

Abstract
Background
Sickle cell disease (SCD) is a complex, chronic condition that impairs health-related quality of life (HRQoL) of affected individuals and their caregivers. As curative therapies emerge, comprehensive cost-effectiveness models will inform their value. These models will require descriptions of health states and their corresponding utility values that accurately reflect HRQoL over the disease trajectory.
Objectives
The objectives of this systematic review were to develop a catalogue of health state utility (HSU) values for SCD, identify research gaps, and provide future directions for preference elicitation.
Methods
Records were identified through searches of PubMed and EMBASE, Tufts Medical Center Cost-Effectiveness Analysis Registry, reference lists of relevant articles, and consultation with SCD experts (2008–2020). We removed duplicate records and excluded ineligible studies. For included studies, we summarize the study characteristics, methods used for eliciting HSUs, and HSU values.
Results
Five studies empirically elicited utilities using indirect methods (EuroQol-5Dimension (n=3) and Short Form-6Dimension (n=2); these represent health states associated with general SCD (n=1), SCD complications (n=2), and SCD treatments (n=3). Additionally, we extracted HSUs from seven quality-adjusted life years (QALY)-based outcome research studies. The HSU among general SCD patients without specifying complications ranged from 0.64 to 0.887. Only 36% of the HSUs used in the QALY-based outcomes research studies were derived from individuals with SCD. No study estimated HSUs in caregivers.
Conclusions
There is a dearth of literature of HSUs for use in SCD models. Future empirical studies should elicit a comprehensive set of HSUs from individuals with SCD, and their caregivers.
Keywords: Health state utility, sickle cell disease, systematic review, cost-effectiveness analysis
Summary of the article
This study comprehensively summarizes and catalogues health state utilities (HSUs) for SCD and its specific complications, and HSUs tied to its treatments.
INTRODUCTION
Sickle cell disease (SCD) is a group of genetically inherited disorders of hemoglobin affecting over 20 million people worldwide.1 In the United States (U.S.), approximately 100,000 people live with SCD; most are of African descent.1 SCD can lead to a number of acute and chronic complications including acute pain episodes, stroke, acute chest syndrome, chronic pain, symptomatic anemia, and increased risk of infections and organ damage; each associated with significantly increased economic burden.1,2 Recent advances in medical care have resulted in a major reduction in SCD-related childhood mortality; SCD has evolved from being a life-threatening disease of childhood to a chronic disease in adults.3
Despite achievements in mortality reduction, the substantial impact on health-related quality of life (HRQoL) of these individuals and their caregivers warrants attention. Previous studies have demonstrated the significant relationship between experiencing SCD complications and deterioration in HRQoL among both children and adults.4 Vaso-occlusive crisis (VOC) is a typical example, which can cause pain, and impair functioning and wellbeing.5 Further, treatment related complications such as iron overload and the need for chelation with transfusions, and graft-versus-host disease (GVHD) with allogeneic hematopoietic stem cell transplantation (AlloHSCT) can significantly impair HRQoL.6
HRQoL is best captured by defining the relevant health states, eliciting values that represent the utility of each of these states, then multiplying each of these by the time spent in each health state to estimate quality adjusted life years (QALYs). Health state utility (HSU) values can be elicited directly through empirical data collection or indirectly through administration of survey instruments that exist for this purpose. The four direct elicitation methods are: visual analogue scale (VAS), time trade-off (TTO), standard gamble (SG) and discrete choice experiment (DCE).7 The surveys most often employed when using the indirect method are the EuroQol-5 Dimensions (EQ-5D), Short Form-6 Dimensions (SF-6D) or the Health Utilities Index (HUI).8 Development of these instruments is grounded in the multi-attribute utility theory.8
HSUs are also used to estimate QALYs gained by a new intervention over the standard of care in cost-effectiveness analysis (CEA). Estimating the value of emerging therapies using CEAs will be particularly important in SCD as genetic therapies are pursued, as these may prove curative for SCD patients. Indeed, the Cure Sickle Cell Initiative funded by the National Heart Lung and Blood Institute (NHLBI;; curesickle.org)9 is a large collaborative research effort intended to accelerate the development of genetic therapies to cure SCD. However, high up-front costs are associated with genetic therapies.10 An existing genetic therapy for beta-thalassemia, another hemoglobin disorder, costs approximately US$1.8 million per treatment.11 Yet, the expense of one-time administration may be offset by the alternative of repeated administration of standard therapies that accumulate large expenses over the lifetime.12 CEA methods will be useful in valuing the impact of potentially curative therapies on the complexities of SCD experienced over a patient’s lifetime.
As members of the NHLBI Cure Sickle Cell Initiative, to inform the future QALY-based CEA models, we conducted a systematic review of the published literature, and created a catalogue of HSUs for SCD-specific comorbidities and treatment complications. Specifically, we summarize the main characteristics, designs and results of studies that estimate HSUs for the SCD population. We identify current research gaps and close by providing directions for future HSU research for SCD.
METHODS
Search Methods and Sources
We conducted a systematic review following the methods of the Cochrane Collaboration and the Agency for Healthcare Research and Quality guidance for systematic reviews, and adopted the population, intervention, comparator, outcomes, timing and setting/study design (PICOTS) framework to establish eligibility criteria.13,14 The adopted PICOTS framework reflects deliberations and decisions made over a three month period in late 2019 by an expert panel that included a molecular biologist, clinicians who care for patients with SCD, health economists, evidence synthesis scientists, and librarians. (Supplementary Table S1) These same investigators decided on the search terms. We searched the PubMed, EMBASE and the Tufts Medical Center CEA Registry databases using a pre-specified protocol (search terms are displayed in Supplementary Table S2). We also identified articles through screening the reference lists of relevant systematic reviews and consultation with experts.
