

Abstract
Polyethyleneglycol (PEG) lipid nanoparticles (LNPs) spontaneously assemble in water forming uniform sized nanoparticles incorporating drugs with prolonged blood clearance over drugs alone. Previously, DSPE-PEG2000 and several drug adducts, including doxorubicin, were analyzed by a combination of physical and molecular dynamic (MD) studies. In this study a complete chemical shift assignment of DSPE-PEG2000 plus or minus doxorubicin was achieved using NMR 1D-selNOESY, NOESY, COSY, TOCSY, HSQC and HSQC-TOCSY. Chemical shift perturbation, titration, relaxation enhancement and NOESY analysis combined with MD reveal detailed structural information at the atomic level including location of doxorubicin in the micelle, its binding constant, the hydrophilic shell organization and the mobility of PEG2000 tail, demonstrating NMR spectroscopy can characterize drug-DSPE-PEG2000 micelles with molecular weights above 180kDa. The MD study revealed that an initial spherical organization led to a more disorganized oblate structure in an aqueous environment, and agreed with the NMR study in details of the fine structure where methyl group(s) of the stearic acid in the hydrophobic core of the micelle are in contact with the phosphate head group of the lipid. Although the molecular size of the LNP drug complex is about 180 kDa, atomic resolution can be achieved by NMR based methods that reveal distinct features of the drug- lipid interactions. Since many drugs have unfavorable blood clearance that may benefit from incorporation into LNPs, a thorough knowledge of their physical-chemical properties is essential to moving them into a clinical setting. This study provides an advanced basic approach that can be used to study a wide range of drug LNP interactions.
Keywords: lipid nanoparticle, doxorubicin, nuclear magnetic resonance, molecular dynamics, polyethylene glycol
Graphical Abstract
INTRODUCTION
Most clinical drugs used in chemotherapy are limited by their rapid blood clearance and high systemic toxicity as exemplified by Doxorubicin (DOX), a front line drug in the treatment of breast cancer1–3. DOX is usually formulated in saline in the pH range of 2–4, presenting a challenge for injections at physiological pH. The liposomal formulation of DOX, Doxcil, extends the blood clearance of the drug, leading to better tumor uptake in animal models and widespread use in the clinic4. The main issue with liposomal formulations is the high cost and effort of preparing liposomes of uniform size. An alternate lipid formulation is the use of micelles of 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycerol)-2000] or DSPE-PEG2000, a composite lipid that not only binds DOX, but spontaneously assembles into uniform sized micelles in water5–6. The lipid portion, DSPE, was chosen for its favorable biodistributions and physical properties, while the covalently attached PEG2000 (polyethylene glycol, Mave= 2000), is a polymeric micelle with a low critical micelle concentration7–8 that makes the micelle stable even during the dilutions that occur upon injection into blood. The hydrophobic core composed of phosphoethanolamine lipid can solubilize a wide range of hydrophobic drugs. The hydrophilic PEG2000 at the outer micelle surface inhibits micelle uptake by the mononuclear phagocyte system, prolonging its circulation half-life9. The polymeric micelles can accumulate in tumor tissue preferentially through the enhanced permeability and retention (EPR) effect10. These features make DSPE-PEG2000 a favorable vehicle for drug delivery in cancer therapy. There are many studies exploring the applications of polymeric micelles for improved drug efficacy and reduced drug toxicity11–12, and also see recent reviews13–17. On the other hand, there were only a few studies addressing the structure and physical properties of DSPE-PEG2000 micelles in the presence or absence of drugs8, 18–20.
Recently, Liang and coworkers6, 21 have extensively studied the incorporation of DOX-HCl into DSPE-PEG2000 using isothermal titration calorimetry (ITC), transmission electron microscopy (TEM), dynamic light scattering (DLS), static light scattering (SLS), small-angle X-ray scattering (SAXS), molecular dynamics (MS) and NMR spectroscopy. An important finding of their study was the observation that DOX was mainly held in the micelle by an electrostatic interaction involving the negative charge of the phosphate head group and the positive charge of the amino group on DOX at neutral pH with a further binding energy contribution from the hydrophobic interaction between the anthracycline ring of DOX and the lipid core. Through a variety of physical tools, Liang and coworkers6, 21 showed the average diameter of the DOX-micelles ranged from 16.5 to 20nm with an average of 90 block lipid-PEG2000 units per micelle. The diameter of the inner hydrophobic core was 4.3nm, the length of two lipid molecules. The hydrophobic core is surrounded by a 2nm thick shell composed of the hydrophilic lipid head group and about ten units of PEG19, 21. Outside this shell is a corona of more flexible glycol units. Liang and coworkers21 showed that the embedding of DOX did not change the aggregation number and the size of the micelle. In addition, the embedded DOX makes the micelle more stable and more uniform in size. SAXS studies revealed the location of DOX at the interface between the hydrophobic core and hydrophilic shell, in agreement with the observed electrostatic interaction between the negative charge of the phosphate head group and the positive charge of the amino group on the DOX6. The same group also performed molecular dynamic (MD) simulations on a system with 90 DSPE-PEG2000 and 90 DOX molecules in an aqueous environment, showing that the initial extended terminal PEG head groups rapidly move in towards the hydrophobic core reducing the initial size of the micelle from 36 to 17.5nm. Based on the MD simulations, they concluded that the anthracycline ring of DOX resides within the hydrophobic core of the micelle. Based on this model, the distance between the D ring of DOX to the methyl group of lipid is about 10 Å assuming the lipid fatty acyl chain adopts the trans configuration in the micelle. In their earlier NMR study6, the same group showed there are NOE cross peaks between the lipid methyl group with the D ring of DOX. The presence of the NOE cross peak is intriguing considering the distances of these two parties exceed 5Å. Possible explanations are either the NOE cross peak may come from a spin diffusion effect considering the NOE mixing time used in their study is 500ms, or there might be a structural arrangement in micelle such that the methyl group of lipid is much closer to the D ring of DOX than 10Å.
In a series of comprehensive studies Onyuksel and coworkers22–25 have shown that drugs and peptides can be incorporated into DSPE-PEG micelles to enhance their in vivo delivery. More recently, they performed MD simulations of DSPE-PEG2000 micelles with Nagg =90, the experimentally measured Nagg, and found the initial donut shaped micelle collapsed to an oblate shape after as little as 7ns of simulation in an aqueous environment8. They identified three regions of the micelle as the core, the ionic interface and the PEG corona, with the latter exhibiting the greatest flexibility8. They conclude that the highly fluctuating corona makes these micelles suitable for a wide range of interactions with small molecules.
Given the exciting possibility that DOX-Micelle mixtures can be used as a simpler method for longer lived, slowly releasing DOX formulations, and the DSPE-PEG micelle is a promising drug delivery platform, we have re-investigated the DSPE-PEG2000 micelle and its complex with DOX using both NMR and MD simulations. In this study, we provide atomic-resolution structural insights of the micelle and its complex with DOX by fully exploring the power of NMR spectroscopy in combination with MD simulation. Through heat-ice cycles during the micelle preparation, and carrying out the NMR experiments at the physiological relevant temperature 37°C, the NMR spectrum resolution was greatly enhanced. A complete assignment of both 1H/13C chemical shifts of DOX and DSPE-PEG2000 was achieved in their free state, and for DSPE-PEG2000 in complex state with DOX. 1H chemical shifts of DOX in complex were also assigned. Based on the chemical shifts assignment, NOESY data provided greater details of the structural organization of the free micelle and its complex with DOX. In addition, chemical shift perturbation and DOX titration to DSPE-PEG2000 was used to locate the binding site and affinity of DOX to the micelle. The NOESY pattern of DSPE-PEG2000 with and without loading of DOX showed that the embedding of DOX did not change the micelle structure. Combining the NMR and MD studies, an explanation was provided for the observed NOE cross peak between the lipid methyl group and the D ring of DOX. Furthermore Gd-DOTA relaxation enhancement studies demonstrated that the access of Gd-DOTA to the lipid head group region is limited. This study showed that NMR spectroscopy is a powerful tool for the structural characterization at atomic resolution level for the DSPE-PEG2000 micelle system even though the molecular weight is about 180kDa. The complete assigned chemical shifts of DSPE-PEG2000 obtained in this work should enable future studies of its interaction with other drugs using NMR.
RESULTS
Chemical shift assignments of free DOX and DOX in the presence DSPE-PEG2000.
The assignment of 1H and 13C of free DOX was straightforward through the analysis of COSY, TOCSY, HSQC and HSQC-TOCSY. The 1H assignment of DOX has been reported before in D2O at temperature of 24°C, 52°C and 82°C26 and at 25°C27. The proton chemical shifts of DOX assigned in this study at 37°C are in agreement with the reported values obtained at 24°C26 and 25°C27. The chemical shifts of 1H/1C (See Figure 1A for DOX numbering) and 3H/3C are very close for both 1H and 13C. The assignment of these two protons was achieved from a series of selective 1H NOESY with refocusing pulse selectively irradiating 4-OCH328 (Bruker selnogp sequence). In these 1H selective NOESY experiments, the NOE peak from 4-OCH3 to 3H started to show up when mixing time was as short as 25 ms due to their short distances, and the peak intensity became stronger as the mixing time increased. The NOE peak from 4-OCH3 to 2H started to appear when the mixing time reached 400ms. The NOE peak to 1H did not appear even when the mixing time was set to 700ms. Thus, the chemical shifts of 1H/1C and 3H/3C were assigned through these series of 1H selective NOE and 1H-13C HSQC. The two protons at position 14 resolved and appeared as an AB splitting pattern in the 1H spectrum (see the inset of Figure 1A). The two protons at position 14 were not resolved in the earlier literature26–27, either due to the lower magnetic field or different experimental conditions used in their study. The AB splitting pattern also showed up in HSQC, where there are three resolved peaks, the middle two strong peaks are merged as single strong peak due to the lower resolution compared to one-dimension 1H spectrum. We labeled these two resolved peaks as 14Ha and 14Hb. No further efforts were made to assign the axial and equatorial protons for position of 8, 10 and 2′ since they can be obtained by comparing the chemical shifts of the corresponding protons with those reported in the literature26–27. Similarly, the 1H-1H J couplings have been fully characterized before26–27, and thus they are not discussed in this study.
Figure 1. 1H-NMR 1D spectra of DSPE-PEG2000, DOX and the mixture.
