

Abstract
Background & Aims
Substitution of lysine for glutamic acid at residu 167 in Transmembrane 6 superfamily member 2 (TM6SF2) is associated with fatty liver disease and reduced plasma lipid levels. Tm6sf2-/- mice replicate the human phenotype but were not suitable for detailed mechanistic studies. As an alternative model, we generated Tm6sf2-/- rats to determine the subcellular location and function of TM6SF2.
Methods
Two lines of Tm6sf2-/- rats were established using gene editing. Lipids from tissues and from newly secreted very low density lipoproteins (VLDLs) were quantified using enzymatic assays and mass spectrometry. Neutral lipids were visualized in tissue sections using Oil Red O staining. The rate of dietary triglyceride (TG) absorption and hepatic VLDL-TG secretion were compared in Tm6sf2-/- mice and in their wild-type littermates. The intracellular location of TM6SF2 was determined by cell fractionation. Finally, TM6SF2 was immunoprecipitated from liver and enterocytes to identify interacting proteins.
Results
Tm6sf2-/- rats had a 6-fold higher mean hepatic TG content (56.1 ± 28.9 9 vs 9.8 ± 3.9 mg/g; P < .0001) and lower plasma cholesterol levels (99.0 ± 10.5 vs 110.6 ± 14.0 mg/dL; P = .0294) than their wild-type littermates. Rates of appearance of dietary and hepatic TG into blood were reduced significantly in Tm6sf2-/- rats (P < .001 and P < .01, respectively). Lipid content of newly secreted VLDLs isolated from perfused livers was reduced by 53% (TG) and 62% (cholesterol) (P = .005 and P = .01, respectively) in Tm6sf2-/- mice. TM6SF2 was present predominantly in the smooth endoplasmic reticulum and endoplasmic reticulum–Golgi intermediate compartments, but not in Golgi. Both apolipoprotein B-48 and acyl-CoA synthetase long chain family member 5 physically interacted with TM6SF2.
Conclusions
TM6SF2 acts in the smooth endoplasmic reticulum to promote bulk lipidation of apolipoprotein B–containing lipoproteins, thus preventing fatty liver disease.
Keywords: Fatty Liver, VLDL, Triglycerides, Liver Perfusion
Abbreviations used in this paper: ACSL5, acyl-CoA synthetase long chain family member 5; Apo, apolipoprotein; CE, cholesteryl ester; CRISPR/Cas9, clustered regularly interspaced short palindromic repeats and CRISPR-associated protein 9; ER, endoplasmic reticulum; ERGIC, endoplasmic reticulum–Golgi intermediate compartment; FLD, fatty liver disease; IP, immunoprecipitation; KO, knockout; LC-MS/MS, liquid chromatography–mass spectrometry; mAb, monoclonal antibody; PBS, phosphate-buffered saline; PC, phosphatidylcholine; PCR, polymerase chain reaction; PE, phosphatidylethanolamine; PFA, paraformaldehyde; PI, protease inhibitor cocktail; RER, rough endoplasmic reticulum; RIPA, radioimmunoprecipitation assay buffer; S2, second supernatant; SDS, sodium dodecyl sulfate; SER, smooth endoplasmic reticulum; TM6SF2, transmembrane 6 superfamily 2; VLDL, very low density lipoprotein; TG, triglyceride; UTSW, University of Texas Southwestern Medical Center; WT, wild-type
Summary.
In this article we report a new rodent model of fatty liver disease generated by inactivating Tm6sf2 in rats. TM6SF2 was localized to a pre-Golgi compartment and plays a key role in the intracellular lipidation of very low density lipoproteins.
Fatty liver disease (FLD) is the most common form of liver disease in the Western world.1 The 2 major causes of FLD are obesity (nonalcoholic FLD) and excess alcohol intake (alcohol-associated FLD). Similar genetic factors confer susceptibility to both forms of FLD, consistent with the notion that these 2 disorders share key mechanistic factors.2,3 The 2 strongest genetic risk factors for FLD are missense variants in patatin phospholipase-like domain containing protein 3 (PNPLA3)4 and in transmembrane 6 superfamily member 2 (TM6SF2).5 Both variants are associated with the full spectrum of nonalcoholic and alcohol-associated FLD: hepatic steatosis, steatohepatitis/alcoholic hepatitis, cirrhosis, and hepatocellular carcinoma.5,6
TM6SF2 is a polytopic membrane protein that is required for bulk lipidation of very low density lipoprotein (VLDL).5,7,8 The E167K substitution destabilizes TM6SF2, conferring a loss of function.5 Tm6sf2-/- mice recapitulate the human phenotype and have reduced hepatic triglyceride (TG) secretion, a major pathway for clearance of hepatic TGs.9 VLDL synthesis is initiated with translation and translocation of apolipoprotein (Apo)B into the endoplasmic reticulum (ER) lumen. Phospholipids, TGs, and cholesteryl esters (CEs) are added to the particle before it is secreted into the blood. It has been proposed that lipidation of VLDL occurs in 2 stages.10 First, phospholipids, TGs, and CE are transferred to nascent ApoB.11 Defects in this first lipidation step result in degradation of poorly lipidated ApoB.12,13 Most neutral lipids likely are added to VLDL in a second, bulk lipidation step.10 Previously, we showed that inactivation of TM6SF2 decreases secretion of VLDL-TG, but not VLDL-ApoB; therefore, the amount of TG secreted per particle is reduced.8,9 The preservation of ApoB secretion in Tm6sf2-/- mice indicates that TM6SF2 is not required for the first lipidation step in VLDL biogenesis.
The proteins required for bulk lipidation of VLDL and the sites at which this step occurs have not been defined clearly.14, 15, 16 TM6SF2 has been localized by biochemical and imaging studies to the ER, ER–Golgi intermediate compartment, and Golgi complex.7,9 To further define the sites of action of TM6SF2 in rat liver and its role in lipid metabolism, we generated Tm6sf2-/- rats to take advantage of established cell fractionation protocols.17
We show that Tm6sf2-/- rats, similar to Tm6sf2-/- mice, replicate the major features of the human phenotype and provide an alternative animal model to study the role of TM6SF2 in both dietary lipid absorption and hepatic lipid secretion.
Results
Generation of Tm6sf2-/- Rats
Twenty-one rats carrying frameshift mutations were generated in the UTSW Transgenic Core in Tm6sf2 were generated using clustered regularly interspaced short palindromic repeats and CRISPR-associated protein 9 (CRISPR-Cas9) technology. Lines were generated from 2 of these rats. In line 1, a T inserted in exon 1 introduces a stop codon in exon 2 (Figure 1A). A map of the CRISPR/Cas9-mediated disruption of Tm6sf2 in line 1 is shown in Figure 1A. Mean levels of TM6SF2 messenger RNA (mRNA) were decreased 38% and 85% in rats heterozygous and homozygous, respectively, for the single base insertion (Figure 1B). No TM6SF2 protein was detected in tissues from Tm6sf2-/- rats (Figure 1C, left). Rats heterozygous for the T insertion (line 1) had an intermediate amount of TM6SF2 protein (54% of wild-type rats [WT]). In the intestines, TM6SF2 expression was restricted to the small intestine, where lipids are absorbed (Figure 1C, right).
Figure 1.

Generation and characterization of Tm6sf2-/-rats using CRISPR/Cas9 technology. (A) Diagram of CRISPR/Cas9-targeted disruption of rat Tm6sf2 by insertion of T in exon 1 (c.80_81insT). Sequences of the guide RNA (gRNA) binding site, protospacer adjacent motif (PAM), and introns (green) are indicated. (B) TM6SF2 mRNA levels in female rat livers (n = 3–5/group, 3–4 wk) were determined using real-time PCR and normalized to levels of cyclophilin B mRNA. (C) Immunoblot of liver lysates (left) and enterocytes (right) from 4- to 5-week-old rats. (D) Body weights, liver weights, and hepatic lipids of chow-fed male rats (n = 11–19/group, 3–4 wk) after a 4-hour fast. (E) Plasma lipid levels were measured in the same rats as used in panel D (left). Plasma samples from WT and Tm6sf2-/- male rats (4/group, 5–6 wk) were pooled and size-fractionated by fast performance liquid chromatography. Cholesterol and TG levels were measured in each fraction (right). (F) Plasma (1 μL) was size-fractionated by 4%–15% gradient SDS–polyacrylamide gel electrophoresis. Levels of APOB were determined by immunoblot analysis using a rabbit polyclonal antibody (ab20737). Bars indicate means ± SD. Differences between groups were analyzed using the Student unpaired 2-tailed t test. ∗P < .05, ∗∗P < .01, and ∗∗∗P < .001, compared with the WT group. All experiments were repeated twice, and results were similar. C, colon; CANX, calnexin; D, duodenum; FN1, fibronectin; HDL, high-density lipoprotein; I, ileum; J, jejunum.
Tm6sf2-/- rats were fertile and offspring of Tm6sf2+/- matings were born at the expected Mendelian and sex ratios (Table 1). All rats used in the experiments described in this article were from line 1 (Figure 1A), and the major findings were replicated in line 2. Line 2 carries a 73–base pair deletion in exon 1 that causes premature termination of translation in exon 2 (Figure 2A). A map of the CRISPR/Cas9-targeted disruption of Tm6sf2 in line 2 is shown in Figure 2A. No TM6SF2 protein was detected in tissues from Tm6sf2-/- rats of line 2 (Figure 2B).
Table 1.
Genotypes of Offspring From Matings of Tm6sf2+/- Rats (n = 56)
| Genotype | Males n = 487 (52%) |
Females n = 453 (48%) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Observed |
Expected |
P value | Observed |
Expected |
P value | |||||
| n | % | n | % | n | % | n | % | |||
| +/+ | 131 | 26.9 | 122 | 25 | .56 | 115 | 25.4 | 113 | 25 | .94 |
| +/- | 222 | 45.6 | 243 | 50 | .20 | 215 | 47.5 | 227 | 50 | .46 |
| -/- | 134 | 27.5 | 122 | 25 | .42 | 123 | 27.2 | 113 | 25 | .50 |
Figure 2.

Generation of Tm6sf2-/-rats (line 2) using CRISPR/Cas9 technology. (A) Diagram of CRISPR/Cas9 targeted disruption of rat Tm6sf2 by introducing a deletion of 73 nucleotide into exon 1 (c.11_83del). Exonic and intronic nucleotides are shown in black and green, respectively. The guide RNA (gRNA) binding site and protospacer adjacent motif (PAM) sequence are indicated (top). Genotyping was performed as described in the legend to Figure 1. The amplified PCR fragment was size-fractionated on an agarose gel (bottom). (B) Immunoblot analysis of lysates from livers of 8-week-old female WT and KO rats. CANX, calnexin.
