

ABSTRACT
Candida albicans (C. albicans) is an opportunistic pathogen causing infections ranging from superficial to life-threatening disseminated infections. In a susceptible host, C. albicans is able to translocate through the gut barrier, promoting its dissemination into deeper organs. C. albicans hyphae can invade human epithelial cells by two well-documented mechanisms: epithelial-driven endocytosis and C. albicans-driven active penetration. One mechanism by which host cells protect themselves against intracellular C. albicans is termed autophagy. The protective role of autophagy during C. albicans infection has been investigated in myeloid cells; however, far less is known regarding the role of this process during the infection of epithelial cells. In the present study, we investigated the role of autophagy-related proteins during the infection of epithelial cells, including intestinal epithelial cells and gut explants, by C. albicans. Using cell imaging, we show that key molecular players of the autophagy machinery (LC3-II, PI3P, ATG16L1, and WIPI2) were recruited at Candida invasion sites. We deepened these observations by electron microscopy analyses that reveal the presence of autophagosomes in the vicinity of invading hyphae. Importantly, these events occur during active penetration of C. albicans into host cells and are associated with plasma membrane damage. In this context, we show that the autophagy-related key proteins ATG5 and ATG16L1 contribute to plasma membrane repair mediated by lysosomal exocytosis and participate in protecting epithelial cells against C. albicans-induced cell death. Our findings provide a novel mechanism by which epithelial cells, forming the first line of defense against C. albicans in the gut, can react to limit C. albicans invasion.
KEYWORDS: Candida albicans, autophagy, epithelial cells, plasma membrane damage, lysosomal exocytosis
Introduction
Candida albicans (C. albicans) is a normal inhabitant of the human commensal microbiota; however, it is also the most common fungal species responsible for opportunistic infections, causing candidiasis that can range from superficial to life-threatening invasive infections in debilitated patients.1Host-related conditions that confer high risk for C. albicans-associated infections include immune dysfunctions such as neutropenia, damage to the mucosal barrier, and dysbiosis of the resident bacterial microbiota.2 Invasive candidiasis occurs through three main stages including (i) the translocation of C. albicans through mucosal barriers in the bloodstream, (ii) its survival into the blood reservoir, and (iii) its escape from the bloodstream to secondarily infect deep-seated organs.3 Invasive candidiasis is associated with high mortality rates ranging from 20 to 49%.1Despite the fact that C. albicans colonizes various mucosal surfaces, such as the mouth or the vagina, most studies consider the gastrointestinal tract as the main portal of entry for C. albicans into the bloodstream.3,4 Indeed, molecular-based studies have identified the gut mycobiota as the main origin of disseminating C. albicans isolates into the blood.5,6
C. albicans is a polymorphic fungus that can grow in either yeast or hyphal morphology, and its ability to switch between these morphologies is considered as an important virulence trait.7 Additional morphotypes, including the gastrointestinally induced transition, the opaquea/α and the gray morphotypes, may also support fungal adaptation to specific host niches.7 Beyond these morphological transitions, C. albicans promotes its pathogenicity not only by the expression of virulence factors comprising adhesins (e.g. Als or Hwp proteins families), invasins (e.g. Als3 and Ssa1), secreted hydrolases (e.g. the Sap protein family), and a toxin (candidalysin) but also by the formation of biofilm, sophisticated nutrient acquisition strategies, and a high metabolic flexibility.1,8
On the host side, innate defenses allow host cells to recognize and respond to fungal infections. The immune system continuously monitors the resident microbiota, and several mechanisms protect the gut from pathogenic microbes such as the production of antimicrobial peptides and immunoglobulin A, the secretion of mucus, and the presence of highly specialized phagocytic cells (macrophages, neutrophils, and dendritic cells).9 Detection of potentially pathogenic fungi and initiation of an innate immune response are achieved through Pattern Recognition Receptors (PRRs) expressed by various cell types in the gut, as exemplified by Dectin-1, which specifically recognizes β-glucans exposed at the fungal surface.10 Autophagy is an intracellular membrane-trafficking process that degrades cytosolic components by sequestrating them into double membrane vesicles termed autophagosomes that eventually fuse with lysosomes. Autophagy program activation requires the regulated and sequential involvement of more than 30 ATG (autophagy-related genes) proteins and notably includes two ubiquitin-like conjugation systems represented by the LC3 (ATG8) and ATG5/ATG12/ATG16L1 protein complexes.11 Early autophagosomal structures emanate from phosphatidyl-inositol-3-phosphate (PI3P)-enriched subdomains of the endoplasmic reticulum membrane (termed omegasomes) that allow the recruitment of PI3P-binding proteins such as WIPI2, which, in turn, promote autophagosomal membrane elongation and recruitment and stabilization of the ubiquitin-like conjugation systems.12 Of note, LC3 protein represents a standard marker for monitoring autophagy since the cytosolic form of LC3 (LC3-I) is conjugated to phosphatidylethanolamine (PE) on the surface of nascent autophagosomes. Autophagy is crucial to protect cells against various stresses and represents a key component of the innate immunity, especially by contributing to pathogen clearance and by modulating the inflammatory response in the gut.13,14 Autophagy has also been described as a protective mechanism against fungal infections.15 However, conflicting data exist on the role of autophagy in vivo during C. albicans infections. Indeed, it was suggested that mice deficient for autophagy (lacking functional Atg5 or Atg7 genes) in the myeloid cell lineage were more susceptible to C. albicans infections than wild-type mice,16,17 whereas no differences were observed in another related study.18 Most of the in vitro studies explored the role of autophagy during C. albicans infections in the context of immune cells.15 Studies using macrophages and dendritic cells have indeed demonstrated that C. albicans infections induce, in a Dectin-1 dependent manner, LC3-associated phagocytosis (LAP).19 LAP is a noncanonical form of autophagy, based on the recruitment of some components of the autophagy machinery to the phagosomal membrane that enhances acidification and killing efficiency of phagosomes.20 Of note, patients bearing a polymorphism, Y238X, in the Dectin-1 encoding gene (CLEC7A) displayed an increased oral and gastrointestinal colonization by C. albicans, compared to control patients.21
Much less is known regarding the role of autophagy during epithelial cell infection by C. albicans, with no study exploring its potential protective role in the gut context. Indeed, only two studies described autophagy activation during the infection of vaginal epithelial cells by C. albicans, an event that improved vaginal cell survival upon infection.22,23 Mucosal epithelial cells are at the frontline of host-microbe interactions and represent initial sentinels and responders to pathogens, thereby contributing to the efficiency of the innate immune response.24 In the gut, a single layer of epithelial cells confers a physical (e.g. tight junctions between cells) and a chemical (e.g. production of antimicrobial factors such as defensins) barrier between the host and its luminal microbiota. Autophagy has been described to contribute to multiple features of the protective role of epithelial cells, including tight junctions’ maintenance and cell death mitigation.1,14 Here, we explored in depth the role of autophagy and autophagy-related proteins in human epithelial cells during C. albicans infection, focusing on intestinal epithelial cells as the main portal of entry for C. albicans-disseminated infections. We showed that key autophagy-related proteins are recruited at C. albicans entry sites. This recruitment occurs only during active penetration by the fungi but not along the epithelial-driven endocytosis of C. albicans into epithelial cells. The recruitment of autophagy-related proteins to C. albicans entry sites is associated with plasma membrane damage and contributes to lysosomal membrane exocytosis, a process involved in plasma membrane repair. Finally, we showed that autophagy limits invasion and protects epithelial cells from C. albicans-induced cell death at early times of infection. This work provides new mechanistic insights into how host cells facing C. albicans invasion recruit components of the autophagic machinery.
Results
The autophagy-related protein LC3 is recruited at C. albicans entry sites
To determine whether a specific autophagic response is induced in host epithelial cells during infection by C. albicans, we first measured the autophagic flux by measuring LC3-II turnover by Western blot using the autophagy flux inhibitor Bafilomycin A1.25 During the activation of the autophagic process, cytosolic LC3 (termed LC3-I) is conjugated with PE and the amount of this lipidated form of LC3, termed LC3-II, correlates with the number of autophagosomes. Bafilomycin A1 prevents autophagosome-lysosome fusion and allows us to measure properly LC3-II accumulation upon stimulation. Immunoblot analysis of total protein extracts showed that infection of HeLa cells by C. albicans did not induce an increase in the steady state level of LC3-II at 2 and 4 h after infection, compared to uninfected cells (Figure 1a,b). Moreover, the inhibition of the autophagic flux by Bafilomycin A1 treatment did not allow us to visualize a significant increase of the LC3 lipidation upon infection. This apparent lack of marked global autophagy activation measured by immunoblot in the epithelial cell monolayer infected by C. albicans might be due to the fact that only a small subpopulation of host cells (around 10%, Sup Figure S1a) are invaded by the yeast at these time points. In order to explore autophagy activation only in infected cells, we used fluorescence microscopy to visualize the intracellular distribution of LC3. For this purpose, HeLa cells that stably express GFP-LC3 were infected for 1, 2, or 4 h with C. albicans (Figure 1c,d). At 1 and 2 h postinfection, only a few and isolated LC3 puncta, likely representing autophagosomes, were detected in the vicinity of invading C. albicans cells within epithelial cells, whereas at 4 h postinfection, a massive local recruitment of LC3 was observed in 47.9% of the C. albicans penetration sites (Figure 1d). This recruitment expressed as a “punctiform” to a “continuous” LC3 staining around the penetration sites likely corresponds to membrane-associated LC3 (i.e. LC3-II conjugated form) (Figure 1c). To confirm that LC3 observed at invasion sites is under its lipidated form, we transfected HeLa cells with the GFP-tagged lipidation defective mutant LC3 G120A26 and compared its recruitment with the wild-type GFP-LC3. Recruitment of this mutant form of LC3 is markedly reduced at C. albicans invasion sites, indicating that lipidation of LC3 is required for this cellular event (Sup Figure S1b,c). This finding was confirmed in cells expressing a dominant negative form of Atg4B (C74A), described to totally abrogate the lipidation of LC3,27 in which the recruitment of the endogenous LC3 was lost compared to control cells (Sup Figure S1d,e). We also used a special permeabilization protocol before cell fixation, which removes soluble LC3 and allows the staining of membrane-associated GFP-LC3. As shown in supplemental Figure 1e, LC3 recruitment at invasion sites confirmed the fact that LC3 was membrane-bound and might correspond to LC3-conjugated forms (Sup Figure S1f). The recruitment of endogenous LC3 was also observed in C. albicans-infected intestinal epithelial cells HCT116 (Figure 1e,f). Finally, ileal explant of GFP-LC3 transgenic mice was infected ex vivo by C. albicans for 4 h and LC3 accumulation was also noticed at entry sites of C. albicans hyphae into intestinal epithelial cells (Figure 1g). Altogether, these results indicate that C. albicans induces the recruitment of a component of the autophagy machinery, i.e. LC3, at its invasion sites in vitro and ex vivo in intestinal epithelial cells.
Figure 1.
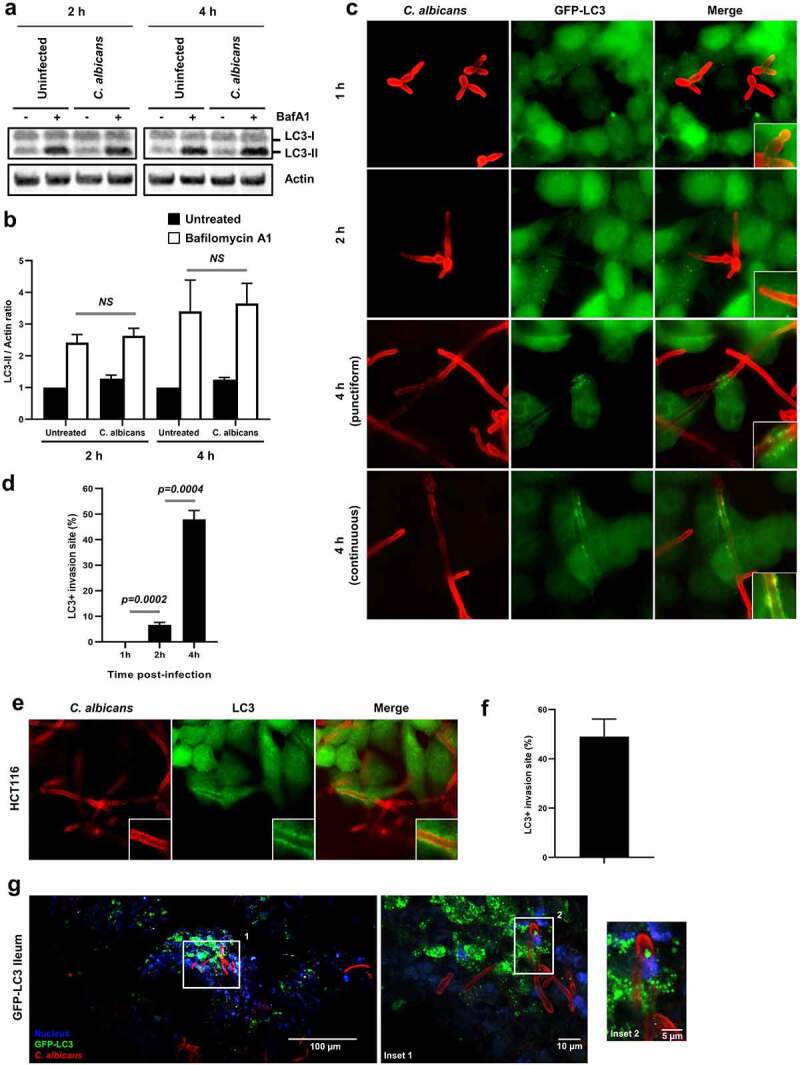
The autophagy-related protein LC3 is recruited at the C. albicans entry sites.
