

Abstract
Background & Aims:
Only a minority of excess alcohol drinkers develop cirrhosis. We developed and evaluated risk stratification scores to identify those at highest risk.
Methods.
Three cohorts (GenomALC-1: n=1690, GenomALC-2: n=3037, UK Biobank: relevant n=6898) with a history of heavy alcohol consumption (≥80 g/day (men), ≥50 g/day (women), for ≥10 years) were included. Cases were participants with alcohol-related cirrhosis. Controls had a history of similar alcohol consumption but no evidence of liver disease. Risk scores were computed from up to eight genetic loci identified previously as associated with alcohol-related cirrhosis and three clinical risk factors. Score performance for the stratification of alcohol-related cirrhosis risk was assessed and compared across the alcohol-related liver disease spectrum, including hepatocellular carcinoma (HCC).
Results:
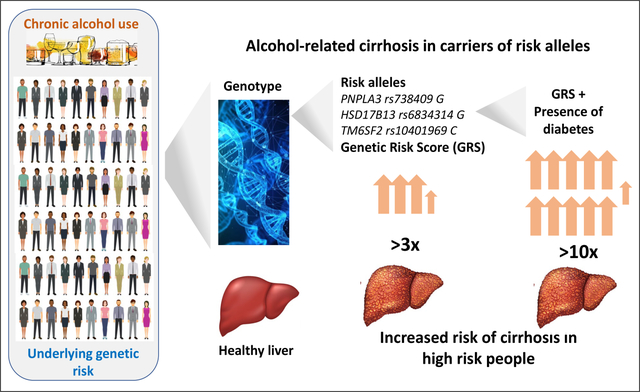
A combination of three single nucleotide polymorphisms (SNPs) (PNPLA3:rs738409, SUGP1-TM6SF2:rs10401969, HSD17B13:rs6834314) and diabetes status best discriminated for cirrhosis risk. The odds ratio (OR) and 95% confidence intervals (CI) for the extreme score quintiles (Q1-Q5) of the 3-SNP score, based on independent allelic effect size estimates, were 5.99 (4.18;8.60) (GenomALC-1); 2.81 (2.03;3.89) (GenomALC-2); and 3.10 (2.32;4.14) (UK Biobank). Patients with diabetes and high-risk score, compared to those without diabetes and a low-risk score, had ORs increased to 14.7 (7.69;28.1) (GenomALC-1) and 17.1 (11.3;25.7) (UK Biobank). Patients with cirrhosis and HCC had significantly higher mean risk scores than patients with cirrhosis alone (0.76±0.06 versus 0.61±0.02, p=0.007). Score performance was not significantly enhanced by information on additional genetic risk variants, body mass index or coffee consumption.
Conclusions:
A risk score based on three genetic risk variants and diabetes status can provide meaningful risk stratification for cirrhosis in excess drinkers, allowing earlier prevention planning including intensive intervention.
Keywords: Hepatocellular carcinoma, risk stratification, chronic alcohol use, genome wide association, single nucleotide polymorphism, coffee
LAY SUMMARY
Excessive chronic drinking leads to liver cirrhosis in some people, but so far there is no way to identify those at high risk of developing this debilitating disease. Our study has developed a genetic risk score (GRS) test that can identify patients at high risk and shows that the risk of cirrhosis is increased >10-fold with just two risk factors - diabetes and high GRS. Risk assessment using this test has potential for early and personalised management of this disease in high-risk patients.
Graphical Abstract
INTRODUCTION
Although the risk for developing cirrhosis is positively associated with alcohol consumption, only a minority of people with high-risk alcohol intake develop cirrhosis. The prevalence can vary between 7–16%1,2 with some reports suggesting the prevalence to be as low as 2%3,4. The risk threshold for what is considered high-risk intake has changed over time5–7. Long-term consumption of 80 grams per day (g/d) or more is associated with increased risk of cirrhosis8,9, but the threshold for liver harm is below this level, especially for women10,11. The 80 g/d (men) and 50 g/d (women) cut-offs were set at a relatively high level to ensure both the cirrhosis and control groups were exposed to a substantial level of alcohol-related risk. We used this threshold to define “heavy drinking” in this study.
Primary prevention of alcohol-related liver disease (ALD) would involve decreasing alcohol intake of the whole population but achieving this remains challenging. Focused intervention through the identification of people with high alcohol intake or more specifically through stratification of individuals within this population at risk for developing cirrhosis depends on identification of those at high risk. Evidence from clinical trials12 suggests that informing excessive drinkers that they have abnormal liver function tests/hepatic fibrosis can motivate them to reduce their alcohol intake. A number of both constitutional13–16, and genetic17–20 risk factors for the development of alcoholic-related cirrhosis have been identified, but no attempt appears to have been made, to date, to bring these together to provide an integrated measure of risk. Thus, the aim of this study was to devise risk scores for the stratification of cirrhosis risk and evaluate them in heavy drinkers from three independent cohorts.
MATERIALS/PATIENTS AND METHODS
Information on disease status, genotypes and clinical risk factors was available for three cohorts: i) GenomALC-1 and ii) GenomALC-2 from the GenomALC consortium, and iii) the UK Biobank. Details of the recruiting and contributing sites, with numbers of patients by diagnosis and by country are given below and in Supplementary Table 1. Cohort characteristics of the cases and controls from each source are described in Supplementary Table 2.
GenomALC-1
The GenomALC-1 cohort was recruited according to a pre-designed protocol between 2012 and 2017 in Australia, France, Germany, Switzerland, the UK, and the USA. The recruitment criteria and the data collection protocol were detailed previously21. Briefly, all participants had a history of heavy drinking (≥80 g/d (men) and ≥50 g/d (women) for ≥10 years). For cases, cirrhosis had been diagnosed by a combination of clinical criteria, laboratory variables and/or liver elastography (Fibroscan®), with liver biopsy if clinically indicated. Clinical features defining the severity of cirrhosis are shown in Supplementary Table 2. Other liver diseases (hepatitis B or C, haemochromatosis, Wilson’s disease, and autoimmune hepatitis) were excluded by laboratory testing or clinical criteria. For controls, liver disease was excluded through a combination of clinical history and measurement of liver function tests (bilirubin, albumin, ALT). For both cases and controls, HIV infection was an exclusion criterion. The study was approved by appropriate Ethics Committees or Institutional Review Boards at each site and conformed to the ethical guidelines of the 1975 Declaration of Helsinki. Participants were provided with explanations of the study and gave written informed consent. Genotyping was performed at Erasmus University Medical Centre, Rotterdam using the Illumina GSA genotyping array, as described20.
