

Abstract
Variably protease-sensitive prionopathy (VPSPr) is a recently described sporadic prion disease with distinctive clinical and histopathological features. We report the clinical, imaging, and neuropathological features of VPSPr in a 46-year-old right-handed man who presented with progressive cognitive decline, behavior disturbances, and a 50-pound weight loss over 6 months. The initial evaluation revealed severe cognitive impairment with no focal neurologic deficits. His cognitive, psychiatric, and behavior symptoms progressed rapidly, and he died 12 months after the initial visit. Throughout his disease course, workup for rapid progressive dementia was unremarkable except that brain MRI diffusion-weighted imaging showed persistent diffuse cortical and thalamic signal abnormalities. Sporadic Creutzfeldt-Jakob disease was highly suspected; however, two EEGs (8 months apart) demonstrated only nonspecific cerebral dysfunction. The patient’s CSF 14-3-3 protein was negative at the initial visit and again 8 months later. His CSF real-time quaking-induced conversion and total tau level were normal. An autopsy of his brain was performed, and the neuropathological findings confirmed VPSPr. Our case underlines the importance of considering VPSPr in the spectrum of prion disease phenotypes when evaluating individuals with rapidly progressive dementia.
Keywords: variably protease-sensitive prionopathy, rapidly progressive dementia, prion disease
Rapidly progressive dementia (RPD) is a unique set of disorders that is secondary to a variety of etiologies including prion disease, neurodegenerative dementia (eg, Alzheimer disease, dementia with Lewy bodies, frontotemporal dementia), and dementia due to various reversible/treatable causes (eg, infectious, autoimmune/paraneoplastic, toxic/metabolic, vasculitic, psychiatric) (Geschwind, 2016). Although RPD is considered rare, no large population-based epidemiology study has been conducted to estimate its true prevalence and incidence. A recent study reported 3.7% RPD cases among 1648 individuals who had been admitted to the neurology unit of a tertiary hospital in Brazil during a 3-year period (Studart Neto et al, 2017).
Clinical presentations of RPD are highly heterogeneous depending on the underlying etiologies. Cognitive decline such as memory loss, visuospatial and language deficits, and executive dysfunction is often the most prominent presentation. However, other neuropsychiatric symptoms such as behavior disturbance, personality change, mood disorders, psychosis, sleep disturbance, alternation of alertness/awareness, involuntary movement such as tremor and myoclonus, gait disturbance, seizure-like activities, ataxia, and parkinsonian features are also commonly seen in individuals with RPD (Geschwind, 2016). Due to its relatively infrequent incidence and the heterogeneity of its clinical presentation and progression, RPD represents one of the most challenging groups of diseases facing clinicians.
The most common underlying etiology of RPD is prion disease (Geschwind, 2016). Although phenotypically diverse, prion spectrum disorders are a set of disorders that are pathologically characterized by the accumulation and aggregation of abnormally folded prion proteins (PrPsc) (Baldwin and Correll, 2019; Kim and Geschwind, 2015). Compared with the typical α-helices that are seen in the normal form of the prion protein (PrPc), PrPsc have a high number of β-pleated sheets. This structure makes the protein partially resistant to proteases and acts as a template for further misfolding of the normal PrPc to the PrPsc (Baldwin and Correll, 2019; Kim and Geschwind, 2015).
The most common human prion disease is sporadic Creutzfeldt–Jakob disease (sCJD), which typically occurs between the ages of 50 and 80 years and has a mean survival rate of 6 months (Baldwin and Correll, 2019). The typical clinical presentation of sCJD is RPD, ataxia and myoclonus, neuropsychiatric behavioral changes, visual impairment, spasticity, weakness, and akinetic mutism. Characteristic diagnostic testing findings include periodic sharp-wave complexes on EEG; abnormal restricted diffusion and FLAIR hyperintensities in the cortex, caudate, and/or putamen of the brain MRI; and an elevated CSF 14-3-3 protein (Baldwin and Correll, 2019; Kim and Geschwind, 2015). The more recently available test of real-time quaking-induced conversion of the CSF PrPc has shown high specificity of 99–100% for sCJD (Baldwin and Correll, 2019).
