss summarized it perfectly in his 1982 history describing them as “richly dramatic”, both for the “medical miracle” of resurrecting people near death by a “magical elixir of life” and for the incredible scientific journey that ended with the successful extraction of pancreatic insulin and its rapid clinical use1,2. The story’s dramatic arc is one woven together by stubborn determination, numerous experimental failures, recurrent serendipity and, ultimately, disputed academic credit. At its center is a pair of unlikely protagonists, Frederick Banting, a surgeon with no apparent formal research experience, and Charles Best, a medical student who won a coin toss for the assignment to work with Banting on a summer research project. After reading an article on the pancreas, Banting appealed to and ultimately received support and advice from J. J. R. Macleod, a Professor of Physiology at the University of Toronto, to begin a project with a simple premise. He proposed to perform surgical ligation of the canine pancreatic duct to isolate the organ’s internal secretions3–8. He aimed to use these secretions for the treatment of diabetes.
At the time Banting and Best began their experiments in May 1921, diabetes was understood to be a disease of the pancreas. The name ‘diabetes’ was coined by Demetrius of Apamea around the first century bc based on the Greek term diabainein meaning ‘siphon’ due to the symptoms of polyuria and polydipsia9. In the 1600s, ‘mellitus’ was added to indicate that urine sweetness differentiated this condition from other causes of polyuria, with the idea that this sweetness might be linked to a similar finding in the blood10. However, it took nearly another century to link the polyuria and polydipsia of diabetes mellitus with excessive glucose in both the blood and urine11. The first working evidence that the pancreas controlled carbohydrate metabolism would not come until 1889, when German scientists Oskar Minkowski and Joseph von Mering performed pancreatectomies on dogs who then developed hyperglycemia and diabetes12. Almost 20 years before Minkowski and von Mering’s seminal work, the first detailed histologic studies of the pancreas were published by Paul Langerhans, as a medical student. His meticulous work described nine different cell types that formed numerous “cell heaps” scattered throughout the gland13. The French scientist G. E. Laguesse would revisit pancreas histology in 1893 and name these collections the “îlots de Langerhans”14,15. The term ‘insulin’ was subsequently coined in 1909 by the Belgian scientist J. de Meyer to describe the still-speculative internal secretion of the pancreas thought to be capable of regulating blood glucose16. At the time experiments were beginning in Toronto in the summer of 1921, a handful of other scientists throughout the world were already pursuing the goal of harnessing this mysterious substance for therapeutic use4.
Whereas others failed or, in the case of the Romanian scientist N. C. Paulesco, would have their work interrupted by World War I (ref. 4), the Toronto group in a mere 9 months successfully isolated insulin from the pancreas. They would go on to prove that the pancreatic extract regulated blood glucose levels and urinary glucose excretion by reinjecting it into pancreatectomized dogs, while keeping the longest living of these dogs, Marjorie, alive for more than 70 days. James Collip, a biochemist from the University of Alberta, on sabbatical at the University of Toronto, joined the team late in the fall of 1921 and played a critical role in developing methods to reliably isolate insulin from the pancreas using alcohol extraction. The first documented patient to receive insulin was 14-year-old Leonard Thompson. He received his first injection in January 1922, at a time when he, by all accounts, was near death. As reports of his treatment spread throughout North America, the team in Toronto received a growing number of desperate appeals from patients and their physicians for the new therapy. They struggled to scale up the production to reach this growing demand. Ultimately this problem was solved through a partnership with the pharmaceutical corporation Eli Lilly and Company in Indianapolis, Indiana. Scientists at Lilly optimized methods of isoelectric precipitation enabling the extraction of large quantities of insulin from porcine pancreata, allowing it to be purified for commercial distribution.
The capability for insulin purification quickly spread to physicians and scientists beyond North America. At a private dinner in 1922, Elliot Joslin shared the news with the Nobel Prize-winning Danish scientist August Krogh and his wife Marie Krogh, who had recently been diagnosed with adult-onset diabetes. The Kroghs extended their trip by several days to visit Macleod in Toronto, obtained a license to bring the team’s insulin purification protocol to Europe, and immediately began production of insulin following their return to Copenhagen (serving as the starting foundation for what eventually became Novo Nordisk)17.
By the end of this incredible journey, the team in Toronto would be deeply fractured by conflict over who deserved scientific credit for the discovery of insulin. However, to ensure access of this lifesaving drug to patients with diabetes, the team agreed to sell their patents back to the University of Toronto for the price of CAN$1. Ultimately Banting and Macleod were awarded the 1923 Nobel Prize in Physiology or Medicine, with Banting sharing his portion of the award with Best, and Macleod doing the same with Collip4,18.
Advances in the understanding of diabetes pathophysiology.
The transformative discovery of insulin, in part, represented an inevitable culmination of a body of work performed by many investigators over many years (Fig. 1). The evolution of our understanding of diabetes pathophysiology has similarly occurred due to the collective observations of numerous clinicians and researchers. Before the clinical availability of insulin, astute observers delineated subgroups of affected individuals based on age of presentation, body habitus and survival on low-carbohydrate diets19. Once insulin therapy was available, clinicians related these differences to insulin requirements, with insulin-insensitive patients usually presenting with symptoms later in life, in association with obesity and a more insidious presentation20,21. Those who were more sensitive to insulin often presented at younger ages and required smaller doses of insulin to suppress urine glucose and become hypoglycemic22. Direct comparison of forearm arteriovenous glucose gradients after simultaneous glucose and insulin administration showed differences in lean, young patients with diabetes compared to older, overweight patients23. These findings suggested that differences in glucose gradients may be related to forearm muscle resistance to insulin action and that those with diabetes could be separated into distinct subgroups—the first with disease resulting from insulin insufficiency and the second with disease occurring due to insulin insensitivity23.
Fig. 1 |. A timeline of key discoveries in our understanding of insulin and diabetes pathophysiology.

Shown in the main branch of the timeline are key discoveries in our understanding of insulin as a central contributor to diabetes pathophysiology10,12,13,18–22,24–26,42,143,144. Included in the left branch of the timeline are important milestones that have enabled the understanding of type 2 diabetes as a disease of impaired insulin secretion and action22,23,39–41,46,47,67,72–75,80,145–147. The right branch highlights notable discoveries that have led to the understanding of type 1 diabetes as an autoimmune disease2,3,5–8,29–31,34–38,43–45,64–66,126,127,148–150.
In the 1950s, the ability to quantify circulating insulin allowed for confirmation of insulin deficiency in certain groups of patients. Initial work used bioassays demonstrating that compared to human plasma from older obese females with nonketotic hyperglycemia, human plasma from young, ketotic patients with diabetes was unable to lower blood glucose values when injected into diabetic rats24. Similarly, the extractable insulin content of pancreata was tested for its ability to induce mouse seizures. These experiments showed that pancreatic insulin was almost undetectable in young people with diabetes. This was in contrast to the nearly ~50% reduction observed in older people with diabetes relative to nondiabetic controls25. Rosalyn Yalow and Solomon Berson’s development of a reliable radioimmunoassay allowed for direct measurement of insulin levels, allowing for the separation of insulin-deficient versus insulin-insensitive diabetes based on measurement of circulating insulin26. Yalow was awarded the 1977 Nobel Prize in Physiology or Medicine for this seminal work, becoming only the second woman to earn this award.
In search of a simple binary classification system, multiple naming iterations would be trialed, including groups I and II; types I and II; insulin sensitive and insensitive; insulin dependent and noninsulin dependent; and diabetes gras (fat) and diabetes maigre (thin)3,27,28. Still the actual etiologic basis for these different disease types remained unclear. Not until the 1950s, following the discovery of an autoimmune basis for other endocrine diseases, did researchers begin to consider autoimmunity as an etiology of insulin-deficient diabetes29–31. Patients with diabetes and an insulin-deficient phenotype were noted to frequently have detectable autoantibodies associated with other autoimmune diseases, including thyroid and gastric antibodies32,33. In animals, injections of anti-insulin serum, or homogenized pancreatic or islet tissues, resulted in development of islet immune lesions, supporting the idea that islets could generate an immune response34–36. Early reports examining small numbers of pancreatic sections from individuals with diabetes had only rarely identified examples of immune cell infiltration into the islet (that is, insulitis)37. However, in 1965, Willy Gepts analyzed a larger number of pancreatic samples obtained from children who died near the time of clinical diagnosis and showed islets with lymphocytic infiltrates in the majority of autopsy specimens, suggesting a clearer link to an immunologic origin of disease38. These findings in postmortem tissue were ultimately validated by key studies showing autoimmunity using blood samples from living donors. Leukocyte migration assays demonstrated that individuals with type 1 diabetes exhibited evidence of anti-pancreatic cell-mediated immunity2. In a now famous ‘eureka’ moment, in 1974, Franco Bottazzo, a research fellow in Deborah Doniach’s laboratory in London, was the first to successfully visualize islet cell antibodies using indirect immunofluorescence, thereby confirming the presence of antibodies reactive to the islet. During experiments originally designed to support his thesis work on Addison’s disease, he observed that pancreatic islets “lit up” after incubation with sera from some patients with polyendocrine autoimmunity, most of whom had or would go on to develop diabetes3,5. These findings would quickly be confirmed by multiple groups around the world3.