(a) Autophagy flux assay. HeLa cells were infected or not with C. albicans for 2 or 4 h, in the presence or absence of Bafilomycin A1 (BafA1) at 100 nM. Immunoblot analysis was performed using anti-LC3B and anti-actin antibodies. (b) The ratio of LC3-II signal relative to actin was measured and normalized to untreated cells without BafA1 (back bars: untreated, white bars: BafA1-treated). NS: not significant. (c) Representative images of C. albicans-infected GFP-LC3 HeLa cells after 1, 2, or 4 h infection. Samples were processed for C. albicans (red) and GFP-LC3 (green) staining. (d) Quantification of the percentage of GFP-LC3 + C. albicans invasion sites at 1, 2, or 4 h postinfection. Data are mean ± SEM of six independent experiments. (e) Representative images of C. albicans-infected HCT116 intestinal epithelial cells after 4 h of infection. Samples were processed for C. albicans (red) and endogenous LC3 (green) staining. (f) Quantification of the percentage of LC3 + C. albicans invasion sites at 4 h postinfection in HCT116 cells. Data are mean ± SEM of three independent experiments. (g) Representative images of ileal explants of GFP-LC3 mice infected for 4 h with C. albicans. Images were acquired using a two-photon excitation microscope. The white square indicates the inset area. Samples were processed for C. albicans (red), GFP-LC3 (green), and nuclei (blue) staining.
Key components of the autophagy machinery are mobilized in the vicinity of C. albicans entry sites
We next examined by immunofluorescence microscopy whether other key components of the autophagy machinery were recruited at the invasion sites of filamentous forms of C. albicans during infection of human epithelial cells. We first focus our attention on ATG16L1 and WIPI2, which participate in the initiation and elongation steps of the autophagic process. Interestingly, ATG16L1 and WIPI2 were markedly recruited at the C. albicans invasion sites at 4 h postinfection and partially colocalized with GFP-LC3 (Figure 2a–d). In addition, we observed the presence of cytosolic PI3P (the key lipid in autophagy initiation)-positive structures in the vicinity of C. albicans invasion sites (Sup Figure S2a).
Figure 2.
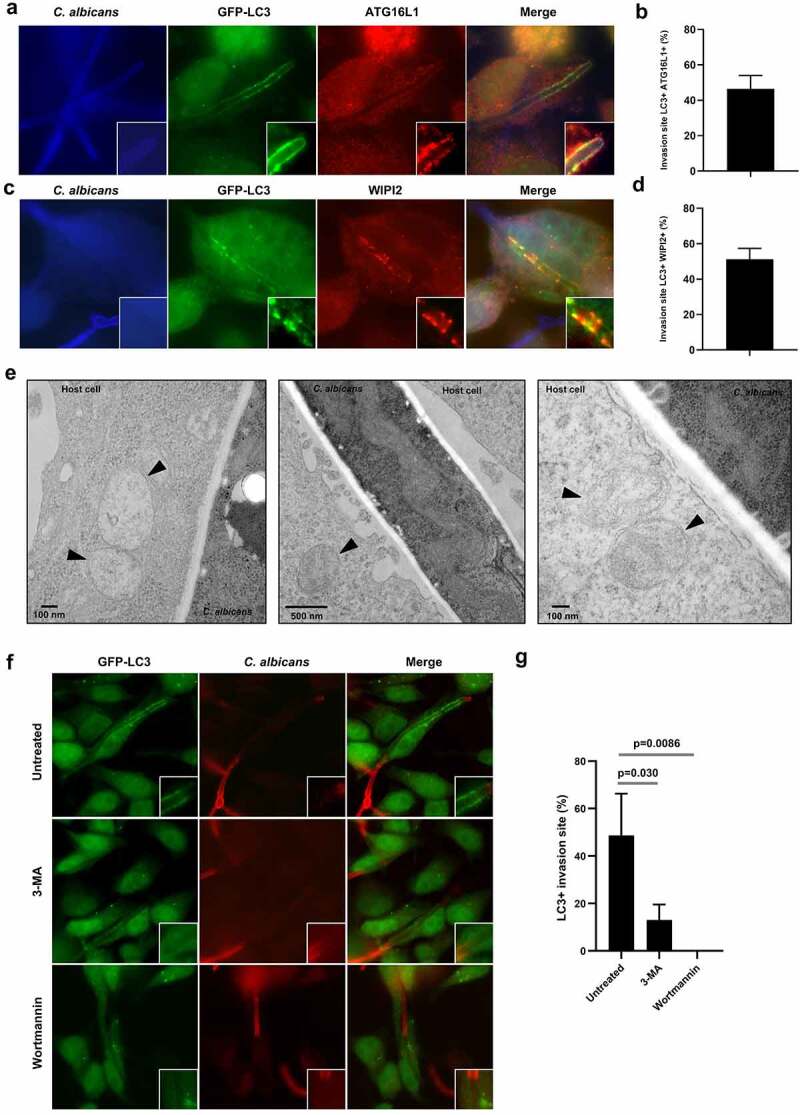
Key components of the autophagy machinery are mobilized in the vicinity of the C. albicans entry sites.
(a and c) Representative images of C. albicans-infected GFP-LC3 HeLa cells after 4 h infection. Samples were processed for C. albicans (blue), GFP-LC3 (green), and (a) ATG16L1 (red) or (c) WIPI2 (red) staining. (b and d) Quantification of the percentage of (b) GFP-LC3+/ATG16L1+ or (d) GFP-LC3+/WIPI2 + C. albicans invasion sites at 4 h postinfection. Data are mean ± SEM of three independent experiments. (e) Ultrastructural analysis by transmission electron microscopy of HeLa cells infected with C. albicans for 4 h showing the presence of intracellular vacuoles with autophagosome features (indicated by black arrowheads) near Candida invasion sites. (f) Representative images of GFP-LC3 HeLa cells untreated or treated with the PI3K inhibitors 3-MA (5 mM) or Wortmannin (100 nM) and infected with C. albicans for 4 h. Samples were processed for C. albicans (red) and GFP-LC3 (green) staining. (g) Quantification of the percentage of GFP-LC3 + C. albicans invasion sites at 4 h postinfection. Data are mean ± SEM of three independent experiments.
The recruitment of ATG16L1 to the C. albicans invasion sites was dependent on its N‐terminal domain (Sup Figure S2b), which associates with ATG5 and ATG12 to promote LC3 lipidation (Sup Figure S2c) and its targeting to membranes during canonical and noncanonical autophagy.28–30 Indeed, an ATG16L1 protein truncated for the ATG5 binding domain (ATG16L1 ΔN), which lacks the ability to rescue LC3 lipidation in HeLa ATG16L1 KO cells (Sup Figure S2c), was no longer recruited at C. albicans invasion sites (Sup Figure S2d). By contrast, a mutant form of ATG16L1 lacking the C‐terminal WD40 domain (ATG16L1 ΔC) was able to partially rescue LC3 lipidation and was still mobilized at the C. albicans entry sites (Sup Figure S2c,d). In line with this observation, the large majority of LC3 + C. albicans invasion sites were positive for the phospho-ATG16L1 immunostaining (70.1%) (Sup Figure S2e,f). Interestingly, the Ser278 phosphorylated form of ATG16L1 has been recently identified as a marker of newly forming autophagosomes.31 Altogether, these observations suggest an active formation of autophagosomes at C. albicans invasion sites.
Given the important role of the Endoplasmic Reticulum (ER) membrane as an assembly platform in promoting the recruitment of autophagy-related proteins in the context of autophagosome biogenesis, especially at contact sites with the plasma membrane (PM),32 we next investigated whether ER markers also colocalized at C. albicans entry sites. We transfected HeLa cells with GFP-taggn tethering the ER to the PM33 and leading to the recruitment of key autophagy machinery components.32 Similarly, to LC3, GFP-E-Syt1 and GFP-E-Syt2 colocalized at the C. albicans penetration sites (Sup Figure S3a,b). Mobilization of ER-related membranes at C. albicans entry sites was confirmed by positive immunostaining for the endogenous ER-protein calreticulin in infected HeLa cells (Sup Figure S3c). By contrast, we were not able to notice at the invasion sites in HeLa cells an enrichment in mitochondria, an organelle also proposed as an assembly platform for autophagosome biogenesis,34 as indicated by immunostaining of the mitochondrial protein TOM20 (Sup Figure S3d).
The observed enrichment in these extended synaptotagmins 1 and 2 (E-Syt1 and E-Syt2) and two ER protein essentials in various autophagy-related proteins at the invasion sites of C. albicans prompted us to investigate the presence of autophagosomal vacuoles by transmission electron microscopy (TEM) in infected cells. We confirmed the presence of multiple intracellular vacuoles, presenting autophagosome features (i.e. double-membrane structures enclosing cytoplasmic content), close to the C. albicans entry sites in HeLa cells (Figure 2e), that corroborate, at least in part, the positive immunostaining for autophagy-related proteins observed at the invasion sites (see Figure 1c, 2a,c).
Since autophagy machinery activation relies on phosphoinositide 3-kinases (PI3Ks) class III signaling,12 we investigated the ability of PI3K inhibitors, i.e. 3-methyladenine (3-MA) and Wortmannin, to inhibit the recruitment of LC3 at C. albicans invasion sites. C. albicans infection concomitant treatment of cells with both inhibitors abrogated GFP-LC3 recruitment to the entry sites in GFP-LC3 HeLa cells (Figure 2f,g), supporting a role for the mobilization of the autophagy machinery in response to the stress induced by C. albicans penetration.
Finally, we measured local autophagosome maturation by infecting HeLa cells stably expressing a mRFP-GFP-LC3 construct with C. albicans. Autophagosome maturation was evaluated by discriminating early autophagic vacuoles (GFP+/mRFP+) from acidic autolysosomes (mRFP+/GFP-, since GFP is sensitive to acidic quenching).25 Although a slight increase in the proportion of mature autolysosomes was observed in the vicinity of C. albicans penetration sites in comparison to uninfected cells, the difference remained nonsignificant (Sup Figure S4a,b). Of note, the presence of some acidic compartments in the vicinity of Candida invasion sites was also confirmed by the accumulation of the lysotracker dye, a probe staining acidic organelles, in GFP-LC3 + C. albicans entry sites (Sup Figure S4c).
Altogether our results suggest that key players of the autophagy machinery are recruited at the invasion sites of C. albicans into host epithelial cells, along with the presence of intracellular vacuoles presenting autophagosome features. We hypothesize that stresses triggered at the host cell surface by the penetration of C. albicans filamentous forms mobilized the autophagy machinery.
Autophagy machinery mobilization is associated with active penetration and not epithelial-driven endocytosis of C. albicans into epithelial cells
C. albicans has developed defined strategies to invade epithelial cells, including two different well-described mechanisms that are dependent on the yeast-to-hyphae transition: (i) epithelial-driven endocytosis of hyphal forms and (ii) C. albicans-driven active penetration.35–37 To elucidate whether the recruitment of autophagy-related proteins at C. albicans entry sites during epithelial invasion was triggered by one of these two invasion mechanisms or both these invasion mechanisms, we used HeLa epithelial cells and blocked active penetration by treating C. albicans hyphae with thimerosal. This antifungal drug kills the fungus and thus selectively inhibits active penetration without affecting epithelial-driven endocytosis of the fungus.38 Pretreatment of C. albicans hyphae by thimerosal resulted in a total abrogation of C. albicans invasion in HeLa cells (Sup Figure S5a), confirming previous data that identified active penetration as the main invasion mechanism in HeLa cells during early phases of C. albicans invasion.38 Thus, no LC3 recruitment was observed at contact sites between thimerosal-treated C. albicans hyphae and HeLa cells (Sup Figure S5b). We performed additional experiments with the human intestinal epithelial cell line HCT116, a cell line that can be invaded by C. albicans via either active penetration or epithelial-driven endocytosis due to a low degree of differentiation.39,40 As a result, active penetration inhibition by pretreating C. albicans hyphae with thimerosal leads to a significant decrease in the percentage of Candida invasion (18.1%) compared to untreated hyphae (62.9%) (Figure 3a). Residual invasion (18.1%) of thimerosal-treated C. albicans was totally blocked by pretreatment of HCT116 cells with cytochalasin D, an endocytosis inhibitor, confirming that the residual invasion was related to epithelial-driven endocytosis of C. albicans (Sup Figure S5c). Collectively, these observations confirmed the occurrence of both invasion mechanisms in this cell line. Although thimerosal-treated C. albicans hyphae were still able to invade HCT116 cells through endocytosis, LC3 was no longer recruited at C. albicans invasion sites (Figure 3b,c). These results suggest that the recruitment of the autophagy-related protein LC3 at the plasma membrane is a cellular response specifically associated with active penetration of C. albicans hyphal forms rather than their endocytosis by epithelial cells.
Figure 3.
