

Abstract
Although continued total sleep deprivation is fatal, the function of sleep remains a mystery. Shorter durations of sleep deprivation are followed by rebound increases in non-rapid eye movement (non-REM) sleep, suggesting a homeostatic control. Measurements of the power spectrum of the electroencephalograph (EEG) suggest that a more accurate marker of the homeostasis may be δ frequency power, because it most closely reflects the duration of the preceding sleep deprivation. Several lines of evidence suggest a link with complex metabolic processes. These include a local homeostatic factor, adenosine, that inhibits neuronal activity in response to increases in the ratio of energy demand to metabolite availability. Other evidence derives from the relationship of circadian genes, NPAS2 and Clock, to metabolism. Additionally, at a systems level, hypocretin/Orexin may coordinate motor activity with feeding. Aloss of hypocretin neurons or a mutation of the genes controlling this peptide system can result in the sleep disorder narcolepsy.
Finally, evidence for a role of non-REM sleep in developmental central nervous system (CNS) plasticity, as well as learning and memory, is discussed.
The function and control of sleep in animals remains one of the most elusive biological enigmas. Because the behavioral manifestations of sleep that characterize this state, including motor inactivity, decreased or delayed responsiveness to sensory stimuli, and rapid return to normal waking levels of these behavioral parameters with arousal (Shaw et al., 2000; Shaw and Franken, 2003), are dependent on altered central nervous system (CNS) activity in vertebrates and probably invertebrates as well, sleep is an enigma of neurobiology. In higher vertebrates, this characteristic behavioral change is correlated with a change in the activity pattern of the majority of neurons in the CNS, as suggested by an altered electroencephalograph (EEG) waveform. The waking EEG shows predominantly low-amplitude but high-frequency components compared to a sleeping EEG (the non-rapid eye movement [non-REM]), with predominantly high-amplitude components, especially in the 0.5–5 Hz range. The other neuronal characteristic of non-REM sleep is a slowing of the CNS action potential firing rates compared to waking levels (Evarts, 1967). In addition, mammals also have CNS state transitions from non-REM to REM sleep. REM sleep is similar to waking regarding the firing rates of most neuronal cell types, as reflected by the EEG. REM is dramatically different, because in this stage, “waking-like” EEG activity is associated with a profound somatomotor inhibition and a reduced responsiveness to sensory stimuli. Neither the reason for these changes (i.e., the function subserved by them) nor the neurobiological mechanism(s) responsible are understood, but some potentially important clues constraining the answers to these questions have emerged from recent studies that follow.
Sleep Deprivation
The traditional approach employed in the investigation of the function of sleep has been to examine the outcome of sleep deprivation, but even this seemingly straightforward approach is not without controversy. Much of it stems from the phenomenological definition of non-REM sleep as a group of temporally associated phenomena that usually includes an EEG with predominant slow wave activity and reduced sensory and motor activity, as indicated by a less active EMG and reduced responsiveness to sensory stimuli. Although this group of phenomena is distinct between normal or average behavioral states of waking and non-REM sleep, when perturbations from the norm occur, then the distinctions break down. This happens with severe sleep deprivation, brain lesions, or pharmacological manipulations (e.g., the intrusion of δ wave activity into the waking EEG). Furthermore, these distinctions cannot be directly applied to non-mammalian species, because the EEG and electromyogram (EMG) are qualitatively different for waking and nonwaking behavioral states.
One important indicator for sleep function is uggested by the rebound increase in sleep that follows sleep deprivation. Although the relationship between sleep and the preceding sleep deprivation is variable, the relationship of the slow wave activity during sleep after sleep deprivation (as measured using a fast Fourier analysis of the EEG power in the δ frequency range of 0.5–4 Hz) is correlated with the duration of sleep deprivation (Borbely, 1982; Franken et al., 1991; Tobler and Borbely, 1986). This relationship is even more robust if one focuses entirely on the delta power deprivation and rebound (Franken et al., 1991). The δ power increases results from a combination of increased activity and synchronization of CNS electrical activity within the δ frequency range. At a cellular level, this probably reflects a coordinated burst-pause firing in the thalamus and cortices. The burst-pause oscillation is intrinsic to many thalamocortical neurons and is mediated by an interaction of the hyperpolarization-activated current (Ih), the transient calcium current (It), and a resting membrane potential in the absence of Ih of approx −80 mV(McCormick and Bal, 1997; McCormick and Pape, 1990). Whether this is only an indicator of sleep homeostasis or rather a mechanism that serves an integral role remains to be answered. Predictably, the burst-pause activity will be associated with a characteristic oscillation at the δ frequency of intracellular calcium concentration. It is conceivable that this particular intracellular calcium dynamic signals or at least modulates gene expression and/or second messenger activity.
What happens with prolonged total sleep deprivation? Rats develop a progressively more debilitated appearance, skin lesions, and increased energy expenditure until 11–32 d of deprivation, at which point they die (Everson et al., 1989). Although the energy expenditure is increased, the food intake is also increased and overall energy use was not significantly changed and neither are glucocorticoid nor thyroid hormonal levels (Bergmann et al., 1989). Gene array analysis shows an altered expression of numerous genes, mainly those involved in intermediate carbohydrate metabolism, protein metabolism, and mitochondrial organogenesis and function (Cirelli, 2002; Cirelli and Tononi, 2000; Terao et al., 2003). Surprisingly, with prolonged sleep deprivation, most of the upregulated gene expression returns to baseline or is slightly depressed with the exception of Arylsulfotransferase in the mammal or its homolog in Drosophila (Cirelli, 2002; Cirelli and Tononi, 2000; Tranque et al., 1996). Furthermore, there is no evidence from the TUNEL technique of cellular degeneration after 14 d of sleep deprivation (Cirelli et al., 1999), although increased amino cupric silver staining occurs, which is indicative of a loss of membrane integrity(Eiland et al., 2002).