GenomALC-2
The biological samples and data were donated by research groups who had independently collected them for other studies. Some of the GenomALC-2 samples were included in a previous GWAS17; therefore, for the purposes of this study, overlapping samples were removed from the analysis. Clinical diagnosis of cases and controls was similar to GenomALC-1 criteria but detailed clinical information was limited for this cohort. Patients had given informed consent and the studies were approved by the appropriate Ethics Review Boards. DNA from these participants’ samples was also genotyped as outlined above for GenomALC-1.
Genotypes in the GenomALC-1 and GenomALC-2 cohorts were 22 cleaned using a widely used quality control pipeline, the GWASTools package https://bioconductor.org/packages/devel/bioc/manuals/GWASTools/man/GWASTools.pdf and imputed to 1000 Genomes reference using the Michigan Imputation Server (MIS)22
UK Biobank
The UK Biobank23 includes approximately 500,000 volunteers from the UK with a wide range of data including computer-administered questionnaires, physical measurements, laboratory tests, and genotyping. All participants gave informed consent, consistent with the UK Biobank Ethics and Governance Framework. Recruitment and initial assessment occurred between 2006 and 2010 when participants were aged 40 to 69 years. Access to the UK Biobank database was obtained (Application 18870) and relevant data (with diagnoses updated to June 2020) were extracted. For cases, information was restricted to assigned clinical diagnosis (Supplementary Table 2) on hospital admissions and diagnoses, and on causes of death in participants who have subsequently died. Information was available on self-reported alcohol intake at the time of assessment (by beverage type, in drinks per week, or for less frequent drinkers per month), and participants also reported whether this was less than, similar to or more than they had been consuming 10 years previously. The amounts were converted to express the alcohol intake in g/d. Participants who had a recorded diagnosis or cause of death of alcohol-related cirrhosis (ICD-10 K70.3, ‘Alcoholic cirrhosis of liver’) from hospital records or death certificates were included as cases (n=594), and those with reported drinking above the 80 or 50 g/day limits, with similar or greater consumption 10 years before, but with no diagnosed liver disease (either alcohol-related or other causes) were included as controls (n=6304). Exclusion criteria for UK Biobank subjects were similar to GenomALC-1.
UK Biobank also included 758 cases within the spectrum of other alcohol-related liver disease diagnoses (Supplementary Table 1). Genotype data for the relevant UK Biobank participants were downloaded from the server and genotypes for the relevant SNPs were extracted. Data on coffee consumption, body mass index (BMI) and diabetes status were recorded (Supplementary Table 2).
Data curation and statistical analysis
Data management and statistical analyses used IBM SPSS Statistics, version 22 (IBM Corp., New York NY). Binary variables were coded as 0 (absent) or 1 (present). Diabetes status (absent/present), BMI, kg/m2) and coffee consumption (0: not a coffee consumer, 1: coffee consumer) shown in our previous report as associated with cirrhosis16 were also modelled. Genotype data were coded as single nucleotide polymorphisms (SNPs) minor allele dosages, assuming an additive model for allelic effects.
Calculation of risk scores requires coefficients for the effect sizes associated with each risk factor, and assessment of the performance of the risk scores requires testing in independent cohorts not included in the derivation of these coefficients. The scheme shown in Table 1 sets out the basis for the scores and the data-sets which were used for evaluation.
Table 1.
Score construction and validation plan.
| Cohorts available for independent validation | ||||
|---|---|---|---|---|
| GenomALC-1 (N=1690) |
GenomALC-2 (N=3037) |
UK Biobank (N=6898) |
||
| 1 | 3-SNP score, using SNPs and coefficients from initial reports17,18 = (0.7839*PNPLA3 rs738409 G dosage) + (0.5423*SUGP1-TM6SF2 rs10401969 C dosage) − (0.4463*HSD17B13 rs6834314 G dosage) | Yes | Yes | Yes |
| 2 | 3-SNP score as in 1 above, with addition of BMI and coffee = [1] + (0.0709*BMI) − (0.645*Coffee) | No (BMI and coffee coefficients are derived from this cohort) | No (no information of BMI and coffee) | Yes |
| 3 | 3-SNP-M score, using SNPs and coefficients from meta-analysis20 = (0.7274*PNPLA3 rs2294915 T dosage) + (0.3988*SUGP1 rs10401969 C dosage) − (0.2485*HSD17B13 rs10433937 G dosage) | No* | Yes | No* |
| 4 | 5-SNP-M score; as in 3 above but with addition of two GW-significant SNPs from meta-analysis = [3] + (0.6419*SERPINA1 rs28929474 T dosage) − (0.2357*FAF2 rs11134997 C dosage) | No* | Yes | No* |
| 5 | 8-SNP-M score; as in 4 but with three additional SNPs with genome-wide significant associations with alcohol-related liver disease = [4] + (0.1446*MBOAT7 rs641738 T dosage) − (0.2401*MTARC1 rs2642438 A dosage) − (0.1304*HNRNPUL1 rs17251589 T dosage) | No* | Yes | No* |
SNP coefficients are derived from this cohort
SNPs with the lowest p-value at three loci (PNPLA3:rs738409, SUGP1-TM6SF2:rs10401969 and HSD17B13:rs6834314) were selected based on previous association with the risk of alcohol-related cirrhosis17,18, and confirmed at genome-wide significance in our meta-analysis20. Two significantly associated SNPs have been reported at SUGP1-TM6SF2 locus17 which are in near-complete linkage disequilibrium (d’1.00, r2 0.955), and rs10401969 was chosen over rs58542926 because of its stronger association with cirrhosis.
A score based on these three loci (‘3-SNP score’) was computed for each participant in each of the three cohorts. Minor allele counts (‘dosage’) were obtained from direct or imputed genotypes for each SNP, multiplied by the beta coefficients for allelic effect sizes (derived from published odds ratios, calculated as beta = loge(OR)) and summed across SNPs (Table 1). The means for 3-SNP scores were also compared between disease diagnostic groups in the three independent cohorts described in Supplementary Table 1.
Scores based on three, five, and eight loci were also computed for the GenomALC-2 samples using coefficients of loci with significant association from the published meta-analysis20 or other sources17,18 (‘3-SNP-M’, ‘5-SNP-M’ and ‘8-SNP-M’ scores) (Table 1). The 3-SNP-M score was based on the loci mentioned above, the 5-SNP-M score included above three loci, and SERPINA1 and FAF2 identified in our meta-analysis, and the 8-SNP-M score which was derived from the 5-SNP-M score with addition of three reported loci (MBOAT7, MTARC1 [previously MARC1], HNRNPUL1) significantly associated with alcohol-related cirrhosis17,24,25.