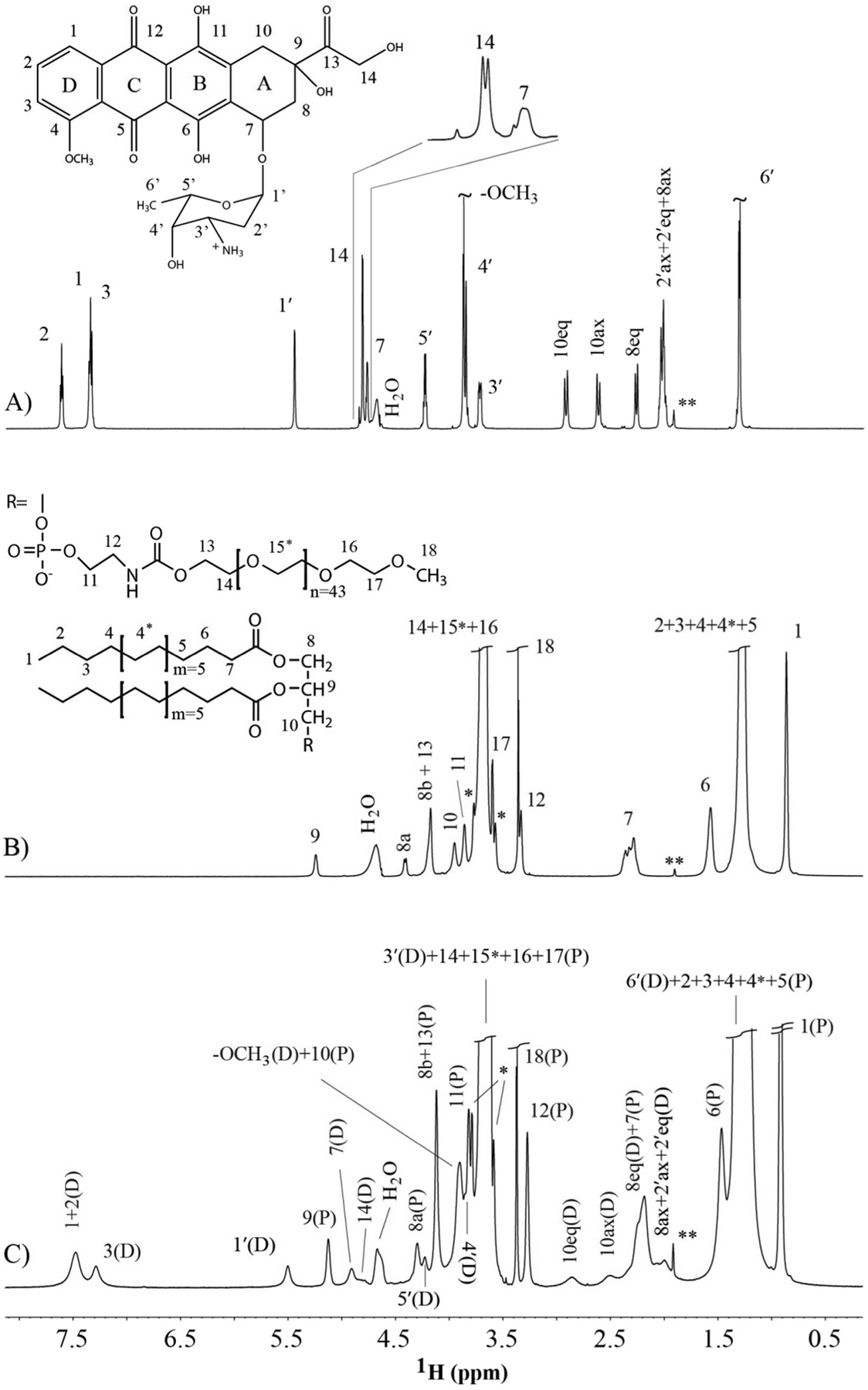
(A) DOX, (B), DSPE-PEG2000 and (C) a 0.96:1 molar ratio of DOX:DSPE-PEG2000 in D2O-based 50mM NaAc-d3 buffer. The structure and numbering of DOX and DSPE-PEG2000 are shown in (A) and (B), respectively. In (C), the protons from DOX are denoted with a letter “D” in parenthesis, and the protons from DSPE-PEG2000 are denoted with a letter “P” in parenthesis. The small peaks denoted with ** are from residual proton of NaAc-d3, and the peaks labeled with * are J coupling splitting between 1H and natural abundance 13C of strong ethylene glycol units. The inset of (A) shows the fine spectrum pattern of two H14 protons.
The assigned proton peaks of DSPE-PEG2000 micelle without DOX and numbering scheme are shown in Figure 1B, and the assigned protons peaks of both DOX and DSPE-PEG2000 in complex are shown in Figure 1C. In the presence of DSPE-PEG2000, the proton peaks of DOX became significantly broadened due to the interaction with DSPE-PEG2000 (Figure 1C). Due to fast transverse relaxation, the COSY, TOCSY, HSQC and HSQC-TOCSY spectra are not very fruitful in the assignment of DOX protons in the complex. However, the proton assignment of DOX in the complex has been achieved using NOESY experiments with different mixing times in comparison with the NOESY spectra of free DOX. The proton 1H of DOX moved significantly downfield in the complex, and merged with proton 2H as shown in Figure 1C. Similar to free DOX, the assignment of 1H, 2H and 3H was achieved using a series of selective NOESY with an array of mixing times with selective irradiation of 4-OCH3 in combination with the titration of DOX to DSPE-PEG2000. By comparison with the NOESY pattern observed from free DOX, the majority of proton assignments for the DOX complex was achieved by analysis of 2D NOESY with mixing time of 50 ms, in which cross peaks from intra-molecules dominated. NOESY spectra with longer mixing times were used to verify a few assignments. 14Ha and 14Hb were assigned from the H1′ NOESY slice with a mixing time of 400ms. Due to the poor sensitivity of HSQC, broadened line width and overlaps, the peaks of 8axH, 2′axH and 2′eqH were not well resolved. There were only two peaks from DOX appeared in the HSQC spectrum acquired on the complex. All the peak assignments of DOX in the presence and absence of DSPE-PEG2000 are listed in Table 1.
Table 1:
Doxorubicin Chemical Shift Assignments at 37°C, pH 5.4a.
| Free DOX | DOX + DSPE-PEG2000 | |||
|---|---|---|---|---|
| Atom No. | 1H(ppm) | 13C(ppm) | 1H(ppm) | 13C(ppm) |
| 1 | 7.36 | 122.39 | 7.48 | |
| 2 | 7.61 | 139.67 | 7.48 | |
| 3 | 7.34 | 122.69 | 7.29 | |
| 4-OCH3 | 3.88 | 59.26 | 3.92 | 59.02 |
| 7 | 4.77 | 71.49 | 4.91 | |
| 8ax | 2.03 | 38.13 | 2.00* | |
| 8eq | 2.27 | 38.13 | 2.31 | |
| 10ax | 2.62 | 34.84 | 2.52 | |
| 10eq | 2.92 | 34.84 | 2.86 | |
| 14a | 4.80 | 67.15 | 4.73 | |
| 14b | 4.83 | 67.18 | 4.83 | |
| 1' | 5.45 | 101.73 | 5.51 | |
| 2'ax | 2.04 | 30.44 | 2.06* | |
| 2'eq | 2.02 | 30.44 | 2.00* | |
| 3' | 3.72 | 49.64 | 3.67 | |
| 4' | 3.85 | 69.07 | 3.85 | |
| 5' | 4.23 | 69.88 | 4.23 | |
| 6' | 1.31 | 18.57 | 1.30 | 18.41 |
The chemical shifts are reference to d4-TSP with its 1H and 13C chemical shifts of methyl groups are at −0.0118 and −0.183 ppm, respectively29.
Due to overlapping and broad linewidths, these assignments are not well defined.
Chemical shift assignments of free DSPE-PEG2000 and DSPE-PEG2000 in the presence of DOX.
The assignment of 1H and 13C of DSPE-PEG2000 in the absence of DOX was achieved using 2D TOCSY, NOESY, 1H-13C-HSQC and 1H-13C-HSQC-TOCSY. The sample dissolved in H2O-based buffer was used for assignment of proton 11H and 12H (see Figure 1B for DSPE-PEG2000 numbering). Since the amide peak is well separated from other peaks, the proton 11H and 12H can be distinguished from other protons through the cross peaks originating from proton 11H/12H to the carbamate NH proton in a 2D TOCSY experiment. The assignment of protons 11H and 12H was achieved by comparing the intensity of the cross peak to the carbamate NH in TOCSY acquired with a mixing time of 15 and 66 ms. The spectrum of TOCSY with a mixing time of 15 ms is shown in Figure 2A, together with the NOESY in the absence of DOX (Figure 2B) and in the presence of DOX (Figure 2C). TOCSY with a different mixing time was used to assign protons 11H and 12H because the COSY spectrum was not useful in the assignment due to interference from strong peaks of water, ethylene glycol and acyl chain methylene groups. There are two chemical shifts from carbamate NH, and one of them is dominated in intensity. The major peak with higher 1H chemical shift value could be due to the hydrogen bond of carbamate NH with an electronegative group in the lipid head group region (see more detail in the discussion). The 13H was assigned through the cross peaks to 11H and 12H from the NOESY spectra acquired on the sample in D2O-based buffer. The 14H was then assigned through the TOCSY correlation with 13H and verified using HSQC-TOCSY. Proton 17H was assigned from the NOESY cross peak with 18H at mixing time of 500ms. The sign of cross peaks between 18H and 17H was opposite to diagonal peaks, indicating the end of the polyethylene glycol chain is very flexible in the micelle. The 16H was assigned from 17H using TOCSY and HSQC-TOCSY experiments. This part of the assignment is highlighted in Figure S1A. The 1H and 13C chemical shifts of other ethylene glycol units are degenerate. The assignment of lipid part was achieved by analyzing the TOCSY, HSQC and HSQC-TOCSY data in combination with comparing to those reported chemical shifts30–31. The HSQC-TOCSY is particularly useful in the assignment of both 1H and 13C chemical shifts of four positions at the end of lipid acyl chain. The assignment of this segment of the lipid acyl chain was obtained from the comparison of HSQC with HSQC-TOCSY as shown in Figure S1B.
Figure 2. TOCSY and NOESY spectra of DSPE-PEG2000.
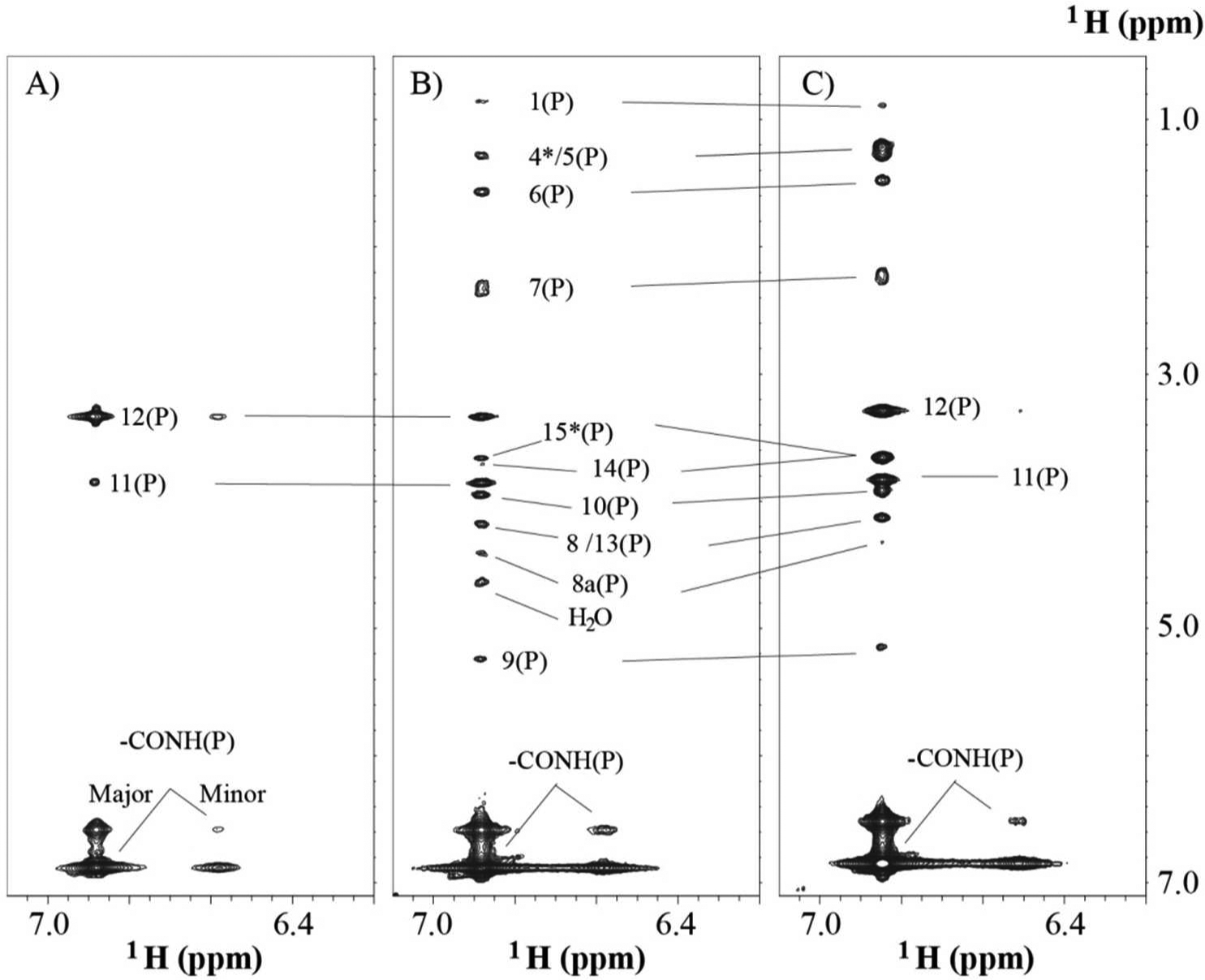
(A) TOCSY 2D slice along carbamate NH of free DSPE-PEG2000. (B) NOESY slice along carbamate NH acquired on free DSPE-PEG2000, and (C) on the complex. There are two peaks from the carbamate NH, in which the minor peak is less than 10% of major one. The letter “P” stands for DSPE-PEG2000.