Chow-Fed Tm6sf2-/- Rats Accumulate Neutral Lipids in Liver and Jejunum
The mean body weight and liver weight did not differ between chow-fed male Tm6sf2-/- and WT littermates (Figure 1D). Compared with Tm6sf2+/+ rats, mean levels of hepatic TG were 6- and 4-fold higher in male and female Tm6sf2-/- rats, respectively (56.1 ± 28.9 vs 9.8 ± 3.9 mg/g, P < .0001; and 53.5 ± 12.2 vs 13.3 ± 2.4 mg/g, P = .0002). The mean hepatic cholesterol and CE levels also were significantly higher in male Tm6sf2-/- rats (3.1 ± 0.4 vs 2.3 ± 0.4 mg/g, P < .0001; and 1.1 ± 0.2 vs 0.3 ± 0.1 mg/g, P < .0001) (Figure 1D). Hepatic TG and cholesterol levels in rats heterozygous for the inactivated allele (Tm6sf2+/- rats) were not different from those of WT littermates. In contrast, Tm6sf2+/- mice had hepatic TG and cholesterol levels that were intermediate between the levels of WT and Tm6sf2-/- mice.9 Levels of hepatic phosphatidylcholine (PC) were slightly but significantly lower in Tm6sf2-/- rats compared with WT littermates (males: 13.6 ± 1.4 vs 15.1 ± 0.8 mg/g, P = .026; females: 13.9 ± 1.3 vs 17.0 ± 1.1 mg/g, P = .001, respectively) (Figure 1D). The mean body weight and liver weight also did not differ between chow-fed female Tm6sf2-/- and WT littermates (Figure 3A). The major findings, including hepatic TG content, observed in line 1 were replicated in line 2 (Figure 3B).
Figure 3.

Body weights, liver weights, and hepatic lipids of Tm6sf2-/-rats (lines 1 and 2). (A) Body weight, liver weight, and hepatic lipids were measured in chow-fed female rats (line 1) after a 4-hour fast (n = 3-5/group, 3–4 wk). (B) Body and liver weights plus hepatic lipid levels in Tm6sf2-/- rats (line 2) and their WT littermates. TGs, cholesterol, CEs, and PCs were measured in chow-fed male rats (n = 3–4/group, 12 wk) and female rats (n = 4–5/group, 3–4 wk) after a 4-hour fast. (C) Plasma aspartate aminotransferase (AST) and alanine transaminase (ALT) were measured in the rats used in the experiment shown in Figure 1. (D) Number and size of lipid droplets in hepatocytes and enterocytes. Bars indicate means ± SD. Differences between groups were analyzed using the Student unpaired 2-tailed t test. ∗P < .05; ∗∗P < .01, and ∗∗∗P < .001. All experiments were repeated once and the results were similar. LD, lipid droplets.
To determine if changes in hepatic lipid levels in knockout (KO) rats were selective for species of lipids, we used liquid chromatography–mass spectrometry (LC-MS/MS) to analyze the fatty acid composition of neutral lipids (TG and CE), and the 2 most plentiful phospholipids, PC, and phosphatidylethanolamine (PE) (Figure 4). Fatty acid species were almost uniformly increased in liver TG and CE of KO rats (Figure 4). Changes in PC and PE species were smaller and less consistent between KO and WT rats.
Figure 4.

Lipidomics of liver from WT and Tm6sf2 KO rats. Livers were collected from chow-fed 6-week-old male rats (n = 3/group) after a 16-hour fast. The fatty acid composition of TGs, CEs, PC, and PE were measured and analyzed by LC-MS as described in the Methods section. Fatty acid content was normalized to the amount of proteins, and presented as fold differences between genotypes. Bars indicate means ± SD. Differences between groups were analyzed using the Student unpaired 2-tailed t test. ∗P < .05, ∗∗P < .01. ∗∗∗P < .001. The experiment was repeated twice and the results were similar.
Tm6sf2-/- Rats Have Decreased Levels of Plasma Cholesterol but Not ApoB
The mean plasma levels of cholesterol, but not TG, were significantly lower in Tm6sf2-/- rats than in littermate controls (99.03 ± 10.5 vs 110.6 ± 14.0 mg/dL; P = .0294) (Figure 1E, left). The 10% decrease in plasma cholesterol level was distributed among all major lipoprotein classes: VLDL, low density lipoprotein, and high-density lipoprotein (Figure 1E, right). Despite little or no reduction in plasma TG levels in Tm6sf2-/-, the TG content of the VLDL fraction was strikingly reduced in these animals (Figure 1E, right).
Plasma levels of ApoB-100 and ApoB-48 did not differ significantly between WT and KO rats (Figure 1F). The mean plasma level of aspartate aminotransferase, but not alanine transaminase, was increased in Tm6sf2-/- rats (Figure 3C). Hepatic ApoB-100 levels did not differ between WT and KO rats, whereas hepatic ApoB-48 levels were slightly lower in Tm6sf2-/- rats (Figure 1G).
Neutral Lipids Accumulate in Cytoplasmic Droplets in Hepatocytes and Enterocytes of Tm6sf2-/- Rats
Tissue sections of chow-fed Tm6sf2-/- and WT rats were stained with Oil Red O to visualize the neutral lipids (Figure 5A). Both the number and size of lipid droplets in hepatocytes and enterocytes were significantly greater in Tm6sf2-/- than WT rats (Figure 3D). The increase in Oil Red O staining in the jejunum was most pronounced in the enterocytes of the villi, the site of uptake of dietary fat.
Figure 5.

Lipid accumulation in tissues of Tm6sf2 KOand WT rats. (A) Liver and jejunal tissue sections from chow-fed male WT and KO rats (n = 4/group, 5–6 wk) after a 4-hour fast were stained with Oil Red O and scanned using a NanoZoomer 2.0-HT, digital slide scanner (DM2000), at 10× and 80× magnification. (B) Chow-fed male rats (n = 4/group, 3–4 wk) were fasted for 16 hours and then gavaged with corn oil (10 μL/g body weight). Blood was collected at the indicated times and plasma TG levels were measured (left). TG absorption was calculated by the area under the curve (right). (C) Fecal lipids were extracted from feces of chow-fed female rats (n = 4/group, 13–14 wk) that were collected for 2 days. All experiments were repeated at least once and the results were similar. (D) Male WT and Tm6sf2-/- rats (n = 4/group, 6 wk) were fasted for 4 hours and then given Triton WR-1339 (500 mg/kg) and 200 μCi of [35S] methionine (1175 Ci/mmol) intravenously and blood was collected at the indicated times and plasma TG was measured. Rats were killed after 120 minutes. To detect [35S] labeled-ApoB, 25 μL plasma was processed as described in the Methods section. Membranes were exposed to Phosphor Screen in Fujifilm BAS cassette 2040 (Fujifilm) for 1 week and signals were quantified using a Storm Imager (PharosFX; Bio-Rad). Circles and bars indicate means ± SD. Differences between groups were analyzed using the Student unpaired 2-tailed t test. ∗∗P < .01, and ∗∗∗∗P < .0001.
To determine if TM6SF2 deficiency reduced absorption of dietary fat, we challenged chow-fed WT and KO rats with corn oil (10 μL/g body weight) and monitored plasma TG levels over the next 5 hours. Plasma TG levels of WT and KO rats were not different at baseline, but diverged after 1 hour: TG levels were lower in Tm6sf2-/- rats and the differences between the strains increased over time (Figure 5B).
The increase in Oil Red O staining in jejunal enterocytes (Figure 5A), taken together with the decrease in plasma TG levels after an oral fat challenge, implicates TM6SF2 in the packaging of dietary lipids into chylomicrons and/or secretion of these particles from enterocytes into bile. No significant differences in levels of fecal lipids, or in body weight, were found between chow-fed Tm6sf2-/- and WT female rats (Figure 5C). Thus, despite the low plasma TG levels over the time course of the study, there was no evidence of fat malabsorption in the Tm6sf2-/- rats. We cannot exclude the possibility that the low postprandial lipemia in the KO rats was the result of accelerated chylomicron clearance, but we believe this is unlikely because liver-derived VLDL and intestine-derived chylomicrons share a common clearance pathway. In this scenario, fasting TG would be expected to be lower in the Tm6sf2-/- animals.
The Secretion Rate of VLDL-TG, but Not VLDL-ApoB, Is Reduced in Tm6sf2-/- Rats
To determine the effect of TM6SF2 inactivation on hepatic secretion of VLDL, we compared rates of hepatic secretion of VLDL-TG and VLDL-ApoB in Tm6sf2-/- and WT male rats (n = 4/group, 6 wk). Rats were injected intravenously with Triton WR-1339 (500 mg/kg) to block lipolysis and 35S-methionine (200 μCi) to label endogenously synthesized proteins.18 TG levels were significantly lower in KO compared with WT littermates over the course of the experiment (Figure 5D, left).
To determine if the reduction in hepatic VLDL-TG secretion in the Tm6sf2-/- rats was the result of a reduction in the number of particles secreted or a decrease in the amount of TG per particle, we compared incorporation of 35S-methionine into VLDL-ApoB in KO and WT rats. At 2 hours after injection, no differences were found in the amount of labeled ApoB-100 or ApoB-48 in the plasma of the 2 strains (Figure 5D, right). Thus, absence of TM6SF2 was associated with a reduction in the amount of TG per VLDL particle, not in the number of VLDL particles that were secreted. This finding resembled what we saw previously in KO mice.9
The decrease in VLDL-TG secretion and associated increase in hepatic TG did not elicit evidence of a compensatory homeostatic response. Expression of hepatic mRNAs encoding key enzymes in fatty acid synthesis was similar in the 2 groups of rats (Figure 6).
Figure 6.
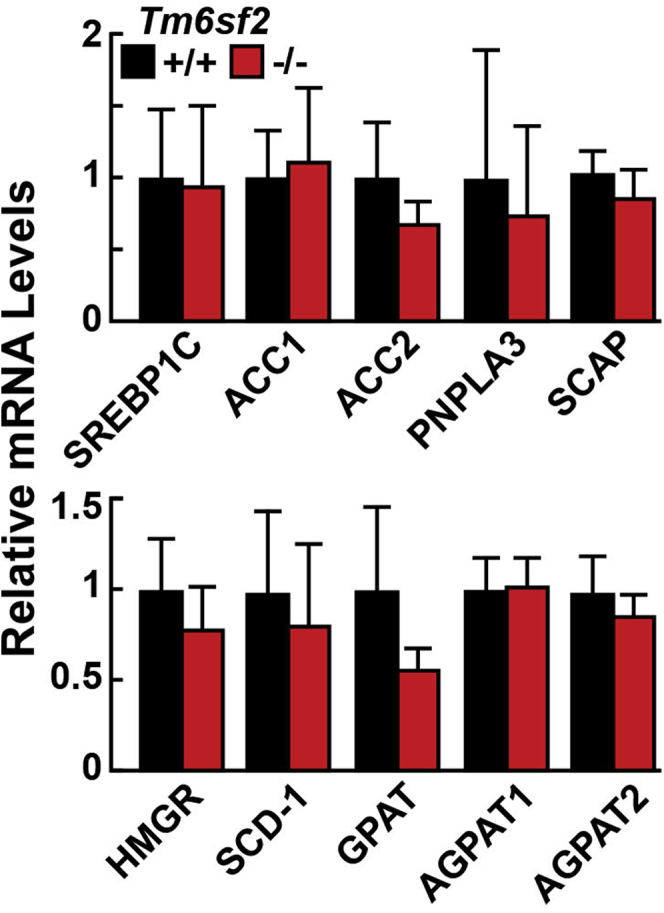
Relative levels of selected mRNAs encoding genes involved in lipid synthesis. Age-matched, chow-fed male WT and Tm6sf2 KO rats (n = 4–5/group, 6–7 wk) were fasted for 4 hours before livers were harvested, and total RNA was isolated as described in the Methods section. Quantitative real-time PCR assays were performed to assay the relative levels of selected mRNAs in livers of the rats. Expression levels were normalized to levels of cyclophilin and compared relative with levels of WT transcript. The official gene symbols were used for all genes. Bars represent means ± SD. The experiment was repeated once and the results were similar.
