

Abstract
Aims
Angiotensin (Ang) II signalling has been suggested to promote cardiac fibrosis in inflammatory heart diseases; however, the underlying mechanisms remain obscure. Using Agtr1a-/- mice with genetic deletion of angiotensin receptor type 1 (ATR1) and the experimental autoimmune myocarditis (EAM) model, we aimed to elucidate the role of Ang II-ATR1 pathway in development of heart-specific autoimmunity and post-inflammatory fibrosis.
Methods and results
EAM was induced in wild-type (WT) and Agtr1a-/- mice by subcutaneous injections with alpha myosin heavy chain peptide emulsified in complete Freund’s adjuvant. Agtr1a-/- mice developed myocarditis to a similar extent as WT controls at day 21 but showed reduced fibrosis and better systolic function at day 40. Crisscross bone marrow chimaera experiments proved that ATR1 signalling in the bone marrow compartment was critical for cardiac fibrosis. Heart infiltrating, bone-marrow-derived cells produced Ang II, but lack of ATR1 in these cells reduced transforming growth factor beta (TGF-β)-mediated fibrotic responses. At the molecular level, Agtr1a-/- heart-inflammatory cells showed impaired TGF-β-mediated phosphorylation of Smad2 and TAK1. In WT cells, TGF-β induced formation of RhoA-GTP and RhoA-A-kinase anchoring protein-Lbc (AKAP-Lbc) complex. In Agtr1a-/- cells, stabilization of RhoA-GTP and interaction of RhoA with AKAP-Lbc were largely impaired. Furthermore, in contrast to WT cells, Agtr1a-/- cells stimulated with TGF-β failed to activate canonical Wnt pathway indicated by suppressed activity of glycogen synthase kinase-3 (GSK-3)β and nuclear β-catenin translocation and showed reduced expression of Wnts. In line with these in vitro findings, β-catenin was detected in inflammatory regions of hearts of WT, but not Agtr1a-/- mice and expression of canonical Wnt1 and Wnt10b were lower in Agtr1a-/- hearts.
Conclusion
Ang II-ATR1 signalling is critical for development of post-inflammatory fibrotic remodelling and dilated cardiomyopathy. Our data underpin the importance of Ang II-ATR1 in effective TGF-β downstream signalling response including activation of profibrotic Wnt/β-catenin pathway.
Keywords: Angiotensin II, Angiotensin II receptor 1, Experimental autoimmune myocarditis, Inflammatory cells, Wnt, TGF-β signalling, Cardiac fibrosis
Graphical Abstract
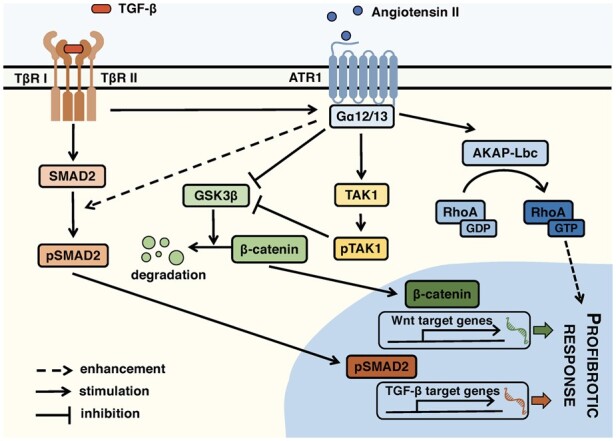
1. Introduction
Myocarditis refers to inflammation of the heart muscle. In humans, infections with cardiotropic viruses or parasite T. cruzi often trigger heart-specific autoimmunity.1 Autoimmune responses often result in ongoing inflammation and heart tissue remodelling, which finally leads to dilated cardiomyopathy (DCM) - a pathogenic condition characterized by fibrotic changes in the myocardium, ventricular dilation, and systolic dysfunction.2 Moreover, myocarditis has been acknowledged as an underestimated cause of sudden death in children and young adults.3
Experimental autoimmune myocarditis (EAM) represents a non-infectious animal model of myocarditis and DCM reflecting key aspects of the human disease.4 In the EAM model, susceptible mice are immunized with alpha myosin heavy chain alpha (αMyHC) in the presence of complete Freund’s adjuvant. Immunized mice develop CD4+ T cell-dependent myocarditis, characterized by an extensive infiltration of cardiac tissue with inflammatory, bone-marrow-derived cells mainly of the myeloid lineage. In EAM, inflammation is replaced by progressive cardiac fibrosis, ventricular dilatation, and systolic dysfunction.5,6
In the body, the renin–angiotensin–aldosterone system (RAAS) regulates blood pressure, sodium balance, and fluid homeostasis. Pharmacological RAAS inhibitors are commonly used as blood pressure lowering drugs and the cornerstone of heart failure treatment in patients with impaired systolic function. The peptide hormone angiotensin II (Ang II) represents the main active component of RAAS. Ang II is known to mediate vasoconstriction, regulate water and sodium retention, promote proinflammatory and profibrotic processes, and therefore has been implicated in pathophysiology of many cardiovascular diseases.7 In animal models, Agtr1a-/- mice that lack Ang II receptor 1 (ATR1) show reduced basal blood pressure and develop polyuria,8,9 whereas continuous infusion of Ang II causes hypertension and cardiac hypertrophy associated with tissue fibrosis and ventricular dysfunction.10
On a molecular level, Ang II can bind to G protein-coupled receptors ATR1 and ATR2. Binding of Ang II to ATR1 triggers G protein-dependent second messenger signalling that activates a number of intracellular protein kinases including protein kinases C (PKC), mitogen-activated protein kinase (MAPKs), or Rho-associated protein kinases (ROCKs).11 In cardiac fibrosis, Ang II-ATR1 signalling has been suggested to activate RhoA bound to A-kinase anchoring protein (AKAP)-Lbc12,13 and to enhance production of profibrotic transforming growth factor beta (TGF-β).14–16 Moreover, Ang II has been implicated in activation of the Wnt signalling pathway.17,18
So far, the role of Ang II signalling in EAM has been addressed with pharmacological RAAS inhibitors. Treatment with ATR1 inhibitors telmisartan, olmesartan, or losartan suppressed proinflammatory cytokine production and ameliorated myocarditis severity in mice and in rats.19–24 Similarly, the angiotensin-converting enzyme (ACE) inhibitor captopril reduced autoreactive responses and cardiac inflammation in EAM.25 Furthermore, in the EAM model, these RAAS inhibitors successfully prevented development of cardiac fibrosis and DCM phenotype administered both pre-25–27 and post-inflammatory.28–30 However, it needs to be noted that conventional RAAS inhibitors display significant off-target activities. Telmisartan, for instance, was shown to act as an agonist of peroxisome proliferator-activated receptor gamma (PPAR-γ) - a ligand-activated transcription factor exerting anti-inflammatory31,32 and antifibrotic33 properties. The ACE inhibitor captopril, on the other hand, likely binds to non-specific targets and suppresses other processes including IL-6 signalling.34 In this work, by using Agtr1a-/- mice, we specifically addressed the role of Ang II-ATR1 signalling in EAM on the genetic level.
