

Abstract
Protein kinase A (PKA) is a central regulator of cardiac performance and morphology. Myocardial PKA activation is induced by a variety of hormones, neurotransmitters, and stress signals, most notably catecholamines secreted by the sympathetic nervous system. Catecholamines bind β-adrenergic receptors to stimulate cAMP-dependent PKA activation in cardiomyocytes. Elevated PKA activity enhances Ca2+ cycling and increases cardiac muscle contractility. Dynamic control of PKA is essential for cardiac homeostasis, as dysregulation of PKA signalling is associated with a broad range of heart diseases. Specifically, abnormal PKA activation or inactivation contributes to the pathogenesis of myocardial ischaemia, hypertrophy, heart failure, as well as diabetic, takotsubo, or anthracycline cardiomyopathies. PKA may also determine sex-dependent differences in contractile function and heart disease predisposition. Here, we describe the recent advances regarding the roles of PKA in cardiac physiology and pathology, highlighting previous study limitations and future research directions. Moreover, we discuss the therapeutic strategies and molecular mechanisms associated with cardiac PKA biology. In summary, PKA could serve as a promising drug target for cardioprotection. Depending on disease types and mechanisms, therapeutic intervention may require either inhibition or activation of PKA. Therefore, specific PKA inhibitors or activators may represent valuable drug candidates for the treatment of heart diseases.
Keywords: Norepinephrine, Isoproterenol, Adenylyl cyclase, Phosphodiesterase, AKAP
1. Introduction of the PKA signalling system
Protein kinases play fundamental roles in nearly every aspect of cell biology and physiology, and thus their malfunctions are frequently associated with diseases. The 3',5'-cyclic adenosine monophosphate (cAMP)-dependent protein kinase [protein kinase A (PKA)], first discovered in 1968,1 has been viewed as the prototype for all protein kinases.2 PKA belongs to the AGC [named after the representative members PKA, 3′,5′-cyclic guanosine monophosphate (cGMP)-dependent protein kinase (PKG) and protein kinase C (PKC)] kinase family, which contains >60 serine/threonine protein kinases. PKA is evolutionarily conserved from fungi to humans and is ubiquitously expressed in all mammalian cell types.2 The PKA holoenzyme, composed of two regulatory (R) and two catalytic (C) subunits, remains in its inactive state as a PKA tetramer (R2C2) in the absence of cAMP. The PKA regulatory (PKA-R) subunits have four isoforms, R1α, R1β, R2α, and R2β (encoded by PRKAR1A, PRKAR1B, PRKAR2A, and PRKAR2B, respectively), whereas three isoforms of the PKA catalytic (PKA-C) subunits have been identified, Cα, Cβ, and Cγ (encoded by PRKACA, PRKACB, and PRKACG, respectively).3 These isoforms differ in their expression patterns and levels and may exhibit alternative splicing. Depending on the R subunit, PKA holoenzyme is divided into type I and II PKA, which bind to R1 and R2, respectively. In general, R1α and Cα1 are the most abundant and ubiquitously expressed PKA-R and PKA-C subunits, respectively.3
The canonical PKA signalling pathway is essential for the cardiac actions of many hormones and neurotransmitters, particularly the catecholamines including norepinephrine secreted by cardiac sympathetic nerve terminals, and epinephrine released by the adrenal medulla (Figure 1).4 Catecholamines bind the transmembrane β-adrenergic receptors (β-ARs), major G-protein-coupled receptors in the heart, leading to release of the stimulatory G-protein α subunit (Gαs) inside the target cells. Gαs then binds and activates adenylyl cyclases (ACs), which convert ATP into cAMP, resulting in a rapid increase in intracellular cAMP levels. The cAMP molecules bind the PKA-R subunits to induce dissociation of the tetrameric PKA holoenzyme, leading to release of the free PKA-C subunits and subsequent PKA activation. PKA phosphorylates a plethora of substrates to regulate cellular activities. It is worth noting that holoenzyme dissociation may not be necessary for PKA activation under certain circumstances.5 PKA can also be activated through non-canonical pathways in a cAMP-independent manner (Figure 1), by a variety of stimuli including reactive oxygen species (ROS),6–8 lipopolysaccharides (LPS)/interleukin-1 (IL-1),9 endothelin-1 (ET-1)/angiotensin II (Ang II),10 transforming growth factor-β (TGF-β),11 sphingosine,12 and peroxynitrite.13 For example, R1α can serve as a redox sensor and undergo oxidant-induced protein degradation, leading to PKA holoenzyme dissociation and kinase activation.6–8 In addition, the PKA-C subunits are also sequestered by the inhibitor of κB (I-κB) proteins within the I-κB-PKA-C complex, and can be activated following I-κB degradation.9,10
Figure 1.

PKA signalling pathway. The canonical cAMP-dependent PKA pathway is initiated by catecholamines, which stimulate β-adrenergic receptor (β-AR) to induce adenylyl cyclase (AC)-mediated cAMP synthesis. The second messenger cAMP then binds the PKA regulatory (PKA-R) subunits, leading to release of the PKA catalytic (PKA-C) subunits and subsequent kinase activation. The non-canonical pathways of PKA activation are cAMP-independent. For example, oxidative stress or TGF-β induces PKA activation through blocking PKA-R-mediated sequestration of PKA-C, whereas ET-1, Ang II, LPS, or IL-1 derepresses I-κB-mediated inhibition of PKA-C. On the other hand, cAMP also exerts its biological functions through direct binding with other effectors beyond PKA, including Epac, PRKX, PRKY, CNG ion channels, HCN ion channels, and POPDC proteins. Ang II, angiotensin II; cAMP, 3',5'-cyclic adenosine monophosphate; CNG, cyclic nucleotide-gated; Epac, exchange protein directly activated by cAMP; ET-1, endothelin-1; HCN, hyperpolarization-activated cyclic nucleotide-gated; I-κB, inhibitor of κB; IL-1, interleukin-1; LPS, lipopolysaccharides; PKA, cAMP-dependent protein kinase, or protein kinase A; PDE, cyclic nucleotide phosphodiesterase; PKI, a PKA inhibitor; POPDC, popeye domain containing; PRKX/Y, protein kinase X/Y-linked; TGF-β, transforming growth factor-β. Arrows indicate activation; bar-headed lines indicate inhibition.
Cellular cAMP levels are determined by the balance between the enzymatic activities of ACs and cyclic nucleotide phosphodiesterases (PDEs). In contrast to ACs, PDEs hydrolyze cAMP to 5′-AMP, thereby reducing cAMP levels and resulting in a decrease in PKA activity (Figure 1). In addition to cAMP, some PDEs also hydrolyze cGMP.14 PKA activity is inhibited by the endogenous PKI protein, a pseudosubstrate of PKA that is capable of sequestering and inactivating the PKA-C subunits.2 Unlike PKA-R, the interaction between PKI and PKA-C is not modulated by the levels of cAMP. A-kinase anchoring proteins (AKAPs) tether PKA holoenzyme to certain subcellular locations, allowing for rapid and precise control of its substrates within the specific compartments.15 For example, short AKAP7 (also known as AKAP15) anchors PKA at the plasma membrane to regulate L-type Ca2+ channel (LTCC, Cav1.2) activity, whereas muscle AKAP (mAKAP, AKAP6) and long AKAP7 (AKAP18) tether PKA at the sarcoplasmic reticulum (SR) to modulate the functions of ryanodine receptors (RyRs) and sarco/endoplasmic reticulum Ca2+-ATPase (SERCA), respectively.15
PKA is a key regulator of cardiac function, structure, and remodelling. Recent studies have uncovered novel roles of PKA in normal and diseased heart, and have provided new insights on the underlying mechanisms. Here, we review our current knowledge of PKA in cardiac physiology and pathophysiology.
2. PKA and cardiac contraction/relaxation
Cardiac muscle contraction and relaxation determine systolic and diastolic functions of the heart. Contraction is initiated by electrical excitation (i.e. membrane depolarization through the action potential), a process termed excitation–contraction coupling.16 Depolarization of the sarcolemma induces cellular Ca2+ entry through LTCC. Elevated cytosolic Ca2+ level triggers Ca2+ release from SR through RyR2 channel, further raising intracellular Ca2+ concentration. Ca2+ binds to cardiac troponin C (cTnC), relieves cardiac troponin I (cTnI)-mediated inhibition of actin-myosin filament interaction, leading to contraction. During cardiac relaxation, Ca2+ is dissociated from cTnC, transported back into the SR via SERCA, and to the extracellular space via the Na+/Ca2+ exchanger (NCX) as well as the sarcolemmal Ca2+-ATPase.
The force (inotropy), rate (chronotropy) of cardiac contraction, and the ability of relaxation (lusitropy) are modulated by the sympathetic (or adrenergic) nervous system through secretion of catecholamines. In the fight-or-flight response, rapid release of catecholamines markedly increases heart rate and contractility through β-AR-dependent activation of the cAMP/PKA signalling pathway. Particularly, catecholamines may bind β-ARs at the sarcolemma and the SR to induce local PKA activation.17 PKA, in turn, phosphorylates target proteins involved in the regulation of contraction/relaxation, including Cav1.2, RyR2, phospholamban (PLN), cTnI, and cardiac myosin-binding protein C (cMyBP-C).16
PKA positively regulates inotropy through phosphorylation of Cav1.2, PLN, and cMyBP-C. Phosphorylation of Cav1.2 or PLN increases Ca2+ availability, whereas phosphorylation of cMyBP-C improves responsiveness of the contractile apparatus to Ca2+. In response to β-adrenergic stimulation, PKA phosphorylates Cav1.2 at S1700, resulting in increased LTCC activity and Ca2+ influx, thereby enhancing contraction.18 The above model is supported by studies in the knock-in mice expressing a non-phosphorylatable mutant of endogenous Cav1.2 (S1700A), which show decreased basal and β-adrenergic-stimulated calcium currents, as well as reduced cardiomyocyte contractility.18 Surprisingly, overexpression of the same Cav1.2 mutant (S1700A) using a transgenic approach does not interfere with Ca2+ current, arguing against a role for S1700 phosphorylation.19 A most recent study further reveals that PKA augments Cav1.2 channel activity indirectly, through phosphorylation and depletion of the Cav1.2 inhibitor Rad.20 Despite the ongoing debate about the mechanisms, there is consensus that LTCC is essential for PKA-dependent Ca2+ influx and cardiac contraction (Table 1). PKA-mediated phosphorylation of PLN at S16 relieves PLN-dependent inhibition of SERCA, resulting in increased SR Ca2+ uptake from the cytosol and consequently increased SR Ca2+ load.25 Therefore, higher levels of Ca2+ can be released from SR for binding to the contractile proteins, leading to more forceful contraction. The physiological significance of PKA-dependent RyR2 phosphorylation has been controversial. Although there is evidence that phosphorylation of RyR2 at S2808 by PKA is necessary for β-adrenergic-induced SR Ca2+ release and cardiac contraction, contradictory findings have been reported.30,31 The discrepancy may be due to the fact that either maximal or minimal RyR2 phosphorylation by PKA favours SR Ca2+ leak.32 Phosphorylation of cMyBP-C at S273, S282, or S302 by multiple kinases including PKA increases myocardial contractility through weakening cMyBP-C-mediated inhibition of myosin,34 increasing force-producing myosin heads,35 or accelerating cross-bridge cycling,36 respectively. Thus PKA-mediated cMyBP-C phosphorylation is also a key regulator of cardiac inotropy.
Table 1.
Myocardial PKA substrates and their biological functions
| Substrate | Phosphorylation site(s) | Molecular/cellular function | Physiological/pathological function |
|---|---|---|---|
| Cav1.2 | Ser1700 | Cav1.2 activation, Ca2+ influx, Ca2+ overload, necrosis | Inotropy,18,19 chronotropy,21,22 heart failure23,24 |
| Rad | Ser25, Ser38, Ser272, Ser300 | Cav1.2 activation, Ca2+ influx | Inotropy20 |
| PLN | Ser16 | SERCA activation, SR Ca2+ uptake | Inotropy,25 chronotropy,21,22,26,27 lusitropy,25 heart failure28,29 |
| RyR2 | Ser2808 | SR Ca2+ release, Ca2+ overload, necrosis | Inotropy,30–32 heart failure28,29,33 |
| cMyBP-C | Ser273, Ser282, Ser302 | Myosin activation, actin-myosin cross-bridge detachment | Inotropy,34–36 lusitropy37,38 |
| cTnI | Ser23, Ser24 | Ca2+-cTnC dissociation | Lusitropy39–41 |
| CcO | CcO activity↓, ROS generation, necrosis | I/R injury42 | |
| HSP20 | Ser16 | Autophagy↑, apoptosis↓, necrosis↓ | I/R injury↓43 |
| eNOS | Ser633, Ser1177 | eNOS activation | I/R injury↓44,45 |
| CREB | Ser133 | CREB-mediated transcription of genes involved in hypertrophy (foetal genes) and apoptosis (ICER,46,47 Bim,48 Bcl-249) | Hypertrophy,49,50 I/R injury→51 |
| GSK-3β | Ser9 | NFAT-mediated transcription of hypertrophic genes↑ | Hypertrophy52,53 |
| NFAT | Ser245, Ser269, Ser294 | NFAT-mediated transcription of hypertrophic genes↓ | Hypertrophy↓54–56 |
| Drp1 | Ser637 | Mitochondrial fission↓ | Hypertrophy↓57 |
| HDAC5 | Ser279 | MEF2-mediated transcription of hypertrophic genes↓ | Hypertrophy↓58,59 |
| HDAC4 | MEF2-mediated transcription of hypertrophic genes↓ | Hypertrophy↓60 | |
| Titin, N2B element | Ser4185, Ser4010 |
Titin compliance↑, cardiomyocyte stiffness↓ |
Diastolic function↑, HFpEF↓61–64 |
| RPN6 | Ser14 | Proteasome activation, clearance of misfolded proteins↑ | Proteinopathy-related HFpEF↓65 |
CcO, cytochrome c oxidase; cMyBP-C, cardiac myosin-binding protein C; CREB, cAMP-response element binding protein; cTnC, cardiac troponin C; cTnI, cardiac troponin I; eNOS, endothelial nitric oxide synthase; GSK-3β, glycogen synthase kinase-3β; HDAC4/5, histone deacetylase 4/5; HFpEF, heart failure with preserved ejection fraction; HSP20, heat shock protein 20; ICER, inducible cAMP early repressor; I/R, ischaemia/reperfusion; MEF2, myocyte enhancer factor 2; NFAT, nuclear factor of activated T cells; PLN, phospholamban; ROS, reactive oxygen species; RyR2, ryanodine receptor 2; SERCA, sarco/endoplasmic reticulum Ca2+-ATPase; SR, sarcoplasmic reticulum; ↑, increase; ↓, decrease; →, no change.