Variably protease-sensitive prionopathy (VPSPr) is another sporadic prion disease with distinctive clinical and histopathological features as well as unique characteristics of the PrPsc (Gambetti et al, 2008; Zou et al, 2010). VPSPr typically has a slower disease progression and a longer survival time than sCJD. Often misdiagnosed as non-Alzheimer dementias, most VPSPr cases have no periodic complexes on EEG, no gray matter signal changes of restricted diffusion by MRI, and a normal CSF 14-3-3 protein (Gambetti et al, 2011; Zou et al, 2013). Pathologically, the PrPsc in VPSPr typically form a ladder-like electrophoretic profile with variable protease resistance (Gambetti et al, 2011; Notari et al, 2018). Clinical features of VPSPr vary depending on the prion protein gene (PRNP gene) codon 129 polymorphisms.
We report the clinical, imaging, and neuropathological features of a VPSPr in a middle-aged man who presented with RPD. We also summarize the clinical and diagnostic features of all VPSPr cases that have been published to date.
CASE REPORT
A 46-year-old right-handed man presented to his primary care provider’s clinic in May 2016 with progressive cognitive decline, behavior disturbances, and a 50-pound weight loss over the previous 6 months. Laboratory tests at the time were normal for complete blood count, electrolytes, kidney and liver function, and thyroid stimulating hormone. A brain MRI without contrast was also normal. The man was started on aripiprazole 5 mg daily for possible bipolar disorder and was referred to a neurologist to further evaluate the underlying cause of his cognitive and behavioral changes. In June 2016, the man presented to the memory disorder clinic at the University of Mississippi Medical Center. He was married, with one adult child, and he had 10 years of education and worked in both construction/house building and farming.
ASSESSMENTS
Evaluation
At the first appointment, we obtained a detailed history from the patient and his wife. According to them, he had a 5-year history of anxiety and depression for which he had been prescribed venlafaxine and lorazepam. He was able to function as a construction worker until ~6 months ago, when his wife observed a drastic change in his memory, mood, and behavior. Symptoms described by his wife included constantly looking for personal belongings, forgetting words in conversation, lacking motivation, ignoring personal hygiene, and losing interest in social activities and personal hobbies. He became irritable much more often than usual and was more talkative, restless, and aggressive. He had lost the ability to perform simple daily tasks such as changing oil for a lawnmower, assembling small projects, measuring and cutting wood, and even operating telephones and TV remotes. His wife had to stop him from driving due to a few reckless events.
His medical history included hypertension (on lisinopril), gout (on allopurinol), and chronic back pain. He also reported a history of insomnia since childhood. His family history was significant for depression and severe insomnia in his father, sister, and son. A review of his social history indicated that he smoked half a pack of cigarettes a day. He used to drink 6 beers a day but quit years ago.
The physical examination revealed normal vital signs and an unremarkable general examination. The neurologic examination was afocal. Initial cognitive screening with the Montreal Cognitive Assessment (Nasreddine et al, 2005) revealed severe cognitive impairment (score of 10 out of 30), with deficits in orientation, attention, executive/visuospatial functioning, verbal fluency, abstraction, and memory function.
Given the rapidly progressive cognitive and behavioral changes exhibited by our patient, we conducted an extensive workup consisting of serum tests, cerebral Tc 99m HMPAO SPECT, brain MRI, EEG, and CSF studies for differentiation of atypical neurodegenerative dementia such as frontotemporal dementia, early onset Alzheimer disease, prion disorders, and other reversible/treatable dementias.
Serum tests for erythrocyte sedimentation rate, antinuclear antibodies, vitamin B12, thyroid stimulating hormone, thyroid peroxidase antibody, rapid plasma reagin, HIV, and paraneoplastic antibodies panel were all normal or negative. The cerebral Tc 99m HMPAO SPECT study reported slightly decreased perfusion in the right temporal and right parietal lobes.
