

Abstract
Gene therapy offers great promises for a cure of hemophilia A resulting from factor VIII (FVIII) gene deficiency. We have developed and optimized a non-viral ultrasound-mediated gene delivery (UMGD) strategy. UMGD of reporter plasmids targeting mice livers achieved high levels of transgene expression predominantly in hepatocytes. Following UMGD of a plasmid encoding human FVIII driven by a hepatocyte-specific promoter/enhancer (pHP-hF8/N6) into the livers of hemophilia A mice, a partial phenotypic correction was achieved in treated mice. In order to achieve persistent and therapeutic FVIII gene expression, we adopted a plasmid (pHP-hF8-X10) encoding an FVIII variant with significantly increased FVIII secretion. By employing an optimized pulse-train ultrasound condition and immunomodulation, the treated hemophilia A mice achieved 25%–150% of FVIII gene expression on days 1–7 with very mild transient liver damage, as indicated by a small increase of transaminase levels that returned to normal within 3 days. Therapeutic levels of FVIII can be maintained persistently without the generation of inhibitors in mice. These results indicate that UMGD can significantly enhance the efficiency of plasmid DNA transfer into the liver. They also demonstrate the potential of this novel technology to safely and effectively treat hemophilia A.
Keywords: ultrasound, microbubble, hemophilia, gene delivery, factor VIII, ultrasound mediated gene delivery, UMGD, nonviral gene transfer, factor VIII variants, persistent gene expression
Graphical abstract
Ultrasound-mediated gene delivery (UMGD) was demonstrated to efficiently transfer factor VIII plasmids driven by a hepatocyte-specific promoter/enhancer into the livers of hemophilia A mice. Persistent and therapeutic levels of factor VIII expression can be achieved, demonstrating the potential for UMGD to safely and effectively treat hemophilia A.
Introduction
Hemophilia A (HA) is an X-linked recessive bleeding disorder caused by a deficiency of blood clotting factor VIII (FVIII; gene F8). According to a classification based on FVIII levels, individuals with less than 1% of normal activity are considered to have severe HA, with 1%–5% classified as moderate, and with 5%–40% as mild. Conventional treatment involves replacement therapy of FVIII by repeated intravenous infusions of plasma-derived or recombinant FVIII concentrates, which are costly and disruptive to normal activities and family life.1, 2, 3 Gene therapy as an alternative approach for hemophilia treatment that allows a wide therapeutic window has drawn significant interest, as even a continuous modest increase of FVIII levels (>1%) can result in a dramatic improvement of the severe bleeding phenotype. It also benefits from some other features of HA disease, including a wide variety of target tissues for FVIII biosynthesis,4, 5, 6, 7 as demonstrated in excellent hemophilia animal models such as mouse,8,9 rat,10 sheep,11 and dog.12 A variety of viral-13, 14, 15 and non-viral-16, 17, 18 gene-transfer-based HA therapies have been reported. In particular, recent HA gene therapy clinical trials19,20 using recombinant adeno-associated viral (rAAV) vectors have shown very promising results. However, significant obstacles remain, preventing treatment for a significant portion of patients, especially those who have high-titer anti-AAV antibodies.21,22 Repeat treatment is also prohibited due to pre-existing anti-AAV antibodies. Clinical applications from viral gene transfer systems were hampered by vector immunogenicity,23,24 the limit of transduced target cells,25 insertional mutagenesis,26 limited gene-carrying capacity, or low transgene expression.14,27
Ultrasound (US)-mediated gene delivery (UMGD) in the presence of microbubbles (MBs) has been considered a promising non-viral gene delivery method. In vitro studies showed that US-induced acoustic cavitation of MBs can transiently increase the cell membrane permeability and enhance cellular uptake of the plasmid.28, 29, 30 UMGD has been applied in vivo to deliver pharmocologics or genetic material into different target organs such as liver,31, 32, 33 brain,34,35 pancreas,36 kidney,37 cornea,38 and others39 for therapeutic treatment. Previously, we showed that therapeutic UMGD enhanced FIX gene transfer by the intrahepatic delivery of an albumin-based US contrast agent, Optison, combined with a FIX plasmid into the mouse liver, which was simultaneously treated with an US.40 These results preliminarily demonstrated the feasibility of applying UMGD in plasmid DNA gene therapy. Subsequently, we developed and optimized the US system, MB composition, and treatment protocol to deliver the luciferase reporter gene plasmid and further improve gene transfer efficiency.41, 42, 43 The following definitive in vivo explorations were performed: (1) injection routes; (2) comparison of focused US, far-field unfocused US, and near-field unfocused US systems; (3) US parameter modifications including peak negative pressure, pulse duration, duty factor, and pulse repetition frequency; (4) pulse-train US exposure regimens; (5) MB composition and concentrations; and (6) liver damage induced by UMGD treatment. Portal vein injection was superior to intrahepatic injection with better tissue distribution, resulting in more efficient gene transfer. US parameters42,43 and MBs44 were optimized, guided by gene expression and liver damage induced by acoustic cavitation as correlates to produce the maximal desired gene transgene expression with minimal liver tissue injury. Moreover, the application of a pulse-train US offered comparable or higher gene transfer efficiency with less liver damage.31,42 Based on these studies, we have scaled up the UMGD system from mouse models to rat models and have continued forth toward large animal models.31,45, 46, 47
All previous achievements provided a platform for pursuing an efficient and safe treatment with UMGD for HA gene therapy. As a first step, we explored UMGD of a therapeutic FVIII plasmid in HA mouse models. The successful results paved the way to deliver FVIII plasmids into large animals and humans.
Results
Distribution of reporter gene expression in mouse liver
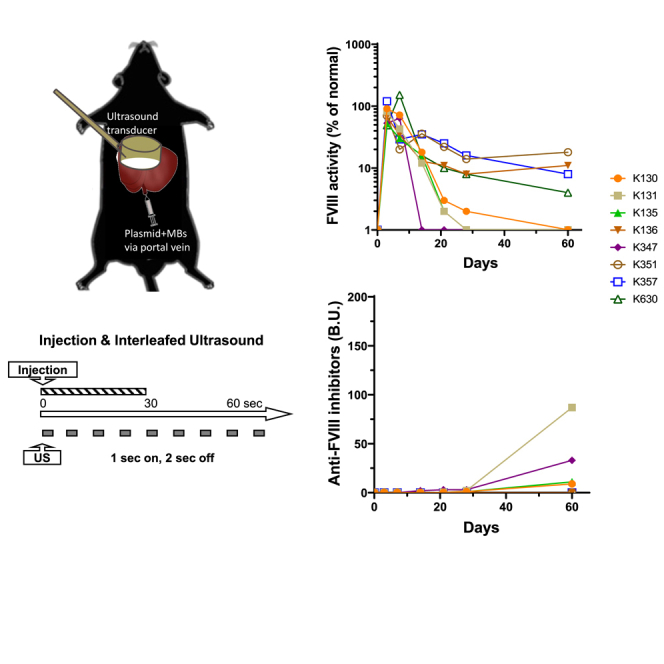
In order to achieve the best therapeutic effect from the delivery of a FVIII plasmid, we first explored the distribution of gene delivery and expression following UMGD of a luciferase reporter construct (pGL4.13 driven by a ubiquitous SV40 promoter) in C57BL/6 mice. A mixture of plasmid and MBs was delivered through a portal vein injection into the mouse liver, and the whole liver was simultaneously treated with an H158 transducer (Figure 1A) using a pulse-train US (1 s on and 2 s off) at 2.0 MPa peak negative pressure (PNP) and 0.018 ms pulse duration (PD) for 60 s under our standard US conditions41,42 (Figures 1B and 1C).
Figure 1.

Setup of the UMGD experiments in mice
(A) Specification and wave form of the US transducer 158 used for the UMGD experiments in mice. (B) Schematics of the portal vein injection of plasmid/MB mixture and simultaneous scanning of the US transducer on the liver surface during UMGD treatment. (C) Pulse-train treatment schedule used in the experiment.