TM6SF2 Localizes to ER and the ER–Golgi Intermediate Compartment, but Not Golgi
To determine where in the secretory pathway TM6SF2 promotes addition of lipids to VLDL, we used 3 different cell fractionation protocols. The first protocol is summarized in Figure 7A (left).19 Binding immunoglobulin protein, an ER resident protein, was present in the heavy (ER) but not light (Golgi) fraction, and GOS28, a Golgi resident protein, was present in the light (Golgi), but not heavy (ER) fraction (Figure 7A, right). ER–Golgi intermediate compartment (ERGIC)-53, a protein that is present in the ERGIC,20 was present in both heavy and light fractions. Thus, this protocol separates Golgi from ER membranes, but does not separate ERGIC from either ER or Golgi membranes. TM6SF2 was detected only in the heavy fraction, which contains both ER and ERGIC membranes (Figure 7A, right).
Figure 7.

Localization of TM6SF2 to the ER and ERGIC using cell fractionation. (A) A diagram of the cell fractionation protocol (left) and immunoblot of fractionated liver lysates (right). Rat liver homogenate (0.5 g) was loaded on a discontinuous sucrose gradient and centrifuged at 100,000g for 1 hour, yielding a light fraction (Golgi) and heavy fraction (ER). Equal proportions (0.8% of total volume) of each fraction were subjected to immunoblot analysis. (B) A diagrammatic scheme of the cell fractionation protocol (left) and immunoblot analysis of cell fractionation of liver lysates (right). Golgi membranes were isolated from livers of WT and Tm6sf2 KO rats after a 16-hour fast. A proportion of each fraction (as indicated) was subjected to immunoblot analysis. All experiments were repeated at least twice and results were similar. ∗Nonspecific band. BIP, binding immunoglobulin protein; CANX, calnexin; COX IV, cytochrome oxidase IV; EEA1, early endosome antigen 1; G, Golgi-enriched fraction; GOS28, Golgi SNAP receptor complex member 1; H, homogenate; I, intermediate fraction; LAMP1, lysosomal-associated membrane protein 1; LDH, lactate dehydrogenase A chain; LSD1, lysine-specific demethylase-1A; Plin2, Perilipin 2; PMP70, 70-kilodalton peroxisomal membrane protein.
To confirm that TM6SF2 is not present in the Golgi, we fractionated membranes using a different protocol (Figure 7B, left).17 TM6SF2 was present in the homogenate and second supernatant (S2), but not in the intermediate or Golgi-enriched fraction (Figure 7B, right). We then compared the lipids/ApoB ratios in the Golgi fractions of KO and WT rats using direct infusion MS/MSALL (Figure 8). The ratio of TG to ApoB was lower in Golgi fractions from Tm6sf2-/- rats (Figure 8), which is consistent with the findings of Davidson and his colleagues,21 who used electron microscopy to show that VLDL particles in Golgi are smaller in liver-specific Tm6sf2-/- than in WT animals. No differences were found between the 2 groups of rats in levels of PC or PE, which likely is owing to contamination with phospholipids not contained in the VLDL.
Figure 8.
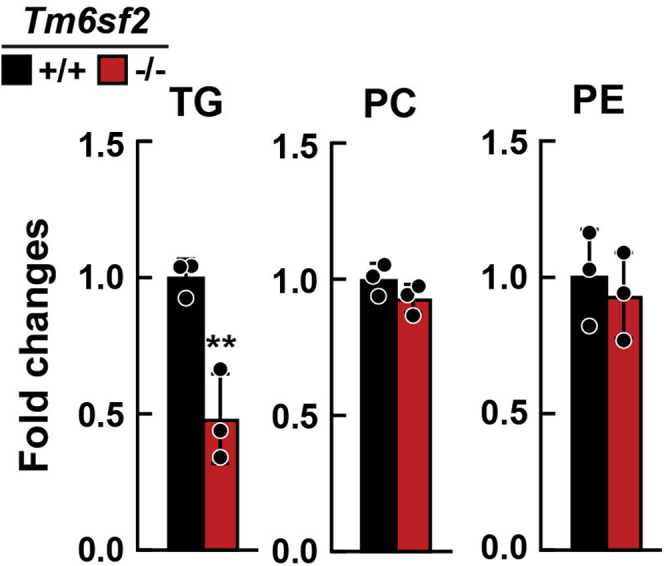
Lipid levels of hepatic Golgi fraction from Tm6sf2-/-and WT littermates. Chow-fed male WT and KO rats (n = 3/group, 6–7 wk) were fasted for 16 hours before livers were harvested. Golgi was isolated from livers as described in the Methods section. Lipids of Golgi were analyzed using direct infusion MS/MSALL. The fatty acid content of each individual species of TG, PC, and PE was summed within each lipid class to provide the TG, PC, and PE contents and normalized to the amount of ApoB determined by immunoblot. Fold changes between genotypes are presented. Bars represent means ± SD. Differences between groups were analyzed using the Student unpaired 2-tailed t test. ∗∗P < .01. The experiment was repeated once and the results were similar.
Finally, we separated Golgi and ER membranes in rat livers (Figure 9A) and cultured rat hepatocytes (Figure 9B) using a continuous Nycondenz (Thermo Fisher Scientific, Waltham, MA) gradient.22 TM6SF2 was present in the ER, but not Golgi fractions, in both of these experiments.
Figure 9.

TM6SF2 is located predominantly in the smooth ER. (A) Liver homogenates (0.25 g) of ad libitum chow-fed male WT and Tm6sf2-/- rats or (B) homogenates from cultured rat hepatocytes (CRL1601 cells) were subjected to Nycodenz continuous density gradient centrifugation as described in the Methods section. Equal proportions (∼1%) of each fraction were subjected to immunoblotting for the indicated organelle markers. (C) Schematic of protocol used to separate rough and smooth ER from primary rat hepatocytes as described in the Methods section (top). Immunoblot analysis was performed using equal proportions (∼1%) of each fraction (bottom). ∗Nonspecific band. All experiments were repeated at least twice, and results were similar. CANX, calnexin; cGN, cis-Golgi; cyt, cytosol; GOS28, Golgi SNAP receptor complex member 1; H, homogenate; LDH, lactate dehydrogenase; PNS, postnuclear supernatant; rER, rough ER; RPN1, ribophorin 1; sER, smooth ER; tGN, trans-Golgi; VTI1A, vesicle transport through interaction with t-SNAREs homolog 1A.
Thus, using 3 different cell fractionation protocols we found that TM6SF2 was present in the ER and ERGIC, but not in the Golgi fraction.
TM6SF2 Localizes to Smooth ER
We used a discontinuous sucrose gradient to fractionate ER membranes into 2 compartments: rough ER (RER) and smooth ER (SER) (Figure 9C, top).23 Both Golgi markers, VTI1A and GOS28, were present in the Golgi fractions, while calnexin, an ER resident protein, was present in the RER and SER fractions (Figure 9C, bottom). Ribophorin 1, which is in the RER, also was present in the SER. Thus, in this gradient, the SER was contaminated with proteins from the RER. TM6SF2, however, was almost exclusively in the SER fraction.
Reduction in Lipid Content of Newly Secreted VLDL in Tm6sf2-/- Mice
To directly measure the rate of VLDL secretion and its lipid composition in the absence of Tm6sf2, livers from Tm6sf2-/- mice were isolated and perfused, and effluent VLDL was examined. Experiments were performed in livers from Tm6sf2-/- mice, which provided practical advantages over perfused rat liver (eg, smaller perfusate volumes, lower flow rates, and ease of maintaining normoxia). Livers from Tm6sf2-/- and WT mice were isolated and perfused in a nonrecirculating mode for 60 minutes with an oxygenated buffer containing a mixture of free fatty acids/albumin (0.8 mmol/L) and other substrates. Perfusate was collected over three 15-minute intervals. VLDL was isolated from the perfusates by ultracentrifugation and proteins subjected to immunoblotting. No differences were apparent in relative levels of ApoE, ApoB-100, or ApoB-48 in VLDL from WT and Tm6sf2-/- mice (Figure 10A). Levels of VLDL-TG, VLDL-cholesterol, VLDL-CE, VLDL-PC, and VLDL-PE were reduced in Tm6sf2-/- mice compared with WT controls, as measured using enzymatic assays (Figure 10B) or LC-MS/MS (Figure 10C). Hepatic O2 consumption did not differ between the strains during liver perfusions (Figure 10D).
Figure 10.

VLDL isolated from liver perfusates of Tm6sf2-/-and WT mice.Chow-fed male Tm6sf2-/- and WT littermates (n = 4/group, 11–13 wk) were anaesthetized and given heparin (4 U/g body weight) before harvesting the livers. Livers were perfused for 60 minutes without recirculation using a buffer containing free fatty acids (0.8 mmol/L). The perfusate was collected over 3 intervals (15–30 min, 31–45 min, and 46–60 min). VLDL was isolated from the samples by ultracentrifugation (d = 1.006 g/mL). (A) Immunoblot analysis of ApoB and ApoE in VLDL (12.5 μL) from Tm6sf2-/- and WT mice. Protein levels were quantified using LI-COR Image Studio Lite (LI-COR Biosciences, Lincoln, NE version 5.2.5) and compared using the Student unpaired 2-tailed t test. (B) VLDL lipids from a 15- to 30-minute time interval were assayed enzymatically and (C) by LC-MS as described in the Methods section. The LC-MS/MS data were analyzed using MultiQuant software (SCIEX). The fatty acid content of each individual species of TG, CE, PC, and PE was summed within each lipid class and normalized to ApoB, as determined by immunoblot. Relative fold changes are presented. (D) Livers were perfused at a rate of 8 mL/min. The concentration of O2 (μmol/L) in perfusate was monitored and hepatic O2 consumption was calculated and normalized to liver weight. Bars indicate means ± SD. Differences between groups were analyzed using the Student unpaired 2-tailed t test. ∗P < .05, ∗∗P < .01. Each independent experiment contains 1 WT and 1 KO mouse (such as WT-1 and KO-1), the experiment was repeated 3 times, all the measurements of samples from 4 independent experiments were performed at the same time.
Thus, we concluded from these studies that the lipid depletion of circulating VLDL in Tm6sf2-/- rodents was owing to intracellular effects of TM6SF2 deficiency in hepatocytes.
TM6SF2 Physically Interacts With ApoB-48 and Acyl-CoA Synthetase Long Chain Family Member 5
To better understand how TM6SF2 promotes lipidation of VLDL, we looked for proteins that interact physically with TM6SF2. We expressed adeno-associated viruses–hTM6SF2–V5 in mouse livers, immunoprecipitated TM6SF2 using V5-agarose beads, then subjected the immunoprecipitate to protein MS. A list of proteins identified is shown in Table 2. The most abundant interacting protein was ApoB and all 101 peptides identified were from the N-terminal half of the protein. We confirmed that ApoB-48 was present in the immunoprecipitate by immunoblotting (Figure 11A, left) and we also showed that TM6SF2 co-immunoprecipitated with ApoB when we used an anti-ApoB antibody (Figure 11A, right). Song and his colleagues24 also identified ApoB as a protein that physically interacts with TM6SF2 in cultured rat hepatocytes.
Table 2.