MRI
While the investigation was ongoing, we received a copy of our patient’s brain MRI from his primary care provider. A cortical signal change consistent with a “cortical ribbon” sign on the diffusion-weighted imaging/apparent diffusion coefficient sequences was noted. At this point, we admitted our patient to the inpatient neurology service for further evaluation. A repeat brain MRI with and without contrast (Figure 1) was obtained, which revealed diffuse abnormal restricted diffusion involving the cerebral cortical gray matter and bilateral thalami, thus raising the suspicion of sCJD.
FIGURE 1.

MRI diffusion-weighted imaging showing persistent, diffuse, symmetric-restricted diffusion in the cortical and thalamic brain regions at both the initial scan and the follow-up scan 8 months later.
EEG and CSF studies
Given the suspicion of sCJD, we obtained an EEG, which showed mild diffuse nonspecific cerebral dysfunction. A lumbar puncture was performed, and CSF studies contained 1 white blood cell/cm3 (normal range [NR] = 0–5/cm3); 10 red blood cells/cm3 (NR = 0/cm3); glucose 65 mg/dl (NR = 40–70 mg/dl); protein 23 mg/dl (NR = 15–45 mg/dL); nonreactive venereal disease research laboratory test (normal = nonreactive); and a normal 14-3-3 protein, paraneoplastic antibodies panel, arbovirus panel, multiple sclerosis panel, Lyme disease antibodies, herpes simplex virus polymerase chain reaction, varicella zoster virus polymerase chain reaction, cytomegalovirus polymerase chain reaction, and angiotensin-converting enzyme level. A chest/abdomen/pelvis CT was done and showed no evidence of occult malignancy.
In summary, our initial evaluation did not identify any treatable/reversable etiology of RPD. While most of our patient’s history and examination would fit a diagnosis of frontotemporal dementia syndrome, his MRI findings of cortical signal abnormality raised concern for sCJD. However, his EEG did not show the characteristic findings that would suggest sCJD, and his CSF 14-3-3 protein was normal.
Follow-up
Nine days after admittance to the memory disorders clinic, our patient was discharged with the following medications: trazodone 50 mg nightly for insomnia, divalproex sodium 500 mg twice a day and risperidone 1 mg twice a day for irritability and hostile behavior, and bupropion 75 mg twice a day for depression. Despite this treatment, his cognitive, psychiatric, and behavior symptoms progressed rapidly.
He presented to our clinic for his first post-hospitalization follow-up 1 month after discharge. His wife reported that during this time, he had developed hallucinations (eg, seeing pine trees outside the porch or animals around the house) and had begun to wander outside their home after midnight. According to his wife, he had also developed postural hand tremor, unsteady gait, and frequent falls. Based on this information, we ordered another brain MRI, which showed persistent abnormal diffuse cortical signal throughout the cerebral hemispheres and the bilateral thalami.
Approximately 6 months later, our patient’s wife called and reported that her husband’s gait had gotten more and more unsteady (eg, he started shuffling), and he started to have urine and bowel incontinence. He was withdrawn, lying in bed most of the day and ignoring basic elements of living such as getting up to eat or to use the bathroom.
Given the rapid progression of our patient’s condition and the fact that no clear diagnosis had been established, we conducted another brain MRI (Figure 1), which again showed abnormal diffuse restricted diffusion involving the cerebral cortical gray matter and bilateral thalami, slightly increased in conspicuity, particularly the temporal lobes and frontal lobes.
Given our initial suspicion of sCJD except for the unremarkable EEG and the normal CSF 14-3-3 protein, we also conducted another EEG, which again showed mild to moderate diffuse nonspecific cerebral dysfunction. We also performed another lumbar puncture, and the CSF studies again showed normal white and red blood cell counts, protein, and glucose. The CSF 14-3-3 protein, total tau, and real-time quaking-induced conversion were all normal. A CSF paraneoplastic panel to rule out autoimmune encephalitis was also sent again, and it was negative. PRNP gene sequencing was done to rule out fatal familiar insomnia, which reported no mutation. A 3-day course of intravenous immunoglobin was given empirically for possible autoimmune encephalitis, but no improvement was seen. A hrain biopsy was discussed but was declined by his family. Our patient was eventually placed in hospice care and died in June 2017, with a total disease duration of 18 months.