On day 1 after gene transfer, we isolated hepatocytes and non-parenchymal cells from treated mouse livers by liver perfusion and centrifuge methods and evaluated the luciferase expression from the two cell populations. Transgene expression was found in both cell types; however, the total luciferase expression levels on a per cell basis in hepatocytes (5.35 × 104 RLU/107 cells) were much higher than in non-parenchymal cells (1.46 × 103 RLU/107 cells; Figure 2A). Furthermore, we delivered a pGFP plasmid driven by a ubiquitous cytomegalovirus (CMV) promoter into the mouse liver by UMGD. Following immunofluorescent staining using anti-GFP, a control liver injected with plasmid only did not show any fluorescence signal, while a UMGD-treated liver showed significant GFP expression (30%–50% GFP+ cells). GFP+ signals were distributed homogeneously and were mostly present in hepatocytes (Figure 2B). These data are consistent with the results obtained from the luciferase reporter construct, indicating that transgenes were predominantly expressed in hepatocytes following UMGD under this standard condition.
Figure 2.

Distribution of gene expression in liver tissue following US/MB-mediated gene delivery
Reporter gene plasmid pGL4 or pGFP mixed with 5% NuvOx MBs were respectively delivered into C57BL/6 mouse liver through portal veil for 30 s with simultaneous 60 s US exposures (1 s on, 2 s off, 1.1 MHz frequency, 20 cycle pulses, 0.018 ms PD, 2.0 MPa PNP, 13.9 Hz PRF). An unfocused H158 transducer was used in these experiments. (A) Luciferase expression levels on a 107 cells basis in hepatocytes and non-parenchymal cells. On day 1 post-treatment, pGL4 plasmid-transferred mouse livers were perfused and isolated into two cell populations, hepatocytes and non-parenchymal cells, and luciferase expressions were evaluated in these two types of cells. The data are presented as the mean ± SD (∗∗p < 0.01). (B) Representative GFP fluorescence staining (green) and nuclei (blue) of UMGD-treated liver and plasmid-only-injected (Control) liver. Mouse liver transferred with pGFP was sectioned and stained to show GFP protein expression.
UMGD of a hepatocyte-specific FVIII plasmid into HA mouse livers
To test the application of UMGD to treat HA, we delivered FVIII plasmids targeting the liver. Since hepatocytes were found to be the primary cell population of gene transfer, we constructed a hepatocyte-specific human FVIII (hFVIII) variant plasmid (pHP-hF8/N6; Figure 3A) under the control of the hepatic locus control region (HCR) and a hepatocyte-specific human α1-anti-trypsin promoter.48 The hF8/N6 gene encodes an FVIII variant that contains a partial B domain deletion, leaving an N-terminal 226 amino acid stretch with 6 intact putative-asparagine-linked glycosylation sites for enhanced secretion efficiency.49 Groups of HA mice were pre-treated with 3 U of hFVIII protein to maintain hemostasis prior to surgery. It should be noted that the HA mice used in the experiments are prone to develop anti-FVIII inhibitory antibodies within 10–14 days. UMGD of pHP-hF8/N6 was carried out in HA mice using an H158 transducer under the same US conditions previously described in the reporter plasmid transfer experiments except using a lower PNP (2.0 MPa) to reduce potential bleeding and liver damage in HA mice. FVIII activities in treated mouse plasma were measured by activated partial thromboplastin time (APTT) assay starting at day 3 post-treatment to avoid measuring residual FVIII activity from FVIII protein pre-treatment. We obtained 5%–20% initial FVIII activity in treated mice (Figure 3B). However, FVIII activities dropped to undetectable levels within 2 weeks following treatment, which coincided with the appearance of anti-FVIII inhibitory antibodies (Figure 3C). Thus, we further pre-treated separate groups of mice with interleukin 2 (IL2)/IL2 monoclonal antibody (mAb) complexes to modulate high-titer antibody responses,50 followed by UMGD treatment. Long-term follow-up showed that 4 out of the 8 treated mice maintained therapeutic levels of FVIII expression without generating high-titer anti-FVIII antibodies, whereas the other 4 mice generated relatively lower titers of anti-FVIII antibodies than did the group without the IL2/IL2 mAb complex treatment, and their FVIII levels dropped to undetectable levels at 2–8 weeks (Figures 3D and 3E).
Figure 3.

UMGD of pHP-hF8/N6 plasmid in HA mice with and without immunomodulation
One hundred μg pHP-hF8/N6 plasmid mixed with 5 vol % NuvOx MB was injected for 30 s with simultaneous pulse-train US exposure (1 s on, 2 s off, 1.1 MHz frequency, 20 cycle pulses, 0.018 ms PD, 2.0 MPa PNP, 13.9 Hz PRF) for 60 s. (A) Structure of the pHP-hF8/N6 plasmid. (B) Long-term FVIII activities in mouse plasma assessed by a modified activated partial thromboplastin time (APTT) assay. (C) Inhibitory antibodies against FVIII were assessed by Bethesda assay. Separate groups of mice were treated the same way except all mice were pretreated with IL-2/IL-2 mAb complexes to modulate immune responses. The treatment schedule is listed below the figure. (D and E) FVIII activities (D) and Inhibitory antibodies (E) against FVIII were examined over time. Each symbol represents data obtained from an individual mouse. Experiments were repeated at least twice.
Correction of the bleeding disorder in HA mice
With relatively low levels of FVIII expression in treated mice, we further evaluated the phenotype correction of HA using a tail-clip bleeding assay in UMGD+IL2/IL2 complex-treated mice. Blood loss during a 10 min period following mouse tail transection was evaluated using hemoglobin levels. Total blood loss was significantly reduced in treated mice compared with in untreated HA mice, although it was still higher than in normal mice (Figure 4A). On average, a 57% therapeutic correction in treated mice was achieved compared with in normal mice as a positive control (100%) and in untreated HA as a negative control (0%), consistent with the results that about half of the treated mice maintained therapeutic levels of FVIII expression following UMGD treatment.
Figure 4.