Area under the ROC curve (AUC) analysis and logistic regressions (with the score as the predictor variable and case/control status as an outcome) were performed. Odds Ratios (ORs) of the score were compared for extreme quintiles (highest Q5 against lowest Q1).
RESULTS
Risk stratification by genetic loci-based scores
Results in the three study cohorts for the 3-SNP score AUCs, logistic regressions and the ORs comparing quintiles Q5 and Q1 of the score, are shown in Table 2. Each of these measures showed better performance of the score in the GenomALC-1 cohort than in either the GenomALC-2 or UK Biobank cohorts, and there was no significant difference in score between men and women (Supplementary Table 3).
Table 2.
Results of ROC curve and logistic regression analyses, and estimated odds ratios for cirrhosis between the lowest (Q1) and highest (Q5) quintiles of scores.
| ROC Curve | Logistic regression | Q1-Q5 Odds Ratio (95% CIs) | |||
|---|---|---|---|---|---|
| AUC | Beta | p-value | |||
| 3-SNP scorei | GenomALC-1 | 0.665 ± 0.014 | 1.092 ± 0.099 | 2.90 × 10−28 | 5.99 (4.18 to 8.60) |
| GenomALC-2 | 0.606 ± 0.014 | 0.669 ± 0.090 | 1.44 × 10−13 | 2.81 (2.03 to 3.89) | |
| UK Biobank | 0.619 ± 0.014 | 0.729 ± 0.080 | 1.06 × 10−19 | 3.10 (2.32 to 4.14) | |
| 3 SNP scorei + BMI, coffee | GenomALC-1 | Not estimatediii | Not estimatediii | Not estimatediii | |
| GenomALC-2 | Not estimatediii | Not estimatediv | Not estimatediv | ||
| UK Biobank | 0.636 ± 0.015 | 0.748 ± 0.073 | 1.77 × 10−24 | 3.37 (2.38 to 4.78) | |
| Comparisons based on coefficients from meta-analysis: | |||||
| 3-SNP-M scoreii | GenomALC-2 | 0.631 ± 0.014 | 0.909 ± 0.103 | 1.17 × 10−18 | 3.65 (2.59 to 5.15) |
| 5-SNP-M scoreii | GenomALC-2 | 0.626 ± 0.014 | 0.813 ± 0.096 | 2.96 × 10−17 | 3.66 (2.62 to 5.12) |
| 8-SNP-M scoreii | GenomALC-2 | 0.633 ± 0.014 | 0.807 ± 0.091 | 6.06 × 10−19 | 3.37 (2.43 to 4.66) |
The results of adding two clinical risk factors (BMI and coffee consumption) to the 3-SNP score are shown in Table 2. Because the beta-coefficients for the two clinical risk factors were derived from the GenomALC-1 cohort, and information on these factors was not available for the GenomALC-2 cohort, this score was only evaluated against the UK Biobank data. A moderate, but not significant, improvement in risk stratification was observed following addition of these clinical risk factors; the Q5-Q1 OR estimate increased from 3.10 to 3.37 but the 95% confidence intervals overlapped. Coffee data did not improve the risk stratification, and nor did BMI (which was non-significant in the UK Biobank group and not available for GenomALC-2) (Table 2). Stratification of risk including the clinical factors in the score showed similar results for men and women (Supplementary Table 3).
The addition of further loci in the 5-SNP-M score (PNPLA3:rs2294915, SUGP1-TM6SF2:rs10401969, HSD17B13:rs10433937, SERPINA1:rs28929474, FAF2:rs11134997)17,24,25 and in the 8-SNP-M score, with MBOAT7:rs641738, MTARC1:rs2642438 and HNRNPUL1:rs17251589 in addition to those in the 5-SNP-M score, did not improve the associations between score and outcome or the risk stratification (Table 2). Because the coefficients for FAF2 and SERPINA1 were obtained from the meta-analysis of the GenomALC-1, Buch study17 and UK Biobank data, the 5-SNP-M and 8-SNP-M scores could only be tested in the GenomALC-2 data. To allow a valid comparison between the multi-SNP scores each was based on the coefficients from our meta-analysis of GWAS results. This resulted in an improvement for the meta-analysis-based 3-SNP-M score compared to the 3-SNP score (Q5-Q1 ORs changed from 2.81 [95% CI 2.03,3.89] to 3.65 [2.59,5.15]). There was also a high correlation between the 3-SNP and 3-SNP-M scores in GenomALC-2 (r = 0.826, n = 3037, p < 10−200; Supplementary Figure 1).
Clinical utility of the risk score
Numerical cut-offs that define or quantify risk are needed if the risk score is to have clinical utility. The 3-SNP scores in the GenomALC-1 cases and controls for the lowest and highest quintile boundaries were close to 0 and 1 (0.033 and 0.964, respectively; Figure 1). Division of the scores into three groups at low, intermediate and high cirrhosis risk was based on the 3-SNP score distribution (Supplementary Figure 2). The final selected scores were, low: <0; intermediate >0 – 0.7 and high risk >0.7. In each study cohort the risk difference between the low- and high-risk groups ranged between 2.5-fold and approximately 5-fold (Table 3). The difference in risk between the high- and low-risk GenomALC-1 groups were similar across the six countries (Figure 2).
Figure 1.