Our patient’s family donated his brain to the US National Prion Disease Pathology Surveillance Center for autopsy. Routine histology demonstrated diffuse spongiform degeneration involving the cerebral cortex and basal ganglia, along with microplaques in the cerebellar molecular layer (Figure 2A, B). An immunohistochemistry study with 3F4, a monoclonal antibody to the prion protein, demonstrated diffuse synaptic staining throughout the neocortex and highlighted cerebellar granular plaques (Figure 2C, D). These pathological findings supported a diagnosis of prion disease.
FIGURE 2.

Our patient’s histology and immunohistochemistry results. A. Sections of the frontal cerebral cortex showing fine, full-thickness spongiform degeneration without prion plaques (H & E × 40). B. H & E stained sections of the cerebellum showing microplaques. C. 3F4 immunohistochemical staining showing a synaptic pattern of immunoreactivity (3F4 IHC × 40). D. 3F4 immunohistochemical staining (H & E, 3F4 IHC × 200) showing highlighted cerebellar granular plaques.
An immunoblot of our patient’s brain homogenates showed multiple bands, including one at 17 kDa (Figure 3) and one at 7 kDa (not included in Figure 3). This ladder-like electrophoretic profile suggested that the PrPsc had variable protease sensitivity, which is a characteristic feature that has been reported previously in VPSPr cases (Zou et al, 2010). PRNP gene codon 129 polymorphism analysis reported heterozygosity (methionine [M]/valine [V]).
FIGURE 3.
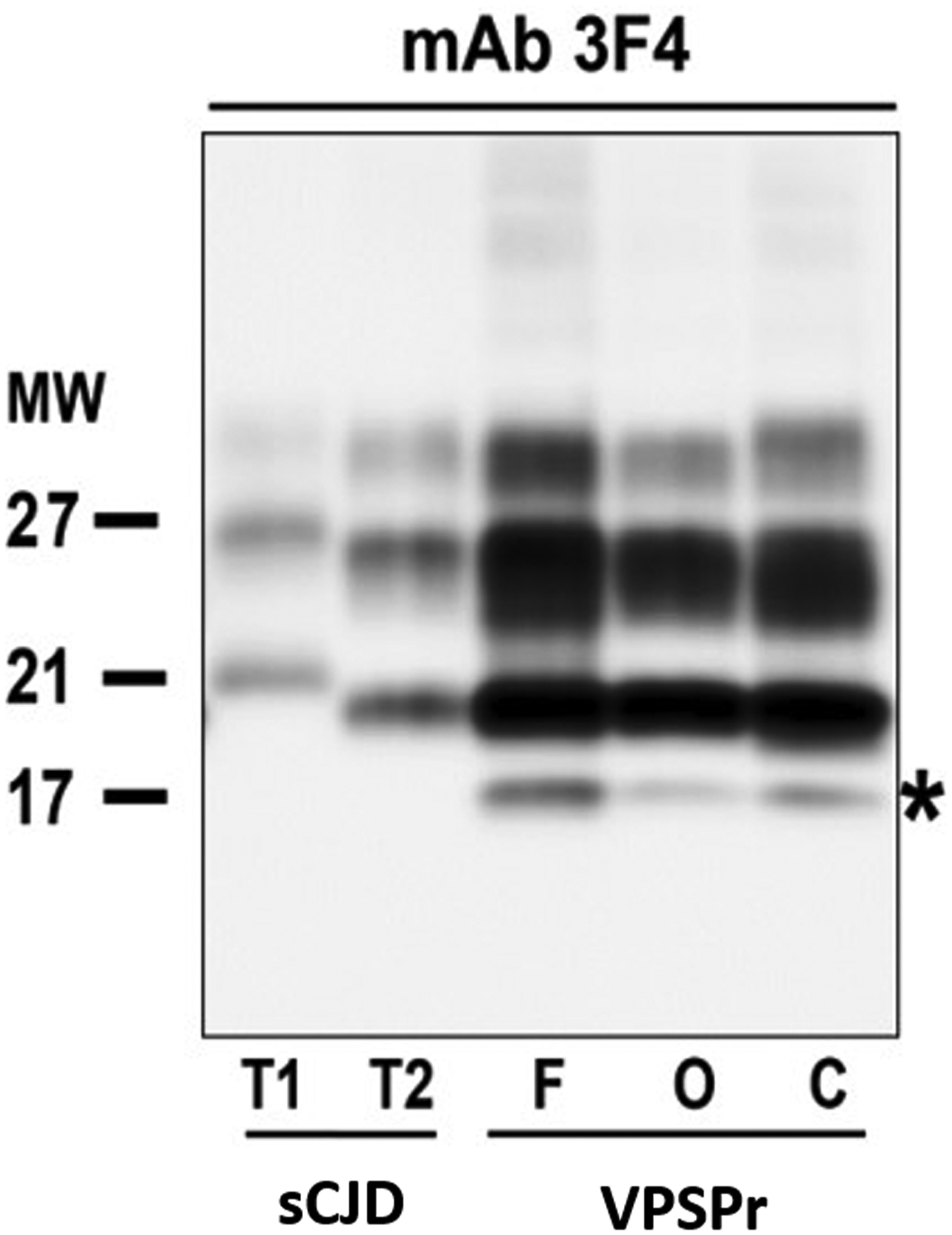
Western blots of brain homogenates probed with mAb 3F4 for the pathogenic form of the human prion protein. F, O, and C cortex of the VPSPr case. Note the typical Type 2 PrPsc pattern with additional 17-kDa band in the VPSPr case. This ladder-like electrophoretic profile suggested that the abnormal prion protein has variable protease sensitivity. C = cerebellar. F = frontal. mAb = monoclonal antibody. MW = molecular weight. O = occipital. PrPsc = abnormally folded prion protein. sCJD = sporadic Creutzfeldt-Jakob disease. T1 = type 1. T2 = type 2. VPSPr = variably protease-sensitive prionopathy.