2. Methods
2.1 Mice
Agtr1a -/- mice8 were originally obtained from the Jackson Laboratory and were back-crossed for at least 10 generations on BALB/c background. Mice were kept under standard laboratory conditions: 12/12 h light/dark cycle, room temperature 20–22°C, humidity 45–55% with ad libitum access to food and water. All experiments were performed in accordance with Swiss and Polish law and were approved by local authorities (license numbers ZH49/2009 and ZH194/2012 for Switzerland and 206/2017 and 235/2019 for Poland). Animal experiment followed the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes.
2.2 EAM induction, generation of bone marrow chimaera, and pharmacological treatment
EAM was induced in 6–9 weeks old WT and Agtr1a-/- BALB/c mice as previously described.6,35 Detailed description is provided in the supplementary method section. Crisscross bone marrow chimaeras were generated by lethal irradiation (2 × 5.5 Gy) and reconstitution with 2 × 106 crude bone marrow cells as described before35 EAM was induced in chimeric mice 6 weeks after bone marrow reconstitution. In certain experiments, telmisartan (10 mg/kg/day; Cayman Chemical) or vehicle control was delivered in drinking water to Agtr1a-/- mice from day 0 until day 21 of EAM. Mice were euthanized by exposure to gradually increasing carbon dioxide concentration or by intraperitoneal injection of anaesthetics ketamine (75 mg/kg) and medetomidine (1 mg/kg).
2.3 Echocardiography
Transthoracic echocardiography was performed under isoflurane (1.5–2 Vol%) anaesthesia using a Vevo 2100 system equipped with 30-MHz transducer (VisualSonics) in WT and Agtr1a-/- mice at day 0 and day 40 of EAM as described previously.36 Detail description is provided in the supplementary method section.
2.4 Hydroxyproline assay
Cardiac tissue was digested in distilled water (100 µL per 10 mg of tissue) using TissueLyser II (Qiagen) for 2 × 2 min at 30 Hz and 100 µL of the homogenate was transferred to 2 mL Teflon capped pressure tight vials. After addition of 100 µL 12 N hydrochloric acid (Sigma), samples were hydrolysed for 3 h at 120°C and centrifuged at 10 000g for 3 min. 10 µL of supernatant was transferred to a 96 well plate and evaporated to dryness at 60°C. Samples were further processed for hydroxyproline assay kit (Sigma) according to manufacturer’s instructions. Absorbance was measured at 560 nm and concentration was determined using the hydroxyproline standard curve.
2.5 Heart inflammatory cell isolation and cell cultures
Isolation of heart inflammatory cells was previously described.6 Briefly, myocarditis-positive hearts were perfused, dissected, and digested with Liberase Blendzyme (Roche) for 45 min at 37˚C and tissue suspensions were passed sequentially through 70 µm and 40 µm cell strainers. Cells were expanded up to 3 passages in the growth medium Iscove`s modified Dulbecco`s medium (IMDM; Corning) supplemented with 20% inactivated FCS (ThermoFisher Scientific); 10 000 U/mL penicillin/streptomycin (Corning); and 0.1 mM 2-mercaptoethanol (Merck). In certain experiments, cultured cells were stimulated with 10 ng/mL recombinant human TGF-β1 (Peprotech). Detail description is provided in the supplementary method section.
2.6 Immunoblotting
For protein expression analysis, a conventional SDS–PAGE followed by protein blotting strategy and the following antibodies anti-ACE (clone 2E2), anti-renin (clone A-1), anti-Gα12 (clone E-12), anti-Gα13 (clone 6F6-B5, all Santa Cruz Biotechnology), anti-GAPDH (clone 14C10), anti-SMAD2 (clone D43B4), anti-pSMAD2 (clone 138D4), anti-TAK1 (polyclonal), anti-pTAK1 (polyclonal, all Cell Signalling), and anti-α-SMA (clone 1A4, Biolegend) were used in the study. Detail description is provided in the supplementary method section.
2.7 Quantitative RT–PCR
For RNA isolation from heart tissue, 25–30 mg of cardiac tissue was lysed in 500 µL of Qiazol (Qiagen) and homogenized using TissueLyser II (Qiagen). RNA from in vitro cell cultures was isolated with Qiazol following manufacturer’s instructions. NG dART RT kit (EurX) was used for cDNA synthesis. Quantitative PCR was performed using SYBR Green qPCR Master Mix (EurX) with a QuantStudio 6 Flex Real-Time PCR System (ThermoFisher Scientific). Primer sequence is provided in the supplementary method section.
2.8 Flow cytometry
Isolation of single cell suspension from EAM heart was previously described.6In vitro expanded cells were prepared by scraping cells from tissue culture plates and passing them through 70 µm and 40 µm cell strainers. Cells were stained with appropriate fluorochrome-conjugated anti-mouse antibodies (see supplementary method section) for 30 min at 4°C and analysed with the FACSCanto 10 Flow Cytometer (BD Biosciences) and the FlowJo v10 software (BD).
2.9 Histopathology and immunohistochemistry
Mouse heart tissues were fixed in 4% formalin and embedded in paraffin. Conventional haematoxylin/eosin and Masson’s trichrome staining were used to assess cardiac inflammation and fibrosis, respectively, as previously described.6 Antigen-specific immunostaining using anti-CD45 (clone A20, BD Biosciences), anti-CD3 (clone SP7, Neomarkers), anti-F4/80 (clone BM6, BMA Biomedicals), anti-periostin (polyclonal), anti-vimentin (clone EPR3776, both Abcam), and anti-β-catenin (clone 14, Transduction Laboratories) antibodies is described in the supplementary method section.
2.10 Immunocytochemistry
Conventional immunocytochemistry using the antibodies used in the study such as anti-α-SMA (clone 1A4, Biolegend), anti-fibronectin (clone FBN11), and anti-β-catenin (clone 15B8, both ThermoFisher Scientific) was performed to visualize cellular localization of the antigens. Detailed description is provided in the supplementary method section.
2.11 Nano-liquid chromatography (LC) with mass spectrometry (MS)
Sample preparation and mass spectrometry measurement were performed as described previously.37 Detail description is provided in the supplementary method section.