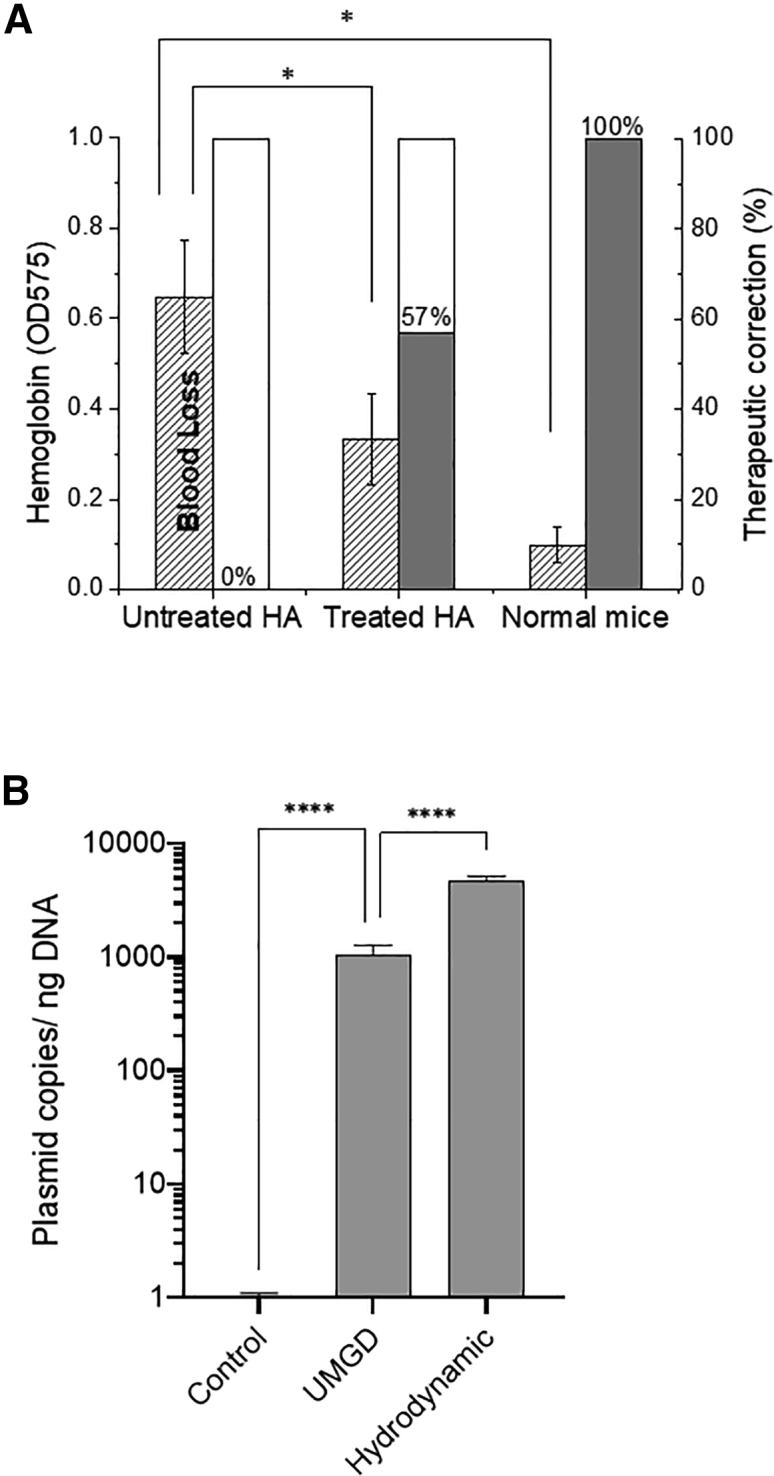
Evaluation of phenotypic correction and vector copy numbers in the liver in UMGD-treated mice
(A) A tail-clip bleeding assay was performed on treated and untreated HA mice as well as on normal mice. Blood loss during a 10 min period following the mouse tail transection was evaluated using hemoglobin levels. Bleeding correction was calculated based on normal mice as the positive control (100%) and untreated HA/Balb/C mice as the negative control (0%). (B) hFVIII plasmid copy numbers in the liver tissue were examined by SYBR-based quantitative real-time PCR in duplicates. Genomic DNA was extracted from the liver tissues. The quantification of hFVIII vector copy numbers in genomic DNA was determined by relating the CT value to the standard curve. The untreated HA mice were used as negative controls, and mice treated with hydrodynamic injections were used as positive controls (n = 3/group). All data are presented as the mean ± S.D. (∗p < 0.05, ∗∗∗∗p < 0.0001).
Evaluation of plasmid copy numbers in mouse liver
Quantification of FVIII-encoding vector copy numbers in the liver tissue was carried out by quantitative real-time PCR using genomic DNA isolated from UMGD-treated mice with untreated HA mice as a negative control and the mice treated with a hydrodynamic injection of FVIII plasmids as a positive control. UMGD-treated mice showed significantly higher copy numbers of vector DNA on day 1 compared with those found in untreated mice (Figure 4B). Compared with mice treated with a hydrodynamic injection, the vector copies were 5- to 8-fold lower. We also examined episomal DNA by Hirt extraction of the liver tissue, and similar results were obtained (data not shown). Hydrodynamic delivery can efficiently disrupt both the endothelial membrane to allow plasmids to enter the extravascular space and the hepatic membrane for plasmid uptake by hepatocyte. In cell culture experiments, UMGD with MBs can achieve an efficient delivery of plasmid DNA into cells.43 However, for in vivo delivery, it is more difficult for both plasmid and μm-sized MBs to penetrate the endothelial membrane. This barrier may be overcome by using nanobubbles as cavitation nuclei combined with UMGD. Nanobubbles with smaller sizes can more easily extravasate from the vasculature to efficiently augment the transfection of hepatocytes (data not shown).
Examination of liver damage in UMGD-treated mice
Transaminase levels (alanine aminotransferase [ALT] and aspartate aminotransferase [AST]) were examined to assess liver functional damages associated with UMGD (Figure 5A). Compared with untreated control mice, treated mice had significantly increased ALT levels (∼1,035 IU/L) and AST levels (∼524 IU/L) on day 1 after treatment. However, both ALT and AST quickly returned to normal levels on day 4 (ALT ∼37 IU/L; AST ∼87 IU/L) and afterward. The liver enzyme levels indicated that the procedure of plasmid/MB portal vein injections and pulse-train acoustic exposure produced transient liver functional damages; however, the damages were repaired rapidly.
Figure 5.

Examination of potential liver damage by UMGD treatment
Blood was collected from the UMGD-treated mouse groups shown in Figure 2. (A) Alanine transaminase (ALT) and aspartate transaminase (AST) levels in UMGD-treated mouse plasma were examined over time. The gray boxes show the normal levels of ALT (18–65 U/L) and AST (20–110 U/L). (B) Representative histological features of liver tissues isolated from UMGD-treated mice. The serial liver sections were harvested on days 1, 7, and 28 after UMGD treatment for H&E staining. Original magnification ×100. Scale bar: 50 μm.
Liver damages were further determined by hematoxylin and eosin (H&E) staining of tissue sections, which were collected from UMGD-treated HA mice on days 1, 7, and 28 after treatment with an untreated mouse liver used as a control (Figure 5B). The representative sections in the figure were intentionally selected to present the types of hepatic damages and the areas with a maximal extent of injury. On day 1 after treatment, there was some focal coagulative necrosis and hemorrhage mostly close to the treated liver surface. The extent of necrosis was variable in sections from different treated liver lobes, ranging from 10%–30% of the total area of the section examined. There was a sharp demarcation between the areas of necrosis and viable tissue, with scattered apoptotic hepatocytes near the boundary. Some extracellular micro-vesicles and dilatation of sinusoid were observed, probably due to plasmid/MB fluid injected through the portal vein. On day 7, liver damages were remarkably diminished and repaired, with only a very small amount of scattered hemorrhage. In a few small areas of the treated surface, granulation and fibrous tissue were present, which were probably caused by the manipulation of US scanning and the application of an US coupling gel. Damages were completely repaired and recovered to normal on day 28 after treatment.
UMGD of high-expressing hFVIII variant constructs in HA mice
From our experience with the non-viral gene transfer of FVIII plasmids in tolerized HA mice, FVIII expression levels usually drop several folds initially then stabilize afterward over time. We seek to increase the levels of FVIII gene expression so that therapeutic levels can be sustained long term. In order to achieve persistent FVIII gene expression for therapeutic treatment, several improvements were pursued.
An FVIII plasmid incorporating a modified FVIII cDNA producing a FVIII protein with higher expression levels, enhanced bioactivity, and a longer half-life would be highly desirable for the successful gene therapy of HA.51 Compared with hFVIII, porcine FVIII52 expresses at higher levels. Recently, a novel hF8 variant cDNA encoding 10 amino acid porcine-FVIII-like substitutions in the A1 domain of the FVIII heavy chain, hF8-X10, significantly improved F8 gene expression levels.53,54 First, we incorporated this hF8 variant cDNA into our liver-specific plasmid vector to generate pHP-hF8-X10 (Figure 6A). When we delivered this plasmid into the HA mice by hydrodynamic delivery, a more than 10-fold higher FVIII gene expression was obtained compared with in the control construct pHP-hF8/N6 (Figure S1). Next, we demonstrated that the therapeutic UMGD pressure threshold can be lowered by increasing PDs. By employing pulse-train US exposure with an acoustic pressure of 1.0 MPa and a PD of 0.4 ms, comparable or higher transgene expression with reduced transient liver damage has been achieved compared with those generated by US exposure at higher pressures (2.0 and 2.7 MPa and a PD of 0.018 ms).43 Furthermore, in order to overcome anti-FVIII immune responses, the HA mice were similarly pretreated with immunomodulation using IL2/IL2 mAb complexes to induce the expansion of regulatory T cells to suppress T helper cell function.
