

ABSTRACT
The tumor microenvironment (TME) plays a critical role in promoting the growth and metastasis of glioblastoma (GBM). Tumor-associated macrophages (TAMs), the most abundant myeloid cells infiltrating in TME, produce proinflammatory cytokines, regulate glioma cell pools, and lead to GBM progression. Understanding the mechanism of GBM-TAMs regulation can help to find new targeted therapeutic strategies against GBM. Based on the CGGA and TCGA GBM cohorts, ARPC1B was defined as the key macrophage-associated gene with prognostic value. Higher ARPC1B expression was associated with progressive malignancy, poor outcomes and TAM infiltration. We demonstrated that macrophage-expressed ARPC1B promoted the migration, invasion, and epithelial–mesenchymal transition of glioma cells. Glioma-intrinsic ARPC1B also maintained the malignant phenotype and promoted macrophage recruitment. Positive feedback signaling between macrophages and glioma cells via ARPC1B was determined to be under control of the IFNγ-IRF2-ARPC1B axis. This study highlights the important role of ARPC1B in GBM malignancy progression and the regulation network between GBM and TAMs, suggesting ARPC1B as a novel biomarker with potential therapeutic implications.
KEYWORDS: ARPC1B, actin cytoskeleton, TAM, macrophage, GBM
Introduction
Glioblastoma (GBM) is the most common malignant brain tumor, which is associated with high recurrence and mortality rates.1 One of the main contributors to the poor prognosis of GBM despite comprehensive treatment is that current treatments mostly target the tumor cells themselves, while ignoring the importance of the tumor microenvironment (TME).2 Malignant GBM is characterized by a complex microenvironment composition, in which the median proportion of tumor cells within glioma tissue is only 74%.3 In recent years, evidence has accumulated to demonstrate that the occurrence and progression of GBM are based on the interactive transformation process of glioma cells and their surrounding cells in the TME.
Tumor-associated macrophages (TAMs) are the most abundant immune cell type in GBM tissues.4 Many research groups, including our group, have demonstrated the crucial role of TAMs in creating an immunosuppressive microenvironment, and thus accelerating the proliferation, angiogenesis and treatment resistance (radiotherapy, chemotherapy, and immunotherapy) of GBM.5–7 Therefore, exploring the interaction mechanism between glioma cells and TAMs can help to resolve the regulatory network in GBM immune microenvironment. This interaction can further guide the development of new TME-targeted therapeutic strategies for GBM.
Accordingly, the aim of this study was to identify key genes associated with TAMs in GBM that are linked to tumor progression, as well as to explore the underlying mechanism. In this study, we reveal ARPC1B (actin-related protein 2/3 complex subunit 1B) as a novel regulator for GBM-TAM regulation. Actin-related protein 2/3 complex (Arp2/3) is an evolutionary conserved molecular machine that generates branched actin networks.8 Over the years, dysregulation of the Arp2/3 regulatory system in cancer has been described that excessive activation of the Arp2/3 complex commonly promotes tumor progression.9–11 ARPC1B is one of the regulatory subunits of Arp2/3 complex, which facilitates assembly and maintenance of the whole complex.12 Mutations in the ARPC1B gene have been found to result in autosomal recessive syndrome of combined immune deficiency, impaired T-cell migration and proliferation and thrombocytopenia.13–15 Meanwhile, ARPC1B is correlated with malignant phenotypes of tumors such as melanoma, osteosarcoma, and oral squamous cell carcinoma.16–18 Unfortunately, there is no comprehensive report on ARPC1B in GBM. Toward this end, we set out to describe ARPC1B function in GBM-TAM regulating network. We further evaluated the impacts and mechanism in vitro using glioma cell lines and in vivo using orthotopic/subcutaneous GBM mouse models. These findings can provide new insight into GBM progression and provide a possible therapeutic target.
Material and methods
Data
Our study covered 1024 gliomas from the Chinese Glioma Genome Atlas (CGGA) RNA-seq cohort and the Cancer Genome Atlas (TCGA) RNA-seq cohort. The CGGA cohort consisted of 325 gliomas. The detailed clinical information of 325 patients was obtained from the CGGA database (http://www.cgga.org.cn). Tumor tissue samples were collected at the time of surgery after informed consent. The histological diagnoses of these samples were confirmed by two neuropathologists according to the 2010 World Health Organization (WHO) classification guidelines. Overall survival (OS) was calculated from the date of diagnosis to death or the end of follow‐up. The point of death was defined by death certification, which could be acquired by local hospitals or police stations. Methods for sequencing, detecting IDH status, and MGMT promoter methylation state were formerly described.3 Another 699 glioma cases were included from the TCGA database (https://tcga-data.nci.nih.gov/tcga/). Information of these patients are available from corresponding website above. GSE55750 database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE55750) was used for discovering the upstream of ARPC1B in glioma cells.
Patients’ samples
The glioma samples for immunohistochemistry examining the expression of ARPC1B (1 case for non-tumor brain tissue as control, 3 cases for grade II, 3 cases for grade III, and 3 cases for grade IV, respectively) were collected at the First Hospital of China Medical University. Patient samples for Western blot were collected at the First Hospital of China Medical University, including 13 samples (12 glioma tissues: 4 cases for grade II, III and IV, respectively, and 1 non-tumor brain tissues from cranial injury internal decompression as control). 30 GBM tissues for immunohistochemistry examining the correlation between ARPC1B and macrophage (IBA1) were collected at the First Hospital of China Medical University. The histological diagnoses of all these samples were confirmed by two neuropathologists according to the 2010 WHO classification guidelines. This study was approved by the Ethics Committee of the First Hospital of China Medical University. Informed consent was obtained from each patient.
Gene Set Enrichment Analysis (GSEA), Gene Set Variation Analysis (GSVA) and Principal Component Analysis (PCA)
GSEA (http://www.broadinstitute.org/gsea/index.jsp) was performed to explore whether the identified sets of genes showed statistical differences between two groups stratified as described above.19 Normalized enrichment score and false discovery rate were used to determine the statistical significance. GSVA R package (http://www.bioconductor.org) was used to calculate the assessment of the underlying pathway activity variation according to the gene sets of defined signaling pathways. PCA were made by R language to distinguish different group information.
Transcription Factors (TFs) prediction
To predict the TFs of ARPC1B, we fist collected the sequencing of ARPC1B gene promoter by UCSC Genome Browser (http://genome.ucsc.edu/). Next, we used PROMO website (http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3) to predict the TFs of ARPC1B gene. The maximum matrix dissimilarity rate was set as zero.
Immunohistochemical staining
The ARPC1B and IBA1 expression in clinical patients’ tissues while ARPC1B, N-cadherin, E-cadherin and IBA1 expression in the tumor tissues extracted from different groups of intracranial and subcutaneous mouse model was detected by immunohistochemistry (IHC). Paraffin sections immunohistochemistry was conducted as mentioned previously.20 These expressions were calculated by German immunohistochemical score (GIS). Percentage of positive cells was graded as 0 (negative), 1 (up to 10%), 2 (11–50%), 3 (51–80%), or 4 (>80% positive cells) and staining intensity as 0 (no staining), 1 (weak), 2 (moderate), or 3 (strong). The final immunoreactive GIS was defined as the multiplication of both grading results (percentage of positive cells * staining intensity).
Immunofluorescence staining
4 μm thick section slides from frozen human tissue were washed three times in PBS. Then the sections were permeabilized with 0.5% Triton X-100 (T8200, Solarbio) for 20 min. After 5% BSA incubation for 1 h, sections were incubated in ARPC1B and glial fibrillary acidic protein (GFAP) antibodies at 4°C overnight. Following incubation with fluorescein (FITC) or rhodamine (TRITC) secondary antibody and 4’,6-diamidino-2-phenylindole (DAPI, C0060, Solarbio), the samples were detected using fluorescence microscope (Leica). The images were merged digitally to monitor the co-localization condition.
Cell lines and culture
Glioma cell lines U87 and human mononuclear macrophage line (THP-1) were purchased from the Shanghai Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Human microglia line HMC3 and murine glioma cell line GL261 were obtained from American Type Culture Collection (Manassas, VA, USA). The extraction of the patient-derived primary glioma cells (PGC28) were extracted as previously described.21 U87 and GL261 were maintained in Dulbecco’s modified Eagle’s medium (DMEM, 10566024, Gibco) supplemented with 10% fetal bovine serum (FBS, 16140071, Gibco) and 1% penicillin/streptomycin (10378016, Gibco). PGC28 and THP-1 were maintained in RPMI-1640 medium (61870036, Gibco) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. THP-1 monocytes were primed with 5 nM PMA (P1585, Sigma) for 48 hours to become THP1-derived macrophages. HMC3 were maintained in Minimum Essential Medium (MEM, 12561056, Gibco) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. All cells were cultured at 37°C with 5% CO2.