TM6SF2-Interacting Proteins Identified by MS
| Protein | Spectral counts |
Spectral index |
||
|---|---|---|---|---|
| Luc | hTM6 | Luc | hTM6 | |
| Apolipoprotein B | – | 281.48 | – | 2.33E-05 |
| Myosin-11 | 52.79 | 2.64E-06 | ||
| ATP-binding cassette subfamily D member 3 | – | 51.00 | – | 1.29E-05 |
| Myosin-1 | 48.39 | 2.39E-06 | ||
| Mitochondrial inner-membrane protein | – | 37.51 | – | 5.55E-06 |
| Myosin-8 | 34.42 | 1.84E-06 | ||
| Long chain fatty acid CoA ligase 5 | – | 28.00 | – | 6.00E-06 |
| Carnitine O-palmitoyltransferase 1 | – | 26.00 | – | 3.21E-06 |
| Bile acyl-CoA synthetase | – | 23.87 | – | 4.80E-06 |
| ATP synthase subunit β | – | 20.00 | – | 3.44E-06 |
| 3-ketoacyl-CoA thiolase | – | 20.00 | – | 4.42E-06 |
| Very-long-chain enoyl-CoA reductase | – | 20.00 | – | 8.62E-06 |
| Myosin light chain 3 | – | 17.02 | – | 2.02E-05 |
| Dolichyl-diphosphooligosaccharide-protein glycosyltransferase subunit 1 | – | 16.00 | – | 3.91E-06 |
| MOSC domain-containing protein 1 | – | 15.50 | – | 8.82E-06 |
| Cytochrome b-c1 complex subunit 2 | – | 15.00 | – | 1.63E-06 |
| Heat shock protein heat shock protein 90-β | – | 13.94 | – | 1.78E-06 |
| Mitochondrial glutamate carrier 1 | – | 13.89 | – | 4.21E-06 |
| Sarcoplasmic/endoplasmic reticulum calcium ATPase 1 | - | 13.00 | - | 1.03E-06 |
| Actin, α | - | 13.00 | - | 3.34E-06 |
NOTE. Proteins with spectral count greater than 13 are listed above. Spectral counts are the number of peptide spectral matches in a sample, where a peptide spectral match is the number of MS/MS spectra that the software was able to match to a peptide from that protein. Spectral index is a value used for comparing protein amounts between samples, and is calculated based on fragment ions that are matched to peptides from the protein, which then are normalized based on protein amount and protein molecular weight. –, undetectable.
ATP, adenosine triphosphate; hTM6, human V5-tagged TM6SF2; Luc, luciferase; MOSC, mitochondrial amidoxine reducing component 1.
Figure 11.

Co-immunoprecipitation of TM6SF2 with ApoB and ACSL5. (A) Hepatic lysates from mice expressing adeno-associated virus (AAV)–human V5-tagged TM6SF2 (hTM6SF2-V5) or AAV-luciferase were precleaned with A/G agarose beads and then (500 μg, input) was incubated with anti-V5 agarose beads (right) or with rabbit anti-mouse ApoB polyclonal antibody (Abcam Cambridge MA) at 4°C overnight. Input (10%) and IP samples were size-fractionated using SDS–polyacrylamide gel electrophoresis (4%–12%) and immunoblotting was performed as described in the Methods section. (B) Mouse intestinal epithelial cells were incubated ± PFA (1%) and then quenched with 1.25 mol/L glycine. Homogenates from PFA-treated cells (PFA-Input) were precleaned with A/G agarose beads and then 500 μg was incubated with mouse anti-mouse TM6SF2 mAb (8B3) (left) or rabbit anti-mouse ACSL5 (25D12) (right). (C) Co-immunoprecipitation of TM6SF2 with ApoB and ACSL5 from rat intestinal epithelial cells. Samples were handled as described in panel A except cells were homogenized in lysis buffer (1% digitonin, 5 mmol/L EDTA, 5 mmol/L ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid plus PI) and a total of 500 μg of the lysate (input) was incubated with rabbit anti-rat TM6SF2 antibody (505E (25 μg) at 4°C overnight. The mixture then was incubated with Dynabead protein G at 4°C for 2 hours. Samples were collected as described in the Methods section. Fraction (10%) were subjected to immunoblot analysis. ∗Nonspecific band. All experiments were repeated at least once, and the results were similar. CANX, calnexin.
To determine if ApoB interacts with TM6SF2 in intestines as well as liver, we repeated the study using mouse enterocytes. We found no proteins that reproducibly co-immunoprecipitated with TM6SF2 in enterocytes unless we treated lysates with a cross-linker (Figure 11B, left). Under these conditions, we identified ApoB as a TM6SF2-interacting protein (Figure 11B, left). We also found that acyl-CoA synthetase long chain family member 5 (ACSL5) interacts with TM6SF2. An anti-ACSL5 monoclonal antibody (mAb) that immunoprecipitated ACSL5 also brought down TM6SF2 (Figure 11B, right).
To confirm that these interactions were not artifacts resulting from overexpression or cross-linking, we repeated the studies in rat enterocytes. Both ApoB-48 and ACSL5 co-immunoprecipitated with rat TM6SF2, although the amount of ApoB linked to TM6SF2 was modest (Figure 11C).
Discussion
We developed a new rodent model of fatty liver disease by inactivating Tm6sf2 in rats. Tm6sf2-/- rats share cardinal features of TM6SF2 deficiency observed in human beings and mice: accumulation of TG in liver and reduced lipid levels in blood.5,9 The establishment of a line of rats lacking TM6SF2 allowed us to take advantage of cell fractionation protocols developed for rat tissues.17,23 Using these protocols we localized TM6SF2 expression to the ER and ERGIC (Figures 7 and 9). No TM6SF2 was found in the Golgi complex. TM6SF2 deficiency was associated with reduced rates of appearance of dietary TG and VLDL-TG in blood (Figure 5B and D, left), but did not interfere with VLDL-ApoB secretion (Figure 5D, right). Consistent with these findings, the ratio of TG to ApoB in Golgi was 50% lower in Tm6sf2-/- rats than in WT littermates (Figure 8). We performed isolated liver perfusion studies that showed that VLDL was lipid-depleted before entering the circulation (Figure 1E). The extent of lipid depletion in nascent VLDL secreted from isolated livers was similar to that seen in the Golgi fraction (∼50%) (Figure 10B and C), yet levels of ApoB-48 and ApoB-100 were similar in the particles from the 2 groups of animals (Figure 10A). Thus, TM6SF2 deficiency reduces lipidation, but not transport, of VLDL in hepatocytes. Finally, we found evidence that TM6SF2 and ApoB physically interact, suggesting that TM6SF2 may chaperone VLDL from the ER to the ERGIC before VLDL are transferred to the Golgi compartment and secreted.
It has been proposed that lipidation of VLDL occurs in the secretory pathway as a 2-step process (Figure 12).10 The first step takes place in the RER as ApoB is translated and translocated into the ER lumen (Figure 12).25 Interruption of this step, as occurs in ApoB or microsomal triglyceride transfer protein deficiency, compromises the integrity of the particle and results in degradation of poorly lipidated, newly synthesized ApoB.10,12,13 Defects in VLDL assembly and secretion are more severe in Mttp-/- mice than in Tm6sf2-/- animals.8 Liver-specific inactivation of Mttp results in almost complete blockade of VLDL secretion and a 70% reduction in circulating TG levels. In contrast, secretion of VLDL-ApoB is preserved in Tm6sf2-/- rodents, while secretion of VLDL-TG is decreased by approximately 50% and plasma TG levels remain unchanged. Boren et al26 reported that human beings homozygous for the E167K variant had a reduced secretion of large VLDL (VLDL1)-ApoB and TG. The basis for the discrepancy between their results and ours is not clear. We found no evidence that TM6SF2 plays a role in the first phase of VLDL synthesis (Figure 12). Rather, TM6SF2 participates in the second step of VLDL lipidation, the so-called bulk lipidation step. The precise location at which this step occurs has not been clearly defined.7, 8, 9 Here, we show that within the ER, TM6SF2 is located predominantly in the SER, a region of the ER lacking polyribosomes. Enzymes involved in lipid synthesis27 and P450-containing enzymes that metabolize lipids and detoxify chemicals are concentrated in the SER.28,29 Although ApoB is synthesized in RER,25 electron microscopic studies fail to find VLDL particles in the RER. In these studies, VLDL localized to the smooth side of the SER/RER interface or to the SER, and was similar in size and appearance to those seen in the Golgi.30 These findings are consistent with the notion that VLDL bulk lipidation occurs predominantly or exclusively in a pre-Golgi compartment, specifically in the SER where TM6SF2 is located.
Figure 12.

VLDL secretion pathway highlighting the role of TM6SF2 as reviewed in the Discussion section. MTTP, microsomal triglyceride transport protein; PLTP, phospholipid transfer protein.
The mechanism by which TG are transferred from cytoplasmic lipid droplets to VLDL is not known. In the absence of TM6SF2, lipidation of VLDL is incomplete and the amount of TG secreted from the liver decreases by 50%. Excess TGs retained in cells must be oxidized or stored in lipid droplets. The expression of mRNAs encoding the major regulators of lipid metabolism was similar in WT and Tm6sf2-/- rats (Figure 6) and mice.9 These findings suggest that the partitioning of TGs between lipid droplets and the secretory pathway is not regulated at the transcriptional level.
TM6SF2 also was found to co-fractionate with ERGIC membranes (Figure 9A), which is consistent with findings by Mahdessian et al,7 who localized TM6SF2 to that compartment. It is of interest that the C-terminus of TM6SF2 contains an ER retrieval motif, KKXX.31 The presence of this sequence in TM6SF2 raises the possibility that TMSF2 escorts VLDL from the ER to ERGIC (Figure 12). TM6SF2 then may be retrieved from the ERGIC via the COP1 machinery and transported back to the ER, while VLDL is transferred to the Golgi and secreted. An attractive hypothesis that warrants testing is that TM6SF2 chaperones VLDL particles that are not fully lipidated back to the ER for further lipidation. If this is true, there must be a mechanism that senses the lipidation state of the particle in the ERGIC. Size or surface tension potentially could be used as indices of the lipidation state of the particle in the ERGIC.
The defect in lipoprotein assembly in both liver and intestine in the Tm6sf2-/- rats was more pronounced than in Tm6sf2-/- mice.9 Despite a greater relative increase in TG accumulation in the livers of Tm6sf2-/- rats (5- to 6-fold in rats vs 2- to 3-fold in mice), we found no indication that Tm6sf2-/- rats were more susceptible to transaminitis than Tm6sf2-/- mice (Figure 3C). A likely explanation for this apparent paradox is that the absolute levels of hepatic TG are similar in Tm6sf2-/- rats and mice. The greater relative increase in liver TGs in Tm6sf2-/- rats than mice is owing to WT rats having lower mean TG levels than mice. Thus, the exposure to increased TG is comparable in TM6SF2 animals from the 2 species. Heterozygous mice (Tm6sf2+/-) had levels of plasma TG and cholesterol that were intermediate between those of WT and homozygous KO mice,9 which is consistent with the human data.5 However, plasma lipid levels of heterozygous rats (Tm6sf2+/-) did not differ from WT littermates. The reasons for this interspecies difference are not clear.
