

Abstract
Objective
To investigate the relationship between hypoxia inducible factor-1α (HIF-1α) and the autophagic response in osteoarthritic chondrocytes (OA), under inflammatory insult as represented by in vitro OA model.
Methods
Human chondrocyte cell line C28/I2 was cultured in both normoxic and hypoxic conditions and treated with interleukin-1β (IL1β) to emulate OA inflammatory insult in vitro. Cellular HIF-1α expression was silenced using siRNA transfection and cellular autophagic (P62/LC3II) response and OA chondrocyte damage (COL2A1/MMP13) related proteins were examined using western blotting. Cellular mitophagic (BNIP3/PINK1/Parkin) and apoptotic (Caspase/Cleaved Caspase 3) were also evaluated to assess mitophagy-mediated cell death due to HIF-1α silencing.
Results
Chondrocyte basal autophagy levels were higher in a HIF-1α elevated environment and was more resistant to IL1β-induced inflammatory insult. Increase in autophagic proteins showed better chondrocyte repair, which resulted a lower level of reactive oxygen species production, and lesser damage to chondrocyte integrity. Silencing HIF-1α activates cellular PINK1/Parkin and BNIP3 mitophagic proteins, which leads to the activation of Caspase/Cleaved Caspase 3 apoptotic cascade.
Conclusion
Our results show that chondrocyte autophagy is dependent on HIF-1α expression, showing the importance of HIF-1α in hypoxic chondrocyte function in OA. Dysregulation of HIF-1α expression results in the activation of mitophagy-mediated apoptosis.
Keywords: osteoarthritis, chondrocyte, hypoxia inducible factor-1α, autophagy, mitophagy
Introduction
The hypoxic nature of articular chondrocytes has been established in previous studies, with the earliest studies reporting that the chondrocyte microenvironment is constantly exposed to oxygen tension as low as 1% in the deepest layers of the growth plate.1,2 Regulation of hypoxia by cellular sensors of oxygen is thus crucial for chondrocyte microenvironment homeostasis. HIF-1α (hypoxia inducible factor-1α) is a transcription factor in mammalian cells sensitive to changes in oxygen levels and is elevated in hypoxic cells, and rapidly degraded when removed from a hypoxic environment. In a previous study by Iyer et al., the conditional knockout of HIF-1α transgenic mice was found to be lethal to gestational mice. 3 Conditional breeding of mice lacking in HIF-1α resulted in smaller mice and still died within a few hours after birth. HIF-1α knockout mice had significantly shorter limbs and gross anomalies in chondrocyte rich–structures such as the ribs and trachea. 4 The partial collapse of HIF-1α knockout mice trachea was mainly responsible for breathing difficulties resulting in death. Reports have also shown the changes in HIF-1α expression in healthy and osteoarthritic (OA) human chondrocytes, inferring the involvement of HIF-1α in OA chondrocyte degeneration. 5
Growing in a hypoxic environment, articular chondrocytes maintain cell turnover different for other oxygen-rich cells.6,7 Articular chondrocytes obtain their needed oxygen and nutrients from the surrounding tissue. Due to the lack of proper cellular nourishment, homeostasis of cell conditions is carried out differently. Chondrocytes maintain proper cell survival and functioning through a constant activation of cell autophagy, a process where cells consume itself through a cascade of processes to maintain energy levels during nutrient stress reflected by the expression of LC3II and P62 proteins.8-10 A decrease in cellular autophagy levels results in the disruption of cell maintenance and plays in important role in age-related degenerative diseases such as OA. 10 Degenerative diseases like OA are characterized by a multifactorial processes which involve a constant expression of degenerative insult and a decrease in self-repairing processes. In OA chondrocytes, the collagen structure is constantly deteriorated showing a low COL2A1 expression and increased MMP13 levels due to excessive oxidative stress. The capacity of adult chondrocytes fundamentally decreases with age, and such results in cell death to the abnormal responses to stressors. 11
The cycle of cellular repair in response to cell ROS (reactive oxygen species), paired with a decreased cellular autophagic response ends up in the activation of chondroptosis, an apoptotic cascade unique to chondrocytes.12-15 During homeostatic conditions, chondroptosis is properly regulated with different activation times in different cells according to their cellular states.16,17 But during pathological conditions, dysregulated chondroptosis results in progressive damage of the articular cartilage layers, disrupting homeostatic balance of the articular structures.12,18,19 The process of OA chondrocyte damage is mediated by the regulation of cellular ROS, which accumulates in the mitochondria. Excessive accumulation of ROS in the mitochondria activates cellular mitophagy, as a mechanism of cell preservation to prevent further cellular damage by recycling damaged mitochondria. Mitophagy activation falls under 2 main mechanisms, through PARKIN (Parkin RBR E3 Ubiquitin Protein Ligase) dependent or independent pathways, which were found to be involved in degenerative diseases.
Proper maintenance of chondrocyte repair is crucial for the regulation of chondrocyte self-repair. Disruption of the repair cycle leads to activation of chondroptosis, which is a prominent characteristic of degenerative changes in OA. This study aims to investigate the effects of cellular oxygen sensor HIF-1α on chondrocyte autophagy and chondroptosis in relation to the progression of OA in articular chondrocytes.