GSC isolation and culture
The gliomasphere (GSC21) used in this study was generated from primary GBM tumors. Briefly, the specimens were cut into small pieces, digested into single cells with Accutase (A6964, Sigma), and red blood cells were lysed by Red Blood Cell Lysis Buffer (R1010, Solarbio). Cell suspensions were then passed through a 70 μm stainless steel mesh and re-cultured in the serum-free stem cell medium. GSC21 was cultured in DMEM/F-12 medium (10565018, Gibco) containing 2% (vol/vol) B27 supplement (17504044, Gibco), epidermal growth factor (EGF, 20 ng/ml, AF-100-15, Peprotech), basic fibroblast growth factor (bFGF, 20 ng/ml, AF-100-18B, Peprotech) and heparin (2.5 μg/ml, H3149, Sigma). Only early passage GSC cells were used for the study. GSC21 were cultured at 37°C with 5% CO2.
The isolation and culture of bone marrow-derived macrophages (BMDMs) in mice
The C57BL/6 mice were euthanized by excessive anesthesia. The femur and the tibia on both legs were removed and the bone marrow was washed out by RPMI-1640 medium. The suspensions were then passed through a 70 μm stainless steel mesh and red blood cells were lysed by Red Blood Cell Lysis Buffer. After centrifugation, cells were cultured in RPMI-1640 medium supplemented with 10% FBS and were primed with 20 ng/ml rmM-CSF (416-ML, R&D) for 2–3 days to become BMDMs. BMDMs were cultured at 37°C with 5% CO2.
RNA interference
Three ARPC1B-targeting siRNAs (siARPC1B), three IRF2-targeting siRNAs (siIRF2) and a negative control siRNA (siNC) synthesized by Sangon Biotech (Shanghai, China) were transfected into U87, PGC28, GSC21 and THP1-derived macrophage cells with Lipofectamine 3000 reagent (L3000015, Invitrogen) according to the manufacturer’s instructions. For transfecting GSC21 cells, the gliomaspheres were dissociated into single cells with Accutase and then seeded in six well plates coated with 0.5% laminin (L2020, Sigma) preparedly in advance. Cells were harvested for the next assay after incubated for 48 h. The siRNA sense and antisense sequences were as follows:
siNC: UUCUCCGAACGUGUCACGUTT and ACGUGACACGUUCGGAGAATT; siARPC1B-467: GCUCUCGUGUGAUCUCCAUTT and AUGGAGAUCACACGAGAGCTT; siARPC1B-731: GGGUACAUGGCGUCUGUUUTT and AAACAGACGCCAUGUACCCTT; siARPC1B-1025: UCCAGAACCUGGACAAGAATT and UUCUUGUCCAGGUUCUGGATT; siIRF2-653: GCGGUCCUGACUUCAACUAUATT and UAUAGUUGAAGUCAGGACCGCTT; siIRF2-767: CCAGACAUUUGCCAAGUUGUATT and UACAACUUGGCAAAUGUCUGGTT; siIRF2-1014: CCUUCGUCACUUCCAACAAACTT and GUUUGUUGGAAGUGACGAAGGTT.
Lentivirus transfection
Lentiviruses carrying ARPC1B knockdown or control vectors were purchased from GeneChem. The lentivirus transduction was performed as the manufacturer’s instructions from GeneChem. For transfecting GSC21 cells, the gliomaspheres were dissociated into single cells with Accutase and then seeded in six well plates coated with 0.5% laminin preparedly in advance. The transfected cells were then selected using 3 μg/ml puromycin (REVG1001, GeneChem) for 15 days. The efficiency of ARPC1B knockdown was assessed at the protein level by Western blotting analysis.
Co-culturing system
Six-well transwells (3450, Corning) were used for creating co-culturing system. For macrophages, we first added 5 nM PMA into THP-1 cells with ARPC1B knockdown or negative control for 48 h in the upper chambers, which could stimulate THP-1 cells to become THP1-derived macrophages. The 6-well transwell inserts with macrophages were then transferred to another 6 well plate which was pre-seeded with different glioma cells and co-cultured for another 48 h. The macrophages in the upper chambers were used for migration assay while the glioma cells in the lower chambers were collected for migration and invasive assay and Western blot test. For microglia, we first planted HMC3 with ARPC1B knockdown or negative control into the upper chambers, and then transferred the 6-well transwell with HMC3 to another 6-well plate which was pre-seeded with different glioma cells and co-cultured for another 48 h. The glioma cells in the lower chambers were collected for migration and invasive assay and Western blot test.
Conditional medium collection
For macrophages, THP-1 cells transfected by ARPC1B knockdown lentivirus previously were induced to THP1-derived macrophages by PMA for 48 h. After removing the supernatant, macrophages were incubated with serum-free RMPI-1640 medium and collected the supernatant after 24 h incubation. Supernatant was mixed with equal volume proportion of RPMI-1640 medium containing 10% FBS to prepare conditional medium (CM). For U87 and PGC28 cells, U87 and PGC28 were transfected by a pool of three ARPC1B-targeting siRNAs for 48 h. After removing the supernatant, U87 and PGC28 were incubated with serum-free RMPI-1640 medium and collected the supernatant after 24 h incubation. Supernatant was mixed with equal volume proportion of RPMI-1640 medium containing 10% FBS to prepare CM.
Cell migration and invasion assay
Transwell inserts with a pore size of 8 µm (3422, Corning) were used for in vitro cell migration and invasion assays. To assess cell migration, U87 were resuspended in DMEM containing 0.2% FBS and seeded into the upper chambers of the transwell insert at a density of 2 × 104 /200 µl. A 600 µl volume of DMEM containing 20% FBS was added to the lower chamber. Macrophages, PGC28 and GSC21 were resuspended in RMPI-1640 containing 0.2% FBS and seeded into the upper chambers of the transwell insert at a density of 2 × 104 /200 µl. A 600 µl volume of RMPI-1640 containing 20% FBS was added to the lower chamber. For the invasion assay, U87 were resuspended as described above and seeded into the upper chamber of the insert that was pre-coated with 500 ng/ml Matrigel solution (356237, Corning) at a density of 4 × 104 /200 µl. A 600 µl volume of DMEM containing 20% FBS was added to the lower chamber. PGC28 and GSC21 were resuspended as described above and seeded into the upper chamber of the insert that was pre-coated with 500 ng/ml Matrigel solution at a density of 4 × 104 /200 µl. A 600 µl volume of RMPI-1640 containing 20% FBS was added to the lower chamber. After 22–24 h, cells on the upper side of the membrane were removed with a cotton swab. The membrane was fixed with methanol and stained with 1% crystal violet solution (G1062, Solarbio). The cells that had migrated to the lower side of the membrane were observed and photographed by the upright Microscope.
Cell proliferation assay
U87 and PGC28 cells were seeded into 96-well plates at a density of 1 × 103 cells in 200 μl medium per well and incubated at 37°C with 5% CO2 for 5 days. The cell growth was measured by adding 20 μl MTS solution (G3581, Promega) into the wells and incubated for 3 h at 37°C. Optical density (OD) values of each well were determined with a microplate reader (VICTOR Nivo Multimode Microplate Reader, PE) at the absorbance of 490 nm.
Protein extraction and Western blotting
Total proteins from tissues and cells were extracted by whole-cell lysis buffer (P0013B, Beyotime) and quantitated as described previously.22 Same micrograms of protein from each sample was loaded onto a lane and electrophoresed using 10% SDS-PAGE followed by transfer to a polyvinylidene difluoride (PVDF) membrane (0.45 μm, Millipore). After being blocked with 5% skimmed milk, the PVDF membranes were incubated with the primary antibody overnight at 4°C. Then, the PVDF membranes were incubated with the secondary antibodies at room temperature for 1 h. Protein expression was visualized with a chemiluminescence ECL reagent (180–5001, Tanon).
Real-time quantitative PCR (RT-PCR)
Total RNA was isolated from cell using MiniBEST Universal RNA Extraction Kit (9767, TaKaRa) according to the manufacturer’s instructions. Total RNA was reversely transcribed into cDNA with Prime-Script RT Master Mix (RR036Q, TaKaRa). qPCR was performed with SYBR Green Master Mix (RR820A, TaKaRa) for triplicate. The following conditions were used: 1 cycle of 95°C for 30s, followed by 40 cycles of a two-step cycling program (95°C for 5s; 60°C for 30s). The mRNA expression of target genes was calculated by the 2−ΔΔCt method and normalized to 18s mRNA expression.
Flow cytometry
The detailed protocol of flow cytometry was done as previously described.23 FACS data were analyzed using FlowJo software (version 10.4).