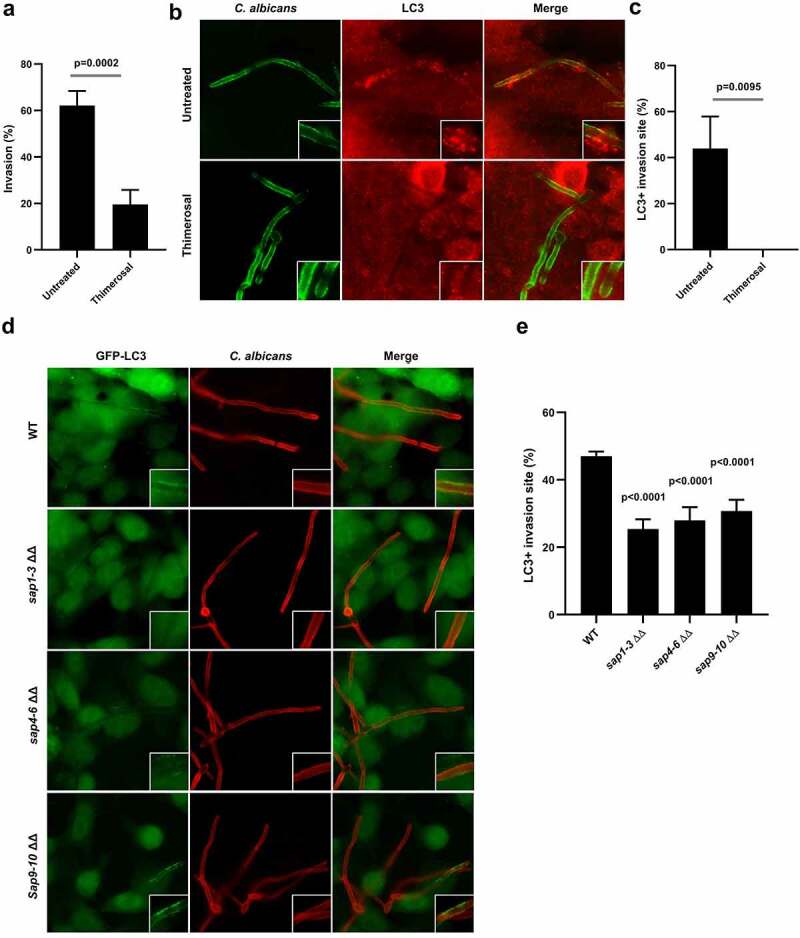
Autophagy machinery recruitment is linked to active penetration of C. albicans into epithelial cells.
(a) Percentage of invasion of untreated or thimerosal-pretreated C. albicans hyphae in HCT116 cells at 4 h postinfection. Each value is the mean of eight independent experiments ± SEM. (b) Representative images of untreated or thimerosal-pretreated C. albicans hyphae in HCT116 cells at 4 h postinfection. Samples were processed for C. albicans (green) and endogenous LC3 (red) staining. (c) Quantification of the percentage of LC3 + C. albicans invasion sites at 4 h postinfection. Data are mean ± SEM of four independent experiments. (d) Representative images of GFP-LC3 HeLa cells infected for 4 h by the wild-type strain of C. albicans (WT) or the corresponding sap 1/2/3 ΔΔ, sap 4/5/6 ΔΔ or sap 9/10 ΔΔ mutants. Samples were processed for C. albicans (red) and GFP-LC3 (green) staining. (e) Quantification of the percentage of LC3 + C. albicans invasion sites at 4 h postinfection. Data are mean ± SEM of four independent experiments.
To confirm these observations, GFP-LC3-expressing HeLa cells were infected with C. albicans mutants lacking genes encoding secreted aspartic proteases (Saps): sap1-3Δ/Δ, sap4-6Δ/Δ, and sap9-10Δ/Δ, that display a reduced ability to invade host cells by active penetration.41–43 For the three SAP-deficient mutants, a significant decrease in the percentage of LC3 + C. albicans invasion sites (sap1-3Δ/Δ: 25.4%, sap4-6Δ/Δ: 28.0%, and sap9-10Δ/Δ: 30.8%) was observed in comparison to cells infected with the wild-type strain (47.0%) (Figure 3d,e). The fact that SAP-deficient mutants, less able to invade host cells by active penetration, failed to recruit the autophagy-related protein LC3 at a level equivalent to that of the wild-type strain confirms the link between active penetration and the recruitment of autophagy-related proteins at C. albicans entry sites. Altogether, these results indicate that recruitment of components of the autophagy machinery is related to active penetration of C. albicans hyphal forms into host epithelial cells, but not to epithelial-driven endocytosis of the fungus.
Recruitment of autophagy-related proteins to C. albicans entry sites is associated with plasma membrane damage
Active penetration of C. albicans into host epithelial cells is likely to occur through molecular mechanisms combining the mechanical pressure exerted by elongating hyphae and the lytic activities of Saps and candidalysin.1 Both mechanisms can act as stressors that affect the host plasma membrane integrity. The recent literature described the contribution of autophagy-related proteins to the repair of damaged membranes, including the plasma membrane and endocytic vacuoles.44–46 Therefore, we hypothesized that the presence of autophagy-related proteins at the C. albicans penetration sites could be linked to plasma membrane damage response. To evaluate the presence of plasma membrane damage at LC3 + C. albicans penetration sites, we first examined the permeability of C. albicans-infected cells to the fluorescent Annexin V. This Ca2+-dependent phospholipid-binding protein has a high affinity for the phosphatidylserine phospholipid that is normally located on the cytoplasmic side of the plasma membrane and therefore not accessible for the binding to Annexin V. Thus, a positive Annexin V staining on cells indicates a loss of membrane integrity. About 53.5% of C. albicans invasion sites were positive for Annexin V staining at 4 h postinfection (Figure 4a,b), with a marked staining at the contact area between C. albicans and the host cell, suggesting the occurrence of plasma membrane damage. This was confirmed by the recruitment of ALIX (ALG-2-interacting protein X), an Endosomal Sorting Complexes Required For Transport (ESCRT) of binding protein described to be mobilized at the injured cell membrane,47 that colocalized with GFP-LC3 in 49.8% of C. albicans entry sites (Figure 4c,d). Additionally, membrane damage was also indicated by the recruitment of the endogenous Galectin-3 protein, colocalizing with GFP-LC3 in 36.9% of C. albicans entry sites (Figure 4e,f). Localization of Galectin-3 at C. albicans invasion sites was confirmed using a GFP-tagged version of this protein (Sup Figure S6a). Galectin is a protein recognizing galactose-containing glycans and well described for its recruitment at damaged membranes concomitantly to the ALIX protein.48,49 Interestingly, the Galectin-3 protein recruitment represents an early event in the recruitment of components of the autophagy machinery in various models of damaged endomembranes.50,51 Codistributions of ALIX and Galectin 3 with GFP-LC3 strongly suggest plasma membrane damage. As illustrated in Figure 4g, plasma membrane alterations were indeed observed by electron microscopy along the C. albicans invasion sites, with clear ruptures in the continuity of the membranes (Figure 4g). Of note, these alterations were not observed in HCT116 cells endocytosing C. albicans hyphae, displaying filopod-like structures and membrane ruffling on their surfaces (Sup Figure S6b). Collectively, these results strengthen the possible link existing between active penetration, plasma membrane damage, and the recruitment of autophagy-related proteins.
Figure 4.
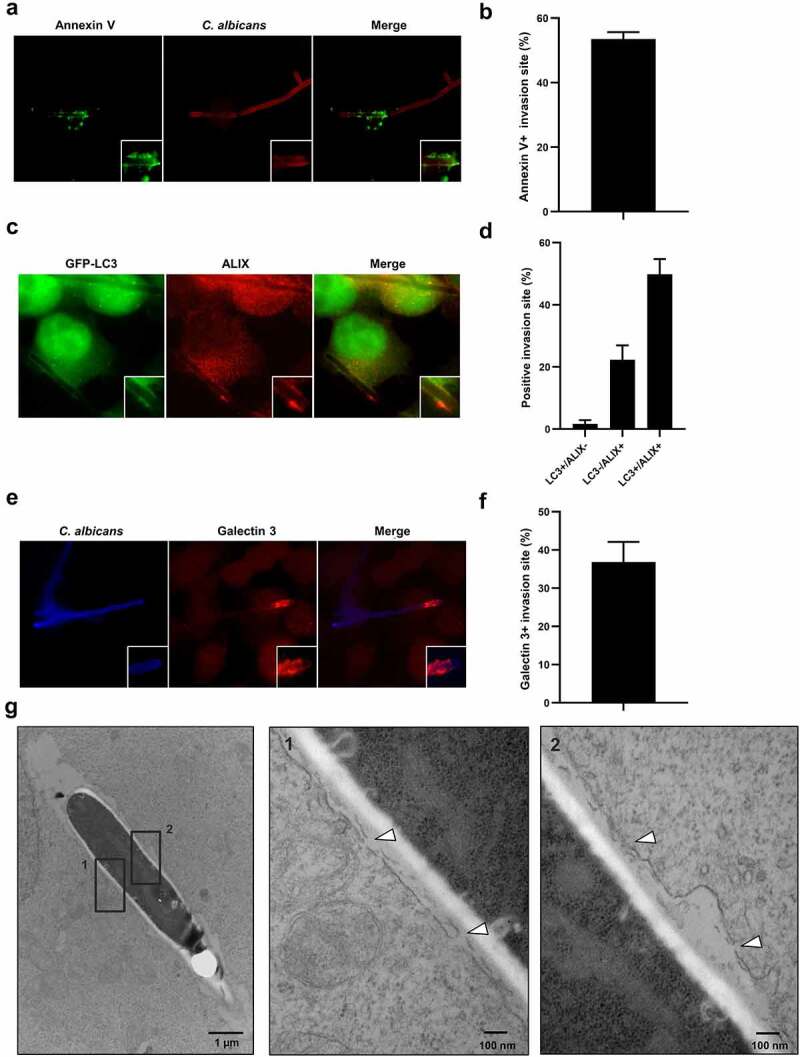
Recruitment of autophagy-related proteins to C. albicans entry sites is associated with plasma membrane damage.
(a) Representative images of C. albicans-infected HeLa cells at 4 h postinfection. Samples were processed for C. albicans (red) and Annexin V (green) staining. (b) Quantification of the percentage of Annexin V + C. albicans invasion sites at 4 h postinfection. Data are mean ± SEM of six independent experiments. (c) Representative images of C. albicans-infected GFP-LC3 HeLa cells at 4 h postinfection. Samples were processed for ALIX (red) and GFP-LC3 (green) staining. (d) Quantification of the percentage of LC3+, ALIX+, or double positive C. albicans invasion sites at 4 h postinfection. Data are mean ± SEM of three independent experiments. (e) Representative images of C. albicans-infected HeLa cells at 4 h postinfection. Samples were processed for C. albicans (blue) and Galectin 3 (red) staining. (f) Quantification of the percentage of Galectin 3 + C. albicans invasion sites at 4 h postinfection. Data are mean ± SEM of three independent experiments. (g) Ultrastructural analysis by transmission electron microscopy of HeLa cells infected with C. albicans for 4 h, showing the presence of plasma membrane damage (indicated by white arrowheads) along the C. albicans invasion sites. Black squares indicate the two inset areas.
The autophagy-related proteins, ATG16L1 and ATG5, contribute to the exocytosis of lysosomes at damaged plasma membranes
In addition to the ESCRT machinery components, Ca2+ influx-driven exocytosis of lysosomes has been reported to contribute to membrane repair and their resealing.52 Recently, it has been proposed that some autophagy-related proteins are required to induce lysosomal membrane exocytosis in the context of cell injuries triggered by the bacterial pore forming toxins pneumolysin from Streptococcus pneumoniae and listeriolysin O from Listeria monocytogenes.46 We therefore investigated whether plasma membrane damage induced by active penetration of C. albicans hyphae correlated with mobilization of lysosomes, in association or not with the recruitment of autophagy-related proteins at damaged sites. For this purpose, we first analyzed the local recruitment of endogenous lysosomal-associated membrane protein 1 (LAMP1) at C. albicans invasion sites. By using immunofluorescence staining, we observed a marked recruitment of LAMP1 at C. albicans invasion sites (Figure 5a), which was confirmed by subcellular distribution of GFP-tag LAMP1, accumulating at most of the C. albicans invasion sites into epithelial cells (Sup Figure S7a). Similarly, lysosomal-associated membrane protein 2A (LAMP2A), another endogenous lysosomal-associated protein, localized at C. albicans invasion sites (Sup Figure S7b). Finally, LAMP1 codistributed with GFP-LC3 in 40% of the invasion sites, whereas 28.7% of entry sites were positive for LAMP1 only (Figure 5b). Collectively, these data suggest that plasma membrane damages promoted by active penetration of C. albicans hyphae into epithelial cells strongly mobilize lysosomal membranes, a phenomenon that is associated with the recruitment of autophagy-related proteins.
Figure 5.
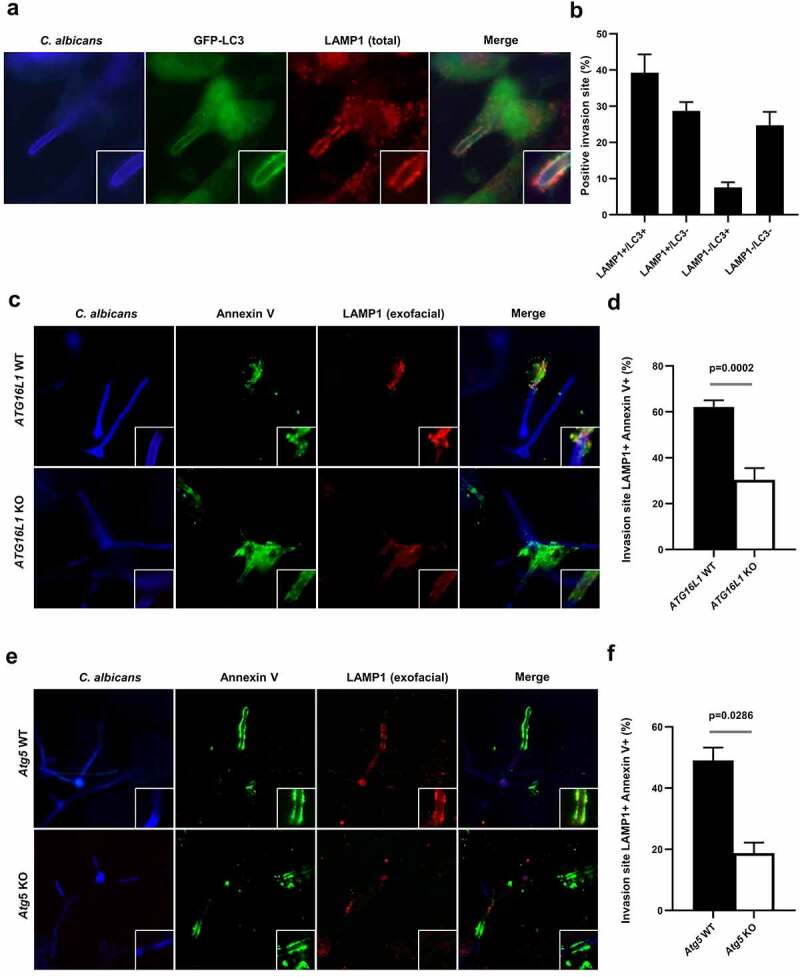
The autophagy-related proteins, ATG16L1 and ATG5, contribute to the exocytosis of lysosomes at the damaged plasma membrane.