The framework was executed as a search strategy in PubMed and EMBASE by the health sciences librarians. The Tufts Medical Center CEA Registry was searched by a health economist. Duplicates were removed and returned studies were screened for eligibility. For studies that met inclusion criteria, relevant data were extracted and synthesized. The content of this report aligns with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) Statement for reporting of systematic reviews.15
Eligibility Criteria
We included English language full-text articles published in peer-reviewed journals from January 2008 through September 2020. We also searched for full-text white papers and limited our search to the time frame of January 2018 through September 2020, reasoning that information provided in the older white papers would not reflect the current research findings. We excluded conference proceedings when full text was not available for assessment and quality rating. We define and place studies into two categories. We define empirically based studies as those that employed the direct elicitation methods (VAS, TTO, SG or DCE) or the indirect methods of multi-attribute utility instruments (e.g. EQ-5D, SF-6D, others). We define QALY-based outcomes research studies as those wherein QALYs were reported; from which we were called to isolate the HSUs that were incorporated. We excluded studies in which the study population was individuals with sickle cell trait. All types of interventions for SCD patients, in any geographic setting globally, were eligible.
Study Selection
Records identified through the databases, found from reference lists of relevant systematic reviews, and in consultation with experts were merged. After duplicate records were removed, one reviewer (B.J.) independently screened the titles and abstracts of all references, excluded those based on the pre-defined criteria, and assessed the full texts of all remaining articles for eligibility. The second reviewer (D.Q.) reviewed 10% of randomly selected references. Discrepancies between reviewers’ judgements were discussed and resolved through consensus.
Data Extraction
For the empirical HSU studies, one reviewer (B.J.) extracted the main characteristics and study designs (publication type, study population, region, study design, sample size, instruments used to elicit HSUs), and HSU estimates and uncertainties. For the QALY-based outcomes studies one reviewer (B.J.) extracted the main characteristics and designs, HSU used, and sources of the HSUs. For the HSUs that were sourced from other published studies we also extracted the main characteristics of the source studies, and instruments used. Sometimes the original HSU values in the source studies differed from those finally included in the outcomes research studies – the authors of the outcome research studies might adjust the original values to better fit their studies. We attempted to replicate, and then describe the adjustment method, when possible. The second reviewer verified the extracted data.
Critical appraisal
We performed a quality assessment for the empirical HSU studies using methods developed by Ara et al. and Brazier et al. in 2017 and 2019. These domains are measurement of variability, response rates to the instrument used, loss to follow-up and handling of missing data.16,17 For the QALY-based outcome research studies, we assessed the relevance of the target population in the source studies16,17 that is, whether the HSUs were derived from studies of individuals with SCD.
RESULTS
Study Selection
Figure 1 presents the PRISMA flow diagram of study selection and reasons for exclusion. Our search identified 636 references. Nineteen additional studies were identified from the reference lists of the literature review, and one was identified in consultation with experts. After removing duplicate articles, we screened the titles and abstracts of the remaining 486 references and included 178 references for full-text assessment. Ten articles met our final inclusion criteria. The schematic diagram in Supplementary Figure S1 shows how these articles were categorized –there were five empirical HSU studies18–22 and seven QALY-based outcomes research studies.19,22–27. Two articles (Arnold et al.19 and Spackman et al.22) contained not only an empirical HSU study, but also a QALY-based cost-utility analyses.
Figure 1.

Preferred Reporting Items for Systematic Reviews and Meta-Analyses flow diagram of studies included in this systematic review and reasons for exclusion.
Overview of Included Studies
The main characteristics of included studies can be found in Table 1. Of the five empirical HSU studies, one was conducted in U.S.,19 three in United Kingdom (U.K.),21,22,24 and one in Nigeria.20 Three were retrospective cohort studies,18,19,21 one was a cross-sectional study20 and one was a randomized control trial (RCT).22 Three studies used the EQ-5D18,19,22 and two studies used SF-6D to elicit HSUs.20,21 No study used direct elicitation methods.
Table 1.
Main characteristics of included studies
| Author, Year | Study Category | Publication Type | Study design | Region | Population | Sample size | Instrument/Method | Intervention Type |
|---|---|---|---|---|---|---|---|---|
| Anie et al. 201218 | • Empirical HSU study | • Journal article | • Retrospective cohort | • U.K. | • Adults with SCD admitted to hospital daycare or inpatient units | • 510 | • EQ-5D | • NA |
| Arnold et al. 201519 | • Empirical HSU study | • Journal article | • Retrospective cohort | • U.S. | • SCD patients | • Intervention group: 26 • Control group: 48 |
• EQ-5D | • AlloHSCT |
| Ojelabi et al. 201520 | • Empirical HSU study | • Journal article | • Cross-sectional study | • Nigeria | • Adults with SCD | • 200 | • SF-6D | • NA |
| Payne et al. 200821 | • Empirical HSU study | • Journal article | • Retrospective cohort | • U.K. | • Patients with β-thalassemia, SCD and myelodysplastic syndromes receiving ICT | • 60 | • SF-6D | • Intervention for SCD complication |
| Spackman et al. 201422 | • Empirical HSU study | • Journal article | • Randomized control trial | • U.K. | • SCD patients undergoing elective surgery | • Intervention group: 18 • Control group: 17 |
• EQ-5D | • Blood transfusion |
| Arnold et al. 201519 | • QALY-based outcomes research study | • Journal article | • Cost-effectiveness analysis | • U.S. | • SCD patients | • NA | • Based on literature | • AlloHSCT |
| Bradt et al., 202023 | • QALY-based outcomes research study | • White paper | • Cost-effectiveness analysis | • U.S. | • SCD patients | • NA | • Based on literature • Assumed by the authors |
• Pharmaceuticals |
| Cherry et al. 201224 | • QALY-based outcomes research study | • Journal article | • Cost-effectiveness analysis | • U.K. | • SCD patients | • NA | • Based on literature • Assumed by the authors |
• Blood transfusion |
| McLeod et al. 200925 | • QALY-based outcomes research study | • Journal article | • Cost-effectiveness analysis | • U.K. | • Patients with beta-thalassemia major or SCD undergoing frequent blood transfusion | • NA | • Based on literature • Assumed by the authors |
• Intervention for SCD complication |
| Spackman et al. 201422 | • QALY-based outcomes research study | • Journal article | • Cost-effectiveness analysis | • U.K. | • SCD patients undergoing elective surgery | • NA | • Based on their own study • Assumed by the authors |
• Blood transfusion |
| O’Brien & Hankins 200926 | • QALY-based outcomes research study | • Journal article | • Comparative effectiveness study | • No specific country | • Children with SCD | • NA | • Based on literature • Clinician opinion |
• AlloHSCT • Blood transfusion • Pharmaceuticals |
| Lubeck et al. 201927 | • QALY-based outcomes research study | • Journal article | • Simulated cohort modeling study | • U.S. | • SCD patients | • NA | • Based on literature | • NA |
Abbreviations: alloHSCT = allogeneic hematopoietic cell transplantation; EQ-5D = EuroQol-5D; HSU = health state utility; NICE = National Institute for Health and Care Excellence; NA = not applicable; SCD = sickle cell disease; SF-6D = short form-6D; U.K.=United Kingdoms, U.S. = United States
Five of the QALY-based outcome research studies were cost-effectiveness analyses. Two of the cost-effectiveness analyses were set in the U.S.,19,23 and three were in the U.K.22,24,25 Two focused on blood transfusion,22,24 one focused on alloHSCT,19 one focused on pharmaceuticals23 and one focused on interventions for SCD treatment complications.25 We also found one study that compared the effectiveness of SCD interventions that did not specify the region;26 and one simulation modeling study that projected outcomes for U.S.-based cohorts with and without SCD.27
The resulting catalogue of HSUs is comprised of three categories: HSUs for SCD without specifying complications (general SCD) (Table 2), HSUs for specific SCD complications (Table 3), and HSUs tied to SCD treatments (Table 4). Supplementary Table S3 presents the main characteristics of source studies for the HSUs used in the QALY-based outcome research studies.