Oral fat tolerance also was much more impaired in Tm6sf2-/- rats than in Tm6sf2-/- mice (Figure 5B).9 Moreover, TG accumulated to a greater extent in enterocytes of KO rats (Figure 5A) than in KO mice.9 Despite the marked reduction in fat tolerance seen in KO animals, the body weights of KO rats did not differ significantly from WT littermates (Figure 1D, left), and levels of fecal lipids were not increased in Tm6sf2-/- rats (Figure 5C). These data indicate that inactivation of TM6SF2 does not cause fat malabsorption (Figure 5B, right). Presumably, the same amount of fat is absorbed but over a longer time span. We cannot rule out the possibility that the differences in plasma TG levels in these experiments are owing to increased TG hydrolysis in the Tm6sf2-/- mice.
We found evidence that TM6SF2 interacts physically with ACSL5 in tissues of rats and mice (Figure 11). ACSL5 is an ER and mitochondrial protein32 that catalyzes conversion of long-chain unsaturated fatty acids to their active acyl-CoA forms.33 If a role of TM6SF2 is to localize ACSL5 to regions of the ER where lipids are added to proteins, inactivation of ACSL5 might be expected to resemble the phenotype of Tm6sf2-/- rodents. Two lines of Acsl5-/- mice have been developed.34,35 These mice, similar to Tm6sf2-/- rodents, have reduced VLDL-TG secretion. However, these mice do not accumulate hepatic TG.35 It is possible that other member of the ACSL family compensate for the absence of ACSL5 in the Acsl5-/- mice. The finding that both ACSL5 and ApoB physically interact with TM6SF2 suggests that TM6SF2 may act as a scaffold in the ER to coordinate the actions of proteins involved in the transfer of lipids from droplets to VLDL before its secretion.
Materials and Methods
Generation of Tm6sf2-/- Rats
Sprague-Dawley rats from Charles River Laboratory (Wilmington, MA) were maintained on a 12-hour light/12-hour dark cycle (lights on from 9:00 AM to 9:00 PM), with free access to chow (2016; Teklad Rodent Diet Envigo, East Millstone, NJ) and water. Unless otherwise indicated, rats were fasted for 4 hours before being anesthetized with isoflurane and killed. All experimental protocols were approved by the University of Texas Southwestern Institutional Animal Care and Research Committee.
Tm6sf2-/- rats were generated using CRISPR/Cas9 technology. The Alt-R CRISPR-Cas9 crisper (cr)RNA (Integated DNA Technologies, Research Triangle Park, NC) (ACUGUGAGAACGCCGAAACUGUUUUAGAGCUAUGCU) was synthesized by Integrated DNA Technologies (Coralville, IA) and annealed to Alt-R tracRNA under standard conditions.36 The annealed sgRNA and Cas9 protein (Integrated DNA Technologies) were microinjected into the cytoplasm of fertilized pronuclear rat eggs, and the surviving eggs were transferred into uteri of pseudopregnant recipient female rats. Genotyping was performed using DNA extracted from ears or tails of 9-week-old N0 rats. Exon 1 and part of intron 1 of Tm6sf2 was polymerase chain reaction (PCR)-amplified using oligonucleotides complementary to the sequences flanking the guide sequence (forward: 5'-ACTTGTGGGCAGAGTCTCGT-3', reverse: 5'-ACCCACTATGGCTCCACATC-3'). The 469–base pair amplified DNA fragments were sequenced using the Sanger method and/or subjected to gel electrophoresis. A total of 21 of 61 rats had 1 or more frameshift mutation(s). Two lines of rats were selected for the studies described in this article: one with an insertion of T in exon 1 (c.80_81insT) and the other with a deletion of 73 nucleotides in exon 1 (c.11_83del). Rats heterozygous for these mutations were outcrossed to WT Sprague-Dawley rats. Offspring of N1 heterozygous (Tm6sf2+/-) mating were used for all experiments described in this article.
Generation of Acsl5-/- Mice
Mouse embryonic stem cells containing 1 inactivated Acsl5 allele were obtained from the Knock-out Mouse Project (Davis, CA). Acsl5-/- mice were generated and genotyped as described previously.9 Offspring of heterozygous (Acsl5+/-) mating were used for all experiments described in this article.
Generation of Antibodies
A polyclonal rabbit anti-rat TM6SF2 (polyclonal antibody 505E) and a monoclonal rabbit anti-mouse ACSL5 (mAb-25D12) were generated against a keyhole limpet hemocyanin-conjugated peptide corresponding to the C-terminal 15 residues of rat TM6SF2 (AAPPSSPQDQGKKQQ) or to residues 668 to 683 (KFFQTQIKSLYESIEE) of ACSL5. The source of all other antibodies used in this article is provided in Table 3.
Table 3.
Antibodies Used in the Experiments Described in This Article
| Antibodies | Species reactivity | Epitope/source | Name or catalogue number | Dilution used |
|---|---|---|---|---|
| Rabbit anti–rTM6SF2 pAba | r | 15 C-terminal residues | pAb-505E | 2 μg/mL |
| Mouse anti–mTM6SF2 mAba | m | 15 C-terminal residues | mAb-8B3 | 5 μg/mL |
| Rabbit anti–mTM6SF2 pAba | m | 15 C-terminal residues | pAb-792 | 5 μg/mL |
| Rabbit anti–mACSL5 mAba | m, r | Residues from 668 to 683 | mAb-25D12 | 5 μg/mL |
| Mouse anti–mACSL5 mAba | m, r | Residues from 668 to 683 | mAb-16C11 | 5 μg/mL |
| Rabbit anti–mDGAT1 pAba | m, r | Gift from Dr Ye Jin, UTSW | pAb-669 | 5 μg/mL |
| Rabbit anti–d calnexin pAb | d, h, m, r | Enzo Life Sciences (Farmingdale NY) | ADI-SPA-860-J | 1:5000 |
| Rabbit anti–hBiP pAb | h, m, r | Cell Signaling Technology (Danvers, MA) | 3183 | 1:1000 |
| Rabbit anti–rGM130 pAb | m, r | Gift from Dr Joachim Seemann, UTSW | 1:1000 | |
| Mouse anti–GOS28 mAb | m, r | Abcam Cambridge, UK | ab82961 | 1:1000 |
| Rabbit anti–hVTI1A pAb | h, m, r | Cell Signaling Technology | 14764 | 1:1000 |
| Rabbit anti–PMP70 mAb | h, m, r | Abcam | ab109448 | 1:2000 |
| Rabbit anti–hLAMP1 pAb | h, m, r | Cell Signaling Technology | 3243 | 1:1000 |
| Rabbit anti–hCOX IV pAb | h, m, r | Cell Signaling Technology | 4844 | 1:1000 |
| Rabbit anti–hLDH mAb | h, m, r | Abcam | ab134187 | 1:1000 |
| Rabbit anti–hLSD1 mAb | h, m, r | Cell Signaling Technology | 2184 | 1:1000 |
| Rabbit anti–PLIN2 mAb | h, m, r | Abcam | ab108323 | 1:2000 |
| Rabbit anti–ATPA1 mAb | h, m, r | Abcam | ab76020 | 1:5000 |
| Rabbit anti-mApoB100/48 pAb | h, m, r | Abcam | ab20737 | 1:1000 |
| Mouse anti-hApoB Rabbit anti-ApoE mAb |
h.m m, r |
Santa Cruz Biotechnology (Dallas TX) Abcam |
Sc-393636 ab183596 | 1:200 1:1000 |
| Rabbit anti–ribophorin I pAb | m, r | Novus Biologicals (Centennial CO) | NBP2-55523 | 0.4 μg/mL |
| Rabbit anti–hERGIC-53 mAb | h, m, r | Abcam | ab125006 | 1:500 |
| Rabbit anti–hEEA1 mAb | h, m, r | Cell Signaling Technology | 3288 | 1:1000 |
h, human; m, mouse; pAb, polyclonal antibody; r, rat.
Generated in house (see the Methods section).
Preparation of Rat and Mouse Intestine Epithelial Cells
Small intestines were isolated and divided into 3 segments of equal length: duodenum, jejunum, and ileum. A 5- to 6-cm section from each segment was flushed with normal saline containing protease inhibitors (PIs) (no. 04693116001; Roche Applied Science, Penzberg, Germany). Luminal epithelial cells were scraped off using glass slides or isolated as described,37 except that the buffer used was phosphate-buffered saline (PBS) plus EDTA (2 mmol/L).
Real-Time PCR Assay of mRNA Expression
Total mRNA was extracted from tissues and real-time PCR were performed.9 Rat cyclophilin B was used as an internal control. Primers for real-time PCR are listed in Table 4.
Table 4.
Primers Used for Reverse-Transcription PCR
| Genes | Forward primer, 5′-3′ | Reverse primer, 5′-3′ |
|---|---|---|
| TM6SF2 | AGGACTGTGGCGATGTCACT | AGCAGACCCAGTACCAGAGC |
| SREBF1 | GACGACGGAGCCATGGATT | GGGAAGTCACTGTCTTGGTTGTT |
| ACC1 | AACATCCTAGATAGTCATGCAGCTACA | CAATGCTCTGGGTGTTCATGAA |
| ACC2 | GCTCGCCCATCTTTCTCATG | TTCCCATACTGATCTGCCAAGAC |
| PNPLA3 | CCAGGTGTGTGCCCGAAT | TCCTTGGATGCTCTGGATCTG |
| SCAP | TGCTCACCGTGGAGATGTCA | TCGGTCCCAGATGTTGATGA |
| HMGR | CGACATCATCATCCTCACGATAA | GCTGACGCAGGTTCTGGAA |
| SCD-1 | AAGATATCCACGACCCCAGCTA | TGCAGCAGGGCCATGAG |
| GPAT | GACGAAGCCTTCCGAAGGA | GACTTGCTGGCGGTGAAGA |
| AGPAT1 | CAGGATGTGAGAGTCTGGGTTTT | GTTTGAAGGGCAGCATGGA |
| AGPAT2 | GGTACACGCAACGACAATGG | GCCTGGATGGCCAAGTAGAA |
| CYCLO B | CGTGGGCTCCGTTGTCTT | TGACTTTAGGTCCCTTCTTCTTATCG |
Levels of Lipids, Lipoproteins, and Plasma Chemistries
Extraction of hepatic lipids, and levels of plasma lipids, lipoproteins, and chemistries were measured as follows. The method of Folch et al38 was used to extract hepatic lipids from liver (∼100 mg). Levels of hepatic TG, cholesterol, and free cholesterol were measured in liver using enzymatic assays (Infinity; Thermo Fisher Scientific, Waltham, MA) and normalized to sample weights. The hepatic lipids are expressed as mg/g liver tissue (wet weight). Blood was collected into tubes containing heparin from the tail veins of the rats. Plasma was isolated by centrifugation at 2000g for 10 minutes at 4°C. To determine the distribution of cholesterol and TG among circulating lipoproteins, plasma from 4 rats was pooled (total volume, 300 μL) and fractionated using fast performance liquid chromatography on a Superose 12 column (GE Healthcare). Cholesterol and TG levels in each fraction were measured enzymatically as described earlier. Aspartate aminotransferase and alanine transaminase were measured using VITROS 250 Microslide Technology (GMI Inc. Ramsey, MN).
Tissue Histology
Thin sections of tissues were fixed in paraformaldehyde (PFA) (4%) for 24 hours at room temperature with gentle shaking, and then equilibrated by sequential, 24-hour incubations in PBS containing 10% (wt/vol) and then 18% (wt/vol) sucrose at 4°C. Oil Red O staining was performed to visualize neutral lipids9 using a NanoZoomer 2.0-HT digital slide scanner (DM2000) (Hamamatsu Photonics, Hamamatsu City, Japan). The size and number of lipid droplets were determined in 3 randomly selected images from each slide (n = 4/group) using ImageJ (National Institutes of Health, Bethesda, MD).