By the end of the 1970s, this work led to the recognition that immune-mediated loss of insulin-secreting cells was the cause of insulin-dependent diabetes3. In parallel, development of techniques to measure insulin-mediated glucose disposal allowed for direct confirmation of insulin resistance in individuals matching a noninsulin-dependent diabetes phenotype39–41. Based on these findings, in 1979 the National Diabetes Data Group proposed classifying diabetes using the terms employed today: type 1 (insulin dependent), type 2 (noninsulin dependent), and ‘other’ denoting forms of disease not fitting into either of these two categories42.
Human cohorts provide a contemporary understanding of diabetes pathophysiology.
New animal models of spontaneous disease43,44 and improvements in immunologic, metabolic and genetic phenotyping in human cohorts have continued to shape our understanding of type 1 and type 2 diabetes over the past half-century. The widely adopted 1986 Eisenbarth model suggested that type 1 diabetes was a chronic autoimmune disease, with genetically predisposed individuals encountering a hypothetical triggering event that activated islet autoimmunity, yielding progressive beta cell destruction and insulin deficiency45. Although a genetic contribution to diabetes was clear based on increased prevalence among family members, analyses of kindreds were limited by lack of a reliable biomarker for ‘pre-diabetes’, as well as a confusing picture based on different inheritance patterns, disease presentations and phenotypes, which also pointed to environmental exposures as contributors6,43–45. A theme of early twin studies indicated >90% concordance of diabetes in those diagnosed at older ages (that is, type 2 diabetes) and approximately 50% concordance of diabetes occurring in children and young adults (that is, type 1 diabetes)8,46,47. The description of the critical role of human leukocyte antigen (HLA) antigen-presentation genes in the transplantation setting48 was followed with the association of these genes with autoimmune diseases including ankylosing spondylitis, multiple sclerosis and type 1 diabetes in the early 1970s (ref. 6). To this day, a standout feature of many autoimmune diseases including type 1 diabetes is that a small number of HLA class 2 alleles, critical for antigen presentation by the immune system, explain a large proportion of disease heritability.
In the 1990s, linkage analysis of sibling pairs affected by type 1 diabetes identified an area on chromosome 11p15 (ref. 49) that was subsequently mapped to a region upstream of the insulin gene50–52 as associated with type 1 diabetes. Subsequent genome-wide linkage and then genome-wide association studies of cases and controls have described more than 60 loci outside the HLA region that also contribute to type 1 diabetes genetic risk53. The majority of variants point towards the role of inflammation and the immune system in type 1 diabetes pathogenesis. More recently, there has been increased focus on whether many of these variants may influence beta cell interactions with the immune system, with over 40% of genes associated with type 1 diabetes being expressed in the islet or beta cell. Coupled with molecular studies in human islets and mouse models of disease, these genetic associations highlight an ongoing dialogue as to whether beta cell or immune system abnormalities are the key driving factor in the pathogenesis of type 1 diabetes54–59, a juxtaposition first described by Bottazzo as the notion of “beta cell homicide or suicide”60.
Contemporary and large natural history studies assembled based on HLA genotypes and family history have shown that type 1 diabetes is a heterogenous disorder and that features beyond autoimmunity, including metabolic factors, exocrine function and environmental exposures, impact progression to clinical disease61. In addition, birth cohort studies have shed light on the timing of autoantibody development, describing a wide range but a surprising peak incidence of islet autoantibody development at 9 months of age62, particularly focused on insulin autoimmunity. A seminal analysis of four different birth cohorts from the USA, Finland and Germany demonstrated that the presence of a single islet autoantibody is associated with a 13% risk of developing type 1 diabetes over 15 years. In contrast, having two or more antibodies is associated with a 70% risk over 10 years of observation and an 84% risk over 15 years63. These and other data led to a proposed modified staging system in 2015. Here, stage 1 diabetes is defined by two or more autoantibodies, while stage 2 diabetes is defined as the presence of multiple autoantibodies and dysglycemia. Stage 3 type 1 diabetes is defined by the progression to overt diabetes based on the American Diabetes Association standards, which include a fasting blood glucose of greater than 7.0 mmol l−1 (1.26 g l−1), a random glucose of >11.1 mmol l−1 (2 g l−1) with symptoms, an abnormal oral glucose tolerance test or a hemoglobin A1C level of >48 mmol mol−1 (6.5%)64. This staging paradigm has provided a regulatory and conceptual framework for efforts focused on disease prevention65,66 and for mechanistic studies focused on developing stage-specific metabolic and immune signatures.
In parallel, beautifully detailed physiologic studies using intravenous and oral glucose tolerance tests and hyperglycemic clamps have provided further insight into the metabolic underpinnings of type 2 diabetes. These studies have demonstrated that to maintain glucose homeostasis, a feedback loop exists in which decreased insulin sensitivity is tightly associated with increased insulin secretion from the beta cell, with this hyperbolic relationship between beta cell responsivity and insulin sensitivity termed the disposition index67,68. Natural history studies of cohorts progressing to type 2 diabetes have demonstrated early impairments in insulin sensitivity, which are evident more than 10 years in advance of diabetes development. Initially, beta cell function is increased, maintaining glucose levels at higher but still normal levels and below the diagnostic threshold for diabetes. However, the ability of the beta cell to maintain this response is finite in some individuals. As the beta cells undergo a process of failure that has been linked with a number of molecular processes, including oxidative and endoplasmic reticulum stress, lipotoxicity and dedifferentiation69–71, beginning around 3 years before the onset of diabetes, decreasing insulin secretion and an accelerated rise in blood glucose levels are observed72–75. However, the temporal relationship between changes in insulin secretion and insulin sensitivity continue to be elucidated, as insulin hypersecretion may also contribute to or exacerbate insulin resistance, and has even been documented before insulin resistance in some individuals76.
Despite the high heritability observed in twin studies46,47, it took longer to begin to identify the genetic loci responsible for the high concordance observed in twin studies of type 2 diabetes. The first associated loci, in genes including TCF7L2, INSR, IRS1, GCK and KCNJ11 (refs.77,78), were originally identified by linkage or candidate gene studies, and since the turn of the century, increasing size and depth of genome-wide association studies have rapidly expanded the list of associated loci in type 2 diabetes to more than 250 with 400 independent signals79. The majority of associated loci are linked to beta cells, supporting the idea that impaired beta cell function is critical to type 2 diabetes pathogenesis78,80. Despite the large number of associated loci, their individual contributions to overall risk are moderate, explaining just under 20% of heritability and highlighting the proportion of ‘missing heritability’ that is still to be fully elucidated. Whereas type 1 diabetes seems to be a discrete entity defined by islet autoimmunity, beta cell destruction and a relatively small group of genes, an outstanding question is whether type 2 diabetes will be resolved into multiple subtypes/clusters defined by genetic associations, mechanisms and phenotype81–83 and whether this approach will improve precision intervention and treatment.
Advances in the molecular characterization of insulin.
Soon after the discovery of insulin and in parallel to its application in clinical medicine, there was a steady march to shed light on the molecular characteristics of the insulin molecule. In 1935, a research fellow, Dorothy Crowfoot Hodgkins, took the first diffraction images of insulin crystals84. She would continue her work on the insulin molecule on and off throughout her career, ultimately solving the crystal structure in 1969 and showing that insulin was a hexamer composed of three heterodimers85. Hodgkins earned the Nobel Prize in Chemistry in 1964 for her pioneering work in crystallography, all while battling her own autoimmune condition, rheumatoid arthritis86. In the early 1950s, Frederick Sanger determined the amino acid sequences of the A and B chains of insulin87–90. By 1955, he would demonstrate the position of the two disulfide bonds linking the A and B chains and the intrachain disulfide bond within the A chain, and in 1958, he was awarded the Nobel Prize in Chemistry91–93. In addition to being the first protein that was successfully sequenced, insulin was the first molecule to be characterized as a prohormone. In another moment of serendipity, Donald Steiner had the opportunity to study an insulinoma tumor removed from a patient at the University of Chicago in 1965. While analyzing extracts of the tumor and in subsequent experiments, Steiner identified proinsulin as the larger single-chain precursor of insulin, established proinsulin as the origin of C-peptide, and showed that insulin and C-peptide were secreted from the beta cell in equimolar ratios94,95. In 1968, Ronald Chance at Lilly Research Laboratories in Indianapolis published the porcine sequence of the proinsulin molecule96.