When DOX was mixed with DSPE-PEG2000, some chemical shifts of DSPE-PEG2000 changed due to their interaction with DOX. On the other hand, the line widths of DSPE-PEG2000 peaks did not increase much (see Figure 1B and 1C). Since the linewidth of DSPE-PEG2000 did not change much in the presence of DOX, the complete 1H and 13C chemical shifts have been achieved using the TOCSY, HSQC and HSQC-TOCSY experiments. The 1H and 13C assignments of DSPE-PEG2000 in the absence and presence of DOX are listed in Table 2.
Table 2:
DSPE-PEG2000 Chemical Shift Assignments at 37°C, pH 5.4a.
| Free DSPE-PEG2000 | DSPE-PEG2000+DOX | |||
|---|---|---|---|---|
| Atom No. | 1H(ppm) | 13C(ppm) | 1H(ppm) | 13C(ppm) |
| 1 | 0.86 | 16.22 | 0.91 | 16.40 |
| 2 | 1.27 | 25.00 | 1.32 | 25.18 |
| 3 | 1.26 | 34.34 | 1.30 | 34.53 |
| 4 | 1.27 | 31.87 | 1.30 | 32.04 |
| 4* | 1.27 | 32.39 | 1.30 | 32.47 |
| 5 | 1.30 | 31.58 | 1.21 | 31.96 |
| 6 | 1.57 | 27.25 | 1.47 | 27.35 |
| 7 | 2.36 | 36.36 | 2.23 | 36.44 |
| 7 | 2.32 | 36.34 | 2.20 | 36.44 |
| 7 | 2.29 | 36.31 | 2.20 | 36.44 |
| 8a | 4.41 | 65.37 | 4.30 | 65.32 |
| 8b | 4.19 | 65.40 | 4.12 | 65.40 |
| 9 | 5.24 | 73.03 | 5.13 | 73.01 |
| 10 | 3.95 | 65.95 | 3.90 | 66.00 |
| 11 | 3.86 | 66.79 | 3.82 | 66.87 |
| 12 | 3.33 | 43.62 | 3.28 | 43.72 |
| 13 | 4.17 | 66.37 | 4.12 | 66.50 |
| 14 | 3.70 | 71.48 | 3.67 | 71.50 |
| 15* | 3.67 | 72.02 | 3.69 | 72.11 |
| 16 | 3.66 | 71.76 | 3.68 | 72.00 |
| 17 | 3.60 | 73.41 | 3.61 | 73.50 |
| 18 | 3.36 | 60.46 | 3.37 | 60.56 |
| NH(major) | 6.88 | 6.85 | ||
| NH(minor) | 6.58 | 6.51 | ||
The chemical shifts are in reference to d4-TSP with its 1H and 13C chemical shifts of methyl groups are at −0.0118 and −0.183 ppm, respectively29.
Chemical shifts changes in the DOX-DSPE-PEG2000 complex.
Upon complex formation, the chemical shifts of both 1H and 13C of DSPE-PEG2000 changed. Since most of the 13C chemical shift changes are smaller than the digital resolution of the spectrum, except for the 13C of atom 5, the analysis was focused on the 1H chemical shift changes. There are five positions, 5–7, 8a, 9, whose 1H chemical shift changes are larger than 2 fold of RMSD of all the measured protons. The protons with significant chemical shift changes indicated they are involved in the interaction with DOX, and all these protons are clustered at or close to the lipid head group. The chemical shift change of methyl proton of lipid tail is smaller than 2 fold of RMSD. As mentioned before, the line width of DSPE-PEG2000 barely changed in the presence of DOX for those resolved peaks (see Figure 1B and C), probably because embedded DOX did neither increase the size of micelle21 nor reduce the flexibility of the micelle. The overlap of HSQC spectra of free DSPE-PEG2000 and its complex with DOX is shown in Figure 3. For DOX, the line widths increased a lot in the presence of micelle. This indicated that DOX was fully embedded in the micelle, and thus its tumbling rate was much slower. In addition to the much slower tumbling rate, another potential contribution to significant linewidth broadening is due to DOX movement within the micelle, which can contribute linewidth broadening by the exchange mechanism. Since only two 13C chemical shifts of DOX are detectable when it forms a complex with DSPE-PEG2000 (Figure 3), therefore, only the chemical shift difference of 1H between free and the complex state was analyzed. There are four protons 1H, 2H, 7H and 10axH whose chemical shift difference are larger than two fold of the RMSD of all observed protons. These four protons are located at rings A and D, the two ends of the four fused rings. There are no observable NMR proton signals at rings B and C when the DOX is dissolved in D2O. Because DOX aggregates when its concentration is greater than 10 μM due to ring stacking32, the chemical shift changes between free DOX and DOX in complex in this study do not necessarily reflect the true chemical environmental changes from DOX monomer to DOX in complex. This may explain why the chemical shift of 3H on ring D did not change significantly while the 1H and 2H of ring D changed a lot.
Figure 3. HSQC overlay spectra of DSPE-PEG2000 and the DOX mixture.
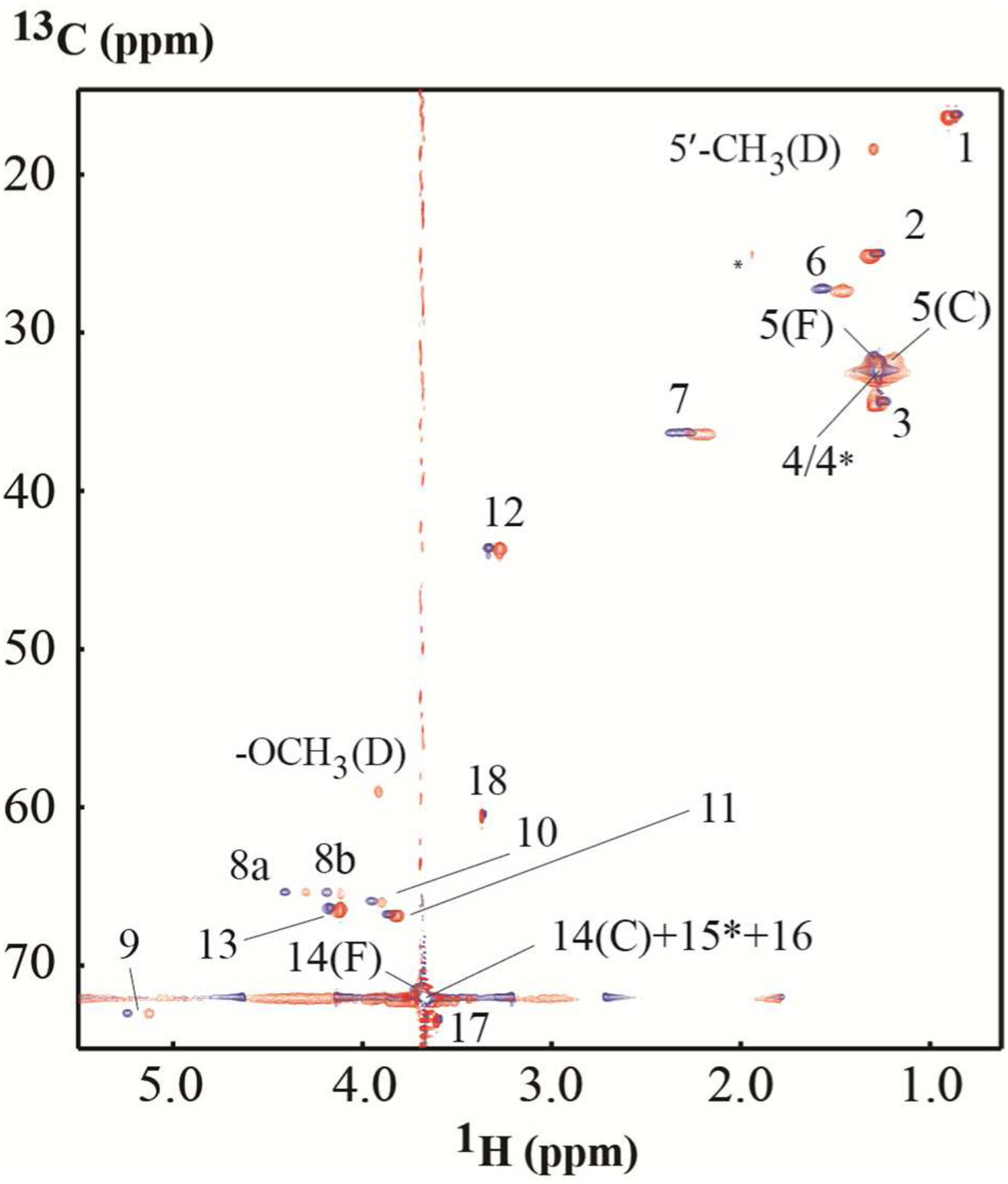
Overlay of HSQC spectra acquired on free DSPE-PEG2000 (blue) and on a mixture of 0.96:1 molar ratio of DOX and DSPE-PEG2000 (red). The ridges were caused by large peaks of the polyethylene glycol units. Due to line broadening, only two methyl groups from DOX appear in the HSQC acquired on the mixture. The letters “C”, “D”, “F” denote the complex, DOX and free DSPE-PEG2000, respectively. The peak denoted with * is from a residual proton of NaAc-d3.
Structural information from NOESY experiments.
To avoid spin diffusion interference in the data analysis, the NOE build-up rate was evaluated using cross peak volumes between 9H and 8Ha of DSPE-PEG2000 versus the mixing time of 50, 150 and 250 ms in D2O-based buffer. The NOE build-up rate maintained a good linearity up to a mixing time of 250 ms with a correlation coefficient of 0.96. The correlation coefficient was 0.91 up to mixing time of 350 ms for the same pair of protons of DSPE-PEG2000 dissolved in H2O-based buffer. So, the following discussion will be focused on the data acquired using mixing time of 250 ms. Figures 2B and 2C present the NOESY slice at the carbamate NH group in the absence and presence of DOX, respectively. To separate the proton numbering of DOX and DSPE-PEG2000, and simplify the discussion, the proton followed by (P) and (D) are used to stand for the proton from DSPE-PEG2000 and DOX, respectively. There are many long range (larger than five bonds connection) intramolecular NOESY cross peaks pointing to the carbamate NH in DSPE-PEG2000 as shown in Figures 2B and 2C. The cross peaks are from all the protons of the lipid glycerol groups, 6H(P), 7H(P) and 1H(P). The cross peak patterns in the absence and presence of DOX were almost the same except for some minor chemical shifts changes in the presence of DOX, and small variations of cross peaks intensities. This observation suggested that the addition of DOX did not change the local structure around the lipid head group. This cross peak network between the carbamate NH with many protons in the proximity of the lipid polar region suggests that the carbamate NH is located close to the lipid head group. An interesting observation is the cross peak of 1H(P) to the carbamate NH. This connection indicates that methyl group of lipid tail could be close to the lipid polar head region.
Shown in Figure 4A are the NOESY slices at 1H/2H(D) and 3H(D) of DOX in the complex with DSPE-PEG2000. There are intermolecular NOESY cross peaks between 1H/2H(D) with 1H, 4*H/5H, 6H, 7H and 8Hb/13H of DSPE-PEG2000. Similarly, cross peaks between 3H(D) and 4*H/5H, 6H, 7H and 8Hb/13H of DSPE-PEG2000 were observed. These cross peaks showed that the D ring of DOX mainly interacts with the acyl chains of DSPE. The distance between 8Hb and 1H(P) would be more than 24Å if the acyl chain adopts the trans configuration in the micelle. Thus, it is difficult to establish a NOESY connection between the D ring of DOX with 8Hb and 1H(P) simultaneously (see discussion for an explanation of this finding). Figure 4B shows NOSEY slices at 1'H(D) and H9(P) acquired on the complex, and Figure 4C shows the slice at H9(P) acquired on free DSPE-PEG2000. There are several intermolecular NOEs identified from Figure 4B, between 1'H(D) and 9H(P), 1'H(D) and 10H(P), between 9H(P) and 7H(D), 9H(P) and 14Ha/14Hb(D). These NOESY cross peaks indicated the close contact between the head group of DSPE-PEG2000 and the sugar ring as well as the anthracycline ring A of DOX. This is in agreement with the location and orientation of DOX determined from SAXS [19]. A comparison of Figure 4B and 4C shows that the addition of DOX to DSPE-PEG2000 did not change the intramolecular NOESY pattern of 9H(P), and thus the local structure around the head group of DSPE-PEG2000 was not altered in the presence of DOX except for a few chemical shift perturbations. Note that there is a cross peak between 1H(P) and 9H(P) in both Figure 4B and 4C. This is another evidence that methyl group of lipid tail is close to the lipid head group.