Absorption and Secretion of TG and VLDL-ApoB
To assess the uptake of dietary TG from the gut into the circulation, chow-fed rats (4 wk) were fasted for 16 hours and then gavaged with corn oil (10 μL/g body weight) (no. C8267; Sigma-Aldrich, St. Louis, MO). Blood was collected from the tail vein at the indicated times and plasma TG levels were measured. To measure rates of hepatic VLDL-TG and VLDL-ApoB secretion, male WT and Tm6sf2-/- rats (n = 4/group, 6-wk) were fasted for 4 hours (9 AM to 1 PM). At time zero, 35S-methionine (200 μCi) and Triton WR133918 were given intravenously.
Blood then was collected at the indicated times. To measure ApoB secretion, blood was collected before and 120 minutes after rats were given 35S-methionine (200 μCi). To detect 35S-labeled ApoB in the plasma, 25 μL plasma was diluted in PBS (1:40) and processed as described.9 Briefly, the diluted plasma was incubated with 100 μL deoxycholate (0.15%) for 10 minutes at room temperature, and then 50 μL trichloroacetic acid was added. After a 30-minute incubation on ice, the mixture was centrifuged at 10,000 × g for 15 minutes at 4°C. The supernatant was discarded, and the pellet was air-dried and then dissolved in a buffer containing 50 μL radioimmunoprecipitation assay (RIPA) buffer, 50 μL membrane protein solubilization buffer (62.5 mmol/L Tris-HCl, pH 6.8, 15% sodium dodecyl sulfate [SDS], 8 mol/L urea, 10% glycerol, and 100 mmol/L dithiothreitol), and 50 μL 6× Laemmli SDS sample buffer (containing β-mercaptoethanol) (no. J61337; Alfa Aesar, Haverhill, MA). Samples were heated for 5 minutes at 95°C, and 20-μL aliquots were subjected to size fractionation by SDS–polyacrylamide gel electrophoresis (5%). Proteins were transferred to a nitrocellulose membrane (Bio-Rad, Hercules, CA) and the membrane was washed twice with PBS and exposed to a Phosphor Screen in a Fujifilm BAS cassette 2040 (Fujifilm, Tokyo, Japan) at -80ºC for 1 week. The Phosphor Screen was read and quantified with a Storm Imager (PharosFX; Bio-Rad).
Fecal Lipid Measurement
Rats (4/group; age, 13–14 wk) were maintained in individual cages for 2 days with free access to chow (2016; Teklad Rodent Diet) and water. Feces were collected, dried completely, and powdered using a porcelain mortar (no. 6325; Cole-Parmer, Vernon Hills, IL). Feces were weighed (total weight) and a total of 300 mg fecal powder was placed into glass tubes to which premixed Folch agent (chloroform/methanol [2:1]) was added. Tubes were vortexed and incubated for 30 minutes at 60°C. The mixture was filtered using a cell strainer (70 μm) (no. 352350; Falcon, Corning, NY) and transferred into clean glass tubes. The volume was brought up to 6 mL before 1.5 mL H2O was added. The tubes were vortexed and spun at 1500 × g for 10 minutes at room temperature. The organic phase was transferred to a preweighed glass tube and dried at 37°C. The tubes were placed in a vacuum oven at 70°C for 30 minutes before they were weighed again. The weight of the tubes before and after addition of the organic phase represented the lipid weight in 300 mg feces, and the concentration of fecal lipids was calculated (mg/g). To calculate the fecal lipids per day, the concentration of fecal lipids was multiplied by the total weight of feces and then divided by 2.
Isolated Liver Perfusion
Mice (n = 4/group; age, 11–13 wk) were fed a chow diet ad libitum, and then anesthetized with ketamine/xylazine via intramuscular injection and treated with heparin (4 U/g body weight) by intraperitoneal injection. Livers were harvested and perfused without recirculation for 60 minutes as described previously.39 Briefly, mice were anesthetized, intraperitoneal heparin was administered, and the portal vein was cannulated and perfused using a modified Krebs–Henseleit buffer containing 1.5/0.15 mmol/L lactate/pyruvate and an 0.8 mmol/L long-chain fatty acid mixture bound to albumin. Livers were isolated in a double-jacketed vessel warmed to 37ºC. Hepatic O2 consumption was monitored during the 60-minute perfusion using dual oxygen electrodes (PreSens Precision Sensing Regensburg, Germany) placed in the efferent and afferent perfusion lines. O2 uptake was corrected for atmospheric loss by subtracting the O2 gradient during perfusion in the absence of a liver. A VLDL-enriched fraction was isolated from the liver perfusate as follows. The density of the perfusate (25 mL) was adjusted to 1.006 g/mL by adding KBr (no. P0838; Sigma-Aldrich). Perfusate was placed in a 23-mL centrifuge tube (PC Oak Ridge, no. 314348; Thermo Scientific) and centrifuged at 378,000g (61,000 rpm) in a T865 rotor for 4 hours. After ultracentrifugation, the top 4 mL was collected (VLDL-enriched fraction) and PI (40 μL) (100× PI: no.04693116001; Roche Applied Science) was added. Lipids in VLDL were measured using high-sensitivity fluorometric assays for PC, cholesterol, and TG (no. CS0001, MAK043, and MAK264; Sigma-Aldrich).
Membrane Fractionation
Buffers used for membrane fractionation included the following: buffer B (50 mmol/L Tris-HCl, pH 7.5, 150 mmol/L NaCl), buffer C, buffer D, buffer E, and buffer F with 15%, 45%, 30%, and 7.5% sucrose (wt/vol), respectively. Heavy and light membranes were separated by serial centrifugation of rat liver homogenates as described previously with the following modifications.19 Briefly, liver tissue (∼0.5 g) was homogenized in ice-cold buffer-C using a 2-mL Dounce homogenizer (15 strokes) and then centrifuged at 2000g for 10 minutes at 4°C. The layer of fat at the top of the tube and the pelleted material at the bottom of the tube were discarded. The supernatant was collected and recentrifuged using identical conditions. The supernatant was diluted in buffer C to a volume of 3 mL. Discontinuous sucrose gradients were generated in 12-mL ultracentrifuge tubes (no. 344059; Beckman Coulter Life Sciences, Indianapolis, IN). A total of 2 mL buffer D was placed at the bottom of the tube and then 4 mL buffer E, 3 mL of diluted supernatant, and 2 mL of buffer F were added sequentially. The gradients were centrifuged at 100,200g (24,200 rpm) in a TH-641 rotor (Thermo Fisher Scientific, Waltham, MA) at 4ºC for 60 minutes. The brake was not used at the end of the centrifugation. Two bands were visible: the upper band (the heavy membrane fraction) contained the ER and the lower band (the light membrane fraction) contained Golgi. The 2 bands were collected separately using Pasteur pipettes. Protein levels were measured using Bicinchoninic Acid (BCA) kits (no. 23224; Thermo Fisher) before aliquoting and then snap-freezing the samples in liquid N2. Samples were stored at -80ºC.
Golgi membranes were isolated from rat livers as described previously with the following modifications: liver homogenates were centrifuged at 500g for 10 minutes at 4°C and the fat pad and pellet were discarded. The supernatant was collected (S1) and spun at 1500g for 10 minutes to obtain the supernatant (S2). S2 was subjected to sucrose gradient fractionation as described previously.17 Aliquots were snap-frozen in liquid N2 and stored at -80ºC.
Membranes also were fractionated using Nycodenz continuous gradients.22 Liver (0.25 g) or CRL1601 cells (three 10-cm dishes) were homogenized in 1 mL buffer G (10 mmol/L triethanolamine acetic acid, pH 7.4, 0.25 mol/L sucrose, 1 mmol/L sodium EDTA plus PI) using a Dounce homogenizer (no. KT885300-0002; Avantor, Radnor, PA) (15 strokes) or by passage through a 25-gauge needle × 13. Homogenates were centrifuged at 2000g for 10 minutes at 4°C to obtain the postnuclear supernatant. The postnuclear supernatant (600 μL) was subjected to Nycodenz continuous gradient centrifugation as described previously.22 The gradients were centrifuged at 234,200g (37,000 rpm) in a TH-641 rotor for 2 hours and then at 175,200g (32,000 rpm) for 16 hours (no brake). After centrifugation, 12 fractions (0.883 mL/fraction) were collected from each gradient. The bottom fraction (∼0.5 mL), which contained insoluble aggregates, was discarded.
SER and RER were separated from primary hepatocytes as described previously,23 except that the homogenates were spun at 1000g instead of 600g for 10 minutes at 4°C to obtain the postnuclear supernatant.
Co-immunoprecipitation of TM6SF2 From Rodent Cells and Tissues
Chow-fed male C57Bl/6J mice (10 wk) were infected with adeno-associated viruses 8 (3.0 × 1011) expressing luciferase or human V5-tagged TM6SF2 (hTM6SF2) under control of the thyroxine binding globulin promotor. After 14 days, mice were killed and their livers were collected. Liver tissue (∼30 mg) was homogenized in 1 mL RIPA buffer plus PI (RIPA+ buffer) and then the proteins were extracted and adjusted to a concentration of 1 μg/μL (input).9 Input (500 μg) was precleared using A/G agarose beads and then incubated with Invitrogen (Waltham, MA) V5 agarose beads (no. S190-120; Thermo Scientific) at 4ºC for 2 hours. The beads were washed with RIPA+ PI buffer and the supernatant was collected by spinning 1000g for 5 minutes at room temperature. Beads were washed 7× before they were incubated at 37°C in 200 μL of dissolving buffer (40 μL membrane protein solubilization buffer, 100 μL RIPA+ buffer, 60 μL 6× Laemmli SDS sample buffer) for 1 hour with gentle vortexing every 10 minutes. The beads were pelleted (1000g for 5 min) and the supernatant was collected (immunoprecipitation [IP] samples). To detect proteins that co-IP with TM6SF2, 10% of the sample was size fractionated by SDS–polyacrylamide gel electrophoresis (10%) and proteomics MS was used (University of Texas Southwestern Medical Center [UTSW] Proteomics Core). One-tenth of the sample was given to the UTSW Proteomics Core for gel protein extraction, trypsin digestion, and peptide identification.