(a) Representative images of C. albicans-infected GFP-LC3 HeLa cells at 4 h postinfection. Samples were processed for C. albicans (blue), LAMP1 (red), and GFP-LC3 (green) staining. (b) Quantification of the percentage of GFP-LC3+, LAMP1+, or double positive C. albicans invasion sites at 4 h postinfection. Data are mean ± SEM of four independent experiments. (c) Representative images of C. albicans-infected HeLa cells depleted for ATG16L1 (ATG16L1 KO) or C. albicans-infected ATG16L1 KO HeLa cells expressing the wild-type ATG16L1 (ATG16L1 WT) at 4 h postinfection. Samples were processed for C. albicans (blue), Annexin V (green), and exofacial LAMP1 (red) staining. (d) Quantification of the percentage of Annexin V + C. albicans also positive for LAMP1 staining at 4 h postinfection in ATG16L1 WT (black bar) or ATG16L1 KO (white bar) HeLa cells. Data are mean ± SEM of seven independent experiments. (e) Representative images of C. albicans-infected wild-type MEFs (Atg5 WT) or Atg5-deficient MEFs (Atg5 KO) at 4 h postinfection. Samples were processed for C. albicans (blue), Annexin V (green), and exofacial LAMP1 (red) staining. (f) Quantification of the percentage of Annexin V + C. albicans invasion sites also positive for LAMP1 staining at 4 h postinfection in Atg5 WT (black bar) or Atg5 KO (white bar) MEFs. Data are mean ± SEM of four independent experiments.
Next, to confirm lysosomal membrane mobilization at the surface of C. albicans-infected cells, we investigated LAMP1 protein delivery to the extracellular side of the plasma membrane as previously described.46 Without prior permeabilization, cells were stained alive on ice with an antibody recognizing an exofacial epitope of LAMP1, which allowed selective visualization of the pool of LAMP1 proteins exposed at the cell surface, as a consequence of lysosomal membrane exocytosis. The exofacial LAMP1 staining was only present along the penetration sites of the C. albicans hyphae (Figure 5c, upper panels), confirming the occurrence of lysosomal exocytosis following C. albicans invasion into epithelial cells. Exofacial LAMP1+ invasion sites were present in 62.2% of Annexin V+ epithelial cells, suggesting a strong association of lysosomal exocytosis with damaged plasma membranes (Figure 5c,d). Finally, since lysosomal membrane exocytosis is a calcium-triggered process, cells were treated with the calcium-chelating agent BAPTA-AM before infection to prevent elevation in the intracellular calcium concentration. As a consequence, we observed a significant decrease in the percentage of LC3 and LAMP1 signals at C. albicans invasion sites in BAPTA-AM-treated cells (8.0%) compared to untreated cells (45.1%) (Sup Figure S7c,d), indicating that calcium signaling contributes to the recruitment of LC3 and LAMP1 to the invasion sites.
Interestingly, a significant decrease in lysosomal exocytosis was observed in CRISPR/Cas9-mediated ATG16L1 gene knock-out HeLa cells as only 30.4% of C. albicans invasion sites were positive for exofacial LAMP1 in these cells staining compared to 62.2% in HeLa cells expressing WT ATG16L1 (Figure 5c, lower panels, and D). Similar results were obtained using murine Atg5-deficient cells, with a significant decrease in the percentage of exofacial LAMP1 + C. albicans invasion sites in Atg5 knock-out cells (18.8%) compared to wild-type cells (49%) (Figure 5e,f). Altogether, these results showed that (i) lysosomal membrane exocytosis occurs at C. albicans penetration sites in epithelial cells and (ii) autophagy-related proteins, including ATG16L1 and ATG5, contribute to this process.
The recruitment of autophagy-related proteins limits invasion and protects cells from C. albicans-induced epithelial cell death
Since autophagy-related proteins are actively recruited during active penetration of C. albicans and seem to contribute to plasma membrane repair by favoring lysosomal exocytosis, we questioned whether autophagy-related proteins are associated with the frequency of invasion events. We therefore, compared the percentage of C. albicans invasion between wild-type and the autophagy-deficient ATG16L1 KO HeLa cells. A differential staining protocol allowing discrimination between extracellular (red + green) and intracellular (only green) fungal hyphae was performed to measure the percentage of internalized hyphae35(Figure 6a). After 4 h of infection, invasion of the ATG16L1-deficient cells by C. albicans was slightly, but significantly increased (50.8%) compared to control cells (39.5%) (Figure 6a,b). Following invasion, the infection of epithelial cells is characterized by a loss of epithelial integrity associated with the death of host cells invaded by C. albicans invasion.53 This cell death is potentially attributed to plasma membrane damage caused by mechanical elongation of invading hyphae and the expression of C. albicans virulence factors such as the Saps proteases and/or the candidalysin toxin.38,53,54 Therefore, we tested whether ATG16L1-deficient cells were more sensitive to C. albicans-induced cell death. Viability of epithelial cells was measured by staining cells with propidium iodide (PI), a cell-death marker. At early times postinfection (4 and 6 h), higher percentage of cell death was observed in ATG16L1-deficient cells compared to wild-type cells (Figure 6c,d). At later time postinfection (20 h), percentage of dead cells increased dramatically compared to early time points (4 and 6 h) in ATG16L1 wild-type or deficient cells, with a slight, but yet significant difference between the two genotypes (Figure 6e). Very similar results were obtained when comparing C. albicans-induced cell death in Atg5-deficient cells with the corresponding wild-type cells (Figure 6f–h). These results strongly suggest that autophagy-related proteins (ATG16L1 and ATG5) participate in cells’ protection against C. albicans-induced cell death at early time points postinfection. However, this protective role was restricted to early time points and less effective at later stages of the infection process.
Figure 6.

The recruitment of autophagy-related proteins limits invasion and protects cells from C. albicans-induced epithelial cell death.
(a) Representative images of C. albicans invasion assay performed in ATG16L1 WT or ATG16L1 KO HeLa cells at 4 h postinfection. Samples were processed for nuclei (blue), extracellular C. albicans (red), and total C. albicans (green) stainings. (b) Percentage of C. albicans invasion in ATG16L1 WT (black bar) or ATG16L1 KO (white bar) HeLa cells. Data are mean ± SEM of eight independent experiments. (c, d, and e) Percentage of cell death as measured by propidium iodide incorporation (PI+ cells) in ATG16L1 WT (black bar) or ATG16L1 KO (white bar) HeLa cells infected by C. albicans for (c) 4 h, (d) 6 h, or (e) 20 h postinfection. Data are mean ± SEM of six independent experiments. (f, g, and h) Percentage of cell death as measured by propidium iodide incorporation (PI+ cells) in Atg5 WT MEFs (black bar) or Atg5 KO MEFs (white bar) infected by C. albicans for (f) 4 h, (g) 6 h, or (h) 20 h postinfection. Data are mean ± SEM of seven independent experiments.
Discussion
A growing number of studies described autophagy and autophagy-related proteins as key players in epithelial response against pathogens.55–59 In this study, we explored the involvement of the autophagy machinery during host epithelial cell response to C. albicans infections. We showed that several autophagy-related proteins (LC3, ATG16L1, and WIPI2) are recruited to C. albicans invasion sites during infection of epithelial cells, including intestinal epithelial cells (cell line and gut explants). This phenotype was associated with autophagosome biogenesis close to invasion sites since early markers of autophagosome formation (PI3P, WIPI2, and phospho-ATG16L1) are detected in the vicinity of these locations. These events only occur during active penetration of C. albicans, and not during induced endocytosis, and are associated with plasma membrane damage. Finally, we demonstrate that the recruitment of the autophagy-related proteins ATG5 and ATG16L1 contributes to lysosomal exocytosis during C. albicans invasion, a mechanism involved in plasma membrane repair,60 and protects epithelial cells against C. albicans-induced cell death at early stages of infection (schematic representation in Figure 7). To our knowledge, this is the first detailed characterization of autophagy-related proteins implication during invasion of host intestinal epithelial cells by the fungal pathogen C. albicans.
Figure 7.
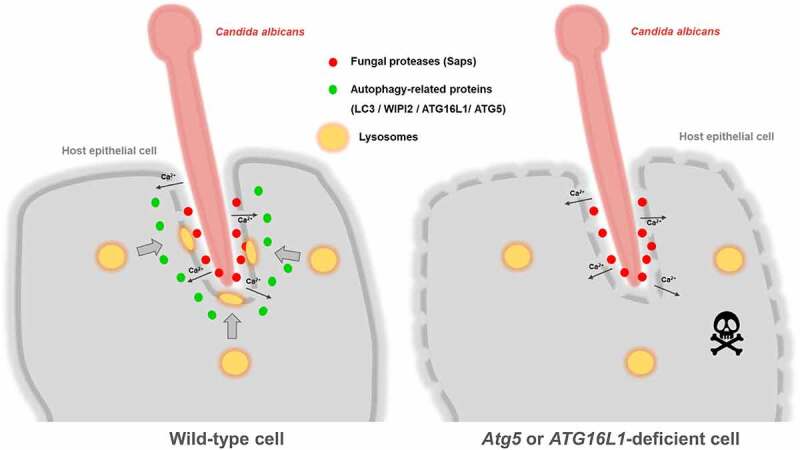
Schematic representation of the hypothetical role of autophagy-related proteins following active penetration of epithelial cells by C. albicans.
Based on the results of our study, we hypothesize that in wild-type cells (left side), active penetration of C. albicans hyphae and secretion of the fungal proteases Saps (red dots) induce plasma membrane damage, causing influx of Ca2+ (black arrows) into infected cells. Several key players of the autophagy machinery (illustrated by green dots), such as LC3, WIPI 2, ATG16L1, and ATG5, are recruited at C. albicans invasion sites and contribute to the exocytosis of lysosomes (yellow rounds), thereby limiting the deleterious effects of C. albicans invasion into host cells. In Atg5- or ATG16L1-deficient cells (right side), the exocytosis of lysosomes is dramatically reduced and cells are more sensitive to C. albicans-induced cell death, especially at early time postinfection.
While the role of autophagy and autophagy-related proteins is well described in bacterial or viral infections, far less is known about their roles in response to eukaryotic pathogens, including fungi, especially in the case of C. albicans that is only partially internalized into host cells. Indeed, C. albicans hyphae are able to penetrate epithelial cells without being fully internalized into host cells, even at late time postinfection.1 Thus, in contrast to C. albicans yeast cells phagocytosed by macrophages or dendritic cell, C. albicans hyphae are rarely fully engulfed by nonprofessional phagocytic host cells and do not end up in full enclosed endocytic or autophagic vacuoles.17,61 This is also in contrast to observations made for some other eukaryotic pathogens. For example, Plasmodium sp. or Toxoplasma gondii or Leishmania amazonensis can be fully internalized into autophagosomes or at least into vacuoles harboring autophagy markers in hepatocytes or macrophages.62–64 Regarding fungal pathogens, a number of studies have reported Cryptococcus neoformans or Aspergillus fumigatus as the inducer of LC3-associated phagocytosis (LAP) in infected macrophages or dendritic cells, known to be specialized phagocytic cells.65 This noncanonical form of autophagy required the recruitment of a part of the proteins of the autophagic machinery (including the PI3KC3 complex, ATG3, ATG5, ATG7, ATG12, and ATG16L1) to microorganisms that are engulfed within a single membrane vacuole (phagosome or endosome).66 We here report that even if C. albicans is rarely fully internalized within host epithelial cells’ cytoplasm, the membranes at invasion sites are massively decorated by several autophagy-related proteins. Additionally, features of active autophagosome biogenesis (as indicated by the presence of LC3 under its conjugated form and positive staining for PI3P, phospho-ATG16L1, and WIPI2) were monitored in close association with this subcellular area, suggesting that the autophagy machinery is mobilized in the immediate vicinity of the plasma membrane following the external stress applied by C. albicans penetration. Our findings in an infectious context are in line with studies demonstrating that the cytosolic side of plasma membrane is an important subcellular compartment for autophagosome biogenesis.32,67 However, since many reports describe a wide range of autophagy-independent roles for ATG proteins, especially in membrane dynamics events,68,69 we cannot rule out the possibility that a part of the ATG proteins recruited at C. albicans entry sites fulfills additional functions, independent of their role in canonical autophagosome biogenesis, in particular by contributing to lysosomal exocytosis.