Methods
Cell Culture
C28/I2 human chondrocyte cell line was cultured in Dulbecco’s modified Eagle’s Medium (DMEM; Keygen Biotech) containing 10% fetal bovine serum (FBS; Sigma-Aldrich) in the incubator at 37 °C under normoxic and hypoxic conditions (1% O2). Culture medium in normoxic and hypoxic conditions were replaced every 3 days and 2 days, respectively.
Cell Viability Assay
C28/I2 chondrocytes were seeded in 96-well plates at a seeding density of 5 × 103 cells and serum starved using DMEM without FBS for 24 hours after adhesion. Cells were then treated with 5, 10, 20 ng/mL of IL1β (Peprotech) for 48 hours in normoxic and hypoxic conditions and cell viability after IL1β treatment was evaluated using Cell Counting Kit-8 solution (Beyotime) for 30 minutes in 37 °C. Cell viability was then measured using a microplate reader at 450 nm absorbance.
Immunofluorescence
Cultured and treated cells were fixed on glass coverslips and blocked using 10% goat serum for 60 minutes. Samples were then probed with primary antibodies for LC3II at 4 °C overnight. Samples were then incubated with the respective fluorochrome-conjugated secondary antibody for 120 minutes in room temperature in total darkness and then cell nuclei were labelled using 4′,6-diamidino-2-phenylindole, dihydrochloride (DAPI) for 5 minutes. Antifade reagent was added prior to mountain glass slides and samples were examined under a fluorescent microscope.
Detection of Cellular ROS and Mitochondrial ROS
Chondrocyte ROS and mitochondrial ROS was measured using 2′7′-dichlorofluorescin diacetate (DCF-DA; Beyotime) and Mito-SOX Red dye (Beyotime). Mito-SOX cell nuclei were also stained with Hoechst (Beyotime) after MitoSox staining. Cells were plated in 24 well plates with 3 duplicate wells and treated with HIF-1α siRNA and IL1β. Cells were treated according to the manufacturer’s instructions and washed with phosphate buffer saline (PBS) 3 times before fluorescent signaling using a fluorescent microscope (Olympus, Japan), and quantified using Image J.
Protein Extraction and Western Blotting
To extract protein from treated chondrocyte samples, chondrocytes were collected and washed 3 times with PBS after removal of culture fluid. Cells were lysed using RIPA buffer containing 1 mM phenylmethanesulfonylfluoride (PMSF). Protein lysate concentration was measured using Bradford Protein Assay Kit (Beyotime). Protein samples were separated using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a 0.45 μM polyvinylidene membrane. The membrane was then probed using antibodies against β-actin, GAPDH, HIF-1α, HIF-2α, PKM2, LC3I/II, P62, MMP13, Col2A1, BNIP3, BAX, Caspase 3/Cleaved Caspase-3, Parkin, Pink1 at 4 °C overnight. The bands were then treated with their respective secondary antibodies at room temperature for 90 minutes. Band detection was done using Super Electrochemiluminescence Fluid (Yeasen) with Image Lab 3.0 software (Bio-Rad) and band quantification was done using Quantity One.
Real-Time Quantitative Polymerase Chain Reaction (PCR)
Treated chondrocytes were PBS washed 3 times after removal of culture fluid and total cell RNA was extracted using Tri Reagent (Sigma-Aldrich). Reverse transcription of cell RNA into cDNA was done using Hifair III 1st Strand cDNA Synthesis SuperMix for qPCR (Yeasen) and quantitative PCR analyses were performed using SYBR Green qPCR master mix (Yeasen) according to the manufacturer’s instructions, using a reaction volume of 10 μL consisting of 5 μL of 2× SYBR Master Mix, 0.4 μL of each primer, and 4.2 μL of diluted cDNA. The reaction was performed using Quantstudio 5 Real-Time PCR (ThermoFisher). RT-qPCR parameters used were 30 seconds at 95 °C followed by 40 cycles of 10 seconds at 95 °C, 20 seconds at 60 °C, 35 seconds at 72 °C. The cycle threshold (Ct) values were collected and normalized to the β-actin expression levels and the results were assessed using the 2-ΔΔCt method. The primer sequences used in the study were the following: for HIF-1α, Forward 5′-GAACGTCGAAAAGAAAAGTCTCG-3′, Reverse 5′-CCTTATCAAGATGCGAACTCACA-3′; for iNOS, Forward 5′-GGAACCTACCAACTGACGGG-3′, Reverse 5′-GTCGATGCACAGCTGAGTGA-3′.
siRNA Transfection
Effective siRNA for HIF-1α and the negative control were designed and purchased from Ribobio (China). Cells were cultured until reaching 40% to 50% confluency, then serum starved for 24 hours. Cells were then transfected using RiboFect Transfection Kit (Ribobio) according to the manufacturer’s instructions. After successful transfection cells were further treated with IL1β.
Live/Dead Assay
Live and dead chondrocytes were measured using Calcein/PI kit (Beyotime) after treatment of HIF-1α siRNA and IL1β in both culture conditions. Cells were plated in 24-well plates with 3 duplicate wells and treated with Calcein/PI solution mix. Ten microliters of Calcein dye (Green) and 10 µL of PI (Red) was mixed in 10 mL PBS. Cells were dyed with the solution mix prepared in 37 °C. After 30 minutes of treatment, cells were washed with PBS 3 times. Detection of live (Ex/Em = 494/517 nm) and dead (Ex/Em = 535/617 nm) was done using a fluorescent microscope.