These structural accomplishments would pave the way for studies describing the interaction of insulin with the insulin receptor97, and would serve as a precursor to our understanding of monogenic forms of diabetes resulting from mutations in the insulin gene, which yield distinct phenotypes based on structural impacts. Altered interaction of structurally abnormal insulin with the insulin receptor leads to altered insulin action, hyperinsulinemia and adult-onset diabetes with autosomal dominant inheritance98. In contrast, recessive mutations impacting insulin biosynthesis result in neonatal diabetes99. Heterozygous mutations can also impair the normal folding of insulin precursors, yielding abnormal molecules that act in a dominant-negative fashion to impair the exit of all proinsulin from the endoplasmic reticulum100. This initially causes insulin deficiency, followed by severe beta cell endoplasmic reticulum stress and apoptosis100,101. Molecular studies defining the biologic impact of mutant INS gene-induced diabetes of youth (MIDY) mutations have also yielded valuable insights into the normal molecular pathways of insulin biosynthesis, precursor processing and transit through the secretory pathway101, recently highlighting how certain conserved residues are critical for normal insulin folding102. Finally, decades after Steiner’s original identification of proinsulin as insulin’s precursor, increased proinsulin secretion relative to insulin or C-peptide is accepted as a serum proxy for beta cell stress and dysfunction and a predictive biomarker for both type 1 and type 2 diabetes103,104.
Advances in the clinical use of insulin.
The molecular characterization of insulin would also dramatically shape diabetes therapy. After the first clinical use of ‘regular’ insulin for patients, the pancreatic extract was further purified, the source of insulin moved to pork and later beef pancreas, and the concentration was increased from the original commercially available U-5 insulin (for example, 5 units ml−1) to U-10, U-20, U-40 and U-80 preparations105. Later, in the early 1970s, the most common insulin preparation became U-100. More concentrated insulins also became available, and were employed for people with severe insulin resistance (U-200, U-300 and U-500); the first was U-500 beef regular insulin, which was developed in 1952.
Although exogenously administered regular insulin was lifesaving, its pharmacokinetics did not mirror that of endogenously produced human insulin. Administered insulin molecules self-associate into hexamers, which must dissociate into dimers and then monomers before entering the circulation, with typical delays of 60–90 min from injection to peak action. This contrasts with the circulating endogenous insulin peak action of approximately 15–30 min after the start of food ingestion. In addition to the delay in action, these first insulins were all short acting (Fig. 2) and required multiple injections per day.
Fig. 2 |. The evolution of improvements in insulin pharmacokinetics.
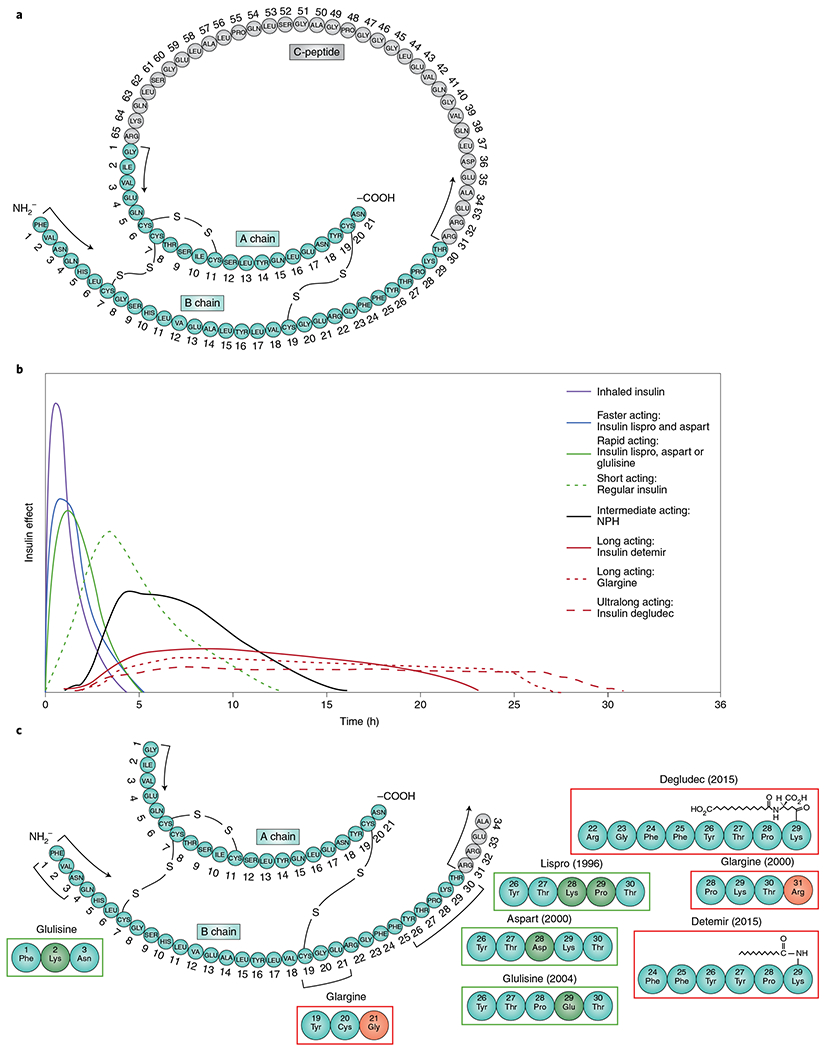
a, The native structure of human proinsulin. b, Representative pharmacokinetic profiles of available insulins administered subcutaneously. c, Structural changes of insulin analogs and years of introduction in the USA including rapid-acting insulin analogs (green boxes) and long-acting insulin analogs (red boxes)96,106,151–154.
Towards a goal of reducing the need for multiple daily injections, the first long-acting insulin was developed in the 1930s by H. C. Hagedorn. It was a suspension protamine zinc insulin that was based on the discovery that insulin action could be prolonged by adding protamine obtained from river trout semen106. The action of protamine zinc insulin lasted 24–36 h. In 1946, Nordisk developed an intermediate-acting neutral protamine Hagedorn insulin that formed microcrystals, could be mixed with regular insulin and lasted 18–24 h. The first ‘peakless’ basal insulin, known as ultralente (belonging to the lente family of insulins), was developed during the 1950s by employing an extended zinc suspension without protamine107. Ultralente was mixed with a semilente (an insulin with a different proportion of zinc and a time–action profile slightly slower than that of regular insulin) to make the intermediate-acting lente. However, because these insulins were suspension based, they had variable day-to-day action, complicating their clinical use, and they still required more than one injection a day to provide a basal coverage.
Before the 1980s, all insulin preparations were derived from animal sources. However, with increased clinical demand and tedious extraction processes (for example, more than 23,500 pancreata were needed to make 1 lb of insulin), available supplies were being outstripped108. Additionally, even with the advent of highly purified monocomponent animal insulin in the 1970s109, many people with diabetes continued to have allergic reactions to the formulations. A pure, scalable insulin source was needed. Just as there had been a race to isolate insulin 50 years earlier, now teams were using what had been learned in the molecular biology renaissance of the previous decade to produce human insulin using recombinant DNA technology.
Insulin cloning in bacteria was a complicated process110. First, the A and B chains needed to be synthesized (the B chain synthesis required cleaving the peptide into two sections). Then the A and B chains needed to be linked together. Finally, the insulin needed to be harvestable for commercial use, which required breaking off the beta-galactosidase required to insert the insulin into Escherichia coli bacteria. In 1978, David Goeddel, Arthur Riggs and their Genentech colleagues working at City of Hope produced the first recombinant DNA human insulin111. Subsequently, Genentech and Lilly agreed to commercialize this new insulin and Humulin R and N insulins came to market in 1983. Novolin R (Novo Nordisk) followed in 1991 and Insuman R (Hoechst) in 1997. Although this represented an improvement in source, these insulins were still zinc-based formulations with slower pharmacokinetic profiles than natively secreted insulin.
The 1993 publication of the Diabetes Control and Complications Trial112 and 1998 United Kingdom Prospective Diabetes Study113–115 demonstrated definitive relationships between glycemic control and microvascular complications and showed that lower A1cs were associated with higher rates of severe hypoglycemia. These observations spurred efforts focused on improving exogenously administered insulin’s pharmacokinetic and pharmacodynamic properties (absorption rate, time to peak and duration of action). This has been accomplished over time (Fig. 2) using recombinant DNA technology and genetic engineering, and adding excipients. Tweaking amino acid sites/composition in the native insulin molecule changed the pharmacokinetics and permitted faster absorption, earlier peak action and faster offset. In 1988 a synthetically designed insulin was produced by replacing the B28 proline with aspartic acid, which favored a molecular conformation leading to rapid dissociation of dimerized insulin chains. The first rapid-acting insulin, insulin lispro (produced by inverting the B29 lysine and B28 proline), came to market in 1996. Next was aspart in 2000, and then glulisine in 2004. An ultrarapid-acting version of insulin aspart was subsequently developed by adding nicotinamide and l-arginine as excipients that improve the insulin’s stability and rate of absorption116. An ultrarapid insulin lispro has also been developed by using a prostacyclin analog to enhance vasodilation and absorption and citrate to enhance local vascular permeability.
