

Abstract
Aims
Cardiac involvement in COVID-19 is associated with adverse outcome. However, it is unclear whether cell-specific consequences are associated with cardiac SARS-CoV-2 infection. Therefore, we investigated heart tissue utilizing in situ hybridization, immunohistochemistry, and RNA-sequencing in consecutive autopsy cases to quantify virus load and characterize cardiac involvement in COVID-19.
Methods and results
In this study, 95 SARS-CoV-2-positive autopsy cases were included. A relevant SARS-CoV-2 virus load in the cardiac tissue was detected in 41/95 deceased (43%). Massive analysis of cDNA ends (MACE)-RNA-sequencing was performed to identify molecular pathomechanisms caused by the infection of the heart. A signature matrix was generated based on the single-cell dataset ‘Heart Cell Atlas’ and used for digital cytometry on the MACE-RNA-sequencing data. Thus, immune cell fractions were estimated and revealed no difference in immune cell numbers in cases with and without cardiac infection. This result was confirmed by quantitative immunohistological diagnosis. MACE-RNA-sequencing revealed 19 differentially expressed genes (DEGs) with a q-value <0.05 (e.g. up: IFI44L, IFT3, TRIM25; down: NPPB, MB, MYPN). The upregulated DEGs were linked to interferon pathways and originate predominantly from endothelial cells. In contrast, the downregulated DEGs originate predominately from cardiomyocytes. Immunofluorescent staining showed viral protein in cells positive for the endothelial marker ICAM1 but rarely in cardiomyocytes. The Gene Ontology (GO) term analysis revealed that downregulated GO terms were linked to cardiomyocyte structure, whereas upregulated GO terms were linked to anti-virus immune response.
Conclusion
This study reveals that cardiac infection induced transcriptomic alterations mainly linked to immune response and destruction of cardiomyocytes. While endothelial cells are primarily targeted by the virus, we suggest cardiomyocyte destruction by paracrine effects. Increased pro-inflammatory gene expression was detected in SARS-CoV-2-infected cardiac tissue but no increased SARS-CoV-2 associated immune cell infiltration was observed.
Keywords: SARS-CoV-2, COVID-19, Cardiac infection, RNA-seq, MACE, Cardiac signature matrix
Graphical Abstract

1. Introduction
To date, more than 4 million people died due to the global pandemic caused by SARS-CoV-2.1 Importantly, effective treatment options remain scarce due to limited knowledge of the exact pathological pathways involved in the disease. In addition to vaccinations, better therapies for patients with COVID-19 are needed. While repurposed antiviral drugs for patients hospitalized with COVID-19 did not reduce the overall mortality,2 immunosuppressant therapies appear to improve outcomes in patients with COVID-19.3,4 Therefore, understanding the immune response in organs that are key to survival may be essential for developing much-needed treatment strategies.
While the virus primarily targets the respiratory system, during the course of the disease, multi organotropism of SARS-CoV-2 was shown.5 This results in thrombotic complications and direct injury of the kidneys and the heart.6 Moreover, cardiac inflammation can be detected by magnetic resonance imaging after COVID-19.7 Furthermore, cardiac dysfunction in patients with COVID-19 predicts a severe course of the disease and premature death,8–10 and additionally increased troponin serum levels are associated with death.9,11 A previous pilot study from our group found SARS-CoV-2 viral RNA in 16/39 (41%) hearts of deceased who died with SARS-CoV-2 infection.12 It remains unclear whether this dangerous cardiac involvement is the result of direct virus infection of the heart or caused by secondary cardiac injury in the context of hypoxia, thrombosis, and multi-organ damage.13
Here, we examined cardiac tissue from 95 COVID-19 autopsy cases and detected SARS-CoV-2 in the heart in 43% of deceased. To unveil pathological consequences of direct cardiac infection with SARS-CoV-2, we compared myocardial tissue from fatal COVID-19 cases with and without cardiac infection utilizing massive analysis of cDNA ends (MACE)-RNA-sequencing and histological investigations. To address immune cell infiltration, a signature matrix based on the ‘Heart Cell Atlas’14 was generated, followed by digital cytometry, which was further confirmed by quantified immunohistological diagnosis. Furthermore, we aimed to identify the main target cell type for virus infection within the cardiac tissue based on fluorescent co-staining and differentially expressed genes (DEGs). To address the reasons for differences in susceptibility for cardiac infection, comorbidities and expression of virus entry factors were compared between cases with and without cardiac SARS-CoV-2 infection.
2. Methods
2.1 Study cohort and tissue sampling
Consecutively, 95 deceased with proven SARS-CoV-2 infection were autopsied at the Institute of Legal Medicine at the University Medical Centre Hamburg-Eppendorf in Germany between April and May 2020. The SARS-CoV-2 infection was confirmed prior to death or post-mortem by quantitative reverse transcription–polymerase chain reaction from pharyngeal swabs15; 88/95 deceased died due to pneumonia. A brief report investigating the SARS-CoV-2 infection of the myocardial tissue for the first 39 cases already described a virus load of more than 1000 copies per µg RNA in 41% of the cases.12 This study was now extended to 95 cases and additional investigations. Furthermore, cases 1–39 were part of an autopsy study of first COVID-19 deaths in Hamburg, Germany, reporting causes of death and accompanying comorbidities.16
Two tissue specimens from the free left ventricular (LV) wall were collected during autopsy and either snap-frozen in liquid nitrogen for RNA-based analyses or fixed in 10% neutral-buffered formalin for histological analyses.
This study was approved by the local ethics committee of the Hamburg Chamber of Physicians (PV7311). In accordance with the Infection Protection Act and the Hamburg Section Act, no individual written consent was necessary and the results may be used anonymously for scientific evaluation. All procedures performed in studies involving human participants were in accordance with ethical standards and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
2.2 RNA isolation and gene expression analyses
Total RNA was isolated using QIAzol (Qiagen, Germany) and subsequently purified with the miRNeasy mini kit (Qiagen, Germany) as described earlier.12,17 RNA was used for TaqMan analyses to quantify virus load and replication as well as gene expression using specific gene expression assays.
These methods are described in detailed in the Supplementary material online.
2.3 MACE as bulk 3′ mRNA-sequencing and Gene Ontology enrichment analyses
For 3′ mRNA sequencing analysis, 10 cases who exhibited a cardiac infection with more than 1000 copies per µg RNA were compared to 10 age-matched cases without cardiac infection selected from our study cohort. MACE is a 3′ mRNA sequencing method based on the analysis of Illumina reads derived from fragments that originate from 3′ mRNA end.18 Using autopsy samples, we opted for this MACE technology rather than regular RNA-seq.19 The RNA samples were processed using the MACE-Kit v.2 according to the manufacturer’s protocol (GenXPro GmbH, Germany). Subsequent Gene Ontology (GO) annotation data are based on ENSEMBL. GO enrichment analyses and their visualization are described in the Supplementary methods.
2.4 Signature matrix and digital cytometry
The signature matrix was generated utilizing the previously published dataset ‘Heart Cell Atlas’14 and can be downloaded in Supplementary material online, Appendix S1. Briefly, LV-derived cells were extracted from the dataset, DEGs for each cell type cluster was calculated with Seurat,20,21 and the resulting gene counts were used to create a signature matrix with CibersortX.22 Detailed methods, as well as the validation of the signature matrix, are described in the Supplementary material online. Next, MACE-RNA-seq data were used to perform digital cytometry to estimate cell fractions with CibersortX.
2.5 Immunohistochemistry and immunofluorescence
LV tissue was fixed in 10% neutral-buffered formalin for at least 48 h at room temperature. Four micrometre-thick formalin-fixed paraffin-embedded (FFPE) tissue sections were rehydrated and used for immunohistochemistry, immunofluorescence, and in situ hybridization as described in the Supplementary methods.17,23
2.6 In situ hybridization—RNAscope
For detection of SARS-CoV-2 plus and minus strand, RNAscope in situ hybridization was performed on FFPE tissue sections of the left ventricle using RNAscope® 2.5 HD Reagent Kit-RED (#322350; Advanced Cell Diagnostics (ACD), USA). For fluorescent staining, RNAscope Multiplex Fluorescent v2 reagent kit (#323100, ACD, USA) was used. Tissue pre-treatment, hybridization of target-specific probe (see Supplementary material online, Table S3), subsequent amplification steps, and signal detection were performed according to the manufacturer’s instructions and described in detail in the Supplementary methods.
2.7 Statistics for case characteristics
Case characteristics were reported as medians and inter-quartile ranges in brackets for continuous variables. Binary variables are shown as counts with frequencies in brackets. The P-values were determined by the comparison to those cases without cardiac infection. Detailed information regarding Kaplan–Meier method and odds ratio is provided in the Supplementary material online.
2.8 General statistics
In general, Mann–Whitney U test was used for continuous variables and chi-squared test for binary variables. However, since different kind of data require specific statistical methods, detailed information about the used test is provided in each methods section in the Supplementary material online as well as in the figure legends.