Statistical Analysis
All data are expressed as the mean ± standard deviation (SD) of measurements repeated 3 times. Raw statistical analyses were processed using GraphPad Prism 8 (USA). Statistical difference was assessed using unpaired t-test when results are shown between 2 groups and one-way ANOVA among multiple groups. A P value of <0.05 was considered statistically significant and represented with a “*”.
Results
IL1β-Induced C28/I2 Human Chondrocyte Induces the OA Characteristics In Vitro
To select a suitable concentration of IL1β interference in normoxia and hypoxia, C28/I2 cells were treated with IL1β for 48 hours using different concentrations (5, 10, 20 ng/mL) and cell viability was measured ( Fig. 1A ). CCK-8 results showed a significant decrease in cell viability with 5 ng/mL IL1β, but cell viability results were only significantly different upon 10 ng/mL treatment in 2 different culture conditions ( Fig. 1B ). ROS production was significantly increased with IL1β treatment, and cellular ROS production was significantly lower in cells cultured in hypoxia compared to cells cultured in normoxia even with IL1β insult ( Fig. 1C and D ). Cellular ROS production was further proven by qPCR results of iNOS showing a lower level of expression in hypoxic cells, even with IL1β inflammatory stress ( Fig. 1E ). A total of 10 ng/mL IL1β was selected as the dosage for cellular treatment for the next part of the study.
Figure 1.

Measurement of inflammatory insult on C28/I2 chondrocytes for cultures in (A) normoxia and hypoxia, with 5, 10, 20 ng/mL IL1β for 48 hours. Cell viability was measured using cell counting kit-8 and a cell viability was compared between 2 culture conditions (B). (C) Cellular oxidative stress following IL1β insult was measured using ROS detection kit, and comparatively quantified (D) in both culture conditions following IL1β treatment. (E) qPCR analysis of iNOS expression verified the increase in cellular ROS production in both culture conditions following IL1β insult. Average values were calculated from 3 individual experiments.
To measure cellular responses to inflammatory IL1β treatment, we determined protein levels from cells lysates ( Fig. 2A and B ). Western blotting of cells cultured in hypoxia showed a higher expression of HIF-1α levels, and was further increased with IL1β treatment, suggesting the role of HIF-1α in cellular inflammatory response to cellular ROS levels. Increase in inflammatory insult also causes an increase in HIF-2α expression, which was significantly lower in a hypoxic environment rich in HIF-1α. A significant increase in LC3II and decrease in P62 expression was seen with IL1β treatment indicates the activation of autophagy with inflammatory stress. Following inflammatory insult, the glycolytic rate-limiting enzyme PKM2 was also increased, showing an increase of dependence of glucose metabolism in the cell. To measure chondrocyte cellular damage, cell lysates were linked with COL2A1 and MMP13 using western blotting. OA chondrocyte degeneration markers COL2A1 and MMP13 was found to be decreased and increased, respectively, with IL1β treatment, showing a degree of impaired chondrocyte structural protein due to an increase in cellular oxidative stress ( Fig. 2D ). Mitophagy and cell apoptotic proteins BNIP3/BAX and Caspase/Cleaved Caspase 3 proteins were not significantly changed with IL1β inflammatory stress, signifying no significant cellular damage leading to the activation of the mitophagy-induced apoptotic cascade. These finding show typical OA cellular changes with 10 ng/mL IL1β treatment of C28/I2 cells in vitro.
Figure 2.

Detection of cellular response to IL1β insult in normoxic and hypoxic conditions. (A) Western blotting of HIF-1α and HIF-2α expression, autophagic markers P62/LC3II, glycolytic enzyme PKM2 and OA chondrocyte degeneration markers COL2A1/MMP13. (B) qPCR of HIF-1α verified the increase of cellular expression in hypoxic conditions and IL1β insult. (C) Immunofluorescence verified the increase autophagic levels when cells were cultured in hypoxia when compared to cells cultured in normoxia. (D) Western blotting of cell mitophagic markers BNIP3/BAX and autophagic markers Caspase 3/Cleaved Caspase 3. Average values were calculated from 3 individual experiments.
Chondroprotective Effect of Autophagy Is Increased in HIF-1α Elevated Environment
Cells cultured in hypoxia had lower levels of cellular ROS production and lower level of iNOS expression compared to normoxic cells even with IL1β treatment ( Fig. 2A and B ). In cells cultured in hypoxia, we examined a higher level of LC3II and lower P62 expression when compared to normoxic cells, indicating a higher level of autophagy expressed in hypoxic cells. IL1β treatment further caused an increase in autophagy levels, and was significantly higher compared to cells in normoxia, even with IL1β treatment. Immunofluorescence of LC3II in both normoxic and hypoxic conditions with IL1β also solidified the findings of increased autophagy in hypoxic conditions ( Fig. 2A ). Cells cultured in a hypoxic environment showed a significantly lower expression of PKM2, indicating a glycolytic shift when cells are exposed to inflammatory insult. Chondrocyte degenerative markers COL2A1 and MMP13 was significantly higher and lower in hypoxic cells compared to cells in normoxia, showing a better degree of chondrocyte repair under inflammatory stress ( Fig. 2D ). Mitophagy proteins BNIP3/BAX and cell apoptotic proteins Caspase 3/Cleaved Caspase 3 did not have any significant change in both normoxic and hypoxic chondrocytes. From the above results, we show that the increase of HIF-1α expression in hypoxic chondrocytes is linked to a higher level of basal autophagy, which is beneficial for chondrocyte regulation, ameliorating IL1β induced chondrocyte inflammatory changes.