Contribution of some autophagy-related proteins (ATG16L1, ATG5, and ATG12) in lysosomal exocytosis was recently highlighted in the context of host cell infection by the Gram-positive pathogenic bacterium Listeria monocytogenes.46 Cells lacking these ATG proteins were unable to trigger lysosomal membrane exocytosis for the repair of membrane damage induced by bacterial pore-forming toxins (LLO and PLY). As a result, an increase in sensitivity of cells to bacterial toxins was observed.46 Interestingly, we report herein similar observations showing that plasma membrane damage induced by C. albicans correlates with (i) a strong decrease in lysosomal membrane exocytosis as observed in ATG16L1- and Atg5-deficient cells and (ii) an increase sensitivity of these deficient cells to C. albicans-mediated cell death. This plasma membrane damage might result from the mechanical stress applied by the fungal active penetration in association with secreted factors (including Saps) already reported to alter the host plasma membrane.1
In the present study, a massive recruitment of ATG16L1, WIPI2, and LC3 (including its conjugated form LC3-II) occurred at C. albicans invasion sites presenting plasma membrane damage that was not observed in plasma membrane damage induced by bacterial pore-forming toxins.46 We hypothesize that, in the case of C. albicans, the plasma membrane damage is focused in a limited area of the plasma membrane (around the active penetration sites) at some stages of infection. This damage is associated with a strong mechanical deformation of the plasma membrane, a stimulus favorable to the recruitment of proteins involved in membrane dynamics regulation.70 This last point offers the intriguing possibility for a role of autophagy machinery, or at least for some autophagy-related proteins, in response to mechanical deformation of the plasma membrane. Since autophagy and its associated proteins are well described as components of stress responsive pathways for mammalian cells, we could assume that autophagy-related structures and/or proteins could play a role following mechanical stress sensed by the plasma membrane. To our knowledge, there is no study describing in depth such a role for autophagy, but it has been recently shown that shear stress in epithelial cells induced by fluid flow is sufficient to mobilize the autophagy machinery.71 Further investigations are thus needed to clearly understand to which extent the autophagy machinery contributes to the response of the plasma membrane to mechanical deformation. In addition, since several hallmarks of autophagosome biogenesis were present at C. albicans invasion sites, we could not exclude that autophagic vesicles contribute directly to plasma membrane repair by providing a pool of membrane available for membrane resealing.
In conclusion, the data presented here describe the contribution of the autophagy machinery at the plasma membrane in response to invasion of C. albicans into epithelial cells. The recruitment of autophagy-related proteins is associated with markers of autophagosomes biogenesis, suggesting that de novo synthesis of autophagic vacuoles occurs in response to C. albicans active penetration into host cells. We assume that this recruitment of ATG proteins is not fully dedicated to canonical autophagosome biogenesis, but participates in broader host cell response mechanisms aimed at coping with C. albicans-imposed mechanical (plasma membrane deformation) and chemical (fungal proteases and toxins) stresses and the subsequent plasma membrane damage. This hypothesis is in perfect line with the growing literature placing autophagy-related proteins as key players of membrane dynamics.68 Functionally, this recruitment of ATG proteins plays a protective role for infected cells by contributing to membrane repair mediated by lysosomal exocytosis and by limiting host cell death at early time postinfection. An interesting challenge in the future will be to explore whether this massive mobilization of lysosomes and vacuoles presenting features of autophagosome at the plasma membrane could release specific cargoes in the extracellular compartment able to limit C. albicans invasion by affecting its integrity, representing thereby an additional protective mechanism to ensure intestinal barrier integrity. Interestingly, such a role of autophagy-related proteins in orchestrating the release of lysosomal material in the extracellular compartment has been described few years ago in the context of osteoclastic bone resorption.72 Some autophagy-related proteins (ATG5, ATG7, ATG4B, and LC3) have been shown to be recruited to the bone-apposed plasma membrane of osteoclasts and to contribute to the exocytosis of lysosomes that release hydrolases and acidify the bone microenvironment to digest the organic matrix of bone.72 Our study, by describing similar findings in the context of C. albicans-apposed plasma membranes of epithelial cells, paves the way for future investigations to decipher whether lysosomes released in this situation by host cells could be detrimental to C. albicans integrity, thereby protecting the host cell.
Materials and methods
Cell lines
Cell lines used in this study are listed in Table 1. HeLa and HCT116 were grown in DMEM with GlutaMax (DMEM-GlutaMax with phenol red, Gibco, 31966–021) supplemented with 10% fetal bovine serum (FBS, PanBiotech, P30-8100) without antibiotics, at 37°C and 5% CO2. GFP-LC3-HeLa (kindly provided by Mathias Faure), mRFP-GFP-LC3-HeLa (kindly provided by David C. Rubinsztein), ATG16L1 WT and KO HeLa cells (both kindly provided by Xavier Ramnik), and HFF and Strawberry-ATG4B-C74A-HFF cells were maintained under the same conditions. The Atg5 Tet-Off MEF m5-7 cells (kindly provided by Noboru Mizushima) were maintained in DMEM with GlutaMax containing 10% FBS with doxycycline (Dox, 20 ng/mL) for one week to fully suppress Atg5 expression.77 All cell lines were routinely tested for mycoplasma contamination using the PCR Mycoplasma Test Kit II (PromoKine).
Table 1.
Cell lines used in this study
| Cell lines |
Description |
Applications |
Origin/Reference |
| HeLa | Human epithelial cells (cervix) | Immunoblot immunofluorescence invasion electron microscopy | ATCC |
| HCT116 | Human epithelial cells (colon) | Immunofluorescence invasion electron microscopy | ATCC |
| GFP-LC3-HeLa | HeLa cells stably expressing GFP-tagged wild-type LC3 | Immunofluorescence | 73 |
| mRFP-GFP-LC3-HeLa | HeLa cells stably expressing mRFP and GFP-tagged wild-type LC3 | Immunofluorescence | 74 |
| ATG16L1 KO HeLa | HeLa cells knocked-out for the ATG16L1 gene using theCRISPR-Cas9 system | Immunofluorescence invasion cell death assay | 75 |
| ATG16L1 WT HeLa | HeLa cells knocked-out for the ATG16L1 gene and complemented with WT ATG16L1 | Immunofluorescence invasion cell death assay | 75 |
| HFF | Human foreskin fibroblasts | Immunofluorescence | 76 |
| Strawberry-ATG4B-C74A-HFF | HFF stably expressing the Strawberry-tagged ATG4B C74A dominant negative | Immunofluorescence | 76 |
| Atg5 Tet-Off MEF m5-7 | Mouse embryonic fibroblasts with a tetracycline-regulated expression of Atg5 | Immunofluorescence cell death assay | 77 |
Plasmid transfection
HeLa cells or ATG16L1 KO HeLa cells were transiently transfected for 24 h using Lipofectamine 2000 (Thermo Fischer Scientific) with the indicated plasmid (listed in Table 2 and detailed in the figure legend) according to the manufacturer’s protocol. During and after transfection, cells were maintained in DMEM with GlutaMax supplemented with 10% FBS, without antibiotics, at 37°C and 5% CO2.
Table 2.
Plasmids used in this study
| Plasmids | Description | Origin/Reference |
|---|---|---|
| pcDNA-mCherry-ATG16L1 WT | mCherry-tagged wild-type ATG16L1 (autophagy-related protein) | 78 |
| pcDNA-mCherry-ATG16L1 ΔC | mCherry-tagged ATG16L1 lacking the C-terminal domain | 78 |
| pcDNA-mCherry-ATG16L1 ΔN | mCherry-tagged ATG16L1 lacking the N-terminal domain | 78 |
| LAMP1-mGFP | GFP-tagged LAMP1 (lysosomal-associated protein) | Addgene (34831) |
| pEGFP-E-Syt1 | GFP-tagged E-Syt1 (ER-associated protein) | 32 |
| pEGFP-E-Syt2 | GFP-tagged E-Syt2 (ER-associated protein) | 32 |
| pEGFP-LC3 | GFP-tagged wild-type LC3 (autophagy-related protein) | 79 |
| pEGFP-LC3 G120A | GFP-tagged lipidation defective mutant of LC3 | 79 |
| pEGFP-Galectin 3 | GFP-tagged Galectin 3 (Lectin binding to the damaged membrane) | 80 |
C. albicans strains and infection
Strains used in this study are listed in Table 3. The C. albicans strain SC5314 was used for all assays except for the assay in Figure 3 (panels D and E), where the C. albicans strain BWP17-CIp30 was used as the parental strains of the SAP-deficient mutants. Fungi were maintained on solid Sabouraud dextrose agar 2%. For experiments, C. albicans was grown in YPD liquid medium (1% yeast extract, 2% peptone, 2% dextrose, and Fischer Bioreagent) at 37°C overnight (ON), in a shaking incubator. Suspension was adjusted at an optical density (OD) of 600 nm equal to 0.3 in fresh YPD medium and incubated for 2 h more for log phase. C. albicans yeast forms were then harvested, sonicated, counted, and adjusted to the desired concentration in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco). For infection, indicated cell lines were infected with 5.104 log phase C. albicans cells for 1 to 4 h and maintained in DMEM with GlutaMax at 37°C and 5% CO2.
Table 3.
Candida strains used in this study
| Strain name | N° interne | Acronym | Control strain | Genotype |
|---|---|---|---|---|
| SC5314 | WT | Clinical isolate from London Mycological Reference Laboratory | ||
| BWP17-CIp30 | M2251 | Parental strain | RPS/rps1::(URA3 HIS1 ARG4) | |
| ∆sap9/∆sap10 + pCIp10 | M1429 | BWP17-CIp30 | sap10∆::hisG/sap10∆::hisG sap9∆::hisG/sap9∆::hisG rps::URA3 | |
| ∆sap1/2/3 + pCIp10 | M1630 | BWP17-CIp30 | sap1∆::hisG/sap1∆::hisG sap2∆::hisG/sap2∆::hisG sap3∆::hisG/sap3∆::hisG rps::URA3 | |
| ∆sap4/5/6 + pCIp10 | M1632 | BWP17-CIp30 | sap6∆::hisG/sap6∆::hisG sap4∆::hisG/sap4∆::hisG sap5∆::hisG/sap5∆::hisG rps::URA3 |
For hyphal growth, 106 yeast forms were seeded on 6-well plates (Costar, VWR) for 4 h in RPMI-1640 medium (Gibco, Life Technologies) at 37°C, 5% CO2. Hyphae were then washed three times in PBS, scraped, and counted. For chemical inactivation, C. albicans yeast forms were incubated in 0.04% Thimerosal-PBS solution for 1 h at room temperature (RT). Candida was then washed in PBS and counted.
Antibodies and reagents
For immunoblotting, we used the following primary antibodies: rabbit polyclonal anti-LC3B (7543) and rabbit polyclonal anti-Actin (A2066), purchased from Sigma, and the secondary antibody anti-rabbit IRDye 680RD Goat anti-Rabbit IgG (926–68071) purchased from Li-Cor. For immunofluorescence experiments, we used the primary antibodies described below. Rabbit monoclonal anti-ATG16L1 (D6D5) and rabbit monoclonal anti-Alix-1 (92880S) were purchased from Cell Signaling Technology. Rabbit polyclonal anti-LC3B (7543) was purchased from Sigma. Rabbit polyclonal anti-Candida albicans (BP-1006) was purchased from Origene. Rabbit polyclonal anti-Calreticulin (10292–1) was purchased from Proteintech. Rabbit polyclonal anti-LAMP2A (ab18528) and rabbit monoclonal anti-phospho-ATG16L1 (Ser278) (EPR19016) were purchased from Abcam. Mouse monoclonal anti-LAMP1 (H4A3) was purchased from DSHB Iowa. Mouse monoclonal anti-WIPI 2 (2A2) was purchased from Millipore. Mouse monoclonal anti-TOM20 (F-10) was purchased from Santa Cruz Biotechnology. Mouse monoclonal anti-Galectin-3 (MAB11541) was purchased from R&D systems. Mouse monoclonal anti-anti β-D-glucans (400–2) was purchased from Biosupplies. Fluorescent secondary antibodies: Alexa Fluor-conjugated anti-rabbit IgG −350, −488, and −568 and Alexa Fluor-conjugated anti-mouse IgG −350, −488, −568 were purchased from Invitrogen (A11046, A11034, A11036, A31552, A11001, A11004, and 1/500), and FITC-conjugated anti-GST antibody was purchased from Rockland.
DAPI for labeling nuclei, thimerosal (T5125, 0.04%) for fungal inactivation, cytochalasine D (500 nM) for endocytosis inhibition, and wortmannin (W1628, 100 nM) and 3-MA (M9281, 5 mM) for inhibiting autophagy were purchased from Sigma. BAPTA-AM (10 μM, 196419, Millipore) was used as a cell-permeant Ca2+ chelator. The acidotropic dye Lysotracker probe DND-99 (100 nM) was purchased from Invitrogen. Bafilomycin A1 (100 nM) for inhibiting autophagy flux was purchased from LC laboratories. FITC-Annexin V (PK-CA707-30017) for detecting plasma membrane damage was purchased from Promokine. Propidium Iodide (#638) for staining dead cells was purchased from Immunochemistry technologies.
Infection of ileal explants from GFP-LC3 mice
Transgenic GFP-LC3 mice were provided by the “RIKEN BioResource Research Center” (RBRC00806; depositor: Noboru Mizushima,81,82 Mice were housed at INRAE facilities, Dijon, France (accreditation number: C21 231 010 EA). All procedures involving animal experimentation were carried out according to the European guidelines for the care and use of laboratory animals. The ileum from GFP-LC3 homozygous mice were collected in prewarmed (37°C) DMEM medium with 1% antibiotics (penicillin/streptomycin) (Eurobio). The ileum was flushed with DMEM to remove fecal contents and opened longitudinally with scissors. Once washed in fresh DMEM medium with 1% antibiotics, the ileum was dissected into 5 mm per 5 mm explants with a sterile scalpel blade and explants were placed with the luminal side up into 6-well culture plates containing prewarmed DMEM medium with 1% antibiotics (2 mL per well). Explants were infected with 5.105 log phase yeast of C. albicans for 4 h at 37°C and 5% CO2.