A major advance in our understanding of sleep function used a Drosophila melanogaster model of sleep (Hendricks et al., 2000; Shaw et al., 2000). Quiet rest (QR) cannot be distinguished from non-REM sleep with traditional means, because the EEG measure of increased slow wave (in the δ frequency range) activity cannot be measured. However, whatever CNS arousal is necessary to sustain the coordinated somatomotor and sensory activity needed to respond to the “gentle tapping or shaking” used to prevent rest is also sufficient to elicit a homeostatic rest rebound, similar to the sleep or δ activity rebound observed in mammals in response to sleep deprivation.
Sleep and Circadian Factors
Shaw and colleagues (2002) have shown that Drosophila with a single mutation of the circadian gene, cycle (cyc; a homolog of the BMAL circadian gene in mammals) or of the heat shock protein (Hsp83) had a strikingly altered response to QR deprivation. The rebound response was greatly exaggerated, and, if the deprivation was prolonged, then the effects were lethal as compared to no lethality in wild-type flies. Finally, preexposure to heat was protective against the lethal effects of sleep deprivation in cyc mutants. Thus, a rest/activity cycle homeostasis was demonstrated for nonvertebrates, controlled by specific genes with vital function, at least regarding the survival of sleep deprivation. Perhaps of even greater import was that these findings suggest a functional interaction between circadian and homeostatic controls of rest/activity.
The interaction between circadian and homeostatic mechanisms of rest/activity control may also be affected by the NPAS2 transcription factor. In the forebrain of mammals, NPAS2 acts as a functional analogue of the circadian gene, Clock, and, either Clock or NPAS2 may bind to form a heterodimer with BMAL1 to activate transcription of downstream circadian gene products, including Per1, Per2, and Cry1 (Reick et al., 2001). However, the activity of either Clock-BMAL1 or NPAS2-BMAL1 heterodimer is under tight control of the redox state of the cell (Rutter et al., 2001). Its activity is greatly facilitated by the reduced state of NADP(H) and NAD(H) and inhibited by the oxidized state. Because the redox state is affected by neuronal activity, prolonged neuronal activity might be expected to feedback onto circadian controlled activities. In fact, sleep deprivation by gentle handling in the absence of increased exercise reset circadian phase in the Syrian Hamster, possibly in relationship to the prolonged increased neuronal activity of waking and an associated altered redox state (Antle and Mistlberger, 2000). Thus, prolonged sleep deprivation might contribute to a disruption of the normal circadian progression of gene expression under the control of the NPAS2/BMAL1 dimer that, in the absence of prolonged waking, occurs in accordance with the expected circadian rest/activity cycles.
Adenosine
Adenosine is another potential factor that might mechanistically link metabolism to neuronal sleep activity. The extracellular adenosine concentration increases whenever the ratio of metabolite demand to metabolite availability increases. The increased concentration can inhibit neuronal activity through adenosine A1 receptors, reducing metabolic demand, and thus providing a negative feedback homeostasis between metabolic demand and neuronal activity (Greene and Haas, 1991). In addition, A1 receptor activity may facilitate δ frequency oscillations (Fig. 1) by (1) a combination of increased G-protein dependent inwardly rectifying potassium (GIRK) channel conductance and decreased activation of the hyperpolarization-activated current, Ih (Pape, 1992), (2) presynaptic inhibition resulting in a functional deafferentation of the thalamocortical circuits that increases the influence of the GIRK and Ih currents to facilitate δ frequency oscillations, and (3) a reduction of cholinergic tone, which inhibits burst-pause firing patterns (McCormick, 1993) by acting at both the cholinergic nuclei (Porkka-Heiskanen et al., 1997; Portas et al., 1997; Rainnie et al., 1994) or locally at the terminal fields of the cholinergic thalamocortical targets (Materi et al., 2000). As noted, this δ frequency activity correlates strongly with sleep homeostatic response to sleep deprivation, and it is conceivable that this kind of neuronal activity is especially facilitated in regions of high adenosine concentration resulting from increased neuronal activity. With sleep deprivation and the associated increase in duration of the neuronal of waking, the extracellular concentration of adenosine increases in the thalamus (Porkka-Heiskanen et al., 1997) and in the cortex (Porkka-Heiskanen et al., 2000). The CNS state of non-REM sleep might be considered permissive to δ wave activity that would be facilitated by the increased adenosine. Others have speculated that adenosine’s role in sleep relates to both δ wave activity (Benington et al., 1995) and its induction of glycogenolysis because glycogen stores may be depleted during prolonged waking (Benington and Heller, 1995; Kong et al., 2002). This is consistent with adenosine’s role in energy metabolism to induce glycogenolysis under conditions of increased neuronal activity (Magistretti et al., 1986).
Fig. 1.