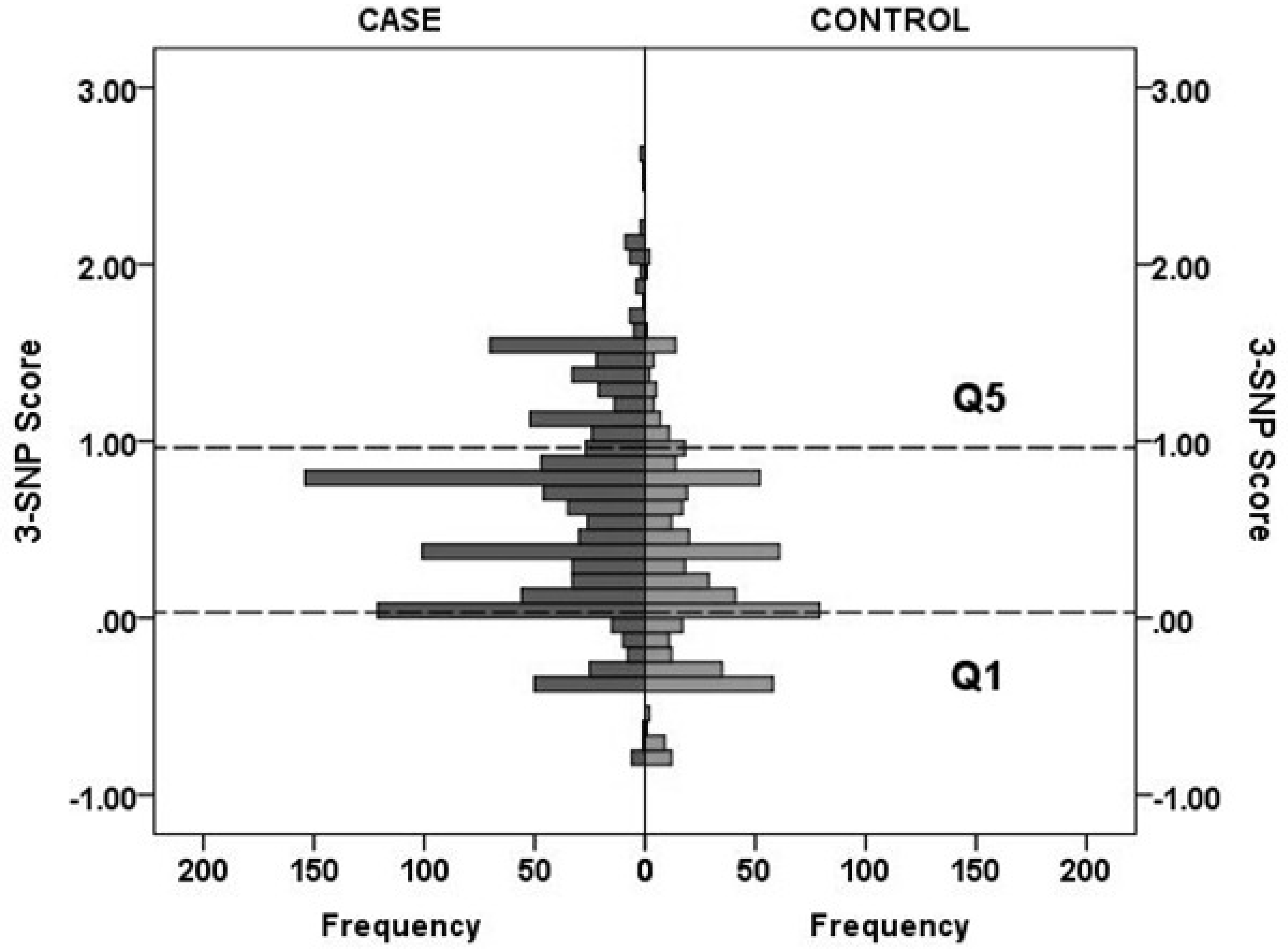
Distribution of 3-SNP scores in cases and controls from the GenomALC-1 data, showing the boundaries of the lowest (Q1) and highest (Q5) quintiles at 0.033 and 0.964, respectively (dotted lines).
Table 3.
Simplification of scoring system into three groups based on numerical values of the 3-SNP score.
| Odds Ratios (95% confidence intervals) | ||||
|---|---|---|---|---|
|
| ||||
| Risk group | score | GenomALC-1 | GenomALC-2 | UK Biobank |
|
| ||||
| Low | ≤ 0 | 1 | 1 | 1 |
| N = 273 (16.2%) | N = 327 (18.5%) | N = 3403 (56.1%) | ||
|
| ||||
| Intermediate | > 0 to 0.70 | 2.13 (1.61 to 2.83) | 1.54 (1.18 to 2.00) | 1.36 (1.04 to 1.77) |
| N = 731 (43.3%) | N = 771 (43.7%) | N = 1207 (19.9%) | ||
|
| ||||
| High | > 0.70 | 4.96 (3.67 to 6.71) | 2.67 (2.02 to 3.53) | 2.654(2.16 to 3.29) |
| N = 686 (40.6%) | N = 668 (37.8%) | N = 1456 (24.0%) | ||
Figure 2.
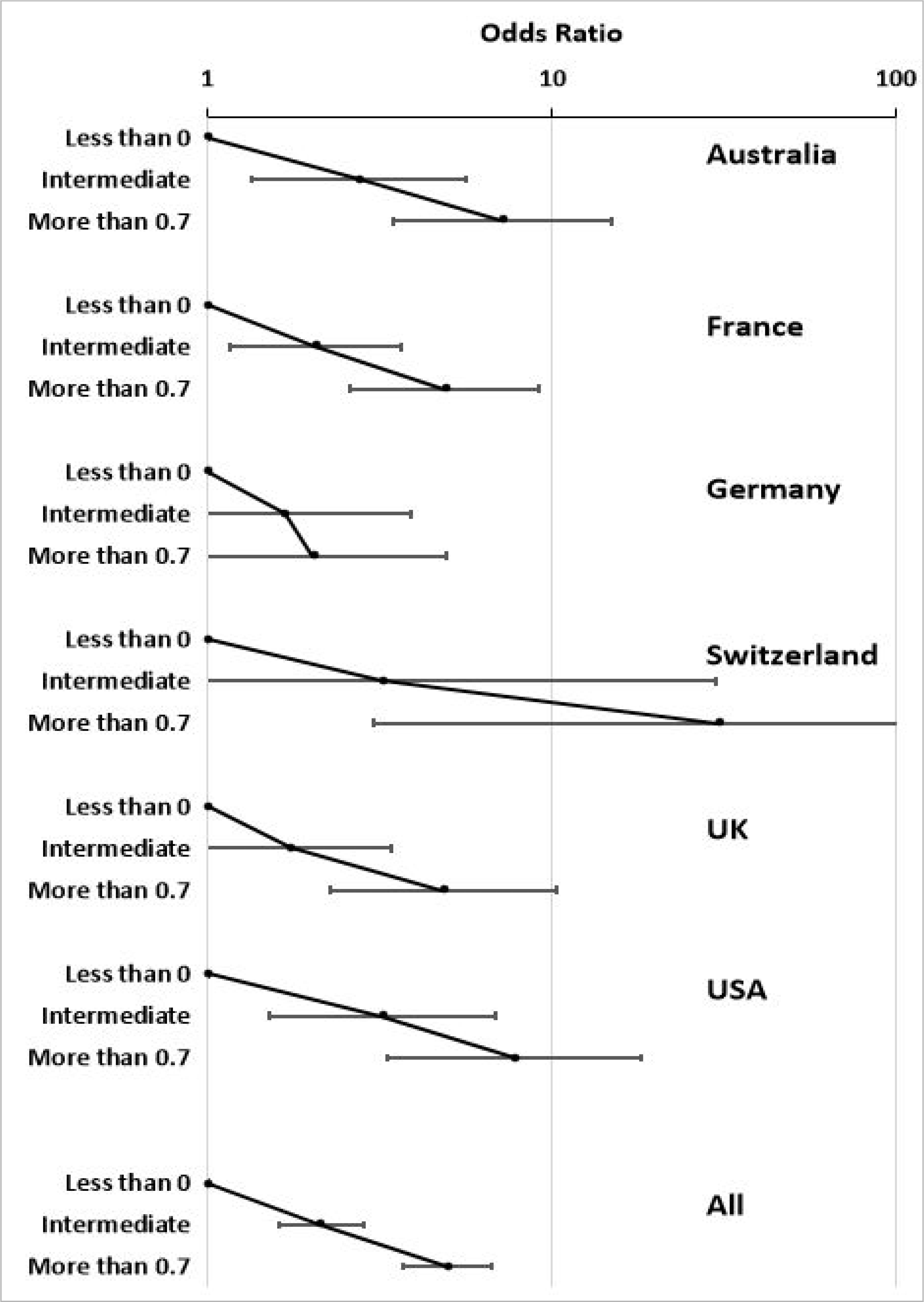
Odds ratios, by country and overall, for the risk of alcohol-related cirrhosis in the GenomALC-1 cohort when results for the 3-SNP score are divided into low (<0), intermediate (0 to 0.7) and high (>0.7) categories. Error bars show 95% confidence intervals.