Table 2.
Health state utilities associated with general sickle cell disease
| Author, Year | Study population, Region | Instrument/Method | Health States | Utility Values |
|---|---|---|---|---|
| Empirical HSU study | ||||
| Ojelabi et al. 201520 | • Adults with SCD • Nigeria |
• SF-6D | • SCD in general | • Utility for SCD in general: 0.65 (SD 0.12) • Utility for SCD in general for males: 0.66 (SD 0.11) • Utility for SCD in general for females: 0.64 (SD 0.12) • Utility decrement due to one year older: 0.023 (SE 0.014) |
| QALY-based cost-effectiveness analysis | ||||
| Bradt et al., 202023 | • SCD patients • U.S. |
• Based on the algorithm reported by Anie et al., which mapped VAS pain score to utility. | • SCD without complication | • Utility for SCD without complication: 0.80 |
| Cherry et al. 201224 | • SCD patients • U.K. |
• Based on literature | • SCD with normal TCD velocity | • Utility for SCD with blood velocity of < 200 cm/second: 0.22 (per three months) |
| QALY-based simulation modeling study | ||||
| Lubeck et al. 201927 | • SCD patients • U.S. |
• Based on the algorithm reported by Anie et al., which mapped VAS pain score to utility. | • SCD in general | • Utility for SCD with no pain: 0.887 • Utility for adult with SCD in general: 0.695 • Utility for children or adolescents with SCD in general: 0.692 |
Abbreviations: HSU = health state utility; NA = not applicable; SCD = sickle cell disease; SD = standard deviation; SF-6D = short form-6D; TCD = Transcranial Doppler; U.K. = United Kingdoms; U.S. = United States; VAS = visual analog scale
Table 3.
Health state utilities associated with complications of sickle cell disease
| Author, Year | Population, Region | Instrument/Method | Health States | Utility Values |
|---|---|---|---|---|
| Pain: Empirical HSU study | ||||
| Anie et al. 2012 | • Adults with SCD admitted to hospital daycare or inpatient units • U.K. |
• EQ-5D | • Pain | • Utility at admission for acute pain: 0.39 (SD 0.40) • Utility at discharge: 0.65 (SD 0.29) • Utility at 1-week follow-up: 0.75 (SD 0.26) |
| Ojelabi et al. 201520 | • Adults with SCD • Nigeria |
• SF-6D | • Pain | • Utility decrement for an increase in frequency of pain: 0.027 (SD: 0.007) |
| Pain: QALY-based cost-effectiveness analysis | ||||
| Bradt et al., 202023 | • SCD patients • U.S. |
• Based on literature | • Pain | • Utility decrement for acute pain crisis (admission): 0.36 • Utility decrement for acute pain crisis (discharge): 0.1 • Utility decrement for two-week pain crisis: 0.23 |
| Cherry et al. 201224 | • SCD patients • U.K. |
• Assumed by the authors | • Pain | • Utility decrement for pain crisis: 0.02 (per three-month) |
| Pain: QALY-based simulation modeling study | ||||
| Lubeck et al. 201927 | • SCD patients • U.S. |
• Based on the algorithm reported by Anie et al., which mapped VAS pain score to utility. | • Pain | • Utility for adults with severe pain: 0.437 • Utility for adults with moderate pain: 0.492 • Utility for adults with mild pain: 0.557 • Utility for children or adolescents with severe pain: 0.270 • Utility for children or adolescents with moderate pain: 0.474 • Utility for children or adolescents with mild pain: 0.703 |
| Stroke: QALY-based cost-effectiveness analysis | ||||
| Bradt et al., 202023 | • SCD patients • U.S. |
• Based on literature | • Stroke | • Utility decrement for minor stroke: 0.16 • Utility decrement for major stroke: 0.57 • Utility decrement for post-stroke on average: 0.30 |
| Cherry et al. 201224 | • SCD patients • U.K. |
• Assumed by the authors | • Stroke | • Utility decrement for mild state post first/second/third stroke: 0.03 (per three-month) • Utility decrement for moderate state post first/second/third stroke: 0.08 (per three-month) • Utility decrement for severe state post first/second/third stroke: 0.13 (per three-month) |
| • TCD > 200 cm/second | • Utility decrement: 0.03 | |||
| Cardiovascular conditions: QALY-based cost-effectiveness analysis | ||||
| Bradt et al., 202023 | • SCD patients • U.S. |
• Assumed by the authors | • Pulmonary hypertension | • Utility decrement: 0.12 |
| • Based on literature | • Myocardial infarction | • Utility decrement for myocardial infarction: 0.13 | ||
| • Based on literature | • Heart failure | • Utility decrement for heart failure: 0.12 | ||
| Acute chest syndrome: QALY-based cost-effectiveness analysis | ||||
| Bradt et al., 202023 | • SCD patients • U.S. |
• Based on literature | • Acute chest syndrome | • Utility decrement for acute chest syndrome: 0.13 |
| Cherry et al. 201224 | • SCD patients • U.K. |
• Assumed by the authors | • Acute chest syndrome | • Utility decrement for acute chest syndrome: 0.06 (per 3-month) |
| Kidney conditions: QALY-based cost-effectiveness analysis | ||||
| Bradt et al., 202023 | • SCD patients • U.S. |
• Based on literature | • AKI/Renal infarction | • Utility decrement for AKI/Renal infarction: 0.14 |
| • SCD patients • U.S. |
• Based on literature | • nephropathy/CKD | • Utility decrement for nephropathy/CKD: 0.14 | |
| Mental health conditions: Empirical HSU study | ||||
| Ojelabi et al. 201520 | • Adults with SCD • Nigeria |
• SF-6D | • Anxiety | • Utility decrement for anxiety: 0.029 (SD 0.014) |
| • Depression | • Utility decrement for depression: 0.037 (SD 0.014) | |||
| Mental health conditions: QALY-based cost-effectiveness analysis | ||||
| Bradt et al., 202023 | • SCD patients • U.S. |
• Based on literature | • Neurocognitive impairment | • Utility decrement for neurocognitive impairment: 0.05 |
| Other complications: Empirical HSU study | ||||
| Ojelabi et al. 201520 | • Adults with SCD • Nigeria |
• SF-6D | • General complications | • Utility decrement for an increase in number of complications: 0.05 (SD: 0.013) |
| Other complications: QALY-based cost-effectiveness analysis | ||||
| Bradt et al., 202023 | • SCD patients • U.S. |
• Based on literature | • Opioid tolerance/dependence. | • Utility decrement for opioid tolerance/dependence: 0.07 |