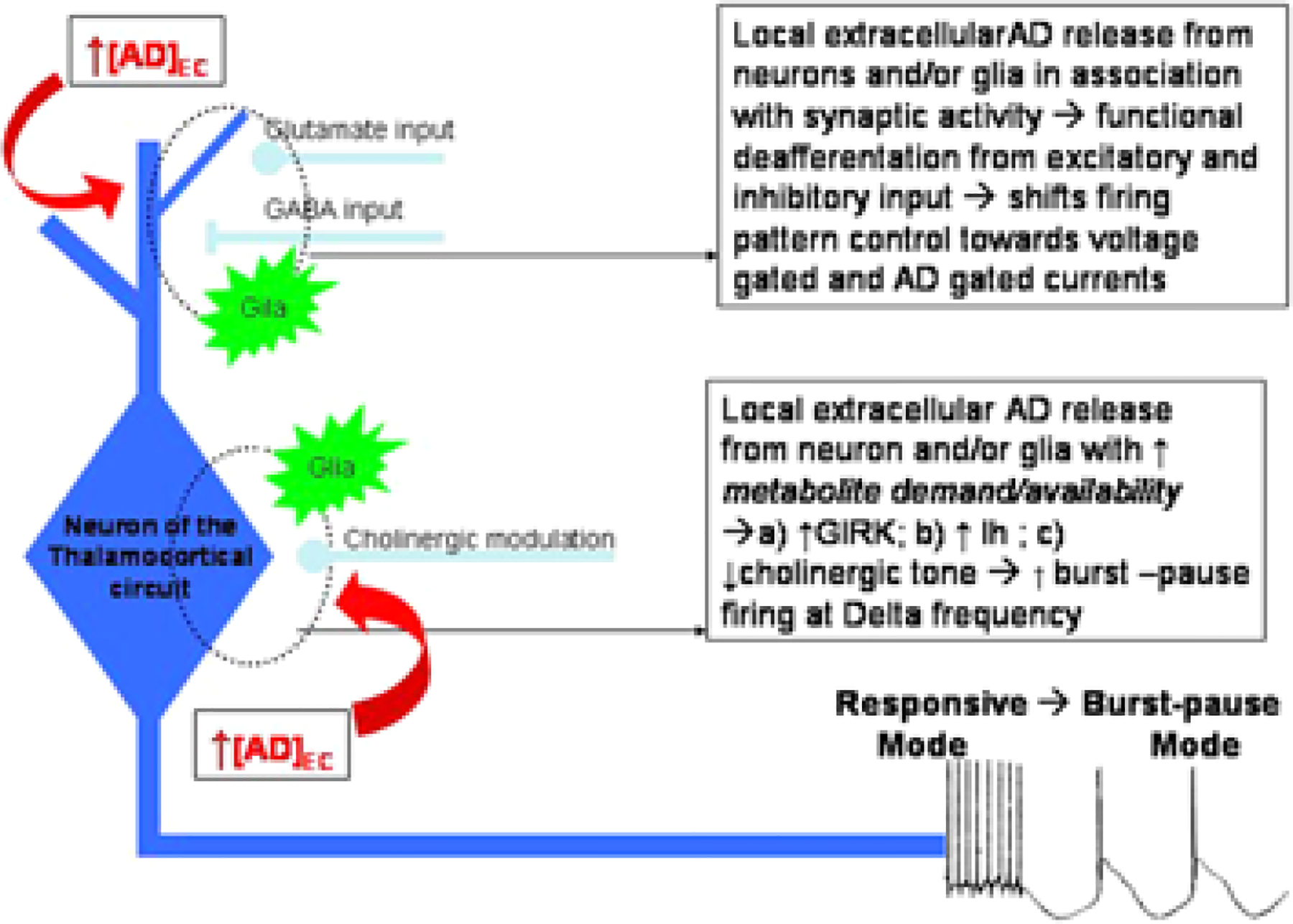
Adenosine facilitates δ frequency activity in thalamocortical circuits.
Hypocretin
Another link between sleep and metabolism is suggested by the fasting-induced arousal observed in mammals. Yamanaka et al. (2003) have reported that hypothalamic hypocretin (Hcrt) (also called Orexin) neurons regulate arousal according to energy balance in studies of Hcrt knockout mice. They find that these mice fail to respond to fasting with increased activity and wakefulness. In prior studies Wu et al. (2002) reported that Hcrt levels were not altered by 48-h food deprivation nor by food consumption in normal dogs. However, when these same dogs were allowed to exercise in a large yard, Hcrt levels increased by an average of 70%. These observations can be reconciled by the hypothesis that Hcrt is more closely linked to aspects of motor activity (and the associated neuronal activity) than to peripheral energy balance per se and that food deprivation in mice under the conditions of Yamanaka et al.’s studies increased motor activity, whereas it did not under the conditions of Wu et al.’s study.
Many of the symptoms of narcolepsy can be mimicked by genetic deletion of the hypocretin or Orexin peptide in mice (Chemelli et al., 1999) or the mutation of the hypocretin or Orexin receptor2 gene in the dog (Lin et al., 1999). In 2001, it was discovered that most human narcolepsy was caused by a loss of hypothalamic cells containing Hcrt (also called Orexin) (Peyron et al., 2000; Thannickal et al., 2000). It was also found that administration of the peptide to genetically narcoleptic dogs reversed symptoms of the disorder (John et al., 2000), suggesting that similar treatment of human narcoleptics could be a uniquely effective treatment for narcolepsy.