Figure 6.

UMGD of high-expressing hFVIII variant plasmids in HA mice
All mice were pretreated with IL-2/IL-2 mAb complexes to modulate immune responses. A 100 μg hFVIII plasmid mixed with 5% RN18 MBs was injected for 30 s with simultaneous pulse-train US exposure (1 s on, 2 s off, 1.1 MHz frequency, 20 cycle pulses, 400 μs PD, 1.1 MPa PNP, 13.9 Hz PRF) for 60 s. (A) Structure of two hFVIII variant plasmids. (B and C) Comparison of FVIII expression levels produced from pHP-hF8/N6 plasmid and pHP-hF8-X10 plasmid assessed by APTT assay on days (B) 3 and (C) 7. All data are presented as the mean ± S.D. (∗p < 0.05). Long-term follow-up of (D) FVIII activities and (E) inhibitory antibodies against FVIII assessed by Bethesda assay following UMGD of pHP-hF8-X10 plasmid. Each symbol represents data obtained from an individual mouse. (F) Transaminase (ALT and AST) levels in mouse plasma over time following UMGD of pHP-hF8-X10 plasmid. The gray boxes show the normal levels of ALT (18–65 U/L) and AST (20–110 U/L).
We then compared FVIII expression levels following UMGD (at 1.1 MPa PNP and 0.4 ms PD) of pHP-hF8/N6 and pHP-hF8-X10 in HA mice (n = 12/group), respectively. In these experiments, we used 5% RN18 MBs to increase UMGD efficiency.44 We obtained 2%–18% versus 30%–77% of FVIII gene expression on day 3 (Figure 6B) and 3%–16% versus 30%–150% on day 7 (Figure 6C) in these two groups of mice. These results indicate that the hF8-X10 variant plasmid can produce high-level FVIII gene expression following UMGD. In experiments for longer-term follow-up, experimental mice were pre-treated with IL2/IL2 mAb complexes to prevent the formation of high-titer inhibitory antibody responses.50 All mice produced initial high levels of FVIII gene expression up to 150%. Long-term follow-up showed that 4 out of the 8 treated mice maintained 8%–20% levels of FVIII expression without generating high-titer anti-FVIII antibodies at day 60, whereas the other 4 mice generated anti-FVIII antibodies and their FVIII levels dropped to undetectable levels at 2–8 weeks (Figures 3C and 3D). Furthermore, a small increase of ALT and AST transaminase levels observed on day 1 and the rapid recovery of these to normal levels on day 4 and afterward indicated that only very minor transient liver damage occurred on day 1 and was repaired quickly and recovered to normal by day 4. Taken together, these data indicated that long-term and therapeutic FVIII gene expression can be achieved following UMGD in HA mice.
Discussion
UMGD has been applied as a non-viral physical gene delivery strategy. We have previously developed and optimized an unfocused US system with MBs to significantly enhance the gene transfer efficiency of reporter plasmids in murine models.31,41,42 In this study, we attempted to set up an efficient non-viral gene delivery method for HA gene therapy by applying UMGD to deliver FVIII-encoding plasmid DNA into the mouse liver.
First, the distribution of reporter gene expression from plasmids delivered by UMGD was investigated. The luciferase gene expression was examined in parenchymal (hepatocytes) and non-parenchymal cells including sinusoidal endothelial, Kupffer, stellate, and other cells. Our results demonstrated that plasmids were delivered and expressed in both parenchymal and non-parenchymal cells. However, the transgene was predominantly expressed in hepatocytes by UMGD at current conditions. We further explored the distribution of transgene expression using a GFP reporter plasmid. The fluorescent staining of treated liver sections showed that 30%–50% of liver cells had GFP expression. Compared with UMGD with the intrahepatic administration of plasmid/MBs mixtures in our previous study,40,41 there was a greater percentage of cells expressing GFP (30%–50% versus 5%–10%). This might be due to the following improvements in the treatment procedure: (1) the delivery route of portal vein injection allowed the mixture of the plasmid and MBs to spread more evenly in the liver tissue through capillary vessels; (2) the treatment regimen of the pulse-train US provided longer quiescent periods between US pulses to allow for the reperfusion of the plasmid and MBs, achieving more sustained distribution in the live tissue; and (3) the unfocused US transducer with a larger footprint could treat a larger tissue volume with a more homogeneous average intensity of US exposure. In addition, GFP+ signals are predominantly present in hepatocytes and homogeneously distributed in spatially localized areas. This is different from hydrodynamic-based gene transfection,48,55,56 which showed a higher intensity but in more concentrated and scattered areas. However, the intensity of GFP expression exhibited regional variability in the whole liver section. This suggests that in the presence of MBs, US-induced inertial cavitation was probably to some extent restricted by liver tissue size, physiological structure, and capillary vessel distribution. These factors would influence the flow distribution of MBs in liver tissue, propagation, scattering and attenuation of the US in tissue, and vessel wall rupture and permeability, leading to regional difference in expression intensity. In any case, the predominance of GFP gene expression in hepatic cells is consistent with what was observed in the luciferase gene transfer study.
Based on this finding of gene expression distribution, we employed a hFVIII plasmid driven by a hepatocyte-specific promoter for therapeutic gene transfer in HA mice. In our previous study,42 gene expression increased as acoustic PNP increased at the range of 0–3.2 MPa and presented a gradual plateau trend at PNP >2 MPa with increased transient liver damage. Although gene transfer efficiency will be enhanced by higher acoustic pressure, it is also critical to minimize any potential liver damage. Therefore, we selected 2 MPa PNP for treatment of HA mice, instead of a higher pressure, to minimize tissue damage. In addition, we have shown previously that the pulse-train US procedure using 1/3 of the US energy could produce comparable or even higher gene transfer efficiency compared with using continuous US pulses.31 In addition, the pulse-train US procedure (1 s on, 2 s off) was employed to allow for a more efficient perfusion of plasmid/MB mixtures in the liver and to further decrease the acoustic-cavitation-induced liver damages. Under these conditions, treated HA mice showed a significant enhancement of hFVIII activity on day 3 compared with that in untreated mice. For long-term follow-up, separate groups of experimental mice were also treated with immunomodulation therapy with IL2/IL2mAb complexes according to the successful protocol in our previous study50 to prevent the FVIII-specific immune responses following gene transfer. Following the initial burst, FVIII activity dropped to undetectable levels with inhibitor formation in treated mice without immunomodulation, whereas a portion of the treated mice with immunomodulation maintained 2%–10% of FVIII levels without inhibitor formation for at least 84 days, and the HA phenotype was corrected as examined by tail-clip assay. These results suggest that effective immunomodulation strategies are required for the inhibitor-prone HA mice to achieve phenotypic correction. However, this strategy can be successfully applied to treat 70%–80% of the HA patients who are not prone to inhibitor formation.
For achieving persistent, therapeutic levels of FVIII expression, several improvements were pursued. We first incorporated a novel FVIII variant cDNA (F8-X10) into the hepatocyte-specific plasmid to increase FVIII gene expression levels. One concern is if the FVIII variant protein will be more immunogenic than BDD-FVIII. We did not find that this is the case, at least in our study. Alternatively, for clinical translations, a codon-optimized BDD-FVIII used in AAV gene therapy trials19,20 can be incorporated in the liver-specific plasmid to produce high-level FVIII gene expression. Second, we demonstrated that the therapeutic UMGD pressure threshold can be lowered by increasing PDs. By employing pulse-train US exposure with an acoustic pressure of 1.0 MPa and a PD of 0.4 ms, comparable or higher transgene expression is achieved compared with those generated by US exposure at higher pressures (>2.0 MPa combined with a PD of 0.018 ms). Most significantly, we observed less transient liver damage evaluated by transaminase levels and histological examination, and the damage was repaired quickly within several days. Third, in order to overcome anti-FVIII immune responses, the HA mice were similarly treated with immunomodulation using IL2/IL2mAb complexes to induce the expansion of regulatory T cells to suppress T helper cell function. With these improvements, a portion of the mice produced persistent, therapeutic levels of FVIII expression and a very mild transient increase in transaminase levels. These exciting results demonstrate that UMGD can achieve safe and effective treatment of HA.