Autophagy flux assay
HeLa cells were pretreated with Bafilomycin A1 at 100 nM for 30 min prior to infection with C. albicans. Bafilomycin A1 was then maintained in the cell culture medium for 2 or 4 h before total protein extraction.
Immunoblot analysis
Whole-cell protein extracts from HeLa cells were prepared from cell monolayer by adding directly 200 µL of 1.25× Laemmli sample buffer. Cell lysates were disrupted by sonication for 5 min, and proteins were denatured by heating at 95°C for 5 min. Protein extracts were clarified by a centrifugation step for 10 min at 4 000 g (room temperature). Equal amounts of protein were subjected to SDS-PAGE (4–15% mini-Protean precast protein gel, Bio-Rad), transferred on the nitrocellulose membrane (Trans-blot turbo, Bio-rad), and then immunoblotted using the indicated primary antibodies. Anti-rabbit and anti-mouse antibodies conjugated with IR800 or IR680 dyes were used as secondary antibodies, and the infrared signal was integrated using an infrared imaging system (LI-COR Odyssey). The band intensities were calculated using the software associated with the Odyssey system (Image studio).
Immunostaining
After fungal infection, cells were washed once with PBS and fixed with 4% paraformaldehyde (PFA) in PBS (0.01 M, pH7.4) for 15 min at room temperature (RT) and washed 3 times in PBS. Cells were permeabilized in 0.1% Triton X-100/PBS for 15 min at RT. Cells were then blocked by 10% goat serum in PBS for 30 min. Cells were incubated with the indicated primary antibodies for 2 h in PBS at RT. Cells were then washed three times with PBS and incubated with the secondary Alexa Fluor-conjugated antibodies for 45 min at RT. Cells were washed three times with PBS and mounted in fluorescence mounting medium (ProLong Gold Antifade, Thermo Fisher Scientific). All images were acquired using a fluorescence microscope (Axiovision Zeiss).
For the staining of membrane-associated GFP-LC3 in HeLa cells, cells were washed once with PBS and gently dipped for 3 s in cold permeabilization buffer (TritonX-100 0.1% in 100 mM KCl, 2 mM MgCl2, 1 mM CaCl2 and 1 mM Hepes, pH 6.9). Then, cells were washed once with PBS and fixed with 4% PFA as described above.
For PI3P staining in GFP-LC3 HeLa cells, cells were washed once with PBS and fixed with 4% in PBS (0.01 M, pH7.4) for 15 min at RT, washed 3 times in PBS and incubated for 1 h with purified FYVE-FYVEGST-recombinant protein (20 μg/mL final concentration), washed with PBS, and labeled with a FITC-conjugated anti-GST antibody as previously described.83
For ileal explants staining, tissues were fixed overnight at 4°C in 4% PFA in PBS (0.01 M, pH7.4). After washing in PBS, explants were permeated/saturated in 1% BSA-PBS, 0.1% Triton X-100, and 0.05% tween 20, for 1 h at room temperature, then incubated overnight at 4°C with rabbit polyclonal anti-C. albicans (BP-1006, Origene), and diluted in saturating buffer. Tissues were rinsed three times in PBS and incubated with the goat anti-rabbit secondary antibody (Alexa fluor 594, A11012, Thermo Fisher scientific) in saturating buffer, for 1 h at room temperature. After washing, tissues were mounted in antifading solution. Cell nuclei were visualized with DAPI. Images were acquired on a two-photon excitation microscope (Nikon A1-MP, DImaCell platform).
Exofacial LAMP1 and Annexin V staining
Exofacial LAMP1 staining was performed as previously described.46 After infection, cells (ATG16L1 WT/KO HeLa cells or Atg5 Tet-Off MEF m5-7) were washed once with cold HBSS with calcium (HBSS+) and then incubated in HBSS+ containing 10% goat serum for 20 min on ice with 1% (v/v) LAMP1 antibody (H4A3), 5% (v/v) FITC-Annexin V to visualize exofacial phosphatidylserine, and C. albicans antibody. Cells were then washed with HBSS+ and fixed in 4% PFA (in PBS+) for 15 min at RT. Cells were washed in HBSS+ and incubated in secondary antibodies Alexa Fluor 568 and 350 conjugated for 30 min at RT. Cells were finally washed in HBSS+, and coverslips were mounted on slides using fluorescence mounting medium.
Invasion assay
Invasion of C. albicans to epithelial cells (ATG16L1 WT/KO HeLa or HCT116 cells) quantification was determined as previously described.35 Briefly, epithelial cells were grown to confluence for 48 h on a coverslip and then infected in DMEM-containing GlutaMax with 5.104 log phase yeast of C. albicans for 1 to 4 h in a humidified incubator (37°C/5% CO2). Cell monolayers were rinsed three times with PBS and fixed with 4% PFA for 10 min at RT. The adherent part of C. albicans (extracellular) was stained for 1 h with the anti-Candida antibody, washed in PBS three times, and then incubated with the Alexa Fluor 568-conjugated secondary antibody. After three washing in PBS, cells were permeabilized with 0.1% Triton X-100/PBS for 15 min. All parts of yeasts (extracellular and intracellular) were stained with the anti-Candida antibody for 1 hour and subsequently with Alexa Fluor 488-conjugated secondary antibody. Coverslips were rinsed three times in PBS and mounted and observed with a fluorescence microscope (Axiovision Zeiss). The percentage of invading C. albicans was determined by dividing the number of totally or partially internalized C. albicans by the total number of adherent C. albicans. At least 100 C. albicans were counted on each coverslip.
Transmission electron microscopy (TEM)
Experiments were carried out in the CellImaP core facility. Cells (HeLa or HCT116), grown on Thermanox coverslips, were fixed for 1 h at 4°C in 4% paraformaldehyde and 2.5% of glutaraldehyde in Sorensen phosphate buffer (0.1 mM, pH 7,3). After fixation, samples were washed by Sorensen phosphate buffer. The postfixation treatment was realized with 1% osmium tetroxide at RT for 1 h. Dehydration and resin impregnation of the samples were performed manually: dehydration was done by increasing degrees of ethyl alcohol (50°, 70°, 95°, and 100°) and substitution was done by three absolute ethanol: Embed-812 resin mixtures and impregnation in Embed-812 resin. The polymerization of samples was performed with a mixture Embed-812: 3% BDMA in the gelatin capsule maintained for 48 h at 60°C. Blocks were cut on an ultramicrotome, and slices (thickness of 60 nm) were deposited on copper/palladium grids. After drying, grids were contrasted with uranyl acetate and lead citrate. TEM observations of cells were realized at the Dimacell imaging facility (Dijon, France) on a HITACHI H-7500 operating at 80 kV.
Cytotoxicity assay
After infection, cells (ATG16L1 WT/KO HeLa cells or Atg5 Tet-Off MEF m5-7) were washed with PBS and incubated with 0.1% propidium iodide in PBS for 15 min on ice. Cells were then fixed with 4% PFA for 15 min at RT and stained for C. albicans as described above. The percentage of cytotoxicity was determined in fungal invaded cells.
Statistical analysis
All experiments were performed at least 3 times. Statistical analyses were performed using GraphPad Prism software (GraphPad Software Inc., San Diego, CA, USA). The nonparametric Mann and Whitney t test was used to compare results between conditions. The p-values ≤ 0.05 were considered as statistically significant.
Supplementary Material
Acknowledgments
We thank David C. Rubinsztein for providing us the mRFP-GFP-LC3-HeLa cells, and Mathias Faure for the GFP-LC3-HeLa cells. We are grateful to Noboru Mizushima for kindly sharing the Atg5 Tet-Off MEF m5-7 cells. We would also like to thank Xavier Ramnik for kindly providing us the ATG16L1/CrisprCas9 knock-out HeLa cells. We also thank Pascale Bomont and Harald Wodrich for kindly sharing m-Cherry-ATG16L1 plasmids and GFP-Galectin-3 plasmid, respectively.
This work was supported by the Regional Council of Bourgogne – Franche Comté, the “Fonds Européen de DEveloppement Régional (FEDER), the Institut Carnot Qualiment, the University of Burgundy and the Hôpital du Bocage, Dijon, France. This work was supported by grants from the Agence Nationale de la Recherche (KANJI, ANR‐08‐MIEN‐ 033‐01) and a BIOASTER‐Sanofi‐Alliance pour les Sciences de la Vie et de la Santé (AVIESAN) joint program (Candida ‐ BAP110) to Frederic Dalle and Christophe d’Enfert.
Funding Statement
This work was supported by the Agence Nationale de la Recherche [KANJI, ANR‐08‐MIEN‐033‐01], European Regional Development Fund [PO FEDER-FSE Bourgogne 2014-2020], BIOASTER‐Sanofi‐Alliance pour les Sciences de la Vie et de la Santé (AVIESAN) [Candida ‐ BAP110], and Institut Carnot Qualiment [Inprobiaus].
Abbreviations
ALIX: ALG-2-interacting protein X
Als: Agglutinin-like sequence
ATG: Autophagy-related gene
C. albicans: Candida albicans
ER: Endoplasmic Reticulum
E-Syt1-2: Extended synaptotagmins 1-2
ESCRT: Endosomal Sorting Complexes Required For Transport
GFP: Green fluorescent protein
Hwp: Hyphal wall protein
LAMP1: Lysosomal-associated membrane protein 1
LC3: Microtubule-associated proteins 1A/1B light chain 3
LLO: Listeriolysin O
PI3K: Phosphoinositide 3-kinase
PI(3)P: Phosphatidylinositol-3-phosphate
PLY: Pneumolysin
PM: Plasma membrane
RFP: Red fluorescent protein
Sap: Secreted aspartyl proteinases
TEM: Transmission electron microscopy
TOM20: Translocase of outer membrane 20
WIPI2: WD repeat domain phosphoinositide-interacting protein 2
Data availability statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
Disclosure statement
There is no potential conflict of interest by the authors.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Basmaciyan L, Bon F, Paradis T, Lapaquette P, Dalle F.. Candida Albicans interactions with the host: crossing the intestinal epithelial barrier. Tissue Barriers. 2019;7(2):1612661. doi: 10.1080/21688370.2019.1612661. PubMed PMID: 31189436; PubMed Central PMCID: PMCPMC6619947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koh AY. Murine models of Candida gastrointestinal colonization and dissemination. Eukaryot Cell. 2013. Nov;12(11):1416–25. doi: 10.1128/EC.00196-13. PubMed PMID: 24036344; PubMed Central PMCID: PMCPMC3837944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sheppard DC, Filler SG. Host cell invasion by medically important fungi. Cold Spring Harb Perspect Med. 2014. Nov 3;5(1):a019687. doi: 10.1101/cshperspect.a019687. PubMed PMID: 25367974; PubMed Central PMCID: PMCPMC4292075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu W, Filler SG. Interactions of Candida albicans with epithelial cells. Cell Microbiol. 2010. Mar;12(3):273–282. doi: 10.1111/j.1462-5822.2009.01412.x. PubMed PMID: 19919567; PubMed Central PMCID: PMCPMC3383095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gouba N, Drancourt M. Digestive tract mycobiota: a source of infection. Med Mal Infect. 2015. Jan-Feb;45(1–2):9–16. doi: 10.1016/j.medmal.2015.01.007. PubMed PMID: 25684583. [DOI] [PubMed] [Google Scholar]
- 6.Saiman L, Ludington E, Pfaller M, Rangel-Frausto S, Wiblin TR, Dawson J, Blumberg HM, Patterson JE, Rinaldi M, Edwards JE. Risk factors for candidemia in Neonatal Intensive Care Unit patients. The National Epidemiology of Mycosis Survey study group. Pediatr Infect Dis J. 2000. Apr;19(4):319–324. doi: 10.1097/00006454-200004000-00011. PubMed PMID: 10783022. [DOI] [PubMed] [Google Scholar]
- 7.Noble SM, Gianetti BA, Witchley JN. Candida albicans cell-type switching and functional plasticity in the mammalian host. Nat Rev Microbiol. 2017. Feb;15(2):96–108. doi: 10.1038/nrmicro.2016.157. PubMed PMID: 27867199; PubMed Central PMCID: PMCPMC5957277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mayer FL, Wilson D, Hube B. Candida albicans pathogenicity mechanisms. Virulence. 2013. Feb 15;4(2):119–128. doi: 10.4161/viru.22913. PubMed PMID: 23302789; PubMed Central PMCID: PMCPMC3654610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drummond RA, Gaffen SL, Hise AG, and Brown, GD. Innate defense against fungal pathogens. Cold Spring Harb Perspect Med. 2014. Nov 10;5(6). doi: 10.1101/cshperspect.a019620. PubMed PMID: 25384766; PubMed Central PMCID: PMCPMC4426252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salazar F, Brown GD. A ntifungal innate immunity: a perspective from the last 10 years. J Innate Immun. 2018;10(5–6):373–397. doi: 10.1159/000488539. PubMed PMID: 29768268; PubMed Central PMCID: PMCPMC6784043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lystad AH, Carlsson SR, Simonsen A. Toward the function of mammalian ATG12-ATG5-ATG16L1 complex in autophagy and related processes. Autophagy. 2019. Aug;15(8):1485–1486. doi: 10.1080/15548627.2019.1618100. PubMed PMID: 31122169; PubMed Central PMCID: PMCPMC6613903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nascimbeni AC, Codogno P, Morel E. Phosphatidylinositol-3-phosphate in the regulation of autophagy membrane dynamics. FEBS J. 2017. May;284(9):1267–1278. doi: 10.1111/febs.13987. PubMed PMID: 27973739. [DOI] [PubMed] [Google Scholar]