Intestinal epithelial cells (∼150 mg) from WT and KO rats were homogenized in lysis buffer (1% high-purity digitonin [300410; Millipore, Burlington, MA], 5 mmol/L EDTA, 5 mmol/L ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid plus PI) and incubated at 4°C for 1 hour with gentle rotation. Lysates were cleared by centrifugation (12,000g) for 15 minutes at 4°C and then adjusted to 1 μg/μL (input). The input (500 μg) was precleaned using protein A/G agarose beads (no. 2003; Santa Cruz Biotechnology, Santa Cruz, CA) at 4ºC for 2 hours. Precleaned protein samples were incubated with rabbit anti-rat TM6SF2 polyclonal antibody (polyclonal antibody 505E) (25 μg) at 4°C overnight with slow rotation. The mixture then was incubated with Dynabeads protein G (100 μL) (Thermo Fisher Scientific, Waltham, MA) at 4°C for 2 hours. Immunoprecipitation also was performed after cross-linking with PFA. Mouse intestinal epithelial cells were incubated in the presence or absence of PFA (1%) (no. 15710; Electron Microscopy Science, Hatfield, PA) in PBS for 20 minutes at room temperature with rotation. Samples were quenched with glycine (no. 50046; Sigma) at a final concentration of 1.25 mol/L for 5 minutes by slow shaking and spun at 1000g for 5 minutes at 4°C. The supernatant was removed and 1 mL RIPA buffer with PI was added to the pellet. Cells then were disrupted with an ULTRA-TURRAX homogenizer system (no. EW-04720-50; Cole-Parmer) and passed through a 23-gauge needle × 10. The homogenate was incubated for 1 hour at 4°C with gentle rotation and then spun at 12,000g for 15 minutes at 4°C. The supernatant was collected (PFA-input). The input (500 μg) was incubated with 70 μg mAb-8B3 or an anti-ACSL5 antibody (mAb-25D12) at 4°C overnight with gentle rotation. The mixture was incubated with 70 μL prewashed protein A/G agarose beads for 2–3 hours. Beads were washed 5 times with RIPA+ buffer and then incubated at 37°C for 1 hour in dissolving buffer (300 μL 4SB buffer [4% SDS, 50 mmol/L Tris; 5 mmol/L EDTA, pH7.4] plus 70 μL 6× Laemmli SDS sample buffer), vortexing every 10 minutes. The samples were spun at 1000g for 5 minutes and the supernatant was collected (IP samples).
Immunoblotting
Liver tissue or intestinal epithelial cells (∼100 mg) were homogenized in 1 mL RIPA+ buffer and immunoblotting was performed as described.9 Extracted proteins were mixed with 2× or 6× Laemmli SDS sample buffer (containing β-mercaptoethanol) (no. J61337; Alfa Aesar). Membrane samples were combined with 1/6 volume of 6× Laemmli SDS sample buffer. Plasma samples were diluted 4× in RIPA buffer supplemented with PI and then mixed with 2× Laemmli SDS sample buffer. All the samples were incubated at 37°C for 1 hour to denature the proteins except the plasma samples, which were incubated at 95°C for 5 minutes. Samples were processed further and incubated with antibodies (Table 3) as described, with any modifications included in the Figure legends.
Lipid Analysis by LC-MS/MS
All solvents were from Sigma-Aldrich (St Louis, MO) and all lipid extractions were performed in 16 × 100-mm glass tubes with Polytetrafluoroethylene-lined caps (Fisher Scientific). Glass Pasteur pipettes and solvent-resistant plasticware tips (Mettler-Toledo, Columbus, OH) were used to minimize leaching of polymers and plasticizers. To analyze VLDL lipids from liver perfusates and liver homogenates, LC-MS/MS was performed as follows. Briefly, VLDLs (80 μL) were transferred to fresh glass tubes for liquid–liquid extraction. Samples were extracted using a modification of the Bligh and Dyer40,41 method: 1 mL each of dichloromethane, methanol, and water was added along with the sample. The mixture was vortexed and centrifuged at 2671g for 5 minutes, resulting in 2 distinct liquid phases. The organic phase (lower phase) was placed in a fresh glass tube with a Pasteur pipette and dried under N2. Samples were resuspended in 400 μL hexane.
Lipids were analyzed by LC-MS/MS using a SCIEX QTRAP 6500+ (SCIEX, Framingham, MA) equipped with a Shimadzu LC-30AD (Shimadzu, Columbia, MD) high-performance liquid chromatography system and a 150 × 2.1 mm, 5-μm Supelco Ascentis silica column (Supelco, Bellefonte, PA). Samples were injected at a flow rate of 0.3 mL/min at 2.5% solvent B (methyl tert-butyl ether) and 97.5% solvent A (hexane). Solvent B was increased to 5% over 3 minutes and then to 60% over 6 minutes. Solvent B was decreased to 0% during 30 seconds while solvent C (90:10 vol/vol isopropanol-water) was set at 20% and increased to 40% during the following 11 minutes. Solvent C was increased to 44% over 6 minutes and then to 60% over 50 seconds. The system was held at 60% solvent C for 1 minute before re-equilibration at 2.5% of solvent B for 5 minutes at a 1.2 mL/min flow rate. Solvent D (95:5 vol/vol acetonitrile-water with 10 mmol/L ammonium acetate) was infused postcolumn at 0.03 mL/min. The column oven temperature was 25°C. Data were acquired in positive and negative ionization mode using multiple reaction monitoring. The LC-MS/MS data were analyzed using MultiQuant software (SCIEX). The fatty acid content of each individual species of TG, CE, and phospholipid was summed within each lipid class to provide the TG, CE, PC, and PE contents.
Direct-infusion MS/MSALL lipid profiling was performed to analyze lipids in Golgi samples isolated from WT and KO rats. Golgi samples (1/3 of total volume) were extracted as described earlier. Samples were resuspended in 600 μL dichloromethane-methanol-isopropanol (2:1:1, vol/vol/vol) containing 8 mmol/L NH4F and infused into a SCIEX TripleTOF 6600+ MS using a custom-configured LEAP PAL HTS-xt autosampler with dynamic load and wash (Raleigh, NC). Samples were infused into the MS for 3 minutes at a flow rate of 10 μL/min through the electrospray port of a DuoSpray ionization source. The MS/MSALL data were obtained by acquiring product-ion spectra at each unit mass between 200 and 1200 daltons. Sample sets were infused in positive ionization mode. Electrospray voltage was set to 5500 V, curtain gas was set to 20, gas 1 and 2 were set to 25 and 55, respectively, and temperature was set to 300°C. Declustering potential and collision energy were set to 120 V and 40 eV, respectively. Gas 1 and 2 as well as source gas were zero-grade air, and curtain gas and collisional activated dissociation gas were nitrogen. Processing the lipidomic data was performed with in-house script. The fatty acid content of each individual species of TG, PC, and PE was summed within each lipid class to provide the TG, PC, and PE contents. Protein concentrations (of liver homogenate) or relative ApoB levels (VLDL and Golgi samples) were determined before the addition of a SPLASHMIX internal standard (Avanti Polar Lipids, Alabaster, AL).
Statistical Methods
Differences between groups were analyzed using the Student unpaired 2-tailed t test as implemented in GraphPad Prism version 8.1.1 (GraphPad Software, San Diego, CA). Values are presented as means ± SD. A P value less than .05 was considered statistically significant.
All authors had access to the study data and reviewed and approved the final manuscript.
Acknowledgments
The authors wish to thank Christina Zhao, Fang Xu, Tommy Hyatt, Kaitlyn Eckert, and Maritza Thomas for excellent technical assistance; Linda Donnelly and Angela Carroll for antibody production; John Shelton for assistance with the histologic studies; Andrew Lemoff in the University of Texas Southwestern Medical Center Proteomics Core for his assistance; and Joachim Seemann, Shimeng Xu, and Youngah Jo for helpful discussions. We also thank Dr. Robert Hammer and the UTSW Transgenic Core for the generation of the Tm6sf2 KO rats.
CRediT Authorship Contributions
Fei Luo, MD, PhD (Data curation: Lead; Formal analysis: Lead; Investigation: Equal; Methodology: Equal; Project administration: Equal; Writing – original draft: Lead)
Eriks Smagris, MD, PhD (Conceptualization: Supporting; Data curation: Equal; Formal analysis: Equal; Investigation: Supporting; Methodology: Supporting; Writing – original draft: Supporting)
Sarah Martin, PhD (Conceptualization: Supporting; Data curation: Equal; Formal analysis: Equal; Investigation: Supporting; Methodology: Supporting; Writing – original draft: Supporting)
Goncalo Vale, PhD (Data curation: Supporting; Formal analysis: Supporting; Investigation: Supporting; Methodology: Supporting; Writing – original draft: Supporting)
Jeffrey G McDonald, PhD (Conceptualization: Supporting; Data curation: Equal; Formal analysis: Equal; Investigation: Supporting; Methodology: Supporting; Writing – original draft: Supporting)
Justin A Fletcher, PhD (Conceptualization: Supporting; Data curation: Supporting; Formal analysis: Supporting; Investigation: Supporting; Methodology: Supporting; Writing – original draft: Supporting)
Shawn C Burgess, PhD (Data curation: Supporting; Formal analysis: Supporting; Funding acquisition: Lead; Investigation: Supporting; Methodology: Supporting; Project administration: Lead; Supervision: Lead; Validation: Lead; Visualization: Lead; Writing – original draft: Supporting)
Helen H. Hobbs, MD (Conceptualization: Lead; Funding acquisition: Lead; Investigation: Lead; Methodology: Lead; Project administration: Lead; Resources: Lead; Supervision: Lead; Validation: Lead; Visualization: Lead; Writing – original draft: Lead)
Jonathan Cohen, PhD (Conceptualization: Lead; Funding acquisition: Lead; Investigation: Lead; Methodology: Lead; Project administration: Lead; Resources: Lead; Supervision: Lead; Validation: Lead; Visualization: Lead; Writing – original draft: Lead)
Footnotes
Data Transparency Statement Data, analytic methods, and study materials will be made available upon reasonable request.
Conflicts of interest The authors disclose no conflicts.
Funding Supported by grants RO1DK090056, F32DK112529, Robert A. Welch Foundation I-1804 (SCB), and PO1 HL20948 from the National Institutes of Health and by the Howard Hughes Medical Institute. Also supported in part by the Chinese Scholarship Council (F.L.).
Contributor Information
Helen H. Hobbs, Email: Helen.hobbs@utsouthwestern.edu.
Jonathan C. Cohen, Email: Jonathan.Cohen@utsouthwestern.edu.