Diabetes
Diabetes is known to have a large effect on cirrhosis risk. Inclusion of diabetes status with genetic risks in a combined risk score led to a bimodal distribution and difficulty in defining score quintiles. Thus, to see the effect of genetic risk score in the context of diabetes status, the 3-SNP score was subdivided by the diabetes status and is presented separately (Table 4).
Table 4.
Risk of alcohol-related cirrhosis by diabetes status, and comparison of risk in the low- and high-risk of the 3-SNP score stratified by diabetes status. For GenomALC-1, diabetes status was at time of recruitment and for UK Biobank at the time of (baseline) assessment. Information on diabetes was not available for the GenomALC-2 group. Only those participants with information on diabetes, and a 3-SNP score, are included.
| Predictor | Group | Contrast | Odds Ratios (95% Confidence Intervals) | |
|---|---|---|---|---|
| GenomALC-1 | UK Biobank | |||
|
| ||||
| Diabetes | Diabetes versus no diabetes | 3.82 (2.67 to 5.47) | 5.62 (4.33 to 7.28) | |
|
| ||||
| 3-SNP score | No diabetes | ≤0 versus >0.7 in non-diabetics | 4.77 (3.45 to 6.58) | 2.37 (1.86 to 3.03) |
| Diabetes | ≤0 (diabetes) versus >0.7 (diabetes) | 5.32 (2.06 to 13.7)1 | 3.74 (2.16 to 6.48)2 | |
| ≤0 (no-diabetes) versus >0.7 (diabetes) | 14.7 (7.69 to 28.1) | 17.1 (11.3 to 25.7) | ||
Breslow-Day test for homogeneity of Odds Ratios in Non-Diabetes and Diabetes groups, GenomALC-1 χ2 = 0.05 p = 0.830.
Breslow-Day test for homogeneity of Odds Ratios in Non-Diabetes and Diabetes groups, UK Biobank χ2 = 2.20 p = 0.138.
People with diabetes showed a substantial increase in the risk of cirrhosis in both the GenomALC-1 (OR 3.82, 95% CI 2.67;5.47) and the UK Biobank (OR 5.62, 95% CI 4.33;7.28) cohorts. The genetic score effects were similar for people with and without diabetes, both in the GenomALC-1 (logistic regression coefficients ± SE, no diabetes: 1.055 ± 0.105; diabetes: 1.276 ± 0.338) and the UK Biobank data (no diabetes: 0.653 ± 0.093; diabetes: 0.735 ± 0.181). Tests for genetic score-diabetes interaction, either by including a (score x diabetes) term in the logistic regression or by testing for heterogeneity of Odds Ratios between those with and without diabetes, showed no evidence for interaction effects in either cohort (Table 4). The combined effects of having diabetes and a high genetic risk score resulted in a >10-fold increased risk in people with diabetes and a high risk 3-SNP score against people without diabetes and a low-risk score, for both GenomALC-1 (OR 14.7, 95% CI 7.69;28.1) and the UK Biobank (OR 17.1, 95% CI 11.3,25.7) (Table 4).
Genetic loci-based risk scores across alcohol-related liver diseases
The mean values for the 3-SNP score varied across groups defined by alcohol intake and by the diagnostic categories for alcohol-related liver disease for both GenomALC-1 and the UK Biobank cohorts (Supplementary Figure 3). Post hoc comparisons showed similar trends of mean 3-SNP risk score increasing with disease severity for the GenomALC-2 cohort that included excessive drinkers with no liver disease and significantly differed between cases with severe alcoholic hepatitis and alcohol-related cirrhosis (p = 0.011) (Supplementary Table 4). Mean 3-SNP score increased with severity of liver disease (Supplementary Figure 3), including when comparing cirrhosis with HCC against cirrhosis without HCC, both for GenomALC-1 (0.757 ± 0.057 versus 0.613 ± 0.019) and UK Biobank (0.717 ± 0.102 versus 0.396 ± 0.031); see also Supplementary Table 5.
DISCUSSION
This study shows that a genetic score based on three lead SNPs associated at genome-wide significance with the risk for developing alcohol-related cirrhosis, can risk-stratify people drinking at potentially harmful levels.
Development of score for risk stratification
The performance of 3-SNP score improved considerably when used in conjunction with information on diabetes status, providing a powerful tool for identifying patients at high risk for developing advanced alcohol-related liver diseases. Higher scores were also associated with other severe liver injuries, including alcoholic hepatitis and HCC.