Tumor xenografts transplantation
Six-week-old male BALB/c nude mice were purchased from Beijing Vital River Laboratory Animal Technology. For determining the malignant effects of ARPC1B in macrophages, 3 µl cell suspension (shNC/shARPC1B-THP1 derived macrophages 1 × 105 with U87 1 × 105; shNC/shARPC1B-THP1 derived macrophages 1 × 105 with GSC21 2 × 105) were injected into the mice brains as previously described.24 For eliminating the faded role of intrinsic macrophages in mice, we injected clophosomes (clodronate liposomes, Liposoma BV) intraperitoneally into BALB/c nude mice every 2 days for 2 weeks to deplete the intrinsic macrophages in mice. Next, we co-transplanted macrophages and U87 cells (shNC/shARPC1B-THP1 derived macrophages 1 × 105 with U87 1 × 105) into BALB/c nude mice treated with clophosomes previously. For detecting the malignant effects of glioma-intrinsic ARPC1B, 3 µl cell suspension (shNC/shARPC1B-U87: 1 × 105; shNC/shARPC1B-GSC21: 2 × 105) were injected into the mice brains. Mice were sacrificed at same time for IHC analysis or observed until death for survival analysis. The section with the largest tumor cross-sectional area was selected for tumor size measurement in intracranial glioma models. For subcutaneous tumor models, 3 × 106 of ARPC1B knockdown U87 cells in 200 µl of PBS were subcutaneously injected into the left flank of BALB/c nude mice. The tumor size was measured every 4 days with a vernier caliper. The following formula was used to calculate the tumor volume: V = (length × width2)/2. The mice were sacrificed by cervical dislocation at day 35 after implantation, and the tumors were photographed.
Four-to-six-week-old male C57BL6 mice were purchased from Beijing Vital River Laboratory Animal Technology. For determining the malignant effects of ARPC1B in macrophages, 3 µl cell suspension (shNC/shARPC1B-BMDMs 2 × 105 with GL261 2 × 105) were injected into the mice brains. Mice were sacrificed at same time for IHC analysis or observed until death for survival analysis. All mice were resided in specific pathogen-free (SPF) conditions.
Ethics statement
The experimental protocol was approved by the ethics committee of The First Hospital of China Medical University. Animal experiments were conducted in accordance with the China Medical University Animal Care and Use Committee guidelines and approved by the Institutional Review Board of The First Hospital of China Medical University.
Statistical analysis
GraphPad Prism 8, and R 4.0.2 (https://www.r-project.org/) software were used for statistical analysis. Pearson correlation was used to calculate correlations. Univariate Cox regression analysis was performed by R Survival package. Patients were divided into high expression group and low expression group based on their median ARPC1B expression. The prognostic difference was evaluated by Kaplan–Meier survival analysis and log-rank test. Date were presented as mean ± standard deviation. Student’s t-test was used to assess differences and statistical significance was defined as a 2-tailed P value < .05.
Results
ARPC1B is the key macrophage-associated gene with prognostic value in GBM
As TAMs played a vital role in GBM progression, we sought to identify key factors that facilitate TAMs (Supplementary Figure S1). We first compiled a “macrophage” gene set as previously reported (Supplementary Table 1),25 and then GSVA algorithm was used to calculate a “macrophage score” based on the gene set above. The CGGA and TCGA GBM cohorts were analyzed for screening. First, we calculated the correlation between gene expression level and the macrophage score in GBM by Pearson correlation analysis. The top 250 score-correlated genes according to the Pearson correlation coefficient were, respectively, filtered out in CGGA and TCGA GBM cohorts. One hundred and thirty-three genes were positively correlated with the macrophage score in both CGGA and TCGA GBM cohorts (Supplementary Table 2). Second, Cox regression analysis indicated that 13 of the 133 genes had prognostic significance (P < .05) in both cohorts (Supplementary Table 3). Third, the Kaplan–Meier curve and log-rank analysis narrowed down the 13 candidates to five genes for which high expression robustly conferred reduced overall survival (OS) in both GBM cohorts (P < .05). Since the role of ARPC1B, one of these five genes, in glioma has not been investigated, we focused on ARPC1B for further exploration.
ARPC1B is highly expressed and strongly associated with TAMs in GBM
To further investigate the clinical significance of ARPC1B in glioma, we analyzed its expression in CGGA and TCGA cohorts. The expression level of ARPC1B was significantly higher in grade IV (GBM) than that of grade II and III tumors (Figure 1a). These results were further verified using immunohistochemistry (Figure 1b and Supplementary Figure 2a) and Western blotting (Figure 1d and Supplementary Figure 2b) of clinical tumor tissues with diverse grades. In addition, ARPC1B expression was significantly higher in IDH-wild-type tumors compared with IDH-mutant tumors, no matter in all grades or GBM (Supplementary Figure 2c).
Figure 1.

The expression profile of ARPC1B and its relationship between TAMs in GBM. (a) Expression of ARPC1B differed by grade in CGGA and TCGA cohorts (Student’s t test). (b) IHC staining of ARPC1B in clinical tissues with different grades. (c) Western blotting of ARPC1B in clinical tissues with different grades. (d) IHC staining of ARPC1B and IBA1(a marker of macrophages) in clinical GBM tissues (n = 30). (e) Statistical graph of IHC staining of ARPC1B and macrophage infiltration (IBA1) in clinical GBM tissues (Student’s t test, n = 30). (f) Kaplan-Meier survival analysis identified ARPC1B as a stable unfavorable prognostic factor in CGGA and TCGA GBM cohorts (log-rank test). (* means P < .05, *** means P < .001, **** means P < .0001).
To further investigate the relationship between ARPC1B and TAMs, immunohistochemistry staining of ARPC1B and IBA1 (a macrophage marker) was conducted with 30 clinical GBM samples (Figure 1d). We found a strongly positive correlation between ARPC1B expression and macrophage infiltration (Supplementary Figure 2d, r = 0.706, P < .0001), which indicated that higher ARPC1B levels were associated with more TAMs infiltration in GBM (Figure 1e). Consistently, ARPC1B expression was significantly correlated with the macrophage score in both CGGA and TCGA GBM cohorts (Supplementary Figure 2e). These results suggested that ARPC1B is a malignant indicator correlated with TAMs.
Considering the role of ARPC1B in higher tumor malignancy and regulating TAMs, the prognostic value of ARPC1B in GBM was next evaluated in both CGGA and TCGA cohorts. Kaplan–Meier curves and log-rank tests indicated that GBM patients with higher ARPC1B expression generally had shorter OS than those with lower expression (Figure 1f). The same trend of prognosis was found in gliomas of all grades (Supplementary Figure 2f). There was a significant difference in OS between GBM patients with high and low ARPC1B expression among those with IDH wild-type, whereas no such difference was found among GBM patients with IDH-mutant (Supplementary Figure 2g and h). Together, these results suggest that ARPC1B is associated with GBM progression and TAMs infiltration, which lead to unsatisfactory outcomes.
ARPC1B in macrophages promotes motility and epithelial–mesenchymal transition (EMT) of glioma cells
Our results from clinical tissue samples and public databases motivated us to further investigate the in-depth mechanisms involved in the interconnection among ARPC1B, TAMs, and glioma cells. THP-1 cells were differentiated into macrophages, followed by co-culture with glioma cells for 48 h in a transwell co-culture system (Figure 2a). We found that ARPC1B knockdown in macrophages significantly decreased their recruiting ability (Supplementary Figure S3a-c). Subsequently, we reconnoitered the effects of ARPC1B in macrophages on the phenotypes of glioma cells. The number of migrating and invasive glioma cells, both U87 and PGC28 (a primary glioma cell line derived from GBM specimen) cells, were significantly decreased after co-culture with ARPC1B-knockdown macrophages (Figure 2b–d). But there had no effects on the proliferation of glioma cells (Supplementary Figure S3d and e). These alterations suggested that ARPC1B knockdown in macrophages might alleviate the EMT status of glioma cells after co-culture. Indeed, the reduced EMT status of glioma cells after co-culture with ARPC1B-knockdown macrophages was confirmed by decreased protein expression of N-cadherin and vimentin, but increased expression of E-cadherin (Figure 2e). The same effects on the migration, invasion, and EMT status of glioma cells were found after treatment with the conditioned medium (CM) of ARPC1B-knockdown macrophages (Supplementary Figure S3f-j). We further isolated bone marrow-derived macrophages (BMDMs) in mice to corroborate the effect of ARPC1B in macrophages on glioma cells. GL261 were co-cultured with ARPC1B-knockdown BMDMs or negative control BMDMs (Supplementary Figure S4a). The migration and invasion abilities of GL261 co-cultured with ARPC1B-knockdown BMDMs were diminished and the EMT status of these cells was also restrained (Supplementary Figure S4b-e).
Figure 2.