PKA drives cardiac chronotropy primarily through phosphorylation of SR Ca2+ cycling proteins in cardiac pacemaker cells (i.e. sinoatrial nodal cells, a group of specialized cardiac myocytes).21 Pacemaker cells are unique in their high basal cAMP level and PKA activity, which are necessary and sufficient for the generation of rhythmic internal Ca2+ store oscillations and spontaneous beating, even in the absence of β-adrenergic stimulation. In response to β-AR stimulation, however, PKA-mediated phosphorylation of PLN at S16 speeds up SR Ca2+ uptake and contributes to the acceleration of heart rate.21 PKA-dependent generation of rhythmic action potentials also requires coupling between spontaneous local Ca2+ release and the membrane ion channels including NCX and hyperpolarization-activated cyclic nucleotide-gated (HCN) channels.66 Cardiac-specific overexpression of AC8 increases AC activity in the sinoatrial node tissue, which is accompanied by a marked increase in heart rate.67 Pharmacological inhibition of PDE3A and PDE4B, the major PDE subtypes expressed in pacemaker cells, synergistically increase the spontaneous beating rate.26 PDE2 inhibition with Bay 60–7550 also accelerates heart rate.22 Conversely, cardiac-specific PDE2 overexpression results in a decrease in phospho-PLN(S16) level, which is associated with reduced resting and maximal heart rate.22 Cardiac-specific PKI transgenic mice show lower PKA activity, reduced PLN phosphorylation at S16, and slower heart rate following β-AR stimulation.27 Intriguingly, basal contractility is enhanced in the transgenic heart overexpressing either PDE2 or PKI, likely due to reduced SR Ca2+ leak, increased myocyte Ca2+ transient and enhanced myofilament Ca2+ sensitivity.22,27 Aberrant PKA activation can cause Ca2+ cycling protein dysfunction and lead to cardiac arrhythmias. For example, overexpression of PDE2 decreases arrhythmia occurrence following myocardial infarction,22 but PDE4D gene deficiency induces PKA-dependent RyR2 hyperphosphorylation, which enhances SR Ca2+ leak and promotes arrhythmias.68
PKA enhances cardiac lusitropy through phosphorylation of PLN, cTnI, and cMyBP-C. Myocardial relaxation depends on efficient cytosolic Ca2+ removal. Phosphorylation of PLN by PKA accelerates SERCA-mediated transport of cytosolic Ca2+ into SR, resulting in faster relaxation.25 PKA-dependent phosphorylation of cTnI and cMyBP-C augments lusitropy primarily through reducing Ca2+ sensitivity. The N-terminal region of cTnI binds cTnC to increase its Ca2+ affinity, which determines myofibrillar Ca2+ sensitivity and contractile activation.39 Phosphorylation of cTnI at the N-terminal residues S23/24 by PKA weakens cTnI–cTnC interaction, thereby promoting dissociation of Ca2+ from cTnC and accelerating myofibril relaxation.40 Importantly, monophosphorylation at S24 is sufficient to reduce Ca2+ sensitivity.41 Phosphorylation of cMyBP-C also represents a major lusitropic mechanism during β-adrenergic stimulation. Concurrent non-phosphorylatable mutations of all PKA target sites on cMyBP-C (S273A, S282A, and S302A) impair diastolic function without altering intracellular Ca2+ handling, suggesting that cMyBP-C phosphorylation enhances relaxation likely through increasing the rates of actin-myosin cross-bridge detachment.37,38 Conversely, phosphomimetic mutations of all three serine residues of cMyBP-C (S273D, S282D, and S302D) increase peak myocardial relaxation velocity, indicating enhanced lusitropy.37 However, the specific role of each individual site remains to be determined.
Although required by the positive inotropic, chronotropic, and lusitropic effects of β-adrenergic stimulation, PKA activity is not necessary for the maintenance of basal heart function.27 Thus, pharmacological PKA inhibition might be a safe and viable strategy for clinical use. Since PKA inhibition with PKI preferentially reduces PKA activity in the SR and myofilament but not the sarcolemma and nuclei, it is possible that basal heart function is maintained by sarcolemmal or nuclear PKA. Nonetheless, long-term PKA inhibition reduces cardiac reserve and impairs exercise capability.27
In summary, PKA plays an essential role in neurohormonal regulation of cardiac function, through phosphorylation of its substrates including Cav1.2, RyR2, PLN, cTnI, and cMyBP-C. Although loss of PKA activity does not diminish cardiac function at resting conditions, it can limit the heart’s ability to cope with stress.
3. PKA and ischaemic heart disease
Ischaemic heart disease, also known as coronary artery disease, occurs when blood flow to the heart muscle is reduced or blocked, most commonly due to atherosclerosis. The biological functions of PKA in atherosclerosis pathogenesis have been described elsewhere.69 This review will summarize our current knowledge of PKA in myocardial ischaemia, with a particular focus on cardiac myocytes. Myocardial ischaemia is characterized by tissue hypoxia (i.e. oxygen deprivation due to insufficient blood supply). Restoration of blood supply by reperfusion results in tissue re-oxygenation but paradoxically also causes ischaemia/reperfusion (I/R) injury. During myocardial ischaemia, interstitial catecholamine concentration dramatically increases by 100–1000 fold (Figure 2),70,71 due to hyperactivation of the sympathetic nervous system.72 Accordingly, both cAMP level73,74 and PKA activity75,76 are increased in the ischaemic myocardium. Following reperfusion, myocardial catecholamine level rapidly declines and returns to baseline within 120 min.70,71 However, cardiac PKA activity remains elevated after reperfusion,76,77 due at least in part to R1α loss as a result of oxidation and dimerization.8,77
Figure 2.
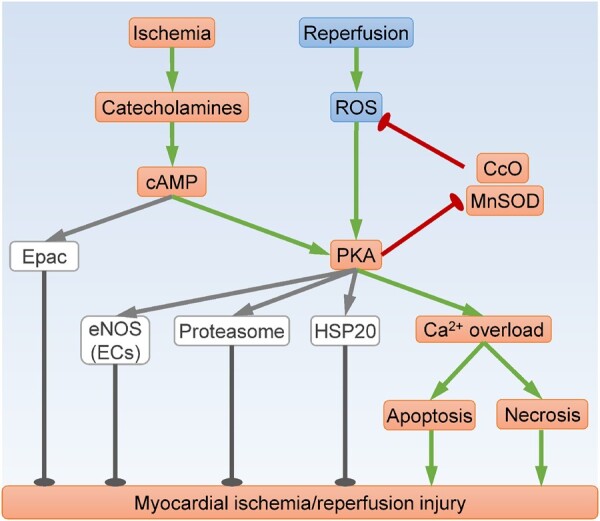
PKA signalling in myocardial ischaemia/reperfusion injury. Both ischaemia and reperfusion induce PKA activation, which can contribute to oxidative stress and Ca2+ overload, leading to cardiac cell death and myocardial damage. However, cAMP-induced activation of Epac, eNOS, proteasome, and HSP20 appear to be cardioprotective. CcO, cytochrome c oxidase; ECs, endothelial cells; eNOS, endothelial nitric oxide synthase; Epac, exchange protein directly activated by cAMP; HSP20, heat shock protein 20; MnSOD, manganese superoxide dismutase; ROS, reactive oxygen species. Arrows indicate activation; bar-headed lines indicate inhibition.
At physiological levels, catecholamines mediate the fight-or-flight response to rapidly increase contractility and heart rate. At extremely high concentrations, however, catecholamines can cause myocardial tissue damage. Administration of a single, high dose of the synthetic catecholamine isoproterenol in rats induces PKA-dependent RyR2 hyperphosphorylation, leading to SR Ca2+ leakage and subsequent myocyte death via apoptosis or necrosis (Figure 2).33 Moreover, isoproterenol induces PKA-mediated phosphorylation of cAMP-response element binding protein (CREB) at S133, leading to enhanced binding of CREB with the cAMP-response element in the promoter region, and subsequent transcription of pro-apoptotic genes including inducible cAMP early repressor (ICER)46,47 and Bim.48 It is noteworthy that CREB deficiency has no impact on apoptosis following I/R injury,51 likely because CREB also mediates transcription of the anti-apoptotic gene Bcl-2 in certain context.49 In addition, acute β-adrenergic stimulation with isoproterenol in mice transiently increases myocyte membrane permeability, an important marker of necrosis.78 Intriguingly, acute catecholamine injury appears to be fully reversible.33,78 By contrast, sustained activation of the PKA substrate LTCC in mice results in cardiac dysfunction and premature death, which is exacerbated by isoproterenol infusion.23 Mechanistically, isoproterenol exposure augments LTCC activity and enhances Ca2+ influx, leading to Ca2+ overload and myocyte necrosis.23
Activation of β-AR/cAMP signalling during the ischaemic phase contributes to myocardial I/R injury. Overexpression of PDE3A1 reduces myocardial cAMP levels and attenuates I/R-induced myocyte apoptosis, possibly due to decreased ICER and increased Bcl-2 expression.79 Moreover, treatment with the β1-AR inhibitor CGP-20712A80 or the PKA inhibitor H89/PKI 42,81 diminishes I/R-induced myocardial necrosis. In the ischaemic heart, excessive cAMP induces PKA-dependent phosphorylation and inhibition of cytochrome c oxidase (CcO), resulting in augmented ROS production.42 In turn, oxidative stress prompts R1α loss to activate PKA,8,77 leading to further CcO inhibition and ROS generation.7 Therefore, a rapid rise in cAMP level may trigger a vicious cycle of oxidative stress through activation of PKA (Figure 2).
Reduced cAMP level during ischaemia, caused by ischaemic preconditioning, is associated with marked protection against myocardial I/R injury.73,74 Interestingly, ischaemic preconditioning initially increases cAMP level during intermittent ischaemia (i.e. the preconditioning phase), but later reduces cAMP accumulation during sustained ischaemia (i.e. the main ischaemia phase) possibly due to β-AR desensitization or compensatory activation of PDEs.73,74 Myocardial I/R injury is attenuated by pre-ischaemic cAMP up-regulation using isoproterenol (a β-AR agonist), forskolin (an AC agonist),74,81–83 milrinone/olprinone/amrinone (PDE3 inhibitors),84,85 trapidil (a non-selective PDE inhibitor),86 or dibutyryl-cAMP (a cell-permeable cAMP analogue).75,84 Germline ablation of PDE3B increases cardiac cAMP level at baseline, reduces infarct size, and improves cardiac function following I/R.87 The cardioprotective effect of PDE3B ablation is blocked by treatment with KT5720, a PKA inhibitor.87 Intramyocardial injection of PKA-Cα siRNAs before ischaemia increases infarct size and impairs systolic function after I/R, possibly due to NF-κB activation and superoxide production.88 Together these studies suggest that pre-ischaemic cAMP/PKA activation represses PKA activity during ischaemia, thereby leading to cardioprotection.
Emerging evidence suggests that PKA could be a better drug target than β-AR, because PKI is more effective than the β1-AR blocker metoprolol in alleviating myocyte apoptosis and cardiac dysfunction following myocardial infarction.89 Mechanistically, β-AR and cAMP induce PKA activation to cause cardiotoxicity but stimulate exchange protein directly activated by cAMP (Epac) to mediate cardioprotection (Figure 2).89 In this regard, cardiac cell death following simulated I/R is exaggerated by inhibition of type 10 soluble AC (sAC) with gene silencing, KH7 treatment, or bicarbonate withdrawal, but mitigated by overexpression of sAC or treatment with the PDE2 inhibitor Bay 60–7550.90 Moreover, incubation with the cAMP inhibitor Rp-cAMP abolishes glucagon-like peptide 1 (GLP-1)-induced protection against I/R Injury.91 Interestingly, the beneficial effects of GLP-1 are mediated by sAC-dependent cAMP/PKA activation, and subsequent endothelial nitric oxide synthase (eNOS) phosphorylation.44,45 PKA has also been shown to protect against I/R injury through proteasome activation,92 as well as phosphorylation of heat shock protein 20.43 Therefore, although believed to be detrimental during myocardial I/R injury, cAMP and PKA may also activate certain protective molecules in some contexts.