The first long-acting once-daily basal insulin, glargine, was approved in 2000 (ref. 117). It was designed to have an extended duration of action through amino acid modifications in both chains (A chain A21 asparagine substituted by glycine and B chain elongated by adding two arginines). These changes achieved a prolonged duration by shifting the isoelectric point to make the insulin soluble at an acidic pH but precipitate at the injection site at a pH of 7.4, allowing for slow dissociation. The next long-acting basal insulin, detemir, was approved in 2005 (ref. 118). Detemir has a fatty-acid (myristic acid) side chain bound to position B29 that facilities self-association and an affinity for albumin allowing for prolonged duration of action without peaks. Insulin degludec followed a decade later; degludec forms a depot of soluble multihexamers at the injection site giving it an ultralong (>42 h) glucose-lowering effect119.
Improvements in insulin therapy have also been realized by changes in the method of delivery. Initially, insulin was available only through administration via vials and syringes. In 1985 the first insulin pen was launched by Novo Nordisk120. More recently developed ‘smart’ insulin pens allow for tracking of insulin dosing, and integration with smartphone applications to provide reminders, integrate with blood glucose data and provide dosing recommendations120. Advances in insulin delivery have also included the development of inhaled insulin with a faster onset of action and offset of effect than any of the injected insulins121. The first, Exubera, came to market in 2006, but was rapidly withdrawn due to poor market uptake. Inhaled technosphere insulin, Afrezza, was launched commercially in the next decade by Mannkind, although cost, limited dosing flexibility and continued concern about pulmonary effects have limited its clinical uptake and use.
Arguably, the most impactful technology-driven advances in insulin delivery have revolved around the technology of continuous subcutaneous insulin infusion using insulin pumps122. The first closed-loop insulin pump that incorporated automatic blood glucose sensing was designed by Arnold Kadish in 1963 (ref. 123). It was large (like an “army backpack”) and impractical for daily use. The first bedside computer-controlled closed-loop system, the Biostator, was invented by Miles Laboratory (Elkhart IN) in 1974 (ref. 124). During the late 1970s, rigorous testing of insulin pumps began in earnest—leading to the first wearable systems, including the ‘big blue brick.’ By the 1980s, continuous subcutaneous insulin infusion had become a viable alternative means of delivering insulin122. In 1983, MiniMed brought the first commercial pump to market. Improvements over the next several decades have included the emergence of new pump models by multiple manufacturers, including tubeless patch pump models, the ability to modify the timing/duration of insulin bolus delivery and improvements in device usability. The development of reliable and accurate continuous glucose monitors allowed for the possibility of integration of glucose data with pump insulin delivery and sparked a flurry of interest to develop safe and effective algorithms for closed-loop systems, notably championed by do-it-yourself movements from the diabetes community itself122,125. Now, increasingly, many pumps employ hybrid closed-loop technologies with automatic insulin dosing by the pump based on continuous glucose readings and trends.
What does the future hold?
In the 100 years since the discovery of insulin, there has been remarkable progress in our ability to treat type 1 and type 2 diabetes, facilitated by an improved understanding of the pathophysiology of the disease and improvements in insulin formulation and delivery. This progress is captured in an impressive series of scientific accomplishments summarized in this Review and shown in Fig. 1, several of them recognized by the most prestigious awards in Medicine, Physiology and Chemistry. However, the true impact of these achievements is best illustrated by the voices of patients who have seen dramatic changes in the management of their type 1 diabetes (Fig. 3).
Fig. 3 |. Advances in diabetes management viewed through the lens of individuals with type 1 diabetes.
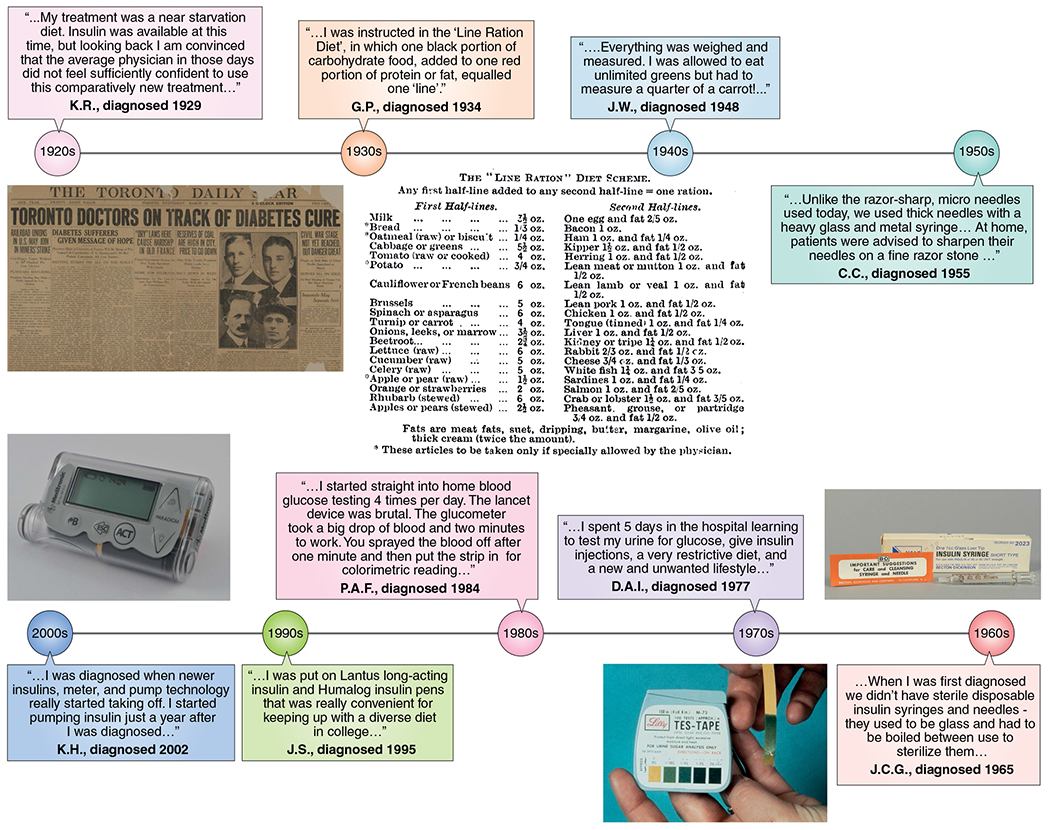
The full names of contributing individuals for the years 1965–2006 are included in the Acknowledgements. The quotes from K.R. (diagnosed 1929) and G.P. (diagnosed 1934) are from ref. 155. The quote from J.W. (diagnosed 1948) is from ref. 156. The quote from C.C. (diagnosed 1955) is from ref. 157.
What do the next 100 years hold for insulin and those who depend on it (Fig. 4)? Furthermore and importantly, will treatment with exogenous insulin therapy become another note in the history books? For type 1 diabetes, this goal is a centerpiece of clinical trials testing disease-modifying interventions, including work that is ongoing in several large networks such as Type 1 Diabetes TrialNet, the Immune Tolerance Network and INNODIA. In 2019, following a nearly three-decade search for successful disease prevention, the Type 1 Diabetes TrialNet study of the anti-CD3 antibody teplizumab showed that a single 14-day course of drug could delay the onset of clinical diabetes (that is, stage 3 type 1 diabetes) by a median of 32.5 months in high-risk multiple-autoantibody-positive individuals with dysglycemia (that is, stage 2 type 1 diabetes)126,127. Results from this seminal study have underscored the importance of identifying the correct therapeutic window for intervention, but have also raised the practical question of how to identify at-risk individuals outside a research setting. In this regard, population-based screening is now being increasingly performed in several countries and regions, and is based on autoantibody measurement and, in some cases, assessment of genetic risk. Genetic risk stratification has focused on assessment of HLA risk or more recently calculation of polygenic genetic risk scores that sum the effects of a large number of variants128. The education and anticipatory guidance provided as part of these programs have been shown to significantly reduce the risk of ketoacidosis at the onset of stage 3 type 1 diabetes129. However, additional research will be needed to identify the ideal timing and frequency of screening and how to prioritize at-risk individuals for interventions. For type 2 diabetes, complementary disease-modifying therapies that may reduce or eliminate the need for insulin administration have also represented a rapidly expanding field of interest130–132.