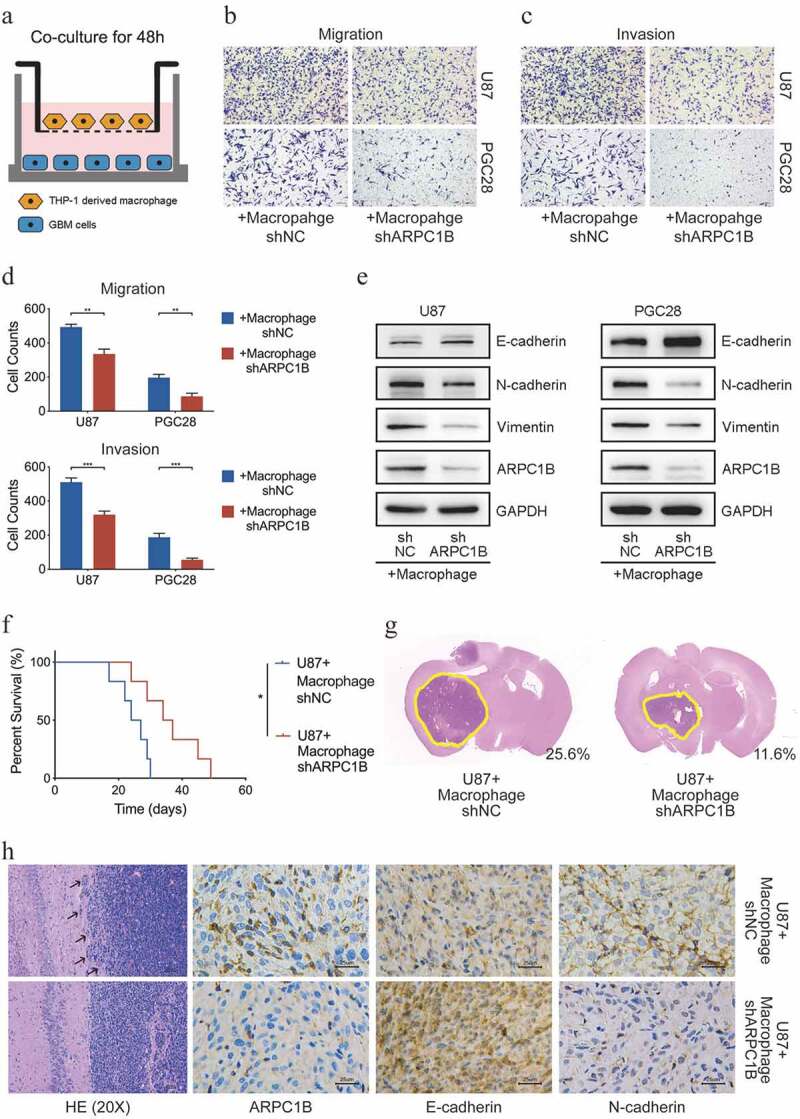
ARPC1B in TAMs regulated the migration, invasion and EMT status of glioma cells. (a) The schematic illustration of co-culturing glioma cells and macrophages. (b-d) The migration and invasion ability of glioma cells after co-culturing with ARPC1B knockdown macrophages for 48 h (Student’s t test, n = 3). (e) The EMT status of glioma cells after co-culturing with ARPC1B knockdown macrophages for 48 h. (f) The percent survival of tumor-bearing mice after different cells implantation intracranially (log-rank test, n = 6). (g) HE staining of tumors in tumor-bearing mice after different cells implantation intracranially (numbers on image means the ratio of tumor area in whole mice brain area) (h) HE and IHC staining of tumors in tumor-bearing mice after different cells implantation intracranially. (* means P < .05, ** means P < .01, *** means P < .001).
Microglia, as the brain-intrinsic myeloid cells, played similar role as macrophages in driving tumor progression.26 Therefore, we also explore the role of ARPC1B in microglia on malignant phenotype of glioma cells. HMC3 cells (a human microglia cell line) were transfected by lentiviruses to knockdown ARPC1B expression and co-cultured with glioma cells (U87 and PGC28) for 48 h (Supplementary Figure S5a). We found that the number of migrating and invasive U87 and PGC28 cells were unchanged after co-cultured with ARPC1B-knockdown microglia (Supplementary Figure S5b-d). The EMT status of U87 and PGC28 cells were also examined, showing there were no difference in EMT status of glioma cells after co-cultured with ARPC1B-knockdown microglia (Supplementary Figure S5e). These results declared that the ARPC1B in macrophages facilitated the malignant phenotype of glioma cells, but not microglia.
We next tried to determine the malignant effects of macrophage-expressed ARPC1B in intracranial xenograft models. Results revealed that mice co-transplanted with U87 and ARPC1B-knockdown macrophages had significantly prolonged survival time and smaller tumor size compared with the control group (Figure 2f,g). HE staining of tumors revealed a clear boundary between the tumor and normal tissue in mice co-transplanted with U87 and ARPC1B-knockdown macrophages compared with the control group, indicating that the migration and invasion ability of tumors was reduced (Figure 2h). Immunohistochemistry showed that co-transplantation of U87 cells and ARPC1B-knockdown macrophages resulted in an attenuated EMT status, characterized by a decrease in N-cadherin expression and an increase in E-cadherin expression (Figure 2h). Same experiments were performed in C57BL6 mice co-transplanted with GL261 and BMDMs for further verification, which also indicated that co-transplantation with GL261 and ARPC1B-knockdown BMDMs prolonged mice survival time, accompanied by smaller tumor size and diminished EMT status compared with the control group (Supplementary Figure S6a-c).
Considering the role of ARPC1B in intrinsic macrophages of mice, clophosome (clodronate liposomes; 100 μl) was intraperitoneally injected into BALB/c nude mice every 2 days for 2 weeks to deplete the intrinsic macrophages in mice (Supplementary Figure S6d). Then we co-transplanted macrophages and U87 cells in the mice treated with clophosome previously. Similar conclusions were confirmed that mice co-transplantated with U87 and ARPC1B-knockdown macrophages owned prolonged survival time. The tumor size in this group was also reduced compared with the control group (Supplementary Figure S6e and f). Taken together, these results indicated that ARPC1B in macrophages facilitated the malignant phenotype of glioma cells.
Glioma-intrinsic ARPC1B mediates migration, invasion, and EMT alterations of themselves
When Western blot analysis was performed to examine the EMT status of glioma cells after co-cultured with macrophages, we surprisingly observed that the ARPC1B expression in glioma cells was also reduced after co-culture with ARPC1B-knockdown macrophages (Figure 2e and Supplementary Figure S3j). We hypothesized that glioma-intrinsic ARPC1B also promoted the malignant phenotype of themselves. Immunofluorescence analysis of clinical GBM samples showed that there were cells expressing both ARPC1B and glial fibrillary acidic protein (GFAP), a glioma cell marker (Figure 3a). It supported a glioma-intrinsic ARPC1B expression. To examine the functions of ARPC1B in glioma cells in greater detail, we used two ARPC1B-specific siRNAs to knock down ARPC1B expression in U87 and PGC28 cells (Figure 3b), which significantly reduced the migratory and invasive abilities of both cell lines (Figure 3c–f). Moreover, after ARPC1B silencing, the expression of N-cadherin and vimentin was reduced, while E-cadherin expression was upregulated in glioma cells (Figure 3b). These data indicated that ARPC1B regulates the migration, invasion, and EMT status of glioma cells.
Figure 3.

Glioma-intrinsic ARPC1B promoted the malignant phenotypes of themselves. (a) Dual Immunofluorescence of ARPC1B and GFAP in clinical GBM samples. (b) The ARPC1B expression and EMT status of glioma cells after two ARPC1B-siRNAs’ transfections. (c-d) The migration ability of glioma cells after ARPC1B knockdown by two ARPC1B-siRNAs (Student’s t test, n = 3). (e-f) The invasion ability of glioma cells after ARPC1B knockdown by two ARPC1B-siRNAs (Student’s t test, n = 3). (g) The percent survival of tumor-bearing mice after U87 with ARPC1B knockdown or negative control implanted intracranially (log-rank test, n = 5). (h) HE and IHC staining of tumors in tumor-bearing mice after U87 with ARPC1B knockdown or negative control implanted intracranially. (* means P < .05, ** means P < .01, *** means P < .001, **** means P < .0001).
To determine the malignant effects of glioma-intrinsic ARPC1B in vivo, we intracranially transplanted U87 cells with ARPC1B knockdown into BALB/c nude mice (Supplementary Figure S7a). ARPC1B knockdown led to a prolonged survival and smaller tumor size (Figure 3g and Supplementary Figure S7b). HE staining revealed a clearer tumor border in mice of the ARPC1B-knockdown group (Figure 3h). Furthermore, immunohistochemical analyses demonstrated that ARPC1B and N-cadherin expression was reduced while E-cadherin was enriched in ARPC1B knockdown group (Figure 3h). Subcutaneously transplanted ARPC1B-knockdown U87 cells significantly decreased subcutaneous tumor growth and their EMT status (Supplementary Figure S7c-e). These data highlighted the important role of glioma-intrinsic ARPC1B in promoting the tumorgenicity of glioma cells.