Figure 4. NOSEY spectra of the DSPE-PEG2000 and complex with DOX.
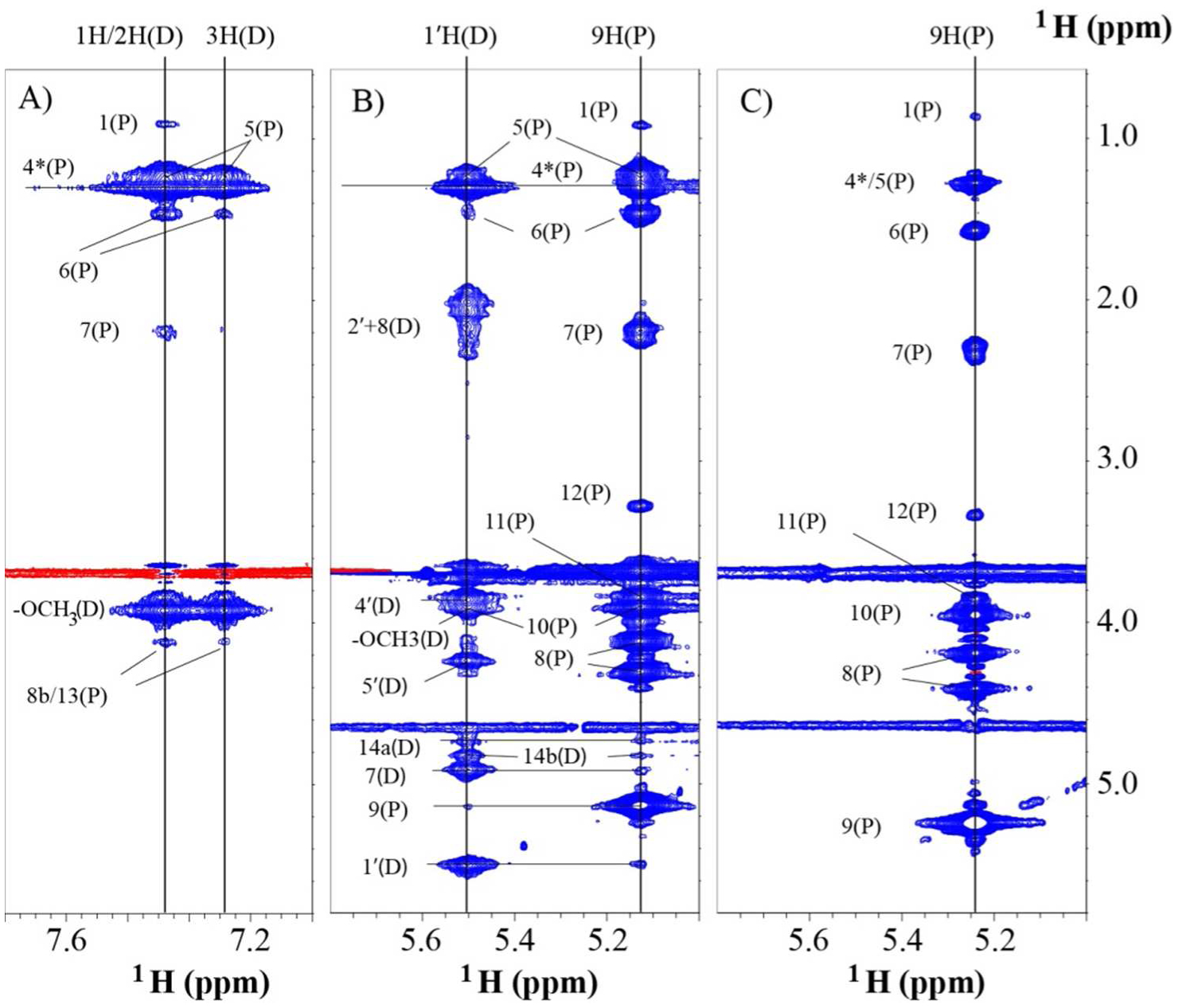
(A) A slice along DOX 1H/2H(D) and 3H(D) from NOESY acquired on the complex. (B) A slice along DOX 1'H(D) and DSPE-PEG2000 9H(P) from NOESY acquired on the complex. (C) Slice of DSPE-PEG2000 9H(P) from NOESY acquired on free DSPE-PEG2000.
Since 1H(P) is located at the distal end of the acyl chains, and there are still NOESY cross peaks between 1H(P) and the carbamate NH, 9H(P), 11H(P) as well as 12H(P) (see Table 3), it is worthwhile to investigate the structural basis for this observation. Figure S2A and S2B present the 1D slices at the position of 6H(P) and 1H(P) along the direct dimension of 2D NOESY data acquired on free DSPE-PEG2000. Although the cross peaks between 6H(P) and other protons are much stronger than those observed from the 1H(P) slice, the protons with cross peaks to both 6H(P) and 1H(P) are exactly the same. These protons include all the lipid glycerol protons, 7H(P), 4*H(P), 11H(P), 12H(P) and 13H(P). In addition to these, there are cross peaks between 1H(P) and 6H(P). Similarly, in the presence of DOX, the NOESY pattern of two 1D slices at position of 6H(P) (Figure S2C) and 1H(P) (Figure S2D) are the same except for the higher intensity of cross peaks to 6H(P) than to 1H(P), and one extra cross peak of 1'(D) to 6H(P). Both 6H(P) and 1H(P) have intermolecular cross peaks with 1H/2H(D), 3H(D), and – OCH3(D), and the intramolecular cross peak patterns are the same compared to those in free DSPE-PEG2000 (Figure S2A and S2B). These observations suggest that 1H(P) is in proximity to 6H(P), leading to a similar NOESY pattern to 6H(P), although with lower intensity.
Table 3:
Representative long range NOEs in DSPE-PEG2000 and intermolecular NOEs between DSPE-PEG2000 and DOX.
| Proton A | Proton B |
|---|---|
| 1H/2H(D) | 1H(P), 4*/5H(P), 6H(P), 7H(P), 8b/13H(P), 9H(P)a, 11H(P)a, 12H(P)a |
| 3H(D) | 1H(P)a, 4*/5H(P), 6H(P), 7H(P)a, 8b/13H(P), 9H(P)a, 11H(P)a |
| 1'H(D) | 4*/5H(P), 6H(P), 9H(P), 10(P) |
| 1H(P) | 1H/2H(D), 3H(D), −OCH3(D), 4*/5H(P), 6H(P), 7H(P), 8aH(P), 8b/13H(P), 9H(P), 10H(P), 11H(P), 12H(P) |
| 6H(P) | 1'H(D), 1H/2H(D), 3H(D), −OCH3(D), 4*/5H(P), 1H(P), 7H(P), 8aH(P), 8b/13H(P), 9H(P), 10H(P), 11H(P), 12H(P) |
| 9H(P) | 1'H(D), 7H(D), 14a/14bH(D), 1H/2H(D)a, 3H(D)a, 1H(P), 4*/5H(P), 6H(P), 7H(P), 11H(P), 12H(P) |
| 11H(P) | −OCH3(D), 1'H(D), 1H/2H(D), 3H(D), 7H(D), 14bH(D), 5'H(D), 2'axH/2'eqH/8axH(D), 1H(P), 4*/5H(P), 6H(P), 7H(P), 8aH(P), 8b/13H(P), 9H(P), 10H(P) |
| 12H(P) | −OCH3(D), 1'H(D), 1H/2H(D), 3H(D), 14bH(D), 5'H(D), 1H(P), 4*/5H(P), 6H(P), 7H(P), 8aH(P), 8b/13H(P), 9H(P), 10H(P) |
| −CONH(P) | 1H(P), 4*/5H(P), 6H(P), 7H(P), 8aH(P), 8b/13H(P), 9H(P), 10H(P) |
Weak peaks identified from a 1D slice along the direct dimension at the proton A position.
Chemical shift perturbations and Kd.
To investigate whether 1H(P) interacts with DOX in a similar manner as 6H(P) and 9H(P), the chemical shifts perturbation of four protons 1H, 6H, 9H and 12H in DSPE-PEG2000 were monitored versus the titration of DOX into DSPE-PEG2000 (Figure 5A and 5B). As shown in Figure 5A (points from the early titration stage) the chemical shift of 1H(P) first moved up-field and then reversed to down-field as the DOX concentration increased. On the other hand, the 6H(P) chemical shift continued moving up-field as the DOXconcentration increased. Figure 5B shows the chemical shift perturbation of four resolved protons of DSPE-PEG2000 versus the titration of DOX where the molar ratio of DOX:DSPE-PEG2000 increased from 0 to 6.4. The 6H(P) started to merge with the large peak of 4*H(P), and thus no more accurate chemical shift values available after the molar ratio reached 3.56. The Kd from global fitting of the chemical shifts of all four protons versus the concentration of DOX was 0.022 ± 0.008 mM with an R2 of 0.988. The Kd from global fitting using data of three protons without 1H(P) was 0.014 ± 0.003 mM with an R2 of 0.995. This Kd value is in good agreement with that obtained using ITC, 0.0084 mM, considering the difference in buffer, pH and temperature used in the ITC study21. The Kd from fitting the chemical shift titration data of only 1H(P) was 0.54 ± 0.28 mM with an R2 of 0.916. Thus, the 1H(P) interaction pattern with DOX was different from the other three protons used to calculate Kd. This is clearly demonstrated in Figure 5B, where the data points of the other three protons fit nicely with the curves, but the data of 1H(P) did not fit the curve. One explanation is that although 1H(P) lies in proximity to 6H(P), and is able to interact with DOX, its distance to DOX is not as well defined as the other three protons. Correspondingly, a smaller chemical shift perturbation amplitude of 1H(P) compared to 6H(P) and 9H(P) was also observed.
Figure 5. Chemical shift changes in DSPE-PEG2000 during DOX titration into the DSPE-PEG2000.