2.12 Rho-binding domain (RBD) pulldown assay and AKAP-Lbc immunoprecipitation
Both assays were performed as described previously.13 Briefly, cells were lysed in RBD lysis buffer. Lysates were incubated with 30 μg of RBD beads for 1 h at 4°C. Beads were then washed with RBD buffer, resuspended in SDS–PAGE sample buffer, separated on acrylamide gels, and electroblotted onto nitrocellulose membranes. Active RhoA was detected on Western blot using a monoclonal anti-RhoA antibody (clone 26C4; Santa Cruz Biotechnology). AKAP-Lbc immunoprecipitation was performed on overnight-dialysed cell culture lysates for 4 h at 4°C with 4 μg of rabbit polyclonal anti-AKAP-Lbc antibodies (Covance) and 20 μl of protein-A-sepharose beads. Following immunoprecipitation, proteins were analysed by SDS–PAGE and immunoblotting. Detail description is provided in the supplementary method section.
2.13 Immunoprecipitation and GSK3β activity measurement
Immunoprecipitation and GSK3β activity measurement was performed as described previously38 with minor modifications. GSK3β activity was measured with the ADP-GLO kinase assay (Promega) according to manufacturer’s protocol. Detail description is provided in the supplementary method section.
2.14 Statistical analysis
For normally distributed data, unpaired Student’s t-test or one-way ANOVA followed by Fisher’s LSD tests was used. For non-parametric data, Mann–Whitney U test or Kruskal–Wallis followed by Dunn’s test was used. Differences were considered statistically significant for p < 0.05. All analyses were performed with the Prism 8 software (GraphPad).
3. Results
3.1 Myocarditis severity does not differ between wild-type and Agtr1a-/- mice at day 21
EAM was induced in wild-type and Agtr1a-/- mice by immunization with αMyHC/CFA. First, we analysed development of αMyHC-reactive CD4+ T cells and severity of myocarditis in immunized animals at the peak of disease (d21). Re-stimulation of splenic CD4+ T cells with αMyHC peptide resulted in similar vigorous proliferation of both wild-type and Agtr1a-/- lymphocytes (FigureS 1). Further, we analysed hearts of immunized mice at day 21 of EAM. Myocarditis severity scores, as well as the amount of CD45+, CD3+, and F4/80+ cells in heart sections showed no differences between analysed groups (Figure 1A, B). Flow cytometry confirmed no significant differences in the number of heart-infiltrating CD11b+ myeloid cells, CD4+ and CD4– T cells, B cells, and NK cells between wild-type and Agtr1a-/- mice at acute stage of EAM (Figure 1C, FigureS2). Of note, our findings were in opposite to previous data showing ameliorated EAM in animals receiving pharmacological ATR1 inhibitors.19–24 To address this discrepancy, we treated one group of Agtr1a-/- mice with ATR1 inhibitor telmisartan starting at day 0 of EAM. Surprisingly, Agtr1a-/- mice receiving telmisartan developed significantly less severe myocarditis compared vehicle-treated counterparts (Figure 1D). Thus, our results point to full susceptibility of Agtr1a-/- mice to EAM and off-target anti-inflammatory activity of telmisartan in this model.
Figure 1.

EAM was induced in wild-type (WT) and in Agtr1a-/- mice by αMyHC/CFA immunization at day 0 and 7. Representative histology (H/E) and immunohistochemistry for CD45, CD3, and F4/80 in hearts of indicated recipients at day 21 (inflammatory phase) are presented in (A). Scale bar = 100 μm. Myocarditis severity scores and quantification of heart-infiltrating CD3+, CD45+, and F4/80+ cells in WT (n = 5) and Agtr1a-/- (n = 5) mice are shown in (B). t-SNE plot presenting cardiac CD45+-gated cell subsets identified by flow cytometry (gating strategy shown in FigureS 2) and quantification of the indicated subsets in hearts of WT (n = 6) and Agtr1a-/- (n = 6) mice at day 21 of EAM are shown in (C). Myocarditis severity scores of vehicle- (control, n = 6) or telmisartan-treated (n = 7) Agtr1a-/- mice at day 21 are presented in (D). Expression of selected profibrotic genes in cardiac tissue at day 21 is shown in (E). p values calculated with Mann–Whitney U test or unpaired Student’s t-test.
Figure 2.

EAM was induced in wild-type (WT) and in Agtr1a-/- mice by αMyHC/CFA immunization at day 0 and 7. Representative Masson’s Trichrome staining and immunohistochemistry for periostin and vimentin in hearts of indicated recipients at day 40 (fibrotic phase) are shown in (A). Scale bar = 100 μm. Quantifications of positive signals for WT (n = 10) and Agtr1a-/- (n = 10) mice are presented in (B). Panel (C) shows hydroxyproline contents in cardiac tissue of WT (n = 7) and Agtr1a-/- (n = 7) mice at day 40. Echocardiography was performed on WT and Agtr1a-/- mice at day 0 and day 40. Panel (D) shows the differences (day 40–day 0) of measured ejection fraction (EF), fractional shortening (FS), and cardiac output (CO) for WT (n = 7) and Agtr1a-/- (n = 7) mice. Heart weight/tibia length (HW/TL) ratio of WT (n = 7) and Agtr1a-/- (n = 7) measured at day 40 are presented in (E). P values for (C–E) calculated with unpaired Student’st-test. Bone marrow (BM) chimeric mice were generated by lethal irradiation of the recipients followed by transplantation of the donor BM. About 6 weeks after BM transplantation, chimeric mice were immunized with αMyHC/CFA, and heart sections were analysed at day 40 of EAM. Panel (F) shows quantifications of Masson’s Trichrome staining and immunopositive signals for periostin and vimentin in the indicated chimeric mice (n = 4–9).p values calculated with one-way ANOVA followed by multiple comparison using the Fisher’s LSD test. *p < 0.05 (post-hoc test).
3.2 Reduced post-inflammatory fibrosis and better systolic function of Agtr1a-/- mice
To better characterize myocarditis in Agtr1a-/- mice, we analysed expression of profibrotic genes in hearts at day 21 of EAM. Cardiac levels of most of the analysed transcripts were lower in Agtr1a-/- mice (Figure 1E). This analysis was performed during the acute phase of inflammation (d21), before the onset of fibrotic phenotype in this model. In order to address the actual consequence of genetic deletion of Agtr1a on cardiac fibrosis, the hearts of wild-type and Agtr1a-/- mice were analysed at day 40 of EAM. At this stage, most of wild-type mice developed cardiac fibrosis. In Agtr1a-/- hearts, we observed a reduced collagen deposition (detected with Masson’s trichrome staining) and significantly less cells immunopositive for myofibroblast marker periostin, but similar number of mesenchymal cells immunopositive for vimentin (Figure 2A, B). Analysis of hydroxyproline content confirmed reduced fibrosis in Agtr1a-/- hearts (Figure 2C). Cardiac fibrosis is associated with systolic heart failure; therefore, we analysed cardiac function of wild-type and Agtr1a-/- mice before disease induction (day 0) and at day 40 of EAM. Along with the disease progression, wild-type mice showed lower ejection fraction suggesting worsening of systolic function (Figure 2D, Table S1). Agtr1a-/- mice were better protected from systolic dysfunction and showed higher ejection fraction at day 40 (Figure 2D, Table S1). This better systolic function was paralleled by lower heart weights of Agtr1a-/- mice (Figure 2E). In line with this finding, we observed lower expression of cardiac hypertrophy markers in hearts of Agtr1a-/- mice (Figure S3). All these data suggest that in the EAM model, Agtr1a-/- mice are protected from developing dilated cardiomyopathy phenotype.