In summary, the majority of studies suggest that activation of PKA during ischaemia contributes to I/R injury through aggravating cardiomyocyte apoptosis and/or necrosis. Ischaemic preconditioning reduces PKA activity during ischaemia and thus attenuates I/R injury. Notably, elevated cAMP levels during ischaemia may also activate protective mechanisms in a PKA-dependent or independent manner.
4. PKA and cardiac hypertrophy
It is well established that chronic stimulation of β1-AR induces cardiac hypertrophy.4 However, whether PKA contributes to β1-AR-mediated hypertrophy remains elusive. Earlier studies reveal that cardiac-specific overexpression of β1-AR,93 Gαs,94 or PKA-Cα,28 markedly increases cardiomyocyte size, but only moderately increases heart weight due to concurrent apoptosis or necrosis. Notably, β-adrenergic stimulation is sufficient to cause apoptosis in adult cardiomyocytes, in a PKA-dependent manner.95 Overexpression of type 5 AC (AC5), a major cardiac isoform, exacerbates apoptosis and only marginally increases cardiomyocyte size following chronic infusion of isoproterenol.96 Mechanistically, AC5-mediated cAMP/PKA activation augments oxidative stress through repression of manganese superoxide dismutase (MnSOD) expression (Figure 2).96 Cardiac-specific transgenic expression of AC6, another major cardiac isoform, does not alter heart weight in mice with ischaemic cardiomyopathy.97 Moreover, overexpression of AC8 fails to increase heart weight, despite four-fold higher PKA activity.98 These studies suggest that PKA activation may primarily provoke myocyte loss, which can eventually lead to compensatory hypertrophy. Given the essential role of PKA in regulating cardiac contractility, it is not surprising that interfering with PKA-dependent phosphorylation can disrupt normal heart function, which may also result in hypertrophic remodelling to compensate for reduced cardiac output. Indeed, mice with non-phosphorylatable mutations of Cav1.2,18,24 PLN,99 cTnI,100 or cMyBP-C37,38 at their PKA sites exhibit cardiac dysfunction and hypertrophy. However, myocardial PKA inactivation by overexpression of PKI does not induce cardiac dysfunction or hypertrophy in mice up to 12 months of age.27 Germline deletion of AC5 in mice does not blunt physiological hypertrophy during postnatal heart development (up to 3–6 months of age),101 or attenuate pathological hypertrophy induced by pressure overload.102 Again, disruption of AC5 primarily suppresses apoptosis, due in part to up-regulation of the anti-oxidant protein MnSOD and the anti-apoptotic molecule Bcl-2.101,102 Collectively, the above in vivo studies do not support a major, direct role of the AC/cAMP/PKA signalling in hypertrophy.
Myocardial hypertrophic growth is not associated with a change in total PKA activity.103 However, in vitro real-time imaging reveals that hypertrophy is accompanied by relocation of PKA and PDE activity within myocytes, resulting in altered cAMP/PKA compartmentalization.104 Upon induction of hypertrophy, expression, and activity of several cAMP-hydrolyzing PDEs, including the pro-hypertrophic PDE1C,105 PDE2,54 and PDE10A106 are increased, whereas PDE3 and PDE4 are down-regulated.107,108 Interestingly, PDE3 or PDE4 appears to be anti-hypertrophic as PDE3/4 inhibition causes spontaneous hypertrophy,54 rendering myocytes insensitive to catecholamine stimulation in vitro54 or pressure overload in vivo.109 Mechanistically, PDE4 suppresses cardiomyocyte hypertrophy through inhibition of nuclear PKA activity.47,50 Since PDEs differ in their subcellular distribution,14 their opposing roles in hypertrophy could be due to differential modulation of PKA activity within specific subcellular compartments.
Indeed, nuclear PKA activation is associated with augmented cardiac hypertrophy (Figure 3). Overexpression of nuclear-targeted PKA-C increases cardiomyocyte size, whereas overexpression of cytosolic-targeted PKA-C enhances Ca2+ transients and cardiac contractile force without inducing hypertrophy.110 β-Adrenergic stimulation or pressure overload-induced hypertrophy requires sAC, which is known to mediate nuclear cAMP synthesis.111 Interestingly, activation of β1-AR increases nuclear PKA activity,47 and results in hypertrophy.93 By contrast, stimulation of β2-AR does not induce nuclear PKA activation,47 and fails to cause hypertrophy.112 Mechanistically, PKA phosphorylates CREB at S133 in the nuclei of cardiomyocytes, thereby initiating CREB-mediated transcription of hypertrophy-related genes.49,50 Since β1-AR-mediated nuclear PKA activation is abolished by PKI,47 cardiac-specific PKI transgenic mice are resistant to isoproterenol-induced CREB phosphorylation at S13327 and subsequent pathological hypertrophy.89 Moreover, overexpression of a non-phosphorylatable CREB mutant (S133A) attenuates isoproterenol-induced hypertrophy.46 Collectively, these studies suggest that CREB-mediated transcription is a critical pro-hypertrophic mechanism downstream of nuclear PKA. β-AR-mediated nuclear PKA activation requires the scaffold protein mAKAP.47 Interestingly, mAKAP facilitates calcineurin-dependent activation of the pro-hypertrophic transcription factor nuclear factor of activated T cells (NFAT), which binds GATA4 or myocyte enhancer factor 2 (MEF2) to stimulate expression of hypertrophy-related genes.113 Glycogen synthase kinase-3β (GSK-3β), which phosphorylates NFAT to trigger its nuclear export and inactivation,52 is repressed by PKA via PKA-dependent phosphorylation at S9.53 Therefore, nuclear PKA may promote hypertrophy in a synergistic manner with calcineurin, through relieving GSK-3β-mediated inhibition of NFAT (Figure 3). In line with these findings, global PKA-Cβ null mice are resistant to Ang II-induced cardiac hypertrophy.114 Intriguingly, overexpression of PKA-Cα in cardiac fibroblasts also induces ventricular dilation and cardiomyocyte hypertrophy, possibly via a paracrine mechanism.115 Taken together, PKA mediates cardiac hypertrophy while localized in the nuclei of cardiomyocytes or in cardiac fibroblasts.
Figure 3.
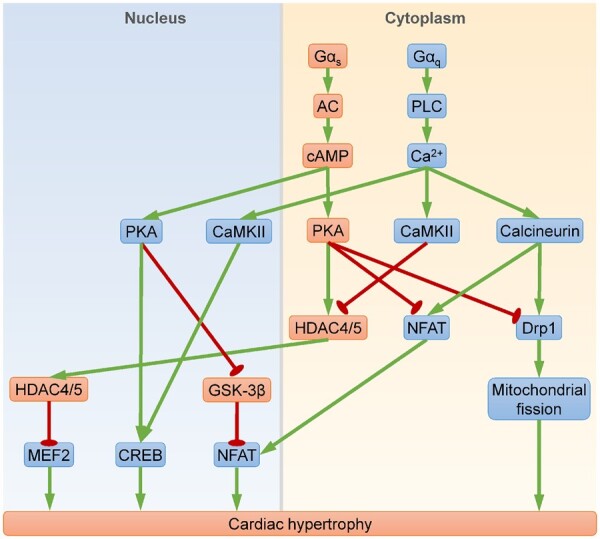
PKA signalling in cardiac hypertrophy. Key molecular events contributing to cardiac hypertrophy include MEF2-, CREB-, NFAT-mediated gene transcription, or Drp1-mediated mitochondrial fission. Gαs stimulates synthesis of the second messenger cAMP, which activates nuclear PKA to induce hypertrophy and cytoplasmic PKA to inhibit hypertrophy. Nuclear PKA promotes hypertrophy through: (i) activating CREB; and (ii) alleviating GSK-3β-mediated NFAT repression. Cytoplasmic PKA inhibits hypertrophy through: (i) invoking HDAC4/5-mediated MEF2 repression; (ii) inactivating NFAT; and (iii) suppressing Drp1. By contrast, Gαq acts through the second messenger Ca2+ to provoke Ca2+/CaMKII- and calcineurin-mediated hypertrophy. CaMKII activates MEF2 and CREB, whereas calcineurin activates NFAT and Drp1. Nuclear PKA may act cooperatively with CaMKII and calcineurin to induce hypertrophy, but cytoplasmic PKA antagonizes CaMKII- and calcineurin-dependent hypertrophy. AC, adenylyl cyclase; CaMKII, Ca2+/calmodulin-dependent protein kinase type II; CREB, cAMP-response element binding protein; GSK-3β, glycogen synthase kinase-3β; HDAC4/5, histone deacetylase 4/5; MEF2, myocyte enhancer factor 2; NFAT, nuclear factor of activated T cells; PLC, phospholipase C. Arrows indicate activation; bar-headed lines indicate inhibition.
In contrast, accumulating evidence suggests that cytoplasmic PKA may inhibit hypertrophy. In response to PDE2 inhibition, an increased local pool of cAMP induces PKA-dependent phosphorylation of NFAT at S245/S269/S294, resulting in cytoplasmic retention of NFAT and inhibition of hypertrophy.54 Cytoplasmic NFAT retention requires mitochondrial localization of PKA, as disruption of AKAP1 (also known as AKAP121, which targets PKA to mitochondria) reduces mitochondrial PKA activity, leading to augmented nuclear NFAT accumulation and aggravated hypertrophy both in vitro and in vivo.55,56 These results suggest that cytoplasmic PKA may directly repress NFAT (Figure 3). Since cytoplasmic PKA activity is restricted by R1α,116 we recently delete the R1α-encoding gene PRKAR1A in mouse heart to assess the impact of cytoplasmic PKA activation on physiological cardiac hypertrophy during development.57 Because homozygous PRKAR1A ablation inhibits cardiomyocyte proliferation and results in embryonic lethality,117 we use a heterozygous deletion approach and show that PRKAR1A deficiency suppresses hypertrophic heart growth, likely through inhibition of mitochondrial fission via PKA‐dependent Drp1 phosphorylation at S637 (Figure 3). Importantly, PRKAR1A mutations/deletions in humans are also associated with reduced left ventricular mass.57 In addition, germline PRKAR2B ablation attenuates age-related cardiac hypertrophy, possibly through a non-cardiomyocyte-autonomous mechanism because R2β is predominantly expressed in non-cardiac tissues.118
Although stimulation of either Gαs-coupled receptors (e.g. β1-AR) or Gαq-coupled receptors (e.g. α1-AR, Ang II type 1 receptor, ET A receptor) can lead to cardiomyocyte hypertrophy, Gαs signalling appears to antagonize Gαq-mediated hypertrophy. For example, treatment with forskolin (an AC agonist),57 or cAMP58 attenuates α1-AR-mediated hypertrophy in vitro. Moreover, Gαq-mediated cardiac foetal gene expression, a common hypertrophy marker, is blocked by isoproterenol, forskolin, or a selective PKA activator 8-CPT-6-Phe-cAMP, but augmented by a PKA inhibitor H89.119 Mechanistically, Gαs-dependent PKA activation induces histone deacetylase 5 (HDAC5) phosphorylation at S279 58,59 or dephosphorylation at S259/S498,120 resulting in translocation of HDAC5 from the cytoplasm to the nucleus, where it represses MEF2-mediated transcription of the hypertrophic gene program (Figure 3). PKA also induces nuclear import of HDAC4 and subsequent MEF2 repression.60 By contrast, Gαq stimulates phospholipase C to increase intracellular Ca2+ level, leading to activation of Ca2+/calmodulin-dependent protein kinase type II (CaMKII) and calcineurin (i.e. protein phosphatase 2B). Intriguingly, CaMKII induces HDAC4 nuclear export to provoke MEF2-mediated pathological cardiac hypertrophy.121 On the other hand, calcineurin dephosphorylates NFAT at S245/S269/S294 and Drp1 at S637, the same sites phosphorylated by cytoplasmic PKA.122,123 Therefore, cytoplasmic PKA may counteract CaMKII- and calcineurin-mediated hypertrophy (Figure 3). It is worth noting that CaMKII also phosphorylates CREB at S133,124 suggesting that CaMKII and nuclear PKA may promote CREB-dependent hypertrophy in a redundant manner.
In summary, PKA differentially modulates cardiac hypertrophy depending on its subcellular localization. Nuclear PKA induces cardiomyocyte hypertrophy through activation of CREB and NFAT-mediated transcription of hypertrophic genes, whereas cytoplasmic PKA inhibits hypertrophy via repression of MEF2 and NFAT-mediated transcription, as well as inhibition of Drp1-mediated mitochondrial fission. In cardiomyocytes, Ca2+ and cAMP are major second messengers downstream of Gαq and Gαs, respectively. Ca2+-dependent activation of CaMKII or calcineurin provokes hypertrophy, which is likely augmented by cAMP-dependent activation of nuclear PKA, but attenuated by cytoplasmic PKA. These points need to be further investigated with approaches that specifically target PKA in various intracellular compartments.
5. PKA and heart failure
Heart failure occurs when the heart is unable to supply adequate blood to meet the metabolic needs of the body. Decreased cardiac output induces compensatory activation of the sympathetic nervous system, which increases circulating catecholamines in an attempt to restore heart function. Although acute neurohormonal activation maintains cardiac output, chronic β-AR stimulation impairs myocardial β-AR responsiveness through receptor down-regulation (via β-arrestin-mediated internalization) and desensitization (via uncoupling from G-proteins).4 Decreased β-AR responsiveness blunts catecholamine-induced PKA activation, thereby reducing myocardial contractility. Thus β-AR antagonism with β-blockers has been a mainstay of heart failure therapy.