HIF-1α Is a Necessity for the Activation of Chondrocyte Autophagy under Inflammatory Stress
To determine the dependency of chondrocyte autophagy to cellular HIF-1α cellular expression, we used siRNA to knockdown cellular HIF-1α expression. Efficacy of the siRNA kit was determined using cell lysates and qPCR of HIF-1α expression levels ( Fig. 3A ). Silencing cellular HIF-1α expression also resulted in the subsequent increase in HIF-2α expression. siHIF-1α cells were treated with IL1β to induce inflammatory stress and cell responses were detected. Cellular ROS production was significantly increased with HIF-1α knockdown regardless of IL1β treatment, increasing ROS expression more than 2-fold ( Fig. 3B ). Increase of cellular ROS was also proven by qPCR of iNOS expression, showing a similar trend with HIF-1α knockdown. Determination of cell autophagy was done using WB of cell lysates following HIF-1α knockdown with and without IL1β inflammatory stress ( Fig. 3C ). We found that LC3II levels were significantly decreased following knockdown of HIF-1α, and was not further reduced with IL1β treatment. P62 expression was decreased when compared to non-siRNA treated cells, but was relatively higher compared to cells with only IL1β treatment, indicating a loss of relation to LC3II function. Loss of P62 regulation was characteristic of dysregulation of autophagy, which was a response to HIF-1α knockdown. Thus, cells lost their autophagic function without proper HIF-1α expression.
Figure 3.

Silencing HIF-1α inhibits cellular autophagic functions. C28/I2 chondrocyte cells were transfected with siRNA, and the subsequent (A) HIF-1α expression was evaluated using Western Blotting and qPCR of the treated cells. (B) The resulting cell ROS production due to silencing HIF-1α and IL1β insult was measured and quantified. (C) HIF-2α expression, glycolytic rate-limiting enzyme PKM2 expression, cell autophagic markers P62/LC3II and OA chondrocyte degeneration markers COL2A1/MMP13 were evaluated using Western blotting after silencing HIF-1α expression, and IL1β insult. (D) Mitophagic markers BNIP3/BAX and PINK/Parkin and apoptotic markers Caspase 3/Cleaved Caspase 3 were evaluated using Western Blotting in cells after HIF-1α silencing and IL1β insult. Average values were calculated from 3 individual experiments.
Inhibition of HIF-1α Induces the Activation of Mitophagy and Cell Death under Inflammatory Stress
To detect the activation of mitophagy with IL1β treatment and HIF-1α knockdown, we measured mitochondrial oxidation using MitoSox (Red; Fig. 4 ). There was a significant increase in fluorescence with HIF-1α knockdown in both normoxic and hypoxic conditions. HIF-1α silenced cells in normoxia showed a significant increase in mitochondrial oxidation when treated with IL1β. Cells in hypoxia, however, showed a significant increase in mitochondrial oxidation even without an inflammatory insult, as cells are unable to properly regulate oxygen levels ( Fig. 3C ). WB results showed a significant increase of OA chondrocyte degeneration markers, a significant drop in COL2A1 and elevated MMP13 after silencing HIF-1α expression, showing poor chondrocyte repair. Silencing cellular HIF-1α expression also resulted in a significant increase in glycolytic dependence, showed by a further increase in cellular PKM2 expression ( Fig. 3D ). Mitochondrial oxidative damage BNIP3 was increased and mitochondrial autophagy regulator BAX was increased with HIF-1α knockdown. Cellular apoptotic protein Caspase and Cleaved Caspase 3 showed typical changes of apoptosis activation. Increase of BNIP3/BAX and apoptotic Caspase and Cleaved Caspase 3 showed the induction of cell apoptosis through excessive mitochondrial damage leading to activation of mitophagy. To detect the mitophagic pathway activated with HIF-1α knockdown, we tested cell lysates for PINK1/Parkin expression. Parkin dependent mitophagy PINK1/Parkin expression was significantly increased after silencing HIF-1α expression, which was further aggravated with IL1β treatment.
Figure 4.