3. Results
3.1 Cardiac infection in fatal COVID-19 cases
We investigated 95 deceased who died with a confirmed SARS-CoV-2 infection. As shown in Table 1, the median age was 84 years (IQR: 76–87) and 54% of them were female. The majority of patients died in hospitals (36% normal care and 26% intensive care) or nursing homes (27%, Supplementary material online, Table S4); 88/95 cases died due to pneumonia. In some cases, sepsis (11 cases) or pulmonary embolism (6 cases) was diagnosed as an additional cause of death. Cardiac tissue was collected during autopsy with a median post-mortem interval of 4 days (IQR: 3–7).
Table 1.
Baseline characteristics for all 95 deceased and subsequently defined groups of cases sorted by virus load in cardiac tissue
| All cases (n = 95) | Cases grouped by cardiac virus load |
Cases analysed by MACE-RNA-seqa |
|||||
|---|---|---|---|---|---|---|---|
| No cardiac infection (n = 46) | Cardiac infection (>1000 copies)b (n = 41) | P-valuec | No cardiac infection (n = 10) | Cardiac infection (>1000 copies) (n = 10) | P-valuec | ||
| Female n (%) | 51 (54) | 21 (46) | 24 (59) | 0.32 | 5 (50) | 7 (70) | 0.65 |
| Age (years) | 84 (76–87) | 81 (74–86) | 84 (78–88) | 0.27 | 82 (78–84) | 84 (79–86) | 0.34 |
| Post-mortem interval (days) | 4.0 (3.0–7.0) | 4.0 (2.0–6.0) | 4.0 (3.0–8.0) | 0.38 | 4.0 (1.5–4.0) | 3.5 (2.2–4.8) | 0.79 |
| Time, diagnosis until death (days) | 9.0 (3.0–18.0) | 12.5 (7.2–20.2) | 8.0 (2.0–10.0) | 0.082 | 12.0 (5.5–19.5) | 8.0 (2.0–10.0) | 0.43 |
| Comorbidities, n (%) | |||||||
| HTN | 44 (46) | 24 (52) | 17 (42) | 0.43 | 4 (40) | 5 (50) | 1.00 |
| CAD | 58 (61) | 31 (67) | 22 (54) | 0.28 | 5 (50) | 4 (40) | 1.00 |
| HF | 49 (52) | 23 (50) | 22 (54) | 0.90 | 4 (40) | 5 (50) | 1.00 |
| Sum of cardiac comorbidities ≥2, n (%) | 48 (51) | 26 (57) | 18 (44) | 0.34 | 2 (20) | 4 (40) | 0.63 |
| DM | 18 (19) | 11 (24) | 5 (12) | 0.26 | 3 (30) | 1 (10) | 0.58 |
| Stroke | 16 (17) | 7 (15) | 7 (17) | 1.00 | 2 (20) | 1 (10) | 1.00 |
| COPD | 49 (52) | 23 (50) | 24 (59) | 0.56 | 2 (20) | 5 (50) | 0.35 |
| Cancer | 29 (31) | 13 (28) | 14 (34) | 0.72 | 3 (30) | 7 (70) | 0.18 |
| Renal failure | 34 (36) | 16 (35) | 17 (42) | 0.67 | 2 (20) | 4 (40) | 0.63 |
| Sum of non-cardiac comorbidities ≥2 | 48 (51) | 22 (48) | 23 (56) | 0.58 | 3 (30) | 7 (70) | 0.18 |
| Sum of comorbidities ≥2 | 88 (93) | 42 (91) | 38 (93) | 1.00 | 8 (80) | 10 (100) | 0.46 |
| BMI (kg/m2) | 24.4 (20.7–28.7) | 24.4 (20.9–28.4) | 23.2 (19.1–28.7) | 0.58 | 27.4 (25.2–28.6) | 22.4 (20.9–31.7) | 0.57 |
| Heart weight (g) | 450 (361–520) | 455 (384–510) | 420 (334–530) | 0.40 | 453 (408–518) | 460 (348–534) | 0.76 |
For continuous variables median (25th–75th percentile) are given. For binary variables absolute numbers and relative frequencies (%) are given. The data availability for all variables is shown in Supplementary material online, Table S4.
BMI, body mass index; CAD, coronary artery disease; COPD, chronic obstructive pulmonary disease; DM, diabetes mellitus; HF, heart failure; HTN, hypertension.
For MACE-RNA-seq, patients died in hospital and with post-mortem intervals of maximal 5 days were chosen from the groups defined by virus load in the cardiac tissue.
Since 1000 copies per µg RNA was deemed as clinically relevant, 8 out of 95 cases <1000 copies per µg RNA were excluded from the group with cardiac infection.
The presented P-values were determined by the comparison to those cases without cardiac infection. Mann–Whitney U test was used for continuous variables and chi-squared test for binary variables.
Virus load in all 95 cardiac tissue specimens was determined from isolated RNA by reverse transcription followed by quantitative PCR. As shown in Figure 1A, SARS-CoV-2 was not present in cardiac tissue in 46 out of 95 cases. A virus load of 1000 copies per µg RNA was deemed as clinically relevant.12 In 41 out of 95 cases (43%), a relevant virus load of SARS-CoV-2 RNA was detected in the heart, while 8 cases did not exceed the clinically relevant number and were therefore excluded from comparisons between cases with and without cardiac infection. The group with cardiac infection revealed a median virus load of 7952 copies per µg RNA (IQR: 2507–32 005). Quantified individual virus load for each case is plotted in Supplementary material online, Figure S1A. To visualize virus, in situ hybridization was performed on tissue sections (n = 4). As shown in Figure 1B, SARS-CoV-2 virus genome (plus strand) was detected either as clusters representing high abundance of virus or as single dots shown in the magnification. Observing the same area in the consecutive section, SARS-CoV-2 intermediate strand for replication (minus strand) was detected to a lower extent. In this representative case, the virus genome was apparently detectable in non-cardiomyocyte structures.
Figure 1.
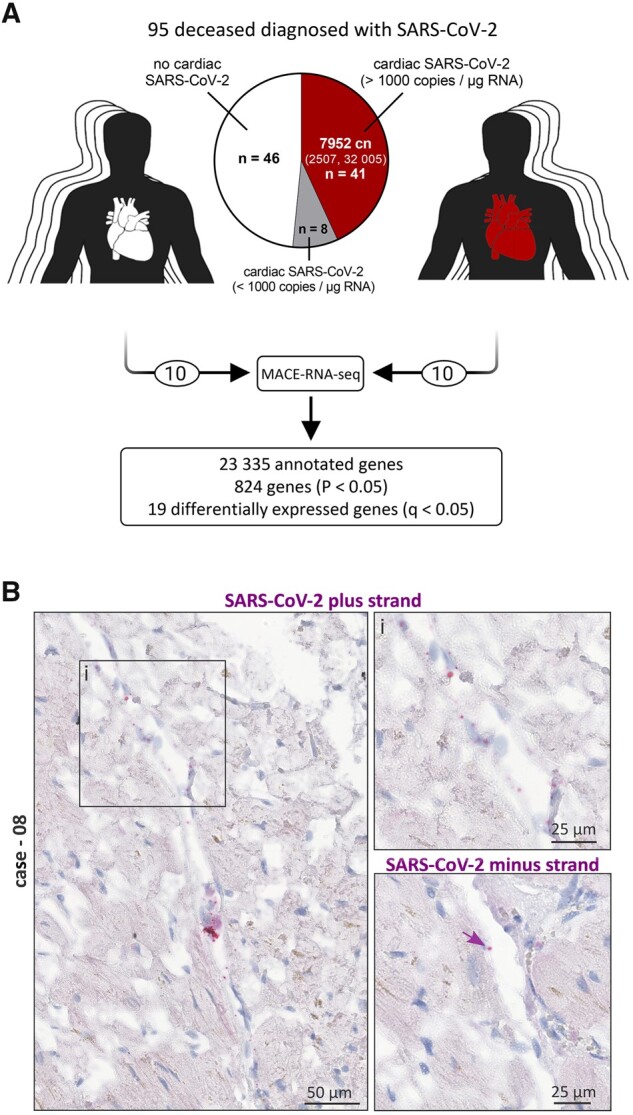
Presence of SARS-CoV-2 in cardiac left ventricular tissue of fatal COVID-19 cases. (A) Presence of SARS-CoV-2 RNA was examined in the cardiac tissue of 95 SARS-CoV-2-positive deceased. Virus load in 1 µg RNA was quantified by reverse transcription followed by quantitative polymerase chain reaction (RT–PCR). Despite the SARS-CoV-2 diagnosis, 46 out of 95 cases revealed no SARS-CoV-2 in the cardiac tissue. A copy number >1000 copies per µg RNA in the heart was deemed as clinically relevant and was detected in 41 cases, while 8 cases did not exceed the relevant number and were therefore excluded from further analyses. Cases without cardiac infection are depicted in white, whereas cases with cardiac infection are depicted in red. The median copy number (cn) per µg RNA was 7952 (IQR: 2507–32 005). Virus load for each case individually is plotted as heatmap in Supplementary material online, Figure S1A. MACE-RNA-seq identified 19 differentially expressed genes (DEGs) comparing cardiac tissue with (n = 10) and without (n = 10) cardiac infection. (B) In situ hybridization was used to visualize SARS-CoV-2 RNA on tissue specimens. Hybridized probes are specific either for the plus strand representing the viral genome or the minus strand representing the intermediate strand for replication. Representative images of Case 08 are displayed. Negative and positive controls for chromogenic in situ hybridization are shown in Supplementary material online, Figure S2.