| • Based on literature | • Fatigue | • Utility decrement for fatigue: 0.12 | ||
| Other complications: QALY-based comparative effectiveness study | ||||
| O’Brien & Hankins 200926 | • Children with SCD • No specific country |
• Clinician opinion • Based on literature |
• VOC | • Utility for patients with severe SCD due to recurrent VOC receiving no treatment: 0.70 (range 0.50–0.90) |
Abbreviations: AKI = acute kidney injury; CI = confidence interval; CKD = chronic kidney disease; EQ-5D = EuroQol-5D; HSU = health state utility; NA = not applicable; SCD = sickle cell disease; SD = standard deviation; SF-6D = short form-6D; TCD = transcranial Doppler; VAS = visual analog scale; VOC = vaso-occlusive crisis; U.K. = United Kingdoms; U.S. = United States
Table 4.
Health state utilities associated with treatments for sickle cell disease
| Author, Year | Population, Region | Instrument/Method | Treatment | Utility Values |
|---|---|---|---|---|
| alloHSCT: Empirical HSU study | ||||
| Arnold et al. 201519 | • SCD patients • U.S. |
• EQ-5D | • alloHSCT | • Utility for post-alloHSCT patients (six years post-alloHSCT): 0.87 • Utility for patients with documented HLA typing and/or alloHSCT consultation but without alloHSCT: 0.91 |
| alloHSCT: QALY-based cost-effectiveness analysis | ||||
| Arnold et al. 201519 | • SCD patients • U.S. |
• Based on literature | • alloHSCT | • Utility at days +45 post-alloHSCT: 0.71 • Utility at days +90 post-alloHSCT: 0.75 • Utility at days +180 post-alloHSCT: 0.79 • Utility at days +365 post-alloHSCT: 0.84 |
| alloHSCT: QALY-based comparative effectiveness study | ||||
| O’Brien & Hankins 200926 | • Children with SCD • No specific country |
• Clinician opinion • Based on literature |
• alloHSCT | • Utility for post-alloHSCT patients with graft failure: 0.55 (range 0.35–0.75) • Utility for post-alloHSCT patient with chronic GVHD and with no graft failure: 0.65 (range 0.45–0.85) • Utility for post-alloHSCT patient with no graft failure and no chronic GVHD: 0.95 (range 0.75–1.0) |
| Blood transfusion: Empirical HSU study | ||||
| Payne et al. 200821 | • Patients with β-thalassemia, SCD and myelodysplastic syndromes receiving ICT • U.K. |
• SF-6D | • Chronic blood transfusion • ICT |
• Utility: 0.66 (range 0.37 to 0.95) |
| Spackman et al. 201422 | • Patients with SCD undergoing elective surgery • U.K. |
• EQ-5D | • Preoperative transfusion | Utility for patients who received no preoperative transfusion • At baseline: 0.793 (SD 0.298) • At follow-up: 0.864 (SD 0.190) Utility for patients who received preoperative transfusion • At baseline: 0.760 (SD 0.236) • At follow-up: 0.854 (SD 0.166) |
| Blood transfusion: QALY-based cost-effectiveness analysis | ||||
| Cherry et al. 201224 | • SCD patients • U.K. |
• Assumed by the authors | • Chronic transfusion • ICT |
• Utility decrement for pre-stroke patients on simple, exchange or combined transfusion: 0.02 (per three months) • Utility decrement for patients on injection chelation: 0.04 (per three months) • Utility gain for patients on oral chelation: 0.03 (per three months) |
| McLeod et al. 200925 | • Patients with beta-thalassemia major or SCD undergoing frequent blood transfusion • U.K. |
• Assumed by the authors | • Chronic blood transfusion • ICT |
• Mean utility for deferoxamine/desferrioxamine administered via a balloon infuser: 0.70 • Mean utility for patients receiving deferiprone: 0.76 |
| Spackman et al. 201422 | • Patients with SCD undergoing elective surgery • U.K. |
• Derived from their own empirical study • Assumed by the authors |
• Preoperative transfusion | • Same values from their own empirical study (see above) • Utility decrement for transfusion complication (e.g. hepatitis B, HIV, hemolytic transfusion reaction, post-transfusion purpura, variant Creutzfeldt-Jakob disease, hepatitis A or malaria): 0.05 |
| Blood transfusion: QALY-based comparative effectiveness study | ||||
| O’Brien & Hankins 200926 | • Children with SCD • No specific country |
• Clinician opinion • Based on literature |
• Chronic blood transfusion | • Utility for patients on chronic transfusion with severe disease and iron overload: 0.55 (range 0.35–0.75) • Utility for patients on chronic transfusion with no severe disease and with iron overload: 0.75 (range 0.55–0.95) • Utility for patients on chronic transfusion with severe disease and with no iron overload: 0.60 (range 0.40–0.80) • Utility for patients on chronic transfusion with no severe disease and with no iron overload: 0.80 (range 0.60–1.00) |
| Pharmaceuticals | ||||
| O’Brien & Hankins 200926 | • Children with SCD • No specific country |
• Clinician opinion • Based on literature |
• Hydroxyurea | • Utility for patients on hydroxyurea with severe disease: 0.65 (range 0.45–0.85) • Utility for patients on hydroxyurea with no severe disease: 0.85 (range 0.65–1.00) |
Abbreviations: alloHSCT = allogeneic hematopoietic cell transplantation; EQ-5D = EuroQol-5D; HSU = health state utility; GVHD = graft versus host disease; ICT = iron chelation therapy; NA = not applicable; SCD = sickle cell disease; SD = standard deviation; SF-6D = short form-6D; U.K. = United Kingdoms; U. S. = United States
General sickle cell disease (Table 2)
One empirical HSU study20 and three QALY-based outcomes research studies reported HSUs for the general SCD without specifying complications.23,24,27 Utilities range from 0.64 (general female patients) to 0.887 (patients without pain). The empirical study provided HSUs stratified by sex, as well as utility decrements associated with each one-year increase in age.20 The three outcome research studies include HSUs for patients without complications,23 for patients with TCD velocity< 200 cm/second,24 and for patients without pain and patients on average,27 respectively. Two of the outcome research studies calculated the HSUs based on the algorithm from Anie et al.’s study18 that mapped the visual analog scale (VAS) pain scores to utilities.23,27 Only one of the outcome research studies included HSUs separately for adults and children/adolescents.27