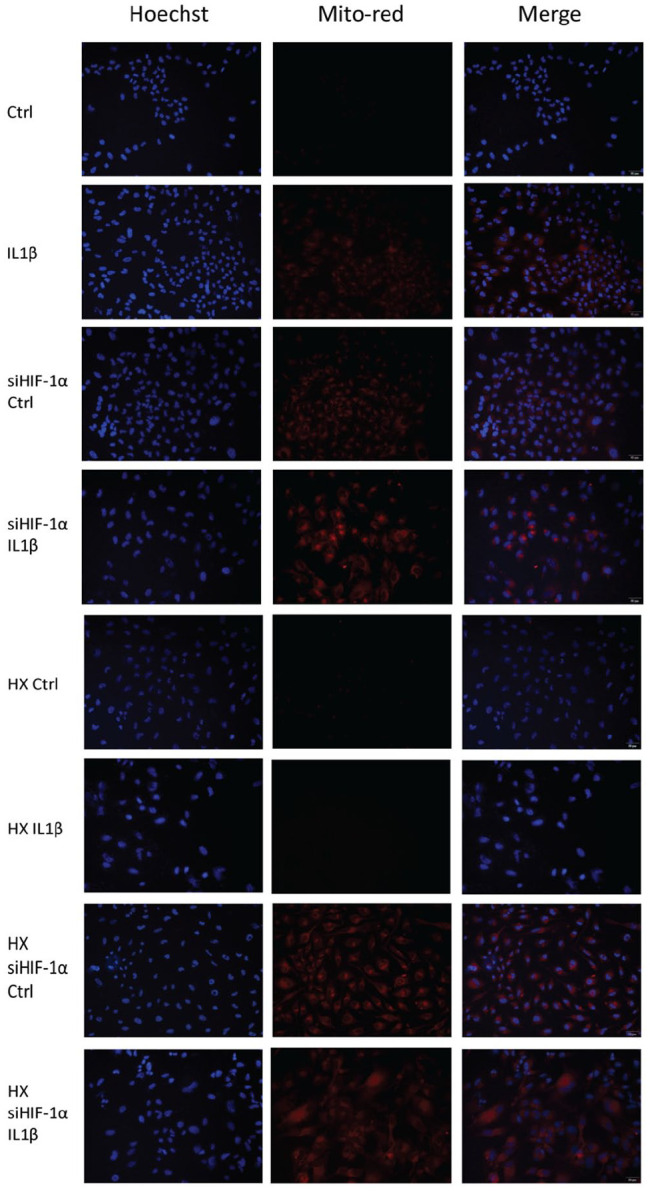
Mitochondrial oxidation detection and nuclei staining using MitoSox (Red) and Hoechst (Blue). HIF-1α silenced cells were cultured in both normoxic and hypoxic conditions and IL1β insult was added for 48 hours. Cell nuclei were stained blue and indications of mitochondrial oxidation are stained red. Mitochondrial oxidation signifies a change in mitochondrial membrane potential, signifying the activation of cellular mitophagy.
Cell death was measured using Live/Dead assay, confirming the increase of the rate of dead chondrocytes following HIF-1α knockdown shown by Live (Green) and Dead (Red) cells ( Fig. 5 ). In both normoxic and hypoxic conditions, treatment of IL1β did not further induce cell death. But after silencing cellular HIF-1α expression, there was a significant increase in the number of dead cells. Cells in normoxia did not undergo cell death with only HIF-1α knockdown, but further IL1β treatment resulted in a significant increase in dead cells. Chondrocytes grown in hypoxia, however, showed a massive increase in the number of dead cells with only the inhibition of HIF-1α expression. The dependency of HIF-1α in hypoxic conditions explains the significant difference in both culture conditions ( Fig. 6 ).
Figure 5.
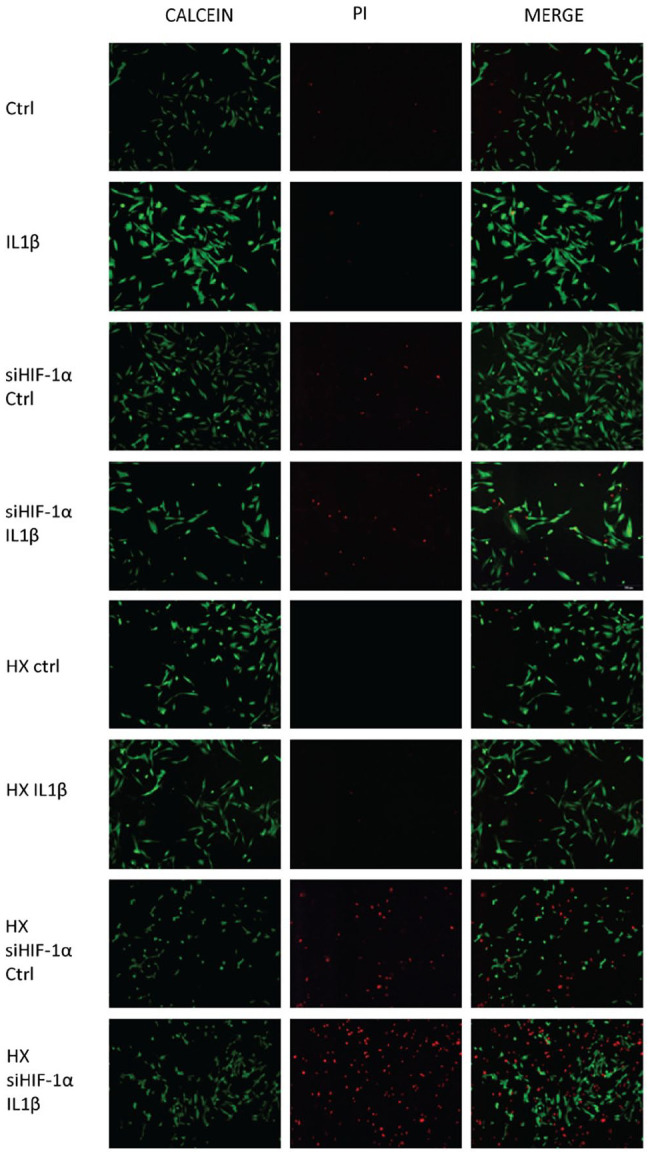
Assessment of dead cells following HIF-1α silencing and IL1β insult. HIF-1α silenced cells were cultured in both normoxic and hypoxic conditions and IL1β insult was added for 48 hours. Live cells were stained with Calcein (Green) and dead cells were stained with PI (Red).