3.3 ATR1 signalling on the bone marrow compartment is critical for post-inflammatory fibrosis
In EAM model, cardiac fibrosis is driven by inflammation. Consequently, we specifically addressed the role of the bone marrow-derived cellular compartment. To this aim, using wild-type and Agtr1a-/- mice, we generated crisscross bone marrow chimaera (in which inflammatory cells originated from donor bone marrow) and immunized them with αMyHC/CFA. Analysis of heart sections at day 40 of EAM showed that chimeric mice with intact ATR1 in inflammatory compartment developed more extensive cardiac fibrosis and showed increased amount of periostin-positive myofibroblasts (Figure 2F). Altogether, these data suggest that Ang II-ATR1 signalling in bone marrow-derived inflammatory cells promotes cardiac fibrosis in EAM.
3.4 Heart-inflammatory cells produce Ang II
In order to study ATR1 signalling in heart-inflammatory cells, we isolated and expanded inflammatory cells from hearts of wild-type and Agtr1a-/- mice with myocarditis. Flow cytometry analysis showed co-expression of pan-leucocyte (CD45) myeloid (CD11b), progenitor (CD133), and stromal/mesenchymal cell (gp38, CD29) markers, but not mesenchymal stem cell marker (CD140a) in expanded wild-type and Agtr1a-/- cells (Figure S4). Next, we asked whether these inflammatory cells were able to produce Ang II under steady-state and pro-fibrotic (+TGF-β) conditions. Heart-inflammatory cells indeed expressed angiotensinogen (Agt), genes encoding enzymes converting angiotensinogen to angiotensin II (Ren1, Ren2, and Ace) and ATR1 (Agtr1a) on the mRNA level in the presence and absence of TGF-β (Figure 3A). Stimulation with TGF-β did not affect Agt expression (Figure 3B) but up-regulated intracellular protein levels of renin and ACE enzymes (Figure 3C, D). Using mass spectroscopy analysis, we confirmed the presence of Ang II in cell lysates and in supernatants. Interestingly, 1 hour following TGF-β treatment extracellular angiotensin II levels dropped, while intracellular levels remained unchanged (Figure 3E).
Figure 3.

Inflammatory cells were isolated from hearts of wild-type mice at day 17–21 of EAM, expanded, and stimulated with TGF-β1 (10 ng/mL). Panel (A) shows expression of genes involved in Ang II synthesis in unstimulated cells and treated with TGF-β for 24 h (n = 4). A kinetic of angiotensinogen (Agt) expression is shown in panel (B) (n = 3–4). p value calculated with one-way ANOVA. Panel (C) shows immunoblots of enzymes involved in Ang II production. Quantifications of the respective protein levels (normalized to GAPDH) are presented in panel (D) (n = 7–8). *p < 0.05 (vs. control) calculated with Kruskal–Wallis followed by Dunn’s test. Levels of Ang II measured with mass spectroscopy in cell lysate (n = 4–5) and in supernatants (n = 5) are shown in panel (E). p values calculated with one-way ANOVA followed by multiple comparison using the Fisher’s LSD test. *p < 0.05 (post-hoc test).
3.5 Reduced TGF-β-mediated profibrotic response of Agtr1a-/- heart-inflammatory cells
TGF-β is a master profibrotic cytokine in the EAM model. We have previously demonstrated that in response to TGF-β, heart-inflammatory cells acquired myofibroblast phenotype.6 In line with our previous data, we showed a rapid up-regulation of genes (Acta2, Col1a1) and proteins (α-SMA) characteristic for myofibroblasts and activation of Wnt pathway in TGF-β-activated inflammatory cells (Figure 4 and Figure S5). Such stimulation activated also the classical TGF-β-dependent pathways SMAD and TAK as well as slightly up-regulated intracellular G proteins Gα12 and Gα13 (Figure 4). In the next step, we analysed whether the profibrotic TGF-β-dependent response involved ATR1 signalling. Activated Agtr1a-/- heart-inflammatory cells showed a clearly reduced number of α-SMA-positive filaments and fibronectin (Figure 5A). Transcriptional analysis of profibrotic genes (including selected Wnts) confirmed impaired response to TGF-β by Agtr1a-/- cells (Figure 5B) suggesting involvement of ATR1 signalling in TGF-β-dependent fibrotic processes.
Figure 4.

Inflammatory cells were isolated from hearts of wild-type mice at day 17–21 of EAM, expanded, and stimulated with TGF-β1 (10 ng/mL) for up to 72 h. Panel (A) shows immunoblots of profibrotic α-SMA, TGF-β downstream molecules (Smad, pSmad, TAK1, pTAK1), and G proteins (Gα12 and Gα13). Quantifications of protein levels (normalized to GAPDH) of α-SMA (n = 6–8), pSmad/Smad2 (n = 7–8), pTAK1/TAK1 (n = 7), Gα12 (n = 9–10), and Gα13 (n = 10–11) are shown in panel (B). *p < 0.05 (vs. control) calculated with Kruskal–Wallis followed by Dunn’s test.
Figure 5.

Inflammatory cells were isolated from hearts of wild-type (WT) and Agtr1a-/- mice at day 17–21 of EAM, expanded, and stimulated with TGF-β1 (10 ng/mL). Representative immunofluorescences of α-SMA and fibronectin of wild-type and Agtr1a-/- cells treated with TGF-β for 14 days are presented in panel (A). Data are representative of three independent experiments. Scale bar = 20 µm. Panel (B) shows transcriptional response to TGF-β (24 h) of WT (white, n = 4) and Agtr1a-/- (black, n = 5) cells. p values calculated with unpaired Student’s t-test, * p < 0.05. Panel (C) shows immunoblots of indicated proteins in wild-type and Agtr1a-/- cells unstimulated or stimulated with TGF-β. Quantifications of protein levels (normalized to GAPDH or β-tubulin) of pSmad/Smad2 (n = 4–5), pTAK1/TAK1 (n = 6), and α-SMA (n = 6) are presented in panel (D). p values calculated with unpaired Student’s t-test.