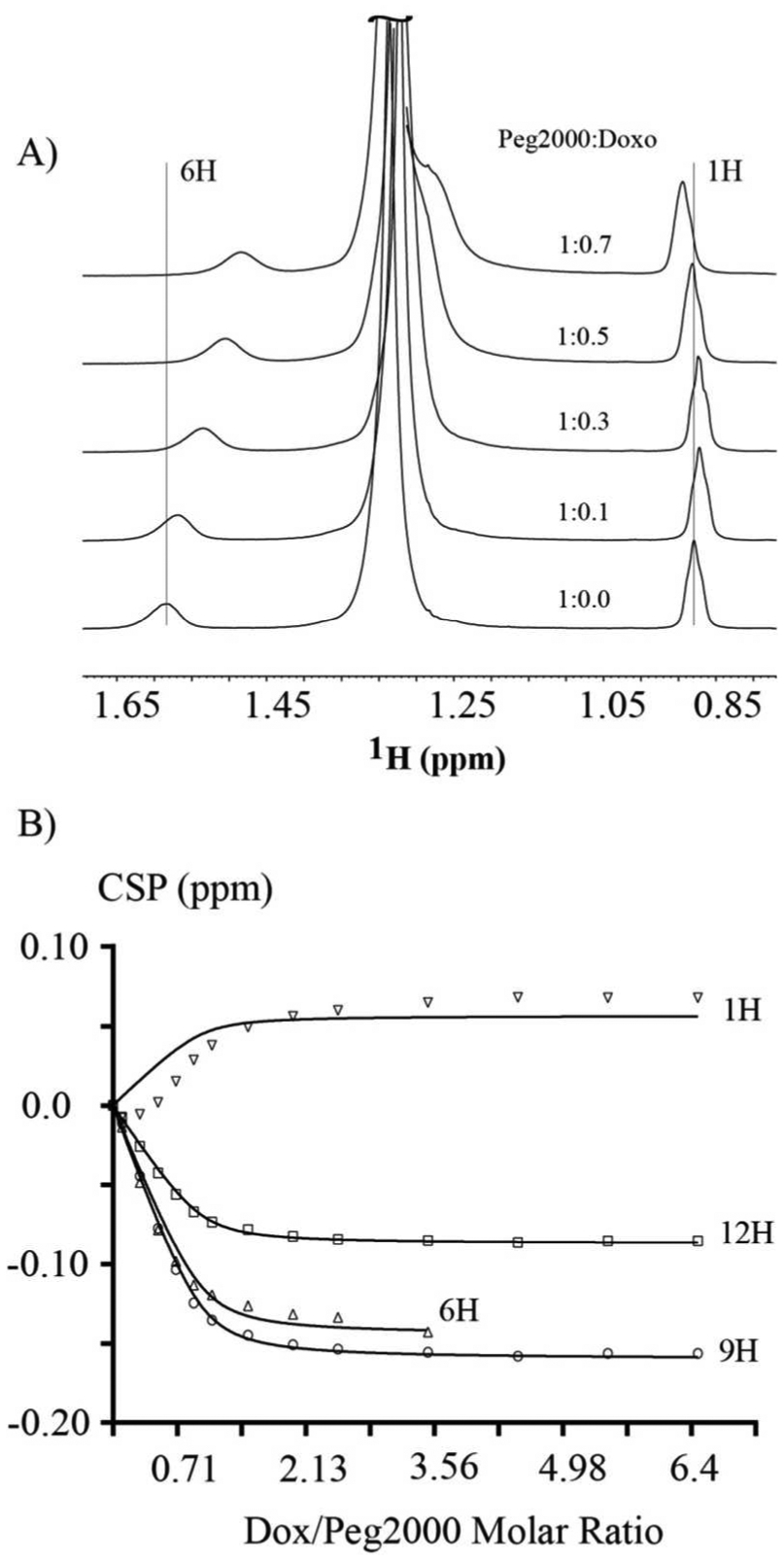
(A) Chemical shifts movement of 1H(P) and 6H(P) of DSPE-PEG2000 versus the several initial titration points of DOX. (B) Global curve fitting of chemical shift perturbation of four protons of DSPE-PEG2000 versus the titration of DOX. The 6H(P) peak started to merge with 4*H(P), and thus no more data was available after molar ratio of DOX:DSPE-PEG2000 was larger than 3.56.
Distance information from paramagnetic relaxation enhancement measurements.
To determine the relative location of 1H(P) to the glycerol group and 6H(P), Gd-DOTA was used to study the paramagnetic relaxation enhancement effect on three well-resolved protons, 1H(P), 6H(P) and 9H(P). Figure 6 shows the line width of these peaks at half height versus the concentration of Gd-DOTA. The relaxation enhancement factor ε was obtained from data fitting using equation (1) given in33. The ε of 9H(P) is the largest (0.48 ± 0.05 Hz/mM) among the three protons. The coefficient of 1H(P) (0.34 ± 0.05 Hz/mM) and 6H(P) (0.38 ± 0.07 Hz/mM) are very close, although the value of 6H(P) is slightly larger. Since Gd(DOTA) is a very polar reagent, and carries a negative charge, it is located in the aqueous phase, and outside the micelle core composed of DSPE lipid. The paramagnetic relaxation enhancement is a function of r−6, where r is the distance between the affected spin and the paramagnetic center34. The observed ε could be explained, qualitatively, that the 9H(P) is closest to Gd-DOTA, and 6H(P) is slightly more exposed to Gd-DOTA than 1H(P). Furthermore, based on ε values of three protons it is reasonable to deduce that the distance difference between 9H(P) and 6H(P) to Gd-DOTA is larger than the distance difference between 6H(P) and 1H(P) to Gd-DOTA.
Figure 6. Line width changes of three protons in DSPE-PEG2000 versus the concentration of Gd-DOTA.
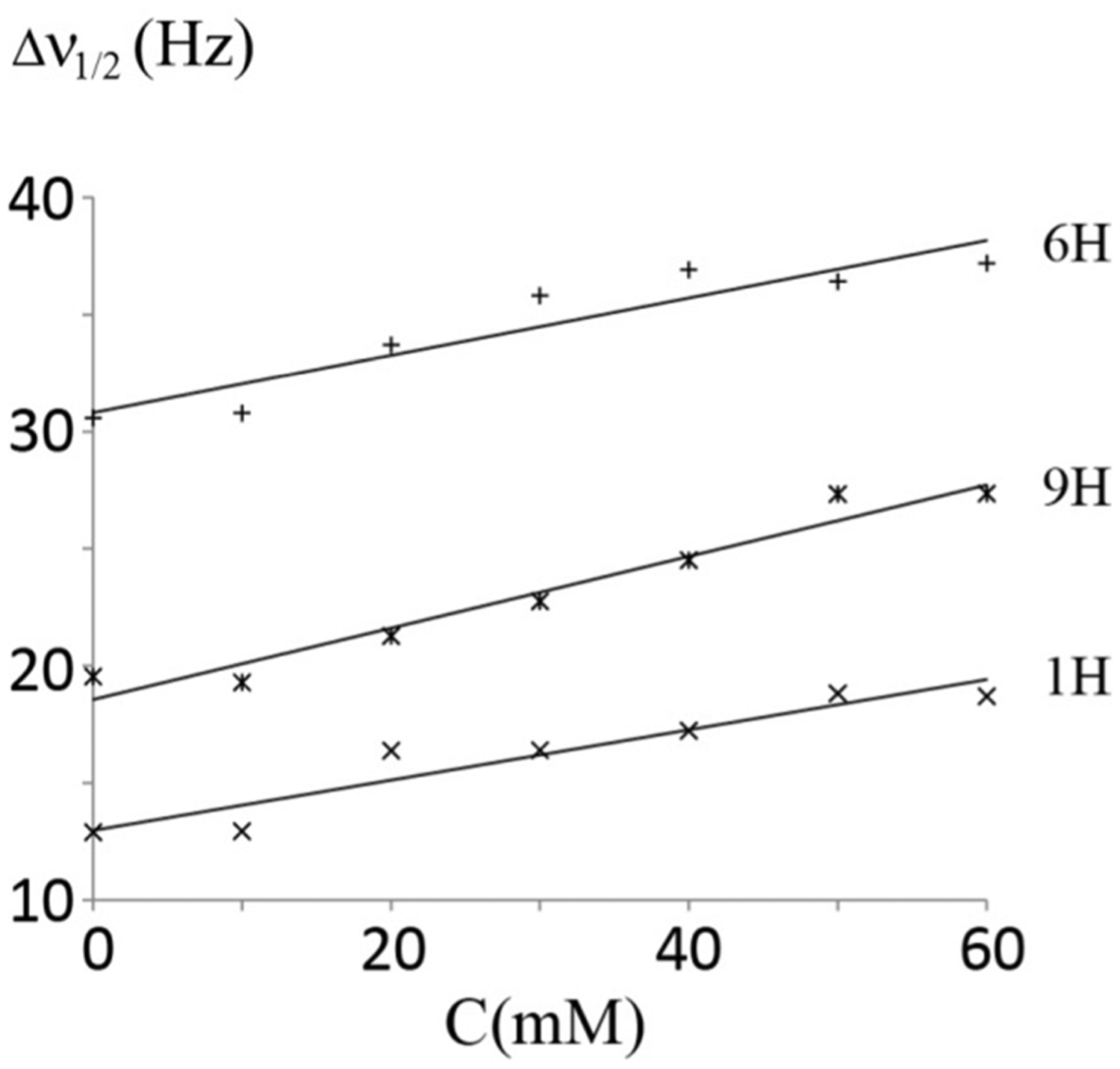
The relaxation enhancement factors of 1H(P), 6H(P) and 9H(P) in DSPE-PEG2000 were obtained by the linear fitting of line width at half height versus the concentration of Gd-DOTA. The ε of 1H(P), 6H(P) and 9H(P) are 0.34 ± 0.05 Hz/mM, 0.38 ± 0.07 Hz/mM and 0.48 ± 0.05 Hz/mM, respectively.
Molecular Dynamics Simulations.
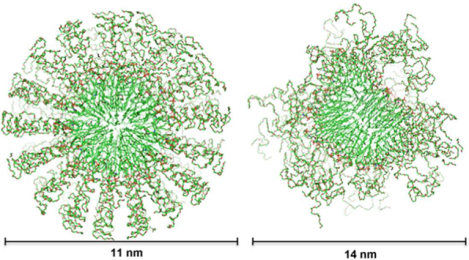
The finding that the terminal methyl group(s) of the stearic acid in the hydrophobic core of the micelle is in contact with the phosphate head group of the lipid suggests that, on the average, one fatty acid can either curl up towards the phosphate head group, or that the units of the micelle stagger in such a way that tails of fatty acid at a time can contact the phosphate head group. Presumably the driving force for this interaction is the presence of the polymer PEG2000 in an aqueous environment. One way to explore the two options considered is to perform MD simulations on the DSPE-PEG2000 micelle embedded in water. At the same time, we would like to investigate the carbamate NH hydrogen bond acceptor in lipid head region through the MD study. The DSPE-PEG2000 micelles structure in aqueous media, the effects of the buffer, mimic bloodstream environment, DSPE-PEG2000 concentration, and the embedding of DOX on the DSPE-PEG2000 micelles structure, size and stability have been reported before8, 19, 21. Based on these earlier studies and the specific issues we would like to address, the MD simulations in this study used 90 DSPE-PEG2000 molecules per micelle with a diameter of 18nm as a starting point. An initial sphere with a radius of 9 nm was constructed with 90 cones, each cone encompassing one spiral DSPE-PEG45 molecule (a single PEG size of n= 45 was used to simplify the simulations with a spiral shape to fill in space as the volume of the cone increased from the inner hydrophobic core to the aqueous environment). In order to maintain an initial spherical shape, one of the fatty acid chains of each unit was curled up towards the phosphate head group (Figure 7A).
Figure 7. MD simulation of DSPE-PEG45 in an aqueous environment.