For gene therapy of HA mice, liver damage has a more important impact on gene transfer efficiency than in normal mice, which could lead to serious bleeding, hepatic apoptosis, or even possible mortality of HA mice. As mentioned above, pulse-train US exposure at 2 MPa or later at 1 MPa was employed for treatment to minimize liver damage induced by acoustic cavitation. Transaminase levels presented a rapid increase on day 1 but decreased very soon back to normal levels within 4 days after treatment, indicating a similar pattern of acute liver functional damage compared with that in a previous normal mouse study.31,40 Consistently, under histological examination, hepatic damage types and patterns of tissue repair were similar to what was found in normal mouse and rat studies: the appearance of coagulative necrosis with hemorrhage and inflammation in the process of tissue repair. However, it was noted that treated HA mouse livers sustained a small amount of hemorrhage until day 7 after treatment. In comparison, hemorrhaging at day 7 was rare in treated normal mice or rats. We believe that the initial mild bleeding from the mechanical tissue damage caused by injection/US was extended and prolonged in HA mice during tissue repair due to their FVIII deficiency. This may be further managed by treating the mice with recombinant FVIII post-surgery. Despite this marginal histological difference between HA mice and normal mice or rats, treated livers of HA mice similarly revealed rapid and significant recovery and repaired completely within 7–28 days post-treatment. In all sections, there was no thermal denaturation observed, indicating there was no thermal effect at current US conditions as previously shown.31,40 Comparing transaminase levels with the histology, despite showing slightly more hemorrhage, HA mice presented a pattern of liver damage similar to normal mice, which was considered to be induced by an acoustic-cavitation-mediated mechanism. Most importantly, the transient liver damage was further reduced when we used the US at a lower acoustic pressure (1 Mpa) and a longer PD (0.4 ms). It may be of concern that there is a potential for hemorrhaging in severe HA patients during the UMGD procedure. However, the bleeding risks can be effectively managed by treatment with recombinant FVIII pre- and post-procedure, as seen in our mouse and preliminary HA dog experiment (data not shown).
In conclusion, based on our reporter gene transfer studies, we explored the feasibility of applying UMGD in the setting of therapeutic plasmid gene transfer. Mouse livers were surgically exposed and injected with plasmid/MBs through the portal vein and treated with US. This treatment method was a direct treatment procedure restricted for use in our mouse models. A minimally invasive procedure has been developed instead for proof of clinical application and transference in large animal models. Plasmid/MBs were infused into the liver via percutaneous access to the internal jugular vein using an interventional radiology approach, and the liver was subsequently treated with US transcutaneously by UMGD.47 The transcutaneous treatment without open surgery further reduced the procedure burden and transient tissue damage, which will also be carefully evaluated in HA large animal models in future studies. The current report constitutes a proof-of-principle study and demonstrates the successful transfer of a FVIII plasmid into an HA mouse model by UMGD to achieve significant and persistent FVIII expression levels. This study provides a promising strategy for the efficient and safe treatment of HA, which is the first step toward the clinical application of UMGD and sustainable treatment of HA.
Materials and methods
Animals
All mice were maintained at a specific-pathogen-free (SPF) vivarium in accordance with the guidelines for animal care of both the National Institutes of Health and Seattle Children's Research Institute. Animal protocols were approved by the Institutional Animal Care and Use Committee of Seattle Children's Research Institute. Eight-week-old C57BL/6 normal mice were purchased from Charles River Laboratories International (Wilmington, MA, USA) and housed under SPF conditions for at least 3 days before the experiments. Eight- to sixteen-week-old FVIII exon 16 knockout HA mice were used for the studies.
Plasmids and MBs
Large preparation of plasmids was carried out by GenScript (Piscataway, NJ, USA) including a luciferase reporter plasmid (pGL4.13 [luc2/SV40]; Promega), a reporter pGFP plasmid driven by a CMV promoter (pGFP), and hepatocyte-specific FVIII plasmids. A hepatocyte-specific hFVIII plasmid (pHP-F8/N6) carrying an hF8/N6 cDNA driven by the hepatic control region (HCR) and a liver-specific α1-antrypsin (HP) promoter55 was constructed. The hFVIII/N6 molecule contains a partial B domain deletion, leaving an N-terminal 226 amino acid stretch containing 6 intact putative-asparagine-linked glycosylation sites to increase secretion of FVIII.49 We further replaced hF8/N6 cDNA with a novel hF8 variant cDNA (hF8-X10) encoding 10 amino acid porcine FVIII-like substitutions (I86V, Y105F, A108S, D115E, Q117H, F129L, G132K, H134Q, M147T, L152P in the FVIII-X10) in the A1 domain of the FVIII heavy chain in the hepatocyte-specific plasmid (pHP-hF8-X10) to obtain significantly increased F8 gene expression levels.53,54 NuvOx MBs (NuvOx Pharma, Tucson, AZ, USA) or RN18 MBs are lipid-shelled US contrast agents composed of encapsulated octafluoropropane gas with a concentration of ∼1 × 1010 MBs/mL and a mean diameter of ∼2 μm or ∼1 μm, respectively. MBs were reconstituted by shaking the vial for 45 s with a Vialmix agitator (Lantheus Medical Imaging, North Billerica, MA, USA) immediately before use.
US system
The US system, as described previously,31,42 was composed of a separate pulse generator and a high-power radio frequency pulse amplifier, which is capable of generating up to 1,000 W of electrical power into a 50 Ω load (model RFG-1000; JJ&A Instruments, Duvall, WA, USA). A customized single-element, 1.1 MHz, 16 mm diameter transducer (model H158; Sonic Concepts, Bothell, WA, USA), driven by the US system, was used to directly apply acoustic pressure to the surgically exposed liver surface via enhanced coupling with sterile US transmission gel.
UMGD of reporter plasmids into mice
Mice were anesthetized by continuous inhalation of isoflurane during treatment. The liver and portal vein were exposed by midline incision. For each mouse, 50 μg reporter plasmid DNA per mouse mixed with 5 vol % NuvOx MBs in 400 μL PBS were co-injected into the liver through the portal vein using a 24G catheter for 30 s. The liver was simultaneously treated by pulse-train US (1 s on, 2 s off, 1.1 MHz frequency, 20 cycle pulses, 2.7 MPa, 0.018 ms PD, 13.9 Hz pulse repetition frequency [PRF]) for a total exposure duration of 60 s. With immediate hemostasis and suturing, the mice recovered from anesthesia within 30 min following treatment. The mice were euthanized for liver harvest on day 1 after gene transfer for reporter gene analysis or maintained for blood collection through experimental duration for therapeutic gene analysis.
Immunofluorescent staining of GFP
Five μm frozen cryosection of mouse liver collected on day 1 was fixed, permeabilized, and blocked with 10% serum in PBS for 30 min. The section was then stained by using anti-GFP-Alexa Fluor conjugates (Life Technologies, Grand Island, NY, USA) at 1:200 dilution in PBS. A liver section from a plasmid-injected mouse without US treatment was also stained as a negative control. All sections were counterstained with UltraCruz Mounting Medium containing 4′,6-diamidino-2-phenylindole (DAPI) (Santa Cruz Biotechnology, Dallas, TX, USA) to visualize cell nuclei. Following staining, all liver sections were examined by the Leica DM6000 B fluorescence microscope system.
Luciferase expression in liver parenchymal and non-parenchymal cells
On day 1 after US/MB-mediated gene transfer, hepatocytes and non-parenchymal cells were isolated from treated livers by liver perfusion and centrifuge methods. A luciferase assay was performed using the Luciferase Assay System (Promega) and a luminometer (Victor 3; PerkinElmer, Wellesley, MA, USA). Luciferase activity was normalized to the total cell number or protein mass of either cell population and was expressed as RLU/107 cells or RLU/mg protein.