3.6 TGF-β triggers ATR1-dependent formation of RhoA-AKAP-Lbc complex
To elucidate the underlying molecular mechanism, we analysed activation of key TGF-β downstream signalling molecules. Agtr1a-/- cells showed a slightly reduced phosphorylation of SMAD2 and TAK1 (Figure 5C, D). Furthermore, TGF-β like Ang II activates RhoA GTPase by stimulating formation of the active RhoA-GTP form. Treatment of heart-inflammatory cells with TGF-β for 30 minutes triggered formation of RhoA-GTP in wild-type and in Agtr1a-/- cells to the same extents (Figure 6A, C). In contrast to the short-term stimulation, RhoA-GTP levels in Agtr1a-/-cells treated with TGF-β for 24 hours were significantly lower than in wild-type cells (Figure 6A, C). In cells stimulated with Ang II, activated RhoA forms a complex with AKAP-Lbc.12 Immunoprecipitation experiments showed enhanced interaction of RhoA with AKAP-Lbc also in wild-type cells stimulated with TGF-β for 24 hours (Figure 6B, C). In Agtr1a-/- cells, interaction of RhoA with AKAP-Lbc was, however, largely impaired (Figure 6B, C). Thus, these data confirmed TGF-β-mediated activation of the ATR1-dependent mechanism in heart-inflammatory cells.
Figure 6.

Inflammatory cells were isolated from hearts of wild-type (WT) and Agtr1a-/- mice at day 17–21 of EAM, expanded, and stimulated with TGF-β1 (10 ng/mL) for 30 min or 24 h. Panel (A) shows anti-RhoA immunoblots of purified GTP-bound Rho protein extracts (RhoA-GTP) and total cell lysates (total RhoA). Panel (B) shows anti-RhoA and anti-AKAP-Lbc immunoblots (IB) of immunoprecipitated (IP) protein extracts with anti-AKAP-Lbc antibodies. Anti-RhoA immunoblots of the whole cell extracts are presented in the bottom panel. Panel (C) shows quantifications of normalized RhoA-GTP levels (normalized to total RhoA, left, n = 4) and amount of RhoA immunoprecipitated with anti-AKAP-Lbc antibodies (normalized to IB AKAP-Lbc, right, n = 3). p values calculated with unpaired Student’s t-test.
3.7 Impaired activation of Wnt/β-catenin pathway in Agtr1a-/- inflammatory cells
In addition to SMAD, TAK, and RhoA, TGF-β also activates a profibrotic Wnt/β-catenin pathway.6,39,40 Under steady-state condition, GSK3β enzyme degrades β-catenin preventing its nuclear translocation and transcriptional activity. In wild-type cells, we observed inhibition of GSK3β activity and nuclear accumulation of β-catenin upon TGF-β treatment (Figure 7A, B). Strikingly, TGF-β failed to reduce GSK3β activity and to trigger nuclear translocation of β-catenin in Agtr1a-/- cells (Figure 7A, B). To validate the relevance of this mechanism in vivo, we analysed β-catenin and expression of Wnts in hearts with myocarditis at day 21. In fact, wild-type hearts showed inflammatory regions positive for β-catenin, whereas in Agtr1a-/- hearts, inflammatory regions were essentially β-catenin-negative (Figure 7C). Furthermore, reduced expression of canonical Wnt1 and Wnt10b was found in hearts of Agtr1a-/- mice (Figure 7D). Taken together, these data indicate that ATR1 controls activation of profibrotic Wnt/β-catenin pathway.
Figure 7.

Inflammatory cells were isolated from hearts of wild-type (WT) and Agtr1a-/- mice at day 17–21 of EAM, expanded, and stimulated with TGF-β1 (10 ng/mL). Panel (A) shows GSK3β activity in response to TGF-β (n = 4–6). * p < 0.05 (vs. control) calculated with Kruskal–Wallis followed by Dunn’s test. Cellular localizations of β-catenin in control and TGF-β-treated (1 h) cells are presented in panel (B). DAPI staining visualizes cell nuclei. Arrows indicate nuclear localization of β-catenin. Scale bar = 10 µm. Data are representative of three independent experiments. Panel (C) shows immunohistochemistry for β-catenin (brown) in hearts at day 21 of EAM (inflammatory phase). Scale bar = 100 μm. Pictures are representative for at least three WT and three Agtr1a-/-mice. Expression of selected Wnt genes in the indicated cardiac tissues at day 21 is shown in (D, n = 4–5). p values calculated with unpaired Student’s t-test.
4. Discussion
Observations from clinical studies and animal models have pointed to a role of Ang II signalling in pathological cardiac remodelling and fibrosis in a number of heart diseases.10,41 In the context of inflammatory heart diseases, published results suggested that Ang II signalling was implicated in development of autoimmune-mediated myocarditis.19,20,23–25 Unexpectedly, following αMyHC/CFA immunization, Agtr1a-/-mice effectively developed heart-specific autoimmunity and were fully susceptible to myocarditis at d21. These findings are contradictory to previous studies showing reduced heart inflammatory responses in mice treated with pharmacological ATR1 inhibitors.20–25 These contradictory findings may be explained with off-target activities of ATR1 antagonists like telmisartan or losartan, such as the activation of anti-inflammatory PPAR-γ signalling pathway.31,32,42 It seems that the cardioprotective effects observed in EAM with previously tested ATR1 inhibitors at acute inflammatory phase were likely independent of the blockade of ATR1 signalling. Our data showing reduced myocarditis in Agtr1a-/- mice treated with telmisartan further strengthen this hypothesis. It is possible that also other RAAS blockers might inhibit inflammatory response independently of Ang II signalling.
Myocarditis is often followed by development of progressive cardiac fibrosis and systolic dysfunction. Results of pharmacological ATR1 blockade in animals with myocarditis pointed to profibrotic Ang II signalling in this model.28–30 However, PPAR-γ signalling that is effectively activated by ATR1 inhibitors telmisartan and losartan has been shown to protect from cardiac fibrosis in various models.33,43 In our experiments, Agtr1a-/-mice showed reduced fibrosis at day 40 confirming profibrotic ATR1-dependent signalling in EAM. Less cardiac fibrosis in Agtr1a-/- mice was paralleled by smaller heart size and better systolic function indicating that these mice are protected from developing dilated cardiomyopathy phenotype. It should be noted that Agtr1a-/- mice showed rather a moderate phenotype. Pharmacological ATR1 inhibitors more effectively prevented fibrosis in EAM, but they target not only ATR1 but also activate anti-inflammatory and antifibrotic PPAR-γ. Although the reduced blood pressure in Agtr1a-/- mice9 could theoretically protect these mice from cardiac fibrosis, our results of the crisscross bone marrow chimaera clearly point to a key role of ATR1 on the bone marrow-derived inflammatory compartment.