In further work in normal animals, Hcrt was released maximally in waking and REM sleep and minimally in non-REM sleep (Kiyashchenko et al., 2002). In waking, its release was linked to certain kinds of motor activity, leading to the hypothesis that release of Hcrt facilitates motor activity during emotionally charged activities of the sort that usually trigger cataplexy (Eiland et al., 2002; Gulyani et al., 2002; Siegel, 2003a; Wu et al., 2002). Even normal individuals experience weakness at these times, which is seen in the “doubling over” that often accompanies laughter or the weakness that can result from sudden-onset, strong emotions. In the absence of the Hcrt-mediated motor facilitation, muscle tone is lost. Many Hcrt neurons also have ascending projections to link cortical arousal to motor and emotional activity. In the absence of Hcrt-mediated facilitation of forebrain arousal centers, waking periods are truncated, resulting in the sleepiness of narcolepsy (Siegel, 2003b; Siegel, 1999; Siegel, 2003a)
Hcrt acts largely by modulating the release of amino acid neurotransmitters (van den Pol et al., 1998). Systemic injection of Hcrt causes a release of glutamate in certain regions innervated by Hcrt, producing a potent postsynaptic excitation (John et al., 2003; Peever et al., 2003). In other regions, it facilitates γ-aminobutyric acid (GABA) release, producing postsynaptic inhibition (Kiyashchenko et al., 2002; Liu et al., 2002). The loss of these competing inhibitory and facilitatory influences leaves brain motor regulatory and arousal systems less stable than the tightly regulated balance that can be maintained in the presence of Hcrt (see Fig. 2). According to this hypothesis, this loss of stability is the underlying cause of narcolepsy, resulting in loss of muscle tone in waking, inappropriate increases of muscle tone during sleep, resulting in a striking increased incidence of REM sleep behavior disorder in narcoleptics (Schenck and Mahowald, 1992). Likewise, although a principal symptom of narcolepsy is intrusions of sleep into the waking period, people with narcolepsy sleep poorly at night with frequent awakenings (Guilleminault and Anognos, 2000; Siegel, 1999; Siegel, 2000). In other words, people with narcolepsy are not simply weaker and sleepier than normals. Rather, their muscle tone and sleep-waking state regulation is less stable than that in normal persons. The authors hypothesize that this results from the loss of the simultaneous inhibitory and excitatory actions of Hcrt.
Fig. 2.
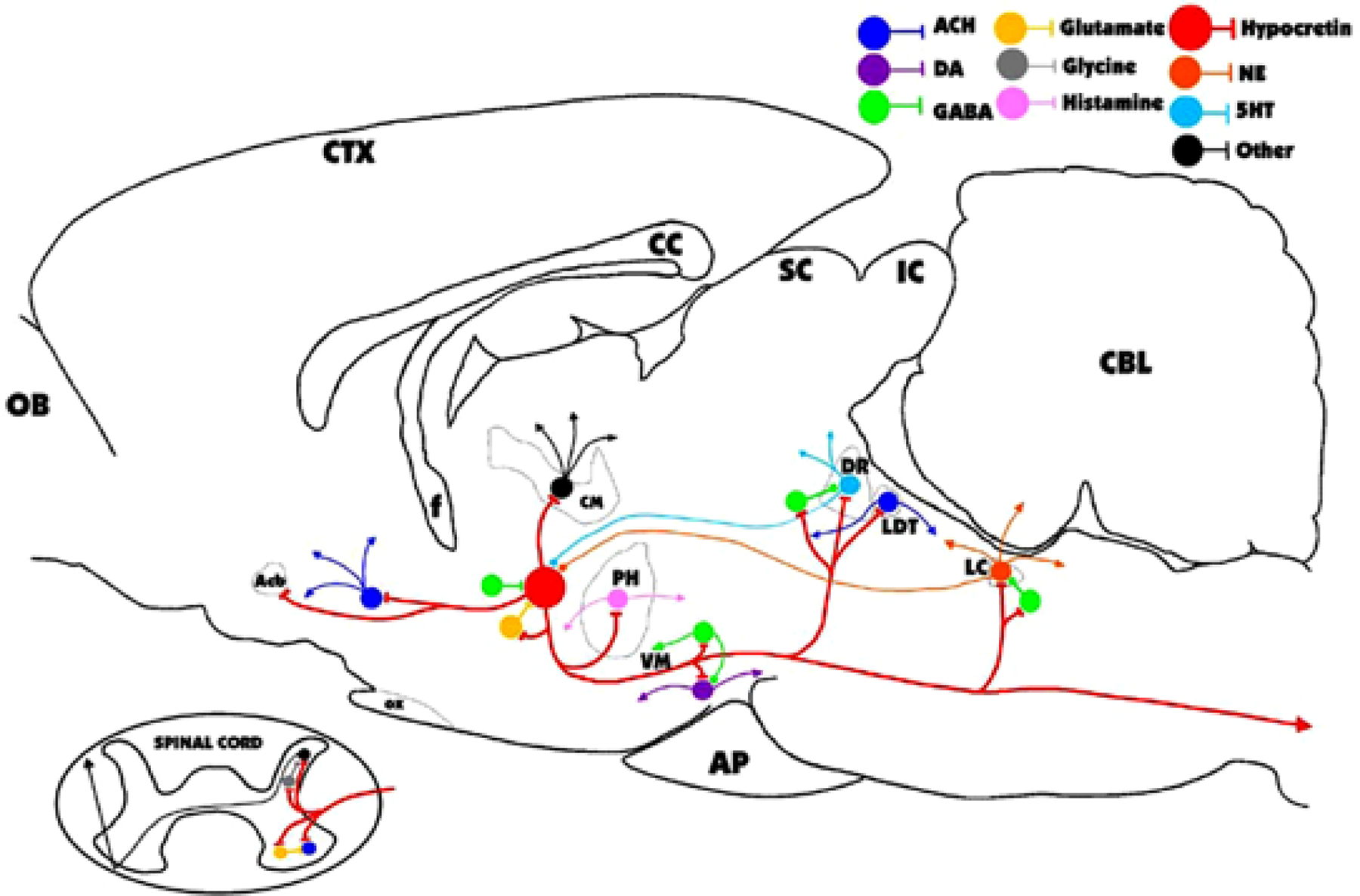
Major identified synaptic interactions of hypocretin (Hcrt) neurons. Lines terminated by perpendicular lines denote excitation; circular terminations indicate inhibition. ACH, acetylcholine; DA, dopamine; NE, norepinephrine; 5HT, serotonin; OB, olfactory bulb; Acb, nucleus accumbens; f, fornix; OX, optic chiasm; CM, centromedian nucleus of the thalamus; PH, posterior hypothalamus; VM, ventral midbrain; AP, anterior pituitary; SC, superior colliculus; IC, inferior colliculus; DR, dorsal raphe; LDT, laterodorsal tegmental and pedunculopontine; LC, locus coeruleus; CBL, cerebellum.
Sleep, Plasticity, and Learning and Memory
Sleep deprivation alters cortical plasticity during development. Occlusion of one eye in the cat for as little as 6 h results in significant plastic changes in the visual thalamocortical circuitry during a critical developmental period for ocular dominance. Sleep deprivation reduces the remodeling induced by the monocular occlusion (Frank et al., 2001). As the authors note, this finding does not imply a particular mechanism for the sleep deprivation effects, nor was it possible to correlate the amount of sleep with the degree of plasticity (altered response). More importantly, it cannot be determined whether the sleep-induced reduction reflects a direct effect on mechanisms responsible for the remodeling or some indirect effect permissive to the remodeling process. For example, this process is depend on protein synthesis(Taha and Stryker, 2002), so that any changes in protein metabolism might have an effect. Similar concerns may apply to the interpretation of the effects of sleep deprivation on consolidation or reconsolidation of a procedural learning and memory task.