Human heart failure is associated with diminished phosphorylation of PLN,125 cTnI,126–128 and cMyBP-C.128,129 Although the cause of discrepancy remains an unsettled debate,130 animal models of heart failure exhibit increased cTnI phosphorylation.131,132 In failing rabbit cardiomyocytes, β-AR-induced PKA activation is diminished at the SR and sarcolemma, but enhanced at the myofilaments, indicating intracellular PKA activity redistribution.132 Accordingly, PKA-mediated phosphorylation of the SR protein PLN is decreased, but phosphorylation of the myofilament protein cTnI is increased in failing myocytes. Myofilament PKA activation in heart failure is due to reduced local PDE activity and enhanced β2-AR signalling. Mechanistically, heart failure is associated with a redistribution of β2-AR and PKA from t-tubules to other sarcolemmal areas owing to reduced expression of caveolin-3, a major structural protein of caveolae.132–134 It is worth noting that total PKA activity following cAMP stimulation is comparable between failing and non-failing human hearts, suggesting that heart failure alters PKA signalling primarily at the level of β-AR.135 Nonetheless, failing human hearts express lower level of R1α,126,136 but opposite findings have also been reported.128
Proper functioning of the heart relies on efficient Ca2+ cycling, which is controlled by PKA-dependent phosphorylation of the Ca2+ handling proteins in a highly dynamic manner. Therefore, prolonged activation or inhibition of PKA as well as its substrates can cause heart failure. For example, either gain-99,137 or loss-of-function PLN mutations138 induce lethal dilated cardiomyopathy. Chronic LTCC activation provokes Ca2+ overload-mediated cardiomyocyte necrosis, eventually leading to heart failure.23 On the other hand, permanent LTCC inactivation also results in heart failure and premature death.24 Constitutive PKA activation induces PLN and RyR2 hyperphosphorylation, leading to reduced contractility, dilated cardiomyopathy and sudden death.28 Although persistent PKA inhibition in mice does not cause functional or morphological abnormalities, depressed PKA activity impairs cardiac adaptation to stress, and may contribute to heart failure morbidity.27
The role of PKA in heart failure is modulated by AKAPs. Failing human hearts exhibit decreased interaction between PKA and AKAP1.136 Ablation of AKAP1 impairs mitochondrial PKA signalling and accelerates heart failure development after pressure overload.55 In addition, heart failure progression is accompanied by myocardial AKAP5 (also known as AKAP150, or AKAP79) down-regulation.29 Loss of AKAP5 abolishes PKA-mediated phosphorylation of PLN and RyR2, disrupts Ca2+ cycling, and predisposes to heart failure.29
Heart failure differentially affects expression of the PDE family members. Human heart failure is associated with decreased levels of PDE3A and PDE4D, which are sufficient to provoke SR Ca2+ leak and cardiomyocyte apoptosis.68,107 Interestingly, failing human hearts express higher levels of PDE1C,105 PDE2,139 and PDE10A,106 which hydrolyze cAMP and may contribute to decreased contractile response. Inhibition of PDE1C augments adenosine A2 receptor (A2R)-dependent cAMP synthesis, thereby providing acute inotropic, lusitropic, and vasodilatory effects in tachypacing-induced heart failure.140 Similarly, inhibition of PDE2141 or PDE10A106 protects against pressure overload-induced heart failure. Since inhibition of PDE1C,105 PDE2,141 or PDE10A106 also increases cardiac cGMP level, their cardioprotective effects are likely mediated by both cAMP-dependent and cAMP-independent mechanisms.
Conventional animal models of heart failure, such as pressure/volume overload or ischaemic/toxic injury, typically develop heart failure with reduced ejection fraction (HFrEF).142 The most common form of heart failure, however, is heart failure with preserved ejection fraction (HFpEF), which is characterized by myocardial stiffness and diastolic dysfunction. At the molecular level, myofilament stiffness is associated with reduced phosphorylation of titin’s N2B element at the PKA/PKG sites S4185/S4010/S4099.61,62 Stimulation with PKA-C reduces passive stiffness of human HFpEF cardiomyocytes in vitro.63 In an HFpEF-like mouse model, administration of metformin augments N2B phosphorylation at the PKA-specific site S4010, leading to increased titin compliance and improved diastolic function.64 In another HFpEF-like model, inhibition of PDE1 enhances PKA-mediated phosphorylation of RPN6 at S14, which augments proteasomal degradation of misfolded proteins and improves diastolic function.65 A new animal model that recapitulates many features of human HFpEF has been developed recently,143 and will likely advance our knowledge of PKA in HFpEF pathogenesis.
In summary, the dynamic, nearly instantaneous control of cardiac contractility by PKA is compromised in heart failure. Although acute PKA activation improves cardiac performance, chronic PKA activation or inhibition can lead to heart failure.
6. PKA and cardiomyopathies
6.1 Diabetic cardiomyopathy
Emerging evidence suggests that catecholamines antagonize insulin-induced cardiac glucose transport.144 Sustained catecholamine stimulation not only contributes to heart failure progression, but also leads to insulin resistance in the heart.145 Mechanistically, overstimulation of β2-AR in cardiomyocytes inhibits insulin-induced translocation of glucose transporter 4 (GLUT4) to the plasma membrane, resulting in reduced glucose uptake in a PKA-dependent manner.145 Treatment with the β-blockers propranolol or metoprolol prevents catecholamine-induced cardiac insulin resistance.145 In adipocytes, catecholamines induce PKA-dependent lipolysis, which blocks insulin-induced, phosphoinositide 3-kinase/Akt/mammalian target of rapamycin-dependent GLUT4 translocation, resulting in decreased glucose uptake.146
Insulin resistance is a common feature of type 2 diabetes mellitus, which can cause cardiac dysfunction through PDE4D-mediated decline in total PKA activity.147 In type 2 diabetes, insulin excess induces PDE4D expression via G-protein-coupled receptor kinase 2 (GRK2)-dependent transactivation of β2-AR/β-arrestin2/extracellular signal-regulated kinase pathway.147 Therefore, treatment with the GRK2 inhibitor paroxetine or the β-blocker carvedilol diminishes PDE4D expression, normalizes cAMP/PKA activity, and prevents type 2 diabetes-associated cardiac dysfunction.147 Interestingly, type 1 diabetes is also associated with reduced cardiac contractility due to decreased PKA activity.148 Since type 1 diabetes is characterized by insulin deficiency, physiological level of insulin may be necessary for myocardial PKA activation. Together, these studies suggest that either excess or deficiency of insulin can diminish myocardial PKA activity and cause diabetic cardiomyopathy.
6.2 Takotsubo cardiomyopathy
Takotsubo cardiomyopathy (also known as stress cardiomyopathy, broken heart syndrome, or apical ballooning syndrome) is a form of acute, transient heart failure that typically occurs after physical or emotional stress. The incidence of takotsubo cardiomyopathy is markedly increased during the coronavirus disease 2019 pandemic, due to either coronavirus infection149 or the pandemic-related psychological stress.150 Although heart function usually recovers within a few weeks, takotsubo cardiomyopathy can cause long-term structural, metabolic changes and a heart failure phenotype.151 Takotsubo cardiomyopathy is associated with elevated circulating and myocardial catecholamine levels, which can be higher than after acute myocardial infarction.152 Excessive catecholamines directly contribute to myocardial contraction band necrosis, a pathological hallmark of takotsubo cardiomyopathy.152 Moreover, catecholamines induce severe lipid accumulation in the heart, resulting in lipotoxicity.153 There is evidence that catecholamine hypersensitivity caused by genetic abnormalities may increase the susceptibility to takotsubo cardiomyopathy, owing to PKA-mediated hyperphosphorylation of RyR2, PLN, cTnI, and Cav1.2.154 Therefore, a genetic predisposition for takotsubo cardiomyopathy is possible, but remains to be further characterized.
6.3 Anthracycline cardiomyopathy
The anthracycline family anticancer drugs, such as doxorubicin, are frequently used in the treatment of various cancers. Unfortunately, their uses are limited by irreversible and dose-dependent cardiotoxicity, which may eventually progress into heart failure. Doxorubicin-induced reduction in cardiac contractility is correlated with decreased Ca2+ transients, likely due to increased RyR2 phosphorylation by PKA.155 Consistent with this, blockade of the β-AR/cAMP/PKA signalling attenuates anthracycline-related cardiotoxicity in cancer patients.156 Interestingly, enhancing PKA-mediated PLN phosphorylation with the PDE3 inhibitor levosimendan maintains cardiac contractility following doxorubicin administration.157 Levosimendan improves cardiomyocyte viability through activation of PKA, as the protective effect is abolished by the PKA inhibitor H89. In addition, PDE1 inhibition with IC86340 or via gene deletion attenuates doxorubicin-induced cardiomyocyte apoptosis, through augmenting A2R-mediated cAMP synthesis.158 Since β-AR-mediated cAMP production provokes apoptosis,95 A2R-derived cAMP likely resides in a different subcellular compartment to protect against apoptosis.158
7. PKA and the sex differences in cardiac health or disease
Biological sex has a profound impact on the heart. Baseline heart function is better in premenopausal women, who are also at lower risk of heart disease compared with age-matched men. Better heart function in females is associated with higher basal PKA activity and distinct gene expression profiles in cardiomyocytes, indicating a fundamental difference between female and male cardiomyocytes.159 Moreover, the female sex hormone oestrogen can activate cAMP/PKA and enhance cardiac contractility under basal conditions.160,161 In response to increasing demand (e.g. exercise, stress, and disease), however, female hearts and myocytes exhibit smaller contractions than males. A possible explanation is that oestrogen antagonizes the function of catecholamines,162 thereby reducing intracellular cAMP levels and suppressing SR Ca2+ release.163,164 Based on the above findings, we speculate that oestrogen counteracts catecholamine-mediated fight-or-flight response via up-regulating cAMP within different intracellular compartments. However, this possibility needs to be confirmed in future studies.
8. Study limitations
Our current knowledge of PKA has been largely based on studies of cAMP, PDEs, AKAPs, and pharmacological PKA inhibitors. Potential limitations of these approaches are summarized below.
8.1 cAMP ≠ PKA
In addition to PKA, cAMP activates other effectors including Epac,165,166 protein kinase X-linked (PRKX)/protein kinase Y-linked (PRKY),167 cyclic nucleotide-regulated cation channels [i.e. cyclic nucleotide-gated (CNG) channels and HCN channels],168,169 and the popeye domain containing (POPDC) proteins (Figure 1).170 Thus cAMP may produce PKA-independent responses.
On the other hand, PKA can also undergo cAMP-independent activation through diverse mechanisms including R1α oxidation,6–8 I-κB degradation,9,10 Smad3/4-PKA-R complex formation,11 and sphingosine-PKA holoenzyme interaction (Figure 1).12 Therefore, cAMP and PKA can be involved in completely different biological processes, and can have functions distinct from each other.
8.2 PDEs may modulate both cAMP and cGMP levels
The role of PKA in myocardial biology has also been investigated using genetic and pharmacological approaches to alter PDE activity. PDEs are divided into 11 super families (PDE1–11), comprising more than 80 different isoforms that modulate the levels of cAMP and/or cGMP.14 As a second messenger, cGMP regulates many aspects of cardiovascular homeostasis and pathophysiology through activation of PKG. Hence, PDEs may have cAMP/PKA-independent biological functions.
8.3 AKAPs may anchor other proteins not related with PKA
As scaffold proteins, AKAPs target PKA to subcellular locations and modulate local substrate function. However, AKAPs also anchor other kinases and regulatory proteins not related with PKA.171 Therefore, AKAP deficiency may cause phenotypes in a PKA-independent manner.
8.4 Specificity of PKA inhibitors
Many previous studies have relied on small molecule inhibitors to control PKA activity. Since pharmacological PKA inhibitors have off-target effects and interfere with a wide range of cellular activities,172 this approach may not accurately demonstrate the role of PKA.
9. Conclusions and perspectives
Preclinical studies over the past decades have greatly advanced our understanding of PKA in the heart. PKA regulates cardiac muscle contraction, relaxation and heart rate through modulating Ca2+ dynamics in cardiac myocytes. Myocardial PKA can be activated by the canonical and the non-canonical pathways. In the canonical PKA pathway, β-AR stimulation by catecholamines induces cAMP-dependent PKA activation. The non-canonical pathways prompt PKA activation in a cAMP-independent manner. Abnormal PKA activity has been observed in a wide range of cardiac pathologies. Therefore, PKA has the potential to serve as a drug target for the treatment of heart diseases.
Despite the great progress in recent years, the precise roles of PKA in heart disease pathogenesis remain not fully understood. Critical areas that warrant further investigation include:
Spatiotemporal regulation of PKA activity in cardiac health and disease
Proteins that determine PKA compartmentalization
Control of cardiomyocyte morphology, fate, and function by PKA
Signalling pathways that regulate apoptosis/necrosis downstream of PKA
Connections between PKA and oxidative stress
Additional PKA substrates in the heart
Role of PKA in the sex differences of heart disease
Heart disease types that require PKA-activating or inhibiting interventions
Adverse effects of PKA activation or inhibition
Development of potent and specific PKA activators and inhibitors.
Our current knowledge of cardiac PKA is mostly obtained using indirect approaches, which have limitations as described above. Strategies that directly alter the PKA holoenzyme should be considered in future research. Specific PKA inhibitors, such as PKI are also valuable tools for studying the biological functions of PKA in cardiac physiology and pathophysiology. Development of specific PKA activators or inhibitors with a satisfactory pharmacokinetic profile, good efficacy, and tolerability would greatly benefit future preclinical and clinical studies.