Exogenous Ang II can induce production of profibrotic factors, like TGF-β or Wnts, and activated phenotype in cardiac fibroblasts, cardiomyocytes, and aortic valve myofibroblasts.14,16,17,44 Here, we demonstrated that TGF-β could trigger ATR1-dependent signalling even in the absence of exogenous Ang II. Our results showed that at least inflammatory myeloid cells were able to produce Ang II and therefore could potentially activate ATR1 in an autocrine or paracrine way. In our experiments, autocrine/paracrine Ang II-ATR1 signalling was not constitutively active but was induced by TGF-β, as demonstrated by ATR1-dependent formation of RhoA-AKAP-Lbc complex and elevated intracellular levels of Gα12 and Gα13 following TGF-β treatment. This hypothesis is further strengthened by previous data showing formation of RhoA-AKAP-Lbc complex in response to exogenous Ang II by the classical Gα12/13-dependent mechanism.12,13 Of note, AKAP-Lbc complex has been also implicated in cardiac fibrosis.12 Thus, in addition to previously described Ang II-induced TGF-β production, our data point to activation of Ang II-ATR1 signalling by TGF-β. It suggests existence of a positive TGF-β-ATR1 feedback loop in fibrotic response.
Our data indicate that Ang II-ATR1 signalling contributes to direct downstream TGF-β-dependent outputs. Although a short-term RhoA activation by TGF-β is Ang II-ATR1 independent, the ATR1 signalling is essential to maintain a long-term RhoA response. It is not surprising, because RhoA is a direct downstream target of Ang II signalling.45 Of note, RhoA-dependent kinases ROCK1 and ROCK2 are not critically involved in cardiac fibrosis in EAM.46 On the other hand, it remains unclear, whether Ang II-ATR1 enhances the canonical Smad response to TGF-β. Agtr1a-/- cells showed a slightly reduced Smad2 phosphorylation, but the difference has not reached statistical significance. Instead, our data clearly pointed to regulation of the Wnt/β-catenin response by ATR1. Previously, we have demonstrated TAK1-dependent activation of profibrotic Wnt/β-catenin by TGF-β in inflammatory myeloid cells and in cardiac fibroblasts in EAM model.6 Here, we show that the intact ATR1 is needed for TGF-β-inducible TAK1 phosphorylation, inhibition of GSK3β activity, and Wnt/β-catenin activation. The ineffective activation of the Wnt/β-catenin pathway can explain reduced profibrotic response of Agtr1a-/- cells. In fact, Wnt/β-catenin signalling has been implicated in fibrogenesis not only in EAM, but also in other cardiac17,39,47 and non-cardiac diseases.48,49 The Wnt/β-catenin signalling is regulated by GSK3β activity and TAK1. Ang II signalling has already been implicated in TAK1 activation,50 but our data specifically underscore its role in TGF-β response. Thus, activation of downstream pathways in TGF-β-mediated profibrotic response is controlled, at least partly, by Ang II-ATR1 signalling.
In conclusion, we delineated for the first time the role of Ang II-ATR1 signalling in myocarditis development and its progression to DCM on the genetic level. We found no evidence for an early proinflammatory role of ATR1 signalling, but proved its importance in fibrotic remodelling during progression of acute myocarditis to an end-stage post-inflammatory phenotype of DCM. Moreover, our results shed new light on the cross-talk between TGF-β and ATR1 signalling pathways in shaping fibrotic responses in the inflamed heart.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Supplementary Material
Acknowledgements
We would like to thank Mrs Marta Bachmann and Mrs Małgorzata Hajduk for excellent technical assistance.
Conflict of interest: none declared.
Funding
This work was supported by the National Science Centre (Poland), (grant number 2014/14/E/NZ5/00175) and by the GZO—Zurich Regional Health Center research fund.
Data availability
The data underlying this article are available in the article and in its online supplementary material.
Translational perspective
Myocardial fibrosis causes impaired cardiac function in inflammatory heart diseases. It has been believed that targeting angiotensin II receptor 1 (ATR1) could not only lower blood pressure, but also effectively block both immune and fibrotic processes in the heart. By using an experimental autoimmune myocarditis model and ATR1-deficient mice, we show that the specific ATR-dependent mechanism is rather limited to control profibrotic, but not inflammatory response. On the molecular level, ATR1 plays a central role in activation of the canonical Wnt pathway in profibrotic response. Thus, these data help to understand how the actual angiotensin II-ATR1 signalling contributes to immunofibrotic heart diseases.
References
- 1. Heymans S, Eriksson U, Lehtonen J, Cooper LT.. The quest for new approaches in myocarditis and inflammatory cardiomyopathy.J Am Coll Cardiol 2016;68:2348–2364. [DOI] [PubMed] [Google Scholar]
- 2. Weintraub RG, Semsarian C, Macdonald P.. Dilated cardiomyopathy. The Lancet 2017;390:400–414. [DOI] [PubMed] [Google Scholar]
- 3. Caforio ALP, Pankuweit S, Arbustini E, Basso C, Gimeno-Blanes J, Felix SB, Fu M, Helio T, Heymans S, Jahns R, Klingel K, Linhart A, Maisch B, McKenna W, Mogensen J, Pinto YM, Ristic A, Schultheiss H-P, Seggewiss H, Tavazzi L, Thiene G, Yilmaz A, Charron P, Elliott PM.. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2013;34:2636–2648. [DOI] [PubMed] [Google Scholar]
- 4. Błyszczuk P. Myocarditis in humans and in experimental animal models.Front Cardiovasc Med 2019;6:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Blyszczuk P, Berthonneche C, Behnke S, Glönkler M, Moch H, Pedrazzini T, Lüscher TF, Eriksson U, Kania G.. Nitric oxide synthase 2 is required for conversion of pro-fibrogenic inflammatory CD133+ progenitors into F4/80+ macrophages in experimental autoimmune myocarditis. Cardiovasc Res 2013;97:219–229. [DOI] [PubMed] [Google Scholar]