Therapeutic FVIII gene transfer in HA mice
For FVIII gene delivery, the mice were anesthetized, and midline incisions were made as described previously. For each mouse, ∼400 μL mixture of 100 μg FVIII plasmid and 5 vol % MBs were injected into the liver through the portal vein using a 24G catheter for 30 s with simultaneous pulse-train US exposure (1 s on, 2 s off, 1.1 MHz frequency, 20 cycle pulses, 13.9 Hz PRF) at either 2.0 MPa PNP, 0.018 ms PD or 1.1 MPa PNP, 0.4 ms PD for a total exposure duration of 60 s. At the end of the procedure of portal injection and US scanning, a GELFOAM sterile compressed sponge (AmerisourceBergen, Chesterbrook, PA, USA) and a Bleed-X hemostatic powder (First Veterinary Supply, Livonia, MI, USA) were applied with direct pressure to the injection site to control bleeding. On days 3 and 7 following treatment, blood samples were collected by retro-orbital bleeding for FVIII activity evaluation.
A long-term study was further performed. For immunomodulation, the experimental HA mice was pretreated with IL-2/IL-2 mAb complexes50 via intraperitoneal injection on days −5, −4, and −3 before gene transfer of the therapeutic FVIII plasmid. IL-2/IL-2mAb complexes were prepared by incubating the mixture of 1 μg recombinant mouse IL-2 (PeproTech, Rocky Hill, NJ, USA) and 5 μg IL-2 mAb (JES6-1A12) (eBioscience, San Diego, CA, USA) at 37°C for 30 min immediately before injection. In addition, to improve the bleeding diathesis, HA mice were pre-treated with 3 U recombinant hFVIII protein before surgery. IL-2/IL-2 mAb complexes + plasma-pre-treated HA and naive HA mice were included as controls. Blood samples were collected at serial time points.
APTT and anti-FVIII antibody assays
FVIII activity was measured by a modified clotting assay using FVIII-deficient plasma and APTT reagents and calculated from a standard curve of pooled normal human plasma.57 The inhibitory antibody against FVIII was measured by Bethesda assay, as previously described.58
Tail-clip bleeding assay
The mouse tail was pre-warmed in 0.9% saline at 37°C for 2 min and subsequently cut at a 3 mm length. The cut tail was then immersed in 14 mL saline at 37°C, and blood was collected for 10 min. The bleeding was terminated by cauterizing the tail. Blood loss was quantified by the hemoglobin level of the collected blood in saline, with the absorbance measured at 560 nm.
Evaluation of FVIII plasmid copy numbers in liver cells
Liver tissues were collected from treated HA mice on day 1 after US-mediated gene transfer. Genomic DNA was extracted using the Wizard Genomic DNA Purification Kit (Promega Corporation, Madison, WI, USA). Genomic DNA was also extracted from an untreated mouse liver and a hydrodynamic-injection-treated mouse liver as the negative and positive controls, respectively. Quantitative real-time PCR of the genomic DNA was performed to measure the copy number of the delivered plasmid by using Applied Biosystems Power SYBR Green PCR Master Mix and 7500 Real-Time PCR system. The primers for the hFVIII plasmid were as follows: 5′-CCAGAGTTCCAAGCCTCCAACA-3′ (forward) and 5′-GGAAGTCAGTCTGTGCTCCAATG-3′ (reverse) (Invitrogen, Carlsbad, CA, USA). The concentration of genomic DNA and the plasmid pHP-F8/N6 were measured using Spectrophotometer (NanoDrop ND-1000), and the corresponding plasmid copy-number concentration was calculated using the following equation:59
A 10-fold serial dilution series of plasmids, ranging from 101 to 108 copies/μL, was used to construct the standard curve. The absolute copy numbers of the FVIII-encoding vector were determined by their CT values using the linear equation defined by the standard curve and normalized by the extracted genomic DNA concentration.
Transaminase assay
Blood samples were collected from treated or untreated control mice on days 1, 4, 7, 14, 21, and 28 after gene delivery for the transaminase assay. ALT and AST levels were measured using a commercial assay kit (Teco Diagnostics, Anaheim, CA, USA).
Histological evaluation
Treated liver tissues were harvested on days 1, 7, and 28 after the portal vein was injected with plasmid/MBs and US treatment. Normal mouse liver was used as control. All samples were fixed in 10% neutrally buffered formalin, embedded in paraffin, and sections were stained with H&E for evaluation of liver damage.
Statistical analysis
All statistical analyses were carried out utilizing GraphPad Prism 7 software. The data were compared using ANOVA or multiple t tests. p values <0.05 were considered statistically significant.
Acknowledgments
This work was supported by grants R01 HL128139 (C.H.M.), R01 HL151077 (C.H.M.), and U54 HL142019 (W.X. and C.H.M.) from the National Heart, Lung, and Blood Institute.
Author contributions
S.S. designed and performed experiments, collected and analyzed data, and wrote the manuscript. M.J.L. performed experiments and collected and analyzed data. M.L.N.-V., D.M.M.-T., and J.H. performed experiments. W.X. provided the hF8-X10 variant cDNA. E.C.U. provided the NuvOx MBs. C.H.M. developed concepts, directed research, performed experiments, and wrote and edited the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.01.006.
Supplemental information
References
- 1.Ljung R.C. Prophylactic infusion regimens in the management of hemophilia. Thromb. Haemost. 1999;82:525–530. [PubMed] [Google Scholar]
- 2.Lofqvist T., Nilsson I.M., Berntorp E., Pettersson H. Haemophilia prophylaxis in young patients--a long-term follow-up. J. Intern. Med. 1997;241:395–400. doi: 10.1046/j.1365-2796.1997.130135000.x. [DOI] [PubMed] [Google Scholar]
- 3.Franchini M., Favaloro E.J., Lippi G. Mild hemophilia A. J. Thromb. Haemost. 2010;8:421–432. doi: 10.1111/j.1538-7836.2009.03717.x. [DOI] [PubMed] [Google Scholar]
- 4.Elder B., Lakich D., Gitschier J. Sequence of the murine factor VIII cDNA. Genomics. 1993;16:374–379. doi: 10.1006/geno.1993.1200. [DOI] [PubMed] [Google Scholar]
- 5.Hollestelle M.J., Thinnes T., Crain K., Stiko A., Kruijt J.K., van Berkel T.J., Loskutoff D.J., van Mourik J.A. Tissue distribution of factor VIII gene expression in vivo--a closer look. Thromb. Haemost. 2001;86:855–861. [PubMed] [Google Scholar]
- 6.Wion K.L., Kelly D., Summerfield J.A., Tuddenham E.G., Lawn R.M. Distribution of factor VIII mRNA and antigen in human liver and other tissues. Nature. 1985;317:726–729. doi: 10.1038/317726a0. [DOI] [PubMed] [Google Scholar]
- 7.Rall L.B., Bell G.I., Caput D., Truett M.A., Masiarz F.R., Najarian R.C., Valenzuela P., Anderson H.D., Din N., Hansen B. Factor VIII:C synthesis in the kidney. Lancet. 1985;1:44. doi: 10.1016/s0140-6736(85)90987-0. [DOI] [PubMed] [Google Scholar]
- 8.Bi L., Lawler A.M., Antonarakis S.E., High K.A., Gearhart J.D., Kazazian H.H., Jr. Targeted disruption of the mouse factor VIII gene produces a model of haemophilia A. Nat. Genet. 1995;10:119–121. doi: 10.1038/ng0595-119. [DOI] [PubMed] [Google Scholar]
- 9.Bril W.S., van Helden P.M., Hausl C., Zuurveld M.G., Ahmad R.U., Hollestelle M.J., Reitsma P.H., Fijnvandraat K., van Lier R.A., Schwarz H.P., et al. Tolerance to factor VIII in a transgenic mouse expressing human factor VIII cDNA carrying an Arg(593) to Cys substitution. Thromb. Haemost. 2006;95:341–347. doi: 10.1160/TH05-08-0559. [DOI] [PubMed] [Google Scholar]