For bulk 3′ mRNA-sequencing (MACE-RNA-seq), 10 cases with high virus load in cardiac tissue were selected and compared to 10 age-matched SARS-CoV-2-positive cases without cardiac infection. As shown in Table 1, the characteristics for both groups analysed by MACE-RNA-seq represent the entire study cohort regarding the listed categories. By MACE-RNA-seq, 23 335 genes were annotated, whereof 824 with a P-value <0.05 and 19 DEGs with a q-value <0.05. MACE-RNA-seq data for all annotated genes are provided in Supplementary material online, Appendix S2.
3.2 SARS-CoV-2 infection of the heart is not associated with increased immune cell infiltration
To investigate immune cell infiltration as a histological sign of virus-induced myocarditis according to the definition published in the position paper of Caforio et al.,24 we performed two analyses: (i) estimating cell fractions from MACE-RNA-seq data by digital cytometry and (ii) immunohistochemical staining to diagnose immune cell infiltration.
Based on the previously published single-cell RNA-sequencing (scRNA-seq) results from the ‘Heart Cell Atlas’,14 a signature matrix was generated comprising only cells from LV tissue as described in detail in the Supplementary material online. Next, the CibersortX-computed signature matrix was validated on two other scRNA-seq datasets from cardiac tissue as described in the Supplementary material online. The signature matrix is shown in Supplementary material online, Figure S5. To utilize the signature matrix for other bulk RNA-seq data from LV tissue, all necessary information is provided in Supplementary material online, Appendix S1.
The cell fractions for each case were individually estimated based on the MACE-RNA-seq data. The mean cell fractions per group were calculated to compare samples with (n = 10) and without (n = 10) cardiac SARS-CoV-2 infection. Cell fraction of endothelial cells (21.3 ± 2.0% vs. 30.3 ± 3.7%; P = 0.0478), neuronal cells (2.0 ± 0.3% vs. 3.0 ± 0.2%; P = 0.0099), and adipocytes (0.14 ± 0.024% vs. 0.32 ± 0.064%; P = 0.0180) is significantly increased in cases with cardiac infection. Box plots of imputed cell fractions are presented in Supplementary material online, Figure S7. Regarding immune cell infiltration, lymphoid (7.8 ± 1.3% vs. 8.8 ± 1.8%; P = 0.6854) and myeloid cells (4.3 ± 0.6% vs. 4.9 ± 0.8%; P = 0.6140) were presented as box plots in Figure 2B.
Figure 2.

Cardiac SARS-CoV-2 infection was not associated with increased immune cell infiltration. (A) The previously published ‘Heart Cell Atlas’14 served as single-cell RNA-sequencing dataset to generate a signature matrix using CibersortX from left ventricular tissue originated cells. Digital cytometry was performed, applying the generated signature matrix to our MACE-RNA-seq data by CibersortX. Cell fractions were estimated for each individual case. Comparing the immune cell fractions between cases with (n = 10) and without (n = 10) cardiac infection, no significant differences were determined using Mann–Whitney U test. Cell fractions are depicted as Tukey-style box plots with median and inter-quartile range without outliers. (B) The number of positively stained immune cells per mm2 is displayed. Cases without cardiac infections (n = 46) are depicted in white, whereas cases with cardiac infection (n = 41) are depicted in red. The patient groups were compared using Mann–Whitney U test revealing no significant differences. Data are depicted as Tukey-style box plots with median and inter-quartile range without outliers. Representative staining of immune cells in cardiac tissue is displayed. On the left panel, the CD45R0 staining for Case 65, quantified as 15.1 cells per mm2, is shown. The middle panel depicts the CD3 staining for Case 21 (7.4 cells/mm2). For Case 47, the CD68 staining is shown on the right panel (14.7 cells/mm2). Magnifications are displayed below for each case.
Subsequently, immune cell infiltration was determined by immunohistochemistry of all 95 cases. Therefore, different T-cell subtypes and macrophages were quantified. The number of positive cells was compared between cases with (n = 41) and without (n = 46) cardiac infection (Figure 2B). No significant differences were determined for CD45R0 (P = 0.61), CD3 (P = 0.17), and CD68 (P = 0.30) between both groups, while CD8 was only detectable in very few cases. Representative staining is shown for three different cases exhibiting increased immune cell numbers of the respective subpopulation. Cases with immune cell infiltrates above the recommended threshold were distributed equally between both groups. In addition, quantified immunohistochemistry is plotted for each case individually in Supplementary material online, Figure S1B.
3.3 DEGs determined by MACE-RNA-seq
Utilizing MACE-RNA-seq, more than 23 000 genes were annotated. As shown in Figure 3A, 824 genes with a P-value <0.05 were identified, 394 genes were upregulated (red), and 430 genes downregulated (blue) in cardiac tissue with cardiac SARS-CoV-2 infection. Next, these 824 genes were restricted by the FDR-adjusted P-value, resulting in 19 DEGs with a q-value <0.05, 11 upregulated (dark red) and 8 downregulated (dark blue). The most significant upregulated gene was APOD (No. 1), while the most significant downregulated gene was NPPB (No. 12). As depicted in the profile plot, both were abundantly expressed (Figure 3B). Interestingly, three interferon-related genes (IFI44L, IFIT3, TRIM25) are upregulated, while the cardiomyocyte marker MB was downregulated. The data and names for all 19 DEGs are plotted for each case separately as heatmap in Figure 3C. To validate the identified DEGs, gene expression of all 19 DEGs was measured by TaqMan. The median fold change is plotted as technical replication next to the median of MACE-RNA-seq results. Individual results per case were plotted in Supplementary material online, Figure S8.
Figure 3.

Identification of 19 differentially expressed genes (DEGs). Gene expression was analysed by MACE-RNA-seq in 10 cases with and 10 cases without cardiac infection. Downregulated genes in SARS-CoV-2-infected cardiac tissue are depicted in blue, while red highlights upregulated genes. FDR-adjusted P-value (q-value) revealed 19 DEGs. (A) Fold change and P-value are displayed in the volcano plot to visualize significant differences in gene expression; 11 upregulated and 8 downregulated DEGs were identified. Gene symbols and full names are displayed in (C) according to here depicted numbers. DEGs were calculated using DESeq2. (B) In the profile plot, fold change and average of normalized counts per 1 million reads are displayed. Red and blue colours highlight genes with P-value <0.05 (small dots) or q-value <0.05 (large dots). Gene symbols and full names are displayed in (C) according to here depicted numbers. (C) The 19 DEGs are displayed for each biological replicate separately. Gene symbols and full names are given. Gene expression is normalized to the mean of the group without cardiac infection and plotted as fold change (log2). As summary, median fold change of each DEG is plotted. Additionally, DEGs were validated using TaqMan analysis and the gene expression is plotted as median fold change (log2). Individual TaqMan values are plotted as heatmap in Supplementary material online, Figure S8. ci, cardiac infection; nci, no cardiac infection.
3.4 Cell type-specific expression of 19 DEGs
To assign the 19 DEGs to specific cardiac cell types, we used LV-derived cells from the scRNA-seq dataset ‘Heart Cell Atlas’14 comprising 14 donors. The average gene expression per cell type and donor of all 19 DEGs are plotted as heatmap with red colour gradient for upregulated and blue colour gradient for downregulated genes in Figure 4A and B, respectively.
Figure 4.

Cellular distribution of differentially expressed genes (DEGs) and cell-specific virus infection. (A and B) The cellular origin of the 19 DEGs was examined in healthy hearts of the published dataset ‘Heart Cell Atlas’.14 Log2 gene expression is plotted, separately for each donor, as the average expression of all cells within the same cell type. Note that data from lymphoid cells are available in 13 of 14 donors and adipocytes in 6 out of 14. (A) The gene expression of the upregulated DEGs is displayed. While APOD is highly expressed in most cell types, its weaker expression in cardiomyocytes is evident. A distinct cluster of the genes IFIT3, IFI44L, ITPR3, and TRIM25 is visible in endothelial cells. IFIT3 is also highly expressed in pericytes. (B) The gene expression of the downregulated DEGs is displayed. The genes AKAP6, MB, MYPN, and NPPB are forming a highly expressed cluster in cardiomyocytes. (C and D) Representative immunofluorescent staining on cardiac tissue from three different cases is depicted. For visualization of the tissue structure, cell membranes and fibrotic tissue are stained with wheat germ agglutinin (WGA, white). (C) Co-staining of SARS-CoV-2 nuclear protein (red) and the endothelial marker intercellular adhesion molecule 1 (ICAM1, green). In all depicted cases, the viral protein is localized within an ICAM1-positive cell. (D) SARS-CoV-2 nuclear protein (red) is not co-localized with α-actinin (green) in all representative cases, but can be found in small cells adjacent to cardiomyocytes.