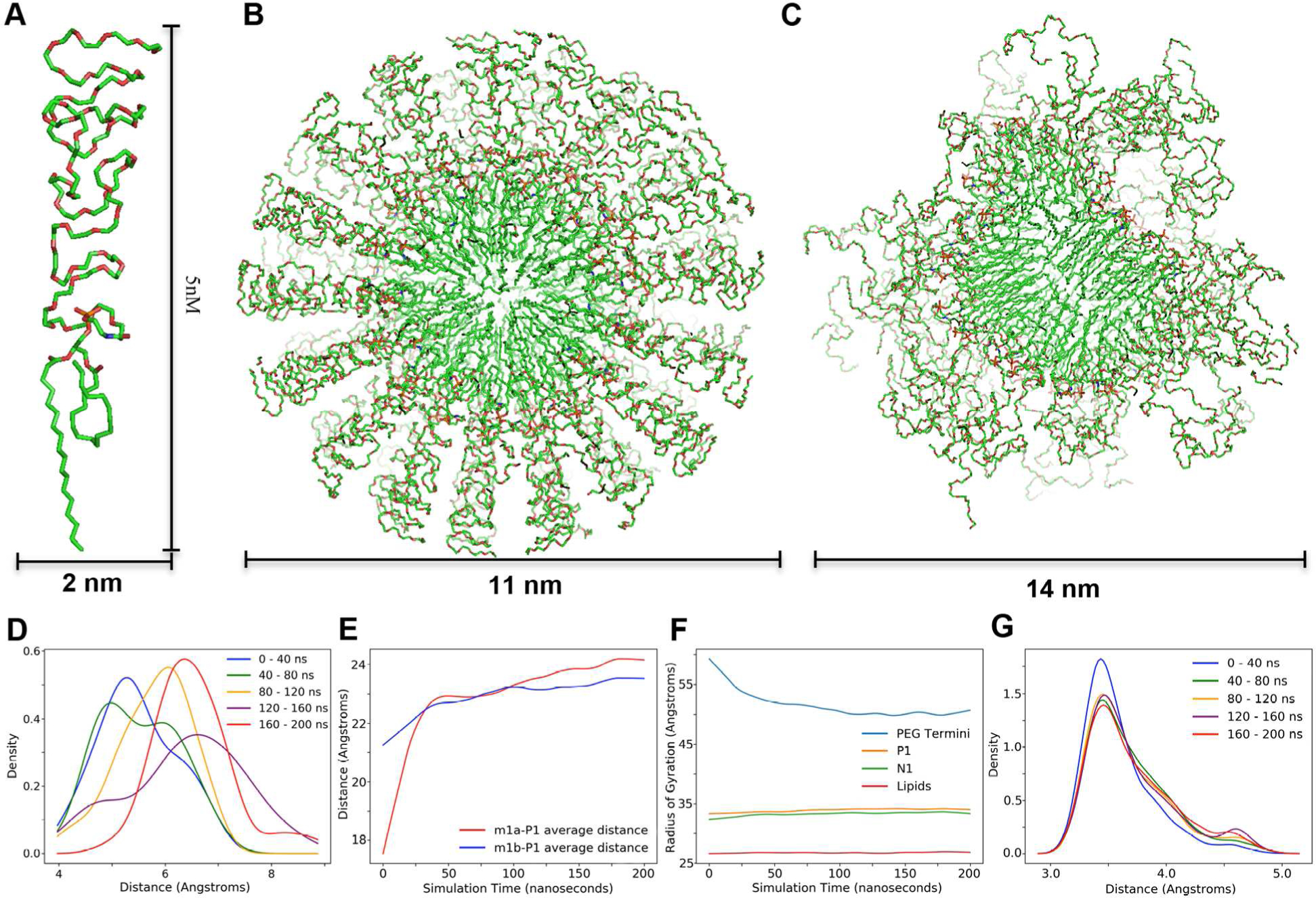
A. Monomer unit with dimensions in nanometers. B. Initial spherical micelle model with dimensions in nanometers. C. Model after MD simulation and energy minimization. D. Distance plot in 40ns periods between phosphate group and the closest lipid methyl group. E. Average distance plot over 200ns between phosphate and intra-lipid methyl group of each tail. F. Radius of gyration of different subgroups in the micelle over 200ns. G. Distance plot in 40ns periods between an oxygen on the phosphodiester and the nitrogen on the carbamate group.
Over the course of the simulation most curled up fatty acid chains straightened back into a parallel orientation to one another resulting in the overall shape of the hydrophobic core transforming from the initial spherical shape (Figure 7B) to an oblate shape (Figure 7C), which is in agreement with the results reported before8, 19, 21. Additionally, the PEG portions unwound from their initial spiral shape and became more fluid interacting with both the lipid head group and the solvent. In the final 200 ns simulation, the minimum distance between the phosphorus and any one of methyl carbon atoms of the lipid are mainly distributed between 6 and 7 Å as shown in Figure 7D. The minimum distance distributions at simulation times greater than 120ns are mainly from intermolecular DSPE-PEG2000 interactions because the intramolecular distance between the phosphorus atom and the two methyl groups is greater than 23Å as shown in Figure 7E. The radius of gyration of PEG termini reduced quickly in the first 30ns of the MD simulation, while the radius of gyration of the lipid core as defined by the phosphorus and carbamate nitrogen atoms did not change much as the simulation time increased. More interestingly, the radius of gyration as defined by the carbamate nitrogen atom is slightly smaller than that defined by the phosphorus atom as shown in Figure 7F. To investigate the potential hydrogen bond between the oxygen of the phosphate group and the NH of the carbamate group, a hydrogen bond was initially built between these two groups. As simulation time increased from 0 ns to 200 ns, the average distance between these two atoms remained constant at around 3.5Å with a minor contribution at 4.6Å (Figure 7G). This suggests that a hydrogen bond between the phosphate and carbamate groups is energetically favored. The small contribution at distance around 4.6Å could be due to a water mediated hydrogen bond, a result in agreement with the NMR data.
DISCUSSION
Although there are many applications using DSPE-PEG2000 or similar derivatives for drug delivery, the mechanism as to how DSPE-PEG2000 forms micelles, the micelle structure and the structure of the complex with ligand are less thoroughly characterized. Earlier NMR applications on the complex of DSPE-PEG2000 with a ligand were mainly focused on monitoring line width changes upon their interaction6, 35. A key NOESY cross peak between the methyl group at the end of the DSPE fatty acyl chain and the D ring of DOX has been used to support the contention that DOX is inserted deep into the micelle core6. In this study, a more comprehensive NMR study was used to investigate the micelle structure of DSPE-PEG2000 and its complex with DOX. Except for chemical shift degeneracy of polyethylene glycol (15*H in PEG, see Figure 1B) and the lipid CH2 group (4*H in DSPE, see Figure 1B), a complete chemical shift assignment of both DOX and DSPE-PEG2000 in free and in the equal molar ratio complex has been achieved. The chemical shift assignment combined with NOESY experiments, chemical shift perturbation, paramagnetic relaxation enhancement study and MD simulation has provided greater details of the structures of the DSPE-PEG2000 micelle and its complex with DOX.
DSPE-PEG2000 micelle structure.
In an aqueous environment, two peaks from the carbamate NH group are observed, in which the minor one is less than 10% of the major peak. These two peaks should arise from the carbamate NH group of the same molecule since there are correlations between these two peaks in a TOCSY experiment, and there are cross peaks between 12H(P) and 11H(P) to these two carbamate NH peaks in TOCSY when the mixing time is 65 msec (result not shown, data with mixing time of 15 msec shown in Figure 2A). The two peaks are at 6.88 and 6.58 ppm, separated by 211 Hz. The line widths at half height are 18.1 and 37.7 Hz for the major and minor peaks, respectively. Based on their relative chemical shifts, the intensity and their line width, the major peak could be due to a hydrogen bond with either the phosphate head group or the ester groups of DSPE. A previous FTIR study showed that while the ester group in the DSPE-PEG2000 micelle did not form a hydrogen bond, it did suggest that the carbamate NH may form a hydrogen bond with water molecules20. To further investigate the hydrogen bond status of the carbamate NH group in the micelle, the distance information between the carbamate NH and phosphate group was investigated by MD. The carbamate NH was first put in a hydrogen bond distance range to the phosphate group in the initial model. After energy minimization, the major average distance distribution between the carbamate N atom and oxygen of phosphate group did not change. This suggested a direct hydrogen bond between these two groups is maintained even after the 200ns MD simulation. There is a minor portion of the molecules in which the distance between two groups is greater than 4.5Å (Figure 7G) which could be due to a water mediated hydrogen bonds. These results are in agreement with the NMR observations.
Furthermore, the minor NH peak which was about 9.1% of the total peak in the free micelle, was reduced to 7% in the DOX loaded micelle. Note that the previous study21 suggested that embedded DOX did neither change the micelle structure nor the aggregation number of DSPE-PEG2000, but instead DOX displaced water molecules in this region. Since the minor peak intensity was not substantially reduced after insertion of DOX from our NMR observation, it suggests that the majority of water molecules still occupy the hydrophilic area even when the molar ratio of DOX and DSPE-PEG2000 is 1:1. Furthermore, these two peaks exchanged in the millisecond time frame since exchange cross peaks were observed in the NOESY experiment as shown in Figure 2B and 2C in the presence or absence of DOX. The finding of two chemical shifts from the same carbamate NH group suggests that this NH group can be used as a handy NMR probe for monitoring changes of the micelle hydrophilic shell caused by interactions with ligand or medium.
It was proposed that about ten ethylene glycol units are in the hydrophilic shell composed of phospholipid and PEG polar groups19, and that this shell is about 2nm thick surrounding the hydrophoblic core19, 21. In our study, NOESY cross peaks of all protons on glycerol, 7H(P), 6H(P), 4*H(P), 5H(P) and 1H(P) to the carbamate NH were observed (see Figure 2B and 2C). Similarly, NOESY cross peaks between these protons to 12H(P) were also observed (see Table 3). These results indicate that the carbamate NH group and 12H(P) reside in the lipid polar region. Since there was no chemical shift dispersion of the ethylene glycol units after position 13H(P), the number of ethylene glycol units in close contact with lipid head group can’t be determined from this NMR study. Importantly, our MD simulation showed that the radius of gyration shell defined by the position of the nitrogen atom of the carbamate NH is closer to the micelle center than the radius of gyration shell defined by the phosphate, a result consistent with our NMR data in that the carbamate NH is embedded in the lipid head group.
The values of relaxation enhancement factor ε in this study are much smaller than the reported ones of the OmpX/DHPC micelle system33, suggesting that the majority of Gd-DOTA is remotely located from the head group of the lipid. This could be due to the restricted access of Gd-DOTA to the lipid head group region because of the presence of a 2nm thick hydrophilic shell outside of the hydrophobic core. When DOX was added to the micelle, the complex was incubated at 65°C for 30 minutes before NMR experiments to ensure that the DOX was fully embedded into micelle. For the Gd-DOTA titration study, the sample was not pre-incubated. This suggests that incubation at higher temperature may loosen up the hydrophilic shell, enhancing the penetration of small molecules through the hydrophilic shell. This might be a required condition for efficiently embedding the therapeutic drugs into DSPE-PEG2000 micelles.
The end of the polyethylene glycol chain is very flexible based on the fact that the sign of NOESY cross peaks between 17H(P) and 18H(P) is opposite to the diagonal peaks. This cross peak did not show up until the NOE mixing time was increased to 500 ms, another indication of high mobility. The presence of DOX did not affect its flexibility since the sign of cross peaks between 17H(P) and 18H(P) was still opposite to the diagonal peaks in the complex. This flexibility was also manifested by the sharp peak of 18H(P) in free DSPE-PEG2000 and in the complex as shown in Figure 1B and 1C. These NMR observations are in agreement with MD results from this and other studies8.
Both NOESY data and paramagnetic relaxation studies showed that the 1H(P) is in proximity to 6H(P). One possible explanation for this finding is that there are significant amount of the trans configurations in the fatty acyl chain, which could make the end of fatty acyl chain flip back, and thus 1H(P) can be in proximity to 6H(P) of the same or other molecules. The FTIR study revealed that there were significant gauche conformers in fatty acyl chain when the temperature is above the phase transition temperature20, which is 12.8°C18. A theoretic study showed that for a n-alkane chain, the all-trans conformer is not energy favored over a hairpin geometry when the n (number of carbons) is larger than 1636. This is because the energy difference between gauche-trans conformers is about 0.56 kcal/mol37, an energy comparable to kT at room temperature. The energy from self-solvation between the two aligned chain segments should overcome this energy barrier easily38 when n is long enough. Thus, the fatty acyl chains of DSPE in micelles could form a hairpin structure as a GGTGG conformer (where G stands for gauche, and T stands for trans), and 1H(P) could reach the lipid interface between hydrophobic and hydrophilic area. Based on this hypothesis, a micelle model with one fatty acyl chain curved up was built within a perfect sphere that accommodates 90 molecules. After MD simulation and energy minimization, both fatty acyl chains are fully relaxed, and both methyl groups are more than 23 Å away from the intra-phosphorus position (Figure 7E). The spherical micelle evolved into an ellipsoid shape with the same amount of molecules. In the ellipsoid micelle, a significant amount of the lipid tail is at right angles to the lipid core, rather than pointing to the center of lipid core. This arrangement brings significant amount of lipid tail within 6–7Å of the lipid phosphate head group (Figure 7D). This micelle organization ensures that some 1H(P)s are in close proximity to lipid head group. Thus, the observed NOE between the fatty acyl methyl group with many protons in the lipid head region, the carbamate NH, as well as the D ring of DOX can be explained by this model. Furthermore, the location of this lipid tail is not well defined within the micelle relative to the polar shell composed of phospholipid head group, which is reflected in the different interaction mechanism between 1H(P) with D ring of DOX versus those of 6H(P) and 9H as shown in Figure 5B.
Interaction of DOX and DSPE-PEG2000.