Acknowledgements
The authors would like to thank Dr Yang K. Xiang (University of California Davis) for critical reading of the manuscript.
Conflict of interest: none declared.
Funding
This work was supported by WSU College of Pharmacy and Pharmaceutical Sciences. Z.C. was supported by the National Heart, Lung, and Blood Institute (NHLBI, R00HL119605, R56HL145034), National Institutes of Health.
References
- 1. Walsh DA, Perkins JP, Krebs EG.. An adenosine 3',5'-monophosphate-dependant protein kinase from rabbit skeletal muscle. J Biol Chem 1968;243:3763–3765. [PubMed] [Google Scholar]
- 2. Taylor SS, Ilouz R, Zhang P, Kornev AP.. Assembly of allosteric macromolecular switches: lessons from PKA. Nat Rev Mol Cell Biol 2012;13:646–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kirschner LS, Yin Z, Jones GN, Mahoney E.. Mouse models of altered protein kinase A signaling. Endocr Relat Cancer 2009;16:773–793. [DOI] [PubMed] [Google Scholar]
- 4. Wang J, Gareri C, Rockman HA.. G-protein-coupled receptors in heart disease. Circ Res 2018;123:716–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Smith FD, Esseltine JL, Nygren PJ, Veesler D, Byrne DP, Vonderach M, Strashnov I, Eyers CE, Eyers PA, Langeberg LK, Scott JD.. Local protein kinase A action proceeds through intact holoenzymes. Science 2017;356:1288–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brennan JP, Bardswell SC, Burgoyne JR, Fuller W, Schroder E, Wait R, Begum S, Kentish JC, Eaton P.. Oxidant-induced activation of type I protein kinase A is mediated by RI subunit interprotein disulfide bond formation. J Biol Chem 2006;281:21827–21836. [DOI] [PubMed] [Google Scholar]
- 7. Srinivasan S, Spear J, Chandran K, Joseph J, Kalyanaraman B, Avadhani NG.. Oxidative stress induced mitochondrial protein kinase A mediates cytochrome c oxidase dysfunction. PLoS One 2013;8:e77129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Haushalter KJ, Schilling JM, Song Y, Sastri M, Perkins GA, Strack S, Taylor SS, Patel HH.. Cardiac ischemia-reperfusion injury induces ROS-dependent loss of PKA regulatory subunit RIalpha. Am J Physiol Heart Circ Physiol 2019;317:H1231–H1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhong H, SuYang H, Erdjument-Bromage H, Tempst P, Ghosh S.. The transcriptional activity of NF-kappaB is regulated by the IkappaB-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell 1997;89:413–424. [DOI] [PubMed] [Google Scholar]
- 10. Dulin NO, Niu J, Browning DD, Ye RD, Voyno-Yasenetskaya T.. Cyclic AMP-independent activation of protein kinase A by vasoactive peptides. J Biol Chem 2001;276:20827–20830. [DOI] [PubMed] [Google Scholar]
- 11. Zhang L, Duan CJ, Binkley C, Li G, Uhler MD, Logsdon CD, Simeone DM.. A transforming growth factor beta-induced Smad3/Smad4 complex directly activates protein kinase A. Mol Cell Biol 2004;24:2169–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ma Y, Pitson S, Hercus T, Murphy J, Lopez A, Woodcock J.. Sphingosine activates protein kinase A type II by a novel cAMP-independent mechanism. J Biol Chem 2005;280:26011–26017. [DOI] [PubMed] [Google Scholar]
- 13. Kohr MJ, Traynham CJ, Roof SR, Davis JP, Ziolo MT.. cAMP-independent activation of protein kinase A by the peroxynitrite generator SIN-1 elicits positive inotropic effects in cardiomyocytes. J Mol Cell Cardiol 2010;48:645–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stangherlin A, Zaccolo M.. Phosphodiesterases and subcellular compartmentalized cAMP signaling in the cardiovascular system. Am J Physiol Heart Circ Physiol 2012;302:H379–H390. [DOI] [PubMed] [Google Scholar]
- 15. Ghigo A, Mika D.. cAMP/PKA signaling compartmentalization in cardiomyocytes: lessons from FRET-based biosensors. J Mol Cell Cardiol 2019;131:112–121. [DOI] [PubMed] [Google Scholar]
- 16. Bers DM. Cardiac excitation-contraction coupling. Nature 2002;415:198–205. [DOI] [PubMed] [Google Scholar]
- 17. Wang Y, Shi Q, Li M, Zhao M, Raghavender Reddy G, Teoh JP, Xu B, Zhu C, Ireton KE, Srinivasan S, Chen SL, Gasser PJ, Bossuyt J, Hell JW, Bers DM, Xiang YK.. Intracellular beta1-adrenergic receptors and organic cation transporter 3 mediate phospholamban phosphorylation to enhance cardiac contractility. Circ Res 2021;128:246–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fu Y, Westenbroek RE, Scheuer T, Catterall WA.. Basal and beta-adrenergic regulation of the cardiac calcium channel CaV1.2 requires phosphorylation of serine 1700. Proc Natl Acad Sci USA 2014;111:16598–16603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang L, Katchman A, Samad T, Morrow J, Weinberg R, Marx SO.. Beta-adrenergic regulation of the L-type Ca2+ channel does not require phosphorylation of alpha1C Ser1700. Circ Res 2013;113:871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu G, Papa A, Katchman AN, Zakharov SI, Roybal D, Hennessey JA, Kushner J, Yang L, Chen BX, Kushnir A, Dangas K, Gygi SP, Pitt GS, Colecraft HM, Ben-Johny M, Kalocsay M, Marx SO.. Mechanism of adrenergic CaV1.2 stimulation revealed by proximity proteomics. Nature 2020;577:695–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vinogradova TM, Lyashkov AE, Zhu W, Ruknudin AM, Sirenko S, Yang D, Deo S, Barlow M, Johnson S, Caffrey JL, Zhou YY, Xiao RP, Cheng H, Stern MD, Maltsev VA, Lakatta EG.. High basal protein kinase A-dependent phosphorylation drives rhythmic internal Ca2+ store oscillations and spontaneous beating of cardiac pacemaker cells. Circ Res 2006;98:505–514. [DOI] [PubMed] [Google Scholar]
- 22. Vettel C, Lindner M, Dewenter M, Lorenz K, Schanbacher C, Riedel M, Lammle S, Meinecke S, Mason FE, Sossalla S, Geerts A, Hoffmann M, Wunder F, Brunner FJ, Wieland T, Mehel H, Karam S, Lechene P, Leroy J, Vandecasteele G, Wagner M, Fischmeister R, El-Armouche A.. Phosphodiesterase 2 protects against catecholamine-induced arrhythmia and preserves contractile function after myocardial infarction. Circ Res 2017;120:120–132. [DOI] [PubMed] [Google Scholar]
- 23. Nakayama H, Chen X, Baines CP, Klevitsky R, Zhang X, Zhang H, Jaleel N, Chua BH, Hewett TE, Robbins J, Houser SR, Molkentin JD.. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J Clin Invest 2007;117:2431–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yang L, Dai DF, Yuan C, Westenbroek RE, Yu H, West N, de la Iglesia HO, Catterall WA.. Loss of beta-adrenergic-stimulated phosphorylation of CaV1.2 channels on Ser1700 leads to heart failure. Proc Natl Acad Sci USA 2016;113:E7976–E7985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kranias EG, Hajjar RJ.. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ Res 2012;110:1646–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vinogradova TM, Sirenko S, Lukyanenko YO, Yang D, Tarasov KV, Lyashkov AE, Varghese NJ, Li Y, Chakir K, Ziman B, Lakatta EG.. Basal spontaneous firing of rabbit sinoatrial node cells is regulated by dual activation of PDEs (phosphodiesterases) 3 and 4. Circ Arrhythm Electrophysiol 2018;11:e005896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang Y, Wang WE, Zhang X, Li Y, Chen B, Liu C, Ai X, Zhang X, Tian Y, Zhang C, Tang M, Szeto C, Hua X, Xie M, Zeng C, Wu Y, Zhou L, Zhu W, Yu D, Houser SR, Chen X.. Cardiomyocyte PKA ablation enhances basal contractility while eliminates cardiac beta-adrenergic response without adverse effects on the heart. Circ Res 2019;124:1760–1777. [DOI] [PubMed] [Google Scholar]
- 28. Antos CL, Frey N, Marx SO, Reiken S, Gaburjakova M, Richardson JA, Marks AR, Olson EN.. Dilated cardiomyopathy and sudden death resulting from constitutive activation of protein kinase A. Circ Res 2001;89:997–1004. [DOI] [PubMed] [Google Scholar]
- 29. Li L, Li J, Drum BM, Chen Y, Yin H, Guo X, Luckey SW, Gilbert ML, McKnight GS, Scott JD, Santana LF, Liu Q.. Loss of AKAP150 promotes pathological remodelling and heart failure propensity by disrupting calcium cycling and contractile reserve. Cardiovasc Res 2017;113:147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dobrev D, Wehrens XH.. Role of RyR2 phosphorylation in heart failure and arrhythmias: controversies around ryanodine receptor phosphorylation in cardiac disease. Circ Res 2014;114:1311–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Houser SR. Role of RyR2 phosphorylation in heart failure and arrhythmias: protein kinase A-mediated hyperphosphorylation of the ryanodine receptor at serine 2808 does not alter cardiac contractility or cause heart failure and arrhythmias. Circ Res 2014;114:1320–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bovo E, Huke S, Blatter LA, Zima AV.. The effect of PKA-mediated phosphorylation of ryanodine receptor on SR Ca(2+) leak in ventricular myocytes. J Mol Cell Cardiol 2017;104:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ellison GM, Torella D, Karakikes I, Purushothaman S, Curcio A, Gasparri C, Indolfi C, Cable NT, Goldspink DF, Nadal-Ginard B.. Acute beta-adrenergic overload produces myocyte damage through calcium leakage from the ryanodine receptor 2 but spares cardiac stem cells. J Biol Chem 2007;282:11397–11409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ponnam S, Sevrieva I, Sun YB, Irving M, Kampourakis T.. Site-specific phosphorylation of myosin binding protein-C coordinates thin and thick filament activation in cardiac muscle. Proc Natl Acad Sci USA 2019;116:15485–15494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McNamara JW, Singh RR, Sadayappan S.. Cardiac myosin binding protein-C phosphorylation regulates the super-relaxed state of myosin. Proc Natl Acad Sci USA 2019;116:11731–11736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mamidi R, Gresham KS, Li J, Stelzer JE.. Cardiac myosin binding protein-C Ser(302) phosphorylation regulates cardiac beta-adrenergic reserve. Sci Adv 2017;3:e1602445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rosas PC, Liu Y, Abdalla MI, Thomas CM, Kidwell DT, Dusio GF, Mukhopadhyay D, Kumar R, Baker KM, Mitchell BM, Powers PA, Fitzsimons DP, Patel BG, Warren CM, Solaro RJ, Moss RL, Tong CW.. Phosphorylation of cardiac myosin-binding protein-C is a critical mediator of diastolic function. Circ Heart Fail 2015;8:582–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gresham KS, Stelzer JE.. The contributions of cardiac myosin binding protein C and troponin I phosphorylation to beta-adrenergic enhancement of in vivo cardiac function. J Physiol 2016;594:669–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hwang PM, Cai F, Pineda-Sanabria SE, Corson DC, Sykes BD.. The cardiac-specific N-terminal region of troponin I positions the regulatory domain of troponin C. Proc Natl Acad Sci USA 2014;111:14412–14417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rao V, Cheng Y, Lindert S, Wang D, Oxenford L, McCulloch AD, McCammon JA, Regnier M.. PKA phosphorylation of cardiac troponin I modulates activation and relaxation kinetics of ventricular myofibrils. Biophys J 2014;107:1196–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Martin-Garrido A, Biesiadecki BJ, Salhi HE, Shaifta Y, Dos Remedios CG, Ayaz-Guner S, Cai W, Ge Y, Avkiran M, Kentish JC.. Monophosphorylation of cardiac troponin-I at Ser-23/24 is sufficient to regulate cardiac myofibrillar Ca(2+) sensitivity and calpain-induced proteolysis. J Biol Chem 2018;293:8588–8599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Prabu SK, Anandatheerthavarada HK, Raza H, Srinivasan S, Spear JF, Avadhani NG.. Protein kinase A-mediated phosphorylation modulates cytochrome c oxidase function and augments hypoxia and myocardial ischemia-related injury. J Biol Chem 2006;281:2061–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Qian J, Ren X, Wang X, Zhang P, Jones WK, Molkentin JD, Fan GC, Kranias EG.. Blockade of Hsp20 phosphorylation exacerbates cardiac ischemia/reperfusion injury by suppressed autophagy and increased cell death. Circ Res 2009;105:1223–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ye Y, Keyes KT, Zhang C, Perez-Polo JR, Lin Y, Birnbaum Y.. The myocardial infarct size-limiting effect of sitagliptin is PKA-dependent, whereas the protective effect of pioglitazone is partially dependent on PKA. Am J Physiol Heart Circ Physiol 2010;298:H1454–H1465. [DOI] [PubMed] [Google Scholar]