- 13.Lapaquette P, Guzzo J, Bretillon L, Bringer M-A. Cellular and molecular connections between autophagy and inflammation. Mediators Inflamm. 2015;2015:398483. doi: 10.1155/2015/398483. PubMed PMID: 26221063; PubMed Central PMCID: PMCPMC4499609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foerster EG, Mukherjee T, Cabral-Fernandes L, Rocha JDB, Girardin SE, Philpott DJ. How autophagy controls the intestinal epithelial barrier. Autophagy. 2021. Apr 27;1–18. doi: 10.1080/15548627.2021.1909406. PubMed PMID: 33906557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kanayama M, Shinohara ML. Roles of autophagy and autophagy-related proteins in antifungal immunity. Front Immunol. 2016;7:47. doi: 10.3389/fimmu.2016.00047. PubMed PMID: 26925060; PubMed Central PMCID: PMCPMC4757664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanayama M, Inoue M, Danzaki K, Hammer G, He Y-W, Shinohara ML. Autophagy enhances NFkappaB activity in specific tissue macrophages by sequestering A20 to boost antifungal immunity. Nat Commun. 2015. Jan 22;6(1):5779. doi: 10.1038/ncomms6779. PubMed PMID: 25609235; PubMed Central PMCID: PMCPMC4304414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nicola AM, Albuquerque P, Martinez LR, Dal-Rosso RA, Saylor C, De Jesus M, Nosanchuk JD, Casadevall A. Macrophage autophagy in immunity to Cryptococcus neoformans and Candida albicans. Infect Immun. 2012. Sep;80(9):3065–3076. doi: 10.1128/IAI.00358-12. PubMed PMID: 22710871; PubMed Central PMCID: PMCPMC3418760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smeekens SP, Malireddi RK, Plantinga TS, Buffen K, Oosting M, Joosten LAB, Kullberg BJ, Perfect JR, Scott WK, van de Veerdonk FL. Autophagy is redundant for the host defense against systemic Candida albicans infections. Eur J Clin Microbiol Infect Dis. 2014. May;33(5):711–722. doi: 10.1007/s10096-013-2002-x. PubMed PMID: 24202731. [DOI] [PubMed] [Google Scholar]
- 19.Ma J, Underhill DM. beta-Glucan signaling connects phagocytosis to autophagy. Glycobiology. 2013. Sep;23(9):1047–1051. doi: 10.1093/glycob/cwt046. PubMed PMID: 23749474; PubMed Central PMCID: PMCPMC3858030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cemma M, Grinstein S, Brumell JH. Autophagy proteins are not universally required for phagosome maturation. Autophagy. 2016. Sep;12(9):1440–1446. doi: 10.1080/15548627.2016.1191724. PubMed PMID: 27310610; PubMed Central PMCID: PMCPMC5082775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Plantinga TS, van der Velden WJ, Ferwerda B, van Spriel, AB, Adema, G, Feuth, T, Donnelly, JP, Brown, GD, Kullberg, BJ, and Blijlevens, NM. et al. Early stop polymorphism in human DECTIN-1 is associated with increased candida colonization in hematopoietic stem cell transplant recipients. Clin Infect Dis. 2009. Sep 1;49(5):724–732. doi: 10.1086/604714. PubMed PMID: 19614557. [DOI] [PubMed] [Google Scholar]
- 22.Shroff A, Reddy KVR. Autophagy gene ATG5 knockdown upregulates apoptotic cell death during Candida albicans infection in human vaginal epithelial cells. Am J Reprod Immunol. 2018. Dec;80(6):e13056. doi: 10.1111/aji.13056. PubMed PMID: 30303264. [DOI] [PubMed] [Google Scholar]
- 23.Shroff A, Sequeira R, Reddy KVR. Human vaginal epithelial cells augment autophagy marker genes in response to Candida albicans infection. Am J Reprod Immunol. 2017. Apr;77(4):e12639. doi: 10.1111/aji.12639. PubMed PMID: 28185353. [DOI] [PubMed] [Google Scholar]
- 24.Larsen SB, Cowley CJ, Fuchs E. Epithelial cells: liaisons of immunity. Curr Opin Immunol. 2020. Feb;62:45–53. doi: 10.1016/j.coi.2019.11.004. PubMed PMID: 31874430; PubMed Central PMCID: PMCPMC7067656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, Abdellatif, M, Abdoli, A, Abel, S, Abeliovich, H, Abildgaard, MH, Abudu, YP, and Acevedo-Arozena, A. et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy. 2021. Jan;17(1):1–382. doi: 10.1080/15548627.2020.1797280. PubMed PMID: 33634751; PubMed Central PMCID: PMCPMC7996087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kabeya Y, Mizushima N, Ueno T, Yamamoto , A, Kirisako, T, Noda , T, Kominami, E, Ohsumi, Y, and Yoshimori, T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000. Nov 1;19(21):5720–5728. doi: 10.1093/emboj/19.21.5720. PubMed PMID: 11060023; PubMed Central PMCID: PMCPMC305793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fujita N, Hayashi-Nishino M, Fukumoto H, Omori H, Yamamoto A, Noda T, Yoshimori T. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol Biol Cell. 2008. Nov;19(11):4651–4659. doi: 10.1091/mbc.E08-03-0312. PubMed PMID: 18768752; PubMed Central PMCID: PMCPMC2575160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fletcher K, Ulferts R, Jacquin E, Veith T, Gammoh N, Arasteh JM, Mayer U, Carding SR, Wileman T, Beale R. The WD40 domain of ATG16L1 is required for its non-canonical role in lipidation of LC3 at single membranes. EMBO J. 2018. Feb 15;37(4). doi: 10.15252/embj.201797840. PubMed PMID: 29317426; PubMed Central PMCID: PMCPMC5813257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell. 2008. May;19(5):2092–2100. doi: 10.1091/mbc.E07-12-1257. PubMed PMID: 18321988; PubMed Central PMCID: PMCPMC2366860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lystad AH, Carlsson SR, de La Ballina LR, Kauffman KJ, Nag S, Yoshimori T, Melia TJ, Simonsen A. Distinct functions of ATG16L1 isoforms in membrane binding and LC3B lipidation in autophagy-related processes. Nat Cell Biol. 2019. Mar;21(3):372–383. doi: 10.1038/s41556-019-0274-9. PubMed PMID: 30778222; PubMed Central PMCID: PMCPMC7032593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tian W, Alsaadi R, Guo Z, Kalinina, A, Carrier, M, Tremblay, ME, Lacoste, B, Lagace, D, and Russell, RC. An antibody for analysis of autophagy induction. Nat Methods. 2020. Feb;17(2):232–239. doi: 10.1038/s41592-019-0661-y. PubMed PMID: 31768061. [DOI] [PubMed] [Google Scholar]
- 32.Nascimbeni AC, Giordano F, Dupont N, Grasso, D, Vaccaro, MI, Codogno, P, and Morel, E. ER-plasma membrane contact sites contribute to autophagosome biogenesis by regulation of local PI3P synthesis. EMBO J. 2017. Jul 14;36(14):2018–2033. doi: 10.15252/embj.201797006. PubMed PMID: 28550152; PubMed Central PMCID: PMCPMC5509996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giordano F, Saheki Y, Idevall-Hagren O, Colombo SF, Pirruccello M, Milosevic I, Gracheva E, Bagriantsev S, Borgese N, De Camilli P, et al. PI(4,5)P(2)-dependent and Ca(2+)-regulated ER-PM interactions mediated by the extended synaptotagmins. Cell. 2013. Jun 20;153(7):1494–1509. doi: 10.1016/j.cell.2013.05.026. PubMed PMID: 23791178; PubMed Central PMCID: PMCPMC3716012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013. Mar 21;495(7441):389–393. doi: 10.1038/nature11910. PubMed PMID: 23455425. [DOI] [PubMed] [Google Scholar]
- 35.Dalle F, Wachtler B, L’Ollivier C, Holland G, Bannert N, Wilson D, Labruã¨re C, Bonnin A, Hube B. Cellular interactions of Candida albicans with human oral epithelial cells and enterocytes. Cell Microbiol. 2010. Feb;12(2):248–271. doi: 10.1111/j.1462-5822.2009.01394.x. PubMed PMID: 19863559. [DOI] [PubMed] [Google Scholar]
- 36.Phan QT, Eng DK, Mostowy S, Park H, Cossart P, Filler SG. Role of endothelial cell septin 7 in the endocytosis of Candida albicans. mBio. 2013. Dec 17;4(6):e00542–13. doi: 10.1128/mBio.00542-13. PubMed PMID: 24345743; PubMed Central PMCID: PMCPMC3870263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zakikhany K, Naglik JR, Schmidt-Westhausen A, Holland, G, Schaller , M, and Hube, B. In vivo transcript profiling of Candida albicans identifies a gene essential for interepithelial dissemination. Cell Microbiol. 2007. Dec 9;9(12):2938–2954. doi: 10.1111/j.1462-5822.2007.01009.x. PubMed PMID: 17645752. [DOI] [PubMed] [Google Scholar]
- 38.Wachtler B, Citiulo F, Jablonowski N, Förster S, Dalle F, Schaller M, Wilson D, Hube B. Candida albicans-epithelial interactions: dissecting the roles of active penetration, induced endocytosis and host factors on the infection process. PLoS One. 2012;7(5):e36952. doi: 10.1371/journal.pone.0036952. PubMed PMID: 22606314; PubMed Central PMCID: PMCPMC3351431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goyer M, Loiselet A, Bon F, L’Ollivier C, Laue M, Holland G, Bonnin A, Dalle F. Intestinal cell tight junctions limit invasion of Candida albicans through active penetration and endocytosis in the early stages of the interaction of the fungus with the intestinal barrier. PLoS One. 2016;11(3):e0149159. doi: 10.1371/journal.pone.0149159. PubMed PMID: 26933885; PubMed Central PMCID: PMCPMC4775037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yeung TM, Gandhi SC, Wilding JL, Muschel R, Bodmer WF. Cancer stem cells from colorectal cancer-derived cell lines. Proc Natl Acad Sci U S A. 2010. Feb 23;107(8):3722–3727. doi: 10.1073/pnas.0915135107. PubMed PMID: 20133591; PubMed Central PMCID: PMCPMC2840416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Albrecht A, Felk A, Pichova I, Naglik JR, Schaller M, de Groot P, MacCallum D, Odds FC, Schäfer W, Klis F. Glycosylphosphatidylinositol-anchored proteases of Candida albicans target proteins necessary for both cellular processes and host-pathogen interactions. J Biol Chem. 2006. Jan 13;281(2):688–694. doi: 10.1074/jbc.M509297200. PubMed PMID: 16269404. [DOI] [PubMed] [Google Scholar]
- 42.Hube B, Sanglard D, Odds FC, Hess D, Monod M, Schäfer W, Brown AJ, Gow NA. Disruption of each of the secreted aspartyl proteinase genes SAP1, SAP2, and SAP3 of Candida albicans attenuates virulence. Infect Immun. 1997. Sep;65(9):3529–3538. doi: 10.1128/IAI.65.9.3529-3538.1997. PubMed PMID: 9284116; PubMed Central PMCID: PMCPMC175503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sanglard D, Hube B, Monod M, Odds FC, Gow NA. A triple deletion of the secreted aspartyl proteinase genes SAP4, SAP5, and SAP6 of Candida albicans causes attenuated virulence. Infect Immun. 1997. Sep;65(9):3539–3546. doi: 10.1128/IAI.65.9.3539-3546.1997. PubMed PMID: 9284117; PubMed Central PMCID: PMCPMC175504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kreibich S, Emmenlauer M, Fredlund J, Rämö P, Münz C, Dehio C, Enninga J, Hardt W-D. Autophagy Proteins Promote Repair of Endosomal Membranes Damaged by the Salmonella Type Three Secretion System 1. Cell Host Microbe. 2015. Nov 11;18(5):527–537. doi: 10.1016/j.chom.2015.10.015. PubMed PMID: 26567507. [DOI] [PubMed] [Google Scholar]
- 45.Lopez-Jimenez AT, Cardenal-Munoz E, Leuba F, Gerstenmaier L, Barisch C, Hagedorn M, King JS, Soldati T. The ESCRT and autophagy machineries cooperate to repair ESX-1-dependent damage at the Mycobacterium-containing vacuole but have opposite impact on containing the infection. PLoS Pathog. 2018. Dec;14(12):e1007501. doi: 10.1371/journal.ppat.1007501. PubMed PMID: 30596802; PubMed Central PMCID: PMCPMC6329560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tan JMJ, Mellouk N, Osborne SE, Ammendolia DA, Dyer DN, Li R, Brunen D, van Rijn JM, Huang J, Czuczman MA. An ATG16L1-dependent pathway promotes plasma membrane repair and limits Listeria monocytogenes cell-to-cell spread. Nat Microbiol. 2018. Dec 3;3(12):1472–1485. doi: 10.1038/s41564-018-0293-5. PubMed PMID: 30478389. [DOI] [PubMed] [Google Scholar]