Our main measure of genetic risk stratification was to compare people who are in the highest quintile for a score against those in the lowest quintile, providing a more practical measure of stratification success than comparing the most extreme of all possible categories, which will usually contain only a small proportion of people26. Substantial Q5-Q1 risk differences were evident for the simple 3-SNP score in each of the cohorts; approximately six-fold in the GenomALC-1 cohort and three-fold in the other cohorts (Table 2). The greater difference in Q5-Q1 risk for GenomALC-1 is likely to be due to a more refined and pre-defined case-control definition for the recruitment protocol in this cohort.
Diabetes status led to a substantial enhancement of the utility of the 3-SNP score, predicting a >10-fold difference in risk between extreme groups (Q5 with diabetes and Q1 non-diabetes). Adding information on further genetic risk variants or BMI and coffee consumption had minimal effect.
Clinical utility of risk-score
Clinical application of a score requires the definition of decision points in numerical terms rather than by reference to population quintiles. However, Q5-Q1 comparisons can be useful for comparison across cohorts, such as in our study, and against genetic scores for other diseases. For clinical application boundaries of 0 and 0.7 were set for the 3-SNP score that provided a potentially useful stratification of risk in each of the three cohorts. As expected, lowering the high-risk threshold (e.g. from 1.0 to 0.7) identified a higher proportion of the cases as being at high risk but the ORs between the high- and low-risk groups decreased. For any classification based on a numerical test or score, changing the cut-off point(s) will alter the trade-off between test sensitivity and specificity and the optimum cut-offs will depend on the use to be made of the test. The prevalence of the condition is also important because this will affect the predictive value of positive or negative results. The AUCs shown in Table 2 were significant but when the desired test sensitivity was set at 80% (to flag nearly all those at high risk) the test specificities were between 30% and 40%. Thus, a substantial number of false positives must be accepted, making the score suitable for risk stratification but not for prediction of outcome in individual patients.
The 3-SNP risk score was also associated with differences across the alcohol-related liver disease spectrum, including HCC. The HCC risk association is consistent with previous information showing that PNPLA3, HSD17B13 and TM6SF2 polymorphisms27–31 are associated with a higher risk for this condition compared to advanced cirrhosis, perhaps suggesting a pro-oncogenic role for these variants.
Scope of risk-score
The loci comprising the current risk score are also implicated in the risk for developing cirrhosis of diverse aetiologies. Using similar polygenic risk scores (PRS) in non-alcoholic fatty liver disease (NAFLD) revealed that combining genetic and clinical features refines the predictive utility of the algorithm for identifying those at higher risk of severe liver disease32–34. Given the many shared genetic and metabolic risks between alcohol-related liver disease and NAFLD, the predictive algorithm defined here may have a wider scope across these diseases for risk stratification of those at higher risk of cirrhosis. Recently Emdin and colleagues30 identified 12 variants, five previously known, including PNPLA3, HSD17B13 and TM6SF, and seven novel, which were associated at genome-wide significance with ‘any cause’ cirrhosis, and aggregated these into a PRS. A high PRS, defined as the top quintile of the distribution, was associated with significantly increased risk of cirrhosis compared with the lowest quintile (OR 2.26; P < .001). Our current study indicates that risk stratification for alcohol-related cirrhosis can be achieved as effectively using fewer genetic markers, and with algorithms based on a smaller base of GWAS information, presumably because the genetic architecture of alcohol-related cirrhosis includes a number of common variants with substantial effects on risk.
Preliminary investigation of adding previously reported risk loci over the 3-SNP score did not significantly improve risk stratification. To develop a robust PRS that incorporates many loci for alcohol-related cirrhosis risk would require a larger population based cohort. Another possible extension, again dependent on the availability of more data, would be to incorporate information on patients’ alcohol consumption in addition to genotyping for genetic variants associated with cirrhosis risk.
The outcome of risk stratification for alcohol-related liver disease can be compared with PRS approaches to other complex diseases, including cardiovascular disease and cancers. A recent study35 showed that for five common diseases (coronary heart disease, type 2 diabetes, atrial fibrillation, breast cancer and prostate cancer), Q1-Q5 differences in PRS were associated with approximately two- to five-fold differences in the cumulative prevalence of diagnosis by age 80. Our 3-SNP score performance was equal to or slightly better than these.
The main strengths of this study were that it employed three large independent cohorts and that the case and control definitions were standardised. The study also had its limitations. First, the included populations were of largely European ancestry so that the finding may not be universally applicable. Second, an unknown proportion of the controls, especially in the UK Biobank cohort, may have undiagnosed alcohol-related liver disease, although it should be recognised that misclassification of some cases as controls would lead to poorer stratification such that the effectiveness of our score would be under-, rather than over-estimated. Finally, the risk scores were derived from groups of heavy drinkers with cirrhosis or without liver disease. However, these were validated in case and control groups selected from the population-based UK Biobank cohort. Application of the risk score to an individual patient should be performed with an understanding that some patients’ outcomes will differ from those predicted by the score. Prospective studies are needed, both to relate score to progression across time in patients who present with early stages of liver disease, and to clarify the relationship between onset of diabetes and of advanced liver disease in patients with excessive alcohol use.
Based on the findings of the present study a 3-SNP score algorithm is proposed for use and interpretation of the risk stratification in heavy drinkers (Box 1).
Box 1. Use of the 3-SNP risk score for alcohol-related cirrhosis.
| Calculate the risk score as: (0.7839*PNPLA3 rs738409 G dosage) + (0.5423*SUGP1-TM6SF2 rs10401969 C dosage) – (0.4463*HSD17B13 rs6834314 G dosage) Assign the patient to the appropriate stratum of risk, as follows: | ||
| Score less than 0 | Score above 0.7 | |
| Low risk | High risk | |
| Relative risk if not diabetic | 1 | 3-fold |
| Relative risk if diabetic | 3-fold or more | Over 10-fold |
| (Patients with scores between 0 and 0.7 are at intermediate risk) | ||
| When making use of this risk information, after appropriate explanation, consent and genotyping, be aware that this is a risk stratification scheme rather than providing individual predictions. Some patients whose score places them in the low-risk group will progress to significant liver disease, especially if they continue to drink excessively. | ||
Conclusions
An algorithm for stratifying the risk of developing alcohol-related cirrhosis among heavy drinkers, based on three genetic loci and information on diabetic status, has been developed and validated. It is intended to identify patients at particularly high risk for developing alcohol-related cirrhosis. In addition to stratifying risk of developing alcohol-related cirrhosis, this algorithm may also stratify risk for developing alcoholic hepatitis and HCC. This risk stratification system could be used to facilitate management of all people at risk for developing significant alcohol-related liver disease.