- 45. Siraj MA, Mundil D, Beca S, Momen A, Shikatani EA, Afroze T, Sun X, Liu Y, Ghaffari S, Lee W, Wheeler MB, Keller G, Backx P, Husain M.. Cardioprotective GLP-1 metabolite prevents ischemic cardiac injury by inhibiting mitochondrial trifunctional protein-alpha. J Clin Invest 2020;130:1392–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tomita H, Nazmy M, Kajimoto K, Yehia G, Molina CA, Sadoshima J.. Inducible cAMP early repressor (ICER) is a negative-feedback regulator of cardiac hypertrophy and an important mediator of cardiac myocyte apoptosis in response to beta-adrenergic receptor stimulation. Circ Res 2003;93:12–22. [DOI] [PubMed] [Google Scholar]
- 47. Bedioune I, Lefebvre F, Lechene P, Varin A, Domergue V, Kapiloff MS, Fischmeister R, Vandecasteele G.. PDE4 and mAKAPbeta are nodal organizers of beta2-ARs nuclear PKA signalling in cardiac myocytes. Cardiovasc Res 2018;114:1499–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lee YY, Moujalled D, Doerflinger M, Gangoda L, Weston R, Rahimi A, de Alboran I, Herold M, Bouillet P, Xu Q, Gao X, Du XJ, Puthalakath H.. CREB-binding protein (CBP) regulates beta-adrenoceptor (beta-AR)-mediated apoptosis. Cell Death Differ 2013;20:941–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Watson PA, Reusch JE, McCune SA, Leinwand LA, Luckey SW, Konhilas JP, Brown DA, Chicco AJ, Sparagna GC, Long CS, Moore RL.. Restoration of CREB function is linked to completion and stabilization of adaptive cardiac hypertrophy in response to exercise. Am J Physiol Heart Circ Physiol 2007;293:H246–H259. [DOI] [PubMed] [Google Scholar]
- 50. Wang L, Burmeister BT, Johnson KR, Baillie GS, Karginov AV, Skidgel RA, O'Bryan JP, Carnegie GK.. UCR1C is a novel activator of phosphodiesterase 4 (PDE4) long isoforms and attenuates cardiomyocyte hypertrophy. Cell Signal 2015;27:908–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Matus M, Lewin G, Stumpel F, Buchwalow IB, Schneider MD, Schutz G, Schmitz W, Muller FU.. Cardiomyocyte-specific inactivation of transcription factor CREB in mice. Faseb J 2007;21:1884–1892. [DOI] [PubMed] [Google Scholar]
- 52. Antos CL, McKinsey TA, Frey N, Kutschke W, McAnally J, Shelton JM, Richardson JA, Hill JA, Olson EN.. Activated glycogen synthase-3 beta suppresses cardiac hypertrophy in vivo. Proc Natl Acad Sci USA 2002;99:907–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fang X, Yu SX, Lu Y, Bast RC Jr, Woodgett JR, Mills GB.. Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc Natl Acad Sci USA 2000;97:11960–11965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zoccarato A, Surdo NC, Aronsen JM, Fields LA, Mancuso L, Dodoni G, Stangherlin A, Livie C, Jiang H, Sin YY, Gesellchen F, Terrin A, Baillie GS, Nicklin SA, Graham D, Szabo-Fresnais N, Krall J, Vandeput F, Movsesian M, Furlan L, Corsetti V, Hamilton G, Lefkimmiatis K, Sjaastad I, Zaccolo M.. Cardiac hypertrophy is inhibited by a local pool of cAMP regulated by phosphodiesterase 2. Circ Res 2015;117:707–719. [DOI] [PubMed] [Google Scholar]
- 55. Schiattarella GG, Boccella N, Paolillo R, Cattaneo F, Trimarco V, Franzone A, D'Apice S, Giugliano G, Rinaldi L, Borzacchiello D, Gentile A, Lombardi A, Feliciello A, Esposito G, Perrino C.. Loss of Akap1 exacerbates pressure overload-induced cardiac hypertrophy and heart failure. Front Physiol 2018;9:558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Abrenica B, AlShaaban M, Czubryt MP.. The A-kinase anchor protein AKAP121 is a negative regulator of cardiomyocyte hypertrophy. J Mol Cell Cardiol 2009;46:674–681. [DOI] [PubMed] [Google Scholar]
- 57. Liu Y, Xia P, Chen J, Bandettini WP, Kirschner LS, Stratakis CA, Cheng Z.. PRKAR1A deficiency impedes hypertrophy and reduces heart size. Physiol Rep 2020;8:e14405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ha CH, Kim JY, Zhao J, Wang W, Jhun BS, Wong C, Jin ZG.. PKA phosphorylates histone deacetylase 5 and prevents its nuclear export, leading to the inhibition of gene transcription and cardiomyocyte hypertrophy. Proc Natl Acad Sci USA 2010;107:15467–15472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chang CW, Lee L, Yu D, Dao K, Bossuyt J, Bers DM.. Acute beta-adrenergic activation triggers nuclear import of histone deacetylase 5 and delays G(q)-induced transcriptional activation. J Biol Chem 2013;288:192–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Backs J, Worst BC, Lehmann LH, Patrick DM, Jebessa Z, Kreusser MM, Sun Q, Chen L, Heft C, Katus HA, Olson EN.. Selective repression of MEF2 activity by PKA-dependent proteolysis of HDAC4. J Cell Biol 2011;195:403–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zile MR, Baicu CF, S. Ikonomidis J, Stroud RE, Nietert PJ, Bradshaw AD, Slater R, Palmer BM, Van Buren P, Meyer M, M. Redfield M, A. Bull D, L. Granzier H, LeWinter MM.. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: contributions of collagen and titin. Circulation 2015;131:1247–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kötter S, Gout L, Von Frieling-Salewsky M, Müller AE, Helling S, Marcus K, Dos Remedios C, Linke WA, Krüger M.. Differential changes in titin domain phosphorylation increase myofilament stiffness in failing human hearts. Cardiovasc Res 2013;99:648–656. [DOI] [PubMed] [Google Scholar]
- 63. Borbély A, Falcao-Pires I, van Heerebeek L, Hamdani N, Édes I, Gavina C, Leite-Moreira AF, Bronzwaer JGF, Papp Z, van der Velden J, Stienen GJM, Paulus WJ, Hypophosphorylation of the Stiff N2B titin isoform raises cardiomyocyte resting tension in failing human myocardium. Circ Res 2009;104:780–786. [DOI] [PubMed] [Google Scholar]
- 64. Slater RE, Strom JG, Methawasin M, Liss M, Gotthardt M, Sweitzer N, Granzier HL.. Metformin improves diastolic function in an HFpEF-like mouse model by increasing titin compliance. J Gen Physiol 2019;151:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhang H, Pan B, Wu P, Parajuli N, Rekhter MD, Goldberg AL, Wang X.. PDE1 inhibition facilitates proteasomal degradation of misfolded proteins and protects against cardiac proteinopathy. Sci Adv 2019;5:eaaw5870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tsutsui K, Monfredi OJ, Sirenko-Tagirova SG, Maltseva LA, Bychkov R, Kim MS, Ziman BD, Tarasov KV, Tarasova YS, Zhang J, Wang M, Maltsev AV, Brennan JA, Efimov IR, Stern MD, Maltsev VA, Lakatta EG.. A coupled-clock system drives the automaticity of human sinoatrial nodal pacemaker cells. Sci Signal 2018;11:eaap7608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Moen JM, Matt MG, Ramirez C, Tarasov KV, Chakir K, Tarasova YS, Lukyanenko Y, Tsutsui K, Monfredi O, Morrell CH, Tagirova S, Yaniv Y, Huynh T, Pacak K, Ahmet I, Lakatta EG.. Overexpression of a neuronal type adenylyl cyclase (type 8) in sinoatrial node markedly impacts heart rate and rhythm. Front Neurosci 2019;13:615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lehnart SE, Wehrens XH, Reiken S, Warrier S, Belevych AE, Harvey RD, Richter W, Jin SL, Conti M, Marks AR.. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell 2005;123:25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hopkins PN. Molecular biology of atherosclerosis. Physiol Rev 2013;93:1317–1542. [DOI] [PubMed] [Google Scholar]
- 70. Schomig A, Dart AM, Dietz R, Mayer E, Kubler W.. Release of endogenous catecholamines in the ischemic myocardium of the rat. Part A: locally mediated release. Circ Res 1984;55:689–701. [DOI] [PubMed] [Google Scholar]
- 71. Lameris TW, de Zeeuw S, Alberts G, Boomsma F, Duncker DJ, Verdouw PD, Veld AJ, van Den Meiracker AH.. Time course and mechanism of myocardial catecholamine release during transient ischemia in vivo. Circulation 2000;101:2645–2650. [DOI] [PubMed] [Google Scholar]
- 72. Graham LN, Smith PA, Stoker JB, Mackintosh AF, Mary DA.. Time course of sympathetic neural hyperactivity after uncomplicated acute myocardial infarction. Circulation 2002;106:793–797. [DOI] [PubMed] [Google Scholar]
- 73. Sandhu R, Thomas U, Diaz RJ, Wilson GJ.. Effect of ischemic preconditioning of the myocardium on cAMP. Circ Res 1996;78:137–147. [DOI] [PubMed] [Google Scholar]
- 74. Lochner A, Genade S, Tromp E, Podzuweit T, Moolman JA.. Ischemic preconditioning and the beta-adrenergic signal transduction pathway. Circulation 1999;100:958–966. [DOI] [PubMed] [Google Scholar]
- 75. Sanada S, Asanuma H, Tsukamoto O, Minamino T, Node K, Takashima S, Fukushima T, Ogai A, Shinozaki Y, Fujita M, Hirata A, Okuda H, Shimokawa H, Tomoike H, Hori M, Kitakaze M.. Protein kinase A as another mediator of ischemic preconditioning independent of protein kinase C. Circulation 2004;110:51–57. [DOI] [PubMed] [Google Scholar]
- 76. Li XD, Yang YJ, Geng YJ, Zhao JL, Zhang HT, Cheng YT, Wu YL.. Phosphorylation of endothelial NOS contributes to simvastatin protection against myocardial no-reflow and infarction in reperfused swine hearts: partially via the PKA signaling pathway. Acta Pharmacol Sin 2012;33:879–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Penela P, Inserte J, Ramos P, Rodriguez-Sinovas A, Garcia-Dorado D, Mayor F Jr. Degradation of GRK2 and AKT is an early and detrimental event in myocardial ischemia/reperfusion. EBioMedicine 2019;48:605–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wallner M, Duran JM, Mohsin S, Troupes CD, Vanhoutte D, Borghetti G, Vagnozzi RJ, Gross P, Yu D, Trappanese DM, Kubo H, Toib A, Sharp TE, Harper SC, Volkert MA, Starosta T, Feldsott EA, Berretta RM, Wang T, Barbe MF, Molkentin JD, Houser SR.. Acute catecholamine exposure causes reversible myocyte injury without cardiac regeneration. Circ Res 2016;119:865–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Oikawa M, Wu M, Lim S, Knight WE, Miller CL, Cai Y, Lu Y, Blaxall BC, Takeishi Y, Abe J, Yan C.. Cyclic nucleotide phosphodiesterase 3A1 protects the heart against ischemia-reperfusion injury. J Mol Cell Cardiol 2013;64:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Spear JF, Prabu SK, Galati D, Raza H, Anandatheerthavarada HK, Avadhani NG.. beta1-Adrenoreceptor activation contributes to ischemia-reperfusion damage as well as playing a role in ischemic preconditioning. Am J Physiol Heart Circ Physiol 2007;292:H2459–H2466. [DOI] [PubMed] [Google Scholar]
- 81. Makaula S, Lochner A, Genade S, Sack MN, Awan MM, Opie LH.. H-89, a non-specific inhibitor of protein kinase A, promotes post-ischemic cardiac contractile recovery and reduces infarct size. J Cardiovasc Pharmacol 2005;45:341–347. [DOI] [PubMed] [Google Scholar]
- 82. Mieno S, Horimoto H, Watanabe F, Nakai Y, Furuya E, Sasaki S.. Potent adenylate cyclase agonist forskolin restores myoprotective effects of ischemic preconditioning in rat hearts after myocardial infarction. Ann Thorac Surg 2002;74:1213–1218. [DOI] [PubMed] [Google Scholar]
- 83. Heinen A, Strothoff M, Schmidt A, Stracke N, Behmenburg F, Bauer I, Hollmann MW, Huhn R.. Pharmacological options to protect the aged heart from ischemia and reperfusion injury by targeting the PKA-BK(Ca) signaling pathway. Exp Gerontol 2014;56:99–105. [DOI] [PubMed] [Google Scholar]
- 84. Sanada S, Kitakaze M, Papst PJ, Asanuma H, Node K, Takashima S, Asakura M, Ogita H, Liao Y, Sakata Y, Ogai A, Fukushima T, Yamada J, Shinozaki Y, Kuzuya T, Mori H, Terada N, Hori M.. Cardioprotective effect afforded by transient exposure to phosphodiesterase III inhibitors: the role of protein kinase A and p38 mitogen-activated protein kinase. Circulation 2001;104:705–710. [DOI] [PubMed] [Google Scholar]