ARPC1B in macrophages regulates phenotype changes of glioma cells via IFNγ-IRF2-ARPC1B axis
Since glioma-intrinsic ARPC1B regulated malignant phenotype of glioma cells themselves and the expression of glioma-intrinsic ARPC1B was also reduced after co-cultured with ARPC1B-knockdown macrophages, we hypothesized that the malignant phenotype alterations of glioma cells after co-culture with ARPC1B-knockdown macrophages were due to the effects on glioma-intrinsic ARPC1B expression. A cytokine array was performed to determine the factors secreted by ARPC1B-knockdown macrophages that could potentially act as signals for regulating the malignant phenotype of glioma cells. Among the 36 cytokines in the array, interferon-gamma (IFNγ) was the most strongly down-regulated in ARPC1B-knockdown macrophages co-cultured with U87 cells compared with the level in the control group (Figure 4a). Therefore, we further focused on IFNγ to determine the regulation mechanism. The PCR results confirmed that IFNγ significantly induced ARPC1B overexpression in U87 and PGC28 cells (Supplementary Figure S8a), and Western blotting results demostrated that IFNγ enhanced ARPC1B expression and EMT status (Figure 4b). We further added IFNγ into the co-culturing system to determine the malignant phenotype alterations of glioma cells. The decreased migration, invasion, and EMT status of glioma cells induced by co-culture with ARPC1B-knockdown macrophages were all rescued by the addition of IFNγ to the co-culture system (Figure 4c–g). These data indicated a role of IFNγ in facilitating the GBM-TAMs network based on ARPC1B.
Figure 4.

ARPC1B regulated the GBM-TAMs networks by IFNγ-IRF2-ARPC1B axis. (a) The expression of several cytokines in ARPC1B knockdown macrophages co-cultured with U87 cells compared to control group (Student’s t test, n = 3). (b) Western blotting of ARPC1B, IRF2 and EMT status after glioma cells treated with different doses of IFNγ. (c-d) The migration ability of glioma cells after co-culturing with ARPC1B knockdown TAMs with or without IFNγ stimulation for 48 h (Student’s t test, n = 3). (e-f) The invasion ability of glioma cells after co-culturing with ARPC1B knockdown macrophages with or without IFNγ stimulation for 48 h (Student’s t test, n = 3). (g) Western blotting of ARPC1B, IRF2 and EMT status of glioma cells after co-culturing with ARPC1B knockdown TAMs with or without IFNγ stimulation for 48 h. (h) The expression correlation of IRF2 and ARPC1B in CGGA GBM cohort (Pearson correlation, n = 138). (i) The expression of IRF2 in high and low ARPC1B group in CGGA GBM cohort (Student’s t test). (j) Western blotting of ARPC1B, IRF2 and EMT status of glioma cells after different doses of IFNγ stimulation with or without IRF2 knockdown. (* means P < .05, ** means P < .01, *** means P < .001, **** means P < .0001).
To further explore the ARPC1B-promoting mechanisms of IFNγ, we predicted the transcription factors (TF) of ARPC1B through the PROMO website, revealing 16 TFs (Supplementary Table S4). GSE55750, a database containing expression profiling of glioma cell after IFNγ stimulation, was next analyzed. 2054 genes showed upregulated expression in glioma cells after IFNγ treatment (Supplementary Table S5). After taking the intersection of the 16 predicted TFs and 2054 genes, four common genes (ATF3, CEBPA, CEBPB and IRF2) were obtained (Supplementary Figure S8b). Among these four genes, only IRF2 was confirmed to be overexpressed in IFNγ-stimulated U87 and PGC28 cells and reduced in both cell lines co-cultured with ARPC1B-knockdown macrophages (Supplementary Figure S8c-e). In CGGA GBM cohort, IRF2 expression was found to be enhanced in high ARPC1B expression group and was positively correlated with ARPC1B expression (Figure 4h,i). Therefore, we further explored the role of IRF2 in the IFNγ-stimulated ARPC1B expression of glioma cells. Western blotting confirmed that IRF2 expression was downregulated in glioma cells after co-culture with ARPC1B-knockdown macrophages, which could be rescued by IFNγ treatment (Figure 4g). IRF2 knockdown with specific siRNAs also decreased the glioma-intrinsic ARPC1B expression level, and drastically attenuated the ability of IFNγ to upregulate ARPC1B expression and EMT status (Figure 4j). Taken together, these data suggested that ARPC1B deficiency in macrophages led to the downregulation of glioma-intrinsic ARPC1B through impairing the IFNγ-IRF2-ARPC1B axis.
Glioma-intrinsic ARPC1B promotes macrophage recruitment
During the progression of GBM, glioma cells can attract macrophages to create an immunosuppressive microenvironment and thus leading to unsatisfied outcome.27 Since we found a co-expression tendency of ARPC1B and IBA1 in clinical GBM samples (Figure 1d), we examine whether glioma-intrinsic ARPC1B promoted macrophage recruitment. GSEA of CGGA GBM data demonstrated significant enrichment in the “macrophage chemotaxis” gene set with high ARPC1B expression (Figure 5a and Supplementary Table S6). PCA also confirmed a distinct “macrophage chemotaxis” phenotype between GBM patients with low and high ARPC1B expression (Supplementary Figure S9a). In vitro experiments showed that incubation of CM from ARPC1B-knockdown glioma cells strongly decreased macrophage migration (Figure 5b,c). The migration of BMDMs was also noticeably decreased when CM from ARPC1B-knockdown GL261 cells was used as a chemoattractant (Supplementary Figure S9b and c). ARPC1B-knockdown glioma cells also weakened IBA1 expression in both intracranial xenograft and subcutaneous mouse models (Figure 3h and Supplementary Figure S7e). We also detected the expression of several chemokines and found that CCL2 and CCL7 were downregulated in ARPC1B-knockdown glioma cells (Figure 5c). Together, these results showed that intrinsic ARPC1B in glioma cells promotes macrophage recruitment, which might be regulated by CCL2 and CCL7.
Figure 5.

Glioma-intrinsic ARPC1B controlled the recruitment of macrophages. (a) “Macrophage Chemotaxis” term enriched in high ARPC1B group in CGGA GBM cohort. (b-c) The migration ability of THP1-derived macrophages decreased under the CM from ARPC1B-knockdown glioma cells (Student’s t test, n = 3). (d) PCR results of several chemokines in ARPC1B-knockdown glioma cells (Student’s t test, n = 3). (e) The illustration of the regulating networks between glioma cells and TAMs based on ARPC1B. (* means P < .05, ** means P < .01, *** means P < .001, **** means P < .0001).
ARPC1B regulates positive feedback between TAMs and glioma cells in patient-derived gliomaspheres
Finally, we evaluated the effects of ARPC1B in patient-derived gliomasphere GSC21 cells, which more faithfully retain the genetic features of primary GBM. The migration, invasion and EMT status of GSC21 cells were reduced by co-culture with ARPC1B-knockdown macrophages and these alterations were all rescued by IFNγ treatment (Figure 6a–c and Supplementary Figure S10a). The ARPC1B expression level and EMT status were also enhanced after treatment with IFNγ (Supplementary Figure S10b). We further co-transplanted GSC21 cells with macrophages into BALB/c nude mice. Results showed that co-transplantation with ARPC1B-knockdown macrophages prolonged the survival time of tumor-bearing mice, decreased tumor growth and dampened EMT status in vivo (Figure 6d,e). The role of glioma-intrinsic ARPC1B was also corroborated in GSC21 cells. After silencing GSC21-intrinsic ARPC1B expression with two siRNAs, the migratory, invasion and EMT status of GSC21 cells were markedly altered (Figure 6f–h and Supplementary Figure S10c). The intracranial xenograft model was further established using GSC21 cells in BALB/c nude mice (Supplementary Figure S10d). High expression of ARPC1B was strongly correlated with a poor prognosis of tumor-bearing mice (Figure 6i). The downregulation of ARPC1B significantly decreased the tumor volume and resulted in a weakened invasive morphology (Figure 6j and Supplementary Figure S10e). Moreover, immunohistochemical analysis indicated that higher ARPC1B expression was accompanied by an enhanced EMT status (Figure 6j). We also discovered a co-expression tendency of ARPC1B and IBA1, indicating that GSC21-intrinsic ARPC1B significantly increased the recruitment of TAMs in vivo (Figure 6j). These data support the hypothesis of positive feedback signaling between TAMs and glioma cells based on ARPC1B during glioma development and progression.