Figure 6.

A diagram representation of the chondrocyte changes in both a normoxic and hypoxic culture environment. Changes in HIF-1α expression levels leads to changes in chondrocyte cellular function and autophagic response to inflammatory insult. Silencing chondrocyte HIF-1α impairs cellular autophagic function which activates cellular mitophagy pathway, leading to chondrocyte cell death.
Discussion
Recent study findings have redefined OA as a multifactor inflammatory disease instead of a purely mechanical degenerative disorder. 20 It was thought that the constant abrasion caused by a defective chondrocyte layer leads to the increase in load-bearing in one pressure point instead of being evenly distributed on the articular interface. 21 Cartilaginous tissue being an avascular tissue having low metabolic activity was thought to be unable of self-repair and was unable to respond to cellular stressors due to its avascular nature. Chondrocytes are the component of adult cartilage, and was thought to only be progressively degraded without any ability of self-repair. The first discovery of inflammatory mediators leading to the production of matric metalloproteinases (MMPs) in chondrocytes have led to the new understanding of OA pathology. Contrary to past studies chondrocytes do possess self-renewing abilities. But different from cells growing in an oxygen-rich environment, chondrocyte cells mainly depend on autophagy instead of cell turnover to maintain a healthy cartilage lining. 10 During the pathogenesis of OA, the functional capabilities of the different structures in the articular joint contributes to the disruption of the hypoxic microenvironment and the overall degeneration of articular chondrocytes. 22 In the recent more studied aspect of OA, the disruption of the protective and disruptive aspects of OA results in a progressive progression of degenerative changes. In this study we aimed to establish the necessity of chondrocyte autophagy and the cellular hypoxic mediator HIF-1α in response to inflammatory insult.
HIF-1α is a hypoxic mediator widely expressed in hypoxic tissue, and was found to play a role in chondrocyte differentiation and metabolism. The regulation of HIF-1α is a complex mechanism involving the HIF-1α subunits on the molecular membrane and the oxygen-dependent prolyl-hydroxylases (PHDs) which are target-specific on HIF-1α. HIF-1α is richly expressed in a hypoxic environment, and is quickly degraded when exposed to an increase in oxygen levels. In an oxygen-rich environment, PHDs interact with the 2 proline molecules within the molecular degradation domain and leads to the degradation of HIF-1α. In a hypoxic environment, the activation of PHDs is inhibited which leads to the accumulation and translocation of HIF-1α into the nucleus and activates specific genes.23-26 During the progression of OA, disruption of the hypoxic environment leads to a change in HIF-1α expression and thus function. In contrast, HIF-2α is readily expressed in normoxic conditions and significantly increased following cellular inflammatory insult.
Under mitochondrial oxidative stress conditions due to improper cellular regulations, the cells activate mitophagy as a component of cell repair process. When cellular ROS levels exceed the cell capacity of ROS detoxification, the oxygen radicals cause damage to mitochondrial DNA integrity and repair capacity, leading to irreversible cell damage. Under stressful conditions such as the increase of MMPs, oxidative stress, and iron starvation, mitophagy is activated to maintain cell homeostasis. Mitophagy is the selective engulfment of damaged mitochondria in response to a change in mitochondrial membrane potential.27,28 Out of the 2 mitophagic pathways, the PINK1/Parkin pathways involve PINK1 targeting damaged mitochondria and facilitates the clustering of depolarized mitochondria. Parkin follows mitochondrial accumulation of PINK1 and promotes mitochondrial engulfment by autophagosomes, indicating mitophagy. The non-Parkin-dependent pathway involves the direct interaction of LC3 and the mitochondrial receptors that activate mitophagy without ubiquitination through the activation of BNIP3.29,30 The involvement of cell mitophagy have been recently found to be involved in the pathogenesis of chondrocyte degeneration and possesses therapeutic value in the targeted treatment of OA.29,31
Current in vitro studies have carried out cellular experiments away from their natural environment, and treated hypoxia as a cellular insult.32,33 In our study, C28/I2 human chondrocyte cell line showed a significantly higher basal autophagic level when cells were cultured in a hypoxia. The increase in autophagic levels protected cells from IL1β insult, resulting in a significantly lower levels of cellular oxidative stress. With a different level of self-repair, chondrocytes would react differently to cellular insult in normoxic and hypoxic conditions. Cellular LC3II was significantly increased in hypoxic cells, coupled with a decrease in P62 expression showing an overall increase in cell autophagy even without an inflammatory insult. With a higher level of autophagy, cells were able to maintain a higher level of COL2A1, representing the higher and stronger collagen structure in articular joints. Lower levels of oxidative stress are reflected by a lower expression of MMP13, and without the activation of HIF-1α targets mitochondrial-oxidative proteins BNIP3 and BAX. With a higher basal level of cell autophagy, studies utilizing human chondrocytes should consider the interference of HIF-1α expression on cellular processes. We hypothesized that HIF-1α plays a role in the management of autophagic processes in chondrocyte cells.