On the basis of the autopsy results, we diagnosed the patient with VPSPr.
DISCUSSION
Because VPSPr is extremely rare, we decided to conduct a literature review to explore the demographic and clinical characteristics, and diagnostic findings, of VPSPr. We searched MEDLINE (PubMed) for existing cases using variably protease-sensitive prionopathy, protease-sensitive prionopathy, and VPSPr as the key words and January 1, 2008, to September 30, 2020, as the time period. The search identified 48 articles, of which 13 reported at least one independent clinical case of pathologically confirmed VPSPr. In total, the 13 articles reported 40 cases of VPSPr.
In 2008, Gambetti et al reported 11 cases of prion disease that were affected by a distinct prionopathy with protease-sensitive PrPsc that differed biochemically and clinically from the typical sporadic prionopathies that have protease-resistant PrPsc. The authors designated this previously unidentified disease as protease-sensitive prionopathy, or PSPr. All 11 cases were homozygous for valine (V/V) at codon 129 of the PRNP gene, which is characterized by a common M/V polymorphism.
In 2010, the same group (Zou et al, 2010) reported 15 new cases of protease-sensitive prionopathy. These cases included codon 129 homozygous V/V (n = 6), homozygous M/M (n = 3), and heterozygous M/V (n = 6), and the M/M and M/V cases demonstrated less sensitivity to protease digestion compared with the V/V cases. The authors renamed the condition as VPSPr. The pathological findings of VPSPr include moderate spongiform degeneration, amyloid microplaques, and a target-like or plaque-like prion protein deposition (Notari et al, 2018). The PrPsc in VPSPr typically form a striking, ladder-like electrophoretic profile of five to seven bands (depending on the antibody) with variable protease resistance depending on the 129 polymorphisms (Notari et al, 2018). Although 30% of the VPSPr cases in the Gambetti et al (2008) and Zou et al (2010) studies had a family history of dementia, there was no mutation found in the PRNP gene of the 26 cases.