Sickle cell disease complications (Table 3)
Pain
Two empirical HSU studies18,20 and three QALY-based outcome research studies23,24,27 reported HSUs associated with pain. The mean HSU ranged from 0.27 (severe pain) to 0.75 (one week after discharging from hospital). One empirical study assessed HSUs in patients at admission for acute pain, at discharge and at one-week follow-up.24 The other empirical study estimated a utility decrement for an increase in frequency of pain.20
Of the three QALY-based outcomes research studies, one applied the same utility values from the first empirical study,23 one included a utility decrement for pain crises without specifying severity,24 and the third used the aforementioned mapping algorithm from Anie et al.18 and calculated HSUs associated with minor, moderate and severe pain for adults and children/adolescents respectively.27
Stroke
Two QALY-based outcomes research studies included HSUs associated with stroke.23,24 The utility decrement ranged from 0.162 (minor stroke) to 0.565 (major stroke). Both studies included utilities for different severity levels. One study specified HSUs for minor and major stroke,23 and the other reported utilities for mild, moderate and severe states post-stroke.24 We also found a second outcome research study that used a utility decrement for pre-stroke patients with TCD scan > 200 cm/second.24
Cardiovascular conditions
One QALY-based outcome research study included utility decrements for pulmonary hypertension, and for myocardial infarction and heart failure.23
Acute chest syndrome
Two QALY-based outcomes research studies provided utility decrements for acute chest syndrome23,24 The utility values ranged from 0.06 (over three months) to 0.13.
Kidney conditions
One QALY-based outcomes research study provided reported a utility decrement of 0.14 for nephropathy, acute kidney injury (AKI) or renal infarction.23.
Mental health conditions
One empirical HSU study20 and one QALY-based outcomes research study23 provided HSUs for mental health conditions. The empirical study estimated utility decrements for anxiety and depression.20 The outcomes research study included a utility decrement due to neurocognitive impairment.23
Other complications
One empirical study reported a utility decrement for an increase in the number of any comorbidities.20 We also found one QALY-based outcome research study that included utility decrements due to fatigue and opioid tolerance/dependence.23 and another study that used a HSU for patients with severe SCD due to recurrent vaso-occlusive crisis26.
Sickle cell disease treatments and treatment complications (Table 4)
Allogeneic hematopoietic stem cell transplantation
One empirical HSU study19 and two QALY-based outcomes research studies19,26 reported HSUs for patients who received alloHCT; these ranged from 0.55 (post-alloHCT patients with graft failure) to 0.95 (post-alloHCT patients without graft failure). The empirical study assessed HSUs at a mean of six years post-alloHSCT.19 One of the outcomes-based research studies included HSUs over the first post-alloHSCT year,19 and the other included HSUs for patients with and without graft failure or with chronic graft versus host disease over the five-year post-alloHCT period.26
Transfusion
Two empirical HSU studies,21,22 and four QALY-based outcome research studies22,24,25 reported HSUs in patients receiving transfusions. The mean HSU value for the empirical studies ranged from 0.55 to 0.854. The 0.55 was reported in the empirical study that measured HSUs for patients who were receiving chronic transfusion and also receiving iron chelation therapy.21The 0.854 was reported in an RCT that investigated whether preoperative transfusion decreases the risk of peri-operative complications in SCD patients undergoing low- or medium-risk surgery..22
In the QALY-based outcomes research studies, various types of utility decrements were included. Two studies included HSUs for the patients on transfusion without transfusion complications.24,26 Three studies included HSUs related to iron overload – one study included HSUs for patients with and without iron overload,26 and two included HSUs associated with treatments for iron overload, such as sub-cutaneous or oral chelation,24 deferoxamine/desferrioxamine, and deferasirox.25 A separate study assumed a utility decrement if patients had any of the following transfusion complications: hepatitis B, HIV, hemolytic transfusion reaction, post-transfusion purpura, variant Creutzfeldt-Jakob disease, hepatitis A or malaria.22
Pharmaceuticals
One QALY-based outcomes research study included HSUs for patients on hydroxyurea with (0.65) and without severe disease (0.85).26
Critical appraisal
The quality assessment for the empirical HSU studies can be found in Supplementary Table S4. Most of the empirical HSU studies reported the variability of their estimates.18,20–22 Three studies reported the response rate to the instruments used;19,21,22 one of the studies had a relatively low response rate (approximately 50%).19 Only one RCT study reported loss-to-follow up, which was less than 20%.22 Finally, two studies provided the methods for handling missing data.20,22
Figure 2 illustrates the distribution of sources of HSUs used in the QALY-based outcomes research studies. Overall, only 36% of the HSUs were elicited from individuals with SCD. Specifically, 83% of the HSUs associated with general SCD were based on studies wherein HSUs were elicited from those with SCD, 50% of HSUs associated with SCD complications relied on studies wherein HSUs were elicited from a non-SCD-specific population, and 52% of the HSUs associated with SCD treatments or treatment complications were based on researchers’ assumptions (including clinician opinion).