In humans, memory can be subdivided into declarative and procedural forms. Declarative memory is fact-based information of the sort that we learn in school or information acquired during daily activities. Declarative memory uses the hippocampus and adjacent temporal lobe structures, and, therefore, these structures have been the focus of physiologically based sleep-learning experimenters, including those observing neuronal unit activity during sleep (Louie and Wilson, 2001). Procedural memory is a nonconscious learning, such as improvement of perceptual or motor skills as might occur in learning to ride a bicycle or to play a musical instrument
Initial sleep learning studies revolved around that concept that dream activity represented a rehearsal of declarative tasks and that REM sleep was critical to declarative memory. However, accumulated evidence demonstrating that neither REM sleep nor total sleep loss interfered with learning when nonstressful sleep deprivation was employed has led critics to question the evidence for a role for sleep in declarative learning (Siegel, 2001; Vertes and Eastman, 2000). Recently, even a leading advocate of a role of sleep in learning has concluded that there is no substantial role for either REM or non-REM sleep in declarative learning (Smith, 2001).
Therefore, most current sleep-learning work has focused on the idea that sleep may have a role in procedural learning. However, evidence for an important role of sleep in procedural learning has also been inconsistent. Studies supporting a role for sleep in the consolidation of human procedural learning have made contradictory claims about similar learning tasks, with some concluding that REM but not non-REM sleep is important, others stating just the reverse, others claiming that both sleep states are essential (Siegel, 2001; Stickgold et al., 2000b), and still others making ad hoc claims, such as that only stage 2 non-REM sleep in the last quarter of the night is important (Walker et al., 2002), which is disputed by procedurally similar studies claiming that REM sleep rather than stage 2 was important (Fischer et al., 2002). Therefore, despite great interest in the area, the evidence for a sleep-learning connection remains weak. Nonetheless, if a subject is allowed to sleep in the hours after training (e.g., 8 h) on certain simple sensory motor tasks the performance improves, despite the absence of any further training (Stickgold et al., 2000a; Walker et al., 2003). Furthermore, performance on this task after reexposure to it can also be affected by subsequent sleep or the lack of it. The mechanisms that might be responsible remain to be examined. This may be a form of consolidation and/or reconsolidation, but the mechanisms are distinct from the consolidation or reconsolidation of either episodic (or in humans, declarative) or emotional, learning and memory, because different neuroanatomical systems are involved with procedural learning and memory. A phenomenon that could involve procedural memory systems is the increased cortical responsiveness observed in non-REM sleep after slow (approx 1 Hz or less) oscillations that lasts for minutes (Steriade and Timofeev, 2003), although the involvement of plasticity lasting for several minutes should not be confused with consolidation, itself. Furthermore, the existence and characterization of consolidation and reconsolidation processes for these kinds of procedural memory is not well established.