Since the 2010 report (Zou et al, 2010), an additional 14 pathologically confirmed VPSPr cases have been reported in the United States, United Kingdom, Netherlands, Austria, and Spain (Aizpurua et al, 2019; Assar et al, 2015; Cannon et al, 2014; Head et al, 2009, 2010, 2013; Jansen et al, 2010; Kim and Geschwind, 2019; Rodríguez-Martínez et al, 2010, 2012; Vicente-Pascual et al, 2018). We analyzed the demographics, clinical characteristics, and diagnostic findings of all 41 VPSPr cases (including our patient) based on available information derived from the original reports (Table 1).
TABLE 1.
Demographics, Clinical Presentation, and Diagnostic Testing Findings of 41 Reported Variably Protease-sensitive Prionopathy Cases
| Codon 129 Genotype | ||||
|---|---|---|---|---|
| Characteristic | M/M (n = 5)† |
M/V (n = 10) |
V/V (n = 26) |
Total (N = 41) |
| Gender‡ (male:female) | 14:12 | |||
| Age of onset, M years (range) | 66 (55–78) | 70 (46–81) | 63 (48–77) | 65 (46–81) |
| Disease duration, M months (range) | 59 (41–78) | 37 (7–72) | 31 (8–120) | 35 (7–120) |
| Cognitive decline | 75% | 70% | 65% | 68% |
| Psychiatric symptoms | 0% | 80% | 69% | 65% |
| Language deficits | 25% | 30% | 50% | 48% |
| Parkinsonian features | 75% | 70% | 42% | 55% |
| Ataxia | 75% | 50% | 46% | 50% |
| Myoclonus | 75% | 20% | 12% | 20% |
| 14-3-3 positivity (positive/total cases tested) | 50% (2/4) | 0% (0/5) | 46% (6/13) | 36% (8/22) |
| Characteristic EEG findings (positive/total cases tested) | 25% (1/4) | 0% (0/5) | 0% (0/21) | 3% (1/30) |
| Characteristic MRI findings (positive/total cases tested) | 0% (0/4) | 10% (1/10) | 8% (2/25) | 8% (3/39) |
One M/M case reported by Zou et al (2010) appeared to be free of clinical symptoms. The case was identified via an aging research project in which the patient participated.
Gender information was reported in only 26 cases.
M = methionine. V = valine.
On the basis of the 41 cases reported so far, VPSPr affects both men and women relatively equally, and the average age of onset is 65 years. The average disease duration is 35 months, which is much longer than the typical disease duration seen in individuals with sCJD (Baldwin and Correll, 2019). Common clinical manifestations of VPSPr include cognitive decline, psychiatric symptoms, language deficits, parkinsonian features, and ataxia. Myoclonus is less common in VPSPr than in sCJD.
VPSPr usually presents with a frontal lobe dementia syndrome, although various antemortem clinical diagnoses such as dementia with Lewy bodies, normal-pressure hydrocephalus, amyotrophic lateral sclerosis, and parkinsonism have also been described (Aizpurua et al, 2019; Cannon et al, 2014; Kim et al, 2019; Vicente-Pascual et al, 2018; Zou et al, 2010). Tests that are commonly used to diagnose sCJD generally are not helpful in diagnosing VPSPr.
Among the 41 cases, the CSF 14-3-3 protein was positive in only eight of the 22 cases that were tested, although three were weak positive, which is considered nondiagnostic in clinical settings. Furthermore, periodic sharp-wave complexes on EEG were reported in only one of the 30 patients who had had an EEG. In addition, only three of the 39 patients who had had a brain MRI showed the cortical restricted diffusion and/or FLAIR cortical ribbon sign as is typically seen in individuals with sCJD.
The clinical features of VPSPr vary depending on the PRNP codon 129 polymorphisms. For example, compared with the other types of carriers, VPSPr is more prevalent in codon 129 V/V carriers (63% of all cases), whereas only 17% of sCJD cases and only 12% of the general population are V/V carriers (Gambetti et al, 2008; Zou et al, 2010). In the 41 cases that we reviewed, the V/V carriers appeared to have a younger age of onset and a shorter disease duration compared with the M/V and M/M carriers. Cognitive decline was common in all three genotypes, whereas psychiatric symptoms were common only in the V/V and M/V carriers and not in the M/M carriers. Language deficits were more common in the V/V carriers, parkinsonian features were more common in the M/M and M/V carriers, and ataxia and myoclonus were more common in the M/M carriers.