Figure 2.
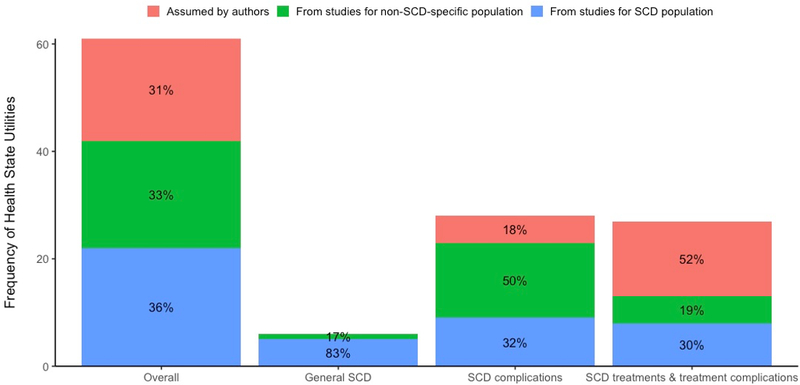
Distribution of sources of the health state utilities used in the outcome research studies. SCD indicates sickle cell disease.
DISCUSSION
To our knowledge, our study is the first to comprehensively summarize and catalogue HSUs for SCD and its specific complications, and HSUs tied to its treatments. We found a sparsity of literature that empirically estimated HSUs in individuals with SCD. In the QALY-based outcomes research studies, only 36% of the HSUs were obtained from the empirical studies conducted in the SCD population.
The limited number of studies we found did not fully address the complexity of the disease – HSUs for many complications were not measured at all, and complications that were represented as HSUs were not assessed in a time-varying fashion. Specifically, we found no U.S.-based empirical HSU study for general SCD or for SCD complications. Most of the empirical studies did not assess HSUs by age. This is a gap, as it will be important to have age-specific estimates because the rate and spectrum of complications can vary throughout a SCD patient’s lifecourse.28 Furthermore, compared to the comprehensive list of complications we established in our PICOTS criteria (Table 1), our analysis suggests that utilities of many SCD complications have not yet been studied empirically (e.g. stroke, infections, priapism, hepatobiliary complications and splenic disease etc.).
Possibly due to the scarcity of empirical HSU data for SCD complications, QALY-based outcomes research studies often relied on assumptions or HSU estimates from non-SCD-specific populations, even for some typical complications. For example, stroke is a common acute complication in the SCD population–historically 10% of children with SCD suffer a symptomatic stroke.29 However, the HSUs associated with stroke were either assumed or obtained from studies that elicited HSUs from individuals with stroke or type 2 diabetes in those without SCD.23,24 Similar to the research gap identified in the empirical studies, the utility loss due to many other SCD complications was not considered in published QALY-based outcomes research studies.
Similarly, the empirical studies related to SCD treatments did not explicitly assess the utility decrements attributable to the treatment complications, such as GVHD, iron overload and alloimmunization (although one study estimated mean HSU among patients receiving iron chelation therapy for treating iron overload21). No empirical studies were found for patients receiving a specific drug therapy. Moreover, the empirical studies did not report HSUs at various time points post treatments. Some complications may occur long after the treatments are administered. In most of the QALY-based outcomes research studies, the utility decrements attributable to the treatment complications were not modeled. Only one CEA and one comparative effectiveness study considered the treatment complications, yet their estimates were based on assumption or clinician opinion.22,26
Our review and findings of HSUs used in CEA models echo the concerns raised by the Good Practices for Outcomes Research Task Force of the International Society of Pharmacoeconomics and Outcomes Research.17 This report states that when HSUs are obtained from the literature for use in CEAs, caution should be exercised to address issues such as relevance of the patient population, sources of the HSUs used, and their method of elicitation.17 The Task Force also suggests that good practice requires a systematic review of existing literature to identify these HSU values.17 Our catalogue identifies the gaps in the existing HSU literature in the context of SCD. As such, it provides a path forward to future empirical work.
Further, the Second Panel on Cost-Effectiveness in Health and Medicine recommends that the spillover effects of disease on family members should be incorporated into CEAs.30 Studies have shown that SCD has a notable impact on the HRQoL of caregivers of SCD patients.31,32 Treatments that help ease the symptoms of SCD patients can also relieve caregiver burden. Neglecting this effect in CEAs may underestimate the value of SCD interventions. Quantifying HSUs among caregivers of SCD patients could be another focus of future empirical HSU studies.
Our study has several key strengths. First, it is comprehensive. By using rigorous methods, we identified not only the HSUs elicited in the empirical studies, but also the HSUs used in outcome research studies, which were either from published literature or based on authors’ assumptions. We made a concerted attempt to track HSUs back to their source, and to validate these by replicating the calculations. Second, we categorized the HSUs as general SCD without specifying complications, SCD complications, and SCD treatments, which are the key clinical inputs necessary for developing a SCD CEA model. This will facilitate the inclusion of HSUs in future QALY-based modeling studies.
Our study also has several limitations. First, our systematic review was limited to articles published in English; and abstracts and conference presentations were not included. Second, we did not include studies published before 2008. However, we found one cost-effectiveness study published in 2009 that relied on HSUs for the thalassemia population25 and one comparative effectiveness study published in 2009 that assumed HSUs based on clinician’s opinion.26 These data suggest that the availability of empirical HSUs studies before 2008 might be sparse. Finally, we did not systematically search the HSU studies for thalassemia, although some of the HSUs were found from the included cost-effectiveness analyses. HSUs associated with thalassemia treatments, such as therapy for transfusion-related iron or gene therapy could be a surrogate, if the data for SCD are not available.
In sum, our findings highlight the dearth of empirical studies that elicited HSUs for SCD. Empirical studies should be conducted to elicit HSUs from individuals with SCD using direct or indirect methods. These studies should capture HSUs that reflect one or more specific complications or receipt of a specific treatment. Estimations of these by age groups and timeframes post treatments would be of value. Moreover, to provide a complete picture of the burden of SCD, HSUs should be elicited from patients’ caregivers. These HSU data can be collected both alongside clinical trials and in cohort studies.33–36 CEA models informed by these newly elicited utilities will more accurately reflect SCD patients’ lifetime experiences and the value of emerging curative therapies.