Neurons located in the hippocampus fire action potentials in correlation with an animal’s location (hence termed place cells). Remarkably, the correlation of the firing of multiple place cells changes with repeated exposure to and exploration of a particular spatial environment such as a maze, and these changed correlations of firing may be observed during sleep (Skaggs and McNaughton, 1996; Wilson and McNaughton, 1994) in conjunction with sharp wave activity in the hippocampus (Lee and Wilson, 2002; Nadasdy et al., 1999; Ylinen et al., 1995). It is noted that the relationship between place cells and spatial learning is not well understood (McHugh et al., 1996) much less the relationship of this kind of correlated firing of multiple place cells. Nevertheless, these kinds of activity are consistent with a permissive or facilitative role of sleep in spatial memory, but the sharp wave activity is not restricted to sleep (Buzsaki et al., 1992). This hippocampal activity does raise the possibility of a role in consolidation of episodic memory traces (Buzsaki, 1998; Csicsvari et al., 2002; Nadasdy et al., 1999). What is clear is that adequate sleep is vital to the performance of a wide range of tasks, whether these tasks are well consolidated or recently learned.
References
- Antle MC, and Mistlberger RE (2000) Circadian clock resetting by sleep deprivation without exercise in the Syrian hamster. J. Neurosci 20, 9326–9332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benington JH, and Heller HC (1995) Restoration of brain energy metabolism as the function of sleep. Prog. Neurobiol 45, 347–360. [DOI] [PubMed] [Google Scholar]
- Benington JH, Kodali SK, Heller HC (1995) Stimulation of A1 adenosine receptors mimics the electroencephalographic effects of sleep deprivation. Brain Res. 692, 79–85. [DOI] [PubMed] [Google Scholar]
- Bergmann BM, Everson CA, Kushida CA, et al. (1989) Sleep deprivation in the rat: V. Energy use and mediation. Sleep 12, 31–41. [DOI] [PubMed] [Google Scholar]
- Borbely AA (1982) Atwo process model of sleep regulation. Hum. Neurobiol 1, 195–204. [PubMed] [Google Scholar]
- Buzsaki G (1998) Memory consolidation during sleep: a neurophysiological perspective. J. Sleep Res 7(suppl 1), 17–23. [DOI] [PubMed] [Google Scholar]
- Buzsaki G, Horvath Z, Urioste R, Hetke J, Wise K (1992) High-frequency network oscillation in the hippocampus. Science 256, 1025–1027. [DOI] [PubMed] [Google Scholar]
- Chemelli RM, Willie JT, Sinton CM, et al. (1999) Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell 98, 437–451. [DOI] [PubMed] [Google Scholar]
- Cirelli C (2002) How sleep deprivation affects gene expression in the brain: a review of recent findings. J. Appl. Physiol 92, 394–400. [DOI] [PubMed] [Google Scholar]
- Cirelli C, Shaw PJ, Rechtschaffen A, Tononi G (1999) No evidence of brain cell degeneration after long-term sleep deprivation in rats. Brain Res. 840, 184–193. [DOI] [PubMed] [Google Scholar]
- Cirelli C, and Tononi G (2000) Gene expression in the brain across the sleep-waking cycle. Brain Res. 885, 303–321. [DOI] [PubMed] [Google Scholar]
- Csicsvari J, Hirase H, Mamiya A, Buzsaki G (2002) Ensemble patterns of hippocampal CA3-CA1 neurons during sharp wave- associated population events. Neuron 28, 585–594. [DOI] [PubMed] [Google Scholar]
- Eiland MM, Ramanathan L, Gulyani S, et al. (2002) Increases in amino-cupric-silver staining of the supraoptic nucleus after sleep deprivation. Brain Res. 945, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evarts EV (1967) Activity of individual cerebral neurons during sleep and arousal. Res. Publ. Assoc. Res. Nerv. Ment. Dis 45, 319–337. [PubMed] [Google Scholar]
- Everson CA, Bergmann BM, Rechtschaffen A (1989) Sleep deprivation in the rat: III. Total sleep deprivation. Sleep 12, 13–21. [DOI] [PubMed] [Google Scholar]
- Fischer S, Hallschmid M, Elsner AL, Born J (2002) Sleep forms memory for finger skills. Proc. Natl. Acad. Sci. USA 99, 11987–11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MG, Issa NP, Stryker MP (2001) Sleep enhances plasticity in the developing visual cortex. Neuron 30, 275–287. [DOI] [PubMed] [Google Scholar]
- Franken P, Dijk DJ, Tobler I, Borbely AA (1991) Sleep deprivation in rats: effects on EEG power spectra, vigilance states, and cortical temperature. Am. J. Physiol 261, R198–R208. [DOI] [PubMed] [Google Scholar]
- Greene RW, and Haas HL (1991) The electro-physiology of adenosine in the mammalian central nervous system. Prog. Neurobiol 36, 329–341. [DOI] [PubMed] [Google Scholar]
- Guilleminault C, and Anognos A (2000) Narcolepsy. In Kryger MH, Roth T, Dement WC (Eds.),Principles and Practice of Sleep Medicine (pp. 676–686). W.B. Saunders, Philadelphia. [Google Scholar]
- Gulyani S, Wu M-F, Nienhuis R, John J, Siegel JM (2002) Cataplexy-related neurons in the amygdala of the narcoleptic dog. Neuroscience 112,355–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendricks JC, Finn SM, Panckeri KA, et al. (2000) Rest in Drosophila is a sleep-like state. Neuron 25, 129–138. [DOI] [PubMed] [Google Scholar]