Figure 6.

ARPC1B regulated positive feedback signaling between TAMs and glioma cells in patient-derived gliomasphere GSC21 cells. (a) The migration ability of GSC21 cells after co-culturing with ARPC1B knockdown macrophages with or without IFNγ stimulation for 48 h (Student’s t test, n = 3). (b) The invasion ability of GSC21 cells after co-culturing with ARPC1B knockdown macrophages with or without IFNγ stimulation for 48 h (Student’s t test, n = 3). (c) Western blotting of ARPC1B, IRF2 and EMT status of GSC21 cells after co-culturing with ARPC1B knockdown macrophages with or without IFNγ stimulation for 48 h. (d) The percent survival of tumor-bearing mice after different cells implantation intracranially (log-rank test, n = 6). (e) HE and IHC staining of tumors in tumor-bearing mice after different cells implantation intracranially (numbers on image means the ratio of tumor area in whole mice brain area). (f) The migration ability of GSC21 cells after ARPC1B knockdown by two ARPC1B-siRNAs (Student’s t test, n = 3). (g) The invasion ability of GSC21 cells after ARPC1B knockdown by two ARPC1B-siRNAs (Student’s t test, n = 3). (h) ARPC1B expression and EMT status of GSC21 cells after two ARPC1B-siRNAs’ transfections. (i) The percent survival of tumor-bearing mice after GSC21 with ARPC1B knockdown or negative control implanted intracranially (log-rank test, n = 6). (j) HE and IHC staining of tumors in tumor-bearing mice after GSC21 with ARPC1B knockdown or negative control implanted intracranially. (* means P < .05, ** means P < .01).
Discussion
Tumor-promoting immune disorder has been regarded as one of the factors contributing to GBM progression.28 An in-depth understanding of the biology of the immunosuppressive TME in GBM may reveal new therapeutic targets. In particular, TAMs comprise nearly 30–50% of the cells in the TME. Our previous study demonstrated the crucial role of TAMs in glioma malignancy, immunosuppressive microenvironment and unfavorable prognosis.3 In the present study, we identified ARPC1B was the key macrophage-associated gene with prognostic value in GBM, which was associated with malignancy and TAMs enrichment. Subsequently, we demonstrated that ARPC1B in macrophages promoted glioma cells migration, invasion and EMT status, whereas glioma-intrinsic ARPC1B also enhanced the migration, invasion and EMT status of themselves. Meanwhile, glioma-intrinsic ARPC1B promoted macrophage chemotaxis. Exploring the role of ARPC1B in GBM-TAMs regulating network (Figure 5d) might expand our understanding of TME and provide us a novel biomarker and therapeutic target in GBM.
Our study was the first time to demonstrate the malignant role of ARPC1B in glioma progression. Recent evidences were presented for proving the tumor-promoting role of ARPC1B. First, the expression of ARPC1B was consistently enhanced along with increasing glioma grade, as confirmed in the bioinformatic analysis and immunohistochemistry or Western blotting of clinical tissues. Second, experiments in vitro and in vivo both illuminated the tumor-promoting role of ARPC1B (migration, invasion and EMT status), no matter in glioma cells or in macrophages. The expression of E-cadherin, a negative regulator of EMT, was found to be negatively correlated with ARPC1B expression. E-cadherin played a role of invasion suppressor to inhibit tumor progression, which further confirmed the tumor-promoting role of ARPC1B.29,30 Third, glioma-intrinsic ARPC1B promoted TAMs infiltration, while TAMs were clearly reported to facilitate glioma progression. These all can explain the positive role of ARPC1B in glioma progression. Several studies had revealed a weak relationship between ARPC1B and tumor progression in other cancer types, which corroborate our results to some extent.16–18 Together, these findings suggested that ARPC1B aggravated glioma malignancy.
For myeloid cells in brain, it consists of brain-resident microglia and infiltrated macrophage from the blood stream.31 Therefore, we respectively examined the role of ARPC1B in macrophage and microglia affecting the malignant phenotype of glioma cells. Our study illustrated that only the ARPC1B in macrophage, not microglia, could promoted the malignancy of glioma cells (migration, invasion and EMT status). As reported, macrophages account for a larger proportion than microglia in glioma. Macrophages derived from bone-marrow accounted for 85% while brain-resident microglia accounted for other 15% of myeloid cells.32 Moreover, the total number of microglia cells does not vary with the grade of malignancy, but macrophages enrich in high-grade gliomas.33 Our previous study also confirmed that macrophage, but not microglia, was identified with prognostic significance in glioma progression.3 These results strengthened the important role of macrophages in glioma progression. Meanwhile, TAMs initiate proinflammatory (M1) or immunosuppressive (M2) function depending on their polarization status.34,35 M2-like macrophage was the main macrophage type that induced immunosuppressive microenvironment and thus accelerated glioma progression. Our results indicated that ARPC1B in macrophages played a tumor-promoting role in glioma malignancy. ARPC1B was also found to create immunosuppressive microenvironment by strongly correlated with immunosuppressors and immune inhibitory checkpoints (PD-1, PD-L1 and TIM3; data not shown). Considering the role of ARPC1B in immunosuppressive microenvironment and glioma progression, we hypothesized that ARPC1B in M2-like macrophage could be upregulated to promote the malignant phenotype of glioma cells more likely. Further studies should be performed to investigate the role of ARPC1B in macrophage polarization status that promote tumor invasion.
Previous studies on genes associated with a poor prognosis of GBM only concentrated on their impact on tumor cells, whereas their effects on non-tumor cells had been largely neglected. Our study demonstrates that ARPC1B in macrophages instructively promoted the metastatic progression (migration, invasion and EMT status) of glioma cells. Previous studies on GBM-TAMs regulating networks indicated that glioma cells can regulate the chemotaxis and polarization of TAMs,36,37 and in turn TAMs can affect the malignant phenotype of glioma cells.26,38,39 Based on this GBM-TAMs network, GBM possessed the characteristics of high recurrence rate and high mortality. Our in vitro and vivo experiments revealed that the same gene (that is, ARPC1B) can act as a driver of malignant progression of tumors whether it is expressed in TAMs or tumor cells, which is similar to the effects of programmed death-1 (PD-1).40,41 Thus, ARPC1B has a tumor-promoting role in both TAMs and glioma cells.
Recent studies of ARPC1B mostly focused on its function in immune cells.12–14 Through a thorough analysis of CGGA and TCGA GBM cohorts, ARPC1B was found to be the key gene that was characterized by strongly positive correlation with TAMs and prognostic value in GBM. We also performed bioinformatic analysis, IHC staining of GBM species, experiments in vitro and vivo to affirm the relationship between ARPC1B and macrophages, which was highly convinced. Moreover, GBM patients with IDH-wildtype exhibited higher ARPC1B expression than patients with IDH-mutant GBM. Compared with IDH-mutant gliomas, patients with IDH-wildtype have been shown to have higher infiltration of immune cells and macrophages.42 We also found that ARPC1B had prognostic significance only for patients with IDH-wildtype and not IDH mutant. These findings suggested that ARPC1B contributes to the poor prognosis of GBM patients by regulating TAMs.
Interferons play a critical role in the immune system process and antitumour immune response. However, analysis of differentially expressed cytokines caused by ARPC1B knockdown in TAMs identified IFNγ as the mediator between glioma cells and TAMs. Moreover, IFNγ could reverse the decreased migration, invasion and EMT status of glioma cells induced by co-culture with ARPC1B-knockdown macrophages. IFNγ also promoted the EMT status in glioma cells. These findings indicate that IFNγ promotes the malignant phenotypes of glioma cells. Indeed, several studies have shown the tumor-promoting and EMT-enhancing role of IFNγ in tumor cells.43–45 We previously developed an interferon risk signature, which was confirmed to be an independent indicator for an unfavorable prognosis in glioma.19 Therefore, our results suggest that IFNγ facilitates the TAMs-GBM network based on ARPC1B to promote the malignancy of glioma cells.
The interferon regulatory factor (IRF) proteins family were the crucial factors in immunoregulation, cell proliferation regulation and cellular response which was involved in tumorigenesis.46,47 Database analysis predicted IRF2, a member of the interferon regulatory factor (IRF) family, as a TF of ARPC1B, which was confirmed to regulate IFNγ-stimulated ARPC1B expression. Western blotting showed that IRF2 deficiency blocked the upregulation of ARPC1B caused by IFNγ treatment, suggesting that IRF2 also plays an oncogenic role in glioma progression. Several studies indicated the potential oncogenic roles of IFNγ through enrichment of IRF2.48 Moreover, IRF2 was associated with a more advanced pathological grade and worse outcomes in glioma patients.49 These results point to a role of IRF2 not only in the IFNγ-mediated regulation of ARPC1B expression but also in glioma progression.