CONCLUSIONS
We developed a comprehensive catalogue of HSUs associated with SCD from the published literature. Our catalogue will benefit future modelers of CEAs of disease-modifying and curative therapies for SCD.
Supplementary Material
Highlights.
While the emerging curative therapies bring hope to patients with sickle cell disease (SCD), concerns about their high costs have been raised. Cost-effectiveness analysis can be used to align the costs of the curative therapies with their expected health benefits.
Our study is the first to comprehensively summarize and catalogue health state utilities (HSUs) for SCD and its specific complications, and HSUs tied to its treatments, which can inform future cost-effectiveness analyses in SCD.
We identify the research gaps, such as the need for more empirical studies to elicit HSUs associated with the SCD and its treatment complications from patients, as well as HSUs from their caregivers.
Acknowledgment:
The authors gratefully acknowledge the following collaborators: N. DiFronzo (NHLBI); C. Henry, K. Johnson, D. Louden, A. Morgan, J. Rich (University of Wasington); W. Wright (Fred Hutchinson Cancer Research Center). The authors also appreciate the valuable insights and suggestions provided by the members of the Clinical and Economic Analysis Initiative Expert Panel.
Funding/Support:
This work was supported by grants from National Heart, Lung and Blood Institute (NHLBI), Cure Sickle Cell initiative (OTA Numbers: OT3HL152448, OT3HL151434)
Role of the Funder/Sponsor:
The funder had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Conflict of Interest Disclosures:
Drs Jiao, Basu, Ramsey, Bender, Quach, and Devine reported receiving a grant from National Heart, Lung, and Blood Institute paid to the University of Washington during the course of this study. Dr Roth is an employee of Genentech. Dr Roth reported receiving a grant from National Heart, Lung, and Blood Institute paid to the University of Washington during the conduct of this study; and consulting fees from BMS, Bayer, and Seattle Genetics outside the submitted work. Dr Devine reported being a member of the Methods and Data Council for Academy Health. Drs Basu and Devine is an editor for Value in Health and had no role in the peer-review process of this article. No other disclosures were reported.
Footnotes
AUTHOR DISCLOSURES [VIH-2021-0324]
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.National Heart, Lung, and Blood Institute. Sickle Cell Disease. Accessed August 2, 2020. https://www.nhlbi.nih.gov/health-topics/education-and-awareness/sickle-cell
- 2.Tanabe P, Spratling R, Smith D, Grissom P, Hulihan M. CE: Understanding the Complications of Sickle Cell Disease. Am J Nurs. 2019;119(6):26–35. doi: 10.1097/01.NAJ.0000559779.40570.2c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chaturvedi S, DeBaun MR. Evolution of sickle cell disease from a life-threatening disease of children to a chronic disease of adults: The last 40 years. Am J Hematol. 2016;91(1):5–14. doi: 10.1002/ajh.24235 [DOI] [PubMed] [Google Scholar]
- 4.Panepinto JA, Bonner M. Health-related quality of life in sickle cell disease: past, present, and future. Pediatr Blood Cancer. 2012;59(2):377–385. doi: 10.1002/pbc.24176 [DOI] [PubMed] [Google Scholar]
- 5.Esham KS, Rodday AM, Smith HP, et al. Assessment of health-related quality of life among adults hospitalized with sickle cell disease vaso-occlusive crisis. Blood Adv. 2020;4(1):19–27. doi: 10.1182/bloodadvances.2019000128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fiuza-Luces C, Simpson RJ, Ramírez M, Lucia A, Berger NA. Physical function and quality of life in patients with chronic graft-versus-host-disease: A summary of preclinical and clinical studies and a call for exercise intervention trials in patients. Bone Marrow Transplant. 2016;51(1):13–26. doi: 10.1038/bmt.2015.195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Webb EJD, O’Dwyer J, Meads D, Kind P, Wright P. Transforming discrete choice experiment latent scale values for EQ-5D-3L using the visual analogue scale. Eur J Health Econ HEPAC Health Econ Prev Care. 2020;21(5):787–800. doi: 10.1007/s10198-020-01173-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marra CA, Woolcott JC, Kopec JA, et al. A comparison of generic, indirect utility measures (the HUI2, HUI3, SF-6D, and the EQ-5D) and disease-specific instruments (the RAQoL and the HAQ) in rheumatoid arthritis. Soc Sci Med 1982. 2005;60(7):1571–1582. doi: 10.1016/j.socscimed.2004.08.034 [DOI] [PubMed] [Google Scholar]
- 9.National Heart, Lung, and Blood Institute. Cure Sickle Cell Initiative. Accessed February 9, 2021. https://curesickle.org/
- 10.Ikawa Y, Miccio A, Magrin E, Kwiatkowski JL, Rivella S, Cavazzana M. Gene therapy of hemoglobinopathies: progress and future challenges. Hum Mol Genet. 2019;28(R1):R24–R30. doi: 10.1093/hmg/ddz172 [DOI] [PubMed] [Google Scholar]
- 11.Harrison C First gene therapy for β-thalassemia approved. Nat Biotechnol. Published online September 8, 2019:1102–1103. [DOI] [PubMed] [Google Scholar]
- 12.Towse A, Fenwick E. Uncertainty and Cures: Discontinuation, Irreversibility, and Outcomes-Based Payments: What Is Different About a One-Off Treatment? Value Health J Int Soc Pharmacoeconomics Outcomes Res. 2019;22(6):677–683. doi: 10.1016/j.jval.2019.03.013 [DOI] [PubMed] [Google Scholar]
- 13.Higgins JPT, Thomas J, Chandler J, et al. Cochrane Handbook for Systematic Reviews of Interventions Version 6.0 (Updated July 2019). Cochrane.; 2019. [Google Scholar]
- 14.Trikalinos TA, Dahabreh IJ, Wallace BC, Schmid CH, Lau J. Towards a Framework for Communicating Confidence in Methodological Recommendations for Systematic Reviews and Meta-Analyses. Agency for Healthcare Research and Quality (US); 2013. Accessed March 2, 2020. http://www.ncbi.nlm.nih.gov/books/NBK164506/ [PubMed] [Google Scholar]