- John J, Wu MF, Siegel JM (2000) Hypocretin-1 reduces cataplexy and normalizes sleep and waking durations in narcoleptic dogs. Sleep 23, A12. [PMC free article] [PubMed] [Google Scholar]
- John J, Wu M-F, Kodama T, Siegel JM (2003) Intravenously administered hypocretin-1 alters brain amino acid release: an in vivo microdialysis study in rats. J. Physiol. (Lond) 548, 557–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyashchenko LI, Mileykovskiy BY, Maidment N, et al. (2002) Release of hypocretin (orexin) during waking and sleep states. J. Neurosci 22, 5282–5286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong J, Shepel PN, Holden CP, Mackiewicz M, Pack AI, Geiger JD (2002) Brain glycogen decreases with increased periods of wakefulness: implications for homeostatic drive to sleep. J. Neurosci 22, 5581–5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AK, and Wilson MA (2002) Memory of sequential experience in the hippocampus during slow wave sleep. Neuron 36, 1183–1194. [DOI] [PubMed] [Google Scholar]
- Lin L, Faraco J, Li R, et al. (1999) The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell 98, 365–376. [DOI] [PubMed] [Google Scholar]
- Liu RJ, van den Pol AN, Aghajanian GK (2002) Hypocretins (orexins) regulate serotonin neurons in the dorsal raphe nucleus by excitatory direct and inhibitory indirect actions. J. Neurosci 22, 9453–9464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louie K, and Wilson MA (2001) Temporally structured replay of awake hippocampal ensemble activity during rapid eye movement sleep. Neuron 29, 145–156. [DOI] [PubMed] [Google Scholar]
- Magistretti PJ, Hof PR, Martin JL (1986) Adenosine stimulates glycogenolysis in mouse cerebral cortex: a possible coupling mechanism between neuronal activity and energy metabolism. J. Neurosci 6, 2558–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Materi LM, Rasmusson DD, Semba K (2000) Inhibition of synaptically evoked cortical acetylcholine release by adenosine: an in vivo microdialysis study in the rat. Neuroscience 97, 219–226. [DOI] [PubMed] [Google Scholar]
- McCormick DA (1993) Actions of acetylcholine in the cerebral cortex and thalamus and implications for function. Prog. Brain Res 98, 303–308. [DOI] [PubMed] [Google Scholar]
- McCormick DA, and Bal T (1997) Sleep and arousal: thalamocortical mechanisms. Annu. Rev. Neurosci 20, 185–215. [DOI] [PubMed] [Google Scholar]
- McCormick DA, and Pape HC (1990) Properties of a hyperpolarization-activated cation current and its role in rhythmic oscillation in thalamic relay neurones. J. Physiol 431, 291–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh TJ, Blum KI, Tsien JZ, Tonegawa S, Wilson MA (1996) Impaired hippocampal representation of space in CA1-specific NMDAR1 knockout mice. Cell 87, 1339–1349. [DOI] [PubMed] [Google Scholar]
- Nadasdy Z, Hirase H, Czurko A, Csicsvari J, Buzsaki G (1999) Replay and time compression of recurring spike sequences in the hippocampus.J. Neurosci 19, 9497–9507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape HC (1992) Adenosine promotes burst activity in guinea-pig geniculocortical neurones through two different ionic mechanisms. J. Physiol 447, 729–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peever JH, Lai YY, Siegel JM (2003) Excitatory effects of hypocretin-1 (orexin-A) in the trigeminal motor nucleus are reversed by NMDA antagonism. J. Neurophysiol [DOI] [PubMed] [Google Scholar]
- Peyron C, Faraco J, Rogers W, et al. (2000) A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat. Med 6, 991–997. [DOI] [PubMed] [Google Scholar]
- Porkka-Heiskanen T, Strecker RE, McCarley RW (2000) Brain site-specificity of extracellular adenosine concentration changes during sleep deprivation and spontaneous sleep: an in vivo microdialysis study. Neuroscience 99, 507–517. [DOI] [PubMed] [Google Scholar]
- Porkka-Heiskanen T, Strecker RE, Thakkar M, et al. (1997) Adenosine: a mediator of the sleep-inducing effects of prolonged wakefulness. Science 276, 1265–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portas CM, Thakkar M, Rainnie DG, Greene RW, McCarley RW (1997) Role of adenosine in behavioral state modulation: a microdialysis study in the freely moving cat. Neuroscience 79, 225–235. [DOI] [PubMed] [Google Scholar]
- Rainnie DG, Grunze HC, McCarley RW, Greene RW (1994) Adenosine inhibition of mesopontine cholinergic neurons: implications for EEG arousal. Science 263, 689–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reick M, Garcia JA, Dudley C, McKnight SL (2001) NPAS2: an analog of clock operative in the mammalian forebrain. Science 293, 506–509. [DOI] [PubMed] [Google Scholar]
- Rutter J, Reick M, Wu LC, McKnight SL (2001) Regulation of clock and NPAS2 DNA binding by the redox state of NAD cofactors. Science 293, 510–514. [DOI] [PubMed] [Google Scholar]