As shown in Figure 4A, APOD is strongly expressed in most cell types, while its weaker expression in cardiomyocytes is evident. However, in endothelial cells, a distinct cluster is visible consisting of the genes IFIT3, IFI44L, ITPR3, and TRIM25. Additionally, IFIT3 is also highly expressed in pericytes. As shown in Figure 4B, a distinct cluster in cardiomyocytes comprising the genes AKAP6, MB, MYPN, and NPPB is evident. Furthermore, LYZ is mainly expressed in immune cells and FN1 in fibroblasts. Taken together, genes upregulated in SARS-CoV-2-infected cardiac tissue originate mainly from endothelial cells, whereas downregulated genes originate mainly from cardiomyocytes.
3.5 Identification of cardiac cell types infected by SARS-CoV-2
To unveil the cardiac cell type infected by SARS-CoV-2, we performed fluorescent in situ hybridization to visualize viral RNA (n = 4) and immunofluorescence staining (n = 4) to detect viral nuclear protein. The fluorescent in situ co-staining of SARS-CoV-2 RNA plus strand (red) and wheat germ agglutinin (WGA, cyan) as marker for cell membranes and fibrotic tissue is depicted in Supplementary material online, Figure S3. In the majority of cases, viral RNA is co-localized with WGA indicating a virus localization in the interstitium or non-myocyte cells. Localization within cardiomyocytes as depicted in Supplementary material online, Figure S3g is rare. To identify specific cell types, immunofluorescence staining was performed with antibodies against ICAM1 or α-actinin together with the nuclear protein of SARS-CoV-2.
As shown in Figure 4C, viral protein (red) was co-localized with ICAM1-stained cells (green), a marker for endothelial cells. In contrast, no co-localization was observed with the cardiomyocyte marker α-actinin (green) and the viral protein (red) shown in Figure 4D. However, the viral protein was detectable in cells surrounding cardiomyocytes, which is in line with the in situ results.
3.6 GO enrichment analysis
GO term analysis of the MACE-RNA-seq data revealed 188 significantly regulated GO terms (q-value <0.05) within all three GO categories. To narrow these GO term analysis, we used the gene ratio calculated by the number of regulated and annotated genes in the respective GO term. A gene ratio of 0.2 means that at least 20% of all annotated genes are regulated in this GO term. In Figure 5, all GO terms with a gene ratio >0.2 and a q-value <0.001 are displayed. Five out of 10 significant GO terms are linked to anti-virus immune response, while the other five are linked to cardiomyocyte structure. The cardiomyocyte-specific GO terms comprise mainly downregulated genes depicted by a negative z score. This is in line with the finding in Figure 4B, that the downregulated genes were mainly assigned to cardiomyocytes. In contrast, the immune-related GO terms revealed a positive z score indicating that these terms mainly consist of upregulated genes.
Figure 5.

Top regulated Gene Ontology (GO) terms are linked to cardiomyocytes and immune response. For each GO term, the gene ratio of regulated genes and annotated genes was calculated. Displayed are GO terms with a gene ratio >0.2 and a q-value <0.001. Whether the majority of genes in this term is up- or downregulated is indicated on the X-axis (z score). The q-value of the respective GO term is displayed by the colour gradient of the respective dot. The size of the dots represents the number of annotated genes. The GO terms are linked to immune response (green) or cardiomyocyte structure (purple). However, cardiomyocyte-specific GO terms are downregulated (negative z score), while GO terms linked to immune response are upregulated (positive z score).
3.7 Replication of SARS-CoV-2 in cardiac tissue is associated with shorter time until death
Besides the virus load, we further investigated whether signs of virus replication are present in the cardiac tissue. Therefore, the presence of the intermediate (–) RNA strand was examined. As shown in Figure 6A, in 28/41 cases with cardiac infection, virus replication was detectable, 14 with weak, 10 with moderate, and 4 with strong virus replication.
Figure 6.

Cardiac replication of SARS-CoV-2 was associated with accelerated death but not with comorbidities. (A) 95 fatal COVID-19 cases were included in this study, whereof 41 exceed a virus load of >1000 copies per µg RNA in the heart. In these cases, virus replication was examined, using a tagged minus strand-specific primer for reverse transcription followed by quantitative PCR of the tagged cDNA. Replication was scored according to the Ct-values (0: no replication/1: weak/2: moderate/3: strong). (B) Kaplan–Meier curves show time between diagnosis and death for cases grouped by cardiac infection and virus replication. Time between diagnosis and death is given by the interval between the first positive respiratory swab for SARS-CoV-2 and date of death. The compared groups were defined using information that was unknown before death. The groups with cardiac infection (red, n = 25) or replication (dark grey, n = 9) were compared to the group without cardiac infection (light grey, n = 26) utilizing log-rank test. To compare multiple survival curves, P-values were adjusted by the Hochberg correction. (C) Expression of four SARS-CoV-2 entry genes was determined using TaqMan analysis. Cases without cardiac infection (n = 46) are depicted in white, whereas cases with cardiac infection (n = 41) are depicted in red. Cases with cardiac infection were constricted to those with virus replication scores >1 (n = 14) and are depicted in dark grey. Gene expression is normalized to the median expression of the group without cardiac infection. Data are depicted as Tukey-style box plots with median and IQR without outliers. Both groups with cardiac infection were separately compared to the group without cardiac infection. Significant differences were determined using Mann–Whitney U test. A P-value <0.05 was considered statistically significant. (D) Comorbidities and cardiac infection (n = 41) or replication (n = 14) were fitted using univariate logistic regression models. Odds ratios (OR) and 95% confidence intervals (CI) were displayed as forest plots. The vertical line at an OR of one is the line of no effect. Due to graphical issues, the CI is clipped indicated by arrows. A P-value <0.05 was considered statistically significant, but was not reached for this analysis. The data availability for all variables is shown in Supplementary material online, Table S4. CAD, coronary artery disease; cCormobidites, cardiac Comorbidities; COPD, chronic obstructive pulmonary disease; DM, diabetes mellitus; HF, heart failure; HTN, hypertension; IQR, inter-quartile range; ncCormobidities, non-cardiac comorbidities.
According to virus load and virus replication of SARS-CoV-2 in cardiac tissue, we defined three groups: (i) without cardiac infection (n = 46), (ii) with cardiac infection higher than 1000 copies (n = 41), which was further constricted to (iii) cases with cardiac virus replication scores higher than 1 (n = 14). Two comparisons were performed: first, cases without cardiac infection (i) to cases with cardiac infection (ii). Second, cases without cardiac infection (i) to cases with cardiac replication (iii). As shown in Supplementary material online, Table S1, both comparisons revealed no significant differences in age, sex, and post-mortem intervals indicating matched groups with respect to typical confounders in post-mortem setting. However, time between diagnosis and death, representing the interval between the first positive respiratory swab for SARS-CoV-2 and date of death, was significantly shorter in cases with cardiac replication (iii) compared to cases without cardiac infection (i) (P = 0.017, Supplementary material online, Table S1). Accordingly, time between diagnosis and death was analysed utilizing Kaplan–Meier Curves shown in Figure 6B and revealed significant differences (P = 0.012). Virus replication in the heart was associated with a significantly shorter survival time.
Next, we examined whether cardiac infection or cardiac replication is associated with expression levels of entry factors described for SARS-CoV-2. Therefore, gene expression of angiotensin-converting enzyme-2 (ACE2), transmembrane serine protease-2 (TMPRSS2), cathepsin-B (CTSB), and cathepsin-L (CTSL) was compared. As depicted in Figure 6C, gene expression of TMPRSS2 was significantly increased in the groups with cardiac infection (P = 0.033, ii) and replication (P = 0.039, iii), whereas gene expression of ACE2, CTSB, and CTSL remained nearly unchanged.
To analyse whether comorbidities promote cardiac infection, odds ratios for the cardiac comorbidities hypertension, coronary artery disease, heart failure (HF), or the sum of cardiac comorbidities revealed no association between comorbidities and cardiac infection or replication (Figure 6D). Additionally, other non-cardiac comorbidities such as diabetes mellitus, stroke, chronic obstructive pulmonary disease, cancer, or the sum of non-cardiac or all comorbidities are not associated with cardiac infection or replication as well.
4. Discussion
Here, in the largest available cardiac autopsy series so far, cardiac tissue with and without SARS-CoV-2 infection of fatal COVID-19 cases was compared. Therefore, MACE-RNA-seq was performed to identify molecular pathomechanisms caused by the local infection of the heart. The key findings of the study are: (i) an increase of immune cell infiltrates due to cardiac SARS-CoV-2 infection was not detected using digital cytometry and immunohistological diagnosis; (ii) MACE-RNA-seq identified increased pro-inflammatory gene expression accompanied by reduced expression of cardiomyocyte-specific genes; (iii) upregulated pro-inflammatory genes mainly originate from endothelial cells; and (iv) fluorescent co-staining revealed that endothelial cells were frequently infected by SARS-CoV-2, whereas infection of cardiomyocytes was rarely detectable.