- 15.Moher D, Liberati A, Tetzlaff J, Altman DG, PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6(7):e1000097. doi: 10.1371/journal.pmed.1000097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ara R, Brazier J, Peasgood T, Paisley S. The Identification, Review and Synthesis of Health State Utility Values from the Literature. PharmacoEconomics. 2017;35(Suppl 1):43–55. doi: 10.1007/s40273-017-0547-8 [DOI] [PubMed] [Google Scholar]
- 17.Brazier J, Ara R, Azzabi I, et al. Identification, Review, and Use of Health State Utilities in Cost-Effectiveness Models: An ISPOR Good Practices for Outcomes Research Task Force Report. Value Health J Int Soc Pharmacoeconomics Outcomes Res. 2019;22(3):267–275. doi: 10.1016/j.jval.2019.01.004 [DOI] [PubMed] [Google Scholar]
- 18.Anie KA, Grocott H, White L, Dzingina M, Rogers G, Cho G. Patient self-assessment of hospital pain, mood and health-related quality of life in adults with sickle cell disease. BMJ Open. 2012;2(4). doi: 10.1136/bmjopen-2012-001274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arnold SD, Jin Z, Sands S, Bhatia M, Kung AL, Satwani P. Allogeneic Hematopoietic Cell Transplantation for Children with Sickle Cell Disease Is Beneficial and Cost-Effective: A Single-Center Analysis. Biol Blood Marrow Transplant J Am Soc Blood Marrow Transplant. 2015;21(7):1258–1265. doi: 10.1016/j.bbmt.2015.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ojelabi AO, Bamgboye AE, Ling J. Preference-based measure of health-related quality of life and its determinants in sickle cell disease in Nigeria. PloS One. 2019;14(11):e0223043. doi: 10.1371/journal.pone.0223043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Payne KA, Rofail D, Baladi J-F, et al. Iron chelation therapy: clinical effectiveness, economic burden and quality of life in patients with iron overload. Adv Ther. 2008;25(8):725–742. doi: 10.1007/s12325-008-0085-z [DOI] [PubMed] [Google Scholar]
- 22.Spackman E, Sculpher M, Howard J, et al. Cost-effectiveness analysis of preoperative transfusion in patients with sickle cell disease using evidence from the TAPS trial. Eur J Haematol. 2014;92(3):249–255. doi: 10.1111/ejh.12232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bradt P, Spackman E, Synnott P, et al. Crizanlizumab, Voxelotor, and L-Glutamine for Sickle Cell Disease: Effectiveness and Value. Institute for Clinical and Economic Review.; 2020. https://icer-review.org/material/sickle-cell-disease-draft-evidence-report/ [Google Scholar]
- 24.Cherry MG, Greenhalgh J, Osipenko L, et al. The clinical effectiveness and cost-effectiveness of primary stroke prevention in children with sickle cell disease: a systematic review and economic evaluation. Health Technol Assess Winch Engl. 2012;16(43):1–129. doi: 10.3310/hta16430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McLeod C, Fleeman N, Kirkham J, et al. Deferasirox for the treatment of iron overload associated with regular blood transfusions (transfusional haemosiderosis) in patients suffering with chronic anaemia: a systematic review and economic evaluation. Health Technol Assess. 2009;13(1):iii–iv, ix–xi, 1–121. doi: 10.3310/hta13010 [DOI] [PubMed] [Google Scholar]
- 26.O’Brien SH, Hankins JS. Decision analysis of treatment strategies in children with severe sickle cell disease. J Pediatr Hematol Oncol. 2009;31(11):873–878. doi: 10.1097/MPH.0b013e3181b83cab [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lubeck D, Agodoa I, Bhakta N, et al. Estimated Life Expectancy and Income of Patients With Sickle Cell Disease Compared With Those Without Sickle Cell Disease. JAMA Netw Open. 2019;2(11):e1915374. doi: 10.1001/jamanetworkopen.2019.15374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Serjeant GR. The Natural History of Sickle Cell Disease. Cold Spring Harb Perspect Med. 2013;3(10). doi: 10.1101/cshperspect.a011783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Centers for Disease Control and Prevention. Complications and Treatments of Sickle Cell Disease. Published June 12, 2019. Accessed March 15, 2020. https://www.cdc.gov/ncbddd/sicklecell/treatments.html
- 30.Sanders GD, Neumann PJ, Basu A, et al. Recommendations for Conduct, Methodological Practices, and Reporting of Cost-effectiveness Analyses: Second Panel on Cost-Effectiveness in Health and Medicine. JAMA. 2016;316(10):1093–1103. doi: 10.1001/jama.2016.12195 [DOI] [PubMed] [Google Scholar]
- 31.Madani BM, Al Raddadi R, Al Jaouni S, Omer M, Al Awa M-I. Quality of life among caregivers of sickle cell disease patients: a cross sectional study. Health Qual Life Outcomes. 2018;16. doi: 10.1186/s12955-018-1009-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van den Tweel XW, Hatzmann J, Ensink E, et al. Quality of life of female caregivers of children with sickle cell disease: a survey. Haematologica. 2008;93(4):588–593. doi: 10.3324/haematol.11610 [DOI] [PubMed] [Google Scholar]
- 33.Farrell AT, Panepinto J, Carroll CP, et al. End points for sickle cell disease clinical trials: patient-reported outcomes, pain, and the brain. Blood Adv. 2019;3(23):3982–4001. doi: 10.1182/bloodadvances.2019000882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McClish DK, Smith WR, Levenson JL, et al. Comorbidity, Pain, Utilization, and Psychosocial Outcomes in Older versus Younger Sickle Cell Adults: The PiSCES Project. BioMed Res Int. 2017;2017:4070547. doi: 10.1155/2017/4070547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dampier C, LeBeau P, Rhee S, et al. Health-related quality of life in adults with sickle cell disease (SCD): a report from the comprehensive sickle cell centers clinical trial consortium. Am J Hematol. 2011;86(2):203–205. doi: 10.1002/ajh.21905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Treadwell MJ, Hassell K, Levine R, Keller S. Adult Sickle Cell Quality-of-Life Measurement Information System (ASCQ-Me). Clin J Pain. 2014;30(10):902–914. doi: 10.1097/AJP.0000000000000054 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.