Our patient’s clinical presentation of rapidly progressive cognitive decline, psychiatric and behavior disturbances, and subsequent parkinsonian and ataxic features—together with the typical brain MRI findings of diffuse, persistent cortical and thalamic restricted diffusion—would meet the diagnostic criteria for sCJD. However, the pathology in our patient revealed VPSPr instead. We also note that at age 46, our case had the youngest age of onset of all reported VPSPr cases.
Our case, and the cases summarized from the literature review, suggests that VPSPr is clinically and pathologically heterogeneous. Because there currently are no diagnostic markers for VPSPr, definitive diagnosis can only be made by demonstrating the ladder-like electrophoretic profile on Western blots of brain homogenates, usually from autopsy.
Study Limitations
Our case report has several limitations. First, our patient had persistent cortical and thalamic signal abnormalities that are consistent with restricted diffusion on brain MRIs obtained early and later in the disease course. MRI restricted diffusion is considered an acute pathophysiological phenomenon secondary to cytotoxic edema. Persistant MRI restricted diffusion has not been well studied in the literature. We do not have a good explanation for the persistence of the MRI restricted diffusion in our case. Second, VPSPr is a very rare prion disease, and clinical diagnostic criteria and consensus have not yet been established. As a result, most of the cases reported in the literature were based on cases that were identified by pathological examination, some of them with limited information regarding the onset of symptoms, clinical presentation, and disease course, which made it difficult to accurately compare the clinical characteristics among cases.
CONCLUSION
Many of the conditions causing RPD are treatable; therefore, prompt and accurate diagnosis is very important. Our case underlines the importance of considering VPSPr in the spectrum of prion disease phenotypes when evaluating individuals with RPD. VPSPr should be considered in the differential diagnosis of a more slowly progressive non-Alzheimer degenerative dementia as well.
ACKNOWLEDGMENTS
The authors thank the patient’s family, the Creutzfeldt-Jakob Disease Foundation, the referring physicians, and, for their technical help and review of data, all of the members of the Case Western Reserve University National Prion Disease Pathology Surveillance Center.
Supported in part by a grant (NU2GCK000434) from the United States Center for Disease Control and a grant (R01 NS103848) from the United States National Institutes of Health.
Glossary
- M
methionine
- NR
normal range
- PRNP gene
prion protein gene
- PrPc
prion protein
- PrPsc
abnormally folded prion proteins
- RPD
rapidly progressive dementia
- sCJD
sporadic Creutzfeldt–Jakob disease
- V
valine
- VPSPr
variably protease-sensitive prionopathy
Footnotes
The authors declare no conflicts of interest.
REFERENCES
- Aizpurua M, Selvackadunco S, Yull H, et al. 2019. Variably protease-sensitive prionopathy mimicking frontotemporal dementia. Neuropathology. 39:135–140. doi: 10.1111/neup.12538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assar H, Topakian R, Weis S, et al. 2015. A case of variably protease-sensitive prionopathy treated with doxycyclin. J Neurol Neurosurg Psychiatry. 86:816–818. letter to the editor [DOI] [PubMed] [Google Scholar]
- Baldwin KJ, Correll CM. 2019. Prion disease. Semin Neurol. 39:428–439. doi: 10.1055/s-0039-1687841 [DOI] [PubMed] [Google Scholar]