Supplementary Material
ACKNOWLEDGEMENTS
We acknowledge Ms Donna Sheedy, Ms Julia Stevens and Prof Jillian Krill for providing access to brain tissue (included in GenomALC-2) from the New South Wales Brain Tissue Resource Centre at the University of Sydney, NSW 2006, Australia (collection of tissues reported in this publication was supported by the National Institute of Alcohol Abuse and Alcoholism of the National Institutes of Health under Award Number R28AA012725. The content is solely the responsibility of the authors and does not represent the official views of the National Institutes of Health). We are grateful to the four Alcoholic Hepatitis Consortia TREAT, InTEAM, DASH and SCAHC (U01 AA021886) from the USA, for providing DNA contributing towards GenomALC-2.
FINANCIAL SUPPORT:
Funding for this study was provided by NIH/NIAAA UO1AA018389 and RO1AA018389 for data collection, analysis, interpretation and patient recruitment. SL: U01 grant from the NIAAA and R01 AA025208, U01 AA026917, and 1I01CX000361. MT: The UK Medical Research Council Stratified Medicine Award (Ref MR/R014019/1), NIHR Imperial Biomedical Research Centre and NIHR Senior Investigator Award (NIHR 200153).
CONFLICT OF INTEREST
NPC has a number of consulting agreements with and research grants from the pharmaceutical industry but they are not significantly or directly related to this paper. MP receives research funding from various organisations including the MRC and NIHR. He has also received partnership funding for the following: MRC Clinical Pharmacology Training Scheme (co-funded by MRC and Roche, UCB, Eli Lilly and Novartis); a PhD studentship jointly funded by EPSRC and Astra Zeneca; and grant funding from Vistagen Therapeutics. He has also unrestricted educational grant support for the UK Pharmacogenetics and Stratified Medicine Network from Bristol-Myers Squibb and UCB. He has developed an HLA genotyping panel with MC Diagnostics, but does not benefit financially from this. He is part of the IMI Consortium ARDAT (www.ardat.org). None of these funding sources were deployed in the undertaking of this study. TRM has conducted clinical research with AbbVie, Genfit, Gilead, and Merck but none of these are related to this manuscript.
REFERENCES
- [1].Askgaard G, Leon DA, Kjaer MS, Deleuran T, Gerds TA, Tolstrup JS: Risk for alcoholic liver cirrhosis after an initial hospital contact with alcohol problems: A nationwide prospective cohort study. Hepatology 2017, 65:929–37. 10.1002/hep.28943. [DOI] [PubMed] [Google Scholar]
- [2].Askgaard G, Kjaer MS, Tolstrup JS: Opportunities to Prevent Alcoholic Liver Cirrhosis in High-Risk Populations: A Systematic Review With Meta-Analysis. Am J Gastroenterol 2019, 114:221–32. 10.1038/s41395-018-0282-6. [DOI] [PubMed] [Google Scholar]
- [3].Hasin DS, Stinson FS, Ogburn E, Grant BF: Prevalence, correlates, disability, and comorbidity of DSM-IV alcohol abuse and dependence in the United States: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch Gen Psychiatry 2007, 64:830–42. 10.1001/archpsyc.64.7.830. [DOI] [PubMed] [Google Scholar]
- [4].Wong T, Dang K, Ladhani S, Singal AK, Wong RJ: Prevalence of Alcoholic Fatty Liver Disease Among Adults in the United States, 2001–2016. JAMA 2019, 321:1723–5. 10.1001/jama.2019.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Pequignot G, Tuyns AJ, Berta JL: Ascitic cirrhosis in relation to alcohol consumption. IntJEpidemiol 1978, 7:113–20. [DOI] [PubMed] [Google Scholar]
- [6].Rehm J, Taylor B, Mohapatra S, Irving H, Baliunas D, Patra J, et al. : Alcohol as a risk factor for liver cirrhosis: a systematic review and meta-analysis. Drug Alcohol Rev 2010, 29:437–45. 10.1111/j.1465-3362.2009.00153.x. [DOI] [PubMed] [Google Scholar]
- [7].Askgaard G, Gronbaek M, Kjaer MS, Tjonneland A, Tolstrup JS: Alcohol drinking pattern and risk of alcoholic liver cirrhosis: a prospective cohort study. J Hepatol 2015, 62:1061–7. 10.1016/j.jhep.2014.12.005. [DOI] [PubMed] [Google Scholar]
- [8].Corrao G, Bagnardi V, Zambon A, Arico S: Exploring the dose-response relationship between alcohol consumption and the risk of several alcohol-related conditions: a meta-analysis. Addiction 1999, 94:1551–73. [DOI] [PubMed] [Google Scholar]
- [9].Lelbach WK: Epidemiology of alcoholic liver disease. ProgLiver Dis 1976, 5:494–515. [PubMed] [Google Scholar]
- [10].Roerecke M, Vafaei A, Hasan OSM, Chrystoja BR, Cruz M, Lee R, et al. : Alcohol Consumption and Risk of Liver Cirrhosis: A Systematic Review and Meta-Analysis. Am J Gastroenterol 2019, 114:1574–86. 10.14309/ajg.0000000000000340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tuyns AJ, Pequignot G: Greater risk of ascitic cirrhosis in females in relation to alcohol consumption. IntJEpidemiol 1984, 13:53–7. [DOI] [PubMed] [Google Scholar]
- [12].Subhani M, Knight H, Ryder S, Morling JR: Does Advice Based on Biomarkers of Liver Injury or Non-Invasive Tests of Liver Fibrosis Impact High-Risk Drinking Behaviour: A Systematic Review With Meta-analysis. Alcohol Alcohol 2021. 10.1093/alcalc/agaa143. [DOI] [PubMed] [Google Scholar]
- [13].Hart CL, Morrison DS, Batty GD, Mitchell RJ, Davey Smith G: Effect of body mass index and alcohol consumption on liver disease: analysis of data from two prospective cohort studies. BMJ 2010, 340:c1240. 10.1136/bmj.c1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Liangpunsakul S, Puri P, Shah VH, Kamath P, Sanyal A, Urban T, et al. : Effects of Age, Sex, Body Weight, and Quantity of Alcohol Consumption on Occurrence and Severity of Alcoholic Hepatitis. Clin Gastroenterol Hepatol 2016, 14:1831–8 e3. 10.1016/j.cgh.2016.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Saab S, Mallam D, Cox GA 2nd, Tong MJ: Impact of coffee on liver diseases: a systematic review. Liver Int 2014, 34:495–504. 10.1111/liv.12304. [DOI] [PubMed] [Google Scholar]