Silencing of HIF-1α function significantly impairs cellular autophagic function. Cellular LC3II levels were decreased regardless of IL1β treatment and resulted in the accumulation of MMP13, and mitochondria damage. The increase in mitochondrial oxidation signifies cellular damage, resulting in the activation of mitophagy-induced cellular apoptosis as shown by the increase in Caspase 3 into its cleaved form. We also found that the activation of PINK1/Parkin and BNIP3/BAX mitophagy proteins to be significantly increased with HIF-1α silenced cells, indicating the activation of the mitophagic pathways leading to cellular damage. Changes in HIF-1α expression was also reflected by a significant change in PKM2 levels, a rate-limiting glycolytic enzyme. This leads to a glycolytic shift following inflammatory insult, from a predominantly glycolytic process to oxidative phosphorylation, resulting in a less efficient cellular energy generation. Due to role of HIF-1α in cellular oxygen regulation in hypoxic conditions, interference with HIF-1α expression in hypoxic conditions can prove to be fatal. This was also shown by the results of the Live/Dead cell showing massive cell death after silencing cellular HIF-1α. But with the similar trend of increased autophagy in HIF-1α elevated hypoxic environment, it is involved in the cellular autophagic cascade. The inhibition of HIF-1α is thus detrimental to cell survivability in response to inflammatory stress, and is crucial for the activation of cell autophagy.
From the results of our study, we conclude that a hypoxic microenvironment for cellular models of OA chondrocytes benefit autophagic responses to inflammatory insult. The increase in autophagic responses relied mainly on the expression of cellular HIF-1α, which was found to be a crucial factor in autophagic activation. The dysregulation of HIF-1α results in the activation of mitophagy-mediated cell death, which plays a major role in OA chondroptosis. In vivo model utilizing targeted HIF-1α modulation would be needed to accurately establish the degree of HIF-1α beneficial to cellular autophagic levels.
Footnotes
Acknowledgment and Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by the National Natural Science Foundation of China (Grant No. 81971308). The authors thank Chenzhong Wang and Zhenqing Wang for their technical assistance.
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval: Ethical approval was not sought for the present study because the study did not involve the usage of any human nor animal specimens.
Informed Consent: Informed consent was not sought for the present study because the study did not involve the usage of any human nor animal specimens.
ORCID iD: Jian Zhang  https://orcid.org/0000-0001-9394-0859
https://orcid.org/0000-0001-9394-0859
References
- 1. Kiaer T, Grønlund J, Sørensen KH. Subchondral pO2, pCO2, pressure, pH, and lactate in human osteoarthritis of the hip. Clin Orthop Relat Res. 1988;(229):149-55. [PubMed] [Google Scholar]
- 2. Najafipour H, Ferrell WR. Comparison of synovial PO2 and sympathetic vasoconstrictor responses in normal and acutely inflamed rabbit knee joints. Exp Physiol. 1995;80(2_suppl):209-20. doi: 10.1113/expphysiol.1995.sp003841 [DOI] [PubMed] [Google Scholar]
- 3. Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998;12(2_suppl):149-62. doi: 10.1101/gad.12.2.149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schipani E, Ryan HE, Didrickson S, Kobayashi T, Knight M, Johnson RS. Hypoxia in cartilage: HIF-1alpha is essential for chondrocyte growth arrest and survival. Genes Dev. 2001;15(21):2865-76. doi: 10.1101/gad.934301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Coimbra IB, Jimenez SA, Hawkins DF, Piera-Velazquez S, Stokes DG. Hypoxia inducible factor-1 alpha expression in human normal and osteoarthritic chondrocytes. Osteoarthritis Cartilage. 2004;12(4):336-45. doi: 10.1016/j.joca.2003.12.005 [DOI] [PubMed] [Google Scholar]
- 6. Villalvilla A, Gómez R, Largo R, Herrero-Beaumont G. Lipid transport and metabolism in healthy and osteoarthritic cartilage. Int J Mol Sci. 2013;14(10):20793-808. doi: 10.3390/ijms141020793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nishida T, Kubota S, Aoyama E, Takigawa M. Impaired glycolytic metabolism causes chondrocyte hypertrophy-like changes via promotion of phospho-Smad1/5/8 translocation into nucleus. Osteoarthritis Cartilage. 2013;21(5):700-9. doi: 10.1016/j.joca.2013.01.013 [DOI] [PubMed] [Google Scholar]
- 8. Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221(1):3-12. doi: 10.1002/path.2697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ravanan P, Srikumar IF, Talwar P. Autophagy: the spotlight for cellular stress responses. Life Sci. 2017;188:53-67. doi: 10.1016/j.lfs.2017.08.029 [DOI] [PubMed] [Google Scholar]