- 47.Scheffer LL, Sreetama SC, Sharma N, Medikayala S, Brown KJ, Defour A, Jaiswal JK. Mechanism of Ca(2)(+)-triggered ESCRT assembly and regulation of cell membrane repair. Nat Commun. 2014. Dec 23;5(1):5646. doi: 10.1038/ncomms6646. PubMed PMID: 25534348; PubMed Central PMCID: PMCPMC4333728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jia J, Claude-Taupin A, Gu Y, Choi SW, Peters R, Bissa B, Mudd MH, Allers L, Pallikkuth S, Lidke KA. Galectin-3 coordinates a cellular system for lysosomal repair and removal. Dev Cell. 2020. Jan 6;52(1):69–87e8. doi: 10.1016/j.devcel.2019.10.025. PubMed PMID: 31813797; PubMed Central PMCID: PMCPMC6997950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johannes L, Jacob R, Leffler H. Galectins at a glance. J Cell Sci. 2018. May 1;131(9). doi: 10.1242/jcs.208884. PubMed PMID: 29717004. [DOI] [PubMed] [Google Scholar]
- 50.Kumar S, Chauhan S, Jain A, Ponpuak M, Choi SW, Mudd M, Peters R, Mandell MA, Johansen T, Deretic V, et al. Galectins and TRIMs directly interact and orchestrate autophagic response to endomembrane damage. Autophagy. 2017. Jun 3;13(6):1086–1087. doi: 10.1080/15548627.2017.1307487. PubMed PMID: 28368693; PubMed Central PMCID: PMCPMC5486367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weng IC, Chen HL, Lo TH, Lin W-H, Chen H-Y, Hsu DK, Liu F-T. Cytosolic galectin-3 and −8 regulate antibacterial autophagy through differential recognition of host glycans on damaged phagosomes. Glycobiology. 2018. Jun 1;28(6):392–405. doi: 10.1093/glycob/cwy017. PubMed PMID: 29800364. [DOI] [PubMed] [Google Scholar]
- 52.Roy D, Liston DR, Idone VJ, Di A, Nelson DJ, Pujol C, Bliska JB, Chakrabarti S, Andrews NW. A process for controlling intracellular bacterial infections induced by membrane injury. Science. 2004. Jun 4;304(5676):1515–1518. doi: 10.1126/science.1098371. PubMed PMID: 15178804. [DOI] [PubMed] [Google Scholar]
- 53.Allert S, Forster TM, Svensson C-M, Richardson JP, Pawlik T, Hebecker B, Rudolphi S, Juraschitz M, Schaller M, Blagojevic M. Candida albicans-Induced epithelial damage mediates translocation through intestinal barriers. mBio. 2018. Jun 5;9(3). doi: 10.1128/mBio.00915-18. PubMed PMID: 29871918; PubMed Central PMCID: PMCPMC5989070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moyes DL, Wilson D, Richardson JP, Mogavero S, Tang SX, Wernecke J, Höfs S, Gratacap RL, Robbins J, Runglall M, et al. Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature. 2016. Apr 7;532(7597):64–68. doi: 10.1038/nature17625. PubMed PMID: 27027296; PubMed Central PMCID: PMCPMC4851236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Benjamin JL, Sumpter R Jr., Levine B, Hooper L. Intestinal epithelial autophagy is essential for host defense against invasive bacteria. Cell Host Microbe. 2013. Jun 12;13(6):723–734. doi: 10.1016/j.chom.2013.05.004. PubMed PMID: 23768496; PubMed Central PMCID: PMCPMC3755484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bretin A, Carriere J, Dalmasso G, Bergougnoux A, B’chir W, Maurin A-C, Müller S, Seibold F, Barnich N, Bruhat A. Activation of the EIF2AK4-EIF2A/eIF2α-ATF4 pathway triggers autophagy response to Crohn disease-associated adherent-invasive Escherichia coli infection. Autophagy. 2016. May 3;12(5):770–783. doi: 10.1080/15548627.2016.1156823. PubMed PMID: 26986695; PubMed Central PMCID: PMCPMC4854551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Conway KL, Kuballa P, Song JH, Patel KK, Castoreno AB, Yilmaz OH, Jijon HB, Zhang M, Aldrich LN, Villablanca EJ, et al. Atg16l1 is required for autophagy in intestinal epithelial cells and protection of mice from Salmonella infection. Gastroenterology. 2013. Dec;145(6):1347–1357. doi: 10.1053/j.gastro.2013.08.035. PubMed PMID: 23973919; PubMed Central PMCID: PMCPMC3840157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lapaquette P, Bringer MA, Darfeuille-Michaud A. Defects in autophagy favour adherent-invasive Escherichia coli persistence within macrophages leading to increased pro-inflammatory response. Cell Microbiol. 2012. Jun;14(6):791–807. doi: 10.1111/j.1462-5822.2012.01768.x. PubMed PMID: 22309232. [DOI] [PubMed] [Google Scholar]
- 59.Munoz-Sanchez S, van der Vaart M, Meijer AH. Autophagy and Lc3-Associated phagocytosis in zebrafish models of bacterial infections. Cells. 2020. Oct 29;9(11):2372. doi: 10.3390/cells9112372. PubMed PMID: 33138004; PubMed Central PMCID: PMCPMC7694021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Andrews NW, Almeida PE, Corrotte M. Damage control: cellular mechanisms of plasma membrane repair. Trends Cell Biol. 2014. Dec;24(12):734–742. doi: 10.1016/j.tcb.2014.07.008. PubMed PMID: 25150593; PubMed Central PMCID: PMCPMC4252702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tam JM, Mansour MK, Khan NS, Seward M, Puranam S, Tanne A, Sokolovska A, Becker CE, Acharya M, Baird MA, et al. Dectin-1-dependent LC3 recruitment to phagosomes enhances fungicidal activity in macrophages. J Infect Dis. 2014. Dec 1;210(11):1844–1854. doi: 10.1093/infdis/jiu290. PubMed PMID: 24842831; PubMed Central PMCID: PMCPMC4271056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Agop-Nersesian C, Niklaus L, Wacker R, Theo Heussler V. Host cell cytosolic immune response during Plasmodium liver stage development. FEMS Microbiol Rev. 2018. May 1;42(3):324–334. doi: 10.1093/femsre/fuy007. PubMed PMID: 29529207; PubMed Central PMCID: PMCPMC5995216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Andrade RM, Wessendarp M, Gubbels MJ, Striepen B, Subauste CS. CD40 induces macrophage anti-Toxoplasma gondii activity by triggering autophagy-dependent fusion of pathogen-containing vacuoles and lysosomes. J Clin Invest. 2006. Sep;116(9):2366–2377. doi: 10.1172/JCI28796. PubMed PMID: 16955139; PubMed Central PMCID: PMCPMC1555650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cyrino LT, Araujo AP, Joazeiro PP. In vivo and in vitro Leishmania amazonensis infection induces autophagy in macrophages. Tissue Cell. 2012. Dec;44(6):401–408. doi: 10.1016/j.tice.2012.08.003. PubMed PMID: 22939777. [DOI] [PubMed] [Google Scholar]
- 65.Evans RJ, Sundaramurthy V, Frickel EM. The interplay of host autophagy and eukaryotic pathogens. Front Cell Dev Biol. 2018;6:118. doi: 10.3389/fcell.2018.00118. PubMed PMID: 30271774; PubMed Central PMCID: PMCPMC6146372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Heckmann BL, Green DR. LC3-associated phagocytosis at a glance. J Cell Sci. 2019. Feb 20;132(5). doi: 10.1242/jcs.222984. PubMed PMID: 30787029; PubMed Central PMCID: PMCPMC6432721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moreau K, Ravikumar B, Renna M, Puri, C, and Rubinsztein, DC. Autophagosome precursor maturation requires homotypic fusion. Cell. 2011. Jul 22;146(2):303–317. doi: 10.1016/j.cell.2011.06.023. PubMed PMID: 21784250; PubMed Central PMCID: PMCPMC3171170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Galluzzi L, Green DR. Autophagy-Independent functions of the autophagy machinery. Cell. 2019. Jun 13;177(7):1682–1699. doi: 10.1016/j.cell.2019.05.026. PubMed PMID: 31199916; PubMed Central PMCID: PMCPMC7173070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nieto-Torres JL, Leidal AM, Debnath J, and Hansen, M. Beyond autophagy: the expanding roles of ATG8 proteins. Trends Biochem Sci. 2021. Feb 5. doi: 10.1016/j.tibs.2021.01.004. PubMed PMID: 33558127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Le Roux AL, Quiroga X, Walani N, Arroyo, M, and Roca-Cusachs, P. The plasma membrane as a mechanochemical transducer. Philos Trans R Soc Lond B Biol Sci. 2019. Aug 19;374(1779):20180221. doi: 10.1098/rstb.2018.0221. PubMed PMID: 31431176; PubMed Central PMCID: PMCPMC6627014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Boukhalfa A, Nascimbeni AC, Ramel D, Dupont, N, Hirsch, E, Gayral, S, Laffargue, M, Codogno, P, and Morel, E. PI3KC2alpha-dependent and VPS34-independent generation of PI3P controls primary cilium-mediated autophagy in response to shear stress. Nat Commun. 2020. Jan 15;11(1):294. doi: 10.1038/s41467-019-14086-1. PubMed PMID: 31941925; PubMed Central PMCID: PMCPMC6962367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.DeSelm CJ, Miller BC, Zou W, Beatty, WL, van Meel, E, Takahata, Y, Klumperman, J, Tooze, SA, Teitelbaum, SL, and Virgin, HW, et al. Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Dev Cell. 2011. Nov 15;21(5):966–974. doi: 10.1016/j.devcel.2011.08.016. PubMed PMID: 22055344; PubMed Central PMCID: PMCPMC3244473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Verlhac P, Gregoire IP, Azocar O, Petkova, DS, Baguet, J, Viret, C, and Faure, M. Autophagy receptor NDP52 regulates pathogen-containing autophagosome maturation. Cell Host Microbe. 2015. Apr 8;17(4):515–525. doi: 10.1016/j.chom.2015.02.008. PubMed PMID: 25771791. [DOI] [PubMed] [Google Scholar]
- 74.Renna M, Schaffner C, Winslow AR, Menzies, FM, Peden, AA, Floto, RA, and Rubinsztein, DC. Autophagic substrate clearance requires activity of the syntaxin-5 SNARE complex. J Cell Sci. 2011. Feb 1;124(Pt3):469–482. doi: 10.1242/jcs.076489. PubMed PMID: 21242315; PubMed Central PMCID: PMCPMC3022001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Begun J, Lassen KG, Jijon HB, Baxt, LA, Goel, G, Heath, RJ, Ng, A, Tam, JM, Kuo, SY, and Villablanca, EJ. et al, et al. Integrated genomics of Crohn’s Disease risk variant identifies a role for CLEC12A in antibacterial autophagy. Cell Rep. 2015. Jun 30;11(12):1905–1918. doi: 10.1016/j.celrep.2015.05.045. PubMed PMID: 26095365; PubMed Central PMCID: PMCPMC4507440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Taisne C, Lussignol M, Hernandez E, Moris, A, Mouna, L, and Esclatine, A. Human cytomegalovirus hijacks the autophagic machinery and LC3 homologs in order to optimize cytoplasmic envelopment of mature infectious particles. Sci Rep. 2019. Mar 14;9(1):4560. doi: 10.1038/s41598-019-41029-z. PubMed PMID: 30872707; PubMed Central PMCID: PMCPMC6418312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hosokawa N, Hara Y, Mizushima N. Generation of cell lines with tetracycline-regulated autophagy and a role for autophagy in controlling cell size. FEBS Lett. 2006. May 15;580(11):2623–2629. doi: 10.1016/j.febslet.2006.04.008. PubMed PMID: 16647067. [DOI] [PubMed] [Google Scholar]
- 78.Scrivo A, Codogno P, Bomont P. Gigaxonin E3 ligase governs ATG16L1 turnover to control autophagosome production. Nat Commun. 2019. Feb 15;10(1):780. doi: 10.1038/s41467-019-08331-w. PubMed PMID: 30770803; PubMed Central PMCID: PMCPMC6377711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Coulon PG, Richetta C, Rouers A, Blanchet, FP, Urrutia, A, Guerbois, M, Piguet, V, Theodorou, I, Bet, A, and Schwartz, O. et al. HIV-Infected dendritic cells present endogenous MHC Class II-Restricted antigens to HIV-Specific CD4+ T Cells. J Immunol. 2016. Jul 15;197(2):517–532. doi: 10.4049/jimmunol.1600286. PubMed PMID: 27288536. [DOI] [PubMed] [Google Scholar]
- 80.Maejima I, Takahashi A, Omori H, Kimura, T, Takabatake, Y, Saitoh, T, Yamamoto, A, Hamasaki, M, Noda, T, and Isaka , Y. et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 2013. Aug 28;32(17):2336–2347. doi: 10.1038/emboj.2013.171. PubMed PMID: 23921551; PubMed Central PMCID: PMCPMC3770333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kuma A, Hatano M, Matsui M, Yamamoto, A, Nakaya, H, Yoshimori, T, Ohsumi, Y, Tokuhisa, T, and Mizushima, N. The role of autophagy during the early neonatal starvation period. Nature. 2004. Dec 23;432(7020):1032–1036. doi: 10.1038/nature03029. PubMed PMID: 15525940. [DOI] [PubMed] [Google Scholar]
- 82.Mizushima N, Yamamoto A, Matsui M, Yoshimori, T, and Ohsumi, Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004. Mar;15(3):1101–1111. doi: 10.1091/mbc.e03-09-0704. PubMed PMID: 14699058; PubMed Central PMCID: PMCPMC363084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nascimbeni AC, Codogno P, Morel E. Local detection of PtdIns3P at autophagosome biogenesis membrane platforms. Autophagy. 2017. Sep 2;13(9):1602–1612. doi: 10.1080/15548627.2017.1341465. PubMed PMID: 28813193; PubMed Central PMCID: PMCPMC5612047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request.