- [16].Whitfield JB, Masson S, Liangpunsakul S, Mueller S, Aithal GP, Eyer F, et al. : Obesity, Diabetes, Coffee, Tea, and Cannabis Use Alter Risk for Alcohol-Related Cirrhosis in 2 Large Cohorts of High-Risk Drinkers. Am J Gastroenterol 2020, 116:106–15. 10.14309/ajg.0000000000000833. [DOI] [PubMed] [Google Scholar]
- [17].Buch S, Stickel F, Trepo E, Way M, Herrmann A, Nischalke HD, et al. : A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat Genet 2015, 47:1443–8. 10.1038/ng.3417. [DOI] [PubMed] [Google Scholar]
- [18].Abul-Husn NS, Cheng X, Li AH, Xin Y, Schurmann C, Stevis P, et al. : A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N Engl J Med 2018, 378:1096–106. 10.1056/NEJMoa1712191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tian C, Stokowski RP, Kershenobich D, Ballinger DG, Hinds DA: Variant in PNPLA3 is associated with alcoholic liver disease. Nat Genet 2010, 42:21–3. 10.1038/ng.488 ng.488 [pii]. [DOI] [PubMed] [Google Scholar]
- [20].Schwantes-An TH, Darlay R, Mathurin P, Masson S, Liangpunsakul S, Mueller S, et al. : Genome-wide association study and meta-analysis on alcohol-related liver cirrhosis identifies novel genetic risk factors. Hepatology 2021, 73:1920–31. 10.1002/hep.31535. [DOI] [PubMed] [Google Scholar]
- [21].Whitfield JB, Rahman K, Haber PS, Day CP, Masson S, Daly AK, et al. : Brief report: genetics of alcoholic cirrhosis-GenomALC multinational study. Alcohol Clin Exp Res 2015, 39:836–42. 10.1111/acer.12693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Das S, Forer L, Schonherr S, Sidore C, Locke AE, Kwong A, et al. : Next-generation genotype imputation service and methods. Nat Genet 2016, 48:1284–7. 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, et al. : UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 2015, 12:e1001779. 10.1371/journal.pmed.1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Emdin CA, Haas ME, Khera AV, Aragam K, Chaffin M, Klarin D, et al. : A missense variant in Mitochondrial Amidoxime Reducing Component 1 gene and protection against liver disease. PLoS Genet 2020, 16:e1008629. 10.1371/journal.pgen.1008629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Innes H, Buch S, Hutchinson S, Guha IN, Morling JR, Barnes E, et al. : Genome-Wide Association Study for Alcohol-Related Cirrhosis Identifies Risk Loci in MARC1 and HNRNPUL1. Gastroenterology 2020, 159:1276–89 e7. 10.1053/j.gastro.2020.06.014. [DOI] [PubMed] [Google Scholar]
- [26].Gellert-Kristensen H, Richardson TG, Davey Smith G, Nordestgaard BG, Tybjaerg-Hansen A, Stender S: Combined Effect of PNPLA3, TM6SF2, and HSD17B13 Variants on Risk of Cirrhosis and Hepatocellular Carcinoma in the General Population. Hepatology 2020, 72:845–56. 10.1002/hep.31238. [DOI] [PubMed] [Google Scholar]
- [27].Salameh H, Raff E, Erwin A, Seth D, Nischalke HD, Falleti E, et al. : PNPLA3 Gene Polymorphism Is Associated With Predisposition to and Severity of Alcoholic Liver Disease. Am J Gastroenterol 2015, 110:846–56. 10.1038/ajg.2015.137. [DOI] [PubMed] [Google Scholar]
- [28].Yang J, Trepo E, Nahon P, Cao Q, Moreno C, Letouze E, et al. : A 17-Beta-Hydroxysteroid Dehydrogenase 13 Variant Protects From Hepatocellular Carcinoma Development in Alcoholic Liver Disease. Hepatology 2019, 70:231–40. 10.1002/hep.30623. [DOI] [PubMed] [Google Scholar]
- [29].Tang S, Zhang J, Mei TT, Guo HQ, Wei XH, Zhang WY, et al. : Association of TM6SF2 rs58542926 T/C gene polymorphism with hepatocellular carcinoma: a meta-analysis. BMC Cancer 2019, 19:1128. 10.1186/s12885-019-6173-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Trépo E, Nahon P, Bontempi G, Valenti L, Falleti E, Nischalke HD, et al. : Association between the PNPLA3 (rs738409 C\textgreaterG) variant and hepatocellular carcinoma: Evidence from a meta-analysis of individual participant data. Hepatology 2014, 59 DOI: 10.1002/hep.26767. [DOI] [PubMed] [Google Scholar]
- [31].Stickel F, Lutz P, Buch S, Nischalke HD, Silva I, Rausch V, et al. : Genetic Variation in HSD17B13 Reduces the Risk of Developing Cirrhosis and Hepatocellular Carcinoma in Alcohol Misusers. Hepatology 2020, 72:88–102. 10.1002/hep.30996. [DOI] [PubMed] [Google Scholar]
- [32].Bianco C, Casirati E, Malvestiti F, Valenti L: Genetic predisposition similarities between NASH and ASH: Identification of new therapeutic targets. JHEP Rep 2021, 3:100284. 10.1016/j.jhepr.2021.100284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bianco C, Jamialahmadi O, Pelusi S, Baselli G, Dongiovanni P, Zanoni I, et al. : Non-invasive stratification of hepatocellular carcinoma risk in non-alcoholic fatty liver using polygenic risk scores. Journal of Hepatology 2021, 74:775–82. 10.1016/j.jhep.2020.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].De Vincentis A, Tavaglione F, Jamialahmadi O, Picardi A, Antonelli-Incalzi R, Valenti L, et al. : A Polygenic Risk Score to Refine Risk Stratification and Prediction for Severe Liver Disease by Clinical Fibrosis Scores. Clin Gastroenterol Hepatol 2021. 10.1016/j.cgh.2021.05.056. [DOI] [PubMed] [Google Scholar]
- [35].Mars N, Koskela JT, Ripatti P, Kiiskinen TTJ, Havulinna AS, Lindbohm JV, et al. : Polygenic and clinical risk scores and their impact on age at onset and prediction of cardiometabolic diseases and common cancers. Nat Med 2020, 26:549–57. 10.1038/s41591-020-0800-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.