This cohort study was performed in the metropolitan area of Hamburg, Germany; 85% of the deceased were aged 70 years or older, which is consistent with COVID-19 deaths in Germany.25 Virus infection of non-respiratory tract organs was documented in COVID-19 autopsy studies earlier.5,8,26 However, we recently showed that SARS-CoV-2 can be detected in the heart of ∼50% of fatal COVID-19 cases.12 By analysing more cases, we confirmed that the heart is targeted by SARS-CoV-2 in ∼40–50% of the COVID-19 autopsy cases. To address virus-associated transcriptomic alterations, 10 cases with and 10 cases without cardiac SARS-CoV-2 infection were analysed utilizing MACE-RNA-seq.
4.1 No increased immune cell infiltrates due to cardiac SARS-CoV-2 infection
To address histological signs of myocarditis, we generated a signature matrix based on the scRNA-seq dataset ‘Heart Cell Atlas’,14 which was used to perform digital cytometry on the determined transcriptome. Cell fractions were estimated, in particular for immune cell fractions, and cases with and without cardiac infection were compared. Based on the transcriptome, no differences in immune cell fractions were determined by digital cytometry between both groups. To validate this result, all 46 cases without cardiac infection and all 41 cases with cardiac infection were diagnosed by immunohistochemistry on tissue sections. For that purpose, different inflammatory cell markers including different T-cell subtypes as well as macrophages were quantified. Again, direct cardiac infection was not accompanied by increased inflammatory cell infiltrates. Case reports of positive cardiac SARS-CoV-2 infection accompanied by histological signs of myocarditis are rare.27,28 However, signs of myocardial inflammation in patients recovered from COVID-19 but without confirmed cardiac SARS-CoV-2 infection were also reported with different incidence rates.7,29,30 Hence, myocarditis might be developed as sequela due to active or prior COVID-19, which does not necessarily include cardiac infection with SARS-CoV-2. Whether myocardial virus activity in the absence of clinically evident myocardial inflammation might result in long-term consequences is unknown.
4.2 Increased pro-inflammatory gene expression in cases with cardiac SARS-CoV-2
Utilizing MACE-RNA-seq, 824 genes with a P-value <0.05 were identified and subsequently narrowed to 19 DEGs with a q-value <0.05. Interestingly, those contained two upregulated apolipoproteins (APOE and APOD) and two upregulated interferon-related genes (IFI44L and IFIT3) as well as upregulated TRIM25 known as interferon expression inducing factor. Interferons are central to antiviral immunity, but impaired interferon activity was described in severe COVID-19 patients.31 In line with this finding, our second top gene IFI44L was described as negative feedback regulator of host antiviral response. IFI44L-silenced cells revealed reduced virus titre indicating that IFI44L supports virus replication.32
Based on the scRNA-seq dataset ‘Heart Cell Atlas’,14 the cellular origin of the 19 DEGs was visualized. While APOD and APOE originate from various cardiac cell types, with the lowest expression in cardiomyocytes, a distinct cluster comprising IFIT3, IFI44L, and TRIM25 was detectable in endothelial cells as main origin. In line with these findings, our fluorescent co-staining revealed co-localization of viral protein and ICAM1 as endothelial marker. Accordingly, we suggest that endothelial cells mainly respond to virus infection in the cardiac tissue. In contrast, the cellular distribution of the downregulated DEGs revealed a distinct cluster comprising AKAP6, MB, MYPN, and NPPB mainly originated from cardiomyocytes.
To go beyond single genes, GO enrichment analysis was performed resulting in overall 188 significant GO terms (q-value <0.05). Subsequently, we selected GO terms with a high ratio between regulated and annotated genes. In line with the identified 19 DEGs, interferon-related GO terms are highly upregulated as well as GO terms which negatively regulate virus replication and life cycle. Additionally, all downregulated GO terms were linked to cardiomyocyte structure suggesting that cardiac SARS-CoV-2 infection leads to destruction of cardiomyocytes. Despite no increased immune cell infiltrates were detected, destruction of cardiomyocytes and pro-inflammatory transcriptomic alterations may fulfil the criteria of for ‘-itis’ as the acronym for inflammation in medical terminology.
Recently, in vitro infection of iPSC-derived cardiac cell types revealed that besides cardiomyocytes, also fibroblasts and endothelial cells are targeted by SARS-CoV-2.33 Visual cytopathic effects are most prevalent in endothelial cells indicating the toxicity of viral exposure.33 In a cardiac tissue sample, SARS-CoV-2 infection of vascular endothelial cells was confirmed recently.34 In line with this finding, our histological analyses revealed viral RNA as well as protein mostly in endothelial or interstitial cells. In several studies, COVID-19 was associated with cardiac endotheliitis without confirmed cardiac endothelial virus infection.35,36 Similarly, pro-inflammatory DEGs were mainly assigned to endothelial cells in our study. Contrarily to in vitro infection of induced pluripotent stem cell-derived (iPSC) cardiomyocytes, in cardiac tissue of the autopsy samples, viral RNA was rarely detectable but not absent in cardiomyocytes.33,37 However, Perez-Bermejo et al.33 reported that myofibrillar disruption in SARS-CoV-2-exposed cardiomyocytes was independent from actively replicating virus. This myofibrillar fragmentation was also observed in cardiomyocytes adjacent to cells with viral dsRNA. Together with our data, this creates evidence that paracrine effects from cells activated by viral exposure may contribute to destruction of cardiomyocytes. This is coherent to other virus infections, which do not target primarily myocytes and still cause myocardial dysfunction.38,39
4.3 Susceptibility of the myocardium for SARS-CoV-2 infection and virus replication
It is unclear which factors determine the susceptibility of the myocardium for SARS-CoV-2 infection. Therefore, gene expression of ACE2, known as receptor of the Spike protein, and the host proteases TMPRSS2, CTSB, and CTSL was investigated.37,40,41 Interestingly, the infection of myocardial cells and replication of SARS-CoV-2 were associated with increased gene expression of the transmembrane protease serine 2 (TMPRSS2). TMPRSS2 is facilitating virus entry into the host cell as well as being essential for virus spread and pathogenicity.41 This is the first report of TMPRSS2 being differently regulated within cardiac tissue and therefore potentially mediating virus infection of cardiac tissue. Together, this might fuel therapeutic application of TMPRSS2 inhibitors for COVID-19, as are being tested in a clinical trial (NCT04321096). Obviously, further studies need to verify this finding, but intriguingly, TMPRSS2 knock-out reduced the inflammatory response after experimental SARS-CoV-2 infection.42 This is coherent to our data, showing less inflammatory response in cases without cardiac SARS-CoV-2 infection. Contrarily, ACE2, as the main receptor for virus uptake, as well as the proteases CTSL and CTSB, known to be involved in viral membrane fusion40 and priming the Spike protein of the virus, were not dysregulated. This suggests that susceptibility to the virus might not be correlated to changes in gene expression of those targets. A previous study showed that ACE2 is increased in cardiomyocytes of patients with HF or aortic stenosis compared to healthy controls.43 In our study, ACE2 is not differentially regulated in the groups with or without cardiac infection, possibly justified by similar cardiac comorbidities in both groups. In our study, 93% of cases suffered from two or more comorbidities which is coherent with other autopsy studies.25,26,44,45 Interestingly, neither cardiac nor other comorbidities were associated with cardiac infection and signs of virus replication in the myocardium were not more frequent in those with comorbidities. This is different in other organs, like the kidney, where cases with organ manifestation in general were more likely to have more comorbidities.5,8 Age might also be a predictor to fuel organotropism of SARS-CoV-2, but all cases included here were of high age making this difficult to assess. In general, autopsy studies need to be interpreted with caution in view of these associations, since survivors of the disease are excluded, which makes this subgroup of COVID-19 patients unique.
In 15% of deceased, relevant virus replication in the myocardium was detected and associated with accelerated death suggesting that active cardiac infection could be a contributor to death. Similarly, SARS-CoV-2 infection of kidney resulted in premature death as well.8 Future studies have to reveal whether replication actively accelerates death or whether more severe COVID-19 patients are prone for cardiac replication.
Although it is already known that cardiac involvement in COVID-19 is associated with adverse outcome, the role of direct myocardial infection is still unclear. It is therefore a clinically important and novel finding, that myocardial SARS-CoV-2 infection is associated with pro-inflammatory transcriptomic alterations. Our study provides new evidence on pro-inflammatory changes in cardiac diseases that could complement the definition of myocarditis in the future. However, the cause of susceptibility for virus infection and replication needs to be elucidated in further studies.
4.4 Limitation
This study has some limitations, including the design as an autopsy study. Elderly age and frequently observed comorbidities of deceased might have influenced the result of cardiac infections and replication. Presumably, in non-autopsy studies, the incidence of virus infection will be lower. Endomyocardial biopsies of patients with COVID-19 are rare and especially no clinical routine so far. As an autopsy study, we have only limited clinical information and no information is present about myocardial biomarkers, which might be upregulated due to the SARS-CoV-2 infection or to systemic inflammation due to COVID-19. Unfortunately, post-mortem biochemistry of cardiac biomarkers in cadaveric serum is not useful even within the first 48 h after death.46 Regarding the observed accelerated death in cases with cardiac virus replication, it should be considered that the diagnosis of SARS-CoV-2 could be performed at different points in progression of the disease. Consequently, time between diagnosis and death could be shorter in cases with cardiac replication without causality. The presented transcriptomic data provide a snapshot at the time of autopsy, whether DEGs are the cause or a consequence of cardiac infection needs to be investigated in the future. Due to the study design, no causality can be established between the transcriptomic alterations and the observed accelerated death. Since all cases were tested positive for SARS-CoV-2 infection, we cannot identify factors mediating an impaired cardiac function due to systemic inflammation caused by COVID-19.