Fig. 4 |. The future of insulin and diabetes therapy.
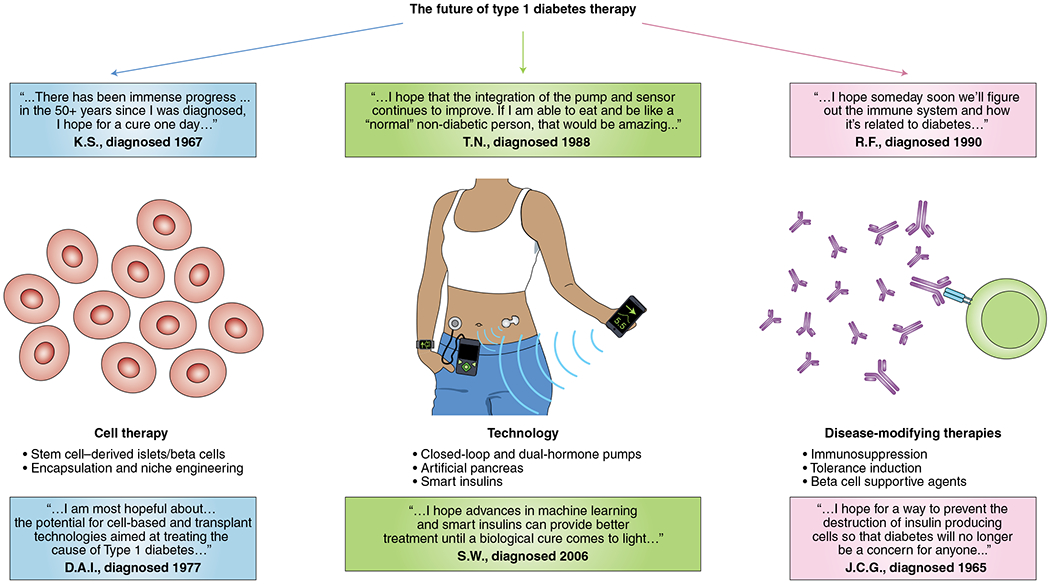
The future of diabetes therapy and prevention includes efforts focused on: the development of a renewable, cellular source of insulin; improvements in technology, including better insulins and novel insulin delivery platforms; and disease-modifying therapies, including immune-modulating therapies and beta cell supportive agents. Quotes are included from individuals who depend on insulin, expressing their hopes surrounding the future of diabetes therapy. Image originally created using biorender.com and AutoDesk SketchBook.
In addition to efforts focused on disease modification, there are continuing efforts to improve insulin therapies and there is still much to be refined in our approach to exogenous insulin delivery. There is a hope for development of better insulins including: insulins with even faster pharmacokinetics, once-weekly insulin, oral insulin and, ultimately, glucose-responsive ‘smart’ insulins that increase circulating concentrations under conditions of hyperglycemia. Additional technological advancements on the horizon include improved algorithms for automated insulin delivery devices, implantable devices and dual-hormonal systems that combine automated delivery of insulin and glucagon133,134. Finally, there is also considerable interest in developing renewable, cellular sources of insulin through the generation of beta-like cells from either induced pluripotent stem cells or embryonic stem cells. While a beta-like cell with behavior that fully recapitulates the physiology of a normal beta cell is yet to be realized, there has been a steady series of improvements to directed differentiation strategies over the past 20 years135–138. In parallel, a large body of work has focused on developing the ideal cellular niche and encapsulation strategies to support normal patterns of hormone secretion while also protecting these cellular implants from autoimmune destruction139,140. Improvements in insulin delivery and monitoring and alternative cell-based sources of insulin have the potential to broadly impact diabetes management and will benefit individuals with type 1 and type 2 diabetes, as well as rarer forms of the disease.
Closing.
In the 1920s, having developed a transformative and lifesaving therapy, the Toronto team faced an almost impossible challenge, and they struggled at the outset to produce enough insulin to meet a rapidly growing demand and to distribute insulin in a fashion that was equitable1. As we celebrate this remarkable centennial anniversary and the subsequent discoveries that have improved life expectancy and quality of life for those with diabetes (Fig. 3), there are continued challenges with accessibility and equity, which have only been exacerbated by advances in diabetes care technology. In a recent analysis of children and adults with type 1 diabetes in the USA, the average cost associated with diabetes totaled nearly US$800 per month, with nearly 50% driven by pharmacy costs141. Even the most basic component of diabetes management, insulin itself, has become unaffordable for many142. From 2012 to 2016, the average list price of insulins increased by 14–17% per year in the USA. These increases are often driven by gaps between the list price and the net price ultimately received by manufacturers, which have been largely attributed to rebates and discounts negotiated between stakeholders in a supply chain with poor transparency143. As members of the Toronto team arranged to sell their patents for insulin back to the University of Toronto for CAN$1, Banting is reported to have remarked, “Insulin belongs to the world, not to me.” Thus, while we envision a future of possibilities for those who require insulin to survive, it is important that we not forget Banting’s altruism and become complacent to this most basic and fundamental challenge of the present. Only once equal access for patients around the globe is established will the remarkable achievements surrounding insulin over the past century truly realize their greatest impact144–150.
Acknowledgements
Research in the laboratory of C.E.-M. is supported by the NIH grants R01 DK093954, R21 DK119800, U01DK127786, R01DK127308 and P30DK097512; the VA Merit Award I01BX001733; and the JDRF grant 2-SRA-2019-834-S-B; as well as gifts from the Sigma Beta Sorority, the Ball Brothers Foundation, and the George and Frances Ball Foundation. E.K.S. is supported by R03 DK117253, R01DK121929 and JDRF 2-SRA-2017-498-M-B. L.A.D. is supported by 1UL1TR002529. We thank the following individuals who willingly shared their personal experiences of living with type 1 diabetes: James C. Garmey (diagnosed 1965); Lis Warren (diagnosed 1965); Karen Stancombe (diagnosed 1967); Debra A. Ignaut (diagnosed 1977); Patrick A. Fueger (diagnosed 1984); Todd Nebesio (diagnosed 1988); Roger Felton (diagnosed 1990); Jason Spaeth (diagnosed 1995) and his wife, Aubrey Spaeth; Kate Haynes (diagnosed 2002); and Staci Weaver (diagnosed 2006).