- 10. Cuervo AM, Bergamini E, Brunk UT, Dröge W, Ffrench M, Terman A. Autophagy and aging: the importance of maintaining “clean” cells. Autophagy. 2005;1(3):131-40. doi: 10.4161/auto.1.3.2017 [DOI] [PubMed] [Google Scholar]
- 11. Loeser RF. Aging and osteoarthritis: the role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthritis Cartilage. 2009;17(8):971-9. doi: 10.1016/j.joca.2009.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Roach HI, Aigner T, Kouri JB. Chondroptosis: a variant of apoptotic cell death in chondrocytes? Apoptosis. 2004;9(3):265-77. doi: 10.1023/b:appt.0000025803.17498.26 [DOI] [PubMed] [Google Scholar]
- 13. Charlier E, Relic B, Deroyer C, Malaise O, Neuville S, Collée J, et al. Insights on molecular mechanisms of chondrocytes death in osteoarthritis. Int J Mol Sci. 2016;17(12):2146. doi: 10.3390/ijms17122146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Millucci L, Giorgetti G, Viti C, Ghezzi L, Gambassi S, Braconi D, et al. Chondroptosis in alkaptonuric cartilage. J Cell Physiol. 2015;230(5):1148-57. doi: 10.1002/jcp.24850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zamli Z, Sharif M. Chondrocyte apoptosis: a cause or consequence of osteoarthritis? Int J Rheum Dis. 2011;14(2_suppl):159-66. doi: 10.1111/j.1756-185X.2011.01618.x [DOI] [PubMed] [Google Scholar]
- 16. Takács-Buia L, Iordachel C, Efimov N, Caloianu M, Montreuil J, Bratosin D. Pathogenesis of osteoarthritis: chondrocyte replicative senescence or apoptosis? Cytometry B Clin Cytom. 2008;74(6):356-62. doi: 10.1002/cyto.b.20428 [DOI] [PubMed] [Google Scholar]
- 17. Hwang HS, Kim HA. Chondrocyte apoptosis in the pathogenesis of osteoarthritis. Int J Mol Sci. 2015;16(11):26035-54. doi: 10.3390/ijms161125943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lotz M, Hashimoto S, Kühn K. Mechanisms of chondrocyte apoptosis. Osteoarthritis Cartilage. 1999;7(4):389-91. doi: 10.1053/joca.1998.0220 [DOI] [PubMed] [Google Scholar]
- 19. Blanco FJ, Guitian R, Vázquez-Martul E, de Toro FJ, Galdo F. Osteoarthritis chondrocytes die by apoptosis. A possible pathway for osteoarthritis pathology. Arthritis Rheum. 1998;41(2_suppl):284-9. doi:10.1002/1529-0131(199802)41:2<284::AID-ART12>3.0.CO;2-T [DOI] [PubMed] [Google Scholar]
- 20. Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 2012;64(6):1697-707. doi: 10.1002/art.34453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Berenbaum F. Osteoarthritis as an inflammatory disease (osteoarthritis is not osteoarthrosis!). Osteoarthritis Cartilage. 2013;21(1):16-21. doi: 10.1016/j.joca.2012.11.012 [DOI] [PubMed] [Google Scholar]
- 22. Pfander D, Cramer T, Swoboda B. Hypoxia and HIF-1alpha in osteoarthritis. Int Orthop. 2005;29(1):6-9. doi: 10.1007/s00264-004-0618-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Semenza GL. HIF-1, O(2), and the 3 PHDs: how animal cells signal hypoxia to the nucleus. Cell. 2001;107(1):1-3. doi: 10.1016/s0092-8674(01)00518-9 [DOI] [PubMed] [Google Scholar]
- 24. Semenza GL. HIF-1 and human disease: one highly involved factor. Genes Dev. 2000;14(16):1983-91. [PubMed] [Google Scholar]
- 25. Zhang FJ, Luo W, Lei GH. Role of HIF-1α and HIF-2α in osteoarthritis. Joint Bone Spine. 2015;82(3):144-7. doi: 10.1016/j.jbspin.2014.10.003 [DOI] [PubMed] [Google Scholar]
- 26. Gelse K, Pfander D, Obier S, et al. Role of hypoxia-inducible factor 1 alpha in the integrity of articular cartilage in murine knee joints. Arthritis Res Ther. 2008;10(5):R111. doi: 10.1186/ar2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sun K, Jing X, Guo J, Yao X, Guo F. Mitophagy in degenerative joint diseases. Autophagy. Published online September 24, 2020. doi: 10.1080/15548627.2020.1822097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shin HJ, Park H, Shin N, Kwon HH, Yin Y, Hwang JA, et al. Pink1-mediated chondrocytic mitophagy contributes to cartilage degeneration in osteoarthritis. J Clin Med. 2019;8(11):1849. doi: 10.3390/jcm8111849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012;2(5):120080. doi: 10.1098/rsob.120080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Eiyama A, Okamoto K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr Opin Cell Biol. 2015;33:95-101. doi: 10.1016/j.ceb.2015.01.002 [DOI] [PubMed] [Google Scholar]
- 31. Schulze-Tanzil G. Experimental therapeutics for the treatment of osteoarthritis. J Exp Pharmacol. 2021;13:101-25. doi: 10.2147/JEP.S237479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Johnson CI, Argyle DJ, Clements DN. In vitro models for the study of osteoarthritis. Vet J. 2016;209:40-9. doi: 10.1016/j.tvjl.2015.07.011 [DOI] [PubMed] [Google Scholar]
- 33. Cope PJ, Ourradi K, Li Y, Sharif M. Models of osteoarthritis: the good, the bad and the promising. Osteoarthritis Cartilage. 2019;27(2_suppl):230-9. doi: 10.1016/j.joca.2018.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]