5. Conclusion
Taken together, SARS-CoV-2 induces transcriptomic alterations by direct infection of the myocardium. While an increase of immune cell infiltrates was not detected, pro-inflammatory response on transcriptomic level was observed. Our findings provide further insights about cell-specific consequences of cardiac infection. We suggest negative paracrine effects on cardiomyocytes. They do not seem to be the main target cell type for virus infection. In contrast, SARS-CoV-2-infected endothelial cells were observed frequently. Moreover, a distinct cluster of upregulated DEGs was detectable in endothelial cells as main origin. Accordingly, we suggest that endothelial cells mainly respond to virus infection in the cardiac tissue. Nevertheless, increasing pathophysiological understanding might allow precise and targeted interventions to prevent early and late subsequent virus-associated myocardial damage.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Authors’ contributions
D.L., H.B., and D.W. designed the research study. A.D.E.F., K.M., C.E., B.O., and K.P. contributed samples. D.L., H.B., G.A., M.S., B.R., S.W., and F.E. collected experimental data. A.D.E.F., C.E., F.B., K.R., S.K., T.B.H., B.O., K.P., and D.W. provided clinical data. D.L., H.B., B.S., G.A., J.W., B.R., F.E., H.-P.S., and D.W. analysed data and performed statistical analysis. D.L., H.B., K.S., P.K., S.B., and D.W. drafted the manuscript. All authors contributed intellectually and approved the manuscript.
Conflict of interest: S.B. has received research funding from Abbott Diagnostics, Bayer, SIEMENS, Singulex, and Thermo Fisher. He received honoraria for lectures from Abbott, Abbott Diagnostics, Astra Zeneca, Bayer, AMGEN, Medtronic, Pfizer, Roche, SIEMENS Diagnostics, SIEMENS, Thermo Fisher, and as member of Advisory Boards and for consulting for Bayer, Novartis, and Thermo Fisher unrelated to the submitted work. P.K. reports research support for basic, translational, and clinical research projects from European Union, British Heart Foundation, Leducq Foundation, Medical Research Council (UK), and German Centre for Cardiovascular Research; support from several drug and device companies active in atrial fibrillation; and has received honoraria from several such companies in the past, but not in the past 3 years. He is listed as inventor on two patents held by the University of Birmingham (Atrial Fibrillation Therapy, WO 2015140571; Markers for Atrial Fibrillation, WO 2016012783), unrelated to the submitted work. S.K. received research support from Ambu, Daiichi Sankyo, ETView Ltd, Fisher & Paykel, Pfizer, and Xenios. He also received lecture fees from Astra, Basilea, C.R. Bard, Baxter, Biotest, Cytosorbents, Fresenius, Gilead, MSD, Pfizer, Philips, and Zoll. He received consultant fees from Baxter, Fresenius, Gilead, MSD, and Pfizer. D.W. reports receiving a speaker’s fee from AstraZeneca, Bayer, and Novartis, unrelated to the submitted work.
Funding
This work was supported by the German Centre for Cardiovascular Research (DZHK) (FKZ 81Z0710108 to D.W. and D.L.), by the grant to D.L. and D.W. from the Deutsche Herzstiftung and by CRC1192 to T.B.H. A.D.E.F., K.M., C.E., K.P., and B.O. were supported by the DEFEAT PANDEMIcs project, which was funded by the German Federal Ministry of Education and Research (BMBF) under grant number 01KX2021. This work was further funded by a grant from the Authorities for Social Welfare, Hamburg, Germany to the Institute of Legal Medicine Hamburg, Germany.
Data availability
The MACE-RNA-seq data underlying this article are available in the article and its Supplementary material online, Appendix S2. The generated signature matrix and underlying data are available in Supplementary material online, Appendix S1.
Supplementary Material
Acknowledgements
We thank the UKE Microscopy Imaging Facility (Umif), University Hospital Centre Hamburg-Eppendorf for providing microscopes and support, and GenXPro for performing MACE analyses and bioinformatics. Parts of figures were created with BioRender.com.
Contributor Information
Hanna Bräuninger, Department of Cardiology, University Heart and Vascular Centre Hamburg, Martinistr. 52, 20246 Hamburg, Germany; DZHK (German Centre for Cardiovascular Research), Partner Site Hamburg/Kiel/Lübeck, 20246 Hamburg, Germany.
Bastian Stoffers, Department of Cardiology, University Heart and Vascular Centre Hamburg, Martinistr. 52, 20246 Hamburg, Germany; DZHK (German Centre for Cardiovascular Research), Partner Site Hamburg/Kiel/Lübeck, 20246 Hamburg, Germany.
Antonia D E Fitzek, Institute of Legal Medicine, University Medical Centre Hamburg-Eppendorf, Martinistr. 52, 20246 Hamburg, Germany.
Kira Meißner, Institute of Legal Medicine, University Medical Centre Hamburg-Eppendorf, Martinistr. 52, 20246 Hamburg, Germany.
Ganna Aleshcheva, Institute for Cardiac Diagnostics and Therapy, Moltkestraße 31, 12203 Berlin, Germany.
Michaela Schweizer, Department of Electron Microscopy, Centre for Molecular Neurobiology, University Medical Centre Hamburg-Eppendorf, Falkenried 94, 20251 Hamburg, Germany.
Jessica Weimann, Department of Cardiology, University Heart and Vascular Centre Hamburg, Martinistr. 52, 20246 Hamburg, Germany.
Björn Rotter, GenXPro GmbH, Frankfurter Innovationszentrum, Biotechnologie (FIZ), Altenhöferallee 3, 60438 Frankfurt am Main, Germany.
Svenja Warnke, Department of Cardiology, University Heart and Vascular Centre Hamburg, Martinistr. 52, 20246 Hamburg, Germany.
Carolin Edler, Institute of Legal Medicine, University Medical Centre Hamburg-Eppendorf, Martinistr. 52, 20246 Hamburg, Germany.
Fabian Braun, III Department of Medicine, University Medical Centre Hamburg-Eppendorf, Martinistr. 52, 20246 Hamburg, Germany.
Kevin Roedl, Department of Intensive Care Medicine, University Medical Centre Hamburg-Eppendorf, Martinistr. 52, 20246 Hamburg, Germany.
Katharina Scherschel, Department of Cardiology, University Heart and Vascular Centre Hamburg, Martinistr. 52, 20246 Hamburg, Germany; Division of Cardiology (cNEP), EVK Düsseldorf, Kirchfeldstrasse 40, 40217 Düsseldorf, Germany; Medical Faculty, Institute of Neural and Sensory Physiology, Heinrich Heine University Düsseldorf, Universitätsstrasse 1, 40225 Düsseldorf, Germany.
Felicitas Escher, Institute for Cardiac Diagnostics and Therapy, Moltkestraße 31, 12203 Berlin, Germany; Department of Cardiology, Charité-Universitaetsmedizin, Augustenburger Platz 1, 13353 Berlin, Germany; DZHK (German Centre for Cardiovascular Research), Partner site Berlin, Berlin, Germany.
Stefan Kluge, Department of Intensive Care Medicine, University Medical Centre Hamburg-Eppendorf, Martinistr. 52, 20246 Hamburg, Germany.
Tobias B Huber, III Department of Medicine, University Medical Centre Hamburg-Eppendorf, Martinistr. 52, 20246 Hamburg, Germany.
Benjamin Ondruschka, Institute of Legal Medicine, University Medical Centre Hamburg-Eppendorf, Martinistr. 52, 20246 Hamburg, Germany.
Heinz-Peter Schultheiss, Institute for Cardiac Diagnostics and Therapy, Moltkestraße 31, 12203 Berlin, Germany.
Paulus Kirchhof, Department of Cardiology, University Heart and Vascular Centre Hamburg, Martinistr. 52, 20246 Hamburg, Germany; DZHK (German Centre for Cardiovascular Research), Partner Site Hamburg/Kiel/Lübeck, 20246 Hamburg, Germany; Institute of Cardiovascular Sciences, University of Birmingham, Birmingham, UK.
Stefan Blankenberg, Department of Cardiology, University Heart and Vascular Centre Hamburg, Martinistr. 52, 20246 Hamburg, Germany; DZHK (German Centre for Cardiovascular Research), Partner Site Hamburg/Kiel/Lübeck, 20246 Hamburg, Germany.
Klaus Püschel, Institute of Legal Medicine, University Medical Centre Hamburg-Eppendorf, Martinistr. 52, 20246 Hamburg, Germany.