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Bliss M The Discovery of Insulin (Univ. Chicago Press, 1982). [Google Scholar]
- 2.Nerup J et al. Cell-mediated immunity in diabetes mellitus. Proc. R. Soc. Med 67, 506–513 (1974). [PMC free article] [PubMed] [Google Scholar]
- 3.Gale EA The discovery of type 1 diabetes. Diabetes 50, 217–226 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Bliss M The history of insulin. Diabetes Care 16, 4–7 (1993). [DOI] [PubMed] [Google Scholar]
- 5.Bottazzo G, Florin-Christensen A & Doniach D Islet-cell antibodies in diabetes mellitus with autoimmune polyendocrine deficiencies. Lancet 304, 1279–1283 (1974). [DOI] [PubMed] [Google Scholar]
- 6.Singal DP, & Blajchman MA Histocompatibility (HL-A) antigens, lymphocytotoxic antibodies and tissue antibodies in patients with diabetes mellitus. Diabetes 22, 429–432 (1973). [DOI] [PubMed] [Google Scholar]
- 7.Nerup J et al. HL-A antigens and diabetes mellitus. Lancet 2, 864–866 (1974). [DOI] [PubMed] [Google Scholar]
- 8.Cudworth AG & Woodrow JC HL-A antigens and diabetes mellitus. Lancet 2, 1153 (1974). [DOI] [PubMed] [Google Scholar]
- 9.Gemmill CL The Greek concept of diabetes. Bull. N. Y. Acad. Med 48, 1033–1036 (1972). [PMC free article] [PubMed] [Google Scholar]
- 10.Allan FN The writings of Thomas Willis, M.D.; diabetes three hundred years ago. Diabetes 2, 74–77 (1953). [DOI] [PubMed] [Google Scholar]
- 11.Dobson M Nature of the urine in diabetes. Med. Obs. Inq 5, 298–310 (1776). [Google Scholar]
- 12.v. Mering J, & Minkowski O Diabetes mellitus nach Pankreasexstirpation. Naunyn Schmiedebergs Arch. Exp. Pathol. Pharmakol 26, 371–387 (1890). [Google Scholar]
- 13.Sakula A Paul Langerhans (1847–1888): a centenary tribute. J. R. Soc. Med 81, 414–415 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laguesse GE Sur la formation des îlots de Langerhans dans le pancréas. C. R. Seances Soc. Biol. Fil 5, 819–820 (1893). [Google Scholar]
- 15.Goet JP Gustave Edouard Laguesse; his demonstration of the significance of the Islands of Langerhans. Diabetes 2, 322–324 (1953). [DOI] [PubMed] [Google Scholar]
- 16.De Meyer J Action de la sécrétion interne du pancréas sur différents organes et en particulier sur la sécrétion rénale. Arch. Fisiol 7, 96–99 (1909). [Google Scholar]
- 17.Lindsten J August Krogh and the Nobel Prize to Banting and Macleod. The Nobel Prize https://www.nobelprize.org/prizes/themes/august-krogh-and-the-nobel-prize-to-banting-and-macleod/ (2 April 2001). [Google Scholar]
- 18.Banting FG, Best CH, Collip JB, Campbell WR & Fletcher AA Pancreatic extracts in the treatment of diabetes mellitus. Can. Med. Assoc. J 12, 141 (1922). [PMC free article] [PubMed] [Google Scholar]
- 19.Lancereaux E Le diabète maigre, ses symptômes, son évolution, son pronostic et son traitement. Union Med. 29, 161 (1880). [Google Scholar]
- 20.Pincus G, Joslin E & White P The age-incidence relations in diabetes mellitus. Am. J. Med. Sci 188, 116–121 (1934). [Google Scholar]
- 21.MacLean H Some observations on diabetes and insulin in general practice. Postgrad. Med. J 1, 73–77 (1926). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Falta W, & Boller R Insulärer und insulinresistenter Diabetes. Klin. Wochenschr 10, 438–443 (1931). [Google Scholar]
- 23.Himsworth HP Diabetes mellitus: its differentiation into insulin-sensitive and insulin insensitive types. Lancet 227, 127–130 (1936). [DOI] [PubMed] [Google Scholar]
- 24.Bornstein J, & Lawrence R Two types of diabetes mellitus, with and without available plasma insulin. Br. Med. J 1, 732 (1951). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wrenshall GA, Bogoch A, & Ritchie R Extractable insulin of pancreas: correlation with pathological and clinical findings in diabetic and nondiabetic cases. Diabetes 1, 87–107 (1952). [DOI] [PubMed] [Google Scholar]
- 26.Yalow RS, & Berson SA Immunoassay of endogenous plasma insulin in man. J. C1in. Invest 39, 1157–1175 (1960). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Himsworth HP Diabetes mellitus: its differentiation into insulin-sensitive and insulin-insensitive types. Int. J. Epidemiol 42, 1594–1598 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Wild SH, & Byrne CD Commentary: sub-types of diabetes–what’s new and what’s not. Int. J. Epidemiol 42, 1600–1602 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Anderson J, Goudie R, Gray K, & Timbury G Auto-antibodies in Addison’s disease. Lancet 269, 1123–1124 (1957). [DOI] [PubMed] [Google Scholar]
- 30.Roitt I, Doniach D, Campbell P & Hudson RV Auto-antibodies in Hashimoto’s disease (lymphadenoid goitre). Lancet 268, 820–821 (1956). [DOI] [PubMed] [Google Scholar]
- 31.Witebsky E, Rose NR, Terplan K, Paine JR & Egan RW Chronic thyroiditis and autoimmunization. J. Am. Med. Assoc 164, 1439–1447 (1957). [DOI] [PubMed] [Google Scholar]
- 32.Irvine W, Clarke B, Scarth L, Cullen D & Duncan L Thyroid and gastric autoimmunity in patients with diabetes mellitus. Lancet 296, 163–168 (1970). [DOI] [PubMed] [Google Scholar]
- 33.Ungar B, Stocks A, Martin F, Whittingham S, & Mackay I Intrinsic-factor antibody in diabetes mellitus. Lancet 290, 77–78 (1967). [DOI] [PubMed] [Google Scholar]
- 34.Renold AE, Soeldner S, & Steinke J in Ciba Foundation Symposium - Aetiology of Diabetes Mellitus and its Complications (Colloquia on Endocrinology) Vol. 15 (eds. Cameron MP, P. M & O’Connor M) 122–139 (Wiley, 1964). [Google Scholar]
- 35.Lacy PE & Wright PH Allergic interstitial pancreatitis in rats injected with guinea pig anti-insulin serum. Diabetes 14, 634–642 (1965). [DOI] [PubMed] [Google Scholar]
- 36.Heydinger DK & Lacy PE Islet cell changes in the rat following injection of homogenized islets. Diabetes 23, 579–582 (1974). [DOI] [PubMed] [Google Scholar]
- 37.Lecompte PM Insulitis in early juvenile diabetes. AMA Arch. Pathol 66, 450–457 (1958). [PubMed] [Google Scholar]
- 38.Gepts W Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes 14, 619–633 (1965). [DOI] [PubMed] [Google Scholar]
- 39.DeFronzo RA, Tobin JD & Andres R Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am. J. Physiol 237, E214–E223 (1979). [DOI] [PubMed] [Google Scholar]
- 40.Reaven GM Insulin resistance in noninsulin-dependent diabetes mellitus. Does it exist and can it be measured?. Am. J. Med 74, 3–17 (1983). [DOI] [PubMed] [Google Scholar]
- 41.Shen SW, Reaven GM & Farquhar JW Comparison of impedance to insulin-mediated glucose uptake in normal subjects and in subjects with latent diabetes. J. Clin. Invest 49, 2151–2160 (1970). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.National Diabetes Data Group. Classification and diagnosis of diabetes mellitus and other categories of glucose intolerance. Diabetes 28, 1039–1057 (1979). [DOI] [PubMed] [Google Scholar]
- 43.Makino S, Muraoka Y, Kishimoto Y & Hayashi Y Genetic analysis for insulitis in NOD mice. Jikken Dobutsu 34, 425–431 (1985). [DOI] [PubMed] [Google Scholar]
- 44.Nakhooda AF, Like AA, Chappel CI, Wei CN & Marliss EB The spontaneously diabetic Wistar rat (the “BB” rat). Studies prior to and during development of the overt syndrome. Diabetologia 14, 199–207 (1978). [DOI] [PubMed] [Google Scholar]
- 45.Eisenbarth GS Type I diabetes mellitus. A chronic autoimmune disease. N. Engl. J. Med 314, 1360–1368 (1986). [DOI] [PubMed] [Google Scholar]
- 46.Gottlieb MS & Root HF Diabetes mellitus in twins. Diabetes 17, 693–704 (1968). [DOI] [PubMed] [Google Scholar]
- 47.Tattersall RB & Pyke DA Diabetes in identical twins. Lancet 2, 1120–1125 (1972). [DOI] [PubMed] [Google Scholar]
- 48.Thorsby E A short history of HLA. Tissue Antigens 74, 101–116 (2009). [DOI] [PubMed] [Google Scholar]
- 49.Davies JL et al. A genome-wide search for human type 1 diabetes susceptibility genes. Nature 371, 130–136 (1994). [DOI] [PubMed] [Google Scholar]
- 50.Bennett ST et al. Susceptibility to human type 1 diabetes at IDDM2 is determined by tandem repeat variation at the insulin gene minisatellite locus. Nat. Genet 9, 284–292 (1995). [DOI] [PubMed] [Google Scholar]
- 51.Vafiadis P et al. Insulin expression in human thymus is modulated by INS VNTR alleles at the IDDM2 locus. Nat. Genet 15, 289–292 (1997). [DOI] [PubMed] [Google Scholar]
- 52.Pugliese A et al. The insulin gene is transcribed in the human thymus and transcription levels correlate with allelic variation at the INS VNTR-IDDM2 susceptibility locus for type 1 diabetes. Nat. Genet 15, 293–297 (1997). [DOI] [PubMed] [Google Scholar]
- 53.Buniello A et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics. Nucleic Acids Res 47, D1005–D1012 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Coomans de Brachène; A et al. IFN-α induces a preferential long-lasting expression of MHC class I in human pancreatic beta cells. Diabetologia 61, 636–640 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marroqui L et al. Interferon-α mediates human beta cell HLA class I overexpression, endoplasmic reticulum stress and apoptosis, three hallmarks of early human type 1 diabetes. Diabetologia 60, 656–667 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Fløyel T et al. CTSH regulates β-cell function and disease progression in newly diagnosed type 1 diabetes patients. Proc. Natl Acad. Sci. USA 111, 10305–10310 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eizirik DL et al. The human pancreatic islet transcriptome: expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet. 8, e1002552 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Eizirik DL, Colli ML & Ortis F The role of inflammation in insulitis and β-cell loss in type 1 diabetes. Nat. Rev. Endocrinol 5, 219–226 (2009). [DOI] [PubMed] [Google Scholar]
- 59.Lee H et al. Beta cell dedifferentiation induced by IRE1α deletion prevents type 1 diabetes. Cell Metab. 31, 822–836 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bottazzo GF Lawrence lecture. Death of a beta cell: homicide or suicide? Diabet. Med 3, 119–130 (1986). [DOI] [PubMed] [Google Scholar]
- 61.Battaglia M et al. Introducing the endotype concept to address the challenge of disease heterogeneity in type 1 diabetes. Diabetes Care 43, 5–12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Krischer JP et al. The 6 year incidence of diabetes-associated autoantibodies in genetically at-risk children: the TEDDY study. Diabetologia 58, 980–987 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ziegler AG et al. Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. J. Am. Med. Assoc 309, 2473–2479 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Insel RA et al. Staging presymptomatic type 1 diabetes: a scientific statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care 38, 1964–1974 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Feutren G et al. Cyclosporin increases the rate and length of remissions in insulin-dependent diabetes of recent onset. Results of a multicentre double-blind trial. Lancet 2, 119–124 (1986). [DOI] [PubMed] [Google Scholar]