- 10.Booth C.J., Brooks M.B., Rockwell S. Spontaneous coagulopathy in inbred WAG/RijYcb rats. Comp. Med. 2010;60:25–30. [PMC free article] [PubMed] [Google Scholar]
- 11.Porada C.D., Sanada C., Long C.R., Wood J.A., Desai J., Frederick N., Millsap L., Bormann C., Menges S.L., Hanna C., et al. Clinical and molecular characterization of a re-established line of sheep exhibiting hemophilia A. J. Thromb. Haemost. 2010;8:276–285. doi: 10.1111/j.1538-7836.2009.03697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lozier J.N., Dutra A., Pak E., Zhou N., Zheng Z., Nichols T.C., Bellinger D.A., Read M., Morgan R.A. The Chapel Hill hemophilia A dog colony exhibits a factor VIII gene inversion. Proc. Natl. Acad. Sci. U S A. 2002;99:12991–12996. doi: 10.1073/pnas.192219599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu C., Lipshutz G.S. AAV-based neonatal gene therapy for hemophilia A: long-term correction and avoidance of immune responses in mice. Gene Ther. 2012;19:1166–1176. doi: 10.1038/gt.2011.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown H.C., Shields J.E., Zhou S.Z., Wright J.F., Spencer H.T., Doering C.B. Bioengineered coagulation factor VIII enables long-term correction of murine hemophilia A following liver-directed adeno-associated viral vector delivery. Blood. 2013;122:14036. doi: 10.1038/mtm.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sarkar R., Tetreault R., Gao G.P., Wang L.L., Bell P., Chandler R., Wilson J.M., Kazazian H.H. Total correction of hemophilia A mice with canine FVIII using an AAV 8 serotype. Blood. 2004;103:1253–1260. doi: 10.1182/blood-2003-08-2954. [DOI] [PubMed] [Google Scholar]
- 16.Roth D.A., Tawa N.E., Jr., O'Brien J.M., Treco D.A., Selden R.F., Factor V.T.T.S.G. Nonviral transfer of the gene encoding coagulation factor VIII in patients with severe hemophilia A. N. Engl. J. Med. 2001;344:1735–1742. doi: 10.1056/NEJM200106073442301. [DOI] [PubMed] [Google Scholar]
- 17.Kren B.T., Unger G.M., Sjeklocha L., Trossen A.A., Korman V., Dietheim-Okita B.M., Reding M.T., Steer C.J. Nanocapsule-delivered Sleeping Beauty mediates therapeutic Factor VIII expression in liver sinusoidal endothelial cells of hemophilia A mice. J. Clin. Invest. 2009;119:2086–2099. doi: 10.1172/JCI34332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dhadwar S.S., Kiernan J., Wen J., Hortelano G. Repeated oral administration of chitosan/DNA nanoparticles delivers functional FVIII with the absence of antibodies in hemophilia A mice. J. Thromb. Haemost. 2010;8:2743–2750. doi: 10.1111/j.1538-7836.2010.04116.x. [DOI] [PubMed] [Google Scholar]
- 19.Rangarajan S., Walsh L., Lester W., Perry D., Madan B., Laffan M., Yu H., Vettermann C., Pierce G.F., Wong W.Y., et al. AAV5-Factor VIII gene transfer in severe hemophilia A. N. Engl. J. Med. 2017;377:2519–2530. doi: 10.1056/NEJMoa1708483. [DOI] [PubMed] [Google Scholar]
- 20.George L.A. Hemophilia gene therapy comes of age. Blood Adv. 2017;1:2591–2599. doi: 10.1182/bloodadvances.2017009878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Colella P., Ronzitti G., Mingozzi F. Emerging issues in AAV-mediated in vivo gene therapy. Mol. Ther. Methods Clin. Dev. 2018;8:87–104. doi: 10.1016/j.omtm.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miesbach W., O'Mahony B., Key N.S., Makris M. How to discuss gene therapy for haemophilia? A patient and physician perspective. Haemophilia. 2019;25:545–557. doi: 10.1111/hae.13769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mingozzi F., High K.A. Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood. 2013;122:23–36. doi: 10.1182/blood-2013-01-306647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rogers G.L., Shirley J.L., Zolotukhin I., Kumar S.R.P., Sherman A., Perrin G.Q., Hoffman B.E., Srivastava A., Basner-Tschakarjan E., Wallet M.A., et al. Plasmacytoid and conventional dendritic cells cooperate in crosspriming AAV capsid-specific CD8(+) T cells. Blood. 2017;129:3184–3195. doi: 10.1182/blood-2016-11-751040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Powell J.S., Ragni M.V., White G.C., Lusher J.M., Hillman-Wiseman C., Moon T.E., Cole V., Ramanathan-Girish S., Roehl H., Sajjadi N., et al. Phase 1 trial of FVIII gene transfer for severe hemophilia A using a retroviral construct administered by peripheral intravenous infusion. Blood. 2003;102:2038–2045. doi: 10.1182/blood-2003-01-0167. [DOI] [PubMed] [Google Scholar]
- 26.Ramezani A., Hawley R.G. Correction of murine hemophilia A following nonmyeloablative transplantation of hematopoietic stem cells engineered to encode an enhanced human factor VIII variant using a safety-augmented retroviral vector. Blood. 2009;114:526–534. doi: 10.1182/blood-2009-01-199653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grieger J.C., Samulski R.J. Packaging capacity of adeno-associated virus serotypes: impact of larger genomes on infectivity and postentry steps. J. Virol. 2005;79:9933–9944. doi: 10.1128/JVI.79.15.9933-9944.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bekeredjian R., Kroll R.D., Fein E., Tinkov S., Coester C., Winter G., Katus H.A., Kulaksiz H. Ultrasound targeted microbubble destruction increases capillary permeability in hepatomas. Ultrasound Med. Biol. 2007;33:1592–1598. doi: 10.1016/j.ultrasmedbio.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 29.Zhou Y., Yang K., Cui J., Ye J.Y., Deng C.X. Controlled permeation of cell membrane by single bubble acoustic cavitation. J. Control. Release. 2012;157:103–111. doi: 10.1016/j.jconrel.2011.09.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sirsi S.R., Borden M.A. Advances in ultrasound mediated gene therapy using microbubble contrast agents. Theranostics. 2012;2:1208–1222. doi: 10.7150/thno.4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song S., Noble M., Sun S., Chen L., Brayman A.A., Miao C.H. Efficient microbubble- and ultrasound-mediated plasmid DNA delivery into a specific rat liver lobe via a targeted injection and acoustic exposure using a novel ultrasound system. Mol. Pharm. 2012;9:2187–2196. doi: 10.1021/mp300037t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Manson M., Zhang F., Novokhodko A., Chen C.-Y., Parker M., Loeb K., Kajimoto M., Storb R., Miao C.H. The XXVII Congress of the International Society on Thrombosis and Haemostasias, Melbourne, Australia, July 6-10, 2019: OC22.22. 2019. Therapeutic levels of factor VIII gene expression were achieved by transcutaneous ultrasound mediated gene delivery into canine liver. [Google Scholar]