Dirk Westermann, Department of Cardiology, University Heart and Vascular Centre Hamburg, Martinistr. 52, 20246 Hamburg, Germany; DZHK (German Centre for Cardiovascular Research), Partner Site Hamburg/Kiel/Lübeck, 20246 Hamburg, Germany.
Diana Lindner, Department of Cardiology, University Heart and Vascular Centre Hamburg, Martinistr. 52, 20246 Hamburg, Germany; DZHK (German Centre for Cardiovascular Research), Partner Site Hamburg/Kiel/Lübeck, 20246 Hamburg, Germany.
Translational perspective
Cardiac injury can be documented in COVID-19, regardless the direct cardiac virus infection and is known to be associated with outcome. However, the direct virus infection of the myocardium leads to transcriptomic alterations and might therefore additionally contribute to pathophysiological processes in COVID-19. Therefore, consequences of cardiac virus infection need to be investigated in future studies, since they might also contribute to long-term effects in case of survival.
References
- 1.GBD 2017 Causes of Death Collaborators. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018;392:1736–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.WHO Solidarity Trial Consortium. Repurposed antiviral drugs for Covid-19—interim WHO solidarity trial results. N Engl J Med 2021;384:497–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.RECOVERY Collaborative Group, Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, Linsell L, Staplin N, Brightling C, Ustianowski A, Elmahi E, Prudon B, Green C, Felton T, Chadwick D, Rege K, Fegan C, Chappell LC, Faust SN, Jaki T, Jeffery K, Montgomery A, Rowan K, Juszczak E, Baillie JK, Haynes R, Landray MJ. Dexamethasone in hospitalized patients with COVID-19. N Engl J Med 2021;384:693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shao Z, Feng Y, Zhong L, Xie Q, Lei M, Liu Z, Wang C, Ji J, Liu H, Gu Z, Hu Z, Su L, Wu M, Liu Z. Clinical efficacy of intravenous immunoglobulin therapy in critical ill patients with COVID-19: a multicenter retrospective cohort study. Clin Transl Immunology 2020;9:e1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Puelles VG, Lutgehetmann M, Lindenmeyer MT, Sperhake JP, Wong MN, Allweiss L, Chilla S, Heinemann A, Wanner N, Liu S, Braun F, Lu S, Pfefferle S, Schroder AS, Edler C, Gross O, Glatzel M, Wichmann D, Wiech T, Kluge S, Pueschel K, Aepfelbacher M, Huber TB. Multiorgan and renal tropism of SARS-CoV-2. N Engl J Med 2020;383:590–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gupta A, Madhavan MV, Sehgal K, Nair N, Mahajan S, Sehrawat TS, Bikdeli B, Ahluwalia N, Ausiello JC, Wan EY, Freedberg DE, Kirtane AJ, Parikh SA, Maurer MS, Nordvig AS, Accili D, Bathon JM, Mohan S, Bauer KA, Leon MB, Krumholz HM, Uriel N, Mehra MR, Elkind MSV, Stone GW, Schwartz A, Ho DD, Bilezikian JP, Landry DW. Extrapulmonary manifestations of COVID-19. Nat Med 2020;26:1017–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Puntmann VO, Carerj ML, Wieters I, Fahim M, Arendt C, Hoffmann J, Shchendrygina A, Escher F, Vasa-Nicotera M, Zeiher AM, Vehreschild M, Nagel E. Outcomes of cardiovascular magnetic resonance imaging in patients recently recovered from coronavirus disease 2019 (COVID-19). JAMA Cardiol 2020;5:1265–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Braun F, Lütgehetmann M, Pfefferle S, Wong MN, Carsten A, Lindenmeyer MT, Nörz D, Heinrich F, Meißner K, Wichmann D, Kluge S, Gross O, Pueschel K, Schröder AS, Edler C, Aepfelbacher M, Puelles VG, Huber TB. SARS-CoV-2 renal tropism associates with acute kidney injury. Lancet 2020;396:597–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Giustino G, Croft LB, Stefanini GG, Bragato R, Silbiger JJ, Vicenzi M, Danilov T, Kukar N, Shaban N, Kini A, Camaj A, Bienstock SW, Rashed ER, Rahman K, Oates CP, Buckley S, Elbaum LS, Arkonac D, Fiter R, Singh R, Li E, Razuk V, Robinson SE, Miller M, Bier B, Donghi V, Pisaniello M, Mantovani R, Pinto G, Rota I, Baggio S, Chiarito M, Fazzari F, Cusmano I, Curzi M, Ro R, Malick W, Kamran M, Kohli-Seth R, Bassily-Marcus AM, Neibart E, Serrao G, Perk G, Mancini D, Reddy VY, Pinney SP, Dangas G, Blasi F, Sharma SK, Mehran R, Condorelli G, Stone GW, Fuster V, Lerakis S, Goldman ME. Characterization of myocardial injury in patients with COVID-19. J Am Coll Cardiol 2020;76:2043–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guzik TJ, Mohiddin SA, Dimarco A, Patel V, Savvatis K, Marelli-Berg FM, Madhur MS, Tomaszewski M, Maffia P, D’Acquisto F, Nicklin SA, Marian AJ, Nosalski R, Murray EC, Guzik B, Berry C, Touyz RM, Kreutz R, Wang DW, Bhella D, Sagliocco O, Crea F, Thomson EC, McInnes IB. COVID-19 and the cardiovascular system: implications for risk assessment, diagnosis, and treatment options. Cardiovasc Res 2020;116:1666–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guo T, Fan Y, Chen M, Wu X, Zhang L, He T, Wang H, Wan J, Wang X, Lu Z. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol 2020;5:811–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lindner D, Fitzek A, Brauninger H, Aleshcheva G, Edler C, Meissner K, Scherschel K, Kirchhof P, Escher F, Schultheiss HP, Blankenberg S, Puschel K, Westermann D. Association of cardiac infection with SARS-CoV-2 in confirmed COVID-19 autopsy cases. JAMA Cardiol 2020;5:1281–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Knowlton KU. Pathogenesis of SARS-CoV-2 induced cardiac injury from the perspective of the virus. J Mol Cell Cardiol 2020;147:12–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Litviňuková M, Talavera-López C, Maatz H, Reichart D, Worth CL, Lindberg EL, Kanda M, Polanski K, Heinig M, Lee M, Nadelmann ER, Roberts K, Tuck L, Fasouli ES, DeLaughter DM, McDonough B, Wakimoto H, Gorham JM, Samari S, Mahbubani KT, Saeb-Parsy K, Patone G, Boyle JJ, Zhang H, Zhang H, Viveiros A, Oudit GY, Bayraktar OA, Seidman JG, Seidman CE, Noseda M, Hubner N, Teichmann SA. Cells of the adult human heart. Nature 2020;588:466–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pfefferle S, Reucher S, Norz D, Lutgehetmann M. Evaluation of a quantitative RT-PCR assay for the detection of the emerging coronavirus SARS-CoV-2 using a high throughput system. Euro Surveill 2020;25:2000152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Edler C, Schröder AS, Aepfelbacher M, Fitzek A, Heinemann A, Heinrich F, Klein A, Langenwalder F, Lütgehetmann M, Meißner K, Püschel K, Schädler J, Steurer S, Mushumba H, Sperhake J-P. Dying with SARS-CoV-2 infection—an autopsy study of the first consecutive 80 cases in Hamburg, Germany. Int J Legal Med 2020;134:1275–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hinrichs S, Scherschel K, Kruger S, Neumann JT, Schwarzl M, Yan I, Warnke S, Ojeda FM, Zeller T, Karakas M, Keller T, Meyer C, Blankenberg S, Westermann D, Lindner D. Precursor proadrenomedullin influences cardiomyocyte survival and local inflammation related to myocardial infarction. Proc Natl Acad Sci U S A 2018;115:E8727–E8736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Muller S, Rycak L, Afonso-Grunz F, Winter P, Zawada AM, Damrath E, Scheider J, Schmah J, Koch I, Kahl G, Rotter B. APADB: a database for alternative polyadenylation and microRNA regulation events. Database (Oxford) 2014;2014:bau076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Boneva S, Schlecht A, Bohringer D, Mittelviefhaus H, Reinhard T, Agostini H, Auw-Haedrich C, Schlunck G, Wolf J, Lange C. 3' MACE RNA-sequencing allows for transcriptome profiling in human tissue samples after long-term storage. Lab Invest 2020;100:1345–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, Hoffman P, Stoeckius M, Papalexi E, Mimitou EP, Jain J, Srivastava A, Stuart T, Fleming LM, Yeung B, Rogers AJ, McElrath JM, Blish CA, Gottardo R, Smibert P, Satija R. Integrated analysis of multimodal single-cell data. Cell 2021;184:3573–3587.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, Satija R. Comprehensive integration of single-cell data. Cell 2019;177:1888–1902.e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, Khodadoust MS, Esfahani MS, Luca BA, Steiner D, Diehn M, Alizadeh AA. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 2019;37:773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Scherschel K, Hedenus K, Jungen C, Lemoine MD, Rubsamen N, Veldkamp MW, Klatt N, Lindner D, Westermann D, Casini S, Kuklik P, Eickholt C, Klocker N, Shivkumar K, Christ T, Zeller T, Willems S, Meyer C. Cardiac glial cells release neurotrophic S100B upon catheter-based treatment of atrial fibrillation. Sci Transl Med 2019;11:eaav7770. [DOI] [PubMed] [Google Scholar]