- 85. Rechtman MP, Van der Zypp A, Majewski H.. Amrinone reduces ischaemia-reperfusion injury in rat heart. Eur J Pharmacol 2000;402:255–262. [DOI] [PubMed] [Google Scholar]
- 86. Sichelschmidt OJ, Hahnefeld C, Hohlfeld T, Herberg FW, Schror K.. Trapidil protects ischemic hearts from reperfusion injury by stimulating PKAII activity. Cardiovasc Res 2003;58:602–610. [DOI] [PubMed] [Google Scholar]
- 87. Chung YW, Lagranha C, Chen Y, Sun J, Tong G, Hockman SC, Ahmad F, Esfahani SG, Bae DH, Polidovitch N, Wu J, Rhee DK, Lee BS, Gucek M, Daniels MP, Brantner CA, Backx PH, Murphy E, Manganiello VC.. Targeted disruption of PDE3B, but not PDE3A, protects murine heart from ischemia/reperfusion injury. Proc Natl Acad Sci USA 2015;112:E2253–E2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zhang Y, Wang XL, Zhao J, Wang YJ, Lau WB, Yuan YX, Gao EH, Koch WJ, Ma XL.. Adiponectin inhibits oxidative/nitrative stress during myocardial ischemia and reperfusion via PKA signaling. Am J Physiol Endocrinol Metab 2013;305:E1436–E1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zhang X, Szeto C, Gao E, Tang M, Jin J, Fu Q, Makarewich C, Ai X, Li Y, Tang A, Wang J, Gao H, Wang F, Ge XJ, Kunapuli SP, Zhou L, Zeng C, Xiang KY, Chen X.. Cardiotoxic and cardioprotective features of chronic beta-adrenergic signaling. Circ Res 2013;112:498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Rinaldi L, Pozdniakova S, Jayarajan V, Troidl C, Abdallah Y, Aslam M, Ladilov Y.. Protective role of soluble adenylyl cyclase against reperfusion-induced injury of cardiac cells. Biochim Biophys Acta Mol Basis Dis 2019;1865:252–260. [DOI] [PubMed] [Google Scholar]
- 91. Bose AK, Mocanu MM, Carr RD, Brand CL, Yellon DM.. Glucagon-like peptide 1 can directly protect the heart against ischemia/reperfusion injury. Diabetes 2005;54:146–151. [DOI] [PubMed] [Google Scholar]
- 92. Asai M, Tsukamoto O, Minamino T, Asanuma H, Fujita M, Asano Y, Takahama H, Sasaki H, Higo S, Asakura M, Takashima S, Hori M, Kitakaze M.. PKA rapidly enhances proteasome assembly and activity in in vivo canine hearts. J Mol Cell Cardiol 2009;46:452–462. [DOI] [PubMed] [Google Scholar]
- 93. Engelhardt S, Hein L, Wiesmann F, Lohse MJ.. Progressive hypertrophy and heart failure in beta1-adrenergic receptor transgenic mice. Proc Natl Acad Sci USA 1999;96:7059–7064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Iwase M, Bishop SP, Uechi M, Vatner DE, Shannon RP, Kudej RK, Wight DC, Wagner TE, Ishikawa Y, Homcy CJ, Vatner SF.. Adverse effects of chronic endogenous sympathetic drive induced by cardiac GS alpha overexpression. Circ Res 1996;78:517–524. [DOI] [PubMed] [Google Scholar]
- 95. Communal C, Singh K, Pimentel DR, Colucci WS.. Norepinephrine stimulates apoptosis in adult rat ventricular myocytes by activation of the beta-adrenergic pathway. Circulation 1998;98:1329–1334. [DOI] [PubMed] [Google Scholar]
- 96. Lai L, Yan L, Gao S, Hu CL, Ge H, Davidow A, Park M, Bravo C, Iwatsubo K, Ishikawa Y, Auwerx J, Sinclair DA, Vatner SF, Vatner DE.. Type 5 adenylyl cyclase increases oxidative stress by transcriptional regulation of manganese superoxide dismutase via the SIRT1/FoxO3a pathway. Circulation 2013;127:1692–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Lai NC, Tang T, Gao MH, Saito M, Takahashi T, Roth DM, Hammond HK.. Activation of cardiac adenylyl cyclase expression increases function of the failing ischemic heart in mice. J Am Coll Cardiol 2008;51:1490–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Lipskaia L, Defer N, Esposito G, Hajar I, Garel MC, Rockman HA, Hanoune J.. Enhanced cardiac function in transgenic mice expressing a Ca(2+)-stimulated adenylyl cyclase. Circ Res 2000;86:795–801. [DOI] [PubMed] [Google Scholar]
- 99. Schmitt JP, Kamisago M, Asahi M, Li GH, Ahmad F, Mende U, Kranias EG, MacLennan DH, Seidman JG, Seidman CE.. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science 2003;299:1410–1413. [DOI] [PubMed] [Google Scholar]
- 100. Dweck D, Sanchez-Gonzalez MA, Chang AN, Dulce RA, Badger CD, Koutnik AP, Ruiz EL, Griffin B, Liang J, Kabbaj M, Fincham FD, Hare JM, Overton JM, Pinto JR.. Long term ablation of protein kinase A (PKA)-mediated cardiac troponin I phosphorylation leads to excitation-contraction uncoupling and diastolic dysfunction in a knock-in mouse model of hypertrophic cardiomyopathy. J Biol Chem 2014;289:23097–23111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Yan L, Vatner DE, O'Connor JP, Ivessa A, Ge H, Chen W, Hirotani S, Ishikawa Y, Sadoshima J, Vatner SF.. Type 5 adenylyl cyclase disruption increases longevity and protects against stress. Cell 2007;130:247–258. [DOI] [PubMed] [Google Scholar]
- 102. Okumura S, Takagi G, Kawabe J, Yang G, Lee MC, Hong C, Liu J, Vatner DE, Sadoshima J, Vatner SF, Ishikawa Y.. Disruption of type 5 adenylyl cyclase gene preserves cardiac function against pressure overload. Proc Natl Acad Sci USA 2003;100:9986–9990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Piddo AM, Sanchez MI, Sapag-Hagar M, Corbalan R, Foncea R, Ebensperger R, Godoy I, Melendez J, Jalil JE, Lavandero S.. Cyclic AMP-dependent protein kinase and mechanical heart function in ventricular hypertrophy induced by pressure overload or secondary to myocardial infarction. J Mol Cell Cardiol 1996;28:1073–1083. [DOI] [PubMed] [Google Scholar]
- 104. Fields LA, Koschinski A, Zaccolo M.. Sustained exposure to catecholamines affects cAMP/PKA compartmentalised signalling in adult rat ventricular myocytes. Cell Signal 2016;28:725–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Knight WE, Chen S, Zhang Y, Oikawa M, Wu M, Zhou Q, Miller CL, Cai Y, Mickelsen DM, Moravec C, Small EM, Abe J, Yan C.. PDE1C deficiency antagonizes pathological cardiac remodeling and dysfunction. Proc Natl Acad Sci USA 2016;113:E7116–E7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Chen S, Zhang Y, Lighthouse JK, Mickelsen DM, Wu J, Yao P, Small EM, Yan C.. A novel role of cyclic nucleotide phosphodiesterase 10A in pathological cardiac remodeling and dysfunction. Circulation 2020;141:217–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Ding B, Abe J, Wei H, Huang Q, Walsh RA, Molina CA, Zhao A, Sadoshima J, Blaxall BC, Berk BC, Yan C.. Functional role of phosphodiesterase 3 in cardiomyocyte apoptosis: implication in heart failure. Circulation 2005;111:2469–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Abi-Gerges A, Richter W, Lefebvre F, Mateo P, Varin A, Heymes C, Samuel JL, Lugnier C, Conti M, Fischmeister R, Vandecasteele G.. Decreased expression and activity of cAMP phosphodiesterases in cardiac hypertrophy and its impact on beta-adrenergic cAMP signals. Circ Res 2009;105:784–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Polidovitch N, Yang S, Sun H, Lakin R, Ahmad F, Gao X, Turnbull PC, Chiarello C, Perry CGR, Manganiello V, Yang P, Backx PH.. Phosphodiesterase type 3A (PDE3A), but not type 3B (PDE3B), contributes to the adverse cardiac remodeling induced by pressure overload. J Mol Cell Cardiol 2019;132:60–70. [DOI] [PubMed] [Google Scholar]
- 110. Yang JH, Polanowska-Grabowska RK, Smith JS, Shields CW, Saucerman JJ.. PKA catalytic subunit compartmentation regulates contractile and hypertrophic responses to beta-adrenergic signaling. J Mol Cell Cardiol 2014;66:83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Schirmer I, Bualeong T, Budde H, Cimiotti D, Appukuttan A, Klein N, Steinwascher P, Reusch P, Mugge A, Meyer R, Ladilov Y, Jaquet K.. Soluble adenylyl cyclase: a novel player in cardiac hypertrophy induced by isoprenaline or pressure overload. PLoS One 2018;13:e0192322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Liggett SB, Tepe NM, Lorenz JN, Canning AM, Jantz TD, Mitarai S, Yatani A, Dorn GW.. Early and delayed consequences of beta(2)-adrenergic receptor overexpression in mouse hearts: critical role for expression level. Circulation 2000;101:1707–1714. [DOI] [PubMed] [Google Scholar]
- 113. Pare GC, Bauman AL, McHenry M, Michel JJ, Dodge-Kafka KL, Kapiloff MS.. The mAKAP complex participates in the induction of cardiac myocyte hypertrophy by adrenergic receptor signaling. J Cell Sci 2005;118:5637–5646. [DOI] [PubMed] [Google Scholar]
- 114. Enns LC, Bible KL, Emond MJ, Ladiges WC.. Mice lacking the Cbeta subunit of PKA are resistant to angiotensin II-induced cardiac hypertrophy and dysfunction. BMC Res Notes 2010;3:307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Imaeda A, Tanaka S, Tonegawa K, Fuchigami S, Obana M, Maeda M, Kihara M, Kiyonari H, Conway SJ, Fujio Y, Nakayama H.. Myofibroblast beta2 adrenergic signaling amplifies cardiac hypertrophy in mice. Biochem Biophys Res Commun 2019;510:149–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Di Benedetto G, Zoccarato A, Lissandron V, Terrin A, Li X, Houslay MD, Baillie GS, Zaccolo M.. Protein kinase A type I and type II define distinct intracellular signaling compartments. Circ Res 2008;103:836–844. [DOI] [PubMed] [Google Scholar]
- 117. Yin Z, Jones GN, Towns WH, Zhang X, Abel ED, Binkley PF, Jarjoura D, Kirschner LS.. Heart-specific ablation of Prkar1a causes failure of heart development and myxomagenesis. Circulation 2008;117:1414–1422. [DOI] [PubMed] [Google Scholar]
- 118. Enns LC, Morton JF, Treuting PR, Emond MJ, Wolf NS, Dai DF, McKnight GS, Rabinovitch PS, Ladiges WC.. Disruption of protein kinase A in mice enhances healthy aging. PLoS One 2009;4:e5963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Patrizio M, Vago V, Musumeci M, Fecchi K, Sposi NM, Mattei E, Catalano L, Stati T, Marano G.. cAMP-mediated beta-adrenergic signaling negatively regulates Gq-coupled receptor-mediated fetal gene response in cardiomyocytes. J Mol Cell Cardiol 2008;45:761–769. [DOI] [PubMed] [Google Scholar]
- 120. Weeks KL, Ranieri A, Karas A, Bernardo BC, Ashcroft AS, Molenaar C, McMullen JR, Avkiran MB.. Adrenergic stimulation induces histone deacetylase 5 (HDAC5) nuclear accumulation in cardiomyocytes by B55alpha-PP2A-mediated dephosphorylation. J Am Heart Assoc 2017;6:e004861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Backs J, Backs T, Neef S, Kreusser MM, Lehmann LH, Patrick DM, Grueter CE, Qi X, Richardson JA, Hill JA, Katus HA, Bassel-Duby R, Maier LS, Olson EN.. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc Natl Acad Sci USA 2009;106:2342–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Cribbs JT, Strack S.. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep 2007;8:939–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Sheridan CM, Heist EK, Beals CR, Crabtree GR, Gardner P.. Protein kinase A negatively modulates the nuclear accumulation of NF-ATc1 by priming for subsequent phosphorylation by glycogen synthase kinase-3. J Biol Chem 2002;277:48664–48676. [DOI] [PubMed] [Google Scholar]
- 124. Ichiki T. Role of cAMP response element binding protein in cardiovascular remodeling: good, bad, or both? ATVB 2006;26:449–455. [DOI] [PubMed] [Google Scholar]
- 125. Dash R, Frank KF, Carr AN, Moravec CS, Kranias EG.. Gender influences on sarcoplasmic reticulum Ca2+-handling in failing human myocardium. J Mol Cell Cardiol 2001;33:1345–1353. [DOI] [PubMed] [Google Scholar]
- 126. Zakhary DR, Moravec CS, Stewart RW, Bond M.. Protein kinase A (PKA)-dependent troponin-I phosphorylation and PKA regulatory subunits are decreased in human dilated cardiomyopathy. Circulation 1999;99:505–510. [DOI] [PubMed] [Google Scholar]
- 127. Messer AE, Jacques AM, Marston SB.. Troponin phosphorylation and regulatory function in human heart muscle: dephosphorylation of Ser23/24 on troponin I could account for the contractile defect in end-stage heart failure. J Mol Cell Cardiol 2007;42:247–259. [DOI] [PubMed] [Google Scholar]