References
- 1.Younossi Z.M., Koenig A.B., Abdelatif D., Fazel Y., Henry L., Wymer M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64:73–84. doi: 10.1002/hep.28431. [DOI] [PubMed] [Google Scholar]
- 2.Cohen J.C., Horton J.D., Hobbs H.H. Human fatty liver disease: old questions and new insights. Science. 2011;332:1519–1523. doi: 10.1126/science.1204265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buch S., Stickel F., Trepo E., Way M., Herrmann A., Nischalke H.D., Brosch M., Rosendahl J., Berg T., Ridinger M., Rietschel M., McQuillin A., Frank J., Kiefer F., Schreiber S., Lieb W., Soyka M., Semmo N., Aigner E., Datz C., Schmelz R., Bruckner S., Zeissig S., Stephan A.M., Wodarz N., Deviere J., Clumeck N., Sarrazin C., Lammert F., Gustot T., Deltenre P., Volzke H., Lerch M.M., Mayerle J., Eyer F., Schafmayer C., Cichon S., Nothen M.M., Nothnagel M., Ellinghaus D., Huse K., Franke A., Zopf S., Hellerbrand C., Moreno C., Franchimont D., Morgan M.Y., Hampe J. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat Genet. 2015;47:1443–1448. doi: 10.1038/ng.3417. [DOI] [PubMed] [Google Scholar]
- 4.Romeo S., Kozlitina J., Xing C., Pertsemlidis A., Cox D., Pennacchio L.A., Boerwinkle E., Cohen J.C., Hobbs H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461–1465. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kozlitina J., Smagris E., Stender S., Nordestgaard B.G., Zhou H.H., Tybjaerg-Hansen A., Vogt T.F., Hobbs H.H., Cohen J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2014;46:352–356. doi: 10.1038/ng.2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Speliotes E.K., Yerges-Armstrong L.M., Wu J., Hernaez R., Kim L.J., Palmer C.D., Gudnason V., Eiriksdottir G., Garcia M.E., Launer L.J., Nalls M.A., Clark J.M., Mitchell B.D., Shuldiner A.R., Butler J.L., Tomas M., Hoffmann U., Hwang S.J., Massaro J.M., O'Donnell C.J., Sahani D.V., Salomaa V., Schadt E.E., Schwartz S.M., Siscovick D.S., Voight B.F., Carr J.J., Feitosa M.F., Harris T.B., Fox C.S., Smith A.V., Kao W.H., Hirschhorn J.N., Borecki I.B. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011;7 doi: 10.1371/journal.pgen.1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mahdessian H., Taxiarchis A., Popov S., Silveira A., Franco-Cereceda A., Hamsten A., Eriksson P., van't Hooft F. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc Natl Acad Sci U S A. 2014;111:8913–8918. doi: 10.1073/pnas.1323785111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Newberry E.P., Hall Z., Xie Y., Molitor E.A., Bayguinov P.O., Strout G.W., Fitzpatrick J.A.J., Brunt E.M., Griffin J.L., Davidson N.O. Liver-specific deletion of mouse Tm6sf2 promotes steatosis, fibrosis, and hepatocellular cancer. Hepatology. 2021;74:1203–1219. doi: 10.1002/hep.31771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smagris E., Gilyard S., BasuRay S., Cohen J.C., Hobbs H.H. Inactivation of Tm6sf2, a gene defective in fatty liver disease, impairs lipidation but not secretion of very low density lipoproteins. J Biol Chem. 2016;291:10659–10676. doi: 10.1074/jbc.M116.719955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olofsson S.O., Stillemark-Billton P., Asp L. Intracellular assembly of VLDL: two major steps in separate cell compartments. Trends Cardiovasc Med. 2000;10:338–345. doi: 10.1016/s1050-1738(01)00071-8. [DOI] [PubMed] [Google Scholar]
- 11.Wetterau J.R., Aggerbeck L.P., Bouma M.E., Eisenberg C., Munck A., Hermier M., Schmitz J., Gay G., Rader D.J., Gregg R.E. Absence of microsomal triglyceride transfer protein in individuals with abetalipoproteinemia. Science. 1992;258:999–1001. doi: 10.1126/science.1439810. [DOI] [PubMed] [Google Scholar]
- 12.Raabe M., Flynn L.M., Zlot C.H., Wong J.S., Veniant M.M., Hamilton R.L., Young S.G. Knockout of the abetalipoproteinemia gene in mice: reduced lipoprotein secretion in heterozygotes and embryonic lethality in homozygotes. Proc Natl Acad Sci U S A. 1998;95:8686–8691. doi: 10.1073/pnas.95.15.8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raabe M., Veniant M.M., Sullivan M.A., Zlot C.H., Bjorkegren J., Nielsen L.B., Wong J.S., Hamilton R.L., Young S.G. Analysis of the role of microsomal triglyceride transfer protein in the liver of tissue-specific knockout mice. J Clin Invest. 1999;103:1287–1298. doi: 10.1172/JCI6576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamaguchi J., Gamble M.V., Conlon D., Liang J.S., Ginsberg H.N. The conversion of apoB100 low density lipoprotein/high density lipoprotein particles to apoB100 very low density lipoproteins in response to oleic acid occurs in the endoplasmic reticulum and not in the Golgi in McA RH7777 cells. J Biol Chem. 2003;278:42643–42651. doi: 10.1074/jbc.M306920200. [DOI] [PubMed] [Google Scholar]
- 15.Gusarova V., Brodsky J.L., Fisher E.A. Apolipoprotein B100 exit from the endoplasmic reticulum (ER) is COPII-dependent, and its lipidation to very low density lipoprotein occurs post-ER. J Biol Chem. 2003;278:48051–48058. doi: 10.1074/jbc.M306898200. [DOI] [PubMed] [Google Scholar]
- 16.Cartwright I.J., Higgins J.A. Direct evidence for a two-step assembly of ApoB48-containing lipoproteins in the lumen of the smooth endoplasmic reticulum of rabbit enterocytes. J Biol Chem. 2001;276:48048–48057. doi: 10.1074/jbc.M104229200. [DOI] [PubMed] [Google Scholar]
- 17.Tang D., Xiang Y., Wang Y. Reconstitution of the cell cycle-regulated Golgi disassembly and reassembly in a cell-free system. Nat Protoc. 2010;5:758–772. doi: 10.1038/nprot.2010.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Millar J.S., Cromley D.A., McCoy M.G., Rader D.J., Billheimer J.T. Determining hepatic triglyceride production in mice: comparison of poloxamer 407 with Triton WR-1339. J Lipid Res. 2005;46:2023–2028. doi: 10.1194/jlr.D500019-JLR200. [DOI] [PubMed] [Google Scholar]
- 19.Xia M., Chandrasekaran P., Rong S., Fu X., Mitsche M.A. Hepatic deletion of Mboat7 (LPIAT1) causes activation of SREBP-1c and fatty liver. J Lipid Res. 2021;62:100031. doi: 10.1194/jlr.RA120000856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schweizer A., Matter K., Ketcham C.M., Hauri H.P. The isolated ER-Golgi intermediate compartment exhibits properties that are different from ER and cis-Golgi. J Cell Biol. 1991;113:45–54. doi: 10.1083/jcb.113.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Newberry E.P., Strout G.W., Fitzpatrick J.A.J., Davidson N.O. Liver-specific deletion of Mttp versus Tm6sf2 reveals distinct defects in stepwise VLDL assembly. J Lipid Res. 2021;62:100080. doi: 10.1016/j.jlr.2021.100080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hammond C., Helenius A. Quality control in the secretory pathway: retention of a misfolded viral membrane glycoprotein involves cycling between the ER, intermediate compartment, and Golgi apparatus. J Cell Biol. 1994;126:41–52. doi: 10.1083/jcb.126.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siddiqi S.A. In vitro analysis of the very-low density lipoprotein export from the trans-Golgi network. Curr Protoc Cell Biol. 2015;67:11211–112117. doi: 10.1002/0471143030.cb1121s67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li B.T., Sun M., Li Y.F., Wang J.Q., Zhou Z.M., Song B.L., Luo J. Disruption of the ERLIN-TM6SF2-APOB complex destabilizes APOB and contributes to non-alcoholic fatty liver disease. PLoS Genet. 2020;16 doi: 10.1371/journal.pgen.1008955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alexander C.A., Hamilton R.L., Havel R.J. Subcellular localization of B apoprotein of plasma lipoproteins in rat liver. J Cell Biol. 1976;69:241–263. doi: 10.1083/jcb.69.2.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boren J., Adiels M., Bjornson E., Matikainen N., Soderlund S., Ramo J., Stahlman M., Ripatti P., Ripatti S., Palotie A., Mancina R.M., Hakkarainen A., Romeo S., Packard C.J., Taskinen M.R. Effects of TM6SF2 E167K on hepatic lipid and very low-density lipoprotein metabolism in humans. JCI Insight. 2020;5 doi: 10.1172/jci.insight.144079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Glaumann H., Dallner G. Lipid composition and turnover of rough and smooth microsomal membranes in rat liver. J Lipid Res. 1968;9:720–729. [PubMed] [Google Scholar]
- 28.Rubin E., Hutterer F., Lieber C.S. Ethanol increases hepatic smooth endoplasmic reticulum and drug-metabolizing enzymes. Science. 1968;159:1469–1470. doi: 10.1126/science.159.3822.1469. [DOI] [PubMed] [Google Scholar]
- 29.Chin D.J., Luskey K.L., Anderson R.G., Faust J.R., Goldstein J.L., Brown M.S. Appearance of crystalloid endoplasmic reticulum in compactin-resistant Chinese hamster cells with a 500-fold increase in 3-hydroxy-3-methylglutaryl-coenzyme A reductase. Proc Natl Acad Sci U S A. 1982;79:1185–1189. doi: 10.1073/pnas.79.4.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Claude A. Growth and differentiation of cytoplasmic membranes in the course of lipoprotein granule synthesis in the hepatic cell. I. Elaboration of elements of the Golgi complex. J Cell Biol. 1970;47:745–766. doi: 10.1083/jcb.47.3.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vincent M.J., Martin A.S., Compans R.W. Function of the KKXX motif in endoplasmic reticulum retrieval of a transmembrane protein depends on the length and structure of the cytoplasmic domain. J Biol Chem. 1998;273:950–956. doi: 10.1074/jbc.273.2.950. [DOI] [PubMed] [Google Scholar]
- 32.Mashek D.G., McKenzie M.A., Van Horn C.G., Coleman R.A. Rat long chain acyl-CoA synthetase 5 increases fatty acid uptake and partitioning to cellular triacylglycerol in McArdle-RH7777 cells. J Biol Chem. 2006;281:945–950. doi: 10.1074/jbc.M507646200. [DOI] [PubMed] [Google Scholar]
- 33.Oikawa E., Iijima H., Suzuki T., Sasano H., Sato H., Kamataki A., Nagura H., Kang M.J., Fujino T., Suzuki H., Yamamoto T.T. A novel acyl-CoA synthetase, ACS5, expressed in intestinal epithelial cells and proliferating preadipocytes. J Biochem. 1998;124:679–685. doi: 10.1093/oxfordjournals.jbchem.a022165. [DOI] [PubMed] [Google Scholar]
- 34.Meller N., Morgan M.E., Wong W.P., Altemus J.B., Sehayek E. Targeting of Acyl-CoA synthetase 5 decreases jejunal fatty acid activation with no effect on dietary long-chain fatty acid absorption. Lipids Health Dis. 2013;12:88. doi: 10.1186/1476-511X-12-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bowman T.A., O'Keeffe K.R., D'Aquila T., Yan Q.W., Griffin J.D., Killion E.A., Salter D.M., Mashek D.G., Buhman K.K., Greenberg A.S. Acyl CoA synthetase 5 (ACSL5) ablation in mice increases energy expenditure and insulin sensitivity and delays fat absorption. Mol Metab. 2016;5:210–220. doi: 10.1016/j.molmet.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miura H., Quadros R.M., Gurumurthy C.B., Ohtsuka M. Easi-CRISPR for creating knock-in and conditional knockout mouse models using long ssDNA donors. Nat Protoc. 2018;13:195–215. doi: 10.1038/nprot.2017.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Engelking L.J., McFarlane M.R., Li C.K., Liang G. Blockade of cholesterol absorption by ezetimibe reveals a complex homeostatic network in enterocytes. J Lipid Res. 2012;53:1359–1368. doi: 10.1194/jlr.M027599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Folch J., Lees M., Sloane Stanley G.H. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- 39.Cappel D.A., Deja S., Duarte J.A.G., Kucejova B., Inigo M., Fletcher J.A., Fu X., Berglund E.D., Liu T., Elmquist J.K., Hammer S., Mishra P., Browning J.D., Burgess S.C. Pyruvate-carboxylase-mediated anaplerosis promotes antioxidant capacity by sustaining TCA cycle and redox metabolism in liver. Cell Metab. 2019;29:1291–1305 e1298. doi: 10.1016/j.cmet.2019.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trinh M.N., Brown M.S., Goldstein J.L., Han J., Vale G., McDonald J.G., Seemann J., Mendell J.T., Lu F. Last step in the path of LDL cholesterol from lysosome to plasma membrane to ER is governed by phosphatidylserine. Proc Natl Acad Sci U S A. 2020;117:18521–18529. doi: 10.1073/pnas.2010682117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bligh E.G., Dyer W.J. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]