- 24. Caforio AL, Pankuweit S, Arbustini E, Basso C, Gimeno-Blanes J, Felix SB, Fu M, Helio T, Heymans S, Jahns R, Klingel K, Linhart A, Maisch B, McKenna W, Mogensen J, Pinto YM, Ristic A, Schultheiss HP, Seggewiss H, Tavazzi L, Thiene G, Yilmaz A, Charron P, Elliott PM; European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2013;34:2636–2648. [DOI] [PubMed] [Google Scholar]
- 25. Menter T, Haslbauer JD, Nienhold R, Savic S, Hopfer H, Deigendesch N, Frank S, Turek D, Willi N, Pargger H, Bassetti S, Leuppi JD, Cathomas G, Tolnay M, Mertz KD, Tzankov A. Postmortem examination of COVID-19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings in lungs and other organs suggesting vascular dysfunction. Histopathology 2020;77:198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Remmelink M, De Mendonca R, D’Haene N, De Clercq S, Verocq C, Lebrun L, Lavis P, Racu ML, Trepant AL, Maris C, Rorive S, Goffard JC, De Witte O, Peluso L, Vincent JL, Decaestecker C, Taccone FS, Salmon I. Unspecific post-mortem findings despite multiorgan viral spread in COVID-19 patients. Crit Care 2020;24:495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Escher F, Pietsch H, Aleshcheva G, Bock T, Baumeier C, Elsaesser A, Wenzel P, Hamm C, Westenfeld R, Schultheiss M, Gross U, Morawietz L, Schultheiss HP. Detection of viral SARS-CoV-2 genomes and histopathological changes in endomyocardial biopsies. ESC Heart Fail 2020;7:2440–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fox SE, Li G, Akmatbekov A, Harbert JL, Lameira FS, Brown JQ, Vander Heide RS. Unexpected features of cardiac pathology in COVID-19 infection. Circulation 2020;142:1123–1125. [DOI] [PubMed] [Google Scholar]
- 29. Joy G, Artico J, Kurdi H, Seraphim A, Lau C, Thornton GD, Oliveira MF, Adam RD, Aziminia N, Menacho K, Chacko L, Brown JT, Patel RK, Shiwani H, Bhuva A, Augusto JB, Andiapen M, McKnight A, Noursadeghi M, Pierce I, Evain T, Captur G, Davies RH, Greenwood JP, Fontana M, Kellman P, Schelbert EB, Treibel TA, Manisty C, Moon JC; COVIDsortium Investigators. Prospective case-control study of cardiovascular abnormalities 6 months following mild COVID-19 in healthcare workers. JACC Cardiovasc Imaging 2021;doi: 10.1016/j.jcmg.2021.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Daniels CJ, Rajpal S, Greenshields JT, Rosenthal GL, Chung EH, Terrin M, Jeudy J, Mattson SE, Law IH, Borchers J, Kovacs R, Kovan J, Rifat SF, Albrecht J, Bento AI, Albers L, Bernhardt D, Day C, Hecht S, Hipskind A, Mjaanes J, Olson D, Rooks YL, Somers EC, Tong MS, Wisinski J, Womack J, Esopenko C, Kratochvil CJ, Rink LD, Simonetti O, Zareba K, Bhatti S, Addison D, Obarski T, Daoud E, Granger M, Smart S, Mayercin-Johnson J, Subramanian P, Glitt J, Mitchell D, Chumita R, Mumford A, Garcia A, Garris L, Liu H, Hatfield B, Zhang Y, Boersma D, Schlader Z, Goodwin S, Port N, Zuidema T, Maldonado J, Eckhardt L, Reeder S, Baker M, Sebastianelli W, Wadlinger R, Millard R, Bosha P, Sunday H, Steele D, Chaudhry A, Smith S, Pfeiffer M, Kellerman J, Billy G, Krystofiak J, Eimer M; Big Ten COVID-19 Cardiac Registry Investigators. Prevalence of clinical and subclinical myocarditis in competitive athletes with recent SARS-CoV-2 infection: results from the big ten COVID-19 cardiac registry. JAMA Cardiol 2021;6:1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, Péré H, Charbit B, Bondet V, Chenevier-Gobeaux C, Breillat P, Carlier N, Gauzit R, Morbieu C, Pène F, Marin N, Roche N, Szwebel T-A, Merkling SH, Treluyer J-M, Veyer D, Mouthon L, Blanc C, Tharaux P-L, Rozenberg F, Fischer A, Duffy D, Rieux-Laucat F, Kernéis S, Terrier B. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020;369:718–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. DeDiego ML, Martinez-Sobrido L, Topham DJ. Novel functions of IFI44L as a feedback regulator of host antiviral responses. J Virol 2019;93:e01159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Perez-Bermejo JA, Kang S, Rockwood SJ, Simoneau CR, Joy DA, Silva AC, Ramadoss GN, Flanigan WR, Fozouni P, Li H, Chen PY, Nakamura K, Whitman JD, Hanson PJ, McManus BM, Ott M, Conklin BR, McDevitt TC. SARS-CoV-2 infection of human iPSC-derived cardiac cells reflects cytopathic features in hearts of patients with COVID-19. Sci Transl Med 2021;13:eabf7872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu J, Li Y, Liu Q, Yao Q, Wang X, Zhang H, Chen R, Ren L, Min J, Deng F, Yan B, Liu L, Hu Z, Wang M, Zhou Y. SARS-CoV-2 cell tropism and multiorgan infection. Cell Discov 2021;7:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Maccio U, Zinkernagel AS, Shambat SM, Zeng X, Cathomas G, Ruschitzka F, Schuepbach RA, Moch H, Varga Z. SARS-CoV-2 leads to a small vessel endotheliitis in the heart. EBioMedicine 2021;63:103182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, Mehra MR, Schuepbach RA, Ruschitzka F, Moch H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020;395:1417–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bojkova D, Wagner JUG, Shumliakivska M, Aslan GS, Saleem U, Hansen A, Luxan G, Gunther S, Pham MD, Krishnan J, Harter PN, Ermel UH, Frangakis AS, Milting H, Zeiher AM, Klingel K, Cinatl J, Dendorfer A, Eschenhagen T, Tschope C, Ciesek S, Dimmeler S. SARS-CoV-2 infects and induces cytotoxic effects in human cardiomyocytes. Cardiovasc Res 2020;116:2207–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Becher PM, Gotzhein F, Klingel K, Escher F, Blankenberg S, Westermann D, Lindner D. Cardiac function remains impaired despite reversible cardiac remodeling after acute experimental viral myocarditis. J Immunol Res 2017;2017:6590609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lindner D, Li J, Savvatis K, Klingel K, Blankenberg S, Tschope C, Westermann D. Cardiac fibroblasts aggravate viral myocarditis: cell specific coxsackievirus B3 replication. Mediators Inflamm 2014;2014:519528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Daniloski Z, Jordan TX, Wessels HH, Hoagland DA, Kasela S, Legut M, Maniatis S, Mimitou EP, Lu L, Geller E, Danziger O, Rosenberg BR, Phatnani H, Smibert P, Lappalainen T, tenOever BR, Sanjana NE. Identification of required host factors for SARS-CoV-2 infection in human cells. Cell 2021;184:92–105.e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hoffmann M, Kleine-Weber H, Pöhlmann S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol Cell 2020;78:779–784.e775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Iwata-Yoshikawa N, Okamura T, Shimizu Y, Hasegawa H, Takeda M, Nagata N. TMPRSS2 contributes to virus spread and immunopathology in the airways of murine models after coronavirus infection. J Virol 2019;93:e01815-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nicin L, Abplanalp WT, Mellentin H, Kattih B, Tombor L, John D, Schmitto JD, Heineke J, Emrich F, Arsalan M, Holubec T, Walther T, Zeiher AM, Dimmeler S. Cell type-specific expression of the putative SARS-CoV-2 receptor ACE2 in human hearts. Eur Heart J 2020;41:1804–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schaller T, Hirschbuhl K, Burkhardt K, Braun G, Trepel M, Markl B, Claus R. Postmortem examination of patients with COVID-19. JAMA 2020;323:2518–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bradley BT, Maioli H, Johnston R, Chaudhry I, Fink SL, Xu H, Najafian B, Deutsch G, Lacy JM, Williams T, Yarid N, Marshall DA. Histopathology and ultrastructural findings of fatal COVID-19 infections in Washington State: a case series. Lancet 2020;396:320–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Woydt L, Bernhard M, Kirsten H, Burkhardt R, Hammer N, Gries A, Dreßler J, Ondruschka B. Intra-individual alterations of serum markers routinely used in forensic pathology depending on increasing post-mortem interval. Sci Rep 2018;8:12811. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MACE-RNA-seq data underlying this article are available in the article and its Supplementary material online, Appendix S2. The generated signature matrix and underlying data are available in Supplementary material online, Appendix S1.