- 128. Han YS, Arroyo J, Ogut O.. Human heart failure is accompanied by altered protein kinase A subunit expression and post-translational state. Arch Biochem Biophys 2013;538:25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Jacques AM, Copeland O, Messer AE, Gallon CE, King K, McKenna WJ, Tsang VT, Marston SB.. Myosin binding protein C phosphorylation in normal, hypertrophic and failing human heart muscle. J Mol Cell Cardiol 2008;45:209–216. [DOI] [PubMed] [Google Scholar]
- 130. Marston SB, de Tombe PP.. Troponin phosphorylation and myofilament Ca2+-sensitivity in heart failure: increased or decreased? J Mol Cell Cardiol 2008;45:603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Dong X, Sumandea CA, Chen YC, Garcia-Cazarin ML, Zhang J, Balke CW, Sumandea MP, Ge Y.. Augmented phosphorylation of cardiac troponin I in hypertensive heart failure. J Biol Chem 2012;287:848–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Barbagallo F, Xu B, Reddy GR, West T, Wang Q, Fu Q, Li M, Shi Q, Ginsburg KS, Ferrier W, Isidori AM, Naro F, Patel HH, Bossuyt J, Bers D, Xiang YK.. Genetically encoded biosensors reveal PKA hyperphosphorylation on the myofilaments in rabbit heart failure. Circ Res 2016;119:931–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Bryant S, Kimura TE, Kong CH, Watson JJ, Chase A, Suleiman MS, James AF, Orchard CH.. Stimulation of ICa by basal PKA activity is facilitated by caveolin-3 in cardiac ventricular myocytes. J Mol Cell Cardiol 2014;68:47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Nikolaev VO, Moshkov A, Lyon AR, Miragoli M, Novak P, Paur H, Lohse MJ, Korchev YE, Harding SE, Gorelik J.. Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 2010;327:1653–1657. [DOI] [PubMed] [Google Scholar]
- 135. Kirchhefer U, Schmitz W, Scholz H, Neumann J.. Activity of cAMP-dependent protein kinase and Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human hearts. Cardiovasc Res 1999;42:254–261. [DOI] [PubMed] [Google Scholar]
- 136. Aye TT, Soni S, van Veen TA, van der Heyden MA, Cappadona S, Varro A, de Weger RA, de Jonge N, Vos MA, Heck AJ, Scholten A.. Reorganized PKA-AKAP associations in the failing human heart. J Mol Cell Cardiol 2012;52:511–518. [DOI] [PubMed] [Google Scholar]
- 137. Haghighi K, Kolokathis F, Gramolini AO, Waggoner JR, Pater L, Lynch RA, Fan G-C, Tsiapras D, Parekh RR, Dorn GW, MacLennan DH, Kremastinos DT, Kranias EG.. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc Natl Acad Sci USA 2006;103:1388–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Haghighi K, Kolokathis F, Pater L, Lynch RA, Asahi M, Gramolini AO, Fan G-C, Tsiapras D, Hahn HS, Adamopoulos S, Liggett SB, Dorn GW, MacLennan DH, Kremastinos DT, Kranias EG.. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest 2003;111:869–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Mehel H, Emons J, Vettel C, Wittköpper K, Seppelt D, Dewenter M, Lutz S, Sossalla S, Maier LS, Lechêne P, Leroy J, Lefebvre F, Varin A, Eschenhagen T, Nattel S, Dobrev D, Zimmermann W-H, Nikolaev VO, Vandecasteele G, Fischmeister R, El-Armouche A.. Phosphodiesterase-2 is up-regulated in human failing hearts and blunts beta-adrenergic responses in cardiomyocytes. J Am Coll Cardiol 2013;62:1596–1606. [DOI] [PubMed] [Google Scholar]
- 140. Hashimoto T, Kim GE, Tunin RS, Adesiyun T, Hsu S, Nakagawa R, Zhu G, O’Brien JJ, Hendrick JP, Davis RE, Yao W, Beard D, Hoxie HR, Wennogle LP, Lee DI, Kass DA.. Acute enhancement of cardiac function by phosphodiesterase type 1 inhibition. Circulation 2018;138:1974–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Baliga RS, Preedy MEJ, Dukinfield MS, Chu SM, Aubdool AA, Bubb KJ, Moyes AJ, Tones MA, Hobbs AJ.. Phosphodiesterase 2 inhibition preferentially promotes NO/guanylyl cyclase/cGMP signaling to reverse the development of heart failure. Proc Natl Acad Sci USA 2018;115:E7428–E7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Riehle C, Bauersachs J.. Small animal models of heart failure. Cardiovasc Res 2019;115:1838–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Schiattarella GG, Altamirano F, Tong D, French KM, Villalobos E, Kim SY, Luo X, Jiang N, May HI, Wang ZV, Hill TM, Mammen PPA, Huang J, Lee DI, Hahn VS, Sharma K, Kass DA, Lavandero S, Gillette TG, Hill JA.. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 2019;568:351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Fu Q, Wang Q, Xiang YK.. Insulin and beta adrenergic receptor signaling: crosstalk in heart. Trends Endocrinol Metab 2017;28:416–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Mangmool S, Denkaew T, Phosri S, Pinthong D, Parichatikanond W, Shimauchi T, Nishida M.. Sustained betaAR stimulation mediates cardiac insulin resistance in a PKA-dependent manner. Mol Endocrinol 2016;30:118–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Mullins GR, Wang L, Raje V, Sherwood SG, Grande RC, Boroda S, Eaton JM, Blancquaert S, Roger PP, Leitinger N, Harris TE.. Catecholamine-induced lipolysis causes mTOR complex dissociation and inhibits glucose uptake in adipocytes. Proc Natl Acad Sci USA 2014;111:17450–17455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Wang Q, Liu Y, Fu Q, Xu B, Zhang Y, Kim S, Tan R, Barbagallo F, West T, Anderson E, Wei W, Abel ED, Xiang YK.. Inhibiting insulin-mediated beta2-adrenergic receptor activation prevents diabetes-associated cardiac dysfunction. Circulation 2017;135:73–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Bockus LB, Humphries KM.. cAMP-dependent protein kinase (PKA) signaling is impaired in the diabetic heart. J Biol Chem 2015;290:29250–29258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Minhas AS, Scheel P, Garibaldi B, Liu G, Horton M, Jennings M, Jones SR, Michos ED, Hays AG.. Takotsubo syndrome in the setting of COVID-19. JACC Case Rep 2020;2:1321–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Jabri A, Kalra A, Kumar A, Alameh A, Adroja S, Bashir H, Nowacki AS, Shah R, Khubber S, Kanaa NA, Hedrick DP, Sleik KM, Mehta N, Chung MK, Khot UN, Kapadia SR, Puri R, Reed GW.. Incidence of stress cardiomyopathy during the coronavirus disease 2019 pandemic. JAMA Netw Open 2020;3:e2014780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Scally C, Rudd A, Mezincescu A, Wilson H, Srivanasan J, Horgan G, Broadhurst P, Newby DE, Henning A, Dawson DK.. Persistent long-term structural, functional, and metabolic changes after stress-induced (takotsubo) cardiomyopathy. Circulation 2018;137:1039–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Pelliccia F, Kaski JC, Crea F, Camici PG.. Pathophysiology of takotsubo syndrome. Circulation 2017;135:2426–2441. [DOI] [PubMed] [Google Scholar]
- 153. Shao Y, Redfors B, Stahlman M, Tang MS, Miljanovic A, Mollmann H, Troidl C, Szardien S, Hamm C, Nef H, Boren J, Omerovic E.. A mouse model reveals an important role for catecholamine-induced lipotoxicity in the pathogenesis of stress-induced cardiomyopathy. Eur J Heart Fail 2013;15:9–22. [DOI] [PubMed] [Google Scholar]
- 154. Borchert T, Hübscher D, Guessoum CI, Lam T-DD, Ghadri JR, Schellinger IN, Tiburcy M, Liaw NY, Li Y, Haas J, Sossalla S, Huber MA, Cyganek L, Jacobshagen C, Dressel R, Raaz U, Nikolaev VO, Guan K, Thiele H, Meder B, Wollnik B, Zimmermann W-H, Lüscher TF, Hasenfuss G, Templin C, Streckfuss-Bömeke K.. Catecholamine-dependent beta-adrenergic signaling in a pluripotent stem cell model of takotsubo cardiomyopathy. J Am Coll Cardiol 2017;70:975–991. [DOI] [PubMed] [Google Scholar]
- 155. Llach A, Mazevet M, Mateo P, Villejouvert O, Ridoux A, Rucker-Martin C, Ribeiro M, Fischmeister R, Crozatier B, Benitah J-P, Morel E, Gómez AM.. Progression of excitation-contraction coupling defects in doxorubicin cardiotoxicity. J Mol Cell Cardiol 2019;126:129–139. [DOI] [PubMed] [Google Scholar]
- 156. Seicean S, Seicean A, Alan N, Plana JC, Budd GT, Marwick TH.. Cardioprotective effect of beta-adrenoceptor blockade in patients with breast cancer undergoing chemotherapy: follow-up study of heart failure. Circ Heart Fail 2013;6:420–426. [DOI] [PubMed] [Google Scholar]
- 157. Efentakis P, Varela A, Chavdoula E, Sigala F, Sanoudou D, Tenta R, Gioti K, Kostomitsopoulos N, Papapetropoulos A, Tasouli A, Farmakis D, Davos CH, Klinakis A, Suter T, Cokkinos DV, Iliodromitis EK, Wenzel P, Andreadou I.. Levosimendan prevents doxorubicin-induced cardiotoxicity in time- and dose-dependent manner: implications for inotropy. Cardiovasc Res 2020;116:576–591. [DOI] [PubMed] [Google Scholar]
- 158. Zhang Y, Knight W, Chen S, Mohan A, Yan C.. Multiprotein complex with TRPC (Transient Receptor Potential-Canonical) channel, PDE1C (Phosphodiesterase 1C), and A2R (Adenosine A2 Receptor) plays a critical role in regulating cardiomyocyte cAMP and survival. Circulation 2018;138:1988–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Trexler CL, Odell AT, Jeong MY, Dowell RD, Leinwand LA.. Transcriptome and functional profile of cardiac myocytes is influenced by biological sex. Circ Cardiovasc Genet 2017;10:e001770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Cao X, Zhou C, Chong J, Fu L, Zhang L, Sun D, Hou H, Zhang Y, Li D, Sun H.. Estrogen resisted stress-induced cardiomyopathy through increasing the activity of beta(2)AR-Galphas signal pathway in female rats. Int J Cardiol 2015;187:377–386. [DOI] [PubMed] [Google Scholar]
- 161. Machuki JO, Zhang HY, Geng J, Fu L, Adzika GK, Wu L, Shang W, Wu J, Kexue L, Zhao Z, Sun H.. Estrogen regulation of cardiac cAMP-L-type Ca(2+) channel pathway modulates sex differences in basal contraction and responses to beta2AR-mediated stress in left ventricular apical myocytes. Cell Commun Signal 2019;17:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. El-Battrawy I, Zhao Z, Lan H, Schunemann JD, Sattler K, Buljubasic F, Patocskai B, Li X, Yucel G, Lang S, Nowak D, Cyganek L, Bieback K, Utikal J, Zimmermann WH, Ravens U, Wieland T, Borggrefe M, Zhou XB, Akin I.. Estradiol protection against toxic effects of catecholamine on electrical properties in human-induced pluripotent stem cell derived cardiomyocytes. Int J Cardiol 2018;254:195–202. [DOI] [PubMed] [Google Scholar]
- 163. Parks RJ, Ray G, Bienvenu LA, Rose RA, Howlett SE.. Sex differences in SR Ca(2+) release in murine ventricular myocytes are regulated by the cAMP/PKA pathway. J Mol Cell Cardiol 2014;75:162–173. [DOI] [PubMed] [Google Scholar]
- 164. Parks RJ, Bogachev O, Mackasey M, Ray G, Rose RA, Howlett SE.. The impact of ovariectomy on cardiac excitation-contraction coupling is mediated through cAMP/PKA-dependent mechanisms. J Mol Cell Cardiol 2017;111:51–60. [DOI] [PubMed] [Google Scholar]
- 165. Lezoualc’h F, Fazal L, Laudette M, Conte C.. Cyclic AMP sensor EPAC proteins and their role in cardiovascular function and disease. Circ Res 2016;118:881–897. [DOI] [PubMed] [Google Scholar]
- 166. Robichaux WG, Cheng X.. Intracellular cAMP sensor EPAC: physiology, pathophysiology, and therapeutics development. Physiol Rev 2018;98:919–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Huang S, Li Q, Alberts I, Li X.. PRKX, a novel cAMP-dependent protein kinase member, plays an important role in development. J Cell Biochem 2016;117:566–573. [DOI] [PubMed] [Google Scholar]
- 168. Biel M. Cyclic nucleotide-regulated cation channels. J Biol Chem 2009;284:9017–9021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. James ZM, Borst AJ, Haitin Y, Frenz B, DiMaio F, Zagotta WN, Veesler D.. CryoEM structure of a prokaryotic cyclic nucleotide-gated ion channel. Proc Natl Acad Sci USA 2017;114:4430–4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Brand T. POPDC proteins and cardiac function. Biochem Soc Trans 2019;47:1393–1404. [DOI] [PubMed] [Google Scholar]
- 171. Diviani D, Reggi E, Arambasic M, Caso S, Maric D.. Emerging roles of A-kinase anchoring proteins in cardiovascular pathophysiology. Biochim Biophys Acta 2016;1863:1926–1936. [DOI] [PubMed] [Google Scholar]
- 172. Murray AJ. Pharmacological PKA inhibition: all may not be what it seems. Sci Signal 2008;1:re4. [DOI] [PubMed] [Google Scholar]