Based on the NOESY cross peaks (see Table 3) and the chemical shift perturbation data, DOX is located close to the phospholipid head group where its anthracycline rings penetrate closer to the fatty acyl chains, and the A ring has an especially strong interaction with the phospholipid head group. Compared to the SAXS and MD study21, NMR could provide more detail information at the atomic resolution level for the interaction between DOX and DSPE-PEG2000 micelle. The curve fitting of chemical shift perturbation versus the DOX titration to DSPE-PEG2000 (Figure 5B) showed that 6H(P), 9H(P) and 12H(P) are involved in the interaction with DOX in the same manner, however, 1H(P) interacts with DOX in a different manner than the other three protons. This could be due to the position of 1H(P) is not as well defined relative to DOX as the other three protons, especially along the vertical distance direction to the core center. As discussed before, the proximity between 1H(P) and 6H(P) is caused by the arrangement of some lipid chains pointing sideways instead of toward the center of the micelle core. Because of these arrangements, the space location between 1H(P) and 6H(P), as well as with DOX is very dynamic due to diffusion of the molecule within the micelle, or a configuration exchange between gauche and trans conformers20. Since the loading of DOX into the lipid did not change the pattern of NOESY cross peaks of protons in DSPE-PEG2000, we conclude the insertion of DOX into DSPE-PEG2000 micelle did not change the overall micelle structure, but instead, a small portion of water in hydrophilic region of micelle was replaced by DOX.
MD simulations of DSPE-PEG2000.
Since the NMR studies showed terminal methyl group(s) of the acyl units in the hydrophobic core of the micelle were in contact with the phosphate head group of the phospholipid, we were prompted to perform MD simulations on a preformed model accommodating this observation. Starting with a spherical model in which one of the two acyl units curled up towards the phosphate head group, we found that the acyl groups rapidly uncurled, and rearranged the overall structure from spherical to oblate. A similar finding was published by Vukovic et al.8 where, starting from a donut model with a large empty space at the center of the micelle, the micelle rapidly rearranged into a collapsed oblate shape. Since the two models had different initial conditions, but gave the same final collapsed oblate structure with similar dimensions that match those of physical studies, we conclude the collapsed oblate structure is preferred. Furthermore, we show that the core structure is stable out to 200ns of simulation when only the PEG corona is still undergoing flexible movements.
Conclusions.
The DSPE-PEG2000 micelle is a very promising drug delivery vehicle for chemotherapeutic applications. The micelle molecular weight is about 180 kDa, a size usually too large for high resolution NMR spectroscopy to characterize. However, in this study, we demonstrated that by preparing a homogeneous micelle and carrying out the NMR experiment at 37°C, complete chemical shift assignments of the micelle with and without inserted DOX can be achieved. Based on the assignments, through the chemical shift perturbation, titration, NOESY pattern analysis and relaxation enhancement studies combined with MD, detailed structural features of the micelle and its complex with DOX have been revealed. This study demonstrates that NMR spectroscopy in conjunction with MD can be a powerful tool in characterization the interaction between DSPE-PEG2000 micelle and the drugs.
EXPERIMENTAL PROCEDURES
Materials.
Gadolinium(III) (1,4,7,10-tetraazacyclododecane)-1,4,7,10-tetraacetate (Gd-DOTA) was purchased from Marcocylics (Dallas, TX). 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycerol)-2000] ammonium salt (DSPE-PEG2000) was purchased from Avanti Polar Lipids, Inc. (Alabaster, AL). Doxorubicin hydrochloride (DOX) was from Sigma (St. Louis, MO). Sodium Acetate-d3, D2O(99.9% atom %D), 3-(Trimethylsilyl) propionic-2,2,3,3,-d4 sodium (TSP) and 3-(Trimethylsilyl)-1-propanesulfonic sodium (DSS) were purchased from Aldrich (St. Louis, MO). The water used in this work was generated from Milli-Q system (Millipore Corporation, Danvers, MA).
Sample Preparation.
DOX was dissolved in D2O and the solution lyophilized. Lyophilized DOX was then dissolved in 50 mM D2O-based NaAc-d3 and adjusted to pH 5.4. The stock DOX concentration used for titration was 16 mM, and the concentration used for NMR study was 4.4mM. For the study in H2O-based 50mM NaAc-d3 buffer, the DOX was dissolved directly into the buffer, and pH was adjusted to 5.4. A stock solution of 200 mM Gd-DOTA was prepared in a similar way to DOX. For the DSPE-PEG2000 sample, the ammonium ion was removed by lyophilization of the DSPE-PEG2000 H2O solution, followed by dissolution in H2O-based 50mM NaAc-d3 buffer, adjusted to pH 5.4. The solution was incubated in a sand-bath at 65°C for 10 minutes, then in an ice-bath for 10 minutes. After three round of heat-cold cycles, the micelles were homogeneous for NMR study with narrower line widths compared to that without heat-cold cycling. The D2O-based DSPE-PEG2000 micelles were prepared in a similar way as that described for DSPE-PEG2000 micelles dissolved in H2O except the DSPE-PEG2000 was first lyophilized with D2O to remove the ammonium ion. The concentration of DSPE-PEG2000 samples for NMR experiments was between 3.8 to 7.3 mM. The mixture of DSPE-PEG2000 and DOX was prepared by addition of the stock solution of DOX to DSPE-PEG2000 to a final concentration 2.6 mM of DOX and 2.7 mM of DSPE-PEG2000 or 3.4mM of DOX and 3.7mM of DSPE-PEG2000. The mixture was incubated at 65°C for 30 minutes before NMR experiments. Internal reference TSP-d4 was used for both concentration determination of DOX and DSPE-PEG2000 and 1H, 13C chemical shifts references29.
NMR spectroscopy.
Most NMR experiments were carried out on Bruker 700 MHz with a few acquired on Bruker 600 MHz spectrometer. Both spectrometers were equipped with TXI-triple resonance inverse cryoprobe. All the NMR experiments were carried out at 37°C. The NMR data was processed with either Bruker topspin 2.1/3.1 or NMRPipe39, and analyzed with Bruker topspin 2.1, NMRView40 or Sparky (Goddard, T.D. and Kneller, D.G. at University of California, San Francisco).
For the DOX 4.4 mM sample, a one-dimension 1H spectrum was acquired using presaturation for water suppression with 16 transients, 65536 complex acquisition points and 20 ppm spectral width. For 2D 1H-1H-TOCSY and NOESY, the spectrum width and complex data points are 12.5 ppm and 4096 for direct dimension, 11 ppm and 300 for indirect dimension. The number of scans for each fid is 8 for TOCSY and 16 for NOESY. TOCSY mixing time was 80 ms. Five NOESY data sets were acquired with mixing time of 50, 100, 250, 400 and 500ms. DQ-COSY data was acquired with 12.5 and 11ppm for direct and indirect dimension, 4096 and 512 complex points were used for direct and indirect dimensions with number of scans of 8 for each fid. For 2D 1H-13C HSQC and HSQC-TOCSY, the spectrum width and complex data points are 13.3 ppm and 2048 for 1H dimension, 90 ppm and 256 for 13C dimension. The number of scans for each fid is 8 for HSQC and 32 for HSQC-TOCSY. Mixing time used in HSQC-TOCSY was 80 ms.
For DSPE-PEG2000, NMR data were acquired on a 3.8mM sample in D2O-based or 7.3mM in H2O/D2O (90%/10%)-based NaAc-d3. The experimental parameters are similar to what aforementioned for the DOX sample except for those described below. For DSPE-PEG2000 dissolved in D2O-based buffer, two 1H-1H TOCSY data with mixing time 35 and 60 ms were acquired; four mixing times of 50, 150 and 250, and 500ms were used for 2D NOESY; three mixing times of 6, 12 and 29ms were used for 1H-13C HSQC-TOCSY. The spectrum width and complex data points of 2D NOESY were 13.9 ppm and 4096 for direct dimension, 11 ppm and 512 for indirect dimension. The spectrum width of 66 ppm was used for 13C-dimension in 1H-13C HSQC and HSQC-TOCSY experiments. For the DSPE-PEG2000 dissolved in H2O-based buffer, two TOCSY spectra with mixing time of 15 and 66 ms, and a set of NOESY spectra with mixing time of 50, 150, 250, 350 and 500ms were acquired.
For the mixture of DOX and DSPE-PEG2000 in D2O-based Na-Ac-d3 buffer, the concentrations of DOX and DSPE-PEG2000 are 2.6 and 2.7 mM, or 3.4 and 3.7 mM, respectively. The TOCSY data was acquired with 30ms mixing time. Four mixing time of 50, 150, 250 and 500ms were used for acquiring NOESY spectra. The spectrum width and complex data points of 2D NOESY are 13.9 ppm and 4096 for direct dimension, 11 ppm and 512 for indirect dimension. The spectrum width of 13C-diemnsion was 66 ppm for 1H-13C-HSQC and HMQC-TOCSY, and the mixing time 25 ms was used for the HMQC-TOCSY. For H2O-based NaAc-d3 buffer sample, the concentrations of DOX and DSPE-PEG2000 are 3.4 and 3.7mM, respectively. The NOESY data were acquired with mixing time of 50, 150, 250 and 350ms.
For the titration of Gd-DOTA to free DSPE-PEG2000, stock Gd-DOTA of 200mM solution or the calculated lyophilized stock solution was gradually added to the 1.36mM DSPE-PEG2000 solution. Line widths of several well resolved peaks of DSPE-PEG2000 were measured. The relaxation enhancement ε of individual protons in the presence of Gd-DOTA was determined through linear regression fitting of line widths versus the concentration of Gd-DOTA as described in33.
The dissociation constant (Kd) of DOX binding to DSPE-PEG2000 was determined through monitoring the chemical shift changes of several resolved DSPE-PEG2000 proton peaks. DOX stock solution (16 mM) was titrated into 0.5mM DSPE-PEG2000 with a molar ratio (DOX:DSPE-PEG2000) over the range 0:1 to 6.5:1. The Kd was determined by curve fitting of the chemical shift perturbation of protons in DSPE-PEG2000 versus the concentration of DOX according to the equation given in41 using GraphPad Prism 6 (GraphPad Software, Inc. La Jolla, CA).
MD simulations.
The molecular dynamics model was built using a sphere with a diameter of 18nm, in which the volume of the sphere was divided by 90 DSPE-PEG2000 units resulting in a volume of approximately 24nm3 for each individual DPSE-PEG2000 unit. Cones were then generated that were approximately 9nm long with a radius of 1.8nm. Based on NMR data and fitting the DPSE molecules to cones in a sphere, it was necessary to bend one of the two fatty acid chains upwards towards the phosphoethanolamine inside the cone, followed by a spiraled PEG2000 portion to occupy space as the volume of each cone grew outwards. The individual DPSE molecules were built in Maestro42 and where minimized using the Optimized Potentials for Liquid Simulations force field (OPLS) in implicit water to a convergence threshold of 0.05. The overall dimension of each unit was a conical shape with a 5nm height and a radius of 1.3nm. We scripted a tool to build micelles by equally spacing a given number of points, in this case 90, around the surface of a sphere, which provides a center line to the surface which we then used to place the individual cones. The micelle generated had gaps between each cone and at the center where water would be expected to pack in. The idea behind this was to allow for movement of the hydrophobic tails and to allow water expulsion during the MD simulation. Once the complete system was generated it was minimized once more using the OPLS force field in implicit water to a convergence threshold of 0.05. Using the Desmond molecular dynamics toolkit43–45 we immersed the micelle in a water box of 15nm3 and neutralized the system. That system was then energy minimized, followed by Nose-Hoover Thermostat MD46 for 0.5ns, then additional 1ns using a Parinello-Rahman barostat47 under isothermal-isobaric condition to equilibrate pressure to 1.05 BAR, followed by 200ns of restraint free MD saving snapshots every 2ps. We scripted another tool that examined the entire molecular dynamic simulation frame by frame and generated the distance between various groups and the radius of gyration of various groups to generate the graphs in Figure 7.
Supplementary Material
Acknowledgements.
The authors gratefully acknowledge the support of the City of Hope Cancer Grant P30CA033572.
Footnotes
Notes: the authors declare no competing financial interest.