- 66.Stiller CR et al. Effects of cyclosporine immunosuppression in insulin-dependent diabetes mellitus of recent onset. Science 223, 1362–1367 (1984). [DOI] [PubMed] [Google Scholar]
- 67.Kahn SE et al. Quantification of the relationship between insulin sensitivity and β-cell function in human subjects. Evidence for a hyperbolic function. Diabetes 42, 1663–1672 (1993). [DOI] [PubMed] [Google Scholar]
- 68.Bergman RN, Phillips LS & Cobelli C Physiologic evaluation of factors controlling glucose tolerance in man: measurement of insulin sensitivity and beta-cell glucose sensitivity from the response to intravenous glucose. J. Clin. Invest 68, 1456–1467 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Evans-Molina C, Hatanaka M & Mirmira RG Lost in translation: endoplasmic reticulum stress and the decline of β-cell health in diabetes mellitus. Diabetes Obes. Metab 15, 159–169 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Butler AE et al. β-cell deficit in obese type 2 diabetes, a minor role of β-cell dedifferentiation and degranulation. J. Clin. Endocrinol. Metab 101, 523–532 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Guo S et al. Inactivation of specific β cell transcription factors in type 2 diabetes. J. Clin. Invest 123, 3305–3316 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lillioja S et al. Impaired glucose tolerance as a disorder of insulin action. Longitudinal and cross-sectional studies in Pima Indians. N. Engl. J. Med 318, 1217–1225 (1988). [DOI] [PubMed] [Google Scholar]
- 73.Martin BC et al. Role of glucose and insulin resistance in development of type 2 diabetes mellitus: results of a 25-year follow-up study. Lancet 340, 925–929 (1992). [DOI] [PubMed] [Google Scholar]
- 74.Saad MF et al. Sequential changes in serum insulin concentration during development of non-insulin-dependent diabetes. Lancet 1, 1356–1359 (1989). [DOI] [PubMed] [Google Scholar]
- 75.Tabak AG et al. Trajectories of glycaemia, insulin sensitivity, and insulin secretion before diagnosis of type 2 diabetes: an analysis from the Whitehall II study. Lancet 373, 2215–2221 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Trico D, Natali A, Arslanian S, Mari A & Ferrannini E Identification, pathophysiology, and clinical implications of primary insulin hypersecretion in nondiabetic adults and adolescents. JCI Insight 3, e124912 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Duggirala R et al. Linkage of type 2 diabetes mellitus and of age at onset to a genetic location on chromosome 10q in Mexican Americans. Am. J. Hum. Genet 64, 1127–1140 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Grant SF et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat. Genet 38, 320–323 (2006). [DOI] [PubMed] [Google Scholar]
- 79.Mahajan A et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet 50, 1505–1513 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McCarthy MI Genomics, type 2 diabetes, and obesity. N. Engl. J. Med 363, 2339–2350 (2010). [DOI] [PubMed] [Google Scholar]
- 81.Udler MS et al. Type 2 diabetes genetic loci informed by multi-trait associations point to disease mechanisms and subtypes: a soft clustering analysis. PLoS Med. 15, e1002654 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ahlqvist E et al. Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol 6, 361–369 (2018). [DOI] [PubMed] [Google Scholar]
- 83.Dennis JM, Shields BM, Henley WE, Jones AG & Hattersley AT Disease progression and treatment response in data-driven subgroups of type 2 diabetes compared with models based on simple clinical features: an analysis using clinical trial data. Lancet Diabetes Endocrinol 7, 442–451 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Crowfoot D X-ray single crystal photographs of insulin. Nature 135, 591–592 (1935). [Google Scholar]
- 85.Adams MJ et al. Structure of rhombohedral 2 zinc insulin crystals. Nature 224, 491–495 (1969). [Google Scholar]
- 86.Howard JA Dorothy Hodgkin and her contributions to biochemistry. Nat. Rev. Mol. Cell Biol 4, 891–896 (2003). [DOI] [PubMed] [Google Scholar]
- 87.Sanger F & Tuppy H The amino-acid sequence in the phenylalanyl chain of insulin. 1. The identification of lower peptides from partial hydrolysates. Biochem. J 49, 463–481 (1951). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sanger F & Tuppy H The amino-acid sequence in the phenylalanyl chain of insulin. 2. The investigation of peptides from enzymic hydrolysates. Biochem. J 49, 481–490 (1951). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sanger F & Thompson E The amino-acid sequence in the glycyl chain of insulin. 1. The identification of lower peptides from partial hydrolysates. Biochem. J 53, 353–366 (1953). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sanger F & Thompson E The amino-acid sequence in the glycyl chain of insulin. 2. The investigation of peptides from enzymic hydrolysates. Biochem. J 53, 366–374 (1953). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Berg P Fred Sanger: a memorial tribute. Proc. Natl Acad. Sci. USA 111, 883–884 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ryle AP, Sanger F, Smith LF & Kitai R The disulphide bonds of insulin. Biochem. J 60, 541–556 (1955). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sanger F Chemistry of insulin; determination of the structure of insulin opens the way to greater understanding of life processes. Science 129, 1340–1344 (1959). [DOI] [PubMed] [Google Scholar]
- 94.Steiner DF Adventures with insulin in the islets of Langerhans. J. Biol. Chem 286, 17399–17421 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Steiner DF & Oyer PE The biosynthesis of insulin and a probable precursor of insulin by a human islet cell adenoma. Proc. Natl Acad. Sci. USA 57, 473–480 (1967). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chance RE, Ellis RM & Bromer WW Porcine proinsulin: characterization and amino acid sequence. Science 161, 165–167 (1968). [DOI] [PubMed] [Google Scholar]
- 97.De Meyts P Insulin and its receptor: structure, function and evolution. Bioessays 26, 1351–1362 (2004). [DOI] [PubMed] [Google Scholar]
- 98.Shoelson S et al. Three mutant insulins in man. Nature 302, 540–543 (1983). [DOI] [PubMed] [Google Scholar]
- 99.Garin I et al. Recessive mutations in the INS gene result in neonatal diabetes through reduced insulin biosynthesis. Proc. Natl Acad. Sci. USA 107, 3105–3110 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Stoy J et al. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc. Natl Acad. Sci. USA 104, 15040–15044 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Liu M et al. Impaired cleavage of preproinsulin signal peptide linked to autosomal-dominant diabetes. Diabetes 61, 828–837 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rege NK et al. Evolution of insulin at the edge of foldability and its medical implications. Proc. Natl Acad. Sci. USA 117, 29618–29628 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sims EK et al. Elevations in the fasting serum proinsulin-to-C-peptide ratio precede the onset of type 1 diabetes. Diabetes Care 39, 1519–1526 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Loopstra-Masters RC, Haffner SM, Lorenzo C, Wagenknecht LE & Hanley AJ Proinsulin-to-C-peptide ratio versus proinsulin-to-insulin ratio in the prediction of incident diabetes: the Insulin Resistance Atherosclerosis Study (IRAS). Diabetologia 54, 3047–3054 (2011). [DOI] [PubMed] [Google Scholar]
- 105.Ovalle F et al. Understanding concentrated insulins: a systematic review of randomized controlled trials. Curr. Med. Res. Opin 34, 1029–1043 (2018). [DOI] [PubMed] [Google Scholar]
- 106.Hirsch IB, Juneja R, Beals JM, Antalis CJ & Wright EE The evolution of insulin and how it informs therapy and treatment choices. Endocr. Rev 41, 733–755 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Murray I & Wilson RB The new insulins—lente, ultralente, and semilente. Br. Med. J 2, 1023–1026 (1953). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fraser L Cloning insulin. Genentech https://www.gene.com/stories/cloning-insulin (7 April 2016). [Google Scholar]
- 109.Lavaux JP, Ooms HA & Christiansen AH Insulin antibodies in insulin-treated patients; a clinical trial with highly purified insulins. Int. Congr. Ser 316, 40–46 (1973). [Google Scholar]
- 110.Institute of Medicine. Sources of Medical Technology: Universities and Industry (National Academies Press, 1995). [PubMed] [Google Scholar]
- 111.Goeddel DV et al. Expression in Escherichia coli of chemically synthesized genes for human insulin. Proc. Natl Acad. Sci. USA 76, 106–110 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.The Diabetes Control and Complications Trial (DCCT) Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med 329, 977–986 (1993). [DOI] [PubMed] [Google Scholar]
- 113.United Kingdom Prospective Diabetes Study (UKPDS). Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet 352, 854–865 (1998). [PubMed] [Google Scholar]
- 114.United Kingdom Prospective Diabetes Study (UKPDS). Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 352, 837–853 (1998). [PubMed] [Google Scholar]
- 115.United Kingdom Prospective Diabetes Study (UKPDS). 13: Relative efficacy of randomly allocated diet, sulphonylurea, insulin, or metformin in patients with newly diagnosed non-insulin dependent diabetes followed for three years. Br. Med. J 310, 83–88 (1995). [PMC free article] [PubMed] [Google Scholar]
- 116.Biester T, Kordonouri O & Danne T Pharmacological properties of faster-acting insulin aspart. Curr. Diabetes Rep 17, 101 (2017). [DOI] [PubMed] [Google Scholar]
- 117.Pieber TR, Eugene-Jolchine I & Derobert E Efficacy and safety of HOE 901 versus NPH insulin in patients with type 1 diabetes. The European Study Group of HOE 901 in type 1 diabetes. Diabetes Care 23, 157–162 (2000). [DOI] [PubMed] [Google Scholar]
- 118.US Food and Drug Administration. Levemir insulin detemir[rDNA origin] injection drug approval package. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2005/021-536_LevemirTOC.cfm (2005).