- 33.Anderson C.D., Walton C.B., Shohet R.V. A comparison of focused and unfocused ultrasound for microbubble-mediated gene delivery. Ultrasound Med. Biol. 2021;47:1785–1800. doi: 10.1016/j.ultrasmedbio.2021.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aryal M., Arvanitis C.D., Alexander P.M., McDannold N. Ultrasound-mediated blood-brain barrier disruption for targeted drug delivery in the central nervous system. Adv. Drug Deliv. Rev. 2014;72:94–109. doi: 10.1016/j.addr.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gorick C.M., Mathew A.S., Garrison W.J., Thim E.A., Fisher D.G., Copeland C.A., Song J., Klibanov A.L., Miller G.W., Price R.J. Sonoselective transfection of cerebral vasculature without blood-brain barrier disruption. Proc. Natl. Acad. Sci. U S A. 2020;117:5644–5654. doi: 10.1073/pnas.1914595117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen J., Chen S., Huang P., Meng X.L., Clayton S., Shen J.S., Grayburn P.A. In vivo targeted delivery of ANGPTL8 gene for beta cell regeneration in rats. Diabetologia. 2015;58:1036–1044. doi: 10.1007/s00125-015-3521-z. [DOI] [PubMed] [Google Scholar]
- 37.Omata D., Munakata L., Kageyama S., Suzuki Y., Maruyama T., Shima T., Chikaarashi T., Kajita N., Masuda K., Tsuchiya N., et al. Ultrasound image-guided gene delivery using three-dimensional diagnostic ultrasound and lipid-based microbubbles. J. Drug Target. 2021:1–8. doi: 10.1080/1061186X.2021.1953510. [DOI] [PubMed] [Google Scholar]
- 38.Aptel F., Lafon C. Therapeutic applications of ultrasound in ophthalmology. Int. J. Hyperthermia. 2012;28:405–418. doi: 10.3109/02656736.2012.665566. [DOI] [PubMed] [Google Scholar]
- 39.Endo-Takahashi Y., Negishi Y. Microbubbles and nanobubbles with ultrasound for systemic gene delivery. Pharmaceutics. 2020;12:964. doi: 10.3390/pharmaceutics12100964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miao C.H., Brayman A.A., Loeb K.R., Ye P., Zhou L., Mourad P., Crum L.A. Ultrasound enhances gene delivery of human factor IX plasmid. Hum. Gene Ther. 2005;16:893–905. doi: 10.1089/hum.2005.16.893. [DOI] [PubMed] [Google Scholar]
- 41.Shen Z.P., Brayman A.A., Chen L., Miao C.H. Ultrasound with microbubbles enhances gene expression of plasmid DNA in the liver via intraportal delivery. Gene Ther. 2008;15:1147–1155. doi: 10.1038/gt.2008.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Song S., Shen Z., Chen L., Brayman A.A., Miao C.H. Explorations of high-intensity therapeutic ultrasound and microbubble-mediated gene delivery in mouse liver. Gene Ther. 2011;18:1006–1014. doi: 10.1038/gt.2011.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tran D.M., Harrang J., Song S., Chen J., Smith B.M., Miao C.H. Prolonging pulse duration in ultrasound-mediated gene delivery lowers acoustic pressure threshold for efficient gene transfer to cells and small animals. J. Control. Release. 2018;279:345–354. doi: 10.1016/j.jconrel.2018.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun R.R., Noble M.L., Sun S.S., Song S., Miao C.H. Development of therapeutic microbubbles for enhancing ultrasound-mediated gene delivery. J. Control. Release. 2014;182:111–120. doi: 10.1016/j.jconrel.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Noble M.L., Kuhr C.S., Graves S.S., Loeb K.R., Sun S.S., Keilman G.W., Morrison K.P., Paun M., Storb R.F., Miao C.H. Ultrasound-targeted microbubble destruction-mediated gene delivery into canine livers. Mol. Ther. 2013;21:1687–1694. doi: 10.1038/mt.2013.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Noble-Vranish M.L., Song S., Morrison K.P., Tran D.M., Sun R.R., Loeb K.R., Keilman G.W., Miao C.H. Ultrasound-mediated gene therapy in swine livers using single-element, multi-lensed, high-intensity ultrasound transducers. Mol. Ther. Methods Clin. Dev. 2018;10:179–188. doi: 10.1016/j.omtm.2018.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tran D.M., Zhang F., Morrison K.P., Loeb K.R., Harrang J., Kajimoto M., Chavez F., Wu L., Miao C.H. Transcutaneous ultrasound-mediated nonviral gene delivery to the liver in a porcine model. Mol. Ther. Methods Clin. Dev. 2019;14:275–284. doi: 10.1016/j.omtm.2019.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miao C.H. A novel gene expression system: non-viral gene transfer for hemophilia as model systems. Adv. Genet. 2005;54:143–177. doi: 10.1016/S0065-2660(05)54007-0. [DOI] [PubMed] [Google Scholar]
- 49.Miao H.Z., Sirachainan N., Palmer L., Kucab P., Cunningham M.A., Kaufman R.J., Pipe S.W. Bioengineering of coagulation factor VIII for improved secretion. Blood. 2004;103:3412–3419. doi: 10.1182/blood-2003-10-3591. [DOI] [PubMed] [Google Scholar]
- 50.Liu C.L., Ye P., Yen B.C., Miao C.H. In vivo expansion of regulatory T cells with IL-2/IL-2 mAb complexes prevents anti-factor VIII immune responses in hemophilia A mice treated with factor VIII plasmid-mediated gene therapy. Mol. Ther. 2011;19:1511–1520. doi: 10.1038/mt.2011.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roberts S.A., Dong B., Firrman J.A., Moore A.R., Sang N., Xiao W. Engineering factor Viii for hemophilia gene therapy. J. Genet. Syndr. Gene Ther. 2011;1:S1–S006. doi: 10.4172/2157-7412.S1-006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Doering C.B., Healey J.F., Parker E.T., Barrow R.T., Lollar P. High level expression of recombinant porcine coagulation factor VIII. J. Biol. Chem. 2002;277:38345–38349. doi: 10.1074/jbc.M206959200. [DOI] [PubMed] [Google Scholar]
- 53.Cao W., Dong B., Horling F., Firrman J.A., Lengler J., Klugmann M., de la Rosa M., Wu W., Wang Q., Wei H., et al. Minimal essential human factor VIII alterations enhance secretion and gene therapy efficiency. Mol. Ther. Methods Clin. Dev. 2020;19:486–495. doi: 10.1016/j.omtm.2020.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang X., Fu R.Y., Li C., Chen C.Y., Firrman J., Konkle B.A., Zhang J., Li L., Xiao W., Poncz M., et al. Enhancing therapeutic efficacy of in vivo platelet-targeted gene therapy in hemophilia A mice. Blood Adv. 2020;4:5722–5734. doi: 10.1182/bloodadvances.2020002479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miao C.H., Ye X., Thompson A.R. High-level factor VIII gene expression in vivo achieved by nonviral liver-specific gene therapy vectors. Hum. Gene Ther. 2003;14:1297–1305. doi: 10.1089/104303403322319381. [DOI] [PubMed] [Google Scholar]
- 56.Ye X., Loeb K.R., Stafford D.W., Thompson A.R., Miao C.H. Complete and sustained phenotypic correction of hemophilia B in mice following hepatic gene transfer of a high-expressing human factor IX plasmid. J. Thromb. Haemost. 2003;1:103–111. doi: 10.1046/j.1538-7836.2003.00024.x. [DOI] [PubMed] [Google Scholar]
- 57.Ye P., Thompson A.R., Sarkar R., Shen Z., Lillicrap D.P., Kaufman R.J., Ochs H.D., Rawlings D.J., Miao C.H. Naked DNA transfer of factor VIII induced transgene-specific, species-independent immune response in hemophilia A mice. Mol. Ther. 2004;10:117–126. doi: 10.1016/j.ymthe.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 58.Kasper C.K., Aledort L., Aronson D., Counts R., Edson J.R., van Eys J., Fratantoni J., Green D., Hampton J., Hilgartner M., et al. Proceedings: a more uniform measurement of factor VIII inhibitors. Thromb. Diath. Haemorrh. 1975;34:612. [PubMed] [Google Scholar]
- 59.Whelan J.A., Russell N.B., Whelan M.A. A method for the absolute quantification of cDNA using real-time PCR. J. Immunol. Methods. 2003;278:261–269. doi: 10.1016/s0022-1759(03)00223-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.