In summary, our study suggests ARPC1B as a novel mediator regulating the GBM-TAMs network, which jointly promotes the malignant progression of GBM. Further research should be carried out to confirm the crucial role of ARPC1B in GBM, which could contribute to improving understanding of the properties and functions of the TME and help to develop new treatment strategies for improving the prognosis of patients with GBM.
Supplementary Material
Acknowledgments
The authors would like to acknowledge all the members in Dr. Wu AH’s laboratory for help with this study.
Funding Statement
This work was supported by the National Natural Science Foundation of China [Nos. U20A20380, 81172409, 81472360, and 81872054 to A. Wu; No. 81872057 to P. Cheng; No. 81902546 to W. Cheng and No. 81672824 to L. Chen]; Liaoning Science and Technology Plan Projects [No. 2011225034 to A. Wu]; Natural Science Foundation of Liaoning Province [No. 20180550063 to P. Cheng]; National Postdoctoral Program for Innovative Talents [No. BX20180384 to W. Cheng]; China Postdoctoral Science Foundation [No. 2019M651169 to W. Cheng]; Liao Ning Revitalization Talents Program [No. XLYC1807255 to W. Cheng].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Authors’ contributions
T.Q.L., C.Z., X.C., W.C., and A.H.W. contributed to the study conceptualization; A.H.W., W.C., P.C. and L.C. secured funding for the project; T.Q.L., C.Z. and X.C. conducted the experiments with assistance from G.F.G., C.Y.Z., J.Q.W. and S.S.; T.Q.L. and W.C. wrote the manuscript. All authors approved final manuscript.
Supplementary materials
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Weller M, van den Bent M, Tonn J, Stupp R, Preusser M, Cohen-Jonathan-Moyal E, Henriksson R, Rhun EL, Balana C, Chinot O, et al. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. 2017;18(6):e315–16. doi: 10.1016/S1470-2045(17)30194-8. [DOI] [PubMed] [Google Scholar]
- 2.Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, deCarvalho AC, Lyu S, Li P, Li Y, et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell. 2017;32(1):42–56.e46. doi: 10.1016/j.ccell.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang C, Cheng W, Ren X, Wang Z, Liu X, Li G, Han S, Jiang T, Wu A.. Tumor purity as an underlying key factor in glioma. Clin Cancer Res. 2017;23(20):6279–6291. doi: 10.1158/1078-0432.CCR-16-2598. [DOI] [PubMed] [Google Scholar]
- 4.Wei J, Chen P, Gupta P, Ott M, Zamler D, Kassab C, Bhat KP, Curran MA, de Groot JF, Heimberger AB, et al. Immune biology of glioma-associated macrophages and microglia: functional and therapeutic implications. Neuro-oncology. 2020;22(2):180–194. doi: 10.1093/neuonc/noz212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akkari L, Bowman R, Tessier J, Klemm F, Handgraaf SM, de Groot M, Quail DF, Tillard L, Gadiot J, Huse JT, et al. Dynamic changes in glioma macrophage populations after radiotherapy reveal CSF-1R inhibition as a strategy to overcome resistance. Sci Transl Med. 2020;12(552). doi: 10.1126/scitranslmed.aaw7843. [DOI] [PubMed] [Google Scholar]
- 6.Saha D, Martuza R, Rabkin S. Macrophage polarization contributes to glioblastoma eradication by combination immunovirotherapy and immune checkpoint blockade. Cancer Cell. 2017;32(2):253–267.e255. doi: 10.1016/j.ccell.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu C, Kros J, Cheng C, Mustafa D. The contribution of tumor-associated macrophages in glioma neo-angiogenesis and implications for anti-angiogenic strategies. Neuro-oncology. 2017;19(11):1435–1446. doi: 10.1093/neuonc/nox081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rocca D, Amici M, Antoniou A, Blanco Suarez E, Halemani N, Murk K, McGarvey J, Jaafari N, Mellor J, Collingridge G, et al. The small GTPase Arf1 modulates Arp2/3-mediated actin polymerization via PICK1 to regulate synaptic plasticity. Neuron. 2013;79(2):293–307. doi: 10.1016/j.neuron.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Semba S, Iwaya K, Matsubayashi J, Serizawa H, Kataba H, Hirano T, Kato H, Matsuoka T, Mukai K. Coexpression of actin-related protein 2 and Wiskott-Aldrich syndrome family verproline-homologous protein 2 in adenocarcinoma of the lung. Clin Cancer Res. 2006;12(8):2449–2454. doi: 10.1158/1078-0432.CCR-05-2566. [DOI] [PubMed] [Google Scholar]
- 10.Chakraborty S, Lakshmanan M, Swa H, Chen J, Zhang X, Ong YS, Loo LS, Akıncılar SC, Gunaratne J, Tergaonkar V, et al. An oncogenic role of Agrin in regulating focal adhesion integrity in hepatocellular carcinoma. Nat Commun. 2015;6(1):6184. doi: 10.1038/ncomms7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yan P, Liu J, Zhou R, Lin C, Wu K, Yang S, Yang S, Zhou J, Xu L, Wang H, et al. LASP1 interacts with N-WASP to activate the Arp2/3 complex and facilitate colorectal cancer metastasis by increasing tumour budding and worsening the pattern of invasion. Oncogene. 2020;39(35):5743–5755. doi: 10.1038/s41388-020-01397-7. [DOI] [PubMed] [Google Scholar]
- 12.Brigida I, Zoccolillo M, Cicalese M, Pfajfer L, Barzaghi F, Scala S, Oleaga-Quintas C, Álvarez-Álvarez JA, Sereni L, Giannelli S, et al. ARPC1BT-cell defects in patients with germline mutations account for combined immunodeficiency. Blood. 2018;132(22):2362–2374. doi: 10.1182/blood-2018-07-863431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Volpi S, Cicalese M, Tuijnenburg P, Tool ATJ, Cuadrado E, Abu-Halaweh M, Ahanchian H, Alzyoud R, Akdemir ZC, Barzaghi F, et al. A combined immunodeficiency with severe infections, inflammation, and allergy caused by ARPC1B deficiency. J Allergy Clin Immunol. 2019;143(6):2296–2299. doi: 10.1016/j.jaci.2019.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Randzavola L, Strege K, Juzans M, Asano Y, Stinchcombe JC, Gawden-Bone CM, Seaman MNJ, Kuijpers TW, Griffiths GM. Loss of ARPC1B impairs cytotoxic T lymphocyte maintenance and cytolytic activity. J Clin Invest. 2019;129(12):5600–5614. doi: 10.1172/JCI129388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kahr W, Pluthero F, Elkadri A, Warner N, Drobac M, Chen CH, Lo RW, Li L, Li R, Li Q, et al. Loss of the Arp2/3 complex component ARPC1B causes platelet abnormalities and predisposes to inflammatory disease. Nat Commun. 2017;8(1):14816. doi: 10.1038/ncomms14816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumagai K, Nimura Y, Mizota A, Miyahara N, Aoki M, Furusawa Y, Takiguchi M, Yamamoto S, Seki N. Arpc1b gene is a candidate prediction marker for choroidal malignant melanomas sensitive to radiotherapy. Invest Ophthalmol Vis Sci. 2006;47(6):2300–2304. doi: 10.1167/iovs.05-0810. [DOI] [PubMed] [Google Scholar]
- 17.Zucchini C, Rocchi A, Manara M, De Sanctis P, Capanni C, Bianchini M, Carinci P, Scotlandi K, Valvassori L. Apoptotic genes as potential markers of metastatic phenotype in human osteosarcoma cell lines. Int J Oncol. 2008;32:17–31. [PubMed] [Google Scholar]
- 18.Auzair L, Vincent-Chong V, Ghani W, Kallarakkal TG, Ramanathan A, Lee CE, Rahman ZAA, Ismail SM, Abraham MT, Zain RB, et al. Caveolin 1 (Cav-1) and actin-related protein 2/3 complex, subunit 1B (ARPC1B) expressions as prognostic indicators for oral squamous cell carcinoma (OSCC). European Archives of Oto-rhino-laryngology: Official Journal of the European Federation of Oto-Rhino-Laryngological Societies (EUFOS): Affiliated with the German Society for Oto-Rhino-Laryngology - Head and Neck Surgery. 2016;273(7):1885–1893. doi: 10.1007/s00405-015-3703-9. [DOI] [PubMed] [Google Scholar]
- 19.Zhu C, Zou C, Guan G, Guo Q, Yan Z, Liu T, Shen S, Xu X, Chen C, Lin Z, et al. Development and validation of an interferon signature predicting prognosis and treatment response for glioblastoma. Oncoimmunology. 2019;8(9):e1621677. doi: 10.1080/2162402X.2019.1621677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng W, Li M, Jiang Y, Zhang C, Cai J, Wang K, Wu A. Association between small heat shock protein B11 and the prognostic value of MGMT promoter methylation in patients with high-grade glioma. J Neurosurg. 2016;125(1):7–16. doi: 10.3171/2015.5.JNS142437. [DOI] [PubMed] [Google Scholar]