Supporting information: supporting information (Figures S1 and S2) is available free of charge on the ACS Publications website.
REFERENCES
- 1.Speth PA; van Hoesel QG; Haanen C, Clinical pharmacokinetics of doxorubicin. Clin Pharmacokinet 1988, 15 (1), 15–31. [DOI] [PubMed] [Google Scholar]
- 2.Chatterjee K; Zhang J; Honbo N; Karliner JS, Doxorubicin cardiomyopathy. Cardiology 2010, 115 (2), 155–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaczmarek A; Brinkman BM; Heyndrickx L; Vandenabeele P; Krysko DV, Severity of doxorubicin-induced small intestinal mucositis is regulated by the TLR-2 and TLR-9 pathways. J Pathol 2012, 226 (4), 598–608. [DOI] [PubMed] [Google Scholar]
- 4.Gabizon A; Shmeeda H; Barenholz Y, Pharmacokinetics of pegylated liposomal Doxorubicin: review of animal and human studies. Clin Pharmacokinet 2003, 42 (5), 419–36. [DOI] [PubMed] [Google Scholar]
- 5.Torchilin VP; Omelyanenko VG; Papisov MI; Bogdanov AA Jr.; Trubetskoy VS; Herron JN; Gentry CA, Poly(ethylene glycol) on the liposome surface: on the mechanism of polymer-coated liposome longevity. Biochim Biophys Acta 1994, 1195 (1), 11–20. [DOI] [PubMed] [Google Scholar]
- 6.Wang YG; Wang RQ; Lu XY; Lu WL; Zhang CL; Liang W, Pegylated Phospholipids-Based Self-Assembly with Water-Soluble Drugs. Pharmaceutical research 2010, 27 (2), 361–370. [DOI] [PubMed] [Google Scholar]
- 7.Ashok B; Arleth L; Hjelm RP; Rubinstein I; Onyuksel H, In vitro characterization of PEGylated phospholipid micelles for improved drug solubilization: effects of PEG chain length and PC incorporation. J Pharm Sci 2004, 93 (10), 2476–87. [DOI] [PubMed] [Google Scholar]
- 8.Vukovic L; Khatib FA; Drake SP; Madriaga A; Brandenburg KS; Kral P; Onyuksel H, Structure and dynamics of highly PEG-ylated sterically stabilized micelles in aqueous media. Journal of the American Chemical Society 2011, 133 (34), 13481–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trubetskoy VS; Torchilin VP, Use of Polyoxyethylene-Lipid Conjugates as Long-Circulating Carriers for Delivery of Therapeutic and Diagnostic Agents. Advanced Drug Delivery Reviews 1995, 16 (2–3), 311–320. [Google Scholar]
- 10.Maeda H; Wu J; Sawa T; Matsumura Y; Hori K, Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release 2000, 65 (1–2), 271–84. [DOI] [PubMed] [Google Scholar]
- 11.Tang N; Du G; Wang N; Liu C; Hang H; Liang W, Improving penetration in tumors with nanoassemblies of phospholipids and doxorubicin. J Natl Cancer Inst 2007, 99 (13), 1004–15. [DOI] [PubMed] [Google Scholar]
- 12.Lu X; Zhang F; Qin L; Xiao F; Liang W, Polymeric micelles as a drug delivery system enhance cytotoxicity of vinorelbine through more intercellular accumulation. Drug Deliv 2010, 17 (4), 255–62. [DOI] [PubMed] [Google Scholar]
- 13.Kwon GS; Kataoka K, Block copolymer micelles as long-circulating drug vehicles. Advanced Drug Delivery Reviews 2012, 64, 237–245. [Google Scholar]
- 14.Torchilin VP, Multifunctional nanocarriers. Advanced Drug Delivery Reviews 2012, 64, 302–315. [DOI] [PubMed] [Google Scholar]
- 15.Jhaveri AM; Torchilin VP, Multifunctional polymeric micelles for delivery of drugs and siRNA. Frontiers in Pharmacology 2014, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan C; Wang Y; Fan W, Exploring polymeric micelles for improved delivery of anticancer agents: recent developments in preclinical studies. Pharmaceutics 2013, 5 (1), 201–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bunker A; Magarkar A; Viitala T, Rational design of liposomal drug delivery systems, a review: Combined experimental and computational studies of lipid membranes, liposomes and their PEGylation. Biochimica et biophysica acta 2016, 1858 (10), 2334–52. [DOI] [PubMed] [Google Scholar]
- 18.Kastantin M; Ananthanarayanan B; Karmali P; Ruoslahti E; Tirrell M, Effect of the lipid chain melting transition on the stability of DSPE-PEG(2000) micelles. Langmuir 2009, 25 (13), 7279–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arleth L; Ashok B; Onyuksel H; Thiyagarajan P; Jacob J; Hjelm RP, Detailed structure of hairy mixed micelles formed by phosphatidylcholine and PEGylated phospholipids in aqueous media. Langmuir 2005, 21 (8), 3279–90. [DOI] [PubMed] [Google Scholar]
- 20.Wu FG; Luo JJ; Yu ZW, Infrared spectroscopy reveals the nonsynchronicity phenomenon in the glassy to fluid micellar transition of DSPE-PEG2000 aqueous dispersions. Langmuir 2010, 26 (15), 12777–84. [DOI] [PubMed] [Google Scholar]
- 21.Wang J; Xing X; Fang X; Zhou C; Huang F; Wu Z; Lou J; Liang W, Cationic amphiphilic drugs self-assemble to the core-shell interface of PEGylated phospholipid micelles and stabilize micellar structure. Philos Trans A Math Phys Eng Sci 2013, 371 (2000), 20120309. [DOI] [PubMed] [Google Scholar]
- 22.Krishnadas A; Rubinstein I; Onyuksel H, Sterically stabilized phospholipid mixed micelles: in vitro evaluation as a novel carrier for water-insoluble drugs. Pharmaceutical research 2003, 20 (2), 297–302. [DOI] [PubMed] [Google Scholar]
- 23.Koo OM; Rubinstein I; Onyuksel H, Camptothecin in sterically stabilized phospholipid micelles: a novel nanomedicine. Nanomedicine : nanotechnology, biology, and medicine 2005, 1 (1), 77–84. [DOI] [PubMed] [Google Scholar]
- 24.Cesur H; Rubinstein I; Pai A; Onyuksel H, Self-associated indisulam in phospholipid-based nanomicelles: a potential nanomedicine for cancer. Nanomedicine : nanotechnology, biology, and medicine 2009, 5 (2), 178–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lim SB; Rubinstein I; Sadikot RT; Artwohl JE; Onyuksel H, A novel peptide nanomedicine against acute lung injury: GLP-1 in phospholipid micelles. Pharmaceutical research 2011, 28 (3), 662–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barthwal R; Srivastava N; Sharma U; Govil G, A 500-Mhz Proton Nmr-Study of the Conformation of Adriamycin. Journal of Molecular Structure 1994, 327 (2–3), 201–220. [Google Scholar]
- 27.Mondelli R; Ragg E; Fronza G; Arnone A, Nuclear-Magnetic-Resonance Conformational Study of Daunomycin and Related Antitumor Antibiotics in Solution - the Conformation of Ring-A. J Chem Soc Perk T 2 1987, (1), 15–26. [Google Scholar]
- 28.Stott K; Stonehouse J; Keeler J; Hwang TL; Shaka AJ, Excitation Sculpting in High-Resolution Nuclear-Magnetic-Resonance Spectroscopy - Application to Selective Noe Experiments. J Am Chem Soc 1995, 117 (14), 4199–4200. [Google Scholar]
- 29.Wishart DS; Bigam CG; Yao J; Abildgaard F; Dyson HJ; Oldfield E; Markley JL; Sykes BD, 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J Biomol NMR 1995, 6 (2), 135–40. [DOI] [PubMed] [Google Scholar]
- 30.Sparling ML; Zidovetzki R; Muller L; Chan SI, Analysis of Membrane-Lipids by 500 Mhz H-1-Nmr. Analytical Biochemistry 1989, 178 (1), 67–76. [DOI] [PubMed] [Google Scholar]
- 31.Fan TWM; Lane AN, Structure-based profiling of metabolites and isotopomers by NMR. Progress in Nuclear Magnetic Resonance Spectroscopy 2008, 52 (2–3), 69–117. [Google Scholar]
- 32.Chaires JB; Dattagupta N; Crothers DM, Self-association of daunomycin. Biochemistry 1982, 21 (17), 3927–32. [DOI] [PubMed] [Google Scholar]
- 33.Hilty C; Wider G; Fernandez C; Wuthrich K, Membrane protein-lipid interactions in mixed micelles studied by NMR spectroscopy with the use of paramagnetic reagents. Chembiochem 2004, 5 (4), 467–473. [DOI] [PubMed] [Google Scholar]
- 34.Battiste JL; Wagner G, Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry 2000, 39 (18), 5355–65. [DOI] [PubMed] [Google Scholar]
- 35.Chandran T; Katragadda U; Teng Q; Tan C, Design and evaluation of micellar nanocarriers for 17-allyamino-17-demethoxygeldanamycin (17-AAG). International Journal of Pharmaceutics 2010, 392 (1–2), 170–177. [DOI] [PubMed] [Google Scholar]
- 36.Thomas LL; Christakis TJ; Jorgensen WL, Conformation of Alkanes in the gas phase and pure liquids. Journal of Physical Chemistry B 2006, 110 (42), 21198–21204. [DOI] [PubMed] [Google Scholar]
- 37.Kint S; Scherer JR; Snyder RG, Raman-Spectra of Liquid Normal-Alkanes .3. Energy Difference between Trans and Gauche Normal-Butane. J Chem Phys 1980, 73 (6), 2599–2602. [Google Scholar]
- 38.Goodman JM, What is the longest unbranched alkane with a linear global minimum conformation? Journal of Chemical Information and Computer Sciences 1997, 37 (5), 876–878. [Google Scholar]
- 39.Delaglio F; Grzesiek S; Vuister GW; Zhu G; Pfeifer J; Bax A, Nmrpipe - a Multidimensional Spectral Processing System Based on Unix Pipes. Journal of Biomolecular Nmr 1995, 6 (3), 277–293. [DOI] [PubMed] [Google Scholar]
- 40.Johnson BA; Blevins RA, NMR View: A computer program for the visualization and analysis of NMR data. J Biomol NMR 1994, 4 (5), 603–14. [DOI] [PubMed] [Google Scholar]
- 41.Williamson MP, Using chemical shift perturbation to characterise ligand binding. Progress in Nuclear Magnetic Resonance Spectroscopy 2013, 73, 1–16. [DOI] [PubMed] [Google Scholar]
- 42.Schrödinger Release 2017–1: Maestro, S, LLC, New York, NY, 2017. [Google Scholar]
- 43.Shivakumar D; Williams J; Wu Y; Damm W; Shelley J; Sherman W, Prediction of Absolute Solvation Free Energies using Molecular Dynamics Free Energy Perturbation and the OPLS Force Field. Journal of chemical theory and computation 2010, 6 (5), 1509–19. [DOI] [PubMed] [Google Scholar]
- 44.Guo Z; Mohanty U; Noehre J; Sawyer TK; Sherman W; Krilov G, Probing the alpha-helical structural stability of stapled p53 peptides: molecular dynamics simulations and analysis. Chemical biology & drug design 2010, 75 (4), 348–59. [DOI] [PubMed] [Google Scholar]
- 45.Bowers Kevin J., C. E, Xu Huafeng, Dror Ron O., Eastwood Michael P., Gregersen Brent A., Klepeis John L., Kolossvary Istvan, Moraes Mark A., Sacerdoti Federico D., Salmon John K., Shan Yibing, and Shaw David E. In Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters, Proceedings of the 2006 ACM/IEEE conference on Supercomputing, Tampa FL, Horner-Miller B, Ed. Tampa, FL, 2017. [Google Scholar]
- 46.Evans DJ; Holian BL, The Nose-Hoover Thermostat. J Chem Phys 1985, 83 (8), 4069–4074. [Google Scholar]
- 47.Parrinello M; Rahman A, Polymorphic Transitions in Single-Crystals - a New Molecular-Dynamics Method. J Appl Phys 1981, 52 (12), 7182–7190. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.