- 6. Blyszczuk P, Müller-Edenborn B, Valenta T, Osto E, Stellato M, Behnke S, Glatz K, Basler K, Lüscher TF, Distler O, Eriksson U, Kania G.. Transforming growth factor-β-dependent Wnt secretion controls myofibroblast formation and myocardial fibrosis progression in experimental autoimmune myocarditis. Eur Heart J 2017;38:1413–1425. [DOI] [PubMed] [Google Scholar]
- 7. Mirabito Colafella KM, Bovée DM, Danser AHJ.. The renin-angiotensin-aldosterone system and its therapeutic targets.Exp Eye Res Elsevier 2019;186:107680. [DOI] [PubMed] [Google Scholar]
- 8. Ito M, Oliverio MI, Mannon PJ, Best CF, Maeda N, Smithies O, Coffman TM.. Regulation of blood pressure by the type 1A angiotensin II receptor gene.Proc Natl Acad Sci 1995;92:3521–3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li XC, Shao Y, Zhuo JL.. AT1a receptor knockout in mice impairs urine concentration by reducing basal vasopressin levels and its receptor signaling proteins in the inner medulla. Kidney Int 2009;76:169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bacmeister L, Schwarzl M, Warnke S, Stoffers B, Blankenberg S, Westermann D, Lindner D.. Inflammation and fibrosis in murine models of heart failure. Basic Res Cardiol 2019;114:19. [DOI] [PubMed] [Google Scholar]
- 11. Kawai T, Forrester SJ, O’Brien S, Baggett A, Rizzo V, Eguchi S.. AT1 receptor signaling pathways in the cardiovascular system. Pharmacol Res 2017;125:4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cavin S, Maric D, Diviani D.. A-kinase anchoring protein-Lbc promotes pro-fibrotic signaling in cardiac fibroblasts. Biochim Biophys Acta Mol Cell Res 2014;1843:335–345. [DOI] [PubMed] [Google Scholar]
- 13. Appert-Collin A, Cotecchia S, Nenniger-Tosato M, Pedrazzini T, Diviani D.. The A-kinase anchoring protein (AKAP)-Lbc-signaling complex mediates α1 adrenergic receptor-induced cardiomyocyte hypertrophy. Proc Natl Acad Sci USA 2007;104:10140–10145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee AA, Dillmann WH, McCulloch AD, Villarreal FJ.. Angiotensin II stimulates the autocrine production of transforming growth factor-β1 in adult rat cardiac fibroblasts. J Mol Cell Cardiol 1995;27:2347–2357. [DOI] [PubMed] [Google Scholar]
- 15. Tomita H, Egashira K, Ohara Y, Takemoto M, Koyanagi M, Katoh M, Yamamoto H, Tamaki K, Shimokawa H, Takeshita A.. Early induction of transforming growth factor-β via angiotensin II type 1 receptors contributes to cardiac fibrosis induced by long-term blockade of nitric oxide synthesis in rats. Hypertension 1998;32:273–279. [DOI] [PubMed] [Google Scholar]
- 16. Schultz JEJ, Witt SA, Glascock BJ, Nieman ML, Reiser PJ, Nix SL, Kimball TR, Doetschman T.. TGF-β1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J Clin Invest 2002;109:787–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhao Y, Wang C, Wang C, Hong X, Miao J, Liao Y, Zhou L, Liu Y.. An essential role for Wnt/β-catenin signaling in mediating hypertensive heart disease.Sci Rep 2018;8:8996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li L, Guleria RS, Thakur S, Zhang C-L, Pan J, Baker KM, Gupta S.. Thymosin β4 prevents angiotensin II-induced cardiomyocyte growth by regulating Wnt/WISP signaling. J Cell Physiol 2016;231:1737–1744. [DOI] [PubMed] [Google Scholar]
- 19. Sukumaran V, Watanabe K, Veeraveedu PT, Ma M, Gurusamy N, Rajavel V, Suzuki K, Yamaguchi K, Kodama M, Aizawa Y.. Telmisartan ameliorates experimental autoimmune myocarditis associated with inhibition of inflammation and oxidative stress. Eur J Pharmacol 2011;652:126–135. [DOI] [PubMed] [Google Scholar]
- 20. Nimata M, Kishimoto C, Yuan Z, Shioji K.. Beneficial effects of olmesartan, a novel angiotensin II receptor type 1 agonist, upon acute autoimmune myocarditis. Mol Cell Biochem 2004;259:217–222. [DOI] [PubMed] [Google Scholar]
- 21. Yuan Z, Nimata M, Okabe T, Shioji K, Hasegawa K, Kita T, Kishimoto C.. Olmesartan, a novel AT 1 antagonist, suppresses cytotoxic myocardial injury in autoimmune heart failure. Am J Physiol Circ Physiol 2005;289:H1147–H1152. [DOI] [PubMed] [Google Scholar]
- 22. Sukumaran V, Watanabe K, Veeraveedu PT, Gurusamy N, Ma M, Thandavarayan RA, Lakshmanan AP, Yamaguchi K, Suzuki K, Kodama M.. Olmesartan, an AT 1 antagonist, attenuates oxidative stress, endoplasmic reticulum stress and cardiac inflammatory mediators in rats with heart failure induced by experimental autoimmune myocarditis. Int J Biol Sci 2011;7:154–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lu H, Zong G, Zhou S, Jiang Y, Chen R, Su Z, Wu Y.. Angiotensin II-C-C chemokine receptor2/5 axis-dependent monocyte/macrophage recruitment contributes to progression of experimental autoimmune myocarditis. Microbiol Immunol 2017;61:539–546. [DOI] [PubMed] [Google Scholar]
- 24. Lu H, Wu Y, Shao X, Zhou S, Jiang Y, Chen R, Zong G, Xu H, Su Z.. ANG II facilitated CD11 + Ly6C hi cells reprogramming into M1-like macrophage through Erk1/2 or p38-Stat3 pathway and involved in EAM. J Leukoc Biol 2018;103:719–730. [DOI] [PubMed] [Google Scholar]
- 25. Bahk TJ, Daniels MD, Leon JS, Wang K, Engman DM.. Comparison of angiotensin converting enzyme inhibition and angiotensin II receptor blockade for the prevention of experimental autoimmune myocarditis. Int J Cardiol 2008;125:85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sukumaran V, Veeraveedu PT, Gurusamy N, Yamaguchi K, Lakshmanan AP, Ma M, Suzuki K, Kodama M, Watanabe K.. Cardioprotective effects of telmisartan against heart failure in rats induced by experimental autoimmune myocarditis through the modulation of angiotensin-converting enzyme-2/angiotensin 1-7/mas receptor axis. Int J Biol Sci 2011;7:1077–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sukumaran V, Veeraveedu PT, Gurusamy N, Lakshmanan AP, Yamaguchi K, Ma M, Suzuki K, Nagata M, Takagi R, Kodama M, Watanabe K.. Olmesartan attenuates the development of heart failure after experimental autoimmune myocarditis in rats through the modulation of ANG 1–7 mas receptor. Mol Cell Endocrinol 2012;351:208–219. [DOI] [PubMed] [Google Scholar]