- Schenck CH, and Mahowald MW (1992) Motor dyscontrol in narcolepsy: rapid-eye-movement (REM) sleep without atonia and REM sleep behavior disorder. Ann. Neurol 32, 3–10. [DOI] [PubMed] [Google Scholar]
- Shaw PJ, Cirelli C, Greenspan RJ, Tononi G (2000) Correlates of sleep and waking in Drosophila melanogaster. Science 287, 1834–1837. [DOI] [PubMed] [Google Scholar]
- Shaw PJ, and Franken P (2003) Perchance to dream: solving the mystery of sleep through genetic analysis. J. Neurobiol 54, 179–202. [DOI] [PubMed] [Google Scholar]
- Shaw PJ, Tononi G, Greenspan RJ, Robinson DF (2002) Stress response genes protect against lethal effects of sleep deprivation in Drosophila. Nature 417, 287–291. [DOI] [PubMed] [Google Scholar]
- Siegel JM (1999) Narcolepsy: a key role for hypocretins (orexins). Cell 98, 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JM (2000) Narcolepsy. Sci. Amer 282, 76–81.11056991 [Google Scholar]
- Siegel JM (2001) The REM sleep-memory consolidation hypothesis. Science 294, 1058–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JM (2003a) Hypocretin (orexin): role in normal behavior and neuropathology. Annu. Rev. Psychol In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JM (2003b) Why we sleep. Sci. Amer 289,92–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaggs WE, McNaughton BL (1996) Replay of neuronal firing sequences in rat hippocampus during sleep following spatial experience. Science 271, 1870–1873. [DOI] [PubMed] [Google Scholar]
- Smith C (2001) Sleep states and memory processes in humans: procedural versus declarative memory systems. Sleep Med. Rev 5, 491–506. [DOI] [PubMed] [Google Scholar]
- Steriade M, and Timofeev I (2003) Neuronal plasticity in thalamocortical networks during sleep and waking oscillations. Neuron 37, 563–576. [DOI] [PubMed] [Google Scholar]
- Stickgold R, James L, Hobson JA (2000a) Visual discrimination learning requires sleep after training. Nat. Neurosci 3, 1237–1238. [DOI] [PubMed] [Google Scholar]
- Stickgold R, Whidbee D, Schirmer B, Patel V, Hobson JA (2000b) Visual discrimination task improvement: A multi-step process occurring during sleep. J. Cogn. Neurosci 12, 246–254. [DOI] [PubMed] [Google Scholar]
- Taha S, and Stryker MP (2002) Rapid ocular dominance plasticity requires cortical but not genicu-late protein synthesis. Neuron 34, 425–436. [DOI] [PubMed] [Google Scholar]
- Terao A, Steininger TL, Hyder K, et al. (2003) Differential increase in the expression of heat shock protein family members during sleep deprivation and during sleep. Neuroscience 116, 187–200. [DOI] [PubMed] [Google Scholar]
- Thannickal TC, Moore RY, Nienhuis R, et al. (2000) Reduced number of hypocretin neurons in human narcolepsy. Neuron 27, 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobler I, and Borbely AA (1986) Sleep EEG in the rat as a function of prior waking. Electroencephalogr. Clin. Neurophysiol 64, 74–76. [DOI] [PubMed] [Google Scholar]
- Tranque P, Crossin KL, Cirelli C, Edelman GM, Mauro VP (1996) Identification and characterization of a RING zinc finger gene (C-RZF) expressed in chicken embryo cells. Proc. Natl. Acad. Sci. USA 93, 3105–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol AN, Gao XB, Obrietan K, Kilduff TS, Belousov AB (1998) Presynaptic and postsynaptic actions and modulation of neuroendocrine neurons by a new hypothalamic peptide, hypocretin/orexin. J. Neurosci 18, 7962–7971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vertes RP, Eastman KE (2000) The case against memory consolidation in REM sleep. Behav. Brain Sci 23, 867–876. [DOI] [PubMed] [Google Scholar]
- Walker MP, Brakefield T, Hobson JA, Stickgold R (2003) Dissociable stages of human memory consolidation and reconsolidation. Nature 425,616–620. [DOI] [PubMed] [Google Scholar]
- Walker MP, Brakefield T, Morgan A, Hobson JA, Stickgold R (2002) Practice with sleep makes perfect: sleep-dependent motor skill learning. Neuron 35, 205–211. [DOI] [PubMed] [Google Scholar]
- Wilson MA, and McNaughton BL (1994) Reactivation of hippocampal ensemble memories during sleep. Science 265, 676–679. [DOI] [PubMed] [Google Scholar]
- Wu MF, John J, Maidment N, Lam HA, Siegel JM (2002) Hypocretin release in normal and narcoleptic dogs after food and sleep deprivation, eating, and movement. Am. J. Physiol. Regul. Integr. Comp. Physiol 283, R1079–R1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka A, Beuckmann CT, Willie JT, et al. (2003) Hypothalamic orexin neurons regulate arousal according to energy balance in mice. Neuron 38, 701–713. [DOI] [PubMed] [Google Scholar]
- Ylinen A, Bragin A, Nadasdy Z, et al. (1995) Sharp wave-associated high-frequency oscillation (200 Hz) in the intact hippocampus: network and intracellular mechanisms. J. Neurosci 15, 30–46. [DOI] [PMC free article] [PubMed] [Google Scholar]