- 21.Zhang L, Guo Q, Guan G, Cheng W, Cheng P, Wu A. Integrin beta 5 is a prognostic biomarker and potential therapeutic target in glioblastoma. Front Oncol. 2019;9:904. doi: 10.3389/fonc.2019.00904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu C, Chen X, Guan G, Zou C, Guo Q, Cheng P, Cheng W, Wu A. IFI30 is a novel immune-related target with predicting value of prognosis and treatment response in glioblastoma. Onco Targets Ther. 2020;13:1129–1143. doi: 10.2147/OTT.S237162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liang Q, Wu J, Zhao X, Shen S, Zhu C, Liu T, Cui X, Chen L, Wei C, Cheng P, et al. Establishment of tumor inflammasome clusters with distinct immunogenomic landscape aids immunotherapy. Theranostics. 2021;11(20):9884–9903. doi: 10.7150/thno.63202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ji J, Ding K, Luo T, Zhang X, Chen A, Zhang D, Li G, Thorsen F, Huang B, Li X, et al. TRIM22 activates NF-κB signaling in glioblastoma by accelerating the degradation of IκBα. Cell Death Differ. 2021;28(1):367–381. doi: 10.1038/s41418-020-00606-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao M, Lin Y, Liu X, Li Y, Zhang C, Wang Z, Wang Z, Wang Y, Guo Z. ISG20 promotes local tumor immunity and contributes to poor survival in human glioma. Oncoimmunology. 2019;8(2):e1534038. doi: 10.1080/2162402X.2018.1534038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu-Ju WC, Chen C, Lin C, Feng L-Y, Lin Y-C, Wei K-C, Huang C-Y, Fang J-Y, Chen P-Y. CCL5 of glioma-associated microglia/macrophages regulates glioma migration and invasion via calcium-dependent matrix metalloproteinase 2. Neuro-oncology. 2020;22(2):253–266. doi: 10.1093/neuonc/noz189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quail D, Joyce J. The microenvironmental landscape of brain tumors. Cancer Cell. 2017;31(3):326–341. doi: 10.1016/j.ccell.2017.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tomaszewski W, Sanchez-Perez L, Gajewski T, Sampson J. Brain tumor microenvironment and host state: implications for immunotherapy. Clin Cancer Res. 2019;25(14):4202–4210. doi: 10.1158/1078-0432.CCR-18-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong A, Gumbiner B. Adhesion-independent mechanism for suppression of tumor cell invasion by E-cadherin. J Cell Biol. 2003;161(6):1191–1203. doi: 10.1083/jcb.200212033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Semb H, Christofori G. The tumor-suppressor function of E-cadherin. Am J Hum Genet. 1998;63(6):1588–1593. doi: 10.1086/302173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hambardzumyan D, Gutmann D, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci. 2016;19(1):20–27. doi: 10.1038/nn.4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Z, Feng X, Herting C, Garcia V, Nie K, Pong W, et al. Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res. 2017;77:2266–2278. doi: 10.1158/0008-5472.CAN-16-2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma J, Chen C, and Li M. Macrophages/Microglia in the glioblastoma tumor microenvironment. Int J Mol Sci. 2021;22 (11): 5775. doi: 10.3390/ijms22115775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Müller S, Kohanbash G, Liu S, Alvarado B, Carrera D, Bhaduri A, Watchmaker PB, Yagnik G, Di Lullo E, Malatesta M, et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017;18(1):234. doi: 10.1186/s13059-017-1362-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dastmalchi F, Deleyrolle L, Karachi A, Mitchell D, Rahman M. Metabolomics monitoring of treatment response to brain tumor immunotherapy. Front Oncol. 2021;11:691246. doi: 10.3389/fonc.2021.691246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yin J, Kim S, Choi E, Oh YT, Lin W, Kim T-H, Sa JK, Hong JH, Park SH, Kwon HJ, et al. ARS2/MAGL signaling in glioblastoma stem cells promotes self-renewal and M2-like polarization of tumor-associated macrophages. Nat Commun. 2020;11(1):2978. doi: 10.1038/s41467-020-16789-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Boeck A, Ahn B, D’Mello C, Lun X, Menon S, Alshehri M, et al. Glioma-derived IL-33 orchestrates an inflammatory brain tumor microenvironment that accelerates glioma progression. Nat Commun. 2020;11(1):4997. doi: 10.1038/s41467-020-18569-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sa J, Chang N, Lee H, Cho HJ, Ceccarelli M, Cerulo L, Yin J, Kim SS, Caruso FP, Lee M, et al. Transcriptional regulatory networks of tumor-associated macrophages that drive malignancy in mesenchymal glioblastoma. Genome Biol. 2020;21(1):216. doi: 10.1186/s13059-020-02140-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Herting C, Chen Z, Maximov V, Duffy A, Szulzewsky F, Shayakhmetov DM, Hambardzumyan D. Tumour-associated macrophage-derived interleukin-1 mediates glioblastoma-associated cerebral oedema. Brain. 2019;142(12):3834–3851. doi: 10.1093/brain/awz331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kleffel S, Posch C, Barthel S, Mueller H, Schlapbach C, Guenova E, Elco C, Lee N, Juneja V, Zhan Q, et al. Melanoma cell-intrinsic PD-1 receptor functions promote tumor growth. Cell. 2015;162(6):1242–1256. doi: 10.1016/j.cell.2015.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li B, Song T, Wang F, Yin C, Li Z, Lin J-P, Meng Y-Q, Feng H-M, Jing T. Tumor-derived exosomal HMGB1 promotes esophageal squamous cell carcinoma progression through inducing PD1 TAM expansion. Oncogenesis. 2019;8(3):17. doi: 10.1038/s41389-019-0126-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Waitkus M, Diplas B, Yan H. Isocitrate dehydrogenase mutations in gliomas. Neuro-oncology. 2016;18(1):16–26. doi: 10.1093/neuonc/nov136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trivanović D, Jauković A, Krstić J, Nikolić S, Okić Djordjević I, Kukolj T, Obradović H, Mojsilović S, Ilić V, Santibanez JF, et al. Inflammatory cytokines prime adipose tissue mesenchymal stem cells to enhance malignancy of MCF-7 breast cancer cells via transforming growth factor-β1. IUBMB Life. 2016;68(3):190–200. doi: 10.1002/iub.1473. [DOI] [PubMed] [Google Scholar]
- 44.Lv N, Gao Y, Guan H, WU D, Ding S, Teng W, Shan Z. Inflammatory mediators, tumor necrosis factor-α and interferon-γ, induce EMT in human PTC cell lines. Oncol Lett. 2015;10(4):2591–2597. doi: 10.3892/ol.2015.3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Imai D, Yoshizumi T, Okano S, Itoh S, Ikegami T, Harada N, Aishima S, Oda Y, Maehara Y. IFN-γ promotes epithelial-mesenchymal transition and the expression of PD-L1 in pancreatic cancer. J Surg Res. 2019;240:115–123. doi: 10.1016/j.jss.2019.02.038. [DOI] [PubMed] [Google Scholar]
- 46.Cui L, Deng Y, Rong Y, Lou W, Mao Z, Feng Y, Xie D, Jin D. IRF-2 is over-expressed in pancreatic cancer and promotes the growth of pancreatic cancer cells. Tumour Biol. 2012;33(1):247–255. doi: 10.1007/s13277-011-0273-3. [DOI] [PubMed] [Google Scholar]
- 47.Wang Y, Liu D, Chen P, Koeffler H, Tong X, Xie D. Involvement of IFN regulatory factor (IRF)-1 and IRF-2 in the formation and progression of human esophageal cancers. Cancer Res. 2007;67(6):2535–2543. doi: 10.1158/0008-5472.CAN-06-3530. [DOI] [PubMed] [Google Scholar]
- 48.Wang Y, Liu D, Chen P, Koeffler H, Tong X, Xie D. Negative feedback regulation of IFN-gamma pathway by IFN regulatory factor 2 in esophageal cancers. Cancer Res. 2008;68(4):1136–1143. doi: 10.1158/0008-5472.CAN-07-5021. [DOI] [PubMed] [Google Scholar]
- 49.Lei J, Zhou M, Zhang F, Wu K, Liu S, Niu H. Interferon regulatory factor transcript levels correlate with clinical outcomes in human glioma. Aging. 2021;13(8):12086–12098. doi: 10.18632/aging.202915. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.