- 28. Sukumaran V, Veeraveedu PT, Gurusamy N, Lakshmanan AP, Yamaguchi K, Ma M, Suzuki K, Kodama M, Watanabe K.. Telmisartan acts through the modulation of ACE-2/ANG 1–7/mas receptor in rats with dilated cardiomyopathy induced by experimental autoimmune myocarditis.Life Sci 2012;90:289–300. [DOI] [PubMed] [Google Scholar]
- 29. Sukumaran V, Watanabe K, Veeraveedu PT, Thandavarayan RA, Gurusamy N, Ma M, Yamaguchi K, Suzuki K, Kodama M, Aizawa Y.. Telmisartan, an angiotensin-II receptor blocker ameliorates cardiac remodeling in rats with dilated cardiomyopathy. Hypertens Res 2010;33:695–702. [DOI] [PubMed] [Google Scholar]
- 30. Sukumaran V, Watanabe K, Veeraveedu PT, Thandavarayan RA, Gurusamy N, Ma M, Yamaguchi K, Suzuki K, Kodama M, Aizawa Y.. Beneficial effects of olmesartan, an angiotensin II receptor type 1 antagonist, in rats with dilated cardiomyopathy. Exp Biol Med (Maywood) 2010;235:1338–1346. [DOI] [PubMed] [Google Scholar]
- 31. Benson SC, Pershadsingh HA, Ho CI, Chittiboyina A, Desai P, Pravenec M, Qi N, Wang J, Avery MA, Kurtz TW.. Identification of telmisartan as a unique angiotensin ii receptor antagonist with selective PPARγ–modulating activity.Hypertension 2004;43:993–1002. [DOI] [PubMed] [Google Scholar]
- 32. Imayama I, Ichiki T, Inanaga K, Ohtsubo H, Fukuyama K, Ono H, Hashiguchi Y, Sunagawa K.. Telmisartan downregulates angiotensin II type 1 receptor through activation of peroxisome proliferator-activated receptor γ. Cardiovasc Res 2006;72:184–190. [DOI] [PubMed] [Google Scholar]
- 33. Maejima Y, Okada H, Haraguchi G, Onai Y, Kosuge H, Suzuki J, Isobe M.. Telmisartan, a unique ARB, improves left ventricular remodeling of infarcted heart by activating PPAR gamma. Lab Invest 2011;91:932–944. [DOI] [PubMed] [Google Scholar]
- 34. Chartier M, Morency L-P, Zylber MI, Najmanovich RJ.. Large-scale detection of drug off-targets: hypotheses for drug repurposing and understanding side-effects. BMC Pharmacol Toxicol 2017;18:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kania G, Siegert S, Behnke S, Prados-Rosales R, Casadevall A, Lüscher TF, Luther SA, Kopf M, Eriksson U, Blyszczuk P.. Innate signaling promotes formation of regulatory nitric oxide-producing dendritic cells limiting T-cell expansion in experimental autoimmune myocarditis. Circulation 2013;127:2285–2294. [DOI] [PubMed] [Google Scholar]
- 36. Zarak-Crnkovic M, Kania G, Jaźwa-Kusior A, Czepiel M, Wijnen WJ, Czyż J, Müller-Edenborn B, Vdovenko D, Lindner D, Gil-Cruz C, Bachmann M, Westermann D, Ludewig B, Distler O, Lüscher TF, Klingel K, Eriksson U, Błyszczuk P.. Heart non-specific effector CD4+ T cells protect from postinflammatory fibrosis and cardiac dysfunction in experimental autoimmune myocarditis. Basic Res Cardiol 2020;115:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Olkowicz M, Chlopicki S, Smolenski RT.. A primer to angiotensin peptide isolation, stability, and analysis by nano-liquid chromatography with mass detection.Methods Mol Biol 2017;1614:175–187. [DOI] [PubMed] [Google Scholar]
- 38. Cole AR, Sutherland C.. Measuring GSK3 expression and activity in cells. Methods Mol Biol 2008;468:45–65. [DOI] [PubMed] [Google Scholar]
- 39. Działo E, Tkacz K, Błyszczuk P.. Crosstalk between the TGF-β and WNT signalling pathways during cardiac fibrogenesis. Acta Biochim Pol 2018;65:341–349. [DOI] [PubMed] [Google Scholar]
- 40. Piersma B, Bank RA, Boersema M.. Signaling in fibrosis: TGF-β, WNT, and YAP/TAZ converge. Front Med (Lausanne) 2015;2:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sayer G, Bhat G.. The renin-angiotensin-aldosterone system and heart failure.Cardiol Clin 2014;32:21–32. [DOI] [PubMed] [Google Scholar]
- 42. Schupp M, Lee LD, Frost N, Umbreen S, Schmidt B, Unger T, Kintscher U.. Regulation of peroxisome proliferator–activated receptor γ activity by losartan metabolites. Hypertension 2006;47:586–589. [DOI] [PubMed] [Google Scholar]
- 43. Qi HP, Wang Y, Zhang QH, Guo J, Li L, Cao YG, Li SZ, Li XL, Shi MM, Xu W, Li BY, Sun HL.. Activation of peroxisome proliferator-activated receptor γ (PPARγ) through NF-κB/brg1 and TGF-β 1 pathways attenuates cardiac remodeling in pressure-overloaded rat hearts. Cell Physiol Biochem 2015;35:899–912. [DOI] [PubMed] [Google Scholar]
- 44. Xie C, Shen Y, Hu W, Chen Z, Li Y.. Angiotensin II promotes an osteoblast-like phenotype in porcine aortic valve myofibroblasts. Aging Clin Exp Res 2016;28:181–187. [DOI] [PubMed] [Google Scholar]
- 45. Carbone ML, Brégeon J, Devos N, Chadeuf G, Blanchard A, Azizi M, Pacaud P, Jeunemaître X, Loirand G.. Angiotensin II activates the RhoA exchange factor Arhgef1 in humans. Hypertens (Dallas, Tex 1979) 2015;65:1273–1278. [DOI] [PubMed] [Google Scholar]
- 46. Tkacz K, Rolski F, Czepiel M, Działo E, Siedlar M, Eriksson U, Kania G, Błyszczuk P.. Haploinsufficient Rock1+/− and Rock2+/− mice are not protected from cardiac inflammation and postinflammatory fibrosis in experimental autoimmune myocarditis. Cells 2020;9:700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lorenzon A, Calore M, Poloni G, Windt LD, Braghetta P, Rampazzo A.. Wnt/β-catenin pathway in arrhythmogenic cardiomyopathy. Oncotarget 2017;8:60640–60655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wei J, Fang F, Lam AP, Sargent JL, Hamburg E, Hinchcliff ME, Gottardi CJ, Atit R, Whitfield ML, Varga J.. Wnt/β-catenin signaling is hyperactivated in systemic sclerosis and induces Smad-dependent fibrotic responses in mesenchymal cells. Arthritis Rheum 2012;64:2734–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang J, Li L, Li L, Yan Q, Li J, Xu T.. Emerging role and therapeutic implication of Wnt signaling pathways in liver fibrosis. Gene 2018;674:57–69. [DOI] [PubMed] [Google Scholar]
- 50. Watkins SJ, Borthwick GM, Oakenfull R, Robson A, Arthur HM.. Angiotensin II-induced cardiomyocyte hypertrophy in vitro is TAK1-dependent and Smad2/3-independent. Hypertens Res 2012;35:393–398. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its online supplementary material.