- Cannon A, Bieniek KF, Lin WL, et al. 2014. Concurrent variably proteasesensitive prionopathy and amyotrophic lateral sclerosis. Acta Neuropathol. 128:313–315. doi: 10.1007/s00401-014-1309-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambetti P, Dong Z, Yuan J, et al. 2008. A novel human disease with abnormal prion protein sensitive to protease. Ann Neurol. 63:697–708. doi: 10.1002/ana.21420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambetti P, Puoti G, Zou WQ. 2011. Variably protease-sensitive prionopathy: a novel disease of the prion protein. J Mol Neurosci. 45:422–424. doi: 10.1007/s12031-011-9543-1 [DOI] [PubMed] [Google Scholar]
- Geschwind MD. 2016. Rapidly progressive dementia. Continuum (Minneap Minn). 22 510–537. doi: 10.1212/CON.0000000000000319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head MW, Knight R, Zeidler M, et al. 2009. A case of protease sensitive prionopathy in a patient in the UK. Neuropathol Appl Neurobiol. 35:628–632. doi: 10.1111/j.1365-2990.2009.01040.x [DOI] [PubMed] [Google Scholar]
- Head MW, Lowrie S, Chohan G, et al. 2010. Variably protease-sensitive prionopathy in a PRNP codon 129 heterozygous UK patient with co-existing tau, α synuclein and Aβ pathology. Acta Neuropathol. 120:821–823. doi: 10.1007/s00401-010-0766-y [DOI] [PubMed] [Google Scholar]
- Head MW, Yull HM, Ritchie DL, et al. 2013. Variably protease-sensitive prionopathy in the UK: a retrospective review 1991–2008. Brain. 136:1102–1115. doi: 10.1093/brain/aws366 [DOI] [PubMed] [Google Scholar]
- Jansen C, Head MW, van Gool WA, et al. 2010. The first case of protease-sensitive prionopathy (PSPr) in the Netherlands: a patient with an unusual GSS-like clinical phenotype. J Neurol Neurosurg Psychiatry. 81:1052–1055. doi: 10.1136/jnnp.2009.175646 [DOI] [PubMed] [Google Scholar]
- Kim MO, Geschwind MD. 2015. Clinical update of Jakob-Creutzfeldt disease. Curr Opin Neurol. 28:302–310. doi: 10.1097/WCO.0000000000000197 [DOI] [PubMed] [Google Scholar]
- Kim SH, Yu MM, Strutt AM. 2019. Variably protease-sensitive prionopathy: a differential diagnostic consideration for dementia. Neurol Clin Pract. 9:145–151. doi: 10.1212/CPJ.0000000000000612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasreddine ZS, Phillips NA, Bédirian V, et al. 2005. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc. 53:695–699. doi: 10.1111/j.1532-5415.2005.53221.x [DOI] [PubMed] [Google Scholar]
- Notari S, Appleby BS, Gambetti P. 2018. Variably protease-sensitive prionopathy. Handb Clin Neurol. 153:175–190. doi: 10.1016/B978-0-444-63945-5.00010-6 [DOI] [PubMed] [Google Scholar]
- Rodríguez-Martínez AB, Garrido JM, Zarranz JJ, et al. 2010. A novel form of human disease with a protease-sensitive prion protein and heterozygosity methionine/valine at codon 129: case report. BMC Neurol. 10:1–10. doi: 10.1186/1471-2377-10-99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Martínez AB, López de Munain A, Ferrer I, et al. 2012. Coexistence of protease sensitive and resistant prion protein in 129VV homozygous sporadic Creutzfeldt–Jakob disease: a case report. Published online October 11. J Med Case Rep. 6:1–5. doi: 10.1186/1752-1947-6-348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studart Neto A, Soares Neto HR, Simabukuro MM, et al. 2017. Rapidly progressive dementia: prevalence and causes in a neurologic unit of a tertiary hospital in Brazil. Alzheimer Dis Assoc Disord. 31:239–243. doi: 10.1097/WAD.0000000000000170 [DOI] [PubMed] [Google Scholar]
- Vicente-Pascual M, Rossi M, Gámez J, et al. 2018. Variably protease-sensitive prionopathy presenting within ALS/FTD spectrum. Ann Clin Transl Neurol. 5:1297–1302. doi: 10.1002/acn3.632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou WQ, Gambetti P, Xiao X, et al. 2013. Prions in variably protease-sensitive prionopathy: an update. Published online July 5. Pathogens. 2:457–471. doi: 10.3390/pathogens2030457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou WQ, Puoti G, Xiao X, et al. 2010. Variably protease-sensitive prionopathy: a new sporadic disease of the prion protein. Ann Neurol. 68:162–172. doi: 10.1002/ana.22094 [DOI] [PMC free article] [PubMed] [Google Scholar]