- 119.Kurtzhals P et al. Multi-hexamer formation is the underlying basis for the ultra-long glucose-lowering effect of insulin degludec. Diabetologia 54, S426 (2011). [Google Scholar]
- 120.Kesavadev J, Saboo B, Krishna MB & Krishnan G Evolution of insulin delivery devices: from syringes, pens, and pumps to DIY artificial pancreas. Diabetes Ther 11, 1251–1269 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Oleck J, Kassam S & Goldman JD Commentary: why was inhaled insulin a failure in the market? Diabetes Spectr 29, 180–184 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kovatchev B A century of diabetes technology: signals, models, and artificial pancreas control. Trends Endocrinol. Metab 30, 432–444 (2019). [DOI] [PubMed] [Google Scholar]
- 123.Kadish AH Automation control of blood sugar. I. A servomechanism for glucose monitoring and control. Am. J. Med. Electron 3, 82–86 (1964). [PubMed] [Google Scholar]
- 124.Albisser AM et al. An artificial endocrine pancreas. Diabetes 23, 389–396 (1974). [DOI] [PubMed] [Google Scholar]
- 125.Kesavadev J, Srinivasan S, Saboo B, Krishna BM & Krishnan G The do-it-yourself artificial pancreas: a comprehensive review. Diabetes Ther 11, 1217–1235 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Herold KC et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N. Engl. J. Med 346, 1692–1698 (2002). [DOI] [PubMed] [Google Scholar]
- 127.Sims EK et al. Teplizumab improves and stabilizes beta cell function in antibody-positive high-risk individuals. Sci. Transl. Med 13, eabc8980 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ferrat LA et al. A combined risk score enhances prediction of type 1 diabetes among susceptible children. Nat. Med 26, 1247–1255 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ziegler AG et al. Yield of a public health screening of children for islet autoantibodies in Bavaria, Germany. J. Am. Med. Assoc 323, 339–351 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Deacon CF Dipeptidyl peptidase 4 inhibitors in the treatment of type 2 diabetes mellitus. Nat. Rev. Endocrinol 16, 642–653 (2020). [DOI] [PubMed] [Google Scholar]
- 131.Rieg T & Vallon V Development of SGLT1 and SGLT2 inhibitors. Diabetologia 61, 2079–2086 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wilding JPH et al. Once-weekly semaglutide in adults with overweight or obesity. N. Engl. J. Med 384, 989–1002 (2021). [DOI] [PubMed] [Google Scholar]
- 133.Russell SJ et al. Outpatient glycemic control with a bionic pancreas in type 1 diabetes. N. Engl. J. Med 371, 313–325 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Haidar A et al. Comparison of dual-hormone artificial pancreas, single-hormone artificial pancreas, and conventional insulin pump therapy for glycaemic control in patients with type 1 diabetes: an open-label randomised controlled crossover trial. Lancet Diabetes Endocrinol 3, 17–26 (2015). [DOI] [PubMed] [Google Scholar]
- 135.D’Amour KA et al. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat. Biotechnol 23, 1534–1541 (2005). [DOI] [PubMed] [Google Scholar]
- 136.Hogrebe NJ, Augsornworawat P, Maxwell KG, Velazco-Cruz L & Millman JR Targeting the cytoskeleton to direct pancreatic differentiation of human pluripotent stem cells. Nat. Biotechnol 38, 460–470 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rezania A et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol 32, 1121–1133 (2014). [DOI] [PubMed] [Google Scholar]
- 138.Evans-Molina C, Vestermark GL & Mirmira RG Development of insulin-producing cells from primitive biologic precursors. Curr. Opin. Organ Transplant 14, 56–63 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Agulnick AD et al. Insulin-producing endocrine cells differentiated in vitro from human embryonic stem cells function in macroencapsulation devices in vivo. Stem Cells Transl. Med 4, 1214–1222 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Nair GG et al. Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived β cells. Nat. Cell Biol 21, 263–274 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Simeone JC et al. Healthcare resource utilization and cost among patients with type 1 diabetes in the United States. J. Manag. Care Spec. Pharm 26, 1399–1410 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Beran D, Lazo-Porras M, Mba CM & Mbanya JC A global perspective on the issue of access to insulin. Diabetologia 64, 954–962 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Cefalu WT et al. Insulin Access and Affordability Working Group: conclusions and recommendations. Diabetes Care 41, 1299–1311 (2018). [DOI] [PubMed] [Google Scholar]
- 144.Sharpey-Schäfer EA An Introduction to the Study of the Endocrine Glands and Internal Secretions: Lane Medical Lectures, 1913 (Stanford Univ. Press, 1914). [Google Scholar]
- 145.Matthews DR et al. Homeostasis model assessment: insulin resistance and β-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28, 412–419 (1985). [DOI] [PubMed] [Google Scholar]
- 146.Goldfine AB et al. Insulin resistance is a poor predictor of type 2 diabetes in individuals with no family history of disease. Proc. Natl Acad. Sci. USA 100, 2724–2729 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Utzschneider KM et al. Oral disposition index predicts the development of future diabetes above and beyond fasting and 2-h glucose levels. Diabetes Care 32, 335–341 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Diabetes Prevention Trial–Type 1 Diabetes Study Group. Effects of insulin in relatives of patients with type 1 diabetes mellitus. N. Engl. J. Med 346, 1685–1691 (2002). [DOI] [PubMed] [Google Scholar]
- 149.Morgan NG & Richardson SJ Fifty years of pancreatic islet pathology in human type 1 diabetes: insights gained and progress made. Diabetologia 61, 2499–2506 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Campbell-Thompson M et al. Network for pancreatic organ donors with diabetes (nPOD): developing a tissue biobank for type 1 diabetes. Diabetes Metab. Res. Rev 28, 608–617 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Al-Tabakha MM Future prospect of insulin inhalation for diabetic patients: the case of Afrezza versus Exubera. J. Control. Release 215, 25–38 (2015). [DOI] [PubMed] [Google Scholar]
- 152.Katzung BGM, Susan B & Trevor AJ Basic & Clinical Pharmacology 12th edn (McGraw Hill, 2012). [Google Scholar]
- 153.Tibaldi JM Evolution of insulin: from human to analog. Am. J. Med 127, S25–S38 (2014). [DOI] [PubMed] [Google Scholar]
- 154.Polonsky KS The past 200 years in diabetes. N. Engl. J. Med 367, 1332–1340 (2012). [DOI] [PubMed] [Google Scholar]
- 155.Gill G, Jones K, Smyth C, Bain S & Barnett A Memories of the early years of insulin treatment in the UK. Pract. Diabetes Int 20, 103–107. [Google Scholar]
- 156.Padmore E Reflections on six decades of living with insulin-treated diabetes. J. Diabetes Nurs 10, 366–370 (2009). [Google Scholar]
- 157.Clothier C Living with diabetes in the 1950s. J. Diabetes Nurs 23, 1–8